Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Characteristics and mechanisms...
Small-molecule modulators of...
Ferroptosis-related signaling...
Ferroptosis regulation in ARDs
Concluding remarks
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2020; 10(26):11976-11997. doi:10.7150/thno.50663 This issue Cite
Review
Novel insights into ferroptosis: Implications for age-related diseases
Ren-Peng Zhou1,2#, Yong Chen1#, Xin Wei1, Bin Yu1, Zhi-Gang Xiong2, Chao Lu1, Wei Hu1 ![]()
1. Department of Clinical Pharmacology, The Second Hospital of Anhui Medical University, Hefei 230601, China.
2. Department of Neurobiology, Morehouse School of Medicine, Atlanta, GA 30310, USA.
#These authors contributed equally to this work and should be considered co-first authors.
Received 2020-7-14; Accepted 2020-9-29; Published 2020-10-26
Abstract

Rapid increase in aging populations is an urgent problem because older adults are more likely to suffer from disabilities and age-related diseases (ARDs), burdening healthcare systems and society in general. ARDs are characterized by the progressive deterioration of tissues and organs over time, eventually leading to tissue and organ failure. To date, there are no effective interventions to prevent the progression of ARDs. Hence, there is an urgent need for new treatment strategies. Ferroptosis, an iron-dependent cell death, is linked to normal development and homeostasis. Accumulating evidence, however, has highlighted crucial roles for ferroptosis in ARDs, including neurodegenerative and cardiovascular diseases. In this review, we a) summarize initiation, regulatory mechanisms, and molecular signaling pathways involved in ferroptosis, b) discuss the direct and indirect involvement of the activation and/or inhibition of ferroptosis in the pathogenesis of some important diseases, and c) highlight therapeutic targets relevant for ARDs.
Keywords: age-related diseases, ferroptosis, lipid peroxidation, iron, reactive oxygen species
Introduction
Over the past several decades, human life expectancy has steadily increased due to benefits from nutrition, technological advances, and improvements in medical care and vaccination [1]. It is estimated that by 2050, the number of people over the age of 60 will reach 2.1 billion [2]. With this steady increase in the number of older adults, age has become a major risk factor for age-related diseases (ARDs). ARDs are characterized by continuous cell loss and deterioration in the quality or function of tissues and organs, resulting in increased susceptibility and vulnerability to certain diseases [3]. Without control measures, this inevitably leads to loss of mobility, aggravation of disease processes, and increased mortality coupled with severe economic and societal burdens [4]. The etiology and pathogenesis of ARDs are complex and remain unclear, greatly limiting clinical diagnosis and treatment of these diseases. Although undetermined factors cause ARDs, the deleterious and progressive changes in multiple organ systems in most ARDs share some basic mechanistic precepts, including oxidative stress, iron accumulation, inflammation, cell injury, and dysfunction [5-7].
Ferroptosis, an iron-dependent non-apoptotic cell death, can be initiated by small molecules that inhibit glutathione biosynthesis or the glutathione-dependent antioxidant enzyme glutathione peroxidase 4 (GPX4), characterized by the iron-dependent accumulation of reactive oxygen species (ROS) and depletion of plasma membrane polyunsaturated fatty acids (PUFAs) [8, 9]. When the intracellular lipid ROS level exceeds the antioxidant activity of GPX4, it leads to the disruption of redox homeostasis [10]. Ferrous iron can trigger harmful peroxidation of PUFAs in membrane phospholipids by forming toxic lipid radicals, eventually leading to cell death [11]. Iron accumulating within several organs during aging, such as brain and muscle, leads to increased oxidative damage and functional decline. Preclinical Alzheimer's disease patients have the highest iron in the cerebral cortex and cerebellum, accompanied by the gradual impairment of cognitive function, suggesting that an imbalance in iron homeostasis is a precursor of neurodegeneration in Alzheimer's disease [12]. Abnormal iron deposition in the substantia nigra, especially in the RII region, can be used as a biomarker to distinguish Parkinson's disease patients from healthy controls and to assess the severity of the disease [13]. Several studies also have revealed that iron accumulation contributes to cell death of organs and tissues in the pathogenesis and progression of amyotrophic lateral sclerosis, Huntington's disease, cardiomyopathy and type 1 diabetes, and therefore the level of iron as a biomarker for the potential role of ferroptosis and an important causative factor for ARDs [14-17]. Recent advances have identified numerous small molecule ferroptosis inducers and inhibitors, including erastin, glutamate, liproxstatin-1 (Lip-1), and ferrostatin-1 (Fer-1). These compounds have been highly valuable for the study of ferroptosis in different diseases in vivo and in vitro, and the data indicated that ferroptosis might be a potential novel target for therapeutic intervention in ARDs [18, 19].
Emerging studies have confirmed that ferroptosis contributes substantially to the pathogenesis of a variety of ARDs, including neurodegenerative and cardiovascular diseases (CVDs). Intervention in ferroptosis pathways effectively inhibits the progression of these disorders, suggesting ferroptosis as a potential treatment target for these diseases. Herein, we provide a summary of the current knowledge on the mechanism of ferroptosis, its functional roles in the development of ARDs, and its potential for pharmacological and therapeutic targeting of ARDs.
Characteristics and mechanisms of ferroptosis
Ferroptosis represents a non-apoptotic programmed cell death
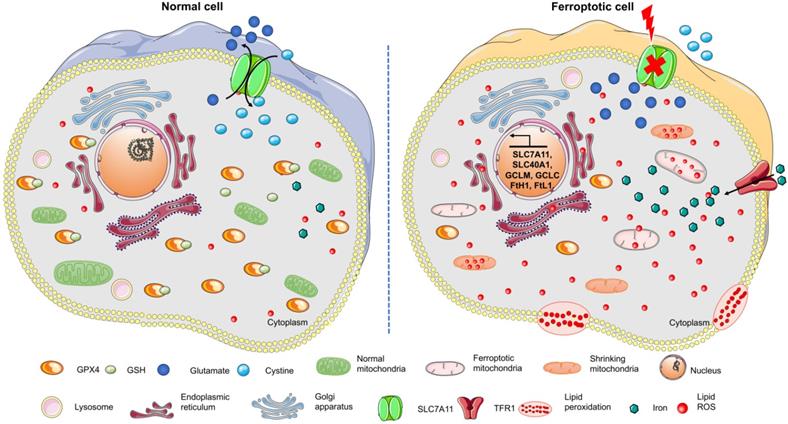
Ferroptosis, a type of cell death induced by erastin or Ras-selective lethal small molecule 3 (RSL3), has attracted much attention in recent years [8]. It is an iron-dependent oxidative cell death that plays a special role in lipid peroxide accumulation in the absence of GPX4. Sensitivity to ferroptosis strongly depends on antioxidant metabolism, lipid homeostasis, and the dynamic equilibrium of iron [20]. As a different form of programmed cell death, ferroptosis plays a critical role in organismal homeostasis and disease pathologies. Unlike the other known forms of programmed cell death, ferroptosis has unique morphological and bioenergy characteristics, including mitochondrial shrinkage, mitochondrial bilateral membrane thickening and rupture, and intracellular NADPH depletion without altering ATP levels (Figure 1). Furthermore, ferroptosis is often accompanied by iron-dependent lipid peroxide accumulation and mitochondrial ROS production. Evidence has confirmed that unlike apoptosis, ferroptosis is not related to caspase activity [21]. Also, necroptosis is accompanied by cellular swelling, disruption of plasma membrane integrity, and the release of intracellular contents, while RIP1/RIP3, the key regulators of necroptosis, are not involved in ferroptosis [22]. The inhibition of autophagy by 3-MA also does not modulate ferroptosis [8]. These findings identify ferroptosis to be a new form of programmed cell death. Given the involvement of ferroptosis in various illnesses, understanding its initiation and underlying regulatory mechanisms may be of great therapeutic significance.
Changes in the morphological and bioenergy characteristics during ferroptosis, including mitochondrial shrinkage, membrane rupture, excess ROS, iron overload, and intracellular GSH depletion. Features of this figure were adapted from Servier Medical Art (http://smart.servier.com/) licensed under a Creative Commons Attribution 3.0 Unported License.
Ferroptosis is induced by cellular iron
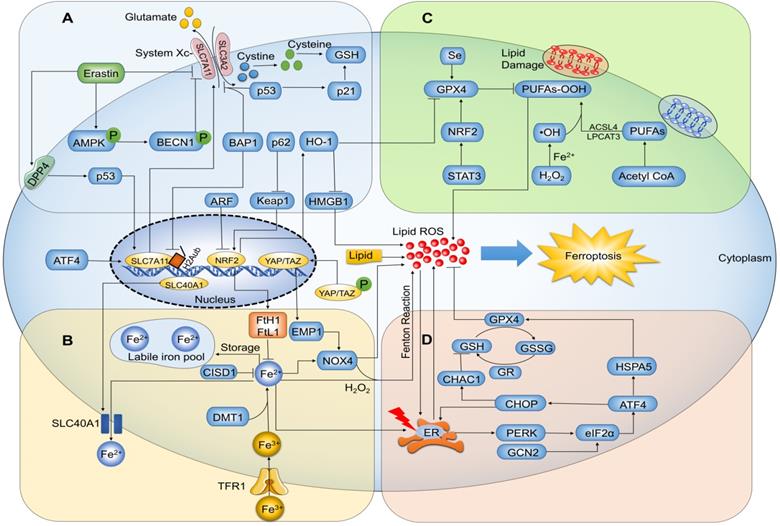
Ferroptosis is a consequence of increased ROS levels caused by elevated intracellular iron concentration, leading to lipid peroxidation and cell death [23] (Figure 2). Iron is a basic component of many enzymes involved in DNA synthesis, heme and iron-sulfur cluster synthesis, and metastasis. As such, it plays a vital role in numerous essential life processes [24]. Free iron is directly linked to ferroptosis because it promotes ROS production via Fenton reactions that cause lipid peroxidation [25]. Ferric iron (Fe3+) bound to transferrin is the main form of circulating iron, which enters cells through membrane protein transferrin receptor 1 (TFR1) and localizes in endosomes, wherein the ferrireductase activity of STEAP3 reduces Fe3+ to ferrous iron (Fe2+). Finally, the divalent metal transporter 1 (DMT1) releases Fe2+ from endosomes into a labile iron pool in the cytoplasm. In general, excess iron is stored in ferritin with ferritin heavy chain 1 (FtH1) and ferritin light chain 1 (FtL1) [26]. Studies have confirmed that ferroptosis-sensitive cells with mutations in Ras display increased expression of TFR1 and reduced expression of ferritin (FtL1 and FtH1) [27]. The iron chelator deferoxamine (DFO) can significantly suppress ferroptosis to protect cells and alleviate ferroptosis-related diseases [28, 29]. Since ferroptosis is mediated by intracellular iron overload, which is regulated by altered iron metabolism, a better understanding of its potential molecular and cellular mechanisms will likely provide novel approaches for ferroptosis regulation.
Ferroptosis-related signaling molecules and signaling pathways. (A) Glutamate exchanges for cystine in a 1:1 ratio through the cystine/glutamate antiporter system Xc-, and inhibition of system Xc- by its core part SLC7A11 induces ferroptosis. (B) Ferric iron (Fe3+) bound to transferrin enters cells via membrane protein transferrin receptor 1 (TFR1) and localizes in endosomes, wherein the ferrireductase activity of STEAP3 reduces Fe3+ to redox-active iron (Fe2+). Finally, divalent metal transporter 1 (DMT1) releases Fe2+ from endosomes into a labile iron pool in the cytoplasm. In general, excess iron is stored in ferritin with ferritin heavy chain 1 (FtH1) and ferritin light chain 1 (FtL1). Under the action of H2O2, Fe2+ catalyzes the production of hydroxyl radical (HO∙) by Fenton reaction, triggering a chain reaction of radical lipid peroxidation and eventually leads to ferroptosis. (C) Ferroptosis is trigged by peroxidation (-OOH) of polyunsaturated fatty acids (PUFAs) and aberrant accumulation of lipid reactive oxygen species (ROS), resulting in membrane destabilization and rupture. Acyl-CoA synthetase long-chain family member 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3) are necessary for ferroptosis to produce the target lipid pool containing arachidonic acid. GPX4 can hydrolyze lipid peroxides into non-toxic lipid alcohols (-OH). (D) Intracellular glutathione exists as oxidized glutathione (GSSG) and reduced glutathione (GSH); GPX4 requires GSH as a cofactor and reduces GSSG to GSH via glutathione reductase (GR). GPX4 inhibits the formation of Fe2+-dependent ROS by converting lipid hydroperoxides into lipid alcohols, and thus inhibits ferroptosis.
Ferroptosis is regulated by mitochondrial iron metabolism
Mitochondria are the primary site of iron utilization, play a major role in regulating oxidative metabolism, and are also the main source of ROS [30]. As the most common metal in mitochondria, iron plays an important role in regulating the physiological function of these organelles [31]. Iron usually crosses both the outer and inner mitochondrial membranes to reach the matrix, where mitochondrial iron metabolism occurs. Iron transport across the inner mitochondrial membrane depends on the membrane transporter mitoferrin 1/2 (Mfrn1/2), the imbalance of which can lead to mitochondrial iron accumulation and oxidative injury [32]. Mfrn1/2 damage has been observed in a variety of neurological diseases associated with ferroptosis [33]. Moreover, mitochondrial iron metabolism is also regulated by the voltage-dependent anion channels, located in the outer mitochondrial membrane [34]. Previous studies demonstrated that erastin-induced voltage-dependent anion channel 2/3 opening was correlated with mitochondrial iron accumulation and ferroptosis [35]. It has also been reported that the accumulation of mitochondrial free iron aggravated erastin-mediated ferroptosis [36]. Physiologically, mitochondrial free iron is strictly controlled by mitochondrial ferritin (FtMt), which can protect cells from the damage caused by mitochondrial ROS [37]. Also, overexpression of FtMt can sequester iron in mitochondria and resist erastin-induced ferroptosis in vivo and in vitro [38].
Ferroptosis is induced by cysteine deprivation
Amino acid metabolism is closely related to ferroptosis regulation, and cysteine is essential for ferroptosis regulation since its availability limits the biosynthesis of glutathione. Cysteine is oxidized to cystine, and an oxidized cysteine dimer linked by a disulfide bridge can be easily transported to mammalian cells as a natural analog of cysteine 2. In cells, cystine is reduced to cysteine, which is an indispensable substrate for the synthesis of biomolecules such as glutathione, proteins, and coenzyme A3 [39, 40]. As a heterodimeric cell surface amino acid antiporter, system Xc- is composed of the 12- transmembrane helix-containing transporter protein SLC7A11 (also known as xCT) linked by a disulfide bridge to the single-pass transmembrane regulatory protein SLC3A2 (4F2hc) [9]. While glutamate can exchange for cystine in a 1:1 ratio via system Xc-, accumulation of extracellular glutamate induces ferroptosis by inhibiting system Xc- [8, 41]. Importantly, erastin can directly inhibit system Xc- function resulting in significant depletion of intracellular glutathione [42]. Cysteine in the culture medium is oxidized to cystine when molecular oxygen levels are high, and reductant levels are low. Therefore, SLC7A11 can protect cultured cells against cell death. Erastin disrupts cystine uptake by SLC7A11 and leads to cell death via inhibiting SLC7A11 activity, resulting in cysteine deficiency, glutathione depletion, and ferroptosis [41, 43]. Thus, compounds or drugs that specifically influence the function of SLC7A11 and thereby modify ferroptosis have been explored in vivo and in vitro for their potential to treat various human diseases.
Ferroptosis is induced by GSH depletion
Glutathione (γ-L-glutamyl-L-cysteinyl glycine) is a tripeptide containing a cysteine unit at its core that plays a key role in protecting against lipid peroxidation in ferroptosis by donating an electron to GPX4 [41]. Intracellular glutathione exists as reduced (GSH) and oxidized glutathione (GSSG), providing the main antioxidant buffer against oxidative stress [44]. Studies have demonstrated that glutamate-cysteine ligase, the first rate-limiting enzyme in the two-step synthesis of glutathione, could be inhibited by buthionine-(S, R)-sulfoximine (BSO), leading to cell death. DFO and a-tocopherol could reverse this effect, but not the necroptosis inhibitor Necrostain-1 or the apoptosis inhibitor zVAD-fmk [9, 45, 46]. Further studies confirmed that erastin induced ferroptosis by GSH down-regulation caused by depletion of intracellular cysteine, whereas p53-p21 signaling delayed ferroptosis by preserving GSH levels, and thereby had a pro-survival effect [47, 48]. As a 12 kDa ubiquitous oxidoreductase, thioredoxin plays an essential role in the thioredoxin antioxidant system, composed of NADPH, thioredoxin, and thioredoxin reductase [49]. In mammalian cells, thioredoxin and glutathione systems can cross-donate electrons and serve as backup systems for each other [50]. Telorack et al. demonstrated that the thioredoxin systems could efficiently compensate deficiency in glutathione biosynthesis in keratinocytes to maintain antioxidant capacity [51]. Therefore, inhibition of ferroptosis induced by glutathione depletion is an essential mechanism preventing oxidative stress and ferroptotic cell death.
