Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2020; 10(11):4749-4761. doi:10.7150/thno.43108 This issue Cite
Research Paper
A COX-2/sEH dual inhibitor PTUPB alleviates lipopolysaccharide-induced acute lung injury in mice by inhibiting NLRP3 inflammasome activation
Hui-Hui Yang1*, Jia-Xi Duan2,3,4*, Shao-Kun Liu2,3,4, Jian-Bing Xiong1, Xin-Xin Guan1, Wen-Jing Zhong1, Chen-Chen Sun1, Chen-Yu Zhang1, Xiao-Qin Luo1, Yan-Feng Zhang1, Ping Chen2,3,4, Bruce D. Hammock5, Sung Hee Hwang5, Jian-Xin Jiang6, Yong Zhou1 ![]() , Cha-Xiang Guan1
, Cha-Xiang Guan1 ![]()
1. Department of Physiology, Xiangya School of Medicine, Central South University, Changsha, Hunan 410078, China;
2. Department of Pulmonary and Critical Care Medicine, the Second Xiangya Hospital, Central South University, Changsha, Hunan 410011, China;
3. Research Unit of Respiratory Disease, Central South University, Changsha, Hunan 410011, China;
4. Hunan Diagnosis and Treatment Center of Respiratory Disease, Central South University, Changsha, Hunan 410011, China;
5. Department of Entomology and Nematology and UC Davis Comprehensive Cancer Center, University of California, Davis, One Shields Avenue, Davis, CA 95616, USA;
6. State Key Laboratory of Trauma, Burns, and Combined Injury, Army Medical University, Chongqing, 400038, China.
*These authors contributed equally to this work
Received 2019-12-16; Accepted 2020-3-8; Published 2020-3-26
Abstract

Rationale: Dysregulation of arachidonic acid (ARA) metabolism results in inflammation; however, its role in acute lung injury (ALI) remains elusive. In this study, we addressed the role of dysregulated ARA metabolism in cytochromes P450 (CYPs) /cyclooxygenase-2 (COX-2) pathways in the pathogenesis of lipopolysaccharide (LPS)-induced ALI in mice.
Methods: The metabolism of CYPs/COX-2-derived ARA in the lungs of LPS-induced ALI was investigated in C57BL/6 mice. The COX-2/sEH dual inhibitor PTUPB was used to establish the function of CYPs/COX-2 dysregulation in ALI. Primary murine macrophages were used to evaluate the underlying mechanism of PTUPB involved in the activation of NLRP3 inflammasome in vitro.
Results: Dysregulation of CYPs/COX-2 metabolism of ARA occurred in the lungs and in primary macrophages under the LPS challenge. Decrease mRNA expression of Cyp2j9, Cyp2j6, and Cyp2j5 was observed, which metabolize ARA into epoxyeicosatrienoic acids (EETs). The expressions of COX-2 and soluble epoxide hydrolase (sEH), on the other hand, was significantly upregulated. Pre-treatment with the dual COX-2 and sEH inhibitor, PTUPB, attenuated the pathological injury of lung tissues and reduced the infiltration of inflammatory cells. Furthermore, PTUPB decreased the pro-inflammatory factors, oxidative stress, and activation of NACHT, LRR, and PYD domains-containing protein 3 (NLRP3) inflammasome in LPS-induced ALI mice. PTUPB pre-treatment remarkably reduced the activation of macrophages and NLRP3 inflammasome in vitro. Significantly, both preventive and therapeutic treatment with PTUPB improved the survival rate of mice receiving a lethal dose of LPS.
Conclusion: The dysregulation of CYPs/COX-2 metabolized ARA contributes to the uncontrolled inflammatory response in ALI. The dual COX-2 and sEH inhibitor PTUPB exerts anti-inflammatory effects in treating ALI by inhibiting the NLRP3 inflammasome activation.
Keywords: acute lung injury, COX-2/sEH dual inhibitor, NLRP3 inflammasome, oxidative stress
Introduction
Acute lung injury (ALI) and acute respiratory distress syndrome (ARDS) are life-threatening diseases characterized by uncontrolled inflammatory responses, elevated penetrability of the alveolar- capillary barrier, and pulmonary edema [1, 2]. Inflammatory response-activated macrophages, and neutrophils infiltrate into the lung. Cytokines are released by infiltrating cells activating local pro-inflammatory networks and reactive oxygen species (ROS) [3, 4]. Also, NACHT, LRR and PYD domain-containing protein 3 (NLRP3) inflammasome plays a pivotal role in the pathogenesis of ALI [5, 6], and could cause irrevocable damage to lung epithelium and endothelial cells [7, 8]. As a major constituent of the outer membrane of gram-negative bacteria, lipopolysaccharide (LPS) induces a diverse spectrum of infections, including life-threatening pneumonia and septicemia. The signaling also actives the downstream nuclear factor kappa-B (NF-κB) and augments inflammatory mediators [9]. Hence, LPS has emerged as a clinically relevant model for ALI [10].
The therapeutic approach to inflammatory diseases has mainly focused on targeting various fatty acids and lipids, such as arachidonic acid (ARA), which could be metabolized into eicosanoids. The cyclooxygenase (COX), lipoxygenase (LOX), and cytochrome p450 (CYP) are the three primary pathways for the production of eicosanoids [11]. Collective evidence suggests that the epoxyeicosatrienoic acids (EETs) derived from the CYP pathway confer anti-inflammatory effects [12, 13]. However, EETs are rapidly metabolized into the corresponding dihydroxyeicosatrienoic acids (DHETs) and diols mainly by the soluble epoxide hydrolase (sEH) [14]. The sEH, a multifunctional protein encoded by the EPHX2 gene, is expressed in numerous tissues, including the lungs [15]. Our laboratory as well as other researchers previously reported that inhibition of sEH or gene knockout exerts protective effects against ALI [16, 17] and pulmonary fibrosis [18]. COX acts as the critical enzyme converting ARA to prostaglandins (PGs), which are generally very low in physiological conditions but increase in acute inflammation [19]. COX-2 accounts for the increased production of PGs during inflammation and immune responses [20]. The expression of COX-2 significantly increases during the development of ALI, and suppressing COX-2 attenuates LPS-induced ALI [21].
Inhibition of a specific biosynthetic ARA pathway may alter the metabolic flux resulting in fatal side effects [22]. sEH inhibitors and EETs can increase the expression of COX-2 [23, 24]. Therefore, it is imperative to develop novel anti-inflammatory strategies with a dual mechanism, which prevent the release of pro-inflammatory PGs and enhance the concentration of EETs. PTUPB (4-(5-phenyl-3-{3-[3-(4-trifluoromethylphenyl)-ureido]-propyl}-pyrazol-1-yl)-benzenesulfonamide), is a novel dual COX-2 and sEH inhibitor, which was developed in our previous study [25]. It reduces the levels of COX-dependent PGs and increases the CYPs-dependent metabolites [26], indicating the suppression of both COX-2 and sEH pathways. Recently, we reported that PTUPB suppresses the growth of glioblastoma [27], reduces kidney injury and sepsis [28, 29], and attenuates pulmonary fibrosis [30]. However, it is not clear whether dual inhibition of COX-2 and sEH exerts any protective effect against ALI. In the present study, we demonstrated dysregulation of CYPs /COX-2 during ALI and effective attenuation of ALI by dual inhibition of COX-2 and sEH with PTUPB.
Materials and Methods
Animals
Adult (6-8 weeks, 18-20 g) male C57BL/6 mice were purchased from Hunan SJA Laboratory Animal Co., Ltd (Hunan, China). All mice were kept in a controlled environment with 24-26 °C, 50%-60% humidity, and 12 h cycle of night and day. Experimental use of mice in the present study was performed according to the guidelines of the National Institutes of Health for live animals.
Animal treatment
Mice were randomly divided into the control, PTUPB, ALI, and ALI + PTUPB groups. ALI was induced as described in our previous study [16]. Briefly, ALI was induced by intratracheal injection of LPS (5 mg/kg, from Escherichia coli O111: B4, Sigma-Aldrich, USA) dissolved in 50 μL sterile saline. Mice in the control and PTUPB groups received 50 μL sterile saline intratracheally. Mice in the PTUPB and PTUPB+ALI group were subcutaneously injected with PTUPB (5 mg/kg) dissolved in PEG400 1 h prior to the intratracheal injection. PEG400 was subcutaneously injected for the control and ALI groups. Mice were sacrificed 12 h after the LPS injection. A lethal dose of LPS (25 mg/kg) was injected into the trachea for survival study. PTUPB (5 mg/kg, subcutaneous) or PEG400 was administered to mice 1 h before or 6 h after the LPS injection. After the LPS injection, the survival rate was tracked every 6 h. All surgical procedures were performed under anesthesia.
