Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
CSC hierarchies
Quiescent subpopulations
Plasticity of the hierarchy and...
Epigenetics role in CSCs
The impact of the tumor...
Therapeutic approach to...
Barriers to targeting CSCs
Conclusion
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2020; 10(7):3083-3098. doi:10.7150/thno.41647 This issue Cite
Review
An evolving paradigm of cancer stem cell hierarchies: therapeutic implications
Alexander J Cole1, Adetunji P Fayomi1, Vivian I Anyaeche2, Shoumei Bai1, Ronald J Buckanovich3 ![]()
1. Department of Internal Medicine and Magee-Womens Research Institute, University of Pittsburgh, Pittsburgh, PA, USA.
2. University of Pittsburgh, School of Medicine, Pittsburgh, PA, USA.
3. Division of Gynecologic Oncology, Department of Obstetrics and Gynecology, Hillman Cancer Center, University of Pittsburgh, Pittsburgh, PA, USA
Received 2019-10-30; Accepted 2020-1-22; Published 2020-2-10
Abstract

Over a decade of research has confirmed the critical role of cancer stem-like cells (CSCs) in tumor initiation, chemoresistance, and metastasis. Increasingly, CSC hierarchies have begun to be defined with some recurring themes. This includes evidence that these hierarchies are 'flexible,' with both cell state transitions and dedifferentiation events possible. These findings pose therapeutic hurdles and opportunities. Here, we review cancer stem cell hierarchies and their interactions with the tumor microenvironment. We also discuss the current therapeutic approaches designed to target CSC hierarchies and initial clinical trial results for CSC targeting agents. While cancer stem cell targeted therapies are still in their infancy, we are beginning to see encouraging results that suggest a positive outlook for CSC-targeting approaches.
Introduction
The concept of adult stem cells initially evolved from landmark studies in the 50's and 60's which demonstrated the ability of transplanted bone marrow cells to rescue irradiated mice by restoring normal blood pathology (1,2). These cells were later termed hematopoietic stem cells (HSCs) and demonstrated to exist in an undifferentiated quiescent state at the peak of a differentiation hierarchy. When stimulated to proliferate, HSCs were shown to yield two distinct cells; one non-dividing (quiescent) stem cell and one actively dividing cell. This phenomenon was termed “asymmetric division”. The proliferating daughter cell was shown to continue to divide and proceed down the hematopoietic hierarchy, from stem cell to progenitor cell, before becoming a fully differentiated mature blood cell. Thus, stem cells, since, have been defined by their ability to self-renew and give rise to a well-differentiated progeny (3). Since these initial studies, multiple types of stem cells have been identified in a wide range of tissue sharing the multipotency characteristics of HSCs.
The first studies suggesting cancer cells may share similar stem cell properties to HSCs were conducted in teratomas, where it was demonstrated that undifferentiated cells preferably gave rise to non-tumorigenic differentiated cells (4). This led researchers to propose the first cancer stem cell hypothesis, that tumors comprise a mixture of malignant stem cells and their benign progeny (5). Shorty following this, a population of leukemia stem cells, which could initiate leukemia in mice, was identified (6). CSCs, defined as cells which can undergo asymmetric division and initiate tumors in mice, have now been identified in a wide variety of tumor types, including melanoma, osteosarcoma, leukemia, breast, colorectal, brain, prostate, pancreatic, ovarian, liver and lung (7). In some cancers, it has not been possible to distinguish CSCs from non-CSCs (8). Such tumors may have a very shallow hierarchy, or a differentiation block at the level of the CSC (8).
In addition to the ability to self-renew and differentiate, CSCs share a number of unique features which set them apart from bulk tumor cells. Epithelial CSCs express many genes/pathways typically associated with normal stem cells, such as SOX2 (9), NANOG (10), OCT3/4 (11), and the WNT/ß-Catenin (12) and Hedgehog pathways (13). In many tumor types, CSCs, or a subset of CSCs, take on an epithelial-to-mesenchymal transition (EMT) profile through the upregulation of genes such as TWIST, SNAIL, and ZEB (14,15). It is therefore unsurprising that CSCs have been demonstrated to drive metastasis in a number of cancer types (16,17). One of the more controversial features of CSCs is innate chemoresistance. While innate chemoresistance is not required to define a CSC, innate therapy resistance has been commonly linked to CSCs. This resistance has been attributed to the ability to become quiescent (18), upregulation of enzymes (such as ALDH) and multidrug resistance pumps to increase chemotherapy elimination from the cell (19), and the upregulation of anti-apoptotic proteins (20). Given their link with tumor initiation and drug resistance, they have been pushed to the forefront of cancer therapy.
The identification of CSCs is based on expression of a variety of cell surface makers, enzyme activity, transcription factors, and efflux pumps. Some are tissue specific, while others relate to pathways known to be essential for the function of normal stem cells. For a summary of these markers, we refer the reader to the review article (21). Here, we will focus our review on the differentiation capacities of CSC populations.
CSC hierarchies
The CSC hypothesis postulates that many heterogenic cancers are organized into hierarchal structures based on differentiation capacity, similarly to HSC organization. The top tier of these CSC hierarchies generally contains the most stem-like cells, capable of self-renewal and differentiation into the less stem-like cells which comprise the lower tiers of the hierarchy (Figure 1). These apex CSCs typically have prodigious tumor initiation capacity and are responsible for driving tumor heterogeneity and composition of the bulk tumor mass and facilitating tumor growth, drug resistance, cancer recurrence, and metastasis. The differentiation hierarchy model of cancer is dependent on the gain and loss of the various markers used to identify the specific CSC populations. As noted above, tissue-specific gene expression results in a wide variety of CSC markers being identified for many different types of cancer and multiple markers for the same cancer type. Consequently, the literature reflects a conglomerate of CSC types, markers, and models. Recently, studies have begun to identify and characterize CSC hierarchies in several cancer types, including ovarian (22), colon (23), breast (24), and brain cancers (25). With the advances in lineage tracing and single-cell sequencing technologies, it is likely CSC hierarchies will be defined in other tumor types. We believe that understanding these hierarchies will identify critical therapeutic targets to improve patient outcomes. Below, we will highlight some defined CSC hierarchies and the role of quiescent CSCs within the hierarchy. We will also discuss dedifferentiation and epigenetic alterations as a source of 'stemness' and the impact of the tumor microenvironment (TME) on CSCs.
Examples of pyramid and linear CSC hierarchies. Unbroken arrows represent differentiation events. Broken arrows represent dedifferentiation events. Two-way arrows represent transitional differentiation events.
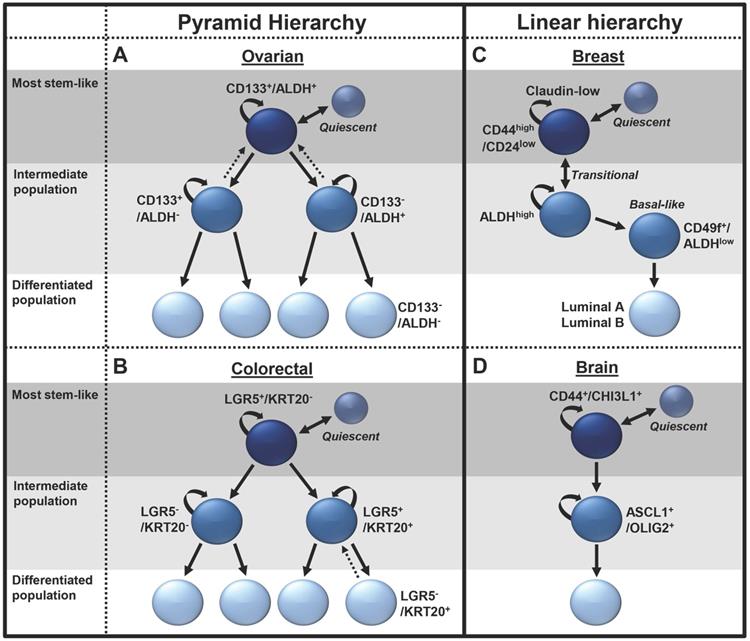
Example stem cell hierarchies
Ovarian cancer: Using the previously identified ovarian CSC markers ALDH and CD133 (26,27), Choi, et al., (2015) defined an ovarian cancer CSC differentiation hierarchy (Figure 1A) (22). This study used single cell-lineage tracing of cell lines and primary human ovarian cancer samples, defining a branched differentiation hierarchy with at least four distinct ovarian cancer cell populations. ALDH+/CD133+ cells sit at the apex of the hierarchy and symmetrically divide to expand CSC numbers in a process enhanced by bone morphogenetic protein 2 (BMP2) secretion from lower-tier cells (21). Alternatively, ALDH+/CD133+ cells can asymmetrically divide to self-renew and produce either ALDH+/CD133- or ALDH-/CD133+ cells (22). These ALDH+/CD133- and ALDH-/CD133+ cells comprise an intermediate CSC population, possessing enhanced tumorigenic potential over the bulk cells, but are less stem-like than the ALDH+/CD133+ cells. ALDH+/CD133- and ALDH-/CD133+ also have the capacity for symmetric division, to expand, or asymmetric division, producing ALDH-/CD133- cells. Interestingly, the ALDH+/CD133- and ALDH-/CD133+ cells each have distinct features. ALDH+ cells are known to preferentially grow in suspension/spheroids and to demonstrate platinum resistance, with ALDH expression regulated by β-catenin (26). In contrast, CD133+ cells are slower growing (22,28) and appear more radiation resistant (29). The bottom tier of the stem cell hierarchy comprise ALDH-/CD133- cells, which make up the majority of the ovarian cancer mass and are generally unable to initiate tumors (26). The lack of stemness in ALDH-/CD133- cells prevent their growth as spheroids and make them sensitive to chemotherapeutics (22,26). One study suggested CD133- cells could initiate tumors. However, this study, like many, is difficult to interpret, as large numbers of cells were used; thus, FACs contamination is problematic (30). However, single-cell studies did observe rare dedifferentiation events wherein ALDH-/CD133- cells could gain expression of CSC markers (22).
Support for this hierarchy also comes from ovarian cancer cell-of-origin studies in mice, where a lineage-tracing study suggested LGR5+, ALDH+/CD133+ ovarian surface-repopulating stem cells were found in the oviductal hila of mice. When mutated, these cells could serve as CSCs (31). This ovarian CSC hierarchy is an example of a classic pyramid of differentiation.
Colorectal cancer: Like ovarian cancer, colorectal cancer has a branched CSC differentiation hierarchy (Figure 1B). This hierarchy recapitulates normal stem cell organization in the colon (23,32,33). Using lineage-tracing in organoids Shimokawa et al., (2017) defined a colorectal CSC hierarchy based on the expression of the CSC marker LGR5 and the differentiation marker KRT20. In this hierarchy, the apex cells LGR5+/KRT20- can self-renew or differentiate into LGR5+/KRT20+ or LGR5-/KRT20+ cells (23). At the bottom of the hierarchy, the LGR5-/KRT20+ exist as terminally differentiated, non-proliferative cells. Similar to the findings in ovarian cancer, in rare incidences, LGR5-/KRT20+ cells could revert into LGR5+/KRT20+, form colonies and proliferate. Interestingly, transplantation of the colorectal organoids in vivo resulted in a recapitulation of the original cancer tissue histology, with the stem-like LGR5 cells localized to the outermost regions of the tumors, surrounded by α-smooth muscle actin-positive fibroblasts and KRT20+ differentiated cells localized to the inner regions (32,33).
Breast cancer: The breast CSC hierarchy has been largely inferred from the well-defined normal breast stem cell hierarchy, with the apex cell proposed as a source of claudin-low/triple-negative breast cancers, bipotent and luminal progenitors driving basal like tumors, and ductal progenitors leading to ER+ luminal A/B tumors (34). While the normal breast stem cell hierarchy is clearly branched, it is not clear that all branches are associated with breast cancer, thus we propose a more linear CSC hierarchy. The top tier of the breast CSC hierarchy is composed of CD24low/CD44high EMT/mesenchymal-like cells, which differentiate into an intermediate stem cell population made up of ALDHhigh epithelial-like cells (Figure 1C) (24). The cells in the mesenchymal state are slower growing and localized to the invasive front of the tumor, while the epithelial-like cells are more rapidly growing and typically located in the center of the tumor mass (24). Making it distinct from the other branched models, the breast CSC model has significantly more plasticity, allowing rapid cell state transitions between the CD24low/CD44high and ALDHhigh stem cell populations. Recent evidence has also suggested a second intermediate breast basal-like CSC population comprising ALDHlow/CD49f+ cells, which are less stem-like than ALDHhigh cells (35). The lowest tier of the hierarchy comprises luminal A/B type cells, which are the most well differentiated and the least stem-like. The enhanced plasticity of this hierarchy and the ability of these cells to transition between the mesenchymal and epithelial states may help them respond to environmental stress. Indeed, the phenotypes for these states may also be more plastic than initially realized, as more recent research has contradicted the roles of these cells, suggesting the CD24low/CD44high population to be the most proliferative and tumorigenic, while the ALDH cells were identified to be more migratory and to promote tumor metastasis (36).