Ferroptosis is prevented by GPX4
GPX4, the only member of the GPX protein subfamily (GPX1-8), can reduce phospholipid hydrogen peroxide. It contains an efficient selenocysteine unit that can increase its peroxidase activity [52, 53]. GPX4 inhibits the formation of Fe2+-dependent ROS by converting lipid hydroperoxides into lipid alcohols. Hence, inhibition of GPX4 leads to an increase of lipid ROS formation and lipid peroxidation, which induces ferroptosis [54]. Evidence has revealed that GPX4 knockdown directly inhibits ferroptosis but does not affect other essential mechanisms [55]. Consistent with this, lack of cysteine diminishes GSH synthesis and reduces GPX4 activity, eventually leading to ferroptosis [56, 57]. RSL3, the first reported effective GPX4 inhibitor identified by chemical screening, has been widely used in the experimental induction of ferroptosis, especially in cancer chemotherapy [27]. Notably, GPX4 ablation in adult mice resulted in embryonic lethality as evidenced by elevated 4-hydroxylnonenal (4-HNE), reduction in the activity of electron transport chain complexes I and IV, and decreased ATP production in mitochondria that eventually led to neuronal loss, suggesting that GPX4 has an essential role in mitochondrial integrity and neuronal survival [58]. Another study confirmed that ferroptosis, rather than apoptosis, is the leading cause of embryonic lethality [45]. Additionally, GPX4 is also involved in T cell immunity, as evidenced by GPX4 levels, which were lower in HIV-infected cell populations than in uninfected cells by using 75Se-labeled human Jurkat T cells [59]. Another study revealed that GPX4-deficient T cells could rapidly accumulate membrane lipid peroxides accompanied by ferroptosis-mediated cell death rather than necroptosis [60]. Since GPX4 can act as an important negative regulatory factor of ferroptosis by scavenging toxic intracellular lipid hydroperoxides, the development of drugs for the regulation of GPX4 is of great practical significance.
Ferroptosis is induced by PUFAs
Excessive PUFA consumption, especially red and processed meat, has been associated with nutritional and environmental health hazards [61]. High PUFA intake indicates an increased risk of ARDs, including cancers, type 2 diabetes, and CVDs, but the specific molecular mechanism remains unclear [62]. Ferroptosis can be driven by excessive peroxidation of PUFAs, characterized by iron-catalyzed excessive peroxidation of PUFA-containing phospholipids [63]. Although PUFAs can increase membrane fluidity and have beneficial effects on human health [64], exposure to excess substrates (iron or glutamate) can trigger enzyme-linked reactions by activating enzymes associated with the biosynthesis and remodeling of PUFAs in the cell membrane. These enzymes include lysophosphatidylcholine acyltransferase 3 (LPCAT3) and acyl-Co synthetase long-chain family member 4 (ACSL4), as well as enzymes that increase intracellular ROS, such as NADPH oxidase [65]. PUFAs are oxidized by intracellular ROS and produce lipid peroxides that lead to ferroptosis [66]. The destruction of cell membranes by lipid peroxidation can cause morphological changes, such as mitochondrial shrinkage and damage. Further, lipid peroxides decompose into reactive derivatives, including 4-HNE and malondialdehyde (MDA), which react with nucleic acids and proteins and thus destroy membrane integrity resulting in cell rupture [67]. A recent study demonstrated that exogenous monounsaturated fatty acids could induce cellular resistance to ferroptosis and reduce the accumulation of lipid peroxides and oxidizable PUFAs [68]. It is noteworthy that close interactions between PUFA metabolism, ROS, GPX4, and ferroptosis may ultimately determine cell survival or death. Hence, the regulatory mechanism of ferroptosis provides a new perspective for cell fate determination.
Small-molecule modulators of ferroptosis
Small molecule inducers of ferroptosis
Ferroptosis was initially defined through a group of small molecules (RAS selective lethal, RSL) that induce selective death of tumor cells carrying RAS mutations [69]. Exploiting the high mutagenicity of the RAS family of small GTPases (NRAS, HRAS, and KRAS), Stockwell and colleagues identified two new RSL small molecules named RSL3 and erastin. Mechanistically, erastin mediates ferroptosis via inhibition of system Xc- and RSL3 functions by inhibiting GPX4; both inhibitors can cause ferroptosis without inducing morphological changes or biochemical processes similar to apoptosis [27, 70]. Other studies have found that sorafenin [71] and glutamate [29] also induced ferroptosis via cystine uptake inhibition by the system Xc-. GSH inhibits ferroptosis by maintaining GPX4 function, essential to prevent harmful phospholipid oxidation. In this respect, a series of small molecules, including RSL3 [55] and FIN56 [72] was found to inactivate GPX4 and induce lipid peroxide accumulation directly, leading to ferroptosis. Other inducers of ferroptosis, such as buthionine sulfoximine [73] and cisplatin [74], may induce synthetic lethality similar to that caused by GSH depletion (Table 1).
Small molecule regulators of ferroptosis
| Regulators | Compounds | Structures | Targets | Model systems |
|---|---|---|---|---|
| Inducers | Erastin | 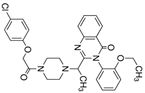 | System Xc- | B16, HT-1080 [228], HT-1080, 143B, BJeLR, Calu-1 [8], Islets [225], MEF, A2780 [74]. |
| Sorafenib | 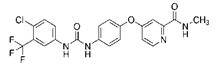 | System Xc- | HepG2, Hep3B, Huh7 [8], Huh7, PLC/PRF5, ACHN, BxPC-3, Caki-1, HCT116, SK-MEL-3, HT-29, NCI-H460, PANC-1 [71]. | |
| Sulfasalazine | 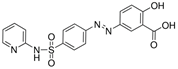 | System Xc- | BJeLR, HT-1080 [8], B16, HT-1080 [228]. | |
| Glutamate | 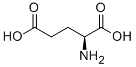 | System Xc- | HT-1080 [8], HT-22 [29]. | |
| (1S,3R)-RSL3 | 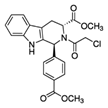 | GPX4 | B16, HT-1080 [228], HT-1080, 143B [8], Islets [225], BJeLR, HT-1080 [55]. | |
| FIN56 | 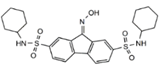 | GPX4 | BJeLR, HT-1080 [72]. | |
| FINO2 | 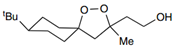 | Iron | HT-1080 [229]. | |
| BAY11-7085 | 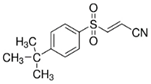 | HO-1 | MDA-MB-231, MDAMB-468, MCF-7, SKBR3, A549, HuH-7, DBTRG-05MG, SKOV3 [230]. | |
| t-BuOOH | 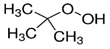 | Lipid peroxidation | NIH3T3, ARPE-19 [81]. | |
| Buthionine Sulfoximine | 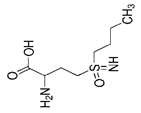 | GSH | B16, HT-1080 [228], HT-1080, BJeLR, DRD [55], HCT116, A549 [73]. | |
| Cisplatin | 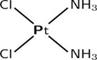 | GSH | MEF, A2780 [74]. | |
| Inhibitors | Ferrostatin-1 | 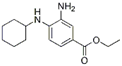 | Lipid ROS | HT-22 [29], ARPE-19 [81], DAUDI, CA-46 [93], HEK-293, HT22, MEF [69], HD brain-slice, oligodendrocytes, HT-1080 [77], PTCs [80]. |
| Liproxstatin-1 | 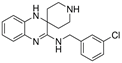 | Lipid ROS | HT-22 [29], HEK-293, HT22, MEF [69], DAUDI, CA-46 [93], RILF [231], intestinal I/R [83], MOR [78]. | |
| SRS16-86 | 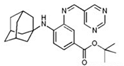 | Lipid ROS | IRI [79]. | |
| Inhibitors | Vitamin E | 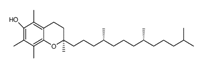 | Lipid ROS | MiaPaCa-2 [232]. |
| Trolox | 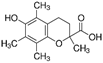 | Lipid ROS | HCC1937, MDAMB-231, Hs 578T [97] | |
| Deferasirox | 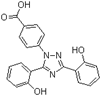 | Iron | AMI [28]. | |
| Deferoxamine | 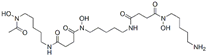 | Iron | ARPE-19 [81], HT-22 [29], DAUDI, CA-46 [93], PTCs [80], MEFs [28]. | |
| Zileuton | 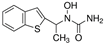 | 5-LOX | HT22 [82]. | |
| N-acetylcysteine | 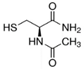 | 5-LOX | Neurons, IHC [187], PTCs [80]. | |
| Rosiglitazone | 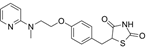 | ACSL4 | Intestinal I/R [83]. |
GPX4, glutathione peroxidase 4, HO-1, heme oxygenase 1, GSH, reduced glutathione, ROS, reactive oxygen species, 5-LOX, 5-lipoxygenase, ACSL4, acyl-Co synthetase long-chain family member 4, “NA”: not application.
Small molecule inhibitors of ferroptosis
Most of the ferroptosis mechanisms were elucidated by the identification of cell death inhibitors, which are classified based on the ferroptosis-triggered lipid peroxidation mechanism. The disruption of redox homeostasis is one of the main causes of ferroptosis; both zileuton and N-acetylcysteine can protect cells from lipid peroxidation by down-regulating 5- lipoxygenase (LOX) [75, 76]. Fat-soluble antioxidants, such as aryl alkylamine compounds Fer-1 and Lip-1, can specifically reduce ROS production to inhibit RSL-induced ferroptosis [77, 78]. A recent study reported that SRS-16-86, a third-generation ferrostatin, has improved plasma and metabolic stability and acts as a ROS scavenger leading to ferroptosis blockage [79]. Also, cellular iron overload is a significant feature in ferroptosis, and commonly used iron-chelating agents, such as deferasirox, deferiprone (DFP), or DFO, can reduce cell death caused by excessive free iron [80, 81]. As a member of the long-chain fatty-acid-coenzyme A ligase family, ACSL4 is a key enzyme that regulates lipid composition. A previous report demonstrated that the expression of ACSL4 was remarkably down-regulated in ferroptosis-resistant cells, such as LNCaP and K562, and might serve as a contributor and biomarker of ferroptosis [82]. Consistent with this observation, ACSL4 inhibitor rosiglitazone could pharmacologically modulate ACSL4 activity to suppress lipid peroxidation and prevent ferroptosis in vivo and in vitro [83].
Regulation of ferroptosis by natural compounds
For decades, natural products were investigated as promising reagents for drug development. Many substances separated from plentiful natural resources are used to prevent and treat various diseases [84]. Numerous roles of natural compounds in iron metabolism and homeostasis have been identified that are relevant to ferroptosis and treating ferroptosis-related diseases. Artemisia annua L. (Asteraceae) has been used in traditional Chinese medicine for 2000 years and is now used as the first-line treatment of malaria throughout Asia and Africa. Interestingly, research in recent years has shown that its biological activity is not limited to malaria treatment. Artemisin and its derivatives (e.g., artesunate, artemisinin, and dihydroartemisinin) have been found to induce ferroptosis in cancer cells through ROS accumulation, an overload of lipid peroxides and iron, and triggering antioxidant stress responses [85-87]. Llabani et al. [49] discovered that ferroptocide is a novel compound that can induce rapid ferroptosis and inhibit thioredoxin in primary cancer cells and immortalized cancer cell lines from patients.
Various natural compounds can induce ferroptosis, while others act as ferroptosis inhibitors to provide new tools for treating other diseases. Baicalein (from Scutellaria baicalensis), a selective inhibitor of arachidonate 12/15-LOX, has been identified through natural product library screening as a ferroptosis inhibitor [88], which exerts neuroprotection against post-traumatic epileptic seizures through ferroptosis suppression [89]. Other natural compounds, such as gastrodin [29] and puerarin [90], are related to renal damage, glutamate-induced cell death, and heart failure through ferroptosis-associated mechanisms or inhibiting ferroptosis signaling pathways. Therefore, inhibition of ferroptotic death using natural products may provide new therapeutic strategies for ferroptosis-related diseases.
Ferroptosis-related signaling molecules and signaling pathways
ATF4 signaling
Activating transcription factor 4 (ATF4), a member of the cAMP response element-binding protein-2 family, participates in regulating multiple signaling pathways including autophagy, translation, oxidative stress, and inflammation, suggesting that it plays a multifaceted role in a variety of pathological processes [91]. Under normal circumstances, ATF4 is constitutively expressed only at low concentrations. However, upon stimulation by microenvironmental stresses, such as anoxia and hypoxia and endoplasmic reticulum stress sensed by upstream eukaryotic translation initiation factor 2α (eIF2α) kinases, expression of ATF4 is elevated to influence development, metabolism, redox balance, and angiogenesis [92]. Also, endoplasmic reticulum stress is related to the activation of ATF4-C/EBP homologous protein (CHOP) pathway, associated with ferroptosis-related diseases such as Burkitt's lymphoma and diabetes myocardial ischemia/reperfusion (I/R) injury [93, 94]. A previous study showed that free uncharged tRNAs could trigger the GCN2-ATF4 axis to mediate a well-characterized transcriptional amino acid response under amino acid deprivation [95]. As an amino acid deficiency sensor, GCN2-ATF4 signaling guides transcriptional control and protein synthesis and degradation [96]. Further, degradation of GSH caused by ATF4 target gene CHAC1 enhanced cystine starvation-induced ferroptosis by the GCN2-eIF2α-ATF4 axis in human triple-negative breast cancer cells [97]. Moreover, heat shock 70 kDa protein 5 (HSPA5), considered to be a molecular chaperone mediating endoplasmic reticulum unfolding of proteins, has been shown to negatively regulate ferroptosis by preventing GPX4 degradation [98]. In glioma cells, dihydroartemisinin induces HSPA5 expression by protein kinase R-like ER kinase (PERK), and up-regulates the activity of ATF4, resulting in increased GPX4 and ferroptosis inhibition [85]. The role of the ATF4-HSPA5-GPX4 axis has also been verified in human pancreatic ductal adenocarcinoma cells in a negative feedback pathway, offering a promising therapeutic strategy for overcoming drug resistance in tumors [99]. As a critical mediator of oxidative and metabolic homeostasis, ATF4 has a dual role in ferroptotic cell death via complex networks of signal regulation and control [100]. Further studies are needed for clarification of the precise regulatory effects of ATF4 on ferroptosis.
NOX4 signaling
NADPH oxidase (NOX) is a major enzyme that transfers electrons from NADPH to molecular oxygen and shuttles electrons across biological membranes to produce superoxide. To date, five NOX genes (NOX1-5) have been identified in the human genome. Abnormal expression of NOX4 affects cell proliferation and apoptosis and is responsible for a variety of pathological processes [101, 102]. Previous reports identified NOX4 to be a novel source of mitochondrial oxidative stress in cardiac myocytes and macrophages. Knockdown of NOX4 inhibits intracellular ROS production, macrophage cytotoxicity, and mitochondrial and DNA damage, implicating NOX4 in oxidative stress-mediated cell injury [101, 103]. In HF rats induced by aortic banding, Toll-like receptor 4 (TLR4) or NOX4 gene knockout can dramatically improve left ventricular remodeling and reduce cardiomyocyte death by suppressing autophagy and ferroptosis [104]. Based on this evidence, pharmacological inhibition or knockdown of NOX4 may partially prevent ferroptosis-induced cell death. Remarkably, ferroptosis is triggered by pseudolaric acid B in glioma cells via activating NOX4 by intracellular Fe2+, resulting in the overproduction of lipid peroxides and H2O2 and can be abolished by DFO, suggesting a novel target for cancer treatment [105]. In summary, NOX4 can mediate various signaling pathways that participate in the induction of ferroptosis, and pharmacological blockade or genetic inactivation of NOX4 may protect against cell death.