Pulmonary function measurement
The testing system of the Buxco pulmonary function (Buxco, Sharon, Connecticut, CT, USA) was employed to detect the pulmonary function of mice, including airway resistance, lung compliance, and pulmonary ventilation, which was described in our previous study [31]. The pulmonary function measurement was carried out in anesthetized mice.
Hematoxylin-Eosin (H&E) staining and inflammatory injury score analysis
Paraffin-embedded left lungs were sliced in 3-μm thickness and then stained with H&E to observe morphologic changes. Lung injury was measured as previously described [32]. Briefly, the severity of the injury was graded from 0 to 4, according to five independent variables: hemorrhage, neutrophils in the alveolar space, hyaline membranes, pertinacious debris filling the airspaces, and septal thickening. The score of 0 means no damage; l, <25% damage; 2, 25 to 50% damage; 3, 50 to 75% damage and 4, > 75% damage. The inflammation score was measured independently by three pathologists blinded to the experiment. The scores from all three were averaged to give a final score.
Bronchoalveolar lavage fluid (BALF) collection and cell counts
BALF was collected as previously described [33]. Briefly, the lungs were lavaged with 0.8 mL ice-cold PBS (pH = 7.4) three times. The recovered fluid was centrifuged at 1500 rpm for 5 min at 4 ℃, and then the supernatant was collected and stored at -80 ℃ for subsequent experiments. The sedimented cell pellets were re-suspended in 0.5 mL PBS, then cells, including macrophages and neutrophils, were counted with a hemocytometer and Wright-Giemsa staining using a light microscope as described in our previous study [16].
Evaluation of oxidative stress
The up-right lung lobes of mice were homogenized in PBS at a ratio of 1:10 (weight: volume). Total activity of superoxide dismutase (SOD), the level of malondialdehyde (MDA), and reactive oxygen species (ROS) in the lungs were assessed by corresponding kits following manufacturer's instructions (Cat# SOD: A001-3; MDA: A003-1; ROS: E004, Jiancheng Bioengineering Institute, Nanjing, China).
Isolation and treatment of primary murine peritoneal macrophages
Primary murine peritoneal macrophages were isolated and cultured as outlined in our previous study [34]. Cells were plated into 12-well plates (1×106 cells/well) for gene and protein detection, 24-well plates (0.5×106 cells/well) for immunofluorescence staining, 6-well plates (2×106 cells/well) for ROS evaluation, and 100 mm culture dishes (1×107 cells/dish) for immunoprecipitation (IP). To estimate the effect of PTUPB on LPS (100 ng/mL)-challenged murine macrophages, a series of concentrations of PTUPB (10, 100, 1000, and 10000 nM) were added 1 h before LPS (100 ng/mL) stimulation; cells in the control group were treated with DMSO. Six hours later, macrophages were harvested for gene detection. Twelve hours later, macrophages were harvested for protein and immunofluorescence staining detection. To evaluate the effect of PTUPB on the NLRP3 inflammasome activation, PTUPB (1000 nM) was added to the cultures 1 h ahead of the LPS treatment (100 ng/mL) for 135 min and adenosine triphosphate (ATP, 2.5 mM) treatment for another 45 min.
Detection of lactate dehydrogenase (LDH) activity
The activity of LDH in serum, BALF, and the supernatant was detected by using the LDH Cytotoxicity Assay Kit (Cat# A020-2, Jiancheng Bioengineering Institute, Nanjing, China).
Real-time quantitative polymerase chain reaction (RT-qPCR)
The extraction of total RNA, generation of cDNA, and RT-qPCR were achieved as described in our previous study [33]. Gene expression was measured by 2-ΔCt, and the relative gene expression was assayed by 2-ΔΔCt according to the previous study [35, 36]. Primers used in this study were synthesized by Sangon Biotech (Shanghai, China) as our previous studies [30, 33], and the sequences are shown in Table 1.
Sequences of the primers used to quantitate gene expression.
| Gene | Forward primer (5′-3′) | Reverse primer (5′-3′) |
|---|---|---|
| Cyp2j9 | AGTCAGTCACCGCCTTTGTG | GTCTCATTGCACGCACTCTC |
| Cyp2j6 | GGTGCCCTTGTTGTTAGCAC | GGCTAACAAGGAGCCGGTAG |
| Cyp2j5 | TGATGGGTTCATCAGCAGGC | CTTGGCTCATCTGGGTTCCAAT |
| Cyp2c29 | CCATGGTTGCAGGTAAACCACAT | TCTGTCCCTGCACCAAAGAG |
| Cyp2c44 | CAAGGTACCCCGAGTGAAGAA | CACGGCATCTGTATAGGGCA |
| Cox-2 | CATCCCCTTCCTGCGAAGTT | CATGGGAGTTGGGCAGTCAT |
| Nox-2 | GACTGCGGAGAGTTTGGAAG | GGTGATGACCACCTTTTGCT |
| Tnf-α | AGCCCCCAGTCTGTATCCTT | CTCCCTTTGCAGAACTCAGG |
| Mcp-1 | GTCCCTGTCATGCTTCTGG | GCGTTAACTGCATCTGGCT |
| Trem-1 | CTGTGCGTGTTCTTTGTC | CTTCCCGTCTGGTAGTCT |
| Nlrp3 | TACGGCCGTCTACGTCTTCT | CGCAGATCACACTCCTCAAA |
| pro-caspase-1 | CACAGCTCTGGAGATGGTGA | CTTTCAAGCTTGGGCACTTC |
| Asc | GACAGTACCAGGCAGTTCGT | AGTCCTTGCAGGTCAGGTTC |
| pro-Il-1β | CAGGCAGGCAGTATCACTCA | AGCTCATATGGGTCCGACAG |
| Nf-κb/p65 | GGAGGCATGTTCGGTAGTGG | CCCTGCGTTGGATTTCGTG |
| β-actin | TTCCAGCCTTCCTTCTTG | GGAGCCAGAGCA GTAATC |
Cytokine detection
The contents of tumor necrosis factor-alpha (TNF-α), monocyte chemotactic protein 1 (MCP-1), and interleukin-1 beta (IL-1β) in BALF, serum, cell culture supernatant, or the lung tissue were measured using ELISA kits (Cat# TNF-α: 88-7324; MCP-1: 88-7391; IL-1β: 88-7013; Invitrogen, Thermo Fisher Scientific, USA). The contents were assayed by comparison of the optical density (450 nm and 570 nm) with the standard curve.
Western blotting
Proteins from the lung tissue or macrophages were extracted and analyzed using Western blotting as described in our previous study [37]. Frozen lung tissue was homogenized and lysed in RIPA buffer (Solarbio, Beijing, China) containing a protease inhibitor (Roche, Mannheim, Germany). The protein concentrations were measured with Pierce™ BCA Protein Assay Kit (Cat# 23225, Thermo Fisher Scientific, Grand Island, NY, USA). Samples mixed with the loading buffer were separated on an SDS-PAGE gel. After transferring to polyvinylidene fluoride membranes (Millipore, Bedford, MA) by electrotransfer, the membranes were blocked with 5% BSA or non-fat milk at room temperature for 1.5 h, incubated with primary antibodies at 4 °C overnight, and then incubated with peroxidase-conjugated secondary antibodies at room temperature for 1 h. The image of protein brands was captured by a gel imaging system. The relative band intensity was measured using the Image Lab Analyzer software (Bio-Rad, Hercules, CA, USA). The antibodies used in the study are shown in Table 2.
