Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Materials and methods
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2020; 10(25):11520-11534. doi:10.7150/thno.44829 This issue Cite
Research Paper
Inactivation of tumor suppressor gene Clusterin leads to hyperactivation of TAK1-NF-κB signaling axis in lung cancer cells and denotes a therapeutic opportunity
Zhipeng Chen1*, Zhenzhen Fan1*, Xiaowei Dou2*, Qian Zhou1*, Guandi Zeng1*, Lu Liu1, Wensheng Chen1, Ruirui Lan1, Wanting Liu1, Guoqing Ru3 ![]() , Lei Yu4
, Lei Yu4 ![]() , Qing-Yu He1
, Qing-Yu He1 ![]() , Liang Chen1
, Liang Chen1 ![]()
1. Key Laboratory of Functional Protein Research of Guangdong Higher Education Institutes and MOE Key Laboratory of Tumor Molecular Biology, Institute of Life and Health Engineering, College of Life Science and Technology, Jinan University, Guangzhou 510632, China.
2. Clinical Research Center, the Affiliated Hospital of Guizhou Medical University, Guiyang 550004, China.
3. Department of Pathology, Zhejiang, Provincial People's Hospital, People's Hospital of Hangzhou, Medical College, Hangzhou 310014, Zhejiang Province, China.
4. Beijing Tongren Hospital, Capital Medical University, 100730 Beijing, China.
*These authors have contributed equally to this work.
Received 2020-2-11; Accepted 2020-9-4; Published 2020-9-16
Abstract

Purpose: Clinical success of precision medicine is severely limited by de novo or acquired drug resistance. It remains a clinically unmet need to treat these patients. Tumor suppressor genes (TSGs) play a critical role in tumorigenesis and impact the therapeutic effect of various treatments. Experimental Design: Using clinical data, in vitro cell line data and in vivo mouse model data, we revealed the tumor suppressive role of Clusterin in lung cancer. We also delineated the signaling cascade elicited by loss of function of CLU in NSCLC cells and tested precision medicine for CLU deficient lung cancers. Results: CLU is a potent and clinically relevant TSG in lung cancer. Mechanistically, CLU inhibits TGFBR1 to recruit TRAF6/TAB2/TAK1 complex and thus inhibits activation of TAK1- NF-κB signaling axis. Lung cancer cells with loss of function of CLU show exquisite sensitivity to TAK1 inhibitors. Importantly, we show that a significant portion of Kras mutation positive NSCLC patients are concurrently deficient of CLU and that TAK1 kinase inhibitor synergizes with existing drugs to treat this portion of lung cancers patients. Conclusions: Combinational treatment with TAK1 inhibitor and MEK1/2 inhibitor effectively shrank Kras mutation positive and CLU deficient NSCLC tumors. Moreover, we put forward a concept that loss of function of a TSG rewires signaling network and thereby creates an Achilles' heel in tumor cells which could be exploited in precision medicine.
Keywords: precision medicine, tumor suppressor gene, non-small cell lung cancer, mouse model.
Introduction
The clinical success of cancer therapies is severely limited by de novo or acquired drug resistance. Accumulating lines of evidence showed that status of tumor suppressor genes significantly impacted the efficacy of precision medicine, including chemotherapy [1], targeting therapy [2] and immunotherapy [3]. It remains an urgent and clinically unmet need to treat those cancer patients who are resistant to otherwise effective therapies in the presence of intact TSGs.
Lung cancer is the leading cause of cancer related deaths globally, with non-small cell lung cancer (NSCLC) the most frequently diagnosed pathological type of lung cancer. Chemotherapy, targeting therapy and immunotherapy are mainstream treatment options for lung cancer patients in clinic. Unfortunately, despite of the advances of multimodality of therapies, the prognosis for lung cancer patients remains disappointingly dismal with overall 5-year survival rate of only around 17% [4]. The difficulties in developing effective therapies for lung cancer patients stem from our limited understanding of tumorigenesis of lung cancer.
Gain-of-function mutations in driver oncogenes and loss-of-functions in tumor suppressor genes (TSGs) are thought to coordinately drive the transformation of lung epithelial cells into tumorigenic cells [5, 6]. Currently, driver oncogenes are relatively well-characterized, with multiple targeting drugs available for lung cancer patients in clinic [7, 8]. However, TSGs remain to be systemically determined for lung cancer.