Brain cancer: Recent advances in single-cell RNA sequencing have overcome the technical barriers for lineage tracing and rendered higher resolution of cancer biology possible (37). Patel, et al., (2014) sequenced 430 cells from five primary glioblastomas. Analysis confirmed subtype classification and revealed significant tumor heterogeneity (38). This study further identified a population of CD133+ quiescent glioblastoma stem cells that were enriched for hypoxia signatures (38). Recently, Wang, et al., (2019) further delineated proliferating glioblastoma CSCs into a linear differentiation hierarchy (Figure 1D). In this model, parallel to the breast cancer model, a mesenchymal CSC population, expressing the glycoprotein markers CD44 and CHI3L1, was the most stem like. These cells were demonstrated to differentiate into pro-neural cells expressing the transcription factors ASCL1 and OLIG2. These intermediate pro-neural progenitors in turn differentiated into a bulk cell population (39).
Quiescent subpopulations
Early CSC studies focused on tumor initiation capacity in animals, and thus by design typically identified rapidly growing CSC populations. However, recent work is beginning to show a critical role for quiescent CSCs in cancer biology and, specifically, in therapeutic resistance. Quiescence describes a reversible state of cellular inactivity, in which a cell has exited the cell cycle into the G0 phase, where it will remain until reentering the cell cycle in response to physiological cell stimuli. Quiescent cells are typically resistant to chemotherapies that target proliferative cells.
Quiescent CSCs likely make up a sub-population of CSCs with enhanced resilience to environmental stresses (18,40). Quiescent CSCs, defined by various markers, have been reported in breast (24), liver (41), melanoma (42), ovarian (40), colon (43), and brain CSCs (38). Importantly, numerous studies across cancer types demonstrate quiescent CSC contribute to chemotherapy resistance and tumor recurrence (44). For example, studies in pancreatic cancer identified a slow-cycling population, enriched for the CSC markers CD24+/CD44+, CD133+ and ALDH, which also had enhanced chemotherapeutic resistance and could recreate the initial heterogeneous tumor cell population (18). Several studies link expression of SOX2 with quiescent CSCs (45,46). We investigated the role of the nuclear factor of activated T-cells (NFAT) family, transcription factors known to regulate quiescence in normal stem cells (28), as regulators of quiescent ovarian CSCs. We found that NFATC4 is enriched in slow proliferating ovarian CSCs and is increased in response to cisplatin therapy, while overexpression of NFATC4 was shown to cause a marked decrease in proliferation and cell size, G0 cell cycle arrest, and chemotherapy resistance (28). Understanding quiescent CSCs thus offers an important opportunity to overcome therapeutic resistance to prevent disease recurrence. Alternatively, the ability to force a quiescent state in residual cancer cells post-therapy would also be of interest to prolong patient progression-free survival. Consistent with this, Chesnokov, et al., (2019) demonstrated that MEK inhibitors induce a G0/G1 cellular arrest with induction of stemness genes SOX2, NANOG, OCT4, and ALDH1A homologs (47).
Plasticity of the hierarchy and dedifferentiation as a source of stemness
Consistent with findings in normal tissues, such as the lung, where cells can undergo lineage switching/dedifferentiation in the face of injury (48), CSC hierarchies are more flexible than originally hypothesized, with significant cellular plasticity. Since initial studies identifying dedifferentiation events of bulk breast cancer cells to breast CSCs both in vitro and in vivo, CSC hierarchies have evolved to incorporate de novo generation of CSCs from what was once believed to be terminally differentiated cells (49). This bidirectional interconversion of non-CSCs to CSCs gives hierarchies the ability to respond to cellular stresses by alternating their differentiation from a mesenchymal to an epithelial phenotype, or de-differentiating up the hierarchy from non-stem-like to stem-like cells to recover an ablated population (23,24). This plasticity poses a therapeutic challenge, but also indicates the critical importance of the stemness. In addition, it offers a potential therapeutic opportunity, as blocking the induction of a state of stemness could represent a means to restrict cancer growth.
The phenomenon of CSC dedifferentiation has been explored in many different tissue models including breast (50), lung (51), melanoma (52), ovarian (22), glioma (53), pancreas (54), and colon (55). However, dedifferentiation studies are a challenge, as experiments using bulk cells often cannot rule out trace contamination of hard-to-detect or previously undefined CSC pools. As such, dedifferentiation studies need to be viewed under a critical lens, with single-cell studies representing the ideal.
Various general mechanisms of dedifferentiation appear to be emerging. One mechanism is EMT driven, where TGF-β or other factors activate the EMT-associated transcription factors (TWIST, SNAIL/SLUG, or ZEB) to assume a stem-like state (50,55-57). However, this may be more a state transition than a true dedifferentiation phenomenon. An alternative mechanism is via induction of core stem cell, 'Yamanaka,' transcription factors, essential for the reprogramming of somatic cells into induced pluripotent stem cells. These factors include; OCT, SOX, MYC and KLF family members, in addition to NANOG and Lin-28. In melanoma, lung, pancreatic, and colon cancers, OCT4 was demonstrated to promote dedifferentiation of bulk cells into CSC (51,52,54,58), while upregulation of NANOG, SOX2, Klf and Lin-28B have also been implicated in this process (51,54,58). Studies in iPS cells suggest that the EMT transcription factors may prime cells for Yamanaka factor induced cellular reprogramming (59). Similar mechanisms may take place in cancer cells (60).
The capacity of CSCs to dedifferentiate under stressful conditions, such as hypoxia, radiation, or stem cell ablation, has also been widely reported (23,61,62), and seems to favor the Yamanaka transcription factor pathways (62-64). In contrast, dedifferentiation events caused by the treatment of bulk cancer cells with chemotherapy seem to utilize not only the SOX2 and OCT3/4 pathways (65), but also stemness genes and CSC markers such as notch and ALDH1 (66). Interestingly, the reactivation of many of these stemness genes have been associated with epigenetic changes such as promoter hypomethylation, and will be discussed below. Various other pathways have also been implicated in dedifferentiation, including; cell cycle activators and developmental genes; however, these mechanism seem to be less common (53).
A new source of dedifferentiation comes from the formation of polyploid giant cancer cells (PGCCs). These cells express the stemness genes OCT4, NANOG and SOX2/4 (67) and have been demonstrated to asymmetrically differentiate to produce cells with increased tumor initiation, immunosuppressive properties, decreased sensitivity to chemotherapeutics and enhanced stemness (68,69). In a study by Zhang, et al., (2014), tumors formed from PGCCs were shown to possess a mesenchymal phenotype and have elevated expression of the CSC markers CD44 and CD133 (69). Interestingly, PGCCs tend to cycle slowly and are thus a potentially important source of quiescent CSC (69).
Epigenetics role in CSCs
Epigenetics is a broad term used to encompass a wide range of mechanisms capable of altering gene expression without altering the DNA sequence. These mechanisms include DNA methylation, histone modifications, noncoding RNAs, and chromatin remodeling. Epigenetics plays a pivotal role in the normal function of embryonic and adult stem cells, controlling their ability to differentiate, self-renew, and maintain pluripotency (70,71). CSCs also rely on a range of epigenetic modulators to maintain and promote their stemness programming. Numerous studies have suggested CSCs possess altered epigenetic landscapes compared with bulk tumor cells (72-74). The CSC marker CD133 has been shown to be hyper-methylated in bulk cells compared to the CSC population (73,74), while CD133 and CD44 were shown to be hypomethylated and subsequently overexpressed in triple-negative breast cancer compared to non-triple-negative (72). In addition to direct effects on CSC markers, epigenetics has been shown to regulate stemness pathways, such as Wnt/β-catenin (75), hedgehog, notch and TGF-β. Recent work by Wang, et al., (2018) demonstrated that the imprint gene ASCL2 is required to maintain Wnt activation in CSCs. In CSCs, ASCL2 is epigenetically regulated by the histone methyltransferase SMYD3, which regulates H3K4me3 status at the ASCL2 locus, promoting it's expression (76). The non-coding RNA lncTCF7 has also been shown to promote Wnt signaling via recruitment of the chromatin-remodeling complex SWI/SNF (77). Hedgehog signaling is another target for epigenetic regulation by CSC. A recent study by Ooki, et al., (2018) demonstrated that in bulk lung cancer populations the promotor of the transcription factor PAX6 was methylated, resulting in its repression, while in CSCs PAX6 was not methylated and promoted transcription of the hedgehog regulator GLI, resulting in an upregulation of SOX2, OCT4, and NANOG, driving cancer cells toward a stem-like state (78). Jin, et al., (2017) confirmed the ability of CSCs to epigenetically regulate notch, demonstrating CSC-specific upregulation of STRAP, which disrupted polycomb repressor complex 2 assembly, preventing the silencing of notch and promoting a stem-like phenotype (79). Several studies have also demonstrated the ability of microRNAs to promote stemness. miR-200c and miR-205, responsible for silencing ZEB1, can be epigenetically silenced, resulting in an upregulation of EMT and CSC phenotypes (80,81).
The impact of the tumor micro-environment on stemness
Over the past decade, cancer research has shifted away from solely targeting the bulk tumor mass, to focusing on the TME. Cells of the TME have been demonstrated to be vital for the regulation of CSC hierarchies. We will focus here on the roles of mesenchymal stem cells (MSCs), cancer-associated fibroblasts (CAFs), tumor associated macrophages (TAMs), suppressive regulatory T cells (Tregs) and vascular cells in promoting cancer stemness (Figure 2).
Tumor microenvironment factors which regulate the CSC hierarchy expansion.
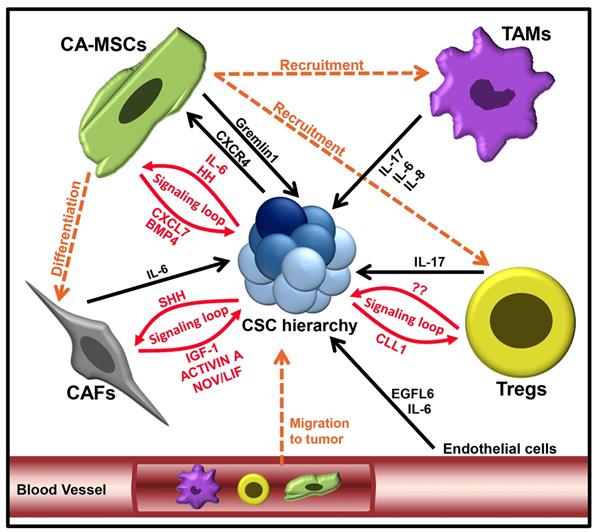
Mesenchymal stem cells are multipotent cells, able to self-renew and differentiate into various cell types, including adipocytes, chondrocytes and osteoblasts (82). In various cancer types, carcinoma associated MSCs (CA-MSCs) have been shown to promote tumor 'stemness', increasing tumor growth, chemotherapy resistance, and metastasis (83,84). One important theme appears to be the establishment of signaling loops between CA-MSCs and CSCs. In ovarian cancer, a BMP4:Hedgehog CA-MSC:CSC signaling loop was found to increase stemness and drive therapeutic resistance (84-86). In breast cancer, an MSC:CSC CXCL7:IL-6 signaling loop increased breast CSC populations, resulting in an overall increase in tumor growth (87). CA-MSCs were also shown to inhibit FOXP2 in breast CSCs, promoting tumor initiation and metastasis (88). Breast CA-MSCs have also been demonstrated to promote breast cancer quiescence and drug resistance (89). In colorectal cancer, CA-MSCs have been shown to promote the dedifferentiation of LGR5- cells through production of Gremlin1 (90). Numerus other studies support the critical link of CA-MSCs and CSCs; for a deeper examination of this area we refer readers to the review (91).
CA-MSCs can also impact CSCs indirectly via the generation of CAFs (92). CAFs are the most abundant cells in the TME and are responsible for regulating the biology of tumor cells through direct contact, paracrine signaling and extra-cellular matrix remodeling (93). A number of studies have demonstrated that CAFs can function similarly to CA-MSCs, via various secreted factors, to enhance CSC expansion and EMT, to promote tumor progression (33,94). For example, CAFs can secrete interleukin (IL)-6, which has been found to activate notch signaling in hepatocellular carcinoma cells, promoting CSC properties (95). Much like those observed for CA-MSCs, CAF:CSC interactions commonly appear to be reciprocal. In a study by Valenti, et al., (2017) CSCs were demonstrated to activate hedgehog signaling in CAFS, while CAFs responded by secreting factors that promoted expansion of CSCs (96). While most studies indicate that CAFs increase tumor stemness, some have suggested that CAF subsets could negatively regulate CSC populations (97). This may, in part, be related to the source of MSCs; although the majority of CAFs are probably derived from the tumor stroma, multiple studies have reported that a substantial number of tumor CAFs are derived from bone marrow MSCs (92,98).