Antibody sources and dilutions.
| Antibodies | Source | Catalog | Dilution ratio |
|---|---|---|---|
| Primary antibodies for Western blotting | |||
| Anti-sEH monoclonal antibody | Abcam | ab155280 | 1: 1000 |
| Anti-COX-2 polyclonal antibody | Proteintech | 12375-1-AP | 1: 2000 |
| Anti-Nrf2 monoclonal antibody | CST | #12721 | 1: 1000 |
| Anti-TREM-1 polyclonal antibody | Proteintech | 11791-1-Ap | 1: 1000 |
| Anti-NLRP3 monoclonal antibody | CST | #15101 | 1: 2000 |
| Anti-IL-1β polyclonal antibody | R&D | AF-401-NA | 1: 2000 |
| Anti-pro-caspase-1/p10/p20 monoclonal antibody | Abcam | Ab179515 | 1: 1000 |
| Anti-I-κBα monoclonal antibody | Abcam | Ab32518 | 1:2500 |
| Secondary antibodies for Western blotting | |||
| Anti-β-actin polyclonal antibody | SAB | #21338 | 1: 7500 |
| Anti-β-tubulin monoclonal antibody | Servicebio | #48885 | 1: 2000 |
| Antibodies for IP assay | |||
| Anti-NLRP3 monoclonal antibody | AdipoGen | AG-20B-0014-C100 | 1:1000 |
| Anti-ASC monoclonal antibody | CST | 67824S | 1:1000 |
IP assay
For the detection of the interaction between NLRP3 and ASC (apoptosis-associated speck-like protein containing a CARD), primary murine peritoneal macrophages were washed with PBS (pH = 7.4) three times, collected and lysed with lysis buffer including complete protease inhibitor PMSF (Solarbio, P0100) and Protease Inhibitor Cocktail (MCE, HY-K0010) on ice for 35 min, then centrifuged at 12,000 rpm for 10 min at 4 °C. The supernatant (100 μL) was transferred to another cold tube and incubated with 55 μL Dynabeads™ Protein G (Invitrogen, 10004D) at 4 °C for 3 h, centrifuged at 500 g for 10 min at 4 °C. Subsequently, the Dynabeads were separated with The DynaMag™-2 (Invitrogen, 12321D). The supernatant (80 μL) was collected and anti-ASC (1:100; CST, 67824S) antibody was added at 4 °C overnight. Then, 30 μL Dynabeads™ Protein G was added to the supernatant and soaked for another 3 h on ice. The beads were washed with the lysis buffer three times and the Dynabeads were separated with The DynaMag™-2. The immunoprecipitated proteins were analyzed by Western blotting.
Statistical analysis
All experiments were independently repeated three times. Results were shown as the mean ± SD values. All data were analyzed with SPSS 22.0 (IBM, Chicago, IL) or GraphPad Prism 7 software (San Diego, CA, USA). A P-value of < 0.05 was regarded as statistically significant. Statistical comparisons between two groups were determined by unpaired t-test. Differences among multiple groups were determined by ANOVA, followed by Bonferroni correction for multiple comparison testing. The data that were not normally distributed were analyzed using nonparametric statistical analysis. The survival rate was assayed by the log-rank test.
Results
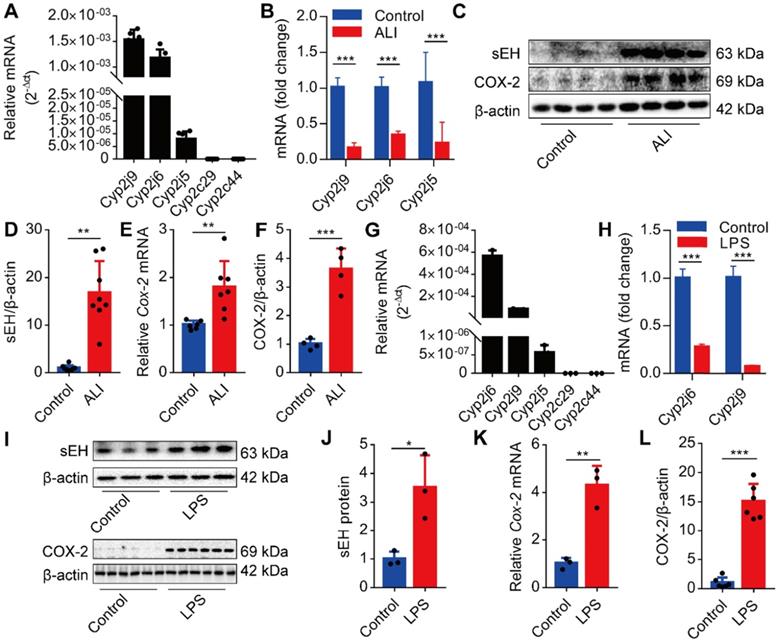
Dysregulation of CYPs /COX-2 metabolism of ARA occurs in the lungs and macrophages under the LPS challenge
We first investigated whether the CYPs/COX-2 metabolism-derived ARA was altered during ALI. We analyzed the expression of Cyp2j9, Cyp2j6, Cyp2j5, Cyp2c29, and Cyp2c44 in the lungs of mice and found that Cyp2j9 and Cyp2j6 were expressed in the lungs, whereas Cyp2c29 and Cyp2c44 mRNAs were undetectable (Figure 1A). Under LPS stimulation, the expression of Cyp2j9, Cyp2j6, and Cyp2j5 mRNAs in the lungs was significantly abated (Figure 1B), while the expression of sEH and COX-2 was considerably increased (Figure 1C-F). Since macrophages have pivotal roles in the initiation of innate immune response at the early stage of ALI, we used primary murine macrophages to evaluate the expression pattern of CYPs/COX-2 metabolism of ARA in vitro. The results were consistent with those of the lungs in vivo (Figure 1G-L). Collectively, these data indicate that the dysregulation of CYPs/COX-2 metabolism of ARA occurs in the lungs and macrophages under the LPS challenge.
Dysregulation of CYPs/COX-2 metabolism of ARA occurs in the lungs and macrophages under the LPS challenge. Cyp2j9 was the most abundant CYP isoform expressed in the lungs, whereas Cyp2c29 and Cyp2c44 mRNA were undetectable (A, n = 6). Cyp2j9, Cyp2j6, and Cyp2j5 mRNAs were robustly decreased at 12 h after LPS administration (5 mg/kg, i.t.) in the lungs (B, n = 6). Western blotting and RT-qPCR results showing increased sEH and COX-2 proteins, and Cox-2 mRNA at 12 h after LPS administration. (C-F, n = 4-8). Cyp2j6 was the most abundant CYP isoform expressed in primary murine macrophages after treatment with LPS (100 ng/mL), whereas Cyp2c29 and Cyp2c44 mRNA were undetectable (G, n = 3). Cyp2j6 and Cyp2j9 mRNA were remarkably decreased at 6 h after LPS administration (H, n = 3). Expression of sEH and COX-2 protein and Cox-2 mRNA were detected by Western blotting and RT-qPCR (I-L, n = 3). Data are expressed as the mean ± SD. * P < 0.05, ** P < 0.01, and *** P < 0.001.
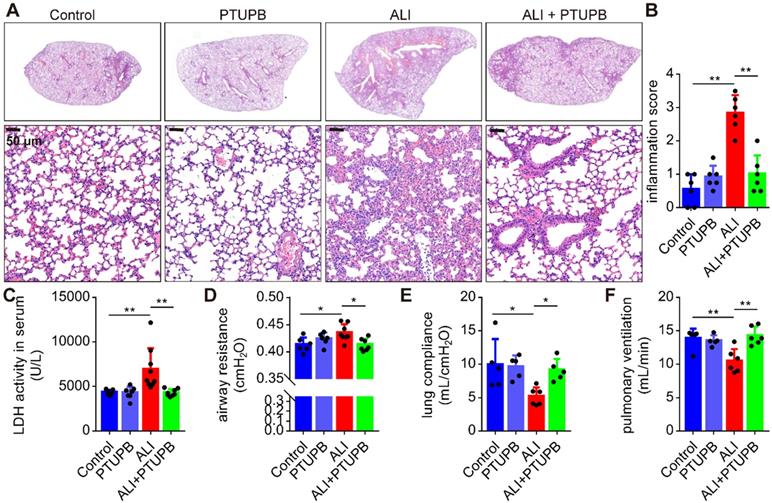
PTUPB attenuates the pathological lung injury and rescues the respiratory function of mice treated with LPS
We used PTUPB for the dual inhibition of COX-2 and sEH to establish the function of CYPs/COX-2 dysregulation in ALI. PTUPB reduced the expression of sEH and COX-2 (Figure S1A-C), but had no effect on the expression of Cyp2j9 and Cyp2j6 (Figure S1D- E), as well as Alox5/12/15 (encoding 5/12/15-LOX, Figure S1F-H). Following LPS stimulation, there was an increase in the thickness of the alveolar wall, interstitial infiltrated inflammatory cells, and the collapse of the alveoli in the lungs of mice (Figure 2A). Remarkably, PTUPB pre-treatment alleviated these pathological changes (Figure 2A-B) and pronounced reduced the LDH activity in the serum (Figure 2C). Additionally, we evaluated the effects of PTUPB on the respiratory function of ALI mice. The results showed that PTUPB pre-treatment significantly reduced airway resistance (Figure 2D), and increased lung compliance and pulmonary ventilation of mice with ALI induced by LPS (Figure 2E-F). These results demonstrate that PTUPB attenuates pathological injury of the lung tissue and enhances the respiratory function of LPS-treated ALI mice.