Clusterin was first identified as a highly conserved glycoprotein [9], encoded by the CLU gene locus on chromosome 8[10]. Functional study revealed CLU as a Golgi chaperone that facilitates the folding of secreted proteins in a manner similar to small heat shock proteins [10-12]. It has been reported to be involved in numerous physiological processes including apoptotic cell death, cell cycle regulation, DNA repair, cell adhesion, tissue remodeling, lipid transportation, membrane recycling, and immune system regulation [13-15].
Emerging evidence supported CLU as a potent oncogene [16], consistent with reports showing its existence in exosomes and helping cancer cells to survive in distant locations [17]. Overexpression of CLU has been reported in bladder cancer [18]. Furthermore, ectopic expression of CLU in primary hepatocellular carcinoma cells increased migration by twofold in vitro and formation of metastatic tumor nodules in liver by eightfold in vivo [19, 20]. Likewise, overexpression of CLU enhances the metastatic ability of human renal cell carcinoma [21] and prostate cancer [22]. On the other hand, tumor suppressor function has also been reported for CLU in neuroblastomas [23], prostate cancer [24], and broadly epithelial cancers [25]. Both tumor promoting or suppressing function have been reported for CLU in lung cancer [26-28]. It, therefore, remains to be clarified CLU's role in lung cancer.
Here we report that CLU is a potent and clinically relevant TSG in lung cancer. CLU inhibits lung cancer cell growth in vitro and tumorigenesis in vivo. Mechanistically, Clusterin inhibits TGFBR1 to recruit TRAF6/TAB2/TAK1 complex and thus inhibits activation of TAK1-NF-κB axis. Analysis of clinical expression data reveals that CLU is reversely correlated with expression of NF-κB target genes. In clinic, a significant portion of Kras mutation positive lung cancer patients concurrently harbored low level of CLU expression. We also show that TAK1 kinase inhibitor synergizes with existing drugs to treat this portion of Kras mutation positive lung cancers. Using lung cancer as a model, we show here that TSG dysfunction creates a targeting opportunity with potential for clinical application. Hereby, we put forward a concept that loss of function of a TSG significantly rewires signaling network and thereby creates an Achilles' heel in cancer cell, which could be exploited in precision medicine.
Results
CLU is an essential tumor suppressor in lung tumorigenesis
In our previous systemic in vivo screening of lung cancer TSGs, we noticed that somatic knockout of CLU in pulmonary epithelia promoted lung cancer development, suggesting CLU to be a TSG in lung cancer [29]. To find out clinical evidence for CLU as a TSG in lung cancer, we compared CLU expression level in lung adenocarcinoma against para-tumoral tissues using GEO data sets (GSE10072 and GSE7670) downloaded from NCBI GEO database and found significantly lower levels of CLU in NSCLS tissues (Figure 1A). We also analyzed CLU mRNA level in lung cancer patients using XENA online tool (http://xena.ucsc.edu/compare-tissue/), which integrated all published comparable data set for expression level analysis, and found significantly lower levels in lung cancers than in normal or para-tumoral lung tissues (Figure S1A and Table S1). Moreover, a higher level of CLU was significantly associated with patients' longer overall survival (Figure 1B). We also noticed similar significant correlation in stage I patients (Figure 1C), indicating that CLU was a clinically relevant TSG in lung cancer and that CLU played an essential role in early stage of lung cancer development.