Components of the immune system also have a direct influence on CSCs. Tumor-associated macrophages (TAMs) are reported to secrete many factors that impact CSCs (99,100). One critical factor produced by TAMs is IL-6. Like CAFs, TAMs can make high levels of IL-6 which has been shown to promote breast CSC self-renewal (101) and to enhance the CD44+ population in hepatoma cancer cells (102), and the CD44+/ALDH1+ populations in pancreatic CSCs (103). In addition to IL-6, TAMs have been demonstrated to secrete IL-8 and IL-17, which increase expansion of ALDH+ breast CSCs (104) and self-renewal of ovarian CD133+ CSCs (105), respectively. Furthermore, IL-8 has been shown enhance CSC self-renewal, sphere formation, migration and expression of stemness-related genes (106). In addition to paracrine signaling, TAMs can also influence CSCs via juxtacrine signaling (107).
Suppressive regulatory T cells, comprising T-regulatory cells (Tregs) and Th17 CD4+ T cells, are known to help cancer evade anti-tumor immunity. Evidence exists that CSCs both recruit and activate these regulatory cells, facilitating crosstalk to enhance CSC stemness and promote their growth. Yang, et al., (2011) demonstrated that Foxp3+ Tregs were capable of promoting colorectal cancer stemness through the secretion of IL-17 (108). Xu, et al., (2018), demonstrated a Treg-CSC signaling loop, whereby breast CSCs secreted the chemokine CCL1, which resulted in the recruitment of Treg cells to the tumor (109). Treg cells were then demonstrated to increased ALDH activity, SOX2 expression and sphere formation in breast cancer cells via paracrine signaling; however, the signaling factors involved were not investigated. Co-engraftment of breast cancer and Tregs resulted in enhanced tumorigenesis, metastasis, and chemoresistance, confirming the supportive role of Tregs in breast cancer. Interestingly, it has also been demonstrated that CSCs can signal MSCs via CXCR4 signaling to induce Treg maturation (110).
Finally, the vascular niche and 'angiokines' can also regulate CSCs. Vascular endothelial growth factor (VEGF) is an important regulator of both vascular cells and CSCs (111). Endothelial cells impact CSCs via secretion of IL-6, promoting their migration (112). Similarly, a vascular factor, EGFL6, has been shown promote CSC asymmetric division, migration, and metastasis (113). For a more in-depth review of the vascular niche, we refer readers to the following review (114).
Therapeutic approach to targeting CSCs
Metabolism targeting therapies
ALDH1 inhibitors: Aldehyde dehydrogenase, primarily the ALDH1A family members, are among the best-supported CSC markers. Multiple preclinical studies indicate that ALDH1A1 or ALDH1A3 knockdown increases chemosensitivity/reverses chemotherapy resistance in ovarian, breast, and lung cancers and melanoma (115-119). Animal studies have confirmed the anti-cancer activity of broad spectrum ALDH inhibitors such as disulfiram and DEAB (120,121). Given the success of these broad-spectrum inhibitors, there has been an attempt to develop more selective ALDH1A family inhibitors. We have developed a pan ALDH1A family inhibitor, 673A, with the advantage being active regardless of the ALDH1A family member expressed. 673A preferentially depletes CD133+ ovarian CSCs, inhibits ovarian tumor initiation in vitro and in vivo, synergizes with chemotherapy against both breast and ovarian cancer cells, and increased tumor eradication in chemotherapy-resistant human-patient-derived xenograft models (29). A selective and potent ALDH1A1 inhibitor, CM37, was found to effectively increase cancer cell reactive oxygen species and DNA damage (122). Similar quinoline-based ALDH1A1 inhibitors showed efficacy in chemosensitization of ALDH1A1-expressing cancer cell lines (123). Newer, more clinically applicable ALDH inhibitors are being developed (124). Indeed, a novel cytotoxic ALDH-targeting pro-drug has shown significant activity in melanoma (125).
While none of these newer compounds have entered clinical trials, several older ALDH inhibitors have been tested in cancer. A phase IIb trial in patients with advanced lung cancer demonstrated that the addition of disulfiram to chemotherapy improved overall survival with ~10% of patients being disease-free at 3 years (126). Similarly, in a small study of high-risk breast cancer patients receiving adjuvant chemotherapy and sodium diethyl dithiocarbamic acid (the primary active metabolite of disulfiram), there was a trend toward increased overall and disease-free survival (127). However, combinatorial studies of disulfiram+copper and temozolomide in temozolomide-resistant glioblastoma showed limited activity (128-130). It is important to note that disulfiram, which was developed as and ALDH2 inhibitor, while active against ALDH1A1, has limited activity against. ALDH1A3, which is prevalent in many cancers. Thus, newer-generation ALDH inhibitors could significantly improve on the encouraging results of disulfiram in lung cancer.
Metformin: Metformin was first shown to target CSCs in breast cancer (131), with subsequent studies demonstrating its ability to target CSCs in many cancers (132-134). Currently, there are at least 14 trials, ongoing or concluded, evaluating the efficacy of metformin in cancer. Initial reports of metformin in pancreatic cancer were disappointing, as they demonstrated no impact on outcomes (135). Similarly, the addition of metformin to chemotherapy for HER2(-) metastatic breast cancer had no impact on progression-free survival (PFS) with overall survival (OS) not reported (136). However, more recent studies are more encouraging. We have completed a phase II study of metformin administered in combination with chemotherapy for non-diabetic patients with advanced stage epithelial ovarian cancer. This study found metformin was associated with a 2.5-fold reduction in ovarian CSCs, and while non-randomized, was associated with a surprisingly long median OS of 57.9 months (137). Significantly, a randomized phase II trial of metformin in combination with an epidermal growth factor receptor-tyrosine kinase inhibitor demonstrated a statistically significant improvement in both PFS and OS, with an impressive 14-month improvement in OS of lung cancer (138).
Antibody therapies targeting CSC surface molecules
CD44: Expressed by many tumors, CD44 is a transmembrane glycoprotein and one of the most researched cell surface targets for CSC therapies (139). CD44 targeting therapies have shown efficacy in preclinical studies (140-142). CD44 antibody-nanoparticle conjugates selectively kill CSCs in head and neck squamous cell carcinoma (140). Anti-CD44 antibody has been shown to facilitate cellular uptake of doxorubicin, inducing chemo-sensitization (143). The first-in-human phase I clinical trial of an anti-CD44 monoclonal antibody (RG7356) for CD44-expressing local advanced or metastatic tumors was recently completed. This trial exhibited safety and efficacy and demonstrated some single-agent activity (144). Dose escalating of this antibody, in combination with various chemotherapies is ongoing. Other anti-CD44 therapies tested in ongoing or completed clinical trials include AMC303 (for solid tumors) and SPL108 (for ovarian epithelial cancer). A number of alternative strategies to target CD44 are being developed; among the most promising is the use of CD44 short binding peptides coupled to toxins, which have been shown to have 4-10 times stronger affinity to CD44 than do antibodies (141,145).
The extracellular domain of CD44 is susceptible to alternative splicing, resulting in the generation of multiple CD44 variant isoforms (CD44v). The expression of splice variant CD44v6 has been demonstrated to correlate with tumor progression and has been shown to be enriched in CSC populations (146). Targeting CD44v6 has advantages as CD44 is expressed by most cells; however, CD44v6 is only expressed in subpopulations of hematopoietic and epithelial cells. Consequently, efforts have been made to design and test CD44v6 target antibody therapies. However, an anti-CD44v6 antibody was tested in clinical trial but was discontinued due to significant skin toxicity (147,148). Another CD44v6 antibody, RO5429083, is undergoing dose escalating trials alone and in combination with various chemotherapies (149).
CD24: The sialoglycoprotein CD24 is expressed on the surface of cells, constitutes a prevalent CSC marker, has roles in cell signaling and has recently been demonstrated to promote tumor immune evasion (150). Preclinical studies with anti-CD24 antibody treatment has impeded tumor growth in hepatocellular carcinoma (151), colorectal and pancreatic adenocarcinoma (152), and reduced CSC populations (153). CD24 has been shown to be a novel 'don't eat me' signal protein, most abundantly expressed in metastatic ovarian cancer and triple-negative breast cancer. CD24 allows tumor cells to evade phagocytosis, by recognizing Siglec-10 on TAMs (150). Genetic ablation of CD24 in tumor cells, or antibodies targeting CD24, or Siglec-10, induces phagocytosis and obrogation of tumor growth. Anti-CD24 antibodies are in preclinical studies of various cancer types (154-156).
CD133 (prominin-1): The pentaspan transmembrane glycoprotein CD133is involved in WNT/β-catenin signaling and is capable of regulating cell differentiation (157,158). Currently there exist a number of CD133-targeting antibodies, such as CD133KDEL which consists of an anti-CD133 single-chain variable fragment (scFv) coupled to pseudomonas exotoxin A (PE38) (159-161). However, most of these agents are still at the preclinical stage. A recent phase I clinical trial by Wang, et al., (2018), treated 23 patients with advanced CD133-positive tumors, using autologous chimeric antigen receptor-modified T-cells (CART) expressing anti-CD133 scFv. Trial results demonstrated ablation of CD133-positive cells in all patients and an increase in disease stability without new metastasis occurring. Toxicities were manageable (162).
Stemness pathway targeted therapeutics
Focal adhesion kinase (FAK) inhibitors: Regulated by OCT-3/4 and NANOG (163,164), FAK plays an important role in CSC self-renewal and tumor progression (165). There are more than 40 clinical trials evaluating the clinical safety or efficacy of FAK inhibitors. Seven studies have demonstrated drug tolerability, but efficacy data are limited. Reports from a phase I trial evaluating the safety of FAK inhibitor PF-04554878 when used in combination with pembrolizumab or gemcitabine in patients with pancreatic ductal adenocarcinoma demonstrated a targeted decrease in FAK phosphorylation in T cells; however, no partial or complete response was observed (166). A phase II trial of PF-04554878 in Merlin-stratified pleural mesothelioma patients after first-line chemotherapy (167) (NCT01870609) did not show an impact on PFS,OS, or quality of life.
Wnt/β-catenin inhibitors: Numerous clinical trials have been initiated to evaluate the safety and/or efficacy of various molecules targeting the Wnt/β-catenin pathway in cancer cells. In general, these drugs have shown limited activity as single agents (168). However, in combination with chemotherapy and other compounds, significant response rates have been reported. A phase Ib study of the Wnt inhibitor ipafricept in combination with nab-paclitaxel/gemcitabine in pancreatic cancer resulted a 34.6% partial response rate, 46.2% having stable disease, for an impressive clinical benefit rate of 80.8% (169). Unfortunately, this study was terminated by the sponsor before completion, possibly related to adverse side effects, though the trial data suggest clinical efficacy for Wnt inhibition.
Notch inhibitors: Monoclonal antibodies that can alter notch ligand-receptor binding and gamma-secretase inhibitors, which can block downstream signaling, are both in clinical development. Presently, there are more than 100 gamma-secretase inhibitors (170), and almost 50 clinical trials have been initiated to evaluate their clinical safety and efficacy. Early trial results indicate that the inhibitors are generally safe but associated with dose-limiting toxicities, predominantly of the gastrointestinal tract (171-173). There are conflicting results on the efficacy of notch inhibitors for cancer treatment, either as a single agent or in combination with other agents. While some studies have reported clinical benefits of notch inhibition, including stability of glioblastoma multiforme or glioma in patients (174), partial response and disease stabilization in patients with pancreatic ductal carcinoma (173), and tumor necrosis and shrinkage in patients with leiomyosarcoma and breast cancer (172), other studies have reported weight loss, higher incidence of skin cancer, deteriorating cognitive ability, and unimpressive clinical outcomes (175).