PTUPB attenuates the pathological lung injury and rescues the respiratory function of ALI mice. C57BL/6 mice were subcutaneously injected with PTUPB 1 h before the LPS administration. Twelve hours after LPS injection (5 mg/kg, i.t.), lung histopathology was performed with H&E staining (A). Inflammation score was measured independently by three pathologists blinded to the experiment (B, n = 6). Activity of LDH in the serum was assayed (C, n = 7-8). Respiratory function was detected by Buxco, including airway resistance (D, n = 5-8), lung compliance (E, n = 5-8), and pulmonary ventilation (F, n = 5-8). Data are mean ± SD. * P < 0.05, ** P < 0.01, and *** P < 0.001.
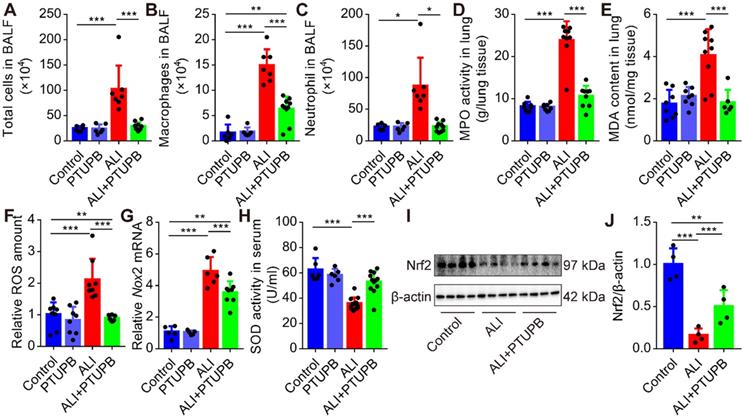
PTUPB reverses the infiltration of inflammatory cells and oxidative stress in ALI mice treated with LPS
Next, we found that pre-treatment PTUPB remarkably reduced the number of total cells (Figure 3A), macrophages (Figure 3B), and neutrophils (Figure 3C) in the BALF, as well as MPO activity in the lungs of ALI mice (Figure 3D). Immunofluorescence results confirmed the inhibitory effects of PTUPB on the accumulation of neutrophil and macrophages (Figure S2). One of the characteristic features of ALI is oxidative stress. Pre-treatment with PTUPB reduced the MDA content and total ROS in the lungs of ALI mice (Figure 3E-F). Furthermore, NADPH oxidases (NOXs) are important sources for the regulated generation of ROS [38] and our results demonstrated that PTUPB suppressed the expression of Nox2 mRNA (Figure 3G). PTUPB pre-treatment also restored the activity of the anti-oxidative enzyme SOD (Figure 3H) in serum, and partially restored the protein expression of nuclear factor-erythroid-2- related-factor-2 (Nrf2) in the lungs, which were both reduced in ALI mice (Figure 3I-J). Collectively, these data reveal that the dual inhibition of COX-2/sEH reverses the accumulation of the inflammatory cells and oxidative stress.
PTUPB reverses the infiltration of inflammatory cells and oxidative stress in ALI mice. C57BL/6 mice received LPS injection (5 mg/kg, i.t.) with or without PTUPB pre-treatment (5 mg/kg) for 1 h. Twelve hours after the LPS injection, the total cells (A), macrophages (B), and neutrophils (C) in the BALF were counted (n = 6-10). MPO activity was detected to quantify the infiltration of neutrophils in the lungs (D, n = 7-10). MDA and total ROS in the lungs were detected 12 h after the LPS administration (E-F, n = 6-9). mRNA expression of Nox2 in the lungs was detected by RT-qPCR (G, n = 5-8). Total SOD activity in serum was detected 12 h after the LPS administration (H, n = 6-10). Protein expression of Nrf2 in the lungs was assayed by Western blotting (I-J, n = 4-8). Data are mean ± SD. * P < 0.05, ** P < 0.01, and *** P < 0.001.
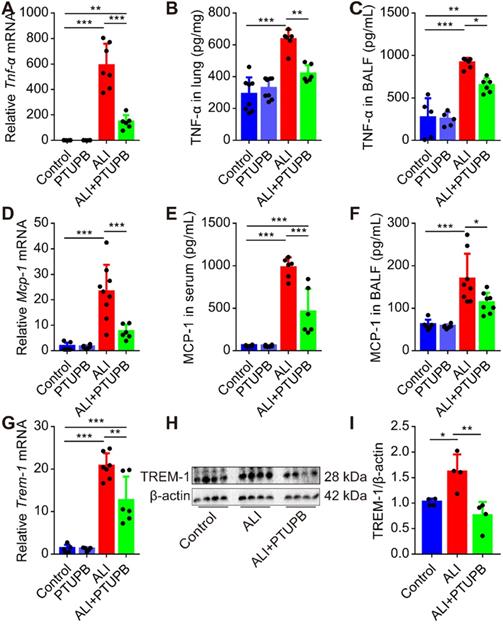
PTUPB reverses the augment of pro-inflammatory factors in the lungs of ALI mice
“Cytokine storms” is well characterized in ALI. TNF-α-targeting aptamer with antagonistic effect on TNF-α attenuates the severity of acute lung injury [39]. We found that the expression of TNF-α and MCP-1 was profoundly up-regulated in the lungs of mice 12 h post LPS treatment (Figure 4A-F). PTUPB pre-treatment remarkably suppressed the increase in TNF-α and MCP-1 expression in the lungs, serum, or BALF of ALI mice (Figure 4A-F). Our pervious study indicated that the triggering receptor expressed on myeloid cells (TREM-1) acted as a pivotal inflammatory amplifier receptor in ALI [37]. Here, we found that PTUPB pre-treatment also down-regulated the mRNA and protein expression of TREM-1 in the lungs of ALI mice (Figure 4G-I). Altogether, our data indicate that blockade of COX-2/sEH reduces the pro- inflammatory factors in ALI mice induced by LPS.
PTUPB attenuates the production of pro-inflammatory factors in the lungs of ALI mice. C57BL/6 mice received LPS injection (5 mg/kg, i.t.) with or without PTUPB pre-treatment (5 mg/kg) for 1 h. Twelve hours after the LPS administration, mRNA expression of Tnf-α (A, n = 6-7), Mcp-1 (D, n = 6-7), and Trem-1 (G, n = 5-7) in the lungs was detected by RT-qPCR. Protein content of TNF-α in lung tissue (B, n = 6-8) and BALF (C, n = 6-8), MCP-1 in serum (E, n = 5-8) and BALF (F, n = 6-8) was assayed by ELISA. Expression of TREM-1 in lung tissue was assayed by Western blotting (H-I, n = 4). Data are expressed as the mean ± SD. * P < 0.05, ** P < 0.01, and *** P < 0.001.