BECN1 signaling
BECN1 is a key macroautophagy regulator that promotes the formation of autophagosomes [21]. A recent report revealed a novel role of BECN1-SLC7A11 complex formation in ferroptosis regulation in cancer cells. Mechanistically, phosphorylation of BECN1 at Ser90/93/96 through AMP-activated protein kinase (AMPK) promotes ferroptosis by binding to SLC7A11 and directly blocking the activity of system Xc-, contributing to cancer cell death [106]. BECN1-SLC7A11-mediated ferroptosis was also observed in SH-SY5Y neuroblastoma cells [107]. Another study found that the BECN1 expression mediated by ELAV-like RNA binding protein 1 (ELAVL1) could also promote ferroptosis by inducing autophagy and ferritinophagy activation in hepatic stellate cells [108]. These findings collectively indicate that BECN1 can regulate both ferroptosis and autophagy induction, but its specific regulatory mechanism and pathophysiological significance remain to be elucidated. In recent years, ferroptosis has been described as autophagy-dependent cell death under specific conditions, since the classic activators of ferroptosis, including erastin and RSL3, can increase autophagy flux in various cells. Excessive autophagy, especially NCOA4-facilitated ferritinophagy, STAT3-induced lysosomal membrane permeabilization, and BECN1-mediated system Xc- inhibition may promote ferroptotic cell death [109, 110]. Therefore, it is essential to measure autophagic activity and flux during ferroptosis to better understand the process and function of autophagy-dependent ferroptosis.
YAP/TAZ signaling
Transcriptional regulators, such as yes-associated protein (YAP), and transcriptional co-activators with PDZ-binding motif (TAZ), known as Hippo signaling cascade effectors, have attracted widespread attention due to their relevance to organ growth, tissue homeostasis, cell proliferation, and cancer. YAP/TAZ are sensors of structural and mechanical cues mediated by the cellular microenvironment, making them exploitable as therapeutic targets in cancer and regenerative medicine [111, 112]. A recent study showed that E-cadherin suppresses ferroptosis by activating the intracellular NF2 and Hippo signaling pathway in epithelial cells, while antagonizing this signaling pathway enables YAP to promote ferroptosis, suggesting that the NF2-YAP axis is responsible for the cancer cells' response to ferroptosis-inducing therapy [113]. Yang et al. demonstrated that the ferroptosis-promoting effect of TAZ was attributed to its ability to regulate ferroptotic cell death through the TAZ-ANGPTL4-NOX2 axis in epithelial ovarian cancer [114]. Moreover, TAZ can also regulate membrane protein 1 and NOX4 levels, resulting in lipid peroxidation and ferroptosis in renal cell carcinoma [115]. Together, these findings indicate that YAP/TAZ and Hippo pathway effectors play a novel role in lipid peroxidation by triggering ferroptosis and have therapeutic potential for epithelial ovarian cancer, renal cell carcinoma, and other TAZ-activated tumors, and might be exploited to modulate ferroptosis.
NRF2 signaling
Nuclear factor erythroid 2-related factor 2 (NRF2), as a transcription factor, participates in the adaptive cellular response following exposure to oxidative and electrophilic stresses. NRF2 binds to antioxidant response elements and promotes a variety of antioxidant gene transcription [116]. NRF2 is mainly complexed with Kelch-like ECH-associated protein 1 (Keap1) and CUL3E3 ubiquitin ligase to maintain its stability via ubiquitin [117]. Mechanistically, NRF2 activation can promote iron storage, reduce iron uptake, limit ROS production, regulate SLC7A11 activity and thus regulate ferroptosis [118, 119]. A recent study demonstrated that the NRF2-Keap1 pathway plays a critical role in cancer cell proliferation and lowering ferroptosis via up-regulating SLC7A11 and amplifying glutamate secretion [120]. Expression of p62 also likely prevents the degradation of NRF2 and enhances subsequent NRF2 nuclear accumulation through Keap1 inactivation, leading to ferroptosis inhibition [121]. On the other hand, alternative reading frame (ARF) tumor suppressor can regulate ferroptotic responses by directly inhibiting the transcriptional role of NRF2 and suppressing its target genes, including SLC7A11. Chen et al. found that ARF inhibited the SLC7A11-activating ability of NRF2, resulting in tumor suppression by inducing ferroptosis in p53 null cells [122]. Shin et al. proposed that NRF2-antioxidant response element (ARE) pathway activation contributed to making head and neck cancer (HNC) cells refractory to GPX4 inhibition via a reduced labile iron pool, leading to ferroptosis resistance [123]. Also, among the iron metabolism proteins associated with iron availability and ferroptosis, both ferritin and heme oxygenase 1 (HO-1) are affected by NRF2 [10]. A recent study reported that HO-1 knockout could promote ferroptosis induced by erastin in kidney cells and hepatocellular carcinoma [121]. Cotreatment with erastin and acetaminophen decreased the expression of HO-1, whereas activation of NRF2 up-regulated HO-1, suggesting that acetaminophen sensitized ferroptosis by regulating the NRF2/HO-1 signaling axis [124]. Hence, NRF2 can act as a key negative regulatory factor of ferroptosis in complex molecular signaling networks and plays a protective role in cell death.
p53 signaling
The tumor suppressor gene p53 inhibits tumorigenesis by initiating apoptosis, cell cycle arrest, and senescence. Recent research has challenged this notion by demonstrating p53 to be a transcriptional repressor of SLC7A11 that can impair cysteine import and promote ferroptosis. Under specific conditions, p53 is activated by various stress stimuli and favors organismal homeostasis by additional mechanisms, including ferroptosis induction. Various signaling pathways participate in ferroptosis regulation by p53 [105, 125, 126]. For example, p53 can indirectly activate the ALOX12 function via SLC7A11, leading to tumor suppression through a distinct ferroptosis pathway [127]. Interestingly, Wang et al. uncovered a previously unappreciated epigenetic mechanism of ferroptosis regulation in which p53 negatively regulates mono-ubiquitination of histone H2B on lys120 (known as an epigenetic mark) by promoting the nuclear translocation of the deubiquitinase USP7, and represses SLC7A11 expression [128]. Furthermore, spermidine/spermine N1-acetyltransferase 1 (SAT1) is a transcription target of p53 and participates in ferroptosis regulation during tumor suppression. In brief, p53-mediated SAT1 activation contributes to ferroptotic responses, and elevated SAT1 expression results in lipid peroxidation and overexpression of 15-LOX, thereby sensitizing cells to undergo ferroptosis during ROS stress [129]. Given the finding that multiple signaling regulators and pathways are involved in ferroptosis, regulating these important signaling molecules and their transduction pathways is of great significance for understanding the pathophysiology of ferroptosis (Table 2).
Ferroptosis-related signaling molecules and signaling pathways
| Signaling molecules | Signaling pathways | Effects of signaling molecules | Mechanisms | References |
|---|---|---|---|---|
| ATF4 | PERK-ATF4-HSPA5 | Inhibition of ferroptosis in glioma cells. | PERK-upregulated ATF4 inducted HSPA5 expression and increased GPX4. | [85, 99]. |
| GCN2-eIF2α-ATF4 | Induction of ferroptosis in human triple negative breast cancer cells. | GCN2 activation increased eIF2α, ATF4 and CHAC1, CHAC1 degraded GSH. | [97]. | |
| ATF4-CHOP-CHAC1 | Induction of ferroptosis in Burkitt's Lymphoma. | The ATF4-CHOP-CHAC1 axis degraded intracellular GSH and up-regulated CHAC1. | [93]. | |
| ATF4-SLC7A11 | Inhibition of ferroptosis in human gliomas. | ATF4 activation elevated SLC7A11. | [100, 233]. | |
| ATF4-CHOP | Induction of ferroptosis in DIR injury. | The activation of ATF4-CHOP produced ERS and interacted with ROS in ferroptosis. | [94]. | |
| NOX4 | TAZ-EMP1-NOX4 | Induction of ferroptosis in renal cell carcinoma. | TAZ up-regulated EMP1, EMP1 increased NOX4 and resulted lipid peroxidation. | [115]. |
| TLR4-NOX4 | Induction of ferroptosis in rats with heart failure. | TLR4 knock-down repressed NOX4, which inhibited cell loss. | [104]. | |
| Fe2+-NOX4-H2O2 | Induction of ferroptosis in glioma cells. | Fe2+ activated NOX4 resulting in H2O2 and lipid peroxides overproduction. | [105]. | |
| EGFR-MAPK-NOX4/GPX4 | Induction of ferroptosis in nonsmall-cell lung cancer cells. | Activated EGFR stimulated MAPK signaling, reduced GPX4 and induced NOX4. | [234]. | |
| BECN1 | AMPK-BECN1-SLC7A11 | Induction of ferroptosis in tumor suppression. | AMPK-Mediated BECN1 phosphorylation blocked SLC7A11. | [106, 107]. |
| ELAVL1/HuR-BECN1-autophagy | Induction of ferroptosis in hepatic stellate cells. | ELAVL1 triggered autophagy and promoted autophagic ferritin degradation by banding to the AREs of the BECN1 mRNA 3'-UTR. | [108]. | |
| YAP/TAZ | E-cadherin-NF2-Hippo-YAP | Induction of ferroptosis in epithelial cells. | E-cadherin activated the intracellular NF2 and Hippo signaling pathway to suppress ferroptosis. | [113]. |
| TAZ-ANGPTL4-NOX2 | Induction of ferroptosis in epithelial ovarian cancer. | TAZ-regulated ANGPTL4 sensitized ferroptosis by activating NOX2. | [114]. | |
| TAZ-EMP1-NOX4 | Induction of ferroptosis in renal cell carcinoma. | TAZ up-regulated EMP1, EMP1 increased NOX4 and resulted lipid peroxidation. | [115]. | |
| NRF2 | NRF2-TGF-β1 | Inhibition of ferroptosis in lung fibrosis. | NRF2 signaling down-regulated TGF-β1 and balanced the ROS level. | [231]. |
| NRF2-HO-1 | Inhibition of ferroptosis in non-small-cell lung cancer. | NRF2 rescued HO-1 downregulation. | [124]. | |
| STAT3-NRF2-GPX4 | Inhibition of ferroptosis in osteosarcoma cells. | Over-activation of STAT3/NRF2 increased GPX4 activity. | [235]. | |
| NRF2-Keap1 | Inhibition of ferroptosis in primary malignant brain tumors. | NRF2-Keap1 signaling upregulated SLC7A11 and amplified glutamate secretion. | [120]. | |
| p62-Keap1-NRF2 | Inhibition of ferroptosis in hepatocellular carcinoma cells. | p62 prevented the degradation of NRF2 and enhanced subsequent NRF2 nuclear accumulation via of Keap1 inactivation. | [121]. | |
| NRF2/p62-ARE | Resistance to ferroptosis in head and neck cancer. | p62-Keap1 interaction activated NRF2, increased ARE resulting in a decreased labile iron pool. | [123]. | |
| ARF-NRF2 | Induction of ferroptosis in tumor suppression. | ARF inhibited NRF2 ability to activate its target genes SLC7A11. | [122]. | |
| p53 | p53-USP7-H2Bub1-SLC7A11 | Sensitizing cells to erastin-induced ferroptosis. | p53 negatively regulated H2Bub1by promoting the nuclear translocation of the deubiquitinase USP7 and repressed the expression of SLC7A11. | [128]. |
| p53-ALOX12 | Induction of ferroptosis in tumor suppression. | p53 activated ALOX12 indirectly by transcriptional repression of SLC7A11. | [127]. | |
| p53-SLC7A11 | Induction of ferroptosis in tumor suppression. | p53 repressed SLC7A11 transcription, reduced cystine uptake, and limited GSH. | [105, 236, 237]. | |
| SOCS1-p53 | Induction of ferroptosis in tumor suppression. | SOCS1 activated p53 via both phosphorylation and stabilization. | [238]. | |
| p53-STAT1-ALOX15 | Induction of ferroptosis in tumor suppression. | p53 directly activated SAT1, and increased the expression of ALOX15. | [129]. |
ATF4, activating transcription factor 4; PERK, protein kinase R-like ER kinase; HSPA5, heat shock 70 kDa protein 5; eIF2α, translation initiation factor 2α; CHOP, C/EBP homologous protein; SLC7A11, solute carrier family 7 member 11; NOX4, NADPH oxidase 4; TAZ, transcriptional coactivator with PDZ-binding motif; EMP, epithelial membrane protein 1; TLR4, Toll-like receptor 4; EGFR, epidermal growth factor receptor; GPX4, glutathione peroxidase 4; AMPK, AMP activated protein kinase; ELAVL1/HuR, ELAV like RNA binding protein 1; YAP, yes-associated protein; NRF2, nuclear factor (erythroid-derived 2)-like 2; TGF-β1, transforming growth factor-β1; HO-1, heme oxygenase-1; STAT3, signal transducer and activator of transcription 3; Keap1, Kelch-like ECH associated protein 1; MAPK, mitogen activated protein kinase; ARE, antioxidant response elements; ARF, alternative reading frame; H2Bub1, monoubiquitination of histone H2B at lysine 120; ALOX12, arachidonate 12-lipoxygenase.
Ferroptosis regulation in ARDs
A growing body of research suggests that ferroptosis contributes to the progression of ARDs, including neurodegenerative and cardiovascular diseases while blocking ferroptosis by pharmacological agents or gene manipulations can inhibit cell injury, prevent disease progression, and improve disease symptoms. It has been reported that treatment with Lip-1 was neuroprotective in vitamin E-deficient diet-fed GPX4BIKO mice [130], and Fer-1 could significantly inhibit lipid peroxidation and ferroptotic cell death in cellular models of Huntington's disease [77]. Furthermore, inhibition of ferroptosis with DFO or NAC treatment significantly reduced iron abundance and the level of oxidative stress, as well as increased cardiomyocyte viability in rat neonatal cardiomyocytes [131] (Table 3).
Signs of ferroptosis and ferroptosis in age-related diseases
| Diseases | Model systems | Biomarkers | Effects of blocking ferroptosis | References |
|---|---|---|---|---|
| NDs | ||||
| AD | P301S Tau transgenic mice | Iron, SOD1, GPX4, xCT, ROS, FPN1, TFR, | Tau phosphorylation↓, iron overload↓, lipid peroxidation↓, inflammation↓, learning ability↗, spatial memory↗. | [139] |
| HDI-treated APP/PS1 mice | FPN, TFR, DMTI, ROS, mitochondria dysfunction | NA | [134] | |
| HT22 cells | GSH, xCT, GR, GCL, GST, ROS. | ROS accumulation↓, Ca2+ influx↓, oxidative stress-induced cell death↓. | [133, 142] | |
| SH-SY5Y cells | Lipid peroxidation | Aβ1-42 aggregation induced toxicity↓, lipid peroxidation↓. | [140] | |
| HT22 cells, BV-2 cells, AD mice model | GSH, NRF2 | ATP loss↓, cell survival↑, neuroinflammation↓, short-term memory↗. | [141] | |
| GPX4BIKO mice | GPX4, lipid peroxidation | Neural protein NeuN↑, Synaptophysin↑, SNAP25↑, neurodegeneration↓, inflammation↓. | [130] | |
| PD | LUHMES cells | Oxidative stress, ROS | New brain cells↑, oxidative stress↓, cell death↓. | [239] |
| GPX4 knockout mice | GPX4, oxidative stress | NA | [154] | |
| SH-SY5Y cells | Lipid peroxidation | ROS/RNS↓, α-syn aggregation↓, cell death↓. | [240, 241] | |
| LUHMES cells, MPTP mice model, OSCs | SLC7A11, GPX4, GSH | MPTP's toxicity↓, dopaminergic neurons loss↓. | [155] | |
| ALS | NSC-34 cells | Oxidative stress, ROS | New brain cells↑, oxidative stress↓, cell death↓. | [239] |
| Plasma samples of patients | Lipid peroxidation, ferritin, iron | NA | [161] | |
| SN4741, N27 cells, primary cortical neurons | Lipid peroxidation, FeII | Lipid peroxidation↓, lipid radicals↓, ferroptotic lethality↓, cell death↓. | [242] | |
| MS | EAE, patients | GSH, GPX4, xCT, γ-glutamylcysteine ligase | NA | [166, 243] |
| EAE | NA | Active EAE disease↓, T-cell function↓, inflammatory cell infiltrates↓, the clinical signs↓. | [167, 168] | |
| HD | mN90Q73 HD mice | ROS | Healthy medium spiny neurons↑. | [77] |
| HD (R6/2) transgenic mice, Human tissue samples, the striatal neurons | Lipid peroxidation | 4-HNE adduct formation↓, ATP generation↗, mitochondrial morphology and function↗, mice lifespan↑. | [179] | |
| HD patients, HD animal model | Lipid peroxidation, GSH, SOD, CAT | NA | [178] | |
| R6/2 HD mice | TFR, FPN, IRPs, iron | Rota-rod endurance↗, lateral ventricles on the treated side↓. | [175] | |
| Stroke | Hippocampal neurons, I/R gerbils | MDA, SOD1, CAT, TFR-1, GPX4, FPN1 | Lipid peroxide↓, cell death↓. | [182] |
| MCAO rats | DMT1, ROS, TFR1, SCL7A11, GPX4, MDA | Iron deposition↓, neurobehavioral scores↓, the numbers of Nissl bodies and visible nuclei↑. | [183] | |
| Cortical neurons, ICH mice and rats | GSH, ALOX5 | Neutralizing toxic lipids↓, neuronal death↓, functional recovery↗. | [187] | |
| Focal cerebral ischemia model | Iron | Cognitive impairment↗, ongoing neuronal damage↓. | [184] | |
| ICH mice, primary cortical neurons, HT22 | NA | Neuronal death↓, hemoglobin-and hemin-induced toxicity↓. | [185] | |
| CVDs | ||||
| Cardiomyopathy | H9c2 cells, I/R rat model | ROS, GPX4, ACSL4, NRF2, MDA, SOD, Fe2+ | Cardiomyocyte death↓, myocardial injury↓, the cardiac function of ischemic cardiomyopathy↗. | [94, 194] |
| H9c2 cells, Nrf2-/- mice, I/R mice model | MDA, NRF2, iron, oxidized lipids | Cardiac hypertrophy Anp, Bnp, and Myh7↓, cardiac function↗, mitochondrial function↗. | [195] | |
| HF | H9c2 cells, aortic banding rats | GPX4, FtH1, iron, NOX4 | Cell viability↑, mitochondrial atrophy↓, striated muscle arrangement↗. | [90] |
| NOX4 knock-down aortic banding rats | GPX4, FtH1 | Myocyte area noted↓, myocyte death↓. | [104] | |
| MI | Cardiomyocytes, I/R mice model | ROS, iron, TFR1, ferritin | Cardiomyocyte death↓. | [200] |
| MI mice model, H9c2 and C2C12 cells, NRVMs | MDA, GSH, GPX4, ACSL4, ROS | Myocardial cell death↓, lipid peroxidation↓. | [203] | |
| AMI mice model, MEFs | SLC7A11, GCLC, FtH1, FtL1, GSH | Cardiomyocyte death↓, the severity of AMI↓. | [28] | |
| Other ARDs | ||||
| DM | DM and DIR model | GPX4, ACSL4, NRF2, MDA, SOD, Fe2+ | The myocardial tissue lesions↓. | [94] |
| MIN6 cells, NaAsO2-exposed rats | GSH, T-SOD, GPX4, MDA, ROS, COX2 | Mitochondrial membrane potential↓, cytochrome c↓, MtROS↑. | [226] | |
| COPD | HBECs, GPX4+/- and GPX4 TG mice | GPX4, iron, ferritin | Lipid peroxidation↓, cell death↓, lung airspace enlargement↓, airway wall thickening↓. | [210] |
ARDs, age-related diseases; NDs, neurodegenerative diseases; AD, Alzheimer's disease; PD, Parkinson disease; HD, Huntington's disease; ALS, amyotrophic lateral sclerosis; MS, multiple sclerosis; CVDs, cardiovascular diseases; HF, heart failure; MI, myocardial infarction; DM, diabetes mellitus; COPD, chronic obstructive pulmonary disease;
“↑”: upregulation; “↓”: downregulation; “↗”: improving; “NA”: not application.