Hedgehog pathway inhibitors (HHi): As discussed above, the hedgehog signaling pathway is essential for maintaining a stem-like state and is therefore exploited by CSC via epigenetic regulation and microenvironment activation. Indeed, a combination of the HHi Daurismo plus low-dose HDAC inhibitor cytarabine vs. cytarabine alone resulted in a doubling of overall survival in elderly patients with acute myeloid leukemia, resulting in FDA approval (176). While HHi have shown significant efficacy in tumors with HHi-driver mutations, including basal cell carcinoma (177) and medulloblastoma (178), the impact as a CSC-modulating drug in HHi wildtype tumors is controversial. There are currently more than 70 phase I-IV clinical trials involving inhibitors of the Hedgehog signaling pathway aimed at eradicating bulk tumors and CSC populations (176). Over 80% of these trials use the inhibitors GDC-449 or LDE225, which bind to and inhibit Smoothened (SMO) activation, preventing downstream hedgehog signaling. Results from early trials have been underwhelming; although most studies show that the HHi are tolerated and significantly inhibit hedgehog signaling, many report no or little effect on CSC populations or improvement in patient outcomes (179-182). Currently, there is a phase III trial (NCT03416179) actively recruiting for Daurismo in combination with intensive or non-intensive chemotherapy.
IL-6/JAK/STAT: Of the secreted TME factors, IL-6 signaling via the JAK/STAT pathway has been a focus. Numerous preclinical studies have demonstrated the ability of IL-6 antibodies to inhibit CSC growth and sensitize them to chemotherapeutics (183,184). An anti-IL-6 receptor antibody has been clinically approved and used for the treatment of rheumatoid arthritis. This antibody is now in multiple clinical trials targeting cancer. Similarly, ruxolitinib, an inhibitor of JAK2 that has been approved for treatment of myeloproliferative neoplasms, is also in clinical trials for the study of several solid tumors. At least one trial, NRG007, has specific translational endpoints, studying CSCs as a target. In a phase I clinical trial non-small cell lung cancer (NSCLC) patients treated with the STAT3 inhibitor OPB-51602 where shown to have a better response to therapy (185). However, a more recent phase I trial of a novel STAT3 inhibitor in patients with advanced hepatocellular carcinoma was less promising (186).
IL-8: is an important cytokine secreted by the TME and bulk tumor cells to promote CSC expansion and stemness. Like IL-6, IL-8 is a target gene of STAT/JAK signaling. Recent studies have shown IL-8-neutralizing antibody or inhibition of the IL-8 receptors CXCR1/2 with the antagonist reparixin, abolishes breast CSCs following chemotherapy withdrawal (187). Currently, an IL-8 antibody is in phase I clinical trials for advanced solid tumors (188). In breast cancer, reparixin was safely combined with paclitaxel, with a 30% response rate and acceptable toxicities (189). Reparixin is currently in phase II clinical trial for breast cancer (NCT02370238).
CXCL12: is a chemokine secreted by CAFs which binds to the receptor CXCR4, expressed on CSCs, to promote metastasis (190). Consequently, several CXCR4 antagonists are now in clinical trial. A phase I trial of the CXCR4 peptide antagonist LY2510924 demonstrated a 20% stable disease rate in patients with advanced cancer. The most promising CXCR4 antagonist is BL-8040, which was recently granted orphan drug status by the FDA. BL-8040 is currently in five phase II trials for multiple cancer types. Early results have suggested BL-8040, in combination with pembrolizumab, is safe and shows a promising OS rate in metastatic melanoma, NSCLC, and bladder cancer (191).
CAF targeted therapeutics
Fibroblast activation protein alpha (FAPα): FAPα is expressed in the CAFs of ~90% of all carcinomas. Substantial efforts have been made to target CAFs using FAPα (192-194), with disappointing single agent results (195). More recent trials have used FAPα inhibitors in combination with other agents; however, these, too, have shown limited clinical efficacy (196,197). Currently, there are three ongoing clinical trials (NCT03875079, NCT03386721, NCT02627274) evaluating the efficacy of the novel bispecific FAP-DR5 antibody RO6874813 as a single agent or in combination therapy. An alternative approach to deplete CAFs is under development which uses a FAPα vaccine. Pre-clinical data demonstrated that FAPα vaccines were able to suppresses tumor growth and metastasis in colon and breast cancer models (198,199). However, no clinical trials have tested these vaccines. A summary of the ongoing clinical trials can be found in Table 1.
Summary of select clinical trial results from the indicated CSC targeting drugs. PFS (Progression free survival), OS (overall survival), PR (partial response), CR (complete response), SD (stable disease), RR (response rate).
| Drug Target | Drug | Clinical Trial | Cancer type | Highlights from Clinical Trials | Remarks | References |
|---|---|---|---|---|---|---|
| ALDH1 | Disulfiram | Phase II (NCT003128 19) | Non-small lung cancer | Statistically significant Improvement in overall survival with some long-term survivors | Combination therapy with cisplatin and vinorelbine | http://theoncologist.alphame dpress.org/content/20/4/366. short |
| Unclear | Metformin | Randomized Phase II (NCT012109 11) | Pancreatic cancer | Metformin treatment did not improve PFS or OS. | Combination therapy with gemcitabine and erlotinib | https://www.ncbi.nlm.nih.go v/pubmed/26067687 |
| Randomized Phase II | Breast Cancer | Metformin treatment did not improve PFS or OS. | Combination with doxorubicin and cytoxan | https://www.ncbi.nlm.nih.go v/pubmed/30536182 | ||
| Randomized Phase II (NCT030717 05) | Lung Adenocarcin oma | Statistically significant improvement in PFS and OS (median OS 31.7 months vs. 17.5 months). | Combination with EGFR tyrosine Kinase inhibitor. | https://www.ncbi.nlm.nih.go v/pubmed/31486833 | ||
| Phase II (NCT015798 12) | Ovarian cancer | Statistically significant reduction in CSC. Median OS was 57.9 months. | Combination therapy with Carboplatin and Taxane | https://ascopubs.org/doi/abs /10.1200/JCO.2017.35.15_su ppl.5556 | ||
| CD44 | RG7356 | Phase I (NCT013589 03) | Solid tumors | Modest single agent activity with 21% stable disease rate | Single agent therapy | https://www.ncbi.nlm.nih.go v/pmc/articles/PMC5346770/ |
| CD133 | CART-133 | Phase I (NCT025413 70) | 23 Patients with metastatic hepatocellul ar, pancreatic or colorectal carcinoma | 3 Patients with PR, 14 SD and median PFS of 5 months. | Single agent therapy | https://www.ncbi.nlm.nih.go v/pmc/articles/PMC5993480/ |
| FAK | VS-6063 | Phase I (NCT025465 31) | Pancreatic ductal adenocarcin oma | Well tolerated but no clinical responses but 54% stable disease | Combination therapy with pembrolizumab and gemcitabine | https://ascopubs.org/doi/abs /10.1200/JCO.2018.36.4_sup pl.380 |
| FAK | VS-6063 | Randomized Phase II (NCT018706 09) | Pleural mesothelio ma | Merlin stratified pleural epithelioma. No PFS or OS improvement. | Single agent therapy vs. Placebo | https://www.ncbi.nlm.nih.go v/pubmed/?term=Maintenan ce+Defactinib+Versus+Placeb o+After+First- Line+Chemotherapy+in+Patie nts+With+Merlin- Stratified+Pleural+Mesothelio ma%3A+COMMAND- A+Double- Blind%2C+Randomized%2C+P hase+II+Study. |
| Wnt/β- catenin | Genistein | Phase I (NCT019857 63) | Colorectal cancer | Well tolerated, adverse events less than grade 4. Partial response in 61.5% | Combination therapy with FOLFOX and/or Bevacizumab | https://link.springer.com/arti cle/10.1007%2Fs00280-019- 03886-3 |
| Wnt/β- catenin | Vantictumab | Phase I (NCT020053 15) | Pancreatic cancer | Partial response in 13 (41.9%) patient. Study terminated due to bone- related adverse events. | Combination therapy with nab-paclitaxel and gemcitabine | https://www.ncbi.nlm.nih.go v/pubmed/31338636 |
| Notch | Gamma secretase Inhibitor | Phase II (NCT019857 63) | Pancreatic adeno- carcinoma | Stable disease was achieved in 25% of (12) and 6-month survival rate in 27.8% of patients. | Single agent therapy | https://www.ncbi.nlm.nih.go v/pubmed/24668033 |
| Phase I (NCT010983 44) | Pancreatic adeno- carcinoma | 68% of patients had stable disease (stage IV pancreatic cancer) with a confirmed partial response in 5% of evaluated patients. | Combination therapy with Gemcitabine | https://www.nature.com/arti cles/bjc2017495 | ||
| Phase I (NCT016950 05) | Metastatic cancer | Anti-tumor activity observed in breast cancer, leiomyosarcoma and cystic carcinoma. | Single agent therapy | https://www.ncbi.nlm.nih.go v/pubmed/30060061 | ||
| Hedgehog | Glasdegib | Phase II (NCT015460 38) | Acute Myeloid Leukemia | 46.4% of 69 patients achieved CR. Median duration to CR is 94 days. Median OS is 14.9 months | Combination therapy with cytarabine and daunorubicin | https://onlinelibrary.wiley.co m/doi/full/10.1002/ajh.25238 |
| IL-6 | Stat 3 inhibitor OPB51602 | Phase I (NCT011848 07) | Non-small lung cancer | PR observed primarily in patients with EGFR mutant lung cancer. | Single agent therapy | https://www.ncbi.nlm.nih.go v/pubmed/25609248 https://ascopubs.org/doi/abs /10.1200/jco.2014.32.15_sup pl.8028 |
| IL-8 | BMS-980253 Anti-IL8 Antibody | Phase I (NCT 02536469) | Solid tumors | 73% stable disease rate with median treatment duration of 25 weeks | Single agent therapy | https://jitc.biomedcentral.co m/articles/10.1186/s40425- 019-0706-x |
| IL-8 | Reparixin | Phase I (NCT023702 38) | HER2 negative Breast cancer | Well tolerated with a 30% response rate | Combined with Paclitaxel | https://www.ncbi.nlm.nih.go v/pubmed/28539464 |
| FAPα | RO6874813 FAP-DR5 bispecific antibody | Phase-I (NCT025581 40) | Solid Tumors | Well tolerated 21% disease control rate (1 PR 6 SD). | Single agent therapy | https://mct.aacrjournals.org/ content/17/1_Supplement/A 092 |
Barriers to targeting CSCs
The primary barrier too many CSC targeting therapies has been toxicity. As CSCs share many markers and regulatory pathways with normal adult stem cells, many of these side effects are 'on target'. For example, notch inhibitors have significant gastrointestinal side effects related to targeting the gut stem cell niche; in clinical trials adverse gastrointestinal events affect up to 50% of patients (173,175). Thrombocytopenia, anemia, and neutropenia are common (172-175). Wnt inhibitors similarly show a number of gastrointestinal side effects and, related to Wnt signaling's role in bone remodeling, bone degradation (168,169). Importantly, the bone side effects could be treated with bisphosphonate therapy (168). Although Hedgehog pathway inhibitors have been commonly associated with a number of adverse events, these are generally of low grade, and management plans have been developed to alleviate these symptoms and limit discontinuation of treatment (200). The most concerning of these side effects are muscle spasms, which can become severe in some patients; however, a recent clinical trial (NCT01893892) demonstrated that levocarnitine could be used to partially alleviate these symptoms. Lastly, as noted above, trials targeting CD44v6 using bivatuzumab mertansine demonstrated serious skin toxicity (147,148). Whether this is drug or class specific remains unknown. However, other approaches, targeting CD44 using the broader-range anti-CD44 humanized antibody demonstrated significantly better tolerance at a >5x high dose than initial anti-CD44v6 trials and did not induced significant skin toxicity; instead, the dose limiting side effects were headaches and febrile neutropenia (144).
Conclusion
Over the past decade, we have learned a significant amount about CSCs and begun to translate this into the clinic. Much like early cancer immunology studies, the earliest attempts at therapeutic approaches targeting CSCs have been disappointing. But the reality is that these attempts are still in their infancy. With increasing studies suggesting the branched nature of CSC hierarchies, it is likely that the best results will be obtained by targeting both arms of the CSC hierarchy. Furthermore, given the potential for dedifferentiation, agents such as chemotherapeutics or tyrosine kinase inhibitors, targeting bulk cells, will still be necessary. And much as better supportive care was needed to improve the tolerability of chemotherapy, supportive approaches for CSC-targeted therapy may be needed to overcome toxicity-related issues.
It is important to note that, even though we are still early in the course of CSC-targeting therapeutic development, we are beginning to see important clinical successes that increase hope that CSC targeting will indeed improve patient outcomes. Positive phase II trials with CSC targeting drugs metformin and disulfiram, showing significant improvements in patient overall survival, should encourage translational scientists to re-double their efforts at targeting CSCs clinically. Like the immune-oncology drugs, we believe that CSC-targeting drugs have an encouraging future.