CLU is an essential tumor suppressor gene in lung cancer. (A) Two published microarray data sets were analyzed to compare CLU expression in normal and tumoral tissues. GEO number and probe set were labeled on the graph. ****P < 0.00001. (B) and (C) Kaplan-Meier survival curve analysis of CLU-high and low lung cancer patients of all pathological stages B or stage I C. (D) Impact of CLU expression on growth rate of Hop62 cell. 800 engineered Hop62 cells were seeded in 96 wells plate and cultured for 5 days. Cell viability was analyzed with CCK8 assay. Statistic with two-tailed t-test on day 5. (E) Impact of CLU expression on 2-D colony formation ability of Hop62 cell. 200 engineered Hop62 cells were seed in 6 well plate and cultured for 7 days. Colonies were fixed and stained with 0.5% crystal violet in methyl alcohol. (F) Quantification of colony numbers of E. Statistic with one-way ANOVA test. (G) Impact of CLU expression on ability of Hop62 cell to form colonies in soft agar culture. 200 cells/well were seeded in 6-well plates and cultured for 14 days before imaging. (H) Quantification of G. shGFP for control knockdown; shCLU for CLU knockdown; sh/+CLU for CLU re-expression in CLU knockdown cells. (I) and (J) 2-D Plate and soft agar colony formation assay of indicated groups in H460 cell. CLU expression was induced with 1 ug/mL Dox. For 2-D colony formation assay, 200 cells/well were seed in 6 well plate and cultured for 7 days. Colonies were fixed with methanol and stained with 0.5% crystal violet. For soft agar colony formation, 200 cells/well were seeded and culture for 14 days before imaging. Two-tailed t-test. (K) Impact of CLU expression on ability of H460 to form xenograft tumor in nude mouse. H460 cells (2 million) were subcutaneously implanted on nude mice followed by Dox-diet treatment. Tumors were harvested 25 days post implantation. Each group n > 6. (L) Weight of tumor in K, two-tailed t-test. (M) Impact of CLU expression on tumor formation in lsl-KrasG12D/+ transgenic mice. Lentivirus of pSECC-sgCLU and pSECC-TdTomato were administered through nasal instillation into lsl-KrasG12D mice to induced lung cancer, designated K-CLU and K-Ctl respectively. Lung tumor formation in K-CLU mice were compared to K-Ctl mice 13 weeks post-infection. Upper panel: Computed tomography (CT) images of lung of KrasG12D/+ mouse; Lower panel: hematoxylin and eosin (H&E) staining of lung sections of KrasG12D/+ mouse. K-Ctl for KrasG12D/+/sgControl; K-CLU is KrasG12D/+/sgCLU, each group n > 6. (N) Quantification of average total tumor number, tumor size and the percentage of G1-G4 tumors for M. (O) Impact of CLU expression on tumor formation in CC10rtTA /Tet-KrasG12D transgenic mice. Upper panel: Computed tomography images of lung of CC10rtTA /Tet-KrasG12D transgenic mice; Lower panel: H&E staining of lung sections of Tet-KrasG12D transgenic mice. CC10rtTA/ Tet-KrasG12D mice infected with lentivirus harboring Tet-mCherry (Tet-Kras*+m, serving as negative control) or lentivirus harboring Tet-CLU (Tet-Kras*+C) were fed with Dox diet for 2 months. Each group n > 6. (P) Quantification of average total tumor number, tumor size and percentage of G1-G4 tumors of O, statistics with two-tailed t-test. All the transgenic mice here were C57BL6 background, about 6-8 weeks old without sex limited. *P < 0.05, **P < 0.005, ***P < 0.0001; Data plotted are mean ± s.e.m.; n = 3.
Two functionally different splicing variants of CLU transcripts have been reported, namely full-length CLU (sCLU) and truncated CLU (nCLU). To find out which variant is downregulated in lung cancer samples, we analyzed their expression in lung cancer samples, para-tumoral tissues and NSCLC cell lines through semi-quantitative RT-PCR with a single pair of primers capable of amplifying both variants and separated the PCR products through high-resolution agarose-gel electrophoresis. Based on the size of the resolved PCR product, we found that only the full-length CLU was expressed in all these samples and that its expression was relatively lower in cancer cells and lung cancer samples in comparison to para-tumoral tissues (Figure S1B). Sanger sequencing confirmed authenticity of sCLU (Figure S1C). sCLU is translated into a protein of around 64 kDa, which is subjected to proteolysis between Arg205 and Ser206 to generates α- and β-chains; α- and β-chains are then linked by five interchain disulfide bonds, which is secreted into extracellular space [9]. Consistently, our western analysis revealed both 64-kDa full-length and 40-kDa CLU variants in total cell lysates and mainly the 40-kDa cleaved fragment in supernatant (Figure S1D). Based on these data, we focused our further experimental efforts on sCLU (the full-length variant, and hereafter designated CLU unless specified) for studying tumor suppressive function in lung cancer.