Neurodegenerative diseases
Alzheimer's disease (AD)
AD is one of the most common neurodegenerative diseases of the central nervous system, characterized by neurofibrillary tangles and amyloid-β (Aβ) plaques in the brain. It is mainly manifested as progressive cognitive dysfunction and impaired behavior in the clinic. With the acceleration in the global aging process, AD affects nearly 44 million people worldwide [132]. Although the scientific community and governments are vigorously promoting the development of new drugs for AD, there is no specific cure at present; symptomatic treatment and delay in disease progression are the only available measures, presenting a major challenge. Abnormal and massive deposition of Aβ plaques is a major pathological mechanism for AD, and drugs that target Aβ are highly sought after. However, in recent years, drugs targeting the Aβ protein have often failed in clinical trials. Thus, attention is being focused on gaining new molecular insights into AD development and seeking new treatment strategies to slow the progress of the disease.
Iron is crucial for the healthy development of the brain as it is used in the synthesis of neurotransmitters, myelin production, myelination, neuronal development, and other cell functions. It is reported that iron levels are elevated in the brain of individuals suffering from clinical AD and contribute to disease progression [12]. Iron accumulation leads to nerve cell damage in patients with AD, likely by potentiating GSH loss [133] and iron deposition results in lipid peroxidation in cells, causing ferroptotic cell death. A recent study demonstrated that following treatment with high dietary iron, expression levels of ferroptosis-related antioxidants, including SLC7A11, GPX4, and superoxide dismutase in the brain, were decreased in APP/PS1 mice (a transgenic mice model of AD), suggesting that iron-induced neuron loss might occur through ferroptosis [134]. Therefore, chelating iron ions may have a therapeutic effect on AD by inhibiting ferroptosis. Notably, the mitochondrial iron storage protein FtMt could prevent mitochondria from iron-induced oxidative injury, and FtMt knockout significantly aggravated the learning and memory impairment in an AD mouse model [135]. Huang et al. reported that Mfrn1 knockdown decreased mitochondrial iron and ROS levels, thus delaying the disease progression in Alzheimer model of C. elegans, characterized by the reduction of paralysis rate and the extension of lifespans [136]. Also, the iron chelator DFO had therapeutic effects on patients with AD [137]. Compared to the control group, DFO treatment at a low dose could slow the clinical progression of AD-related dementia compared with the control group. Moreover, multi-target iron chelators HLA-20 ([5-(4-propargylpiperazin-1-ylmethyl)-8-hydroxyquinoline]) and M30 ([5-(N-methylN-propargylaminomethyl)-8-hydroxyquinoline]) also had potential therapeutic effects on sporadic AD [138]. The iron-chelating action of α-lipoic acid could also suppress ROS production and increase GPX4 and SLC7A11 expressions in P301S mice, suggesting that α-lipoic acid treatment might enhance neuronal survival by regulating ferroptosis [139].
Many recent studies have revealed that some compounds with anti-AD effects have an inhibitory effect on neuronal loss related to ferroptosis. For example, the chalcone derivative 14a-c exhibited a potent anti-ferroptotic cell death activity against RSL3- or erastin-induced ferroptosis by inhibiting lipid peroxidation [140]. Similarly, 7-O-cinnamoyltaxifolin and 7-O-feruloyltaxifolin inhibited ferroptosis induced by RSL3, and the novel oxindole compound GIF-0726-r also prevented ferroptosis induced by erastin in HT22 cells, a murine hippocampal neuronal cell line [141, 142]. These findings indicated that natural product hybrids with suppressive effects on ferroptosis might serve as preventive neuroprotectants for treating neurodegenerative disorders such as AD. In another study, ferroptosis inhibitor Lip-1 treatment improved neurodegeneration in vitamin E-deficient diet-fed GPX4BIKO mice (a mouse model of conditionally deleting GPX4 in forebrain neurons) [130]. Thus, although the precise effects and mechanisms of ferroptosis in the pathogenesis of AD remain still unclear, these results suggest that targeting ferroptosis could provide opportunities for developing novel AD treatments.
Parkinson's disease (PD)
PD is a progressive neurodegenerative disease characterized by dopaminergic neuronal death in the substantia nigra pars compacta (SNc), eventually resulting in rigidity, resting tremors, and other motor symptoms. Although the precise cause of dopaminergic neuronal loss is still unclear, it has been suggested that iron-induced dopaminergic degeneration is a key event in PD pathogenesis [143]. The concentration of iron is elevated in the SNc of deceased and living PD patients and is considered a pathognomonic hallmark of the disease [144]. Iron accumulation induces ferroptosis characteristics such as elevated hydroxyl radicals and lipid peroxidation, likely contributing to the oxidative injury of nigral dopaminergic neurons in PD [145]. Therefore, chelating iron ions can inhibit ferroptosis and protect against neuronal injury in PD. Treatment with iron chelators can prevent dopaminergic neuronal loss in the SNc and rescue motor deficits in PD mouse model [146, 147]. In the MPTP-induced parkinsonian phenotype, FtMt was shown to protect against neuronal damage by inhibiting cellular iron accumulation and subsequent oxidative stress [148]. Importantly, a double-blind, placebo-controlled randomized clinical trial in PD patients has shown that DFP is safe and effective for PD treatment [147], suggesting that inhibition of ferroptosis by iron chelation might also provide therapeutic opportunities for PD patients.
Loss of glutathione in the substantia nigra is a major feature of PD. Depleted glutathione can cause nigral dopaminergic neuronal death and progressive motor imbalance [149]. Clinical trials showed that glutathione level was restored following glutathione administration, indicating its mild therapeutic effect in PD patients [150, 151]. GPX4, an important lipid repair enzyme in the inhibition of ferroptosis, was reduced in the SNc of deceased PD patients, while its up-regulation was associated with neuron density [152]. Ablation of glutathione by depleting intracellular cysteine levels with erastin treatment, suppressing glutathione availability as a substrate for GPX4, could induce ferroptosis [46, 55]. Additionally, GPX4 exerted a protective effect against neurodegeneration by regulating ferroptosis in the PD pathology [130]. Depletion of GPX4 induced ferroptosis in motor neurons, leading to dramatic motor neuron degeneration and paralysis [153]. Furthermore, GPX4 could prevent neuronal dysfunction and PD-like symptoms, and GPX4 loss in dopaminergic neurons induced anxiety behavior and diminished spontaneous locomotor activity [154].
Fer-1 derivatives, as iron chelators, are drug candidates for pharmacologically modulating ferroptosis [155]. Administration of vitamin E, a ferroptosis inhibitor, can delay motor neuron death and paralysis caused by GPX4 depletion. The ferroptosis inhibitor Fer-1 could inhibit 1-methyl-4-phenylpyridinium (MPP+)-induced dopaminergic neuroblastoma cell (SH-SY5Y) death in vitro, a widely used PD model [156]. Recently, Do Van et al. showed that neurotoxins, including erastin and MPP+, commonly used in PD models, could induce ferroptosis in LUHMES cells, a human neuronal precursor cell line [155]. Furthermore, pre-treatment with ferroptosis inhibitors, including Fer-1 and the iron chelator DFP prevented the toxicity of glutathione depletion in LUHMES cells and MPTP toxicity in mice. Overall, these results demonstrate that dopaminergic neuronal loss in PD may partially be due to ferroptosis, indicating that blocking ferroptosis may have a neuroprotective effect on PD.
Amyotrophic lateral sclerosis (ALS)
ALS is a devastating neurodegenerative disease caused by lower and upper motor neuron loss, resulting in progressive paralysis and death. Although its etiology is not fully understood, motor neuron death is considered one of the main causes. Therefore, unveiling the mechanism of motor neuron death and intervening against it may provide a treatment strategy for ALS. The dysregulation of iron metabolism has been shown to play a vital role in ALS pathophysiology [157]. Serum iron and ferritin levels are higher in ALS patients than controls and are associated with lower survival rates in ALS patients. Similar phenomena are observed in animal models of ALS. In a transgenic mouse model of ALS with G37R mutation in superoxide dismutase 1 (SOD1 G37R), iron levels were elevated in ventral motor neurons and glia. After treatment with the iron-selective chelator salicylaldehyde isonicotinoyl hydrazine, the lifespan of SOD1 G37R mice could be extended, spinal motor neuron survival was up-regulated and motor function improved [158]. Similarly, iron accumulated in the spinal cords of SOD1 G93A-transgenic mice, another ALS model [157]. More importantly, treatment with iron chelators, including M30 and VK-28, could delay the disease onset, extend the life of G93A-SOD1 ALS mice, and mitigate motor neuron damage [159, 160]. Recently, a phase III clinical trial identified four biomarkers closely related to ferroptosis [161]. Thus, pharmacological intervention with iron chelators (and ferroptosis inhibitors) can significantly improve the disease symptoms in animal models of ALS. Although the specific mechanism relating ferroptosis to ALS is unknown, these studies indicate that blocking ferroptosis may be a potential treatment for ALS.
Multiple sclerosis (MS)
MS is an autoimmune disease characterized by inflammatory demyelination of the central nervous system, targeting oligodendrocytes and myelin. Although the pathogenesis of MS is still unknown, new insights suggest that oligodendrocyte loss is one of the key pathophysiological events [162]. Several studies have revealed that abnormal iron metabolism and the resulting cytotoxicity contribute to neurodegeneration and hence the pathogenesis and progression of MS [163, 164]. Recent histological and magnetic resonance imaging (MRI) results have shown a high concentration of iron in the brains of MS patients and in an experimental autoimmune encephalomyelitis (EAE) animal model of MS, especially in oligodendrocytes [163-165]. Accumulation of iron contributes to progressive axonal degeneration in MS through increased ROS production and the promotion of iron-mediated oxidative damage [163].
Following oxidative stress, lipid peroxidation and free radicals play an essential role in MS pathogenesis. Iron overload and lipid peroxidation have been observed in MS and EAE, indicating that ferroptosis may occur in MS. GPX4 levels were decreased in MS brains and EAE spinal cords, and GCLc, SLC7A11, and GSH levels were also significantly decreased in EAE mice relative to controls. The levels of lipid peroxidation products, MDA and 4-HNE, were also increased in EAE mice compared with controls and so was the proportion of damaged mitochondria with irregular matrices, disrupted membranes, and degenerated cristae [166]. Furthermore, the chelation of iron using DFO and DFP (also a ferroptosis inhibitor) reduced the severity of EAE [167, 168]. These findings indicate that neuronal damage of EAE shares the common characteristic features of ferroptosis, identifying it as a potential therapeutic target for MS progression.
Huntington's disease (HD)
HD, a neurodegenerative disorder, is caused by an abnormal repetition of the CAG trinucleotide sequence in the huntingtin gene, characterized by motor, behavioral, and cognitive dysfunction [169]. The disease has characteristic features with selective loss of medium-spiny neurons and the formation of intraneuronal protein aggregates. Despite our understanding of HD's genetics, the precise mechanisms of neuronal death are still not completely understood. Hence, there is no effective intervention available to prevent or delay the development of HD. Iron dysregulation and its accumulation in cellular and subcellular sites of the brain are also implicated in HD pathogenesis. Previous studies reported a significant increase in iron levels in HD patients' basal ganglia that occurs early in the disease process [170]. MRI and quantitative susceptibility mapping revealed excessive iron deposition in the occipital cortex, globus pallidum, and putamen in HD patients [171-173]. Ferritin iron and ferroportin accumulation in striatum and cortex was also observed in HD patients compared with healthy controls [174, 175]. Intraventricular administration of DFO or oral DFP (iron-selective chelation) relieved HD symptoms in an R6/2 mouse model [175, 176]. Furthermore, decreased GSH levels occur in HD patients and HD mice induced by 3-nitropropionic acid (3-NP) and, and supplementing with cystamine or cysteamine could restrain 3-NP-induced HD striatal neuronal death by up-regulating GSH levels [177, 178]. Besides, extensive lipid peroxidation was observed in HD patients and R6/2 and mN90Q73 HD mouse models [77, 178, 179]. More importantly, Fer-1 treatment significantly inhibited lipid peroxidation and iron-induced cell death in cellular models of HD [77]. Taken together, these findings suggest that ferroptosis might play a deleterious role in HD development, and inhibiting ferroptosis may provide an important strategy for the treatment of HD.
Stroke
Stroke has become one of the most common causes of morbidity and mortality worldwide and is the leading cause of disability. The incidence of stroke increases with age, doubling each decade after 45, with more than 70% of strokes occurring in individuals above 65. At present, the clinical treatment of stroke is still limited to intervention measures to restore blood flow by drug or mechanical thrombolysis with limited success, and there are no effective measures to protect the brain from ischemic cell death [180]. Research on brain injury after stroke has mainly focused on excitotoxicity, inflammation, oxidative stress, and apoptosis [181]. Oxidative stress has a crucial role in neuropathological lesions, and abundant non-heme iron in the brain triggers membrane lipid peroxidation via Fenton chemistry, leading to brain edema, mitochondrial damage, and functional disorders. Notably, oxidative injury has become a key index for evaluating I/R-induced neuronal injury. However, it is difficult to translate these benefits to the clinic. Therefore, clarification of protective mechanisms and the development of optimized pharmacological blocking agents are imperative [182].
Accumulating evidence revealed that ferroptosis contributes to stroke [183] and its inhibition can significantly ameliorate the disease severity and improve functional recovery. Tuo et al. reported that tau-mediated iron export could protect against ferroptotic injury after ischemic stroke [184], and carvacrol increased GPX4, thereby inhibiting ferroptosis from protecting against hippocampal neuron I/R injury after ischemic stroke in gerbils [182]. Ferroptosis also induced neuronal death after hemorrhagic stroke [185] and a single dose of Se delivered into the brain could drive the expression of GPX4, protect neurons, and improve behavior in a hemorrhagic stroke model [186]. Also, N-acetylcysteine (NAC), a precursor of GSH, targeted ALOX5-derived toxic arachidonic acid and synergistically acted with prostaglandin E2 to inhibit ferroptosis and improve the prognosis of mice after hemorrhagic stroke [187]. Therefore, modulators of ferroptosis are potential pharmacological targets of stroke.