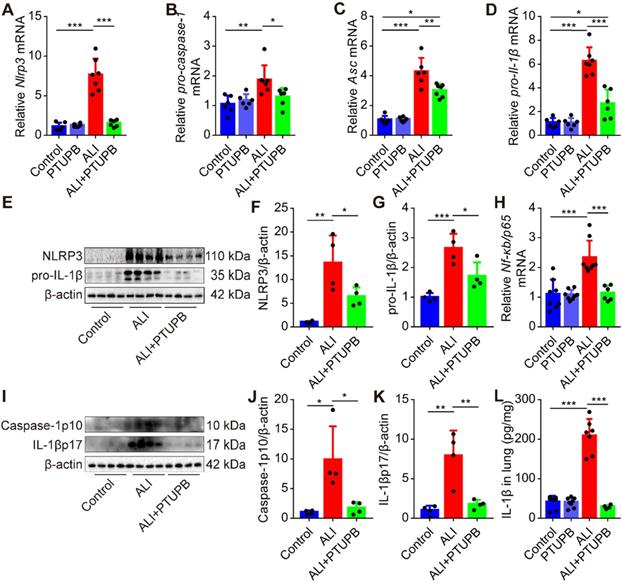
PTUPB inhibits activation of NLRP3 inflammasome in the lungs of ALI mice
A growing body of literature has shown that the NLRP3 inflammasome plays a pivotal role in the pathogenesis of ALI [5, 6]. Here, we found that blockade of COX-2/sEH by PTUPB reversed the up- regulation of components of the NLRP3 inflammasome, including Nlrp3, pro-caspase-1, Asc, and pro-Il-1β mRNAs in the lungs of ALI mice (Figure 5A-D). Besides, PTUPB pre-treatment also strongly reduced the expression of NLRP3 and pro-IL-1β proteins (Figure 5E-G) and NF-κB is known to be involved in the expression of these proteins. We found that PTUPB also reduced the mRNA expression of Nf-κb/ p65 (Figure 5H). Also, caspase-1 p10 is a biomarker for the activation of NLRP3 inflammasome, which cleaves pro-IL-1β into IL-1β p17 [40, 41]. We found that PTUPB pre-treatment markedly blocked LPS-induced expression of caspase-1 p10 and release of IL-1β p17 (Figure 5I-L). Altogether, these data indicate that PTUPB inhibits the activation of NLRP3 inflammasome in the lungs of ALI mice.
PTUPB inhibits the activation of NLRP3 inflammasome in the lung of ALI mice. C57BL/6 mice received LPS injection (5 mg/kg, i.t.) with or without PTUPB pre-treatment (5 mg/kg) for 1 h. mRNA expression of Nlrp3 (A, n = 6-7), pro-caspase-1 (B, n = 6-7), Asc (C, n = 6-7), and pro-Il-1β (D, n = 6-7) in the lungs was detected by RT-qPCR 12 h after the LPS injection. Protein expression of NLRP3 and pro-IL-1β (E-G, n = 4) in the lungs was detected by Western blotting. mRNA expression of Nf-κb/p65 (H, n = 6-9) in the lungs was detected by RT-qPCR 12 h after the LPS injection. Protein expression of caspase-1p10 and IL-1βp17 in the lungs was detected by Western blotting (I-K, n = 4). Concentration of IL-1β in lung tissue was detected by ELISA (L, n = 5-8). Data are expressed as the mean ± SD. * P < 0.05, ** P < 0.01, and *** P < 0.001.
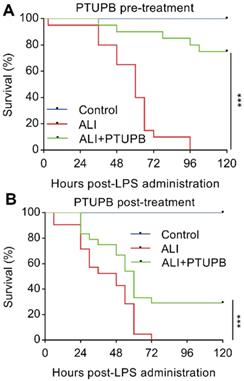
Prophylactic and therapeutic treatments of PTUPB significantly prevent the death of LPS-treated mice
Pre-treatment with PTUPB (5 mg/kg) 1 h before LPS administration significantly prevented the death of mice receiving 25 mg/kg LPS (Figure 6A). We further examined the potential of therapeutic benefits of PTUPB treatment (5 mg/kg) 6 h post LPS administration. Post-treatment with PTUPB significantly prevented the death of LPS-treated mice (Figure 6B). These results indicate that blockade of COX-2/sEH by PTUPB shows prophylactic and therapeutic protection against LPS-induced ALI in mice.
Prophylactic and therapeutic treatment of PTUPB prevents death from LPS administration in mice. PTUPB (5 mg/kg, s.c.) was administered to mice 1 h before (A) or 6 h after (B) induction of ALI by a lethal dose of LPS (25 mg/kg, i.t.). The mortality of the mice was monitored every 6 h, and the percent survival rate was expressed as a Kaplan-Meier survival curve (n = 20 per group). *** P < 0.001.
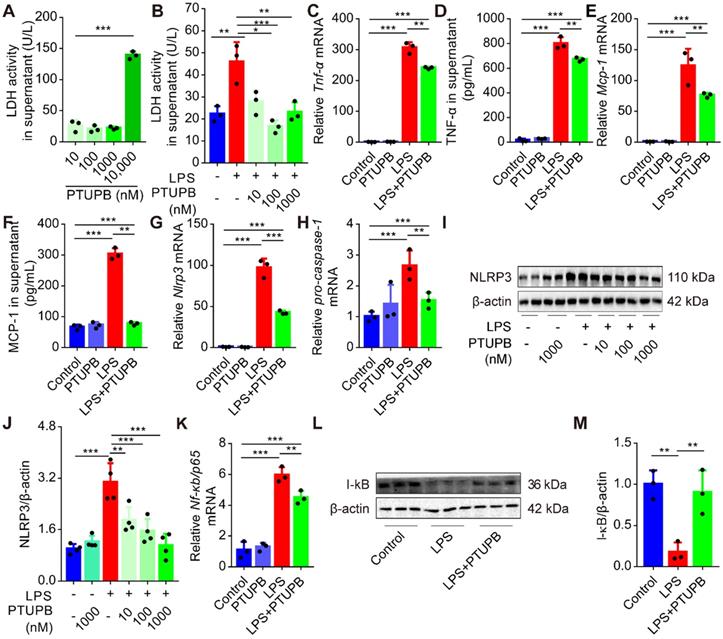
PTUPB reduces priming of NLRP3 inflammasome in primary murine macrophages
Treatment with clodronate-loaded liposomes that deplete macrophages significantly attenuated the lung tissue pathological injury (Figure S3A-C), and reduced the activation of NLRP3 inflammasome (Figure S3D-F). We evaluated the effects of PTUPB on the inflammatory response of primary murine macrophages and found that low-dose PTUPB (≤1000 nM) had no detectable influence on LDH release from macrophages (Figure 7A). However, PTUPB pre-treatment at 10, 100, and 1000 nM concentrations remarkably reduced the LDH activity induced by LPS (100 ng/mL) (Figure 7B). PTUPB also restored the protein expressions of COX-2 and sEH induced by LPS in macrophages (Figure S4). Furthermore, PTUPB (1000 nM) significantly reversed the expression of pro-inflammatory factors induced by LPS, including TNF-α (Figure 7C-D) and MCP-1 (Figure 7E-F). Importantly, PTUPB pre-treatment remarkably suppressed priming of NLRP3 inflammasome characterized by the inhibition of Nlrp3 and pro-caspase-1 mRNAs, and NLRP3 protein (Figure 7G-J). NF-κB plays a vital role in priming of NLRP3 inflammasomes [40]. PTUPB pre-treatment inhibited the expression of Nf-κb/p65 mRNA (Figure 7K), restored the expression of I-κBα protein in the lungs (Figure 7L-M), and inhibited its translocation into the cell nucleus (Figure S5). These results imply that blockade of COX-2/sEH reverses the priming of NLRP3 inflammasome in macrophages in vitro.
PTUPB reduces the inflammation and priming of NLRP3 inflammasome in primary murine macrophages. Primary murine macrophages were treated with serial concentrations of PTUPB (10, 100, 1000, and 10000 nM) for 6 h and the LDH activity in the supernatant of cell culture was detected (A). Macrophages were treated with LPS (100 ng/mL) for 6 h with or without PTUPB pre-treatment (10, 100, and 1000 nM) for 1 h and activity of LDH in the supernatant was detected (B). Expression of Tnf-α (C), Mcp-1 (E), Nlrp3 (G), pro-caspase-1 (H), and Nf-κb/p65 (K) mRNAs in macrophages was detected by RT-qPCR. Concentration of TNF-α (D) and MCP-1 (F) in the supernatant of cell culture was detected by ELISA (n = 3). Protein expression of NLRP3 and IκBα in macrophages was detected by Western blotting (I-J, n = 4; L-M, n = 3). Data are expressed as the mean ± SD. ** P < 0.01, and *** P < 0.001.