In order to validate its TSG function, we sought to ectopically express CLU in lung cancer cell lines with lower baseline expression and knockdown in those with relatively higher expression. For this purpose, we checked CLU expression level in lung cancer cell lines commonly used in cancer research community through qRT-PCR analysis, including two pairs of lung cancer/para-tumoral tissues as references for CLU expression in tumoral and normal lung tissues. We found comparable CLU mRNA level between tumor samples and most of the lung cancer cell lines, which was consistently lower than that of para-tumoral tissues (Figure S1E). Similar pattern was seen in CLU protein level (Figure S1F). We pick A549 and HOP62 as CLU-high and H460 and EKVX CLU-low cells. We then knockdown CLU in Hop62 cells (designated Hop62-shCLU) and found that CLU knockdown significantly promoted growth rate as compared to control knockdown cells (Hop62-shGFP). Moreover, re-expression of shRNA resistant CLU (designated Hop62-sh/+CLU) slowed down the growth rate to the degree comparable to that of Hop62-shGFP (Figure S1G & Figure 1D). We also found that CLU knockdown enhanced the capacity of Hop62 cells to form colonies in 2-D plate and that ectopic expression of shRNA-resistant CLU downregulated this colony-forming ability (Figure 1E-F). We found similar pattern of colony-forming ability in soft agar culture (Figure 1G-H). These effects were repeated on A549 cells (Figure S1H-K), strongly arguing TSG function for CLU in lung cancer. We also generated H460 and EKVX cells for doxycycline (Dox) inducible expression of CLU (designated H460-tet-CLU and EKVX-tet-CLU, Figure S1L). We found that Dox treatment inhibited the growth rate (Figure S1M), ability to form colonies in 2-D plates (Figure 1I & Figure S1N) and soft-agar culture conditions for H460 cells (Figure 1J & Figure S1O). Similar effect was also found using EKVX-tet-CLU cell line (Figure S1P-R). These in vitro data strongly argued that CLU was a potent TSG in lung cancer.
To evaluate the tumor suppressive role of CLU in vivo, we used xenograft tumor model. We found that expression of CLU significantly suppressed growth of xenograft tumor derived from H460-tet-CLU (Figure 1K-L).
Mouse models of autochthonous lung cancer recapitulate the course of tumorigenesis and tumor development more faithfully than xenograft tumor models. We then went on to study the tumor suppressive function of CLU using mouse models of autochthonous lung cancer. Jacks and colleagues have earlier established an efficient method for simultaneous knockout of a target gene and activation of mutant Kras in the lung epithelia of lsl-KrasG12D transgenic mice using recombinant lentivirus co-expressing Cre and CRISPR/CAS9[30]. Following this protocol, we intranasally delivered lentiviruses targeting either TdTomato (serving as negative control) or CLU (CLU knockout efficiency in Figure S1S) into lsl-KrasG12D mice. Lsl-KrasG12D mice infected with sgTdTomato virus (designated K-Ctl for control KrasG12D mice) look rather healthy 13 weeks after treatment. Computed tomography (CT) imaging revealed little tumor burden at this stage (Figure 1M). Consistently, pathological analysis revealed occasional macroscopic lung tumor nodules of lung adenoma (Figure 1M). In stark contrast, mice began panting and exhibited hunched posture post 13 weeks of nasal inhalation of lenti-sgCLU (referred to as K-CLU mice for KrasG12D/CLU-/-), indicative of severe lung disease. CT imaging confirmed relatively heavier tumor burden in K-CLU mice (Figure 1M). Consistently, we detected macroscopic lung tumor nodules in 4 out of 6 mice (Figure 1M). Statistical analyses showed that CLU deletion significantly increased mutant Kras-driven lung tumor numbers, tumor size and amount of stage 4 tumors (Figure 1N).
We also studied the impact of CLU overexpression on tumorigenesis using Dox inducible Tet-KrasG12D/CC10rtTA mice (referred to hereafter as Tet-Kras* mice). Tet-Kras* mice develop lung cancers after feeding with Dox diet for around 3 months, but remain lung cancer free if fed with normal diet [31]. We then infected lung epithelial compartment of Tet-Kras* mice with lentivirus harboring Tet-CLU cassette through intra-nasal instillation (designated Tet-Kras*+C mice) following our earlier protocol [32], such that virus-infected mice started to express KrasG12D and CLU in lung epithelial cells when fed with Dox diet (Figure S1T). In parallel, we also generated a cohort of Tet-Kras* mice infected with lentivirus harboring Tet-mCherry (serving as Control, designated Tet-Kras*+m) (Figure S1T). Tet-Kras*+m mice began panting and exhibited hunched posture 3 months after Dox diet treatment, suggestive of severe lung disease. CT imaging revealed heavy tumor burdens in both lungs of these mice at this stage. Pathological analysis revealed poorly differentiated lung adenocarcinomas with features of diffused bronchial adenocarcinomas (Figure 1O). In stark contrast, Tet-Kras*+C mice looked largely normal at this stage and harbored significantly lower burden of lung cancers (Figure 1O), as well as tumor number and tumor size (Figure 1P). Moreover, significantly lower percentage of malignant tumors were found in Tet-Kras*+C mice model (Figure 1P).
Of note, in vitro and in vivo tumor suppressive functions of CLU were also confirmed on lung cancers driven by two other important oncoproteins, namely mutant EGFR driven and EML4-ALK driven lung cancers (Figure S2A-G).