Cardiovascular diseases
Cardiomyopathy
Cardiomyopathy is a heterogeneous group of myocardial diseases correlated with structural and functional abnormalities caused by the heart's abnormal mechanical and electrical activity, characterized by inappropriate ventricular hypertrophy or dilatation. Severe cardiomyopathy can cause cardiovascular death or progressive HF [188]. Cell loss caused by terminally differentiated cardiomyocyte death is an important cause of cardiomyopathy. Different forms of cell death related to cell loss, such as autophagy, apoptosis, and necrosis, have been confirmed in myocardial injury [189].
The role of ferroptosis in myocardial pathology has also been investigated in recent years. Iron homeostasis plays a critical role in myocardial injury, and an iron overload cardiomyopathy is caused by the accumulation of iron in the myocardium [190]. Moreover, myocardial hemorrhage can contribute to iron deposition in cardiac tissue, resulting in excessive ROS production, triggering pathological events such as inflammation [191]. Doxorubicin has high cardiotoxicity, limiting its clinical application, and iron chelators exert cardioprotective effects against this cardiotoxicity [192]. Ferroptosis, characterized by altered iron status, is also associated with cardiac oxidative stress during cardiac dysfunction. Studies demonstrated that ferroptosis was associated with diabetic myocardial I/R injury, and its inhibition could alleviate the injury [94]. Moreover, the activities of glutathione peroxidase and SOD were decreased in myocardial tissues of diabetic cardiomyopathy rats, while the level of MDA was increased, and inhibition of these changes could protect against oxidative stress and inflammation in myocardial tissue [193]. Another study showed that a lipid kinase ENPP2 involved in lipid metabolism in cardiomyocytes could protect these cells against erastin-induced ferroptosis [194]. Results from Fang et al. strongly supported the idea that ferroptosis may serve as a target for the prevention of cardiomyopathy, and pharmacologically blocking ferroptosis and iron chelation therapy may provide a new strategy for the treatment of fatal heart disease [195].
Heart failure (HF)
Heart failure (HF) is a pathological condition in which the heart fails to pump enough blood to meet the body's need. It can be induced by many reasons, including myocytes' loss caused by cell death during the final stage of CVD. Under hemodynamic stress, such as hypertension or myocardial infarction, compensatory cardiomyocytes lead to myocardial hypertrophy, and if left uncontrolled, this hypertrophic response culminates in ventricular dilatation and progressive cell loss, eventually developing into HF [90]. A previous study [131] demonstrated a significant reduction in FtH in an in vivo mouse model of HF and showed that iron deposition and the resulting increase in oxidative stress in hearts after myocardial infarction contributes to cardiomyocyte death. Moreover, treatment with DFO or NAC could significantly decrease the abundance of iron and the level of oxidative stress, as well as increase the viability in rat neonatal cardiomyocytes harboring an adenoviral vector expressing short hairpin RNA targeted to FtH [131]. Lapenna et al. demonstrated that in contrast to young adult rabbits, aged rabbits possessed higher levels of redox-active catalytic low molecular weight iron, coupled with greater lipid and protein oxidation in the heart tissue. DFO administration could reduce H2O2/iron (Fenton reaction)-dependent damage in perfused hearts of aged rats but not of young adult rats. This suggests that iron status may be responsible for cardiac oxidative stress and hemodynamic dysfunction [196].
Evidence supports the idea that diabetic patients with HF display abnormal myocardial iron status, and DFO could alleviate coronary microvascular adaptation by inhibiting iron-catalyzed oxidative reactions [197]. Interestingly, in HF mice induced by hypobaric hypoxia, treatment with two novel nitronyl nitroxide radicals could reduce oxidant stress via free radical scavenging activity, resulting in increased SOD activity, catalase, and GSH-Px, and reducing MDA and ROS [198]. Liu and colleagues reported that ferroptosis was directly involved in HF, as demonstrated by an elevated labile iron pool and lipid peroxide levels in the HF rat model. Additionally, ferrastin-1 could reverse the decrease in erastin-induced cell viability, while the TLR4/NOX4 pathway promoted myocyte death by ferroptosis and autophagy during HF [90, 104]. To sum up, these findings indicate that ferroptosis is associated with HF pathology and targeting ferroptosis may provide a novel anti-HF strategy.
Myocardial infarction (MI)
MI, known as heart attack, is an irreversible heart muscle death following a prolonged lack of oxygen/ischemia supply. MI is the most common cause of death worldwide, and elucidation of the underlying mechanisms represents a major opportunity and challenge for prevention and treatment [199]. Iron deposition in peri-infarcted and non-infarcted areas has been observed in MI mice following the left coronary artery ligation [131]. One study demonstrated that both ferroptosis inducers (e.g., erastin and RSL3) and excess iron accelerated iron incorporation, lipid ROS generation, and triggered cell death in isolated adult mouse cardiomyocytes. These effects were inhibited by Fer-1, implicating the involvement of ferroptosis [200]. The increase in ROS was attributed to reduced activity of antioxidant enzymes SOD, GPX1, and catalase following MI [201]. Interestingly, supplementation with the cellular antioxidant GSH could enhance myocardial resistance to I/R in vivo and protect the intact heart against oxidative damage, suggesting that oxidative stress was involved in cardiac tissue injury and cardiomyocyte death [202]. Furthermore, Park et al. proposed that MI could induce the reduction of GPX4, which may sensitize cardiac cells to ferroptosis under low GSH conditions [203]. A recent mechanistic study showed that BTB domain and CNC homolog 1 (BACH1), as a regulator of iron and heme metabolism, could promote ferroptosis by coordinating transcriptional regulation of GSH and labile iron metabolism; BACH1-/- mice were more resistant to MI than wild-type mice, and DFO could relieve the severity of ischemic injury [28]. Thus, preventing ferroptosis may provide a new therapy for patients with MI.
Other age-related diseases
Chronic obstructive pulmonary disease (COPD)
COPD is a group of chronic lung disorders, characterized by a slowly progressive irreversible bronchial obstruction. Its main pathological manifestation is pulmonary emphysema, and it often occurs in the elderly. COPD is mainly caused by cigarette smoke (CS), and without a curative treatment, it has become a leading cause of premature death in industrialized countries [204]. Thompson et al. demonstrated that iron concentration was increased by smoking in bronchoalveolar lavage fluid and alveolar macrophages, resulting in disruption of iron homeostasis [205]. The elevated iron concentrations in the lungs increased the risk of pulmonary injury [206]. DeMeo's team identified an important susceptibility gene for COPD, an iron-responsive element-binding protein that could up-regulate mitochondrial iron loading in association with CS-induced inflammation and lung injury [207, 208]. Interestingly, as a key risk factor of COPD, CS could trigger iron-catalyzed oxidative stress and lead to lung injury, suggesting that oxidant/antioxidant balance plays a critical role in COPD [209]. A recent report demonstrated that CS could induce epithelial cell ferroptosis in COPD, indicating accumulated labile iron and enhanced lipid peroxidation, which could be eliminated by GPX4 knockout or Fer-1 treatment [210]. The GSH-based antioxidant protection system plays a key role in oxidant/antioxidant imbalance in patients with COPD, and GSH-Px makes an important contribution to maintain lung function [138, 211]. A clinical study on COPD patients confirmed that doxycycline treatment could dramatically decrease lipid hydroperoxides and overall oxidative stress while increasing GSH-Px, GSH, and total nitrite antioxidant capacity, thereby improving lung function [212]. Given the important roles of iron homeostasis and lipid peroxidation in COPD, targeting ferroptosis might provide a novel opportunity for COPD treatment.
Diabetes mellitus (DM)
DM, a chronic metabolic and degenerative disease is characterized by hyperglycemia due to defective insulin secretion or insulin dysfunction [213]. Dysfunctional islet β-cell secretion and programmed cell death are two associated pathological processes [214, 215]. Islet β-cell death plays a crucial role in the occurrence and development of type 2 DM (T2DM) and suppressing islet β-cell death is a challenging clinical problem. Iron overload is an important factor leading to the deterioration of diabetes [216]. Circulating iron and ferritin levels, a biomarker for increased body iron stores, are significantly elevated in patients with T2DM [217, 218]. Furthermore, excess free reactive Fe2+ can catalyze ROS formation through the Fenton reaction, which induces oxidative stress [218]. Plasma levels of enzymes, such as GSH and SOD, and H2O2 concentrations are reduced in diabetic patients and animal models [219-221]. Additionally, blood levels of lipid peroxidation and MDA are higher in diabetic patients than in healthy individuals [220, 222]. It has been demonstrated that high glucose could increase MDA levels and reduce SOD and GPX4 activities in SRA01/04 cells [223]. Moreover, the functional variant GPX4 (rs713041) regulates the risk of complications in patients with type 1 diabetes [224]. The GPX4 protein abundance was decreased in the DM rat myocardial tissue compared with normal rats. Inhibition of ferroptosis using Fer-1 could reduce DM myocardial I/R injury in vivo and cell injury in vitro [94]. Also, ferroptosis-inducing agents such as RSL3 and erastin could induce human islet death and compromise islet function in vitro, which could be ameliorated by pre-treatment of islets with Fer-1 or DFO [225].
Ferroptosis is also related to arsenic-induced islet β cell dysfunction, and Fer-1 can suppress NaAsO2-induced ferroptotic islet β cell death and pancreatic dysfunction by inhibiting the mitochondrial ROS-autophagy-ferritin pathway [226]. These findings collectively suggest that iron imbalance, oxidative stress, and lipid peroxidation often occur in diabetic patients, resulting in ferroptosis and consequently exacerbating pancreatic function loss, indicating that ferroptosis blockade may provide a potential therapeutic strategy for DM. Nevertheless, further research is required to clarify the role of ferroptosis inhibitors in DM animal models and accurately define the specific biological effects of ferroptosis in this age-related disorder in vitro and in vivo.
Concluding remarks
Since the discovery of ferroptosis, researchers have mainly focused on tumor prevention and treatment. However, given our expanding understanding of the impact of ferroptosis, its role in other age-related diseases has recently received much attention. Human aging is accompanied by a general decline in physiological functions, especially during the later stages. Consequently, there is an increase in the incidence of neurodegenerative diseases, CVDs, and other ARDs. Depleted GPX4 and GSH, elevated iron, and excessive lipid peroxidation are common features in ferroptosis and ARDs. Accumulating evidence has demonstrated that cells that undergo ferroptosis could secrete factors that strongly activate the innate immune system, leading to lipid peroxidation, the root cause of tissue damage and organ failure. This is especially true for neurodegenerative disorders, CVDs, and diabetes, where ferroptosis may underlie neuronal loss and damage to cardiomyocytes and β-cells correlated with these diseases.
Ferroptosis is likely to be a major cause of degenerative diseases, but it is not known whether the pathological mechanisms and signaling pathways in animal models closely resemble those in human patients. Although the regulatory mechanisms and molecular pathways in ferroptosis have been extensively explored using in vivo and in vitro disease models, strong evidence for ferroptosis in human cells and human autopsy tissues is still lacking. Furthermore, ferroptosis simply provides a connection between the phenotype of basic organ dysfunction and the observed accumulation of lipid peroxidation products in human pathology, but the mechanism by which ferroptosis regulates cell and tissue degeneration is still unclear. Previous results have shown that iron accumulates in aging tissues [227]; however, whether ferroptosis is related to cell senescence and tissue aging in ARDs needs further investigation. Despite intriguing questions, there are no clinical trials to directly investigate the effects of ferroptosis-specific inhibitors or activators in age-related degenerative diseases. Therefore, in future studies, targeting ferroptosis as a potential strategy for treating ARDs is clearly promising.
Numerous studies have provided insights into the mechanisms and factors associated with the regulation of ARDs by ferroptosis. In this context, the development of antibodies and drugs against ferroptosis might benefit patients with a broad spectrum of ARD-related diseases. Research in this field is still in its infancy, and much work is needed to elucidate the detailed processes and mechanisms through which ferroptosis participates in ARDs. Future work is expected to provide novel therapeutic strategies for preventing, controlling, and treating ARDs.