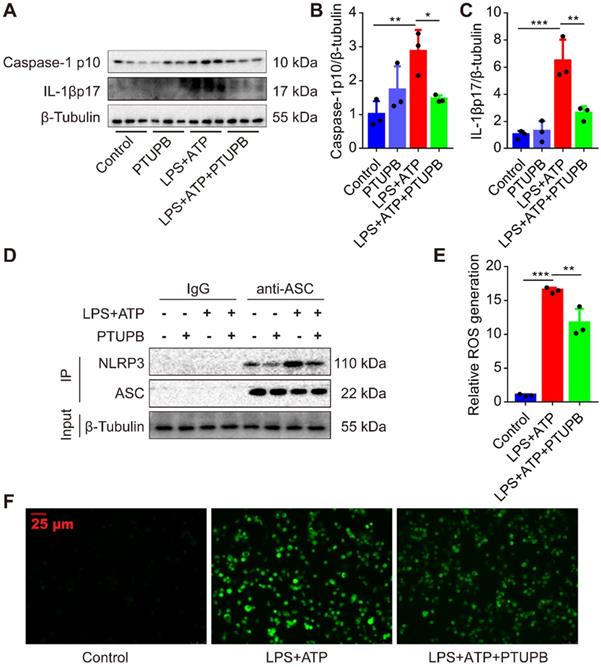
PTUPB inhibits the activation of NLRP3 inflammasome in primary murine macrophages
Lastly, we treated macrophages with LPS plus ATP to activate the NLRP3 inflammasome. We found that PTUPB reduced the protein expression of caspase-1 p10 and IL-1β p17 in macrophages (Figure 8A-C). NLRP3 complex formation is the key step for the activation of NLRP3 inflammasome [42]. IP results showed that PTUPB inhibited the interaction of endogenous NLRP3 and ASC in macrophages (Figure 8D). ROS is one of the most important factors regulating the activation of NLRP3 inflammasome and PTUPB reversed the elevated ROS (Figure 8E-F). These results imply that blockade of COX-2/sEH reverses the activation of NLRP3 inflammasome in macrophages by suppressing ROS generation.
PTUPB inhibits the activation of NLRP3 inflammasome in primary murine macrophages. To evaluate the effects of PTUPB on the activation of NLRP3 inflammasome, PTUPB (1000 nM) was added 1 h before LPS-priming (100 ng/mL) for 135 min. Subsequently, cells were stimulated with ATP (2.5 mM) for 45 min. Protein expression of caspase-1 p10 and IL-1β p17 in macrophages was detected by Western blotting (A-C, n = 3). Interaction between endogenous NLRP3 and ASC was analyzed by IP (D). The ROS was analyzed by a ROS kit (E-F). Data are expressed as the mean ± SD. * P < 0.05, ** P < 0.01, and *** P < 0.001.
Discussion
Our study demonstrated that CYPs/COX-2 dysregulation occurred during the inflammation induced by LPS both in mice and in cultured macrophages, which was evidenced by lower CYPs expression and higher COX-2 expression. Restoring the homeostasis of ARA metabolism by CYPs/COX-2 with PTUPB could alleviate lung tissue pathological injury, reduce oxidative stress and decrease the secretion of pro-inflammatory factors, and inhibit the activation of NLRP3 inflammasome in the lungs of ALI mice. We report here for the first time that dysregulated metabolism of CYPs/COX-2-derived ARA plays a vital role in LPS-induced ALI mice, and the dual inhibition of COX-2 and sEH may be an effective anti-inflammatory strategy in treating ALI.
Proper regulation of the biosynthesis of eicosanoids is critical for homeostasis. Detrimental consequences of the overproduction of COX and LOX pathways have been described in many inflammatory diseases in human beings [43, 44]. For example, COX-2 is expressed in different cell types in the lungs, and its increased expression was observed in the lungs of LPS-induced ALI mice [21]. Similarly, EETs exhibit anti-inflammatory actions in many diseases, such as central nervous system diseases [12], insulin resistance [45], and pulmonary diseases [16, 18, 34]. However, litter is known about the dysregulated metabolism of CYPs/COX-2-derived ARA in LPS-induced ALI. This is the first report showing a decrease of CYP2J/2C and an increase of COX-2, and demonstrating the dysregulation of CYPs/COX-2 in the lungs of LPS-induced ALI mice.
A growing body of studies focuses on designing multiple ligands (DMLs) to augment drug efficacy, improve drug safety, and reduce side effects. Compared with combination drugs or therapies, DMLs have several advantages [22, 46, 47]. We had previously designed PTUPB, which is a novel COX-2 and sEH dual inhibitor [25]. We demonstrated that PTUPB could suppress the growth of glioblastoma, abate kidney injury, and exert a synergistic effect with cisplatin [26-28]. Importantly, we did not observe any acute toxicity in vivo or in vitro [25]. We also found that PTUPB had no effect on the expression of LOXs but significantly reduced the expression of COX-2 and sEH. In this study, we found that dual inhibition of COX-2 and sEH exhibited a powerful therapeutic potential for ALI.
One critical pathophysiological process in ALI/ARDS is manifested by extensive inflammation [48]. Our laboratory as well as other investigators have shown that NLRP3 inflammasome plays a central role in the immense inflammation of ALI [6, 37]. Recently, one study described that monounsaturated and polyunsaturated fatty acids could impede the NLRP3 inflammasome activation in metabolic diseases [49], but it was not clear whether CYPs/COX-2 were involved in the activation of NLRP3 inflammasome. In this study, our findings demonstrated that the dysregulated metabolism of CYPs/COX-2 was associated with the activation of NLRP3 inflammasome in the lungs of ALI mice. It has been reported that COX-2-mediated PGE2 positively regulates the activation of the NLRP3 inflammasome, and inhibition of COX-2 reduces NLRP3 inflammasome-derived IL-1β secretion and pyroptosis in macrophages [50, 51]. We have found that inhibition of sEH suppressed the secretion of IL-1β in LPS-induced ALI mice [16], indicating that EETs could inhibit the activation of NLRP3 inflammasome. In hyperoxia-induced ALI mice, Ephx2-/- mice showed decreased activation of NLRP3 inflammasome [52]. These reports support our findings that dual inhibition of COX-2 and sEH with PTUPB suppresses the activation of NLRP3 inflammasome.
Oxidative stress is another indispensable characteristic of ALI. Our studies show that PTUPB attenuates the oxidative stress in the lungs of ALI mice, and enhances the antioxidative mechanism involving Nrf2 and SOD. It has been well documented that ROS is one of the most important factors regulating the activation of NLRP3 inflammasome [40]. Although one study proposed that Nrf2 was necessary for the NLRP3 inflammasome activation [53], another one indicated that Nrf2 limited the NLRP3 inflammasome activation by down-regulating ROS production [54]. Our findings demonstrated that PTUPB restored the expression of Nrf2 and reduced the oxidative stress in the lungs of LPS-induced ALI mice. Given these results, we infer that PTUPB inhibits the NLRP3 inflammasome activation by restoring the balance of oxidative and anti-oxidative mechanisms during ALI.
There are limitations to the present study. First, we evaluated the gene expression of specific critical enzymes, including Cyp2j/2c. It is possible that whether their mediators contribute to the mechanism of action of PTUPB in LPS-induced mice. Our previous study detected the ARA metabolites of the CYP, COX, and LOX pathways in the serum of LPS-treated mice by LC-MS/MS [55]. Especially, the pro-inflammatory mediator PGE2 had the most significant alteration (~30 fold), and 12,13-DHOME, 9,10-DHOME in the sEH pathway were increased [55]. We also found that PTUPB reduced the level of PGE2 derived from the COX-2 pathway and 12,13-DiHOME derived from the sEH pathway in PDX BL0269 tumor tissues [26]. Thus, it appears that PTUPB treatment affects the profiles of ARA metabolites. And second, the reduction in the expression of sEH and COX-2 by PTUPB might be an indirect effect caused by inflammation, which up-regulates the expressions of sEH and COX-2 [19, 56]. PTUPB blocks the enzyme activities of sEH and COX-2, resulting in low inflammation. So, we propose that PTUPB not only blocks the enzyme activity but also indirectly reduces the enzyme expression.
In conclusion, this is the first report revealing that dysregulation of CYPs/COX-2-derived ARA metabolism contributes to lung injury in LPS-induced ALI mice. PTUPB, a dual COX-2 and sEH inhibitor, exerts an anti-inflammatory response and protects mice against LPS-induced ALI. Therefore, CYPs/ COX-2 dysregulation represents a novel potential target for treating ALI/ARDS.