Acknowledgements
RJB is supported by NIH R01CA203810, R01CA218026, and R01CA214567, AJC is support by an OCRA Ann Schreiber award. We would like to thank Bruce Campbell for editing.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ford CE, Hamerton JL, Barnes DW, Loutit JF. Cytological identification of radiation-chimaeras. Nature. 1956Mar10;177(4506):452-454
2. Siminovitch L, Mcculloch EA, Till JE. The distribution of colony-forming cells among spleen colonies. J Cell Physiol. 1963Dec;62:327-336
3. Doulatov S, Notta F, Laurenti E, Dick JE. Hematopoiesis: a human perspective. Cell Stem Cell. 2012Feb3;10(2):120-136
4. Kleinsmith LJ, Pierce GB. Multipotentiality of single embryonal carcinoma cells. Cancer Res. 1964Oct;24:1544-1551
5. Pierce GB, Speers WC. Tumors as caricatures of the process of tissue renewal: prospects for therapy by directing differentiation. Cancer Res. 1988Apr15;48(8):1996-2004
6. Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997Jul;3(7):730-737
7. Chen W, Dong J, Haiech J, Kilhoffer M-C, Zeniou M. Cancer stem cell quiescence and plasticity as major challenges in cancer therapy. Stem Cells Int. 2016Jun21;2016:1740936
8. Kreso A, Dick JE. Evolution of the cancer stem cell model. Cell Stem Cell. 2014Mar6;14(3):275-291
9. Boumahdi S, Driessens G, Lapouge G, Rorive S, Nassar D, Le Mercier M. et al. SOX2 controls tumour initiation and cancer stem-cell functions in squamous-cell carcinoma. Nature. 2014Jul10;511(7508):246-250
10. Cao J, Zhao M, Liu J, Zhang X, Pei Y, Wang J. et al. RACK1 Promotes Self-Renewal and Chemoresistance of Cancer Stem Cells in Human Hepatocellular Carcinoma through Stabilizing Nanog. Theranostics. 2019Jan24;9(3):811-828
11. Miftakhova RR, Rakhmatullina AR, Mingaleeva RN, Garanina EE, Khaiboullina SF, Rizvanov AA. The expression of pluripotency genes regulates properties of cancer stem cells in MCF-7 breast cancer model. J Clin Oncol. 2017May20;35(15_suppl):e23018-e23018
12. Pandit H, Li Y, Li X, Zhang W, Li S, Martin RCG. Enrichment of cancer stem cells via β-catenin contributing to the tumorigenesis of hepatocellular carcinoma. BMC Cancer. 2018Aug3;18(1):783
13. Zhao C, Chen A, Jamieson CH, Fereshteh M, Abrahamsson A, Blum J. et al. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature. 2009Apr9;458(7239):776-779
14. Oskarsson T, Batlle E, Massagué J. Metastatic stem cells: sources, niches, and vital pathways. Cell Stem Cell. 2014Mar6;14(3):306-321
15. Schmidt JM, Panzilius E, Bartsch HS, Irmler M, Beckers J, Kari V. et al. Stem-cell-like properties and epithelial plasticity arise as stable traits after transient Twist1 activation. Cell Rep. 2015Jan13;10(2):131-139
16. Dieter SM, Ball CR, Hoffmann CM, Nowrouzi A, Herbst F, Zavidij O. et al. Distinct types of tumor-initiating cells form human colon cancer tumors and metastases. Cell Stem Cell. 2011Oct4;9(4):357-365
17. Grillet F, Bayet E, Villeronce O, Zappia L, Lagerqvist EL, Lunke S. et al. Circulating tumour cells from patients with colorectal cancer have cancer stem cell hallmarks in ex vivo culture. Gut. 2017;66(10):1802-1810
18. Dembinski JL, Krauss S. Characterization and functional analysis of a slow cycling stem cell-like subpopulation in pancreas adenocarcinoma. Clin Exp Metastasis. 2009May7;26(7):611-623
19. Chow EK-H, Fan L, Chen X, Bishop JM. Oncogene-specific formation of chemoresistant murine hepatic cancer stem cells. Hepatology. 2012Oct;56(4):1331-1341
20. Konopleva M, Zhao S, Hu W, Jiang S, Snell V, Weidner D. et al. The anti-apoptotic genes Bcl-X(L) and Bcl-2 are over-expressed and contribute to chemoresistance of non-proliferating leukaemic CD34+ cells. Br J Haematol. 2002Aug;118(2):521-534
21. Atashzar MR, Baharlou R, Karami J, Abdollahi H, Rezaei R, Pourramezan F. et al. Cancer stem cells: A review from origin to therapeutic implications. J Cell Physiol. 2019 Jul 8
22. Choi Y-J, Ingram PN, Yang K, Coffman L, Iyengar M, Bai S. et al. Identifying an ovarian cancer cell hierarchy regulated by bone morphogenetic protein 2. Proc Natl Acad Sci USA. 2015Dec15;112(50):E6882-8
23. Shimokawa M, Ohta Y, Nishikori S, Matano M, Takano A, Fujii M. et al. Visualization and targeting of LGR5+ human colon cancer stem cells. Nature. 2017May11;545(7653):187-192
24. Liu S, Cong Y, Wang D, Sun Y, Deng L, Liu Y. et al. Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Rep. 2014Jan14;2(1):78-91
25. Bradshaw A, Wickremsekera A, Tan ST, Peng L, Davis PF, Itinteang T. Cancer stem cell hierarchy in glioblastoma multiforme. Front Surg. 2016Apr15;3:21
26. Silva IA, Bai S, McLean K, Yang K, Griffith K, Thomas D. et al. Aldehyde dehydrogenase in combination with CD133 defines angiogenic ovarian cancer stem cells that portend poor patient survival. Cancer Res. 2011Jun1;71(11):3991-4001
27. Kryczek I, Liu S, Roh M, Vatan L, Szeliga W, Wei S. et al. Expression of aldehyde dehydrogenase and CD133 defines ovarian cancer stem cells. Int J Cancer. 2012Jan1;130(1):29-39
28. Cole AJ, Iyengar M, O'Hayer P, Chan D, Delgoffe G, Aird KM. et al. NFATC4 promotes quiescence and chemotherapy resistance in ovarian cancer. BioRxiv. 2019 Oct 31
29. Chefetz I, Grimley E, Yang K, Hong L, Vinogradova EV, Suciu R. et al. A Pan-ALDH1A Inhibitor Induces Necroptosis in Ovarian Cancer Stem-like Cells. Cell Rep. 2019Mar12;26(11):3061-3075.e6
30. Stewart JM, Shaw PA, Gedye C, Bernardini MQ, Neel BG, Ailles LE. Phenotypic heterogeneity and instability of human ovarian tumor-initiating cells. Proc Natl Acad Sci USA. 2011Apr19;108(16):6468-6473
31. Flesken-Nikitin A, Hwang C-I, Cheng C-Y, Michurina TV, Enikolopov G, Nikitin AY. Ovarian surface epithelium at the junction area contains a cancer-prone stem cell niche. Nature. 2013Mar14;495(7440):241-245
32. Cernat L, Blaj C, Jackstadt R, Brandl L, Engel J, Hermeking H. et al. Colorectal cancers mimic structural organization of normal colonic crypts. PLoS One. 2014Aug11;9(8):e104284
33. Vermeulen L, De Sousa E Melo F, van der Heijden M, Cameron K, de Jong JH, Borovski T. et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat Cell Biol. 2010May;12(5):468-476
34. Visvader JE, Stingl J. Mammary stem cells and the differentiation hierarchy: current status and perspectives. Genes Dev. 2014Jun1;28(11):1143-1158
35. Duru N, Gernapudi R, Lo P-K, Yao Y, Wolfson B, Zhang Y. et al. Characterization of the CD49f+/CD44+/CD24- single-cell derived stem cell population in basal-like DCIS cells. Oncotarget. 2016Jul26;7(30):47511-47525
36. Li W, Ma H, Zhang J, Zhu L, Wang C, Yang Y. Unraveling the roles of CD44/CD24 and ALDH1 as cancer stem cell markers in tumorigenesis and metastasis. Sci Rep. 2017Oct23;7(1):13856
37. Baslan T, Hicks J. Unravelling biology and shifting paradigms in cancer with single-cell sequencing. Nat Rev Cancer. 2017Aug24;17(9):557-569
38. Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H. et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014Jun20;344(6190):1396-1401
39. Wang L, Babikir H, Muller S, Yagnik G, Shamardani K, Catalan F. et al. The phenotypes of proliferating glioblastoma cells reside on a single axis of variation. Cancer Discov. 2019 Sep 25
40. Gao MQ, Choi YP, Kang S, Youn JH, Cho NH. CD24+ cells from hierarchically organized ovarian cancer are enriched in cancer stem cells. Oncogene. 2010May6;29(18):2672-2680
41. Haraguchi N, Ishii H, Mimori K, Tanaka F, Ohkuma M, Kim HM. et al. CD13 is a therapeutic target in human liver cancer stem cells. J Clin Invest. 2010Sep;120(9):3326-3339
42. Roesch A, Fukunaga-Kalabis M, Schmidt EC, Zabierowski SE, Brafford PA, Vultur A. et al. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell. 2010May14;141(4):583-594
43. Vincent Z, Urakami K, Maruyama K, Yamaguchi K, Kusuhara M. CD133-positive cancer stem cells from Colo205 human colon adenocarcinoma cell line show resistance to chemotherapy and display a specific metabolomic profile. Genes Cancer. 2014Jul;5(7-8):250-260
44. Kurtova AV, Xiao J, Mo Q, Pazhanisamy S, Krasnow R, Lerner SP. et al. Blocking PGE2-induced tumour repopulation abrogates bladder cancer chemoresistance. Nature. 2015Jan8;517(7533):209-213
45. Vanner RJ, Remke M, Gallo M, Selvadurai HJ, Coutinho F, Lee L. et al. Quiescent sox2(+) cells drive hierarchical growth and relapse in sonic hedgehog subgroup medulloblastoma. Cancer Cell. 2014Jul14;26(1):33-47
46. Chen J, Li Y, Yu T-S, McKay RM, Burns DK, Kernie SG. et al. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature. 2012Aug23;488(7412):522-526
47. Chesnokov M, Khan I, Park Y, Ezel J, Mehta G, Yousif A. et al. The MEK1/2 pathway as a therapeutic target in high-grade serous ovarian carcinoma. BioRxiv. 2019 Sep 16
48. Kotton DN, Morrisey EE. Lung regeneration: mechanisms, applications and emerging stem cell populations. Nat Med. 2014Aug;20(8):822-832
49. Chaffer CL, Brueckmann I, Scheel C, Kaestli AJ, Wiggins PA, Rodrigues LO. et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc Natl Acad Sci USA. 2011May10;108(19):7950-7955
50. Zhou C, Jiang H, Zhang Z, Zhang G, Wang H, Zhang Q. et al. ZEB1 confers stem cell-like properties in breast cancer by targeting neurogenin-3. Oncotarget. 2017Aug15;8(33):54388-54401
51. Chiou S-H, Wang M-L, Chou Y-T, Chen C-J, Hong C-F, Hsieh W-J. et al. Coexpression of Oct4 and Nanog enhances malignancy in lung adenocarcinoma by inducing cancer stem cell-like properties and epithelial-mesenchymal transdifferentiation. Cancer Res. 2010Dec15;70(24):10433-10444
52. Kumar SM, Liu S, Lu H, Zhang H, Zhang PJ, Gimotty PA. et al. Acquired cancer stem cell phenotypes through Oct4-mediated dedifferentiation. Oncogene. 2012Nov22;31(47):4898-4911
53. Jeon H-M, Jin X, Lee J-S, Oh S-Y, Sohn Y-W, Park H-J. et al. Inhibitor of differentiation 4 drives brain tumor-initiating cell genesis through cyclin E and notch signaling. Genes Dev. 2008Aug1;22(15):2028-2033
54. Herreros-Villanueva M, Zhang JS, Koenig A, Abel EV, Smyrk TC, Bamlet WR. et al. SOX2 promotes dedifferentiation and imparts stem cell-like features to pancreatic cancer cells. Oncogenesis. 2013Aug5;2:e61
55. Nakano M, Kikushige Y, Miyawaki K, Kunisaki Y, Mizuno S, Takenaka K. et al. Dedifferentiation process driven by TGF-beta signaling enhances stem cell properties in human colorectal cancer. Oncogene. 2019;38(6):780-793
56. Mani SA, Guo W, Liao M-J, Eaton EN, Ayyanan A, Zhou AY. et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008May16;133(4):704-715