Acknowledgements
This project was supported by the National Natural Science Foundation of China under grant 81902182, the Natural Science Foundation of Anhui Province under grant 1908085QH317, and the National Natural Science Foundation Incubation Program of The Second Hospital of Anhui Medical University under grant 2019GMFY03.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Dzau VJ, Balatbat CA. Health and societal implications of medical and technological advances. Sci Transl Med. 2018 10
2. Amaya-Montoya M, Perez-Londono A, Guatibonza-Garcia V, Vargas-Villanueva A, Mendivil CO. Cellular Senescence as a Therapeutic Target for Age-Related Diseases: A Review. Adv Ther. 2020;37:1407-24
3. Luo J, Mills K, le Cessie S, Noordam R, van Heemst D. Ageing, age-related diseases and oxidative stress: What to do next? Ageing Res Rev. 2020;57:100982
4. Figueira I, Fernandes A, Mladenovic Djordjevic A, Lopez-Contreras A, Henriques CM, Selman C. et al. Interventions for age-related diseases: Shifting the paradigm. Mech Ageing Dev. 2016;160:69-92
5. Xu J, Knutson MD, Carter CS, Leeuwenburgh C. Iron accumulation with age, oxidative stress and functional decline. PLoS One. 2008;3:e2865
6. Jin H, Randazzo J, Zhang P, Kador PF. Multifunctional antioxidants for the treatment of age-related diseases. J Med Chem. 2010;53:1117-27
7. Franceschi C, Garagnani P, Parini P, Giuliani C, Santoro A. Inflammaging: a new immune-metabolic viewpoint for age-related diseases. Nat Rev Endocrinol. 2018;14:576-90
8. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE. et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060-72
9. Cao JY, Dixon SJ. Mechanisms of ferroptosis. Cell Mol Life Sci. 2016;73:2195-209
10. Kajarabille N, Latunde-Dada GO. Programmed Cell-Death by Ferroptosis: Antioxidants as Mitigators. Int J Mol Sci. 2019 20
11. Lee J, You JH, Shin D, Roh JL. Inhibition of Glutaredoxin 5 predisposes Cisplatin-resistant Head and Neck Cancer Cells to Ferroptosis. Theranostics. 2020;10:7775-86
12. Smith MA, Zhu X, Tabaton M, Liu G, McKeel DW Jr, Cohen ML. et al. Increased iron and free radical generation in preclinical Alzheimer disease and mild cognitive impairment. J Alzheimers Dis. 2010;19:363-72
13. Ghassaban K, He N, Sethi SK, Huang P, Chen S, Yan F. et al. Regional High Iron in the Substantia Nigra Differentiates Parkinson's Disease Patients From Healthy Controls. Front Aging Neurosci. 2019;11:106
14. Chen L, Hua J, Ross CA, Cai S, van Zijl PCM, Li X. Altered brain iron content and deposition rate in Huntington's disease as indicated by quantitative susceptibility MRI. J Neurosci Res. 2019;97:467-79
15. Diez-Lopez C, Comin-Colet J, Gonzalez-Costello J. Iron overload cardiomyopathy: from diagnosis to management. Curr Opin Cardiol. 2018;33:334-40
16. Sheelakumari R, Madhusoodanan M, Radhakrishnan A, Ranjith G, Thomas B. A Potential Biomarker in Amyotrophic Lateral Sclerosis: Can Assessment of Brain Iron Deposition with SWI and Corticospinal Tract Degeneration with DTI Help? AJNR Am J Neuroradiol. 2016;37:252-8
17. Stordal K, McArdle HJ, Hayes H, Tapia G, Viken MK, Lund-Blix NA. et al. Prenatal iron exposure and childhood type 1 diabetes. Sci Rep. 2018;8:9067
18. Conrad M, Pratt DA. The chemical basis of ferroptosis. Nat Chem Biol. 2019;15:1137-47
19. Dong T, Liao D, Liu X, Lei X. Using Small Molecules to Dissect Non-apoptotic Programmed Cell Death: Necroptosis, Ferroptosis, and Pyroptosis. Chembiochem. 2015;16:2557-61
20. Sang M, Luo R, Bai Y, Dou J, Zhang Z, Liu F. et al. Mitochondrial membrane anchored photosensitive nano-device for lipid hydroperoxides burst and inducing ferroptosis to surmount therapy-resistant cancer. Theranostics. 2019;9:6209-23
21. Xu T, Ding W, Ji X, Ao X, Liu Y, Yu W. et al. Molecular mechanisms of ferroptosis and its role in cancer therapy. J Cell Mol Med. 2019;23:4900-12
22. Parisi LR, Morrow LM, Visser MB, Atilla-Gokcumen GE. Turning the Spotlight on Lipids in Non-Apoptotic Cell Death. ACS Chem Biol. 2018;13:506-15
23. Conrad M, Kagan VE, Bayir H, Pagnussat GC, Head B, Traber MG. et al. Regulation of lipid peroxidation and ferroptosis in diverse species. Genes Dev. 2018;32:602-19
24. Ye H, Rouault TA. Human iron-sulfur cluster assembly, cellular iron homeostasis, and disease. Biochemistry. 2010;49:4945-56
25. Latunde-Dada GO. Ferroptosis: Role of lipid peroxidation, iron and ferritinophagy. Biochim Biophys Acta Gen Subj. 2017;1861:1893-900
26. Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X. et al. Ferroptosis: process and function. Cell Death Differ. 2016;23:369-79
27. Yang WS, Stockwell BR. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol. 2008;15:234-45
28. Nishizawa H, Matsumoto M, Shindo T, Saigusa D, Kato H, Suzuki K. et al. Ferroptosis is controlled by the coordinated transcriptional regulation of glutathione and labile iron metabolism by the transcription factor BACH1. J Biol Chem. 2020;295:69-82
29. Jiang T, Cheng H, Su J, Wang X, Wang Q, Chu J. et al. Gastrodin protects against glutamate-induced ferroptosis in HT-22 cells through Nrf2/HO-1 signaling pathway. Toxicol In vitro. 2020;62:104715
30. Battaglia AM, Chirillo R, Aversa I, Sacco A, Costanzo F, Biamonte F. Ferroptosis and Cancer: Mitochondria Meet the "Iron Maiden" Cell Death. Cells. 2020 9
31. Richardson DR, Lane DJ, Becker EM, Huang ML, Whitnall M, Suryo Rahmanto Y. et al. Mitochondrial iron trafficking and the integration of iron metabolism between the mitochondrion and cytosol. Proc Natl Acad Sci U S A. 2010;107:10775-82
32. Paradkar PN, Zumbrennen KB, Paw BH, Ward DM, Kaplan J. Regulation of mitochondrial iron import through differential turnover of mitoferrin 1 and mitoferrin 2. Mol Cell Biol. 2009;29:1007-16
33. Wu JR, Tuo QZ, Lei P. Ferroptosis, a Recent Defined Form of Critical Cell Death in Neurological Disorders. J Mol Neurosci. 2018;66:197-206
34. Tateda C, Kusano T, Takahashi Y. The Arabidopsis voltage-dependent anion channel 2 is required for plant growth. Plant Signal Behav. 2012;7:31-3
35. Maldonado EN, Sheldon KL, DeHart DN, Patnaik J, Manevich Y, Townsend DM. et al. Voltage-dependent anion channels modulate mitochondrial metabolism in cancer cells: regulation by free tubulin and erastin. J Biol Chem. 2013;288:11920-9
36. Strzyz P. Iron expulsion by exosomes drives ferroptosis resistance. Nat Rev Mol Cell Biol. 2020;21:4-5
37. Drysdale J, Arosio P, Invernizzi R, Cazzola M, Volz A, Corsi B. et al. Mitochondrial ferritin: a new player in iron metabolism. Blood Cells Mol Dis. 2002;29:376-83
38. Wang YQ, Chang SY, Wu Q, Gou YJ, Jia L, Cui YM. et al. The Protective Role of Mitochondrial Ferritin on Erastin-Induced Ferroptosis. Front Aging Neurosci. 2016;8:308
39. Sato H, Tamba M, Ishii T, Bannai S. Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J Biol Chem. 1999;274:11455-8
40. Fujii J, Homma T, Kobayashi S. Ferroptosis caused by cysteine insufficiency and oxidative insult. Free Radic Res. 2019 p: 1-12
41. Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ. et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell. 2017;171:273-85
42. Hayano M, Yang WS, Corn CK, Pagano NC, Stockwell BR. Loss of cysteinyl-tRNA synthetase (CARS) induces the transsulfuration pathway and inhibits ferroptosis induced by cystine deprivation. Cell Death Differ. 2016;23:270-8
43. Seib TM, Patel SA, Bridges RJ. Regulation of the system x(C)- cystine/glutamate exchanger by intracellular glutathione levels in rat astrocyte primary cultures. Glia. 2011;59:1387-401
44. Dangol S, Chen Y, Hwang BK, Jwa NS. Iron- and Reactive Oxygen Species-Dependent Ferroptotic Cell Death in Rice-Magnaporthe oryzae Interactions. Plant Cell. 2019;31:189-209
45. Seiler A, Schneider M, Forster H, Roth S, Wirth EK, Culmsee C. et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab. 2008;8:237-48
46. Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ. et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol. 2014;16:1180-91
47. Ursini F, Maiorino M. Lipid peroxidation and ferroptosis: The role of GSH and GPx4. Free Radic Biol Med. 2020;152:175-85
48. Chen Y, Zhu G, Liu Y, Wu Q, Zhang X, Bian Z. et al. O-GlcNAcylated c-Jun antagonizes ferroptosis via inhibiting GSH synthesis in liver cancer. Cell Signal. 2019;63:109384
49. Llabani E, Hicklin RW, Lee HY, Motika SE, Crawford LA, Weerapana E. et al. Diverse compounds from pleuromutilin lead to a thioredoxin inhibitor and inducer of ferroptosis. Nat Chem. 2019;11:521-32
50. Lu J, Holmgren A. The thioredoxin antioxidant system. Free Radic Biol Med. 2014;66:75-87
51. Telorack M, Meyer M, Ingold I, Conrad M, Bloch W, Werner S. A Glutathione-Nrf2-Thioredoxin Cross-Talk Ensures Keratinocyte Survival and Efficient Wound Repair. PLoS Genet. 2016;12:e1005800
52. Brutsch SH, Wang CC, Li L, Stender H, Neziroglu N, Richter C. et al. Expression of inactive glutathione peroxidase 4 leads to embryonic lethality, and inactivation of the Alox15 gene does not rescue such knock-in mice. Antioxid Redox Signal. 2015;22:281-93
53. Melchers J, Diechtierow M, Feher K, Sinning I, Tews I, Krauth-Siegel RL. et al. Structural basis for a distinct catalytic mechanism in Trypanosoma brucei tryparedoxin peroxidase. J Biol Chem. 2008;283:30401-11
54. Ingold I, Berndt C, Schmitt S, Doll S, Poschmann G, Buday K. et al. Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis. Cell. 2018;172:409-22 e21
55. Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS. et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317-31
56. Sun Y, Zheng Y, Wang C, Liu Y. Glutathione depletion induces ferroptosis, autophagy, and premature cell senescence in retinal pigment epithelial cells. Cell Death Dis. 2018;9:753
57. Viktorinova A. Iron-mediated oxidative cell death is a potential contributor to neuronal dysfunction induced by neonatal hemolytic hyperbilirubinemia. Arch Biochem Biophys. 2018;654:185-93
58. Yoo SE, Chen L, Na R, Liu Y, Rios C, Van Remmen H. et al. Gpx4 ablation in adult mice results in a lethal phenotype accompanied by neuronal loss in brain. Free Radic Biol Med. 2012;52:1820-7
59. Gladyshev VN, Stadtman TC, Hatfield DL, Jeang KT. Levels of major selenoproteins in T cells decrease during HIV infection and low molecular mass selenium compounds increase. Proc Natl Acad Sci U S A. 1999;96:835-9
60. Matsushita M, Freigang S, Schneider C, Conrad M, Bornkamm GW, Kopf M. T cell lipid peroxidation induces ferroptosis and prevents immunity to infection. J Exp Med. 2015;212:555-68
61. Neff RA, Edwards D, Palmer A, Ramsing R, Righter A, Wolfson J. Reducing meat consumption in the USA: a nationally representative survey of attitudes and behaviours. Public Health Nutr. 2018;21:1835-44
62. Fogelholm M, Kanerva N, Mannisto S. Association between red and processed meat consumption and chronic diseases: the confounding role of other dietary factors. Eur J Clin Nutr. 2015;69:1060-5
63. Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A. 2016;113:E4966-75
64. Coelho OGL, da Silva BP, Rocha D, Lopes LL, Alfenas RCG. Polyunsaturated fatty acids and type 2 diabetes: Impact on the glycemic control mechanism. Crit Rev Food Sci Nutr. 2017;57:3614-9
65. Ng SW, Norwitz SG, Taylor HS, Norwitz ER. Endometriosis: The Role of Iron Overload and Ferroptosis. Reprod Sci. 2020
66. Su LJ, Zhang JH, Gomez H, Murugan R, Hong X, Xu D. et al. Reactive Oxygen Species-Induced Lipid Peroxidation in Apoptosis, Autophagy, and Ferroptosis. Oxid Med Cell Longev. 2019;2019:5080843
67. Gaschler MM, Stockwell BR. Lipid peroxidation in cell death. Biochem Biophys Res Commun. 2017;482:419-25
68. Magtanong L, Ko PJ, To M, Cao JY, Forcina GC, Tarangelo A. et al. Exogenous Monounsaturated Fatty Acids Promote a Ferroptosis-Resistant Cell State. Cell Chem Biol. 2019;26:420-32 e9
69. Zilka O, Shah R, Li B, Friedmann Angeli JP, Griesser M, Conrad M. et al. On the Mechanism of Cytoprotection by Ferrostatin-1 and Liproxstatin-1 and the Role of Lipid Peroxidation in Ferroptotic Cell Death. ACS Cent Sci. 2017;3:232-43
70. Dolma S, Lessnick SL, Hahn WC, Stockwell BR. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell. 2003;3:285-96
71. Lachaier E, Louandre C, Godin C, Saidak Z, Baert M, Diouf M. et al. Sorafenib induces ferroptosis in human cancer cell lines originating from different solid tumors. Anticancer Res. 2014;34:6417-22
72. Shimada K, Skouta R, Kaplan A, Yang WS, Hayano M, Dixon SJ. et al. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat Chem Biol. 2016;12:497-503
73. Mai TT, Hamai A, Hienzsch A, Caneque T, Muller S, Wicinski J. et al. Salinomycin kills cancer stem cells by sequestering iron in lysosomes. Nat Chem. 2017;9:1025-33
74. Sato M, Kusumi R, Hamashima S, Kobayashi S, Sasaki S, Komiyama Y. et al. The ferroptosis inducer erastin irreversibly inhibits system xc- and synergizes with cisplatin to increase cisplatin's cytotoxicity in cancer cells. Sci Rep. 2018;8:968
75. Liu Y, Wang W, Li Y, Xiao Y, Cheng J, Jia J. The 5-Lipoxygenase Inhibitor Zileuton Confers Neuroprotection against Glutamate Oxidative Damage by Inhibiting Ferroptosis. Biol Pharm Bull. 2015;38:1234-9
76. Codenotti S, Poli M, Asperti M, Zizioli D, Marampon F, Fanzani A. Cell growth potential drives ferroptosis susceptibility in rhabdomyosarcoma and myoblast cell lines. J Cancer Res Clin Oncol. 2018;144:1717-30
77. Skouta R, Dixon SJ, Wang J, Dunn DE, Orman M, Shimada K. et al. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J Am Chem Soc. 2014;136:4551-6
78. Chen X, Zhang B, Liu T, Feng M, Zhang Y, Zhang C. et al. Liproxstatin-1 Attenuates Morphine Tolerance through Inhibiting Spinal Ferroptosis-like Cell Death. ACS Chem Neurosci. 2019;10:4824-33
79. Linkermann A, Skouta R, Himmerkus N, Mulay SR, Dewitz C, De Zen F. et al. Synchronized renal tubular cell death involves ferroptosis. Proc Natl Acad Sci U S A. 2014;111:16836-41
80. Adedoyin O, Boddu R, Traylor A, Lever JM, Bolisetty S, George JF. et al. Heme oxygenase-1 mitigates ferroptosis in renal proximal tubule cells. Am J Physiol Renal Physiol. 2018;314:F702-F14
81. Totsuka K, Ueta T, Uchida T, Roggia MF, Nakagawa S, Vavvas DG. et al. Oxidative stress induces ferroptotic cell death in retinal pigment epithelial cells. Exp Eye Res. 2019;181:316-24
82. Yuan H, Li X, Zhang X, Kang R, Tang D. Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem Biophys Res Commun. 2016;478:1338-43
83. Li Y, Feng D, Wang Z, Zhao Y, Sun R, Tian D. et al. Ischemia-induced ACSL4 activation contributes to ferroptosis-mediated tissue injury in intestinal ischemia/reperfusion. Cell Death Differ. 2019;26:2284-99
84. Ye J, Zhang R, Wu F, Zhai L, Wang K, Xiao M. et al. Non-apoptotic cell death in malignant tumor cells and natural compounds. Cancer Lett. 2018;420:210-27
85. Chen Y, Mi Y, Zhang X, Ma Q, Song Y, Zhang L. et al. Dihydroartemisinin-induced unfolded protein response feedback attenuates ferroptosis via PERK/ATF4/HSPA5 pathway in glioma cells. J Exp Clin Cancer Res. 2019;38:402
86. Eling N, Reuter L, Hazin J, Hamacher-Brady A, Brady NR. Identification of artesunate as a specific activator of ferroptosis in pancreatic cancer cells. Oncoscience. 2015;2:517-32
87. Wang L, Zhang Z, Li M, Wang F, Jia Y, Zhang F. et al. P53-dependent induction of ferroptosis is required for artemether to alleviate carbon tetrachloride-induced liver fibrosis and hepatic stellate cell activation. IUBMB Life. 2019;71:45-56