Abbreviations
ALI: acute lung injury; ARA: arachidonic acid; ARDS: acute respiratory distress syndrome; ASC: apoptosis-associated speck-like protein containing a CARD; ATP: adenosine triphosphate; BALF: bronchoalveolar lavage fluid; COX: cyclooxygenase; CYP: cytochrome P450; DHETs: dihydroxyeicosatrienoic acid; DMLs: designing multiple ligands; EETs: epoxyeicosatrienoic acids; H&E: Hematoxylin-Eosin; IP: immunoprecipitation; IL-1β: interleukin-1 beta; LPS: lipopolysaccharide; LOX: lipoxygenase; MCP-1: monocyte chemotactic protein 1; MDA: malondialdehyde; NF-κB: nuclear factor kappa-B; NLRP3: NACHT, LRR and PYD domains-containing protein 3; NOX-2: NADPH oxidases 2; Nrf2: nuclear factor-erythroid-2-related-factor-2; PGs: prostaglandins; PTUPB: 4-(5-phenyl-3-{3-[3-(4-trifluoromethylphenyl)-ureido]-propyl}-pyrazol-1-yl)-benzenesulfonamide; RT-qPCR: real-time quantitative polymerase chain reaction; ROS: reactive oxygen species; sEH: soluble epoxide hydrolase; SOD: superoxide dismutase; TNF-α: tumor necrosis factor-alpha.
Supplementary Material
Supplementary materials and methods, figures, and tables.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81500065, 81670014, 81530063, 91949110), the Open Project of the State Key Laboratory of Trauma, Burns, and Combined Injury, Army Medical University (SKLKF201702), Hunan Provincial Natural Science Foundation of China (2018JJ3701, 2019JJ70008), Fundamental Research Funds for the Central Universities of Central South University (2018zzts038, GCX20190889Y), and the Open Sharing Fund for the Large-scale Instruments and Equipment of Central South University (CSUZC201944). Partial support was provided by R01ES002710 and P42 ES004699 from NIEHS.
Author Contributions
H.H.Y., J.X.D., J.B.X., X.X.G., W.J.Z., C.C.S., C.Y.Z., X.Q.L., and Y.F.Z. performed the experiments; Y.Z., J.X.D. and H.H.Y. analyzed the data; B.D.H., Y.Z., S.K.L., P.C., J.X.J., and C.X.G. contributed reagents/materials/analysis tools; S.H.H. and B.D.H. designed and synthesized PTUPB; Y.Z., H.H.Y., and J.X.D. wrote the paper; C.X.G. and Y.Z. conceived, designed the experiments, and critically reviewed the manuscript. All authors had final approval of the submitted versions.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Rawal G, Yadav S, Kumar R. Acute Respiratory Distress Syndrome: An Update and Review. J Transl Int Med. 2018;6:74-7
2. Li K, He Z, Wang X, Pineda M, Chen R, Liu H. et al. Apigenin C-glycosides of Microcos paniculata protects lipopolysaccharide induced apoptosis and inflammation in acute lung injury through TLR4 signaling pathway. Free Radic Biol Med. 2018;124:163-75
3. Tao W, Miao QB, Zhu YB, Shu YS. Inhaled neutrophil elastase inhibitor reduces oleic acid-induced acute lung injury in rats. Pulm Pharmacol Ther. 2012;25:99-103
4. Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med. 2000;342:1334-49
5. Ying Y, Mao Y, Yao M. NLRP3 Inflammasome Activation by MicroRNA-495 Promoter Methylation May Contribute to the Progression of Acute Lung Injury. Mol Ther Nucleic Acids. 2019;18:801-14
6. Zhang Y, Li X, Grailer JJ, Wang N, Wang M, Yao J. et al. Melatonin alleviates acute lung injury through inhibiting the NLRP3 inflammasome. J Pineal Res. 2016;60:405-14
7. Li C, Yang D, Cao X, Wang F, Jiang H, Guo H. et al. LFG-500, a newly synthesized flavonoid, attenuates lipopolysaccharide-induced acute lung injury and inflammation in mice. Biochem Pharmacol. 2016;113:57-69
8. Zhao Z, Tang X, Zhao X, Zhang M, Zhang W, Hou S. et al. Tylvalosin exhibits anti-inflammatory property and attenuates acute lung injury in different models possibly through suppression of NF-kappaB activation. Biochem Pharmacol. 2014;90:73-87
9. Kim SR, Kim HJ, Kim DI, Lee KB, Park HJ, Jeong JS. et al. Blockade of Interplay between IL-17A and Endoplasmic Reticulum Stress Attenuates LPS-Induced Lung Injury. Theranostics. 2015;5:1343-62
10. Hu Y, Lou J, Mao YY, Lai TW, Liu LY, Zhu C. et al. Activation of MTOR in pulmonary epithelium promotes LPS-induced acute lung injury. Autophagy. 2016;12:2286-99
11. McReynolds CB, Hwang SH, Yang J, Wan D, Wagner K, Morisseau C. et al. Pharmaceutical Effects of Inhibiting the Soluble Epoxide Hydrolase in Canine Osteoarthritis. Front Pharmacol. 2019;10:533
12. Zarriello S, Tuazon JP, Corey S, Schimmel S, Rajani M, Gorsky A. et al. Humble beginnings with big goals: Small molecule soluble epoxide hydrolase inhibitors for treating CNS disorders. Prog Neurobiol. 2019;172:23-39
13. Shahabi P, Siest G, Meyer UA, Visvikis-Siest S. Human cytochrome P450 epoxygenases: variability in expression and role in inflammation-related disorders. Pharmacol Ther. 2014;144:134-61
14. Cho HJ, Switzer CH, Kamynina A, Charles R, Rudyk O, Ng T. et al. Complex interrelationships between nitro-alkene-dependent inhibition of soluble epoxide hydrolase, inflammation and tumor growth. Redox Biol. 2020;29:101405
15. Tanaka H, Kamita SG, Wolf NM, Harris TR, Wu Z, Morisseau C. et al. Transcriptional regulation of the human soluble epoxide hydrolase gene EPHX2. Biochim Biophys Acta. 2008;1779:17-27
16. Zhou Y, Liu T, Duan JX, Li P, Sun GY, Liu YP. et al. Soluble Epoxide Hydrolase Inhibitor Attenuates Lipopolysaccharide-Induced Acute Lung Injury and Improves Survival in Mice. Shock. 2017;47:638-45
17. Liu LP, Li B, Shuai TK, Zhu L, Li YM. Deletion of soluble epoxide hydrolase attenuates mice Hyperoxic acute lung injury. BMC Anesthesiol. 2018;18:48
18. Zhou Y, Yang J, Sun GY, Liu T, Duan JX, Zhou HF. et al. Soluble epoxide hydrolase inhibitor 1-trifluoromethoxyphenyl-3- (1-propionylpiperidin-4-yl) urea attenuates bleomycin-induced pulmonary fibrosis in mice. Cell Tissue Res. 2016;363:399-409
19. Aoki T, Narumiya S. Prostaglandins and chronic inflammation. Trends Pharmacol Sci. 2012;33:304-11
20. Simmons DL, Botting RM, Hla T. Cyclooxygenase isozymes: the biology of prostaglandin synthesis and inhibition. Pharmacol Rev. 2004;56:387-437
21. Al-Harbi NO, Imam F, Al-Harbi MM, Ansari MA, Zoheir KM, Korashy HM. et al. Dexamethasone Attenuates LPS-induced Acute Lung Injury through Inhibition of NF-kappaB, COX-2, and Pro-inflammatory Mediators. Immunol Invest. 2016;45:349-69
22. P JJ, Manju SL, Ethiraj KR, Elias G. Safer anti-inflammatory therapy through dual COX-2/5-LOX inhibitors: A structure-based approach. Eur J Pharm Sci. 2018;121:356-81
23. Kim HS, Kim SK, Kang KW. Differential Effects of sEH Inhibitors on the Proliferation and Migration of Vascular Smooth Muscle Cells. Int J Mol Sci. 2017 18
24. Park SK, Herrnreiter A, Pfister SL, Gauthier KM, Falck BA, Falck JR. et al. GPR40 is a low-affinity epoxyeicosatrienoic acid receptor in vascular cells. J Biol Chem. 2018;293:10675-91