57. Chen Y-C, Chen Y-W, Hsu H-S, Tseng L-M, Huang P-I, Lu K-H. et al. Aldehyde dehydrogenase 1 is a putative marker for cancer stem cells in head and neck squamous cancer. Biochem Biophys Res Commun. 2009Jul31;385(3):307-313
58. Pang M, Wu G, Hou X, Hou N, Liang L, Jia G. et al. LIN28B promotes colon cancer migration and recurrence. PLoS One. 2014Oct31;9(10):e109169
59. Liu X, Sun H, Qi J, Wang L, He S, Liu J. et al. Sequential introduction of reprogramming factors reveals a time-sensitive requirement for individual factors and a sequential EMT-MET mechanism for optimal reprogramming. Nat Cell Biol. 2013Jul;15(7):829-838
60. Chien C-S, Wang M-L, Chu P-Y, Chang Y-L, Liu W-H, Yu C-C. et al. Lin28B/Let-7 Regulates Expression of Oct4 and Sox2 and Reprograms Oral Squamous Cell Carcinoma Cells to a Stem-like State. Cancer Res. 2015Jun15;75(12):2553-2565
61. Wang P, Wan W-W, Xiong S-L, Feng H, Wu N. Cancer stem-like cells can be induced through dedifferentiation under hypoxic conditions in glioma, hepatoma and lung cancer. Cell Death Discov. 2017Jan23;3:16105
62. Lagadec C, Vlashi E, Della Donna L, Dekmezian C, Pajonk F. Radiation-induced reprogramming of breast cancer cells. Stem Cells. 2012May;30(5):833-844
63. Hadjimichael C, Chanoumidou K, Papadopoulou N, Arampatzi P, Papamatheakis J, Kretsovali A. Common stemness regulators of embryonic and cancer stem cells. World J Stem Cells. 2015Oct26;7(9):1150-1184
64. Ghisolfi L, Keates AC, Hu X, Lee D, Li CJ. Ionizing radiation induces stemness in cancer cells. PLoS One. 2012Aug21;7(8):e43628
65. Hu X, Ghisolfi L, Keates AC, Zhang J, Xiang S, Lee D. et al. Induction of cancer cell stemness by chemotherapy. Cell Cycle. 2012Jul15;11(14):2691-2698
66. Liu L, Yang L, Yan W, Zhai J, Pizzo DP, Chu P. et al. Chemotherapy Induces Breast Cancer Stemness in Association with Dysregulated Monocytosis. Clin Cancer Res. 2018May15;24(10):2370-2382
67. Niu N, Mercado-Uribe I, Liu J. Dedifferentiation into blastomere-like cancer stem cells via formation of polyploid giant cancer cells. Oncogene. 2017Aug24;36(34):4887-4900
68. Diaz-Carballo D, Saka S, Klein J, Rennkamp T, Acikelli AH, Malak S. et al. A distinct oncogenerative multinucleated cancer cell serves as a source of stemness and tumor heterogeneity. Cancer Res. 2018 Feb 12
69. Zhang S, Mercado-Uribe I, Xing Z, Sun B, Kuang J, Liu J. Generation of cancer stem-like cells through the formation of polyploid giant cancer cells. Oncogene. 2014Jan2;33(1):116-128
70. Mu X, Yan S, Fu C, Wei A. The histone acetyltransferase MOF promotes induces generation of pluripotent stem cells. Cell Reprogram. 2015Aug;17(4):259-267
71. Jackson M, Krassowska A, Gilbert N, Chevassut T, Forrester L, Ansell J. et al. Severe global DNA hypomethylation blocks differentiation and induces histone hyperacetylation in embryonic stem cells. Mol Cell Biol. 2004Oct;24(20):8862-8871
72. Kagara N, Huynh KT, Kuo C, Okano H, Sim MS, Elashoff D. et al. Epigenetic regulation of cancer stem cell genes in triple-negative breast cancer. Am J Pathol. 2012Jul;181(1):257-267
73. Gopisetty G, Xu J, Sampath D, Colman H, Puduvalli VK. Epigenetic regulation of CD133/PROM1 expression in glioma stem cells by Sp1/myc and promoter methylation. Oncogene. 2013Jun27;32(26):3119-3129
74. Baba T, Convery PA, Matsumura N, Whitaker RS, Kondoh E, Perry T. et al. Epigenetic regulation of CD133 and tumorigenicity of CD133+ ovarian cancer cells. Oncogene. 2009Jan15;28(2):209-218
75. Rheinbay E, Suvà ML, Gillespie SM, Wakimoto H, Patel AP, Shahid M. et al. An aberrant transcription factor network essential for Wnt signaling and stem cell maintenance in glioblastoma. Cell Rep. 2013May30;3(5):1567-1579
76. Wang T, Wu H, Liu S, Lei Z, Qin Z, Wen L. et al. SMYD3 controls a Wnt-responsive epigenetic switch for ASCL2 activation and cancer stem cell maintenance. Cancer Lett. 2018Aug28;430:11-24
77. Wang Y, He L, Du Y, Zhu P, Huang G, Luo J. et al. The long noncoding RNA lncTCF7 promotes self-renewal of human liver cancer stem cells through activation of Wnt signaling. Cell Stem Cell. 2015Apr2;16(4):413-425
78. Ooki A, Dinalankara W, Marchionni L, Tsay J-CJ, Goparaju C, Maleki Z. et al. Epigenetically regulated PAX6 drives cancer cells toward a stem-like state via GLI-SOX2 signaling axis in lung adenocarcinoma. Oncogene. 2018Jul6;37(45):5967-5981
79. Jin L, Vu T, Yuan G, Datta PK. STRAP promotes stemness of human colorectal cancer via epigenetic regulation of the NOTCH pathway. Cancer Res. 2017Oct15;77(20):5464-5478
80. Tellez CS, Juri DE, Do K, Bernauer AM, Thomas CL, Damiani LA. et al. EMT and stem cell-like properties associated with miR-205 and miR-200 epigenetic silencing are early manifestations during carcinogen-induced transformation of human lung epithelial cells. Cancer Res. 2011Apr15;71(8):3087-3097
81. Chang C-J, Chao C-H, Xia W, Yang J-Y, Xiong Y, Li C-W. et al. p53 regulates epithelial-mesenchymal transition and stem cell properties through modulating miRNAs. Nat Cell Biol. 2011Mar;13(3):317-323
82. Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD. et al. Multilineage potential of adult human mesenchymal stem cells. Science. 1999Apr2;284(5411):143-147
83. Correa D, Somoza RA, Lin P, Schiemann WP, Caplan AI. Mesenchymal stem cells regulate melanoma cancer cells extravasation to bone and liver at their perivascular niche. Int J Cancer. 2016Jan15;138(2):417-427
84. McLean K, Gong Y, Choi Y, Deng N, Yang K, Bai S. et al. Human ovarian carcinoma-associated mesenchymal stem cells regulate cancer stem cells and tumorigenesis via altered BMP production. J Clin Invest. 2011Aug;121(8):3206-3219
85. McCann CK, Growdon WB, Kulkarni-Datar K, Curley MD, Friel AM, Proctor JL. et al. Inhibition of Hedgehog signaling antagonizes serous ovarian cancer growth in a primary xenograft model. PLoS One. 2011Nov29;6(11):e28077
86. Steg AD, Katre AA, Bevis KS, Ziebarth A, Dobbin ZC, Shah MM. et al. Smoothened antagonists reverse taxane resistance in ovarian cancer. Mol Cancer Ther. 2012Jul;11(7):1587-1597
87. Liu S, Ginestier C, Ou SJ, Clouthier SG, Patel SH, Monville F. et al. Breast cancer stem cells are regulated by mesenchymal stem cells through cytokine networks. Cancer Res. 2011Jan15;71(2):614-624
88. Cuiffo BG, Campagne A, Bell GW, Lembo A, Orso F, Lien EC. et al. MSC-regulated microRNAs converge on the transcription factor FOXP2 and promote breast cancer metastasis. Cell Stem Cell. 2014Dec4;15(6):762-774
89. Bliss SA, Sinha G, Sandiford OA, Williams LM, Engelberth DJ, Guiro K. et al. Mesenchymal Stem Cell-Derived Exosomes Stimulate Cycling Quiescence and Early Breast Cancer Dormancy in Bone Marrow. Cancer Res. 2016Oct1;76(19):5832-5844
90. Davis H, Irshad S, Bansal M, Rafferty H, Boitsova T, Bardella C. et al. Aberrant epithelial GREM1 expression initiates colonic tumorigenesis from cells outside the stem cell niche. Nat Med. 2015Jan;21(1):62-70
91. Chandler C, Liu T, Buckanovich R, Coffman LG. The double edge sword of fibrosis in cancer. Transl Res. 2019Feb21;209:55-67
92. Mishra PJ, Mishra PJ, Humeniuk R, Medina DJ, Alexe G, Mesirov JP. et al. Carcinoma-associated fibroblast-like differentiation of human mesenchymal stem cells. Cancer Res. 2008Jun1;68(11):4331-4339
93. Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer. 2016Aug23;16(9):582-598
94. Lau EYT, Lo J, Cheng BYL, Ma MKF, Lee JMF, Ng JKY. et al. Cancer-Associated Fibroblasts Regulate Tumor-Initiating Cell Plasticity in Hepatocellular Carcinoma through c-Met/FRA1/HEY1 Signaling. Cell Rep. 2016May10;15(6):1175-1189
95. Xiong S, Wang R, Chen Q, Luo J, Wang J, Zhao Z. et al. Cancer-associated fibroblasts promote stem cell-like properties of hepatocellular carcinoma cells through IL-6/STAT3/Notch signaling. Am J Cancer Res. 2018Feb1;8(2):302-316
96. Valenti G, Quinn HM, Heynen GJJE, Lan L, Holland JD, Vogel R. et al. Cancer Stem Cells Regulate Cancer-Associated Fibroblasts via Activation of Hedgehog Signaling in Mammary Gland Tumors. Cancer Res. 2017Apr15;77(8):2134-2147
97. Patel AK, Vipparthi K, Thatikonda V, Arun I, Bhattacharjee S, Sharan R. et al. A subtype of cancer-associated fibroblasts with lower expression of alpha-smooth muscle actin suppresses stemness through BMP4 in oral carcinoma. Oncogenesis. 2018Oct5;7(10):78
98. Quante M, Tu SP, Tomita H, Gonda T, Wang SSW, Takashi S. et al. Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell. 2011Feb15;19(2):257-272
99. Sainz B, Martín B, Tatari M, Heeschen C, Guerra S. ISG15 is a critical microenvironmental factor for pancreatic cancer stem cells. Cancer Res. 2014Dec15;74(24):7309-7320
100. Sainz B, Alcala S, Garcia E, Sanchez-Ripoll Y, Azevedo MM, Cioffi M. et al. Microenvironmental hCAP-18/LL-37 promotes pancreatic ductal adenocarcinoma by activating its cancer stem cell compartment. Gut. 2015Dec;64(12):1921-1935
101. Iliopoulos D, Hirsch HA, Struhl K. An epigenetic switch involving NF-kappaB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell. 2009Nov13;139(4):693-706
102. Wan S, Zhao E, Kryczek I, Vatan L, Sadovskaya A, Ludema G. et al. Tumor-associated macrophages produce interleukin 6 and signal via STAT3 to promote expansion of human hepatocellular carcinoma stem cells. Gastroenterology. 2014Dec;147(6):1393-1404
103. Mitchem JB, Brennan DJ, Knolhoff BL, Belt BA, Zhu Y, Sanford DE. et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 2013Feb1;73(3):1128-1141
104. Ginestier C, Liu S, Diebel ME, Korkaya H, Luo M, Brown M. et al. CXCR1 blockade selectively targets human breast cancer stem cells in vitro and in xenografts. J Clin Invest. 2010Feb;120(2):485-497
105. Xiang T, Long H, He L, Han X, Lin K, Liang Z. et al. Interleukin-17 produced by tumor microenvironment promotes self-renewal of CD133+ cancer stem-like cells in ovarian cancer. Oncogene. 2015Jan8;34(2):165-176
106. Jin F, Miao Y, Xu P, Qiu X. IL-8 regulates the stemness properties of cancer stem cells in the small-cell lung cancer cell line H446. Onco Targets Ther. 2018Sep11;11:5723-5731
107. Lu H, Clauser KR, Tam WL, Fröse J, Ye X, Eaton EN. et al. A breast cancer stem cell niche supported by juxtacrine signalling from monocytes and macrophages. Nat Cell Biol. 2014Nov;16(11):1105-1117