Taken together, our data strongly argued that CLU was a clinically relevant and essential TSG in lung cancer.
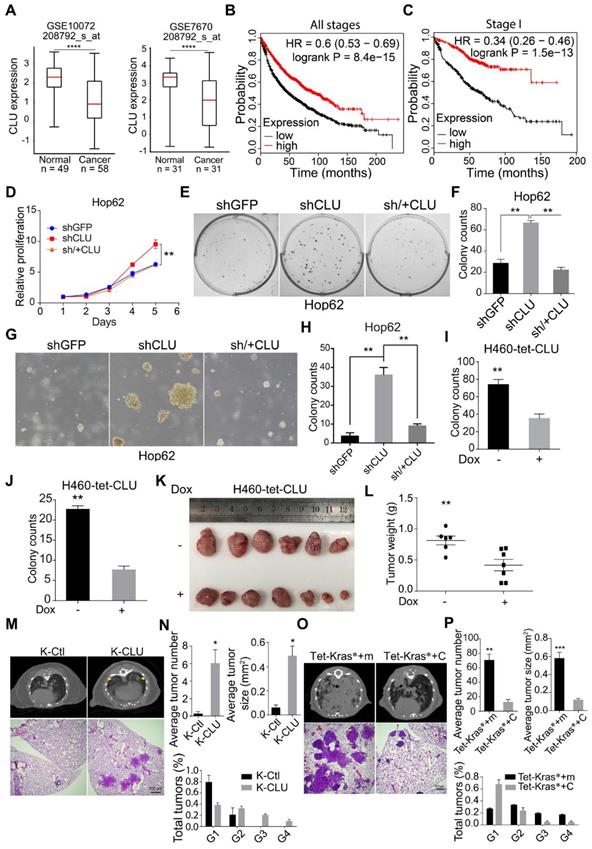
CLU knockdown enhances TAK1 signaling
Having confirmed CLU's growth inhibitory effect on lung cancer cells, we went on to study the underlying mechanism. We tried to find out the signaling pathway responsible for enhanced growth rate caused by CLU knockdown. For this purpose, we checked the impact of inhibitors against various growth promoting pathways on the growth rate of Hop62-shCLU. Our pilot experiment showed that CLU re-expression almost completely neutralized the enhanced ability to form colonies on 2-D plate caused by CLU knockdown, suggesting that 2-D colony forming assay was suitable for evaluating the growth inhibiting ability of a particular signaling pathway (Figure 2A). We then tested inhibitors against signaling pathways frequently involved in cell growth, including TGFBR (SB431542), PI3K (LY294002), AKT (MK2206), NOTCH (RO4929097), JAK (Ruxolitinib), EGFR (Gefitinib), NF-κB (BAY001) and TAK1 (NG25) and found inhibitors against NF-κB and TAK1 were the most potent hits in our screening (Figure 2A-B & Figure S3A-B). As TAK1 is an upstream activator of NF-κB[33], our result suggested that TAK1-NF-κB signaling axis mediated the growth promoting effect caused by CLU knockdown. In line with this, phosphorylation of TAK1 is higher in Hop62-shCLU, but lower when re-expressed CLU (Figure 2C). Interestingly, although TGF-β1 treated Hop62 cells exhibited higher baseline level of TAK1 phosphorylation, we also found the ability of Clusterin to inhibit TAK1 phosphorylation (Figure 2D), suggesting that Clusterin regulated TAK1 activity through a mechanism parallel to TGF-β1 stimulation. Fibronectin (encoded by FN1 gene), COL1A1 and COL4A1 expression have been reported to be positively regulated by TAK1 activity [34, 35]. Consistently, quantitative RT-PCR confirmed that COL1A1 and COL4A1 were negatively regulated by CLU (Figure 2E-F). We also found that CLU knockdown increased and its re-expression downregulated Fibronectin expression in Hop62 cell (Figure 2G). Similar results were found in A549 and EKVX lung cancer cells (Figure S3C-D). Critically, we found that TAK1 knockdown prevented the enhancement of proliferation of Hop62 cells caused by CLU knockdown (Figure S3E & Figure 2H). Similar pattern was observed in 2-D colony forming ability of Hop62 (Figure 2I-J). Moreover, TAK1 knockdown or NG25 treatment decreased FN1, COL1A1 and COL4A1 expression in Hop62-shCLU cells (Figure 2K-Q). Taken together, our data showed that Clusterin negatively regulated TAK1's activity to promote growth of NSCLC cells.