88. Xie Y, Song X, Sun X, Huang J, Zhong M, Lotze MT. et al. Identification of baicalein as a ferroptosis inhibitor by natural product library screening. Biochem Biophys Res Commun. 2016;473:775-80
89. Li Q, Li QQ, Jia JN, Sun QY, Zhou HH, Jin WL. et al. Baicalein Exerts Neuroprotective Effects in FeCl3-Induced Posttraumatic Epileptic Seizures via Suppressing Ferroptosis. Front Pharmacol. 2019;10:638
90. Liu B, Zhao C, Li H, Chen X, Ding Y, Xu S. Puerarin protects against heart failure induced by pressure overload through mitigation of ferroptosis. Biochem Biophys Res Commun. 2018;497:233-40
91. Pitale PM, Gorbatyuk O, Gorbatyuk M. Neurodegeneration: Keeping ATF4 on a Tight Leash. Front Cell Neurosci. 2017;11:410
92. Singleton DC, Harris AL. Targeting the ATF4 pathway in cancer therapy. Expert Opin Ther Targets. 2012;16:1189-202
93. Wang N, Zeng GZ, Yin JL, Bian ZX. Artesunate activates the ATF4-CHOP-CHAC1 pathway and affects ferroptosis in Burkitt's Lymphoma. Biochem Biophys Res Commun. 2019;519:533-9
94. Li W, Li W, Leng Y, Xiong Y, Xia Z. Ferroptosis Is Involved in Diabetes Myocardial Ischemia/Reperfusion Injury Through Endoplasmic Reticulum Stress. DNA Cell Biol. 2020;39:210-25
95. Tang X, Keenan MM, Wu J, Lin CA, Dubois L, Thompson JW. et al. Comprehensive profiling of amino acid response uncovers unique methionine-deprived response dependent on intact creatine biosynthesis. PLoS Genet. 2015;11:e1005158
96. Jiang G, Santos Rocha C, Hirao LA, Mendes EA, Tang Y, Thompson GR 3rd. et al. HIV Exploits Antiviral Host Innate GCN2-ATF4 Signaling for Establishing Viral Replication Early in Infection. mBio. 2017 8
97. Chen MS, Wang SF, Hsu CY, Yin PH, Yeh TS, Lee HC. et al. CHAC1 degradation of glutathione enhances cystine-starvation-induced necroptosis and ferroptosis in human triple negative breast cancer cells via the GCN2-eIF2alpha-ATF4 pathway. Oncotarget. 2017;8:114588-602
98. Bai T, Liang R, Zhu R, Wang W, Zhou L, Sun Y. MicroRNA-214-3p enhances erastin-induced ferroptosis by targeting ATF4 in hepatoma cells. J Cell Physiol. 2020
99. Zhu S, Zhang Q, Sun X, Zeh HJ 3rd, Lotze MT, Kang R. et al. HSPA5 Regulates Ferroptotic Cell Death in Cancer Cells. Cancer Res. 2017;77:2064-77
100. Chen D, Fan Z, Rauh M, Buchfelder M, Eyupoglu IY, Savaskan N. ATF4 promotes angiogenesis and neuronal cell death and confers ferroptosis in a xCT-dependent manner. Oncogene. 2017;36:5593-608
101. Kuroda J, Ago T, Matsushima S, Zhai P, Schneider MD, Sadoshima J. NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. Proc Natl Acad Sci U S A. 2010;107:15565-70
102. Sun X, Yang Y, Shi J, Wang C, Yu Z, Zhang H. NOX4- and Nrf2-mediated oxidative stress induced by silver nanoparticles in vascular endothelial cells. J Appl Toxicol. 2017;37:1428-37
103. Lee CF, Qiao M, Schroder K, Zhao Q, Asmis R. Nox4 is a novel inducible source of reactive oxygen species in monocytes and macrophages and mediates oxidized low density lipoprotein-induced macrophage death. Circ Res. 2010;106:1489-97
104. Chen X, Xu S, Zhao C, Liu B. Role of TLR4/NADPH oxidase 4 pathway in promoting cell death through autophagy and ferroptosis during heart failure. Biochem Biophys Res Commun. 2019;516:37-43
105. Wang Z, Ding Y, Wang X, Lu S, Wang C, He C. et al. Pseudolaric acid B triggers ferroptosis in glioma cells via activation of Nox4 and inhibition of xCT. Cancer Lett. 2018;428:21-33
106. Song X, Zhu S, Chen P, Hou W, Wen Q, Liu J. et al. AMPK-Mediated BECN1 Phosphorylation Promotes Ferroptosis by Directly Blocking System Xc(-) Activity. Curr Biol. 2018;28:2388-99 e5
107. Liu R, Li X, Zhao G. Beclin1-mediated ferroptosis activation is associated with isoflurane-induced toxicity in SH-SY5Y neuroblastoma cells. Acta Biochim Biophys Sin (Shanghai). 2019;51:1134-41
108. Zhang Z, Yao Z, Wang L, Ding H, Shao J, Chen A. et al. Activation of ferritinophagy is required for the RNA-binding protein ELAVL1/HuR to regulate ferroptosis in hepatic stellate cells. Autophagy. 2018;14:2083-103
109. Zhou B, Liu J, Kang R, Klionsky DJ, Kroemer G, Tang D. Ferroptosis is a type of autophagy-dependent cell death. Semin Cancer Biol. 2019
110. Liu J, Kuang F, Kroemer G, Klionsky DJ, Kang R, Tang D. Autophagy-Dependent Ferroptosis: Machinery and Regulation. Cell Chem Biol. 2020;27:420-35
111. Piccolo S, Dupont S, Cordenonsi M. The biology of YAP/TAZ: hippo signaling and beyond. Physiol Rev. 2014;94:1287-312
112. Zanconato F, Cordenonsi M, Piccolo S. YAP/TAZ at the Roots of Cancer. Cancer Cell. 2016;29:783-803
113. Wu J, Minikes AM, Gao M, Bian H, Li Y, Stockwell BR. et al. Intercellular interaction dictates cancer cell ferroptosis via NF2-YAP signalling. Nature. 2019;572:402-6
114. Yang WH, Huang Z, Wu J, Ding CC, Murphy SK, Chi JT. A TAZ-ANGPTL4-NOX2 Axis Regulates Ferroptotic Cell Death and Chemoresistance in Epithelial Ovarian Cancer. Mol Cancer Res. 2020;18:79-90
115. Yang WH, Ding CC, Sun T, Rupprecht G, Lin CC, Hsu D. et al. The Hippo Pathway Effector TAZ Regulates Ferroptosis in Renal Cell Carcinoma. Cell Rep. 2019;28:2501-8 e4
116. Hou Y, Xue P, Bai Y, Liu D, Woods CG, Yarborough K. et al. Nuclear factor erythroid-derived factor 2-related factor 2 regulates transcription of CCAAT/enhancer-binding protein beta during adipogenesis. Free Radic Biol Med. 2012;52:462-72
117. Kurutas EB. The importance of antioxidants which play the role in cellular response against oxidative/nitrosative stress: current state. Nutr J. 2016;15:71
118. Mou Y, Wang J, Wu J, He D, Zhang C, Duan C. et al. Ferroptosis, a new form of cell death: opportunities and challenges in cancer. J Hematol Oncol. 2019;12:34
119. Kerins MJ, Ooi A. The Roles of NRF2 in Modulating Cellular Iron Homeostasis. Antioxid Redox Signal. 2018;29:1756-73
120. Fan Z, Wirth AK, Chen D, Wruck CJ, Rauh M, Buchfelder M. et al. Nrf2-Keap1 pathway promotes cell proliferation and diminishes ferroptosis. Oncogenesis. 2017;6:e371
121. Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R. et al. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology. 2016;63:173-84
122. Chen D, Tavana O, Chu B, Erber L, Chen Y, Baer R. et al. NRF2 Is a Major Target of ARF in p53-Independent Tumor Suppression. Mol Cell. 2017;68:224-32 e4
123. Shin D, Kim EH, Lee J, Roh JL. Nrf2 inhibition reverses resistance to GPX4 inhibitor-induced ferroptosis in head and neck cancer. Free Radic Biol Med. 2018;129:454-62
124. Gai C, Yu M, Li Z, Wang Y, Ding D, Zheng J. et al. Acetaminophen sensitizing erastin-induced ferroptosis via modulation of Nrf2/heme oxygenase-1 signaling pathway in non-small-cell lung cancer. J Cell Physiol. 2020;235:3329-39
125. Kaiser AM, Attardi LD. Deconstructing networks of p53-mediated tumor suppression in vivo. Cell Death Differ. 2018;25:93-103
126. Lei P, Bai T, Sun Y. Mechanisms of Ferroptosis and Relations With Regulated Cell Death: A Review. Front Physiol. 2019;10:139
127. Chu B, Kon N, Chen D, Li T, Liu T, Jiang L. et al. ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nat Cell Biol. 2019;21:579-91
128. Wang Y, Yang L, Zhang X, Cui W, Liu Y, Sun QR. et al. Epigenetic regulation of ferroptosis by H2B monoubiquitination and p53. EMBO Rep. 2019;20:e47563
129. Ou Y, Wang SJ, Li D, Chu B, Gu W. Activation of SAT1 engages polyamine metabolism with p53-mediated ferroptotic responses. Proc Natl Acad Sci U S A. 2016;113:E6806-E12
130. Hambright WS, Fonseca RS, Chen L, Na R, Ran Q. Ablation of ferroptosis regulator glutathione peroxidase 4 in forebrain neurons promotes cognitive impairment and neurodegeneration. Redox Biol. 2017;12:8-17
131. Omiya S, Hikoso S, Imanishi Y, Saito A, Yamaguchi O, Takeda T. et al. Downregulation of ferritin heavy chain increases labile iron pool, oxidative stress and cell death in cardiomyocytes. J Mol Cell Cardiol. 2009;46:59-66
132. Hashemiaghdam A, Mroczek M. Microglia heterogeneity and neurodegeneration: The emerging paradigm of the role of immunity in Alzheimer's disease. Journal of neuroimmunology. 2020;341:577185
133. Maher P. Potentiation of glutathione loss and nerve cell death by the transition metals iron and copper: Implications for age-related neurodegenerative diseases. Free Radic Biol Med. 2018;115:92-104
134. Li LB, Chai R, Zhang S, Xu SF, Zhang YH, Li HL. et al. Iron Exposure and the Cellular Mechanisms Linked to Neuron Degeneration in Adult Mice. Cells. 2019 8
135. Wang P, Wu Q, Wu W, Li H, Guo Y, Yu P. et al. Mitochondrial Ferritin Deletion Exacerbates beta-Amyloid-Induced Neurotoxicity in Mice. Oxid Med Cell Longev. 2017;2017:1020357
136. Huang J, Chen S, Hu L, Niu H, Sun Q, Li W. et al. Mitoferrin-1 is Involved in the Progression of Alzheimer's Disease Through Targeting Mitochondrial Iron Metabolism in a Caenorhabditis elegans Model of Alzheimer's Disease. Neuroscience. 2018;385:90-101
137. Crapper McLachlan DR, Dalton AJ, Kruck TP, Bell MY, Smith WL, Kalow W. et al. Intramuscular desferrioxamine in patients with Alzheimer's disease. Lancet. 1991;337:1304-8
138. Marushchak M, Maksiv K, Krynytska I, Stechyshyn I. Glutathione antioxidant system of lymphocytes in the blood of patients in a setting of concomitant chronic obstructive pulmonary disease and arterial hypertension. Pol Merkur Lekarski. 2019;47:177-82
139. Zhang YH, Wang DW, Xu SF, Zhang S, Fan YG, Yang YY. et al. alpha-Lipoic acid improves abnormal behavior by mitigation of oxidative stress, inflammation, ferroptosis, and tauopathy in P301S Tau transgenic mice. Redox Biol. 2018;14:535-48
140. Cong L, Dong X, Wang Y, Deng Y, Li B, Dai R. On the role of synthesized hydroxylated chalcones as dual functional amyloid-beta aggregation and ferroptosis inhibitors for potential treatment of Alzheimer's disease. Eur J Med Chem. 2019;166:11-21
141. Gunesch S, Hoffmann M, Kiermeier C, Fischer W, Pinto AFM, Maurice T. et al. 7-O-Esters of taxifolin with pronounced and overadditive effects in neuroprotection, anti-neuroinflammation, and amelioration of short-term memory impairment in vivo. Redox Biol. 2020;29:101378
142. Hirata Y, Yamada C, Ito Y, Yamamoto S, Nagase H, Oh-Hashi K. et al. Novel oxindole derivatives prevent oxidative stress-induced cell death in mouse hippocampal HT22cells. Neuropharmacology. 2018;135:242-52
143. Zhou ZD, Lan YH, Tan EK, Lim TM. Iron species-mediated dopamine oxidation, proteasome inhibition, and dopaminergic cell demise: implications for iron-related dopaminergic neuron degeneration. Free Radic Biol Med. 2010;49:1856-71
144. Michaeli S, Oz G, Sorce DJ, Garwood M, Ugurbil K, Majestic S. et al. Assessment of brain iron and neuronal integrity in patients with Parkinson's disease using novel MRI contrasts. Mov Disord. 2007;22:334-40
145. Mohanakumar KP, de Bartolomeis A, Wu RM, Yeh KJ, Sternberger LM, Peng SY. et al. Ferrous-citrate complex and nigral degeneration: evidence for free-radical formation and lipid peroxidation. Ann N Y Acad Sci. 1994;738:392-9
146. Kaur D, Yantiri F, Rajagopalan S, Kumar J, Mo JQ, Boonplueang R. et al. Genetic or pharmacological iron chelation prevents MPTP-induced neurotoxicity in vivo: a novel therapy for Parkinson's disease. Neuron. 2003;37:899-909
147. Devos D, Moreau C, Devedjian JC, Kluza J, Petrault M, Laloux C. et al. Targeting chelatable iron as a therapeutic modality in Parkinson's disease. Antioxid Redox Signal. 2014;21:195-210
148. You LH, Li Z, Duan XL, Zhao BL, Chang YZ, Shi ZH. Mitochondrial ferritin suppresses MPTP-induced cell damage by regulating iron metabolism and attenuating oxidative stress. Brain Res. 2016;1642:33-42
149. Garrido M, Tereshchenko Y, Zhevtsova Z, Taschenberger G, Bahr M, Kugler S. Glutathione depletion and overproduction both initiate degeneration of nigral dopaminergic neurons. Acta Neuropathol. 2011;121:475-85
150. Mischley LK, Leverenz JB, Lau RC, Polissar NL, Neradilek MB, Samii A. et al. A randomized, double-blind phase I/IIa study of intranasal glutathione in Parkinson's disease. Mov Disord. 2015;30:1696-701
151. Hauser RA, Lyons KE, McClain T, Carter S, Perlmutter D. Randomized, double-blind, pilot evaluation of intravenous glutathione in Parkinson's disease. Mov Disord. 2009;24:979-83
152. Bellinger FP, Bellinger MT, Seale LA, Takemoto AS, Raman AV, Miki T. et al. Glutathione Peroxidase 4 is associated with Neuromelanin in Substantia Nigra and Dystrophic Axons in Putamen of Parkinson's brain. Mol Neurodegener. 2011;6:8
153. Chen L, Hambright WS, Na R, Ran Q. Ablation of the Ferroptosis Inhibitor Glutathione Peroxidase 4 in Neurons Results in Rapid Motor Neuron Degeneration and Paralysis. J Biol Chem. 2015;290:28097-106
154. Schriever SC, Zimprich A, Pfuhlmann K, Baumann P, Giesert F, Klaus V. et al. Alterations in neuronal control of body weight and anxiety behavior by glutathione peroxidase 4 deficiency. Neuroscience. 2017;357:241-54
155. Do Van B, Gouel F, Jonneaux A, Timmerman K, Gele P, Petrault M. et al. Ferroptosis, a newly characterized form of cell death in Parkinson's disease that is regulated by PKC. Neurobiol Dis. 2016;94:169-78
156. Ito K, Eguchi Y, Imagawa Y, Akai S, Mochizuki H, Tsujimoto Y. MPP+ induces necrostatin-1- and ferrostatin-1-sensitive necrotic death of neuronal SH-SY5Y cells. Cell Death Discov. 2017;3:17013
157. Lee JK, Shin JH, Gwag BJ, Choi EJ. Iron accumulation promotes TACE-mediated TNF-alpha secretion and neurodegeneration in a mouse model of ALS. Neurobiol Dis. 2015;80:63-9
158. Jeong SY, Rathore KI, Schulz K, Ponka P, Arosio P, David S. Dysregulation of iron homeostasis in the CNS contributes to disease progression in a mouse model of amyotrophic lateral sclerosis. J Neurosci. 2009;29:610-9
159. Wang Q, Zhang X, Chen S, Zhang X, Zhang S, Youdium M. et al. Prevention of motor neuron degeneration by novel iron chelators in SOD1(G93A) transgenic mice of amyotrophic lateral sclerosis. Neurodegener Dis. 2011;8:310-21
160. Kupershmidt L, Weinreb O, Amit T, Mandel S, Carri MT, Youdim MB. Neuroprotective and neuritogenic activities of novel multimodal iron-chelating drugs in motor-neuron-like NSC-34 cells and transgenic mouse model of amyotrophic lateral sclerosis. FASEB J. 2009;23:3766-79
161. Devos D, Moreau C, Kyheng M, Garcon G, Rolland AS, Blasco H. et al. A ferroptosis-based panel of prognostic biomarkers for Amyotrophic Lateral Sclerosis. Sci Rep. 2019;9:2918
162. Langley MR, Yoon H, Kim HN, Choi CI, Simon W, Kleppe L. et al. High fat diet consumption results in mitochondrial dysfunction, oxidative stress, and oligodendrocyte loss in the central nervous system. Biochim Biophys Acta Mol Basis Dis. 2020;1866:165630
163. Stephenson E, Nathoo N, Mahjoub Y, Dunn JF, Yong VW. Iron in multiple sclerosis: roles in neurodegeneration and repair. Nat Rev Neurol. 2014;10:459-68
164. Khalil M, Enzinger C, Langkammer C, Tscherner M, Wallner-Blazek M, Jehna M. et al. Quantitative assessment of brain iron by R(2)* relaxometry in patients with clinically isolated syndrome and relapsing-remitting multiple sclerosis. Mult Scler. 2009;15:1048-54
165. Ge Y, Jensen JH, Lu H, Helpern JA, Miles L, Inglese M. et al. Quantitative assessment of iron accumulation in the deep gray matter of multiple sclerosis by magnetic field correlation imaging. AJNR Am J Neuroradiol. 2007;28:1639-44
166. Hu CL, Nydes M, Shanley KL, Morales Pantoja IE, Howard TA, Bizzozero OA. Reduced expression of the ferroptosis inhibitor glutathione peroxidase-4 in multiple sclerosis and experimental autoimmune encephalomyelitis. J Neurochem. 2019;148:426-39