25. Hwang SH, Wagner KM, Morisseau C, Liu JY, Dong H, Wecksler AT. et al. Synthesis and structure-activity relationship studies of urea-containing pyrazoles as dual inhibitors of cyclooxygenase-2 and soluble epoxide hydrolase. J Med Chem. 2011;54:3037-50
26. Wang F, Zhang H, Ma AH, Yu W, Zimmermann M, Yang J. et al. COX-2/sEH Dual Inhibitor PTUPB Potentiates the Antitumor Efficacy of Cisplatin. Mol Cancer Ther. 2018;17:474-83
27. Li J, Zhou Y, Wang H, Gao Y, Li L, Hwang SH. et al. COX-2/sEH dual inhibitor PTUPB suppresses glioblastoma growth by targeting epidermal growth factor receptor and hyaluronan mediated motility receptor. Oncotarget. 2017;8:87353-63
28. Hye Khan MA, Hwang SH, Sharma A, Corbett JA, Hammock BD, Imig JD. A dual COX-2/sEH inhibitor improves the metabolic profile and reduces kidney injury in Zucker diabetic fatty rat. Prostaglandins Other Lipid Mediat. 2016;125:40-7
29. Zhang YF, Sun CC, Duan JX, Yang HH, Zhang CY, Xiong JB. et al. A COX-2/sEH dual inhibitor PTUPB ameliorates cecal ligation and puncture-induced sepsis in mice via anti-inflammation and anti-oxidative stress. Biomed Pharmacother. 2020;126:109907
30. Zhang CY, Duan JX, Yang HH, Sun CC, Zhong WJ, Tao JH. et al. COX-2/sEH dual inhibitor PTUPB alleviates bleomycin-induced pulmonary fibrosis in mice via inhibiting senescence. FEBS J. 2019
31. Sun GY, Yang HH, Guan XX, Zhong WJ, Liu YP, Du MY. et al. Vasoactive intestinal peptide overexpression mediated by lentivirus attenuates lipopolysaccharide-induced acute lung injury in mice by inhibiting inflammation. Mol Immunol. 2018;97:8-15
32. Duan JX, Zhou Y, Zhou AY, Guan XX, Liu T, Yang HH. et al. Calcitonin gene-related peptide exerts anti-inflammatory property through regulating murine macrophages polarization in vitro. Mol Immunol. 2017;91:105-13
33. Zhong WJ, Yang HH, Guan XX, Xiong JB, Sun CC, Zhang CY. et al. Inhibition of glycolysis alleviates lipopolysaccharide-induced acute lung injury in a mouse model. J Cell Physiol. 2019;234:4641-54
34. Dong L, Zhou Y, Zhu ZQ, Liu T, Duan JX, Zhang J. et al. Soluble Epoxide Hydrolase Inhibitor Suppresses the Expression of Triggering Receptor Expressed on Myeloid Cells-1 by Inhibiting NF-kB Activation in Murine Macrophage. Inflammation. 2016;40:13-20
35. Huang XT, Yue SJ, Li C, Huang YH, Cheng QM, Li XH. et al. A Sustained Activation of Pancreatic NMDARs Is a Novel Factor of β-Cell Apoptosis and Dysfunction. Endocrinology. 2017;158:3900-13
36. Zhang M, Zheng Y, Sun Y, Li S, Chen L, Jin X. et al. Knockdown of NEAT1 induces tolerogenic phenotype in dendritic cells by inhibiting activation of NLRP3 inflammasome. Theranostics. 2019;9:3425-42
37. Liu T, Zhou Y, Li P, Duan JX, Liu YP, Sun GY. et al. Blocking triggering receptor expressed on myeloid cells-1 attenuates lipopolysaccharide-induced acute lung injury via inhibiting NLRP3 inflammasome activation. Sci Rep. 2016;6:39473
38. Hayes P, Knaus UG. Balancing reactive oxygen species in the epigenome: NADPH oxidases as target and perpetrator. Antioxid Redox Signal. 2013;18:1937-45
39. Lai WY, Wang JW, Huang BT, Lin EP, Yang PC. A Novel TNF-alpha-Targeting Aptamer for TNF-alpha-Mediated Acute Lung Injury and Acute Liver Failure. Theranostics. 2019;9:1741-51
40. Swanson KV, Deng M, Ting JP. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol. 2019;19:477-89
41. Zhou Y, Zhang CY, Duan JX, Li Q, Yang HH, Sun CC. et al. Vasoactive intestinal peptide suppresses the NLRP3 inflammasome activation in lipopolysaccharide-induced acute lung injury mice and macrophages. Biomed Pharmacother. 2020;121:109596
42. Huppertz C, Jager B, Wieczorek G, Engelhard P, Oliver SJ, Bauernfeind FG. et al. The NLRP3 inflammasome pathway is activated in sarcoidosis and involved in granuloma formation. Eur Respir J. 2020
43. Ricciotti E, FitzGerald GA. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol. 2011;31:986-1000
44. Mashima R, Okuyama T. The role of lipoxygenases in pathophysiology; new insights and future perspectives. Redox Biol. 2015;6:297-310
45. Yang Y, Dong R, Chen Z, Hu D, Fu M, Tang Y. et al. Endothelium-specific CYP2J2 overexpression attenuates age-related insulin resistance. Aging Cell. 2018 17
46. Bitto A, Minutoli L, David A, Irrera N, Rinaldi M, Venuti FS. et al. Flavocoxid, a dual inhibitor of COX-2 and 5-LOX of natural origin, attenuates the inflammatory response and protects mice from sepsis. Crit Care. 2012;16:R32
47. Rao CV, Janakiram NB, Madka V, Devarkonda V, Brewer M, Biddick L. et al. Simultaneous targeting of 5-LOX-COX and EGFR blocks progression of pancreatic ductal adenocarcinoma. Oncotarget. 2015;6:33290-305
48. Thompson BT, Chambers RC, Liu KD. Acute Respiratory Distress Syndrome. N Engl J Med. 2017;377:562-72
49. Ralston JC, Lyons CL, Kennedy EB, Kirwan AM, Roche HM. Fatty Acids and NLRP3 Inflammasome-Mediated Inflammation in Metabolic Tissues. Annu Rev Nutr. 2017;37:77-102
50. Hua KF, Chou JC, Ka SM, Tasi YL, Chen A, Wu SH. et al. Cyclooxygenase-2 regulates NLRP3 inflammasome-derived IL-1beta production. J Cell Physiol. 2015;230:863-74
51. Kim Y, Gromovsky AD, Brown JM, Chung S. Gamma-tocotrienol attenuates the aberrant lipid mediator production in NLRP3 inflammasome-stimulated macrophages. J Nutr Biochem. 2018;58:169-77
52. Li PS, Tao W, Yang LQ, Shu YS. Effect of Soluble Epoxide Hydrolase in Hyperoxic Acute Lung Injury in Mice. Inflammation. 2018;41:1065-72
53. Sogawa Y, Nagasu H, Iwase S, Ihoriya C, Itano S, Uchida A. et al. Infiltration of M1, but not M2, macrophages is impaired after unilateral ureter obstruction in Nrf2-deficient mice. Sci Rep. 2017;7:8801
54. Liu X, Zhang X, Ding Y, Zhou W, Tao L, Lu P. et al. Nuclear Factor E2-Related Factor-2 Negatively Regulates NLRP3 Inflammasome Activity by Inhibiting Reactive Oxygen Species-Induced NLRP3 Priming. Antioxid Redox Signal. 2017;26:28-43
55. Yang J, Schmelzer K, Georgi K, Hammock BD. Quantitative profiling method for oxylipin metabolome by liquid chromatography electrospray ionization tandem mass spectrometry. Anal Chem. 2009;81:8085-93
56. Wang W, Yang J, Zhang J, Wang Y, Hwang SH, Qi W. et al. Lipidomic profiling reveals soluble epoxide hydrolase as a therapeutic target of obesity-induced colonic inflammation. Proc Natl Acad Sci U S A. 2018;115:5283-8
Author contact
![]() Corresponding authors: Cha-Xiang Guan or Yong Zhou. Department of Physiology, Xiangya Medical School, Central South University, Changsha, Hunan 410078, China. Tel.: +86-731-82355051; Fax: +86-731-82355056; E-mail: guanchaxiangedu.cn or zhouyong421edu.cn
Corresponding authors: Cha-Xiang Guan or Yong Zhou. Department of Physiology, Xiangya Medical School, Central South University, Changsha, Hunan 410078, China. Tel.: +86-731-82355051; Fax: +86-731-82355056; E-mail: guanchaxiangedu.cn or zhouyong421edu.cn