CLU knockdown enhances TAK1 signaling. (A) TAK1 or NF-κB inhibitor specifically reduced colony forming ability of CLU knockdown lung cancer cell. 200 Hop62-shCLU cells were seeded and treated with indicated inhibitors for 7 days; NG25 (2.5 μM), BAY001 (1 μM), SB431542 (2.5 μM). Cell colonies were fixed and stained with 0.5% crystal violet in methyl alcohol. shGFP for control knockdown; shCLU for CLU knockdown; sh/+CLU for CLU re-expression in CLU knockdown cells. (B) Quantification of colony numbers. Statistic with one-way ANOVA test. (C) and (D) Impact of CLU expression on TAK1 activity. Hop62 cells were treated (C) without or (D) with TGF-β1 (5 ng/mL) for 2 h. Phosphor-TAK1, total TAK1 and CLU protein level was detected with indicated antibodies. (E) and (F) RT-PCR analysis of impact of CLU knockdown or re-expression on COL1A1 and COL4A1 expression in Hop62 cells. Total RNA of Hop62 cells with indicated were extracted and COL1A1 and COL4A1 expression was detected with RT-PCR. (G) Impact of CLU expression on Fibronectin expression in lung cancer cells. Fibronectin and CLU levels were assayed through Western analysis. (H) Impact of TAK1 knockdown or re-expression on cell proliferation of CLU knockdown Hop62 (Hop62-shCLU/tet-shTAK1) cells. TAK1 knockdown in Hop62-shCLU cells was induced with 1 μg/mL Dox. Statistic shown on day 5 with two-tailed t-test. (I) Impact of TAK1 knockdown on 2-D colony formation of CLU knockdown Hop62 cells. Cell colonies were fixed and stained with 0.5% crystal violet in methyl alcohol. (J) Quantification of I, one-way ANOVA test. (K-N) RT-PCR analysis of impact of TAK1 knockdown on FN1, COL1A1 and COL4A1 expression in CLU knockdown Hop62 cells. Hop62-shCLU cells were treated with 1 μg/mL Dox for TAK1 inducible knockdown. Statistic with two-tailed t-test. (O-Q) RT-PCR analysis of the impact of TAK1 inhibition with NG25 on FN1, COL1A1 and COL4A1 expression. Hop62-shCLU cells were treated with NG25 (2.5 μM) for 24 h before detecting mRNA level. *P < 0.05, **P < 0.005, ***P < 0.0001; Data plotted are mean ± s.e.m.; n = 3.
Clusterin inhibits TGFBR1 to recruit TRAF6/TAB2/TAK1 complex
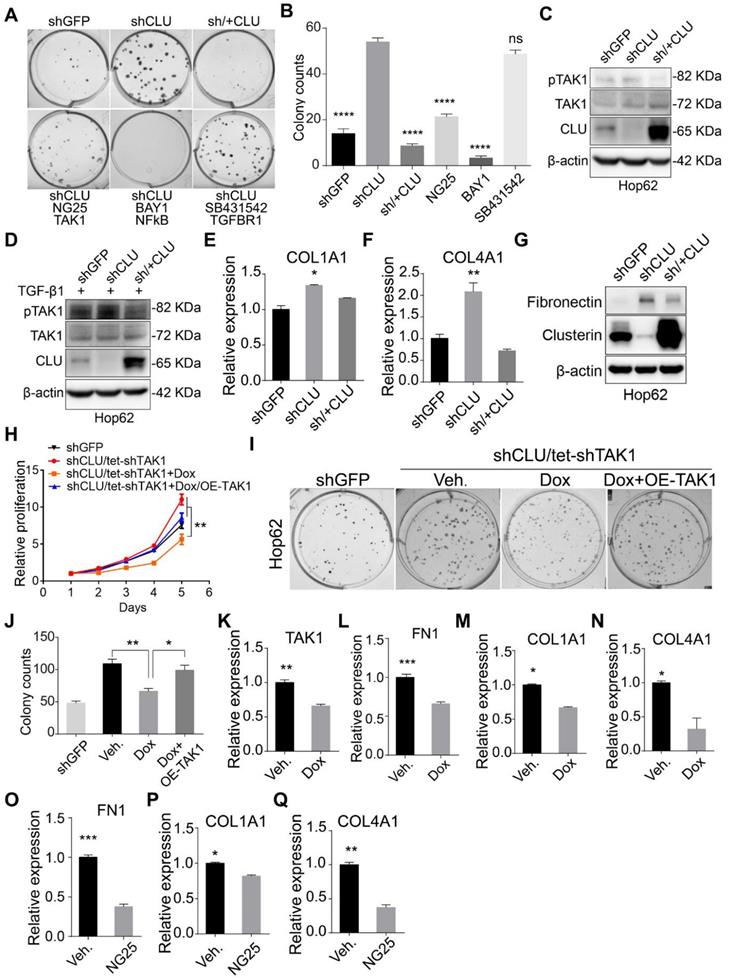
We then asked the molecular mechanism underlying the ability of Clusterin to negatively regulate TAK1 activity. Earlier, genome-wide screening has shown that Clusterin interacted with TGFBR1[36]. We were able to confirm interaction between Clusterin and TGFBR1 when overexpressed in 293T cells (Figure 3A & 3B).