108. Yang S, Wang B, Guan C, Wu B, Cai C, Wang M. et al. Foxp3+IL-17+ T cells promote development of cancer-initiating cells in colorectal cancer. J Leukoc Biol. 2011Jan;89(1):85-91
109. Xu Y, Dong X, Qi P, Ye Y, Shen W, Leng L. et al. Sox2 Communicates with Tregs Through CCL1 to Promote the Stemness Property of Breast Cancer Cells. Stem Cells. 2017;35(12):2351-2365
110. Patel SA, Dave MA, Bliss SA, Giec-Ujda AB, Bryan M, Pliner LF. et al. Treg/Th17 polarization by distinct subsets of breast cancer cells is dictated by the interaction with mesenchymal stem cells. J Cancer Stem Cell Res. 2014 May 29;2014(2)
111. Mercurio AM. Vegf/neuropilin signaling in cancer stem cells. Int J Mol Sci. 2019 Jan 23;20(3)
112. Kim HS, Chen Y-C, Nör F, Warner KA, Andrews A, Wagner VP. et al. Endothelial-derived interleukin-6 induces cancer stem cell motility by generating a chemotactic gradient towards blood vessels. Oncotarget. 2017Nov21;8(59):100339-100352
113. Bai S, Ingram P, Chen Y-C, Deng N, Pearson A, Niknafs YS. et al. EGFL6 regulates the asymmetric division, maintenance, and metastasis of ALDH+ ovarian cancer cells. Cancer Res. 2016Nov1;76(21):6396-6409
114. Ping Y-F, Zhang X, Bian X-W. Cancer stem cells and their vascular niche: Do they benefit from each other? Cancer Lett. 2016Oct1;380(2):561-567
115. Chen M-H, Weng J-J, Cheng C-T, Wu R-C, Huang S-C, Wu C-E. et al. ALDH1A3, the major aldehyde dehydrogenase isoform in human cholangiocarcinoma cells, affects prognosis and gemcitabine resistance in cholangiocarcinoma patients. Clin Cancer Res. 2016Aug15;22(16):4225-4235
116. Landen CN, Goodman B, Katre AA, Steg AD, Nick AM, Stone RL. et al. Targeting aldehyde dehydrogenase cancer stem cells in ovarian cancer. Mol Cancer Ther. 2010Dec;9(12):3186-3199
117. Luo Y, Dallaglio K, Chen Y, Robinson WA, Robinson SE, McCarter MD. et al. ALDH1A isozymes are markers of human melanoma stem cells and potential therapeutic targets. Stem Cells. 2012Oct;30(10):2100-2113
118. Yip NC, Fombon IS, Liu P, Brown S, Kannappan V, Armesilla AL. et al. Disulfiram modulated ROS-MAPK and NFκB pathways and targeted breast cancer cells with cancer stem cell-like properties. Br J Cancer. 2011May10;104(10):1564-1574
119. Li Z, Xiang Y, Xiang L, Xiao Y, Li F, Hao P. ALDH maintains the stemness of lung adenoma stem cells by suppressing the Notch/CDK2/CCNE pathway. PLoS One. 2014Mar26;9(3):e92669
120. Morrison BW, Doudican NA, Patel KR, Orlow SJ. Disulfiram induces copper-dependent stimulation of reactive oxygen species and activation of the extrinsic apoptotic pathway in melanoma. Melanoma Res. 2010Feb;20(1):11-20
121. Kast RE, Belda-Iniesta C. Suppressing glioblastoma stem cell function by aldehyde dehydrogenase inhibition with chloramphenicol or disulfiram as a new treatment adjunct: an hypothesis. Curr Stem Cell Res Ther. 2009Dec;4(4):314-317
122. Nwani NG, Condello S, Wang Y, Swetzig WM, Barber E, Hurley T. et al. A Novel ALDH1A1 Inhibitor Targets Cells with Stem Cell Characteristics in Ovarian Cancer. Cancers (Basel). 2019 Apr 8;11(4)
123. Yang S-M, Martinez NJ, Yasgar A, Danchik C, Johansson C, Wang Y. et al. Discovery of Orally Bioavailable, Quinoline-Based Aldehyde Dehydrogenase 1A1 (ALDH1A1) Inhibitors with Potent Cellular Activity. J Med Chem. 2018Jun14;61(11):4883-4903
124. Huddle BC, Grimley E, Buchman CD, Chtcherbinine M, Debnath B, Mehta P. et al. Structure-Based Optimization of a Novel Class of Aldehyde Dehydrogenase 1A (ALDH1A) Subfamily-Selective Inhibitors as Potential Adjuncts to Ovarian Cancer Chemotherapy. J Med Chem. 2018Oct11;61(19):8754-8773
125. Sarvi S, Crispin R, Lu Y, Zeng L, Hurley TD, Houston DR. et al. ALDH1 Bio-activates Nifuroxazide to Eradicate ALDHHigh Melanoma-Initiating Cells. Cell Chem Biol. 2018Dec20;25(12):1456-1469.e6
126. Nechushtan H, Hamamreh Y, Nidal S, Gotfried M, Baron A, Shalev YI. et al. A phase IIb trial assessing the addition of disulfiram to chemotherapy for the treatment of metastatic non-small cell lung cancer. Oncologist. 2015Apr;20(4):366-367
127. Dufour P, Lang JM, Giron C, Duclos B, Haehnel P, Jaeck D. et al. Sodium dithiocarb as adjuvant immunotherapy for high risk breast cancer: a randomized study. Biotherapy. 1993;6(1):9-12
128. Huang J, Chaudhary R, Cohen AL, Fink K, Goldlust S, Boockvar J. et al. A multicenter phase II study of temozolomide plus disulfiram and copper for recurrent temozolomide-resistant glioblastoma. J Neurooncol. 2019May;142(3):537-544
129. Huang J, Campian JL, Gujar AD, Tran DD, Lockhart AC, DeWees TA. et al. A phase I study to repurpose disulfiram in combination with temozolomide to treat newly diagnosed glioblastoma after chemoradiotherapy. J Neurooncol. 2016Mar10;128(2):259-266
130. Huang J, Campian JL, Gujar AD, Tsien C, Ansstas G, Tran DD. et al. Final results of a phase I dose-escalation, dose-expansion study of adding disulfiram with or without copper to adjuvant temozolomide for newly diagnosed glioblastoma. J Neurooncol. 2018May;138(1):105-111
131. Hirsch HA, Iliopoulos D, Tsichlis PN, Struhl K. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res. 2009Oct1;69(19):7507-7511
132. Coyle C, Cafferty FH, Vale C, Langley RE. Metformin as an adjuvant treatment for cancer: a systematic review and meta-analysis. Ann Oncol. 2016Sep28;27(12):2184-2195
133. Deng J, Peng M, Wang Z, Zhou S, Xiao D, Deng J. et al. Novel application of metformin combined with targeted drugs on anticancer treatment. Cancer Sci. 2019Jan;110(1):23-30
134. Tan W, Tang H, Jiang X, Ye F, Huang L, Shi D. et al. Metformin mediates induction of miR-708 to inhibit self-renewal and chemoresistance of breast cancer stem cells through targeting CD47. J Cell Mol Med. 2019Sep;23(9):5994-6004
135. Kordes S, Pollak MN, Zwinderman AH, Mathôt RA, Weterman MJ, Beeker A. et al. Metformin in patients with advanced pancreatic cancer: a double-blind, randomised, placebo-controlled phase 2 trial. Lancet Oncol. 2015Jul;16(7):839-847
136. Nanni O, Amadori D, De Censi A, Rocca A, Freschi A, Bologna A. et al. Metformin plus chemotherapy versus chemotherapy alone in the first-line treatment of HER2-negative metastatic breast cancer. The MYME randomized, phase 2 clinical trial. Breast Cancer Res Treat. 2019Apr;174(2):433-442
137. Buckanovich RJ, Brown J, Shank J, Griffith KA, Reynolds RK, Johnston C. et al. A phase II clinical trial of metformin as a cancer stem cell targeting agent in stage IIc/III/IV ovarian, fallopian tube, and primary peritoneal cancer. J Clin Oncol. 2017May20;35(15_suppl):5556-5556
138. Arrieta O, Barrón F, Padilla M-ÁS, Avilés-Salas A, Ramírez-Tirado LA, Arguelles Jiménez MJ. et al. Effect of Metformin Plus Tyrosine Kinase Inhibitors Compared With Tyrosine Kinase Inhibitors Alone in Patients With Epidermal Growth Factor Receptor-Mutated Lung Adenocarcinoma: A Phase 2 Randomized Clinical Trial. JAMA Oncol. 2019 Sep 5;e192553
139. Yan Y, Zuo X, Wei D. Concise review: emerging role of CD44 in cancer stem cells: A promising biomarker and therapeutic target. Stem Cells Transl Med. 2015Sep;4(9):1033-1043
140. Su Z, Liu D, Chen L, Zhang J, Ru L, Chen Z. et al. CD44-Targeted Magnetic Nanoparticles Kill Head And Neck Squamous Cell Carcinoma Stem Cells In An Alternating Magnetic Field. Int J Nanomedicine. 2019Sep16;14:7549-7560
141. Cho J-H, Lee S-C, Ha N-R, Lee S-J, Yoon M-Y. A novel peptide-based recognition probe for the sensitive detection of CD44 on breast cancer stem cells. Mol Cell Probes. 2015Dec;29(6):492-499
142. Zhang S, Wu CCN, Fecteau J-F, Cui B, Chen L, Zhang L. et al. Targeting chronic lymphocytic leukemia cells with a humanized monoclonal antibody specific for CD44. Proc Natl Acad Sci USA. 2013Apr9;110(15):6127-6132
143. Arabi L, Badiee A, Mosaffa F, Jaafari MR. Targeting CD44 expressing cancer cells with anti-CD44 monoclonal antibody improves cellular uptake and antitumor efficacy of liposomal doxorubicin. J Control Release. 2015Dec28;220(Pt A):275-286
144. Menke-van der Houven van Oordt CW, Gomez-Roca C, van Herpen C, Coveler AL, Mahalingam D, Verheul HMW. et al. First-in-human phase I clinical trial of RG7356, an anti-CD44 humanized antibody, in patients with advanced, CD44-expressing solid tumors. Oncotarget. 2016Nov29;7(48):80046-80058
145. Park H-Y, Lee K-J, Lee S-J, Yoon M-Y. Screening of peptides bound to breast cancer stem cell specific surface marker CD44 by phage display. Mol Biotechnol. 2012Jul;51(3):212-220
146. Todaro M, Gaggianesi M, Catalano V, Benfante A, Iovino F, Biffoni M. et al. CD44v6 is a marker of constitutive and reprogrammed cancer stem cells driving colon cancer metastasis. Cell Stem Cell. 2014Mar6;14(3):342-356
147. Riechelmann H, Sauter A, Golze W, Hanft G, Schroen C, Hoermann K. et al. Phase I trial with the CD44v6-targeting immunoconjugate bivatuzumab mertansine in head and neck squamous cell carcinoma. Oral Oncol. 2008Sep;44(9):823-829
148. Tijink BM, Buter J, de Bree R, Giaccone G, Lang MS, Staab A. et al. A phase I dose escalation study with anti-CD44v6 bivatuzumab mertansine in patients with incurable squamous cell carcinoma of the head and neck or esophagus. Clin Cancer Res. 2006Oct15;12(20 Pt 1):6064-6072
149. Ma L, Dong L, Chang P. CD44v6 engages in colorectal cancer progression. Cell Death Dis. 2019Jan10;10(1):30
150. Barkal AA, Brewer RE, Markovic M, Kowarsky M, Barkal SA, Zaro BW. et al. CD24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy. Nature. 2019Aug;572(7769):392-396
151. Sun F, Wang T, Jiang J, Wang Y, Ma Z, Li Z. et al. Engineering a high-affinity humanized anti-CD24 antibody to target hepatocellular carcinoma by a novel CDR grafting design. Oncotarget. 2017Aug1;8(31):51238-51252
152. Sagiv E, Kazanov D, Arber N. CD24 plays an important role in the carcinogenesis process of the pancreas. Biomed Pharmacother. 2006Jul;60(6):280-284
153. Overdevest JB, Thomas S, Kristiansen G, Hansel DE, Smith SC, Theodorescu D. CD24 offers a therapeutic target for control of bladder cancer metastasis based on a requirement for lung colonization. Cancer Res. 2011Jun1;71(11):3802-3811
154. Sun F, Wang Y, Luo X, Ma Z, Xu Y, Zhang X. et al. Anti-CD24 Antibody-Nitric Oxide Conjugate Selectively and Potently Suppresses Hepatic Carcinoma. Cancer Res. 2019Jul1;79(13):3395-3405
155. Klapdor R, Wang S, Morgan M, Dörk T, Hacker U, Hillemanns P. et al. Characterization of a Novel Third-Generation Anti-CD24-CAR against Ovarian Cancer. Int J Mol Sci. 2019 Feb 3;20(3)