167. Pedchenko TV, LeVine SM. Desferrioxamine suppresses experimental allergic encephalomyelitis induced by MBP in SJL mice. J Neuroimmunol. 1998;84:188-97
168. Mitchell KM, Dotson AL, Cool KM, Chakrabarty A, Benedict SH, LeVine SM. Deferiprone, an orally deliverable iron chelator, ameliorates experimental autoimmune encephalomyelitis. Mult Scler. 2007;13:1118-26
169. Ceccon A, Tugarinov V, Ghirlando R, Clore GM. Abrogation of prenucleation, transient oligomerization of the Huntingtin exon 1 protein by human profilin I. Proc Natl Acad Sci U S A. 2020;117:5844-52
170. Bartzokis G, Cummings J, Perlman S, Hance DB, Mintz J. Increased basal ganglia iron levels in Huntington disease. Arch Neurol. 1999;56:569-74
171. van Bergen JM, Hua J, Unschuld PG, Lim IA, Jones CK, Margolis RL. et al. Quantitative Susceptibility Mapping Suggests Altered Brain Iron in Premanifest Huntington Disease. AJNR Am J Neuroradiol. 2016;37:789-96
172. Rosas HD, Chen YI, Doros G, Salat DH, Chen NK, Kwong KK. et al. Alterations in brain transition metals in Huntington disease: an evolving and intricate story. Arch Neurol. 2012;69:887-93
173. Di Paola M, Phillips OR, Sanchez-Castaneda C, Di Pardo A, Maglione V, Caltagirone C. et al. MRI measures of corpus callosum iron and myelin in early Huntington's disease. Hum Brain Mapp. 2014;35:3143-51
174. Simmons DA, Casale M, Alcon B, Pham N, Narayan N, Lynch G. Ferritin accumulation in dystrophic microglia is an early event in the development of Huntington's disease. Glia. 2007;55:1074-84
175. Chen J, Marks E, Lai B, Zhang Z, Duce JA, Lam LQ. et al. Iron accumulates in Huntington's disease neurons: protection by deferoxamine. PLoS One. 2013;8:e77023
176. Agrawal S, Fox J, Thyagarajan B, Fox JH. Brain mitochondrial iron accumulates in Huntington's disease, mediates mitochondrial dysfunction, and can be removed pharmacologically. Free Radic Biol Med. 2018;120:317-29
177. Mao Z, Choo YS, Lesort M. Cystamine and cysteamine prevent 3-NP-induced mitochondrial depolarization of Huntington's disease knock-in striatal cells. Eur J Neurosci. 2006;23:1701-10
178. Klepac N, Relja M, Klepac R, Hecimovic S, Babic T, Trkulja V. Oxidative stress parameters in plasma of Huntington's disease patients, asymptomatic Huntington's disease gene carriers and healthy subjects: a cross-sectional study. J Neurol. 2007;254:1676-83
179. Lee J, Kosaras B, Del Signore SJ, Cormier K, McKee A, Ratan RR. et al. Modulation of lipid peroxidation and mitochondrial function improves neuropathology in Huntington's disease mice. Acta Neuropathol. 2011;121:487-98
180. Zhou R, Leng T, Yang T, Chen F, Hu W, Xiong ZG. beta-Estradiol Protects Against Acidosis-Mediated and Ischemic Neuronal Injury by Promoting ASIC1a (Acid-Sensing Ion Channel 1a) Protein Degradation. Stroke. 2019;50:2902-11
181. Iadecola C, Anrather J. Stroke research at a crossroad: asking the brain for directions. Nat Neurosci. 2011;14:1363-8
182. Guan X, Li X, Yang X, Yan J, Shi P, Ba L. et al. The neuroprotective effects of carvacrol on ischemia/reperfusion-induced hippocampal neuronal impairment by ferroptosis mitigation. Life Sci. 2019;235:116795
183. Lan B, Ge JW, Cheng SW, Zheng XL, Liao J, He C. et al. Extract of Naotaifang, a compound Chinese herbal medicine, protects neuron ferroptosis induced by acute cerebral ischemia in rats. J Integr Med. 2020
184. Tuo QZ, Lei P, Jackman KA, Li XL, Xiong H, Li XL. et al. Tau-mediated iron export prevents ferroptotic damage after ischemic stroke. Mol Psychiatry. 2017;22:1520-30
185. Zille M, Karuppagounder SS, Chen Y, Gough PJ, Bertin J, Finger J. et al. Neuronal Death After Hemorrhagic Stroke In vitro and In vivo Shares Features of Ferroptosis and Necroptosis. Stroke. 2017;48:1033-43
186. Alim I, Caulfield JT, Chen Y, Swarup V, Geschwind DH, Ivanova E. et al. Selenium Drives a Transcriptional Adaptive Program to Block Ferroptosis and Treat Stroke. Cell. 2019;177:1262-79 e25
187. Karuppagounder SS, Alin L, Chen Y, Brand D, Bourassa MW, Dietrich K. et al. N-acetylcysteine targets 5 lipoxygenase-derived, toxic lipids and can synergize with prostaglandin E2 to inhibit ferroptosis and improve outcomes following hemorrhagic stroke in mice. Ann Neurol. 2018;84:854-72
188. Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D. et al. Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation. 2006;113:1807-16
189. Chiong M, Wang ZV, Pedrozo Z, Cao DJ, Troncoso R, Ibacache M. et al. Cardiomyocyte death: mechanisms and translational implications. Cell Death Dis. 2011;2:e244
190. Kremastinos DT, Farmakis D. Iron overload cardiomyopathy in clinical practice. Circulation. 2011;124:2253-63
191. Kobayashi M, Suhara T, Baba Y, Kawasaki NK, Higa JK, Matsui T. Pathological Roles of Iron in Cardiovascular Disease. Curr Drug Targets. 2018;19:1068-76
192. Miranda CJ, Makui H, Soares RJ, Bilodeau M, Mui J, Vali H. et al. Hfe deficiency increases susceptibility to cardiotoxicity and exacerbates changes in iron metabolism induced by doxorubicin. Blood. 2003;102:2574-80
193. Ma L, Li XP, Ji HS, Liu YF, Li EZ. Baicalein Protects Rats with Diabetic Cardiomyopathy Against Oxidative Stress and Inflammation Injury via Phosphatidylinositol 3-Kinase (PI3K)/AKT Pathway. Med Sci Monit. 2018;24:5368-75
194. Bai YT, Chang R, Wang H, Xiao FJ, Ge RL, Wang LS. ENPP2 protects cardiomyocytes from erastin-induced ferroptosis. Biochem Biophys Res Commun. 2018;499:44-51
195. Fang X, Wang H, Han D, Xie E, Yang X, Wei J. et al. Ferroptosis as a target for protection against cardiomyopathy. Proc Natl Acad Sci U S A. 2019;116:2672-80
196. Lapenna D, Ciofani G, Pierdomenico SD, Giamberardino MA, Porreca E. Iron status and oxidative stress in the aged rabbit heart. J Mol Cell Cardiol. 2018;114:328-33
197. Nitenberg A, Ledoux S, Valensi P, Sachs R, Antony I. Coronary microvascular adaptation to myocardial metabolic demand can be restored by inhibition of iron-catalyzed formation of oxygen free radicals in type 2 diabetic patients. Diabetes. 2002;51:813-8
198. Jing L, Shao J, Sun W, Lan T, Jia Z, Ma H. et al. Protective effects of two novel nitronyl nitroxide radicals on heart failure induced by hypobaric hypoxia. Life Sci. 2019;248:116481
199. Ward-Caviness CK, Xu T, Aspelund T, Thorand B, Montrone C, Meisinger C. et al. Improvement of myocardial infarction risk prediction via inflammation-associated metabolite biomarkers. Heart. 2017;103:1278-85
200. Baba Y, Higa JK, Shimada BK, Horiuchi KM, Suhara T, Kobayashi M. et al. Protective effects of the mechanistic target of rapamycin against excess iron and ferroptosis in cardiomyocytes. Am J Physiol Heart Circ Physiol. 2018;314:H659-H68
201. Wang Q, Wang XL, Liu HR, Rose P, Zhu YZ. Protective effects of cysteine analogues on acute myocardial ischemia: novel modulators of endogenous H(2)S production. Antioxid Redox Signal. 2010;12:1155-65
202. Ramires PR, Ji LL. Glutathione supplementation and training increases myocardial resistance to ischemia-reperfusion in vivo. Am J Physiol Heart Circ Physiol. 2001;281:H679-88
203. Park TJ, Park JH, Lee GS, Lee JY, Shin JH, Kim MW. et al. Quantitative proteomic analyses reveal that GPX4 downregulation during myocardial infarction contributes to ferroptosis in cardiomyocytes. Cell Death Dis. 2019;10:835
204. Vlahos R, Bozinovski S. Recent advances in pre-clinical mouse models of COPD. Clin Sci (Lond). 2014;126:253-65
205. Thompson AB, Bohling T, Heires A, Linder J, Rennard SI. Lower respiratory tract iron burden is increased in association with cigarette smoking. J Lab Clin Med. 1991;117:493-9
206. Ghio AJ. Disruption of iron homeostasis and lung disease. Biochim Biophys Acta. 2009;1790:731-9
207. DeMeo DL, Mariani T, Bhattacharya S, Srisuma S, Lange C, Litonjua A. et al. Integration of genomic and genetic approaches implicates IREB2 as a COPD susceptibility gene. Am J Hum Genet. 2009;85:493-502
208. Cloonan SM, Glass K, Laucho-Contreras ME, Bhashyam AR, Cervo M, Pabon MA. et al. Mitochondrial iron chelation ameliorates cigarette smoke-induced bronchitis and emphysema in mice. Nat Med. 2016;22:163-74
209. Lee CH, Goag EK, Lee SH, Chung KS, Jung JY, Park MS. et al. Association of serum ferritin levels with smoking and lung function in the Korean adult population: analysis of the fourth and fifth Korean National Health and Nutrition Examination Survey. Int J Chron Obstruct Pulmon Dis. 2016;11:3001-6
210. Yoshida M, Minagawa S, Araya J, Sakamoto T, Hara H, Tsubouchi K. et al. Involvement of cigarette smoke-induced epithelial cell ferroptosis in COPD pathogenesis. Nat Commun. 2019;10:3145
211. Arja C, Surapaneni KM, Raya P, Adimoolam C, Balisetty B, Kanala KR. Oxidative stress and antioxidant enzyme activity in South Indian male smokers with chronic obstructive pulmonary disease. Respirology. 2013;18:1069-75
212. Singh B, Ghosh N, Saha D, Sarkar S, Bhattacharyya P, Chaudhury K. Effect of doxycyline in chronic obstructive pulmonary disease - An exploratory study. Pulm Pharmacol Ther. 2019;58:101831
213. Vantyghem MC, de Koning EJP, Pattou F, Rickels MR. Advances in beta-cell replacement therapy for the treatment of type 1 diabetes. Lancet. 2019;394:1274-85
214. Rojas J, Bermudez V, Palmar J, Martinez MS, Olivar LC, Nava M. et al. Pancreatic Beta Cell Death: Novel Potential Mechanisms in Diabetes Therapy. J Diabetes Res. 2018;2018:9601801
215. Ghosh R, Colon-Negron K, Papa FR. Endoplasmic reticulum stress, degeneration of pancreatic islet beta-cells, and therapeutic modulation of the unfolded protein response in diabetes. Mol Metab. 2019;27(S):60-8
216. Simcox JA, McClain DA. Iron and diabetes risk. Cell Metab. 2013;17:329-41
217. Canturk Z, Cetinarslan B, Tarkun I, Canturk NZ. Serum ferritin levels in poorly- and well-controlled diabetes mellitus. Endocr Res. 2003;29:299-306
218. Altamura S, Kopf S, Schmidt J, Mudder K, da Silva AR, Nawroth P. et al. Uncoupled iron homeostasis in type 2 diabetes mellitus. J Mol Med (Berl). 2017;95:1387-98
219. Thomas B, Rao A, Prasad BR, Kumari S. Serum levels of antioxidants and superoxide dismutase in periodontitis patients with diabetes type 2. J Indian Soc Periodontol. 2014;18:451-5
220. Pieme CA, Tatangmo JA, Simo G, Biapa Nya PC, Ama Moor VJ, Moukette Moukette B. et al. Relationship between hyperglycemia, antioxidant capacity and some enzymatic and non-enzymatic antioxidants in African patients with type 2 diabetes. BMC Res Notes. 2017;10:141
221. Mancino R, Di Pierro D, Varesi C, Cerulli A, Feraco A, Cedrone C. et al. Lipid peroxidation and total antioxidant capacity in vitreous, aqueous humor, and blood samples from patients with diabetic retinopathy. Mol Vis. 2011;17:1298-304
222. Al-Khaldi A, Sultan S. The expression of sirtuins, superoxide dismutase, and lipid peroxidation status in peripheral blood from patients with diabetes and hypothyroidism. BMC Endocr Disord. 2019;19:19
223. Gong W, Zhu G, Li J, Yang X. LncRNA MALAT1 promotes the apoptosis and oxidative stress of human lens epithelial cells via p38MAPK pathway in diabetic cataract. Diabetes Res Clin Pract. 2018;144:314-21
224. Admoni SN, Santos-Bezerra DP, Perez RV, Patente TA, Monteiro MB, Cavaleiro AM. et al. Glutathione peroxidase 4 functional variant rs713041 modulates the risk for cardiovascular autonomic neuropathy in individuals with type 1 diabetes. Diab Vasc Dis Res. 2019;16:297-9
225. Bruni A, Pepper AR, Pawlick RL, Gala-Lopez B, Gamble AF, Kin T. et al. Ferroptosis-inducing agents compromise in vitro human islet viability and function. Cell Death Dis. 2018;9:595
226. Wei S, Qiu T, Yao X, Wang N, Jiang L, Jia X. et al. Arsenic induces pancreatic dysfunction and ferroptosis via mitochondrial ROS-autophagy-lysosomal pathway. J Hazard Mater. 2020;384:121390
227. Masaldan S, Clatworthy SAS, Gamell C, Meggyesy PM, Rigopoulos AT, Haupt S. et al. Iron accumulation in senescent cells is coupled with impaired ferritinophagy and inhibition of ferroptosis. Redox Biol. 2018;14:100-15
228. Wang W, Green M, Choi JE, Gijon M, Kennedy PD, Johnson JK. et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature. 2019;569:270-4
229. Gaschler MM, Andia AA, Liu H, Csuka JM, Hurlocker B, Vaiana CA. et al. FINO2 initiates ferroptosis through GPX4 inactivation and iron oxidation. Nat Chem Biol. 2018;14:507-15
230. Chang LC, Chiang SK, Chen SE, Yu YL, Chou RH, Chang WC. Heme oxygenase-1 mediates BAY 11-7085 induced ferroptosis. Cancer Lett. 2018;416:124-37
231. Li X, Duan L, Yuan S, Zhuang X, Qiao T, He J. Ferroptosis inhibitor alleviates Radiation-induced lung fibrosis (RILF) via down-regulation of TGF-beta1. J Inflamm (Lond). 2019;16:11
232. Daher B, Parks SK, Durivault J, Cormerais Y, Baidarjad H, Tambutte E. et al. Genetic Ablation of the Cystine Transporter xCT in PDAC Cells Inhibits mTORC1, Growth, Survival, and Tumor Formation via Nutrient and Oxidative Stresses. Cancer Res. 2019;79:3877-90
233. Chen D, Rauh M, Buchfelder M, Eyupoglu IY, Savaskan N. The oxido-metabolic driver ATF4 enhances temozolamide chemo-resistance in human gliomas. Oncotarget. 2017;8:51164-76
234. Poursaitidis I, Wang X, Crighton T, Labuschagne C, Mason D, Cramer SL. et al. Oncogene-Selective Sensitivity to Synchronous Cell Death following Modulation of the Amino Acid Nutrient Cystine. Cell Rep. 2017;18:2547-56
235. Liu Q, Wang K. The induction of ferroptosis by impairing STAT3/Nrf2/GPx4 signaling enhances the sensitivity of osteosarcoma cells to cisplatin. Cell Biol Int. 2019;43:1245-56
236. Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H. et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520:57-62
237. Jiang L, Hickman JH, Wang SJ, Gu W. Dynamic roles of p53-mediated metabolic activities in ROS-induced stress responses. Cell Cycle. 2015;14:2881-5
238. Saint-Germain E, Mignacca L, Vernier M, Bobbala D, Ilangumaran S, Ferbeyre G. SOCS1 regulates senescence and ferroptosis by modulating the expression of p53 target genes. Aging (Albany NY). 2017;9:2137-62
239. Gouel F, Do Van B, Chou ML, Jonneaux A, Moreau C, Bordet R. et al. The protective effect of human platelet lysate in models of neurodegenerative disease: involvement of the Akt and MEK pathways. J Tissue Eng Regen Med. 2017;11:3236-40
240. Kabiraj P, Valenzuela CA, Marin JE, Ramirez DA, Mendez L, Hwang MS. et al. The neuroprotective role of ferrostatin-1 under rotenone-induced oxidative stress in dopaminergic neuroblastoma cells. Protein J. 2015;34:349-58
241. Carrazza S, Forte S, Kassabov Z, Rojo J. Specialized minimal PDFs for optimized LHC calculations. Eur Phys J C Part Fields. 2016;76:205
242. Southon A, Szostak K, Acevedo KM, Dent KA, Volitakis I, Belaidi AA. et al. Cu(II) (atsm) inhibits ferroptosis: Implications for treatment of neurodegenerative disease. Br J Pharmacol. 2020;177:656-67
243. Choi IY, Lee SP, Denney DR, Lynch SG. Lower levels of glutathione in the brains of secondary progressive multiple sclerosis patients measured by 1H magnetic resonance chemical shift imaging at 3 T. Mult Scler. 2011;17:289-96
Author contact
![]() Corresponding author: Prof. Wei Hu, Department of Clinical Pharmacology, The Second Hospital of Anhui Medical University, Hefei 230601, China, Tel./Fax: + 86-551 63806057; E-mail: huweiedu.cn.
Corresponding author: Prof. Wei Hu, Department of Clinical Pharmacology, The Second Hospital of Anhui Medical University, Hefei 230601, China, Tel./Fax: + 86-551 63806057; E-mail: huweiedu.cn.