CLU competes against TAK1 for binding TGFBR1. (A) and (B) Reciprocal immunoprecipitation showing interaction between TGFBR1 and CLU. 293T cells were co-transfected with CLU-3×Flag and TGFBR1-myc expressing plasmid, followed by Co-IP with Flag (A) or myc (B) antibody. (C) 293T-tet-CLU cells were co-transferred with TRAF6-HA and TGFBR1-Myc expressing plasmids as indicated. Cell lysates were IPed with Myc antibody followed by western blotting with HA, Myc and CLU antibodies respectively. (D) H460-tet-CLU cells were co-transferred with TRAF6-HA and TGFBR1-Myc expressing plasmids as indicated. Cell lysates were IPed with Myc antibody followed by western blotting with HA, Myc and CLU antibodies respectively. (E) 293T-tet-CLU cell were co-transfected with TAK1-Flag and TGFBR1-Myc expressing plasmids. Cell lysates IPed with Flag antibody followed by western blotting with Myc, Flag and CLU antibodies respectively. (F) H460-tet-CLU cell lysate IPed with TAK1 antibody followed by western blotting with TGFBR1, TAK1 and CLU antibodies. (G) 293T-tet-CLU cells were co-transfected with TAK-Flag and TAB2-Myc expressing plasmids as indicated. Cell lysates were IPed with Myc antibody followed by western blotting with Flag, Myc and CLU antibodies respectively. (H) Schematic diagram of TGFBR1-myc truncation mutation. (I) Co-IP evaluating the interaction between CLU and TGFBR1 or its mutants. 293T cells were co-transfected with CLU-3×Flag and TGFBR1-myc or its muctant expressing plasmid for 24 h before performing Co-IP with Myc antibody and immunoblotting with Flag and Myc tag antibodies.
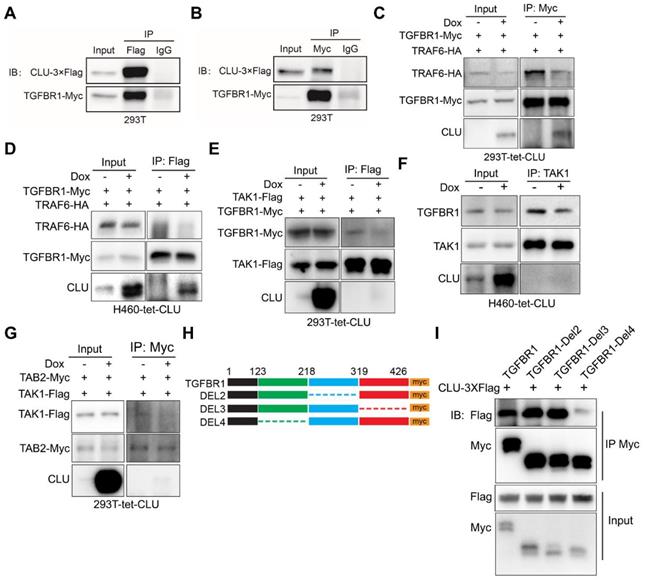
Considering the facts that TGFBR1 directly recruits TRAF6[37] and that TAB2 recruit TAK1 to TRAF6 through direct interaction with both protein [38], we hypothesized that CLU inhibited TGFBR1 to recruit TRAF6/TAB2/TAK1 complex in lung cancer cells, and thereby inhibit the signaling of downstream TAK1-NF-κB signaling pathway.