156. Han Y, Sun F, Zhang X, Wang T, Jiang J, Cai J. et al. CD24 targeting bi-specific antibody that simultaneously stimulates NKG2D enhances the efficacy of cancer immunotherapy. J Cancer Res Clin Oncol. 2019May;145(5):1179-1190
157. Takenobu H, Shimozato O, Nakamura T, Ochiai H, Yamaguchi Y, Ohira M. et al. CD133 suppresses neuroblastoma cell differentiation via signal pathway modification. Oncogene. 2011Jan6;30(1):97-105
158. Mak AB, Nixon AML, Kittanakom S, Stewart JM, Chen GI, Curak J. et al. Regulation of CD133 by HDAC6 promotes β-catenin signaling to suppress cancer cell differentiation. Cell Rep. 2012Oct25;2(4):951-963
159. Zhao L, Yang Y, Zhou P, Ma H, Zhao X, He X. et al. Targeting cd133high colorectal cancer cells in vitro and in vivo with an asymmetric bispecific antibody. J Immunother. 2015Aug;38(6):217-228
160. Damek-Poprawa M, Volgina A, Korostoff J, Sollecito TP, Brose MS, O'Malley BW. et al. Targeted inhibition of CD133+ cells in oral cancer cell lines. J Dent Res. 2011May;90(5):638-645
161. Waldron NN, Barsky SH, Dougherty PR, Vallera DA. A bispecific EpCAM/CD133-targeted toxin is effective against carcinoma. Target Oncol. 2014Sep;9(3):239-249
162. Wang Y, Chen M, Wu Z, Tong C, Dai H, Guo Y. et al. CD133-directed CAR T cells for advanced metastasis malignancies: A phase I trial. Oncoimmunology. 2018May7;7(7):e1440169
163. Ho B, Olson G, Figel S, Gelman I, Cance WG, Golubovskaya VM. Nanog increases focal adhesion kinase (FAK) promoter activity and expression and directly binds to FAK protein to be phosphorylated. J Biol Chem. 2012May25;287(22):18656-18673
164. Kobayashi K, Takahashi H, Inoue A, Harada H, Toshimori S, Kobayashi Y. et al. Oct-3/4 promotes migration and invasion of glioblastoma cells. J Cell Biochem. 2012Feb;113(2):508-517
165. Luo M, Fan H, Nagy T, Wei H, Wang C, Liu S. et al. Mammary epithelial-specific ablation of the focal adhesion kinase suppresses mammary tumorigenesis by affecting mammary cancer stem/progenitor cells. Cancer Res. 2009Jan15;69(2):466-474
166. Wang-Gillam A, Lockhart AC, Tan BR, Suresh R, Lim K-H, Ratner L. et al. Phase I study of defactinib combined with pembrolizumab and gemcitabine in patients with advanced cancer. J Clin Oncol. 2018Feb;36(4_suppl):380-380
167. Fennell DA, Baas P, Taylor P, Nowak AK, Gilligan D, Nakano T. et al. Maintenance Defactinib Versus Placebo After First-Line Chemotherapy in Patients With Merlin-Stratified Pleural Mesothelioma: COMMAND-A Double-Blind, Randomized, Phase II Study. J Clin Oncol. 2019Apr1;37(10):790-798
168. Smith DC, Rosen LS, Chugh R. et al. First-in-human evaluation of the human monoclonal antibody vantictumab (OMP-18R5; anti-Frizzled) targeting the WNT pathway in a phase I study for patients with advanced solid tumors. J Clin Oncol. 2013 no. 15_suppl: 2540
169. Dotan E, Cardin DB, Lenz H-J, Messersmith WA, O'Neil B, Cohen SJ. et al. Phase Ib study of WNT inhibitor ipafricept (IPA) with nab-paclitaxel (Nab-P) and gemcitabine (G) in patients (pts) with previously untreated stage IV pancreatic cancer (mPC). J Clin Oncol. 2019Feb;37(4_suppl):369-369
170. Venkatesh V, Nataraj R, Thangaraj GS, Karthikeyan M, Gnanasekaran A, Kaginelli SB. et al. Targeting Notch signalling pathway of cancer stem cells. Stem Cell Investig. 2018Mar12;5:5
171. Xu R, Shimizu F, Hovinga K, Beal K, Karimi S, Droms L. et al. Molecular and clinical effects of notch inhibition in glioma patients: A phase 0/I trial. Clin Cancer Res. 2016Oct1;22(19):4786-4796
172. Massard C, Azaro A, Soria JC, Lassen U, Le Tourneau C, Sarker D. et al. First-in-human study of LY3039478, an oral Notch signaling inhibitor in advanced or metastatic cancer. Ann Oncol. 2018Sep1;29(9):1911-1917
173. Cook N, Basu B, Smith D-M, Gopinathan A, Evans J, Steward WP. et al. A phase I trial of the γ-secretase inhibitor MK-0752 in combination with gemcitabine in patients with pancreatic ductal adenocarcinoma. Br J Cancer. 2018Mar20;118(6):793-801
174. Krop I, Demuth T, Guthrie T, Wen PY, Mason WP, Chinnaiyan P. et al. Phase I pharmacologic and pharmacodynamic study of the gamma secretase (Notch) inhibitor MK-0752 in adult patients with advanced solid tumors. J Clin Oncol. 2012Jul1;30(19):2307-2313
175. Lee SM, Moon J, Redman BG, Chidiac T, Flaherty LE, Zha Y. et al. Phase 2 study of RO4929097, a gamma-secretase inhibitor, in metastatic melanoma: SWOG 0933. Cancer. 2015Feb1;121(3):432-440
176. Cortes JE, Gutzmer R, Kieran MW, Solomon JA. Hedgehog signaling inhibitors in solid and hematological cancers. Cancer Treat Rev. 2019Jun;76:41-50
177. Axelson M, Liu K, Jiang X, He K, Wang J, Zhao H. et al. U.S. Food and Drug Administration approval: vismodegib for recurrent, locally advanced, or metastatic basal cell carcinoma. Clin Cancer Res. 2013May1;19(9):2289-2293
178. Casey D, Demko S, Shord S, Zhao H, Chen H, He K. et al. FDA approval summary: sonidegib for locally advanced basal cell carcinoma. Clin Cancer Res. 2017May15;23(10):2377-2381
179. Kim EJ, Sahai V, Abel EV, Griffith KA, Greenson JK, Takebe N. et al. Pilot clinical trial of hedgehog pathway inhibitor GDC-0449 (vismodegib) in combination with gemcitabine in patients with metastatic pancreatic adenocarcinoma. Clin Cancer Res. 2014Dec1;20(23):5937-5945
180. Kaye SB, Fehrenbacher L, Holloway R, Amit A, Karlan B, Slomovitz B. et al. A phase II, randomized, placebo-controlled study of vismodegib as maintenance therapy in patients with ovarian cancer in second or third complete remission. Clin Cancer Res. 2012Dec1;18(23):6509-6518
181. Ma Y, Yu W, Shrivastava A, Alemi F, Lankachandra K, Srivastava RK. et al. Sanguinarine inhibits pancreatic cancer stem cell characteristics by inducing oxidative stress and suppressing sonic hedgehog-Gli-Nanog pathway. Carcinogenesis. 2017Oct1;38(10):1047-1056
182. Ko AH, LoConte N, Tempero MA, Walker EJ, Kate Kelley R, Lewis S. et al. A Phase I Study of FOLFIRINOX Plus IPI-926, a Hedgehog Pathway Inhibitor, for Advanced Pancreatic Adenocarcinoma. Pancreas. 2016Mar;45(3):370-375
183. Ogawa H, Koyanagi-Aoi M, Otani K, Zen Y, Maniwa Y, Aoi T. Interleukin-6 blockade attenuates lung cancer tissue construction integrated by cancer stem cells. Sci Rep. 2017Sep26;7(1):12317
184. Ying J, Tsujii M, Kondo J, Hayashi Y, Kato M, Akasaka T. et al. The effectiveness of an anti-human IL-6 receptor monoclonal antibody combined with chemotherapy to target colon cancer stem-like cells. Int J Oncol. 2015Apr;46(4):1551-1559
185. Wong AL, Soo RA, Tan DS, Lee SC, Lim JS, Marban PC. et al. Phase I and biomarker study of OPB-51602, a novel signal transducer and activator of transcription (STAT) 3 inhibitor, in patients with refractory solid malignancies. Ann Oncol. 2015May;26(5):998-1005
186. Yoo C, Kang J, Lim HY, Kim JH, Lee M-A, Lee K-H. et al. Phase I Dose-Finding Study of OPB-111077, a Novel STAT3 Inhibitor, in Patients with Advanced Hepatocellular Carcinoma. Cancer Res Treat. 2019Apr;51(2):510-518
187. Jia D, Li L, Andrew S, Allan D, Li X, Lee J. et al. An autocrine inflammatory forward-feedback loop after chemotherapy withdrawal facilitates the repopulation of drug-resistant breast cancer cells. Cell Death Dis. 2017Jul13;8(7):e2932
188. Bilusic M, Heery CR, Collins JM, Donahue RN, Palena C, Madan RA. et al. Phase I trial of HuMax-IL8 (BMS-986253), an anti-IL-8 monoclonal antibody, in patients with metastatic or unresectable solid tumors. J Immunother Cancer. 2019Sep5;7(1):240
189. Schott AF, Goldstein LJ, Cristofanilli M, Ruffini PA, McCanna S, Reuben JM. et al. Phase Ib Pilot Study to Evaluate Reparixin in Combination with Weekly Paclitaxel in Patients with HER-2-Negative Metastatic Breast Cancer. Clin Cancer Res. 2017Sep15;23(18):5358-5365
190. Zhou W, Guo S, Liu M, Burow ME, Wang G. Targeting CXCL12/CXCR4 axis in tumor immunotherapy. Curr Med Chem. 2019;26(17):3026-3041
191. Ott PA, Govindan R, Naing A, Friedlander TW, Margolin K, Lin JJ. et al. 1127OA personal neoantigen vaccine, NEO-PV-01, with anti-PD1 induces broad de novo anti-tumor immunity in patients with metastatic melanoma, NSCLC, and bladder cancer. Ann Oncol. 2018 Oct 1;29(suppl_8)
192. Adams S, Miller GT, Jesson MI, Watanabe T, Jones B, Wallner BP. PT-100, a small molecule dipeptidyl peptidase inhibitor, has potent antitumor effects and augments antibody-mediated cytotoxicity via a novel immune mechanism. Cancer Res. 2004Aug1;64(15):5471-5480
193. Santos AM, Jung J, Aziz N, Kissil JL, Puré E. Targeting fibroblast activation protein inhibits tumor stromagenesis and growth in mice. J Clin Invest. 2009Dec;119(12):3613-3625
194. Edosada CY, Quan C, Tran T, Pham V, Wiesmann C, Fairbrother W. et al. Peptide substrate profiling defines fibroblast activation protein as an endopeptidase of strict Gly(2)-Pro(1)-cleaving specificity. FEBS Lett. 2006Mar6;580(6):1581-1586
195. Narra K, Mullins SR, Lee H-O, Strzemkowski-Brun B, Magalong K, Christiansen VJ. et al. Phase II trial of single agent Val-boroPro (Talabostat) inhibiting Fibroblast Activation Protein in patients with metastatic colorectal cancer. Cancer Biol Ther. 2007Nov;6(11):1691-1699
196. Eager RM, Cunningham CC, Senzer N, Richards DA, Raju RN, Jones B. et al. Phase II trial of talabostat and docetaxel in advanced non-small cell lung cancer. Clin Oncol (R Coll Radiol). 2009Aug;21(6):464-472
197. Eager RM, Cunningham CC, Senzer NN, Stephenson J, Anthony SP, O'Day SJ. et al. Phase II assessment of talabostat and cisplatin in second-line stage IV melanoma. BMC Cancer. 2009Jul30;9:263
198. Loeffler M, Krüger JA, Niethammer AG, Reisfeld RA. Targeting tumor-associated fibroblasts improves cancer chemotherapy by increasing intratumoral drug uptake. J Clin Invest. 2006Jul;116(7):1955-1962
199. Chen M, Lang JY, Li T, Wang Q, Xu G, Lu S. Antitumor effect of a whole tumor cell vaccine expressing human fibroblast activation protein in murine tumor models. J Clin Oncol. 2017May20;35(15_suppl):e14542-e14542
200. Lacouture ME, Dréno B, Ascierto PA, Dummer R, Basset-Seguin N, Fife K. et al. Characterization and Management of Hedgehog Pathway Inhibitor-Related Adverse Events in Patients With Advanced Basal Cell Carcinoma. Oncologist. 2016Aug10;21(10):1218-1229
Author contact
![]() Corresponding author: Dr. Ronald Buckanovich. Email: buckanovichrjmagee.edu
Corresponding author: Dr. Ronald Buckanovich. Email: buckanovichrjmagee.edu