To test above hypothesis, we generated a 293T cell line for Dox inducible expression of Clusterin (designated 293T-tet-CLU, Figure S4A). Bimolecular fluorescence complementation (BiFC) assay enables efficient visualization of interactions between two proteins in cells by fusing both target proteins to C- and N- terminal half of luciferase respectively [39]. We then conducted BiFC assay in 293T-tet-CLU cell line and found that CLU expression inhibited interaction between TGFBR1 and TRAF6 (Figure S4B). Co-immunoprecipitation (co-IP) further confirmed the ability of CLU to inhibit interaction between ectopically overexpressed TRAF6 and TGFBR1 in 293T-tet-CLU cell (Figure 3C). To test whether this was also true in lung cancer cells, we generated H460 for Dox inducible expression of Clusterin (designated H460-tet-CLU, Figure S1L) and found that CLU expression effectively inhibited recruitment of TRAF6 by TGFBR1 (Figure 3D).
If CLU blocked recruitment of TRAF6 by TGFBR1, TGFBR1/TRAF6/TAB2/TAK1 complex is expected to fall apart in cells expressing CLU. Consistent with our hypothesis, BiFC assay revealed that CLU inhibited interaction between TGFBR1 and TAB2 (Figure S4C); TGFBR1 and TAK1 (Figure S4D); TAK1 and TRAF6 (Figure S4E), and TAK1 and TAB2 (Figure S4F). More importantly, co-IP experiment confirmed that CLU inhibited the interaction between TGFBR1 and TAK1 in 293T-tet-CLU (Figure 3D) and H460-tet-CLU (Figure 3F) or TAK1 and TAB2 in 293T-tet-CLU (Figure 3G). Collectively, our data strongly argued that CLU expression inhibited TGFBR1 to recruit TRAF6/TAB2/TAK1 complex.
To pin down the domain of TGFBR1 important for interacting with Clusterin, we went on to generate a series of truncation mutants of TGFBR1. Intracellular part of TGFBR1 features 2 domains: GS domain of 30 amino acids (aa) and kinase domain of 291 aa. In considering this pattern of unbalanced distribution, we then constructed a series of mutation with approximately 100 aa deletion (Figure 3H). Our co-IP result showed that 123-218aa region of TGFBR1 is critical for binding to Clusterin, as mutant deleted of this region showed very weak interaction with Clusterin (Figure 3I).
TAK1-NF-κB pathway mediates growth-promoting effects in CLU-deficient lung cancer cells
TAK1 signals through MKK3/P38 and NF-κB branches, both of which play important roles in cell growth [34]. We detected no significant impact of p38 inhibitor on the growth of Hop62-shCLU (Figure S5A-B), suggesting that this branch might not play an important role in mediating growth-promoting effect in CLU knockdown NSCLC cells. On the other hand, our inhibitor screening experiment has shown that NF-κB inhibitor BAY001 or TAK1 inhibitor NG25 significantly inhibited colony formation of HOP62-shCLU (Figure 2A), suggesting that CLU inhibited lung cancer cells by negatively regulating TAK1-NF-κB signal axis. Consistently, we detected a significant increase in mRNA expression of NF-κB transcriptional targets in HOP62-shCLU cells, including IL-1β, TNF and Jun, which were inhibited by re-expression of CLU (Figure 4A). Likewise, overexpression of CLU inhibited the increased P65 phosphorylation caused by TGF-β1 treatment in H460. Of note, this effect was also seen in A549 (Figure 4B). Importantly, knockdown the expression of TAK1 prevented the activation of P65 in CLU knockdown H460 cells (Figure 4C).
TAK1-NF-κB pathway mediates growth-promoting effects in CLU-deficient lung cancer cells. (A) RT-PCR evaluation of expression of NF-κB target genes in CLU knockdown or replenished Hop62 cells. Total RNA of engineered Hop62 cells were extracted. Expression of indicated genes was assayed with RT-PCR. (B) Influence of CLU expression on phosphorylation of NF-κB proteins in lung cancer cells. Lung cancer cell lines were treated with TGF-β1 with or without Dox. Total proteins were separated by SDS-PAGE following immunoblotting with antibodies against indicated NF-κB proteins. (C) Western blot evaluating the impact of TAK1 on P65 activation in CLU knockdown cells. Cells were treated with TGF-β1 (5 ng/mL) for 4 h. (D) and (E) Western blot evaluating the impact of CLU on cellular localization of NF-κB proteins. CLU expression in engineered lung cancer cells was induced with Dox. Cytoplasmic and nuclear fractions were separated from the lung cancer cells, followed by western blot with indicated antibodies. (F) Lung cancer cells were treated with TGF-β1 (5 ng/mL) for 2 hours before cytoplasmic and nuclear fraction were separated. Western blot was performed with indicated antibodies. Loading control: GAPDH for cytoplasmic; LaminB1 for nuclear. *P < 0.05, **P < 0.005, ***P < 0.0005, n = 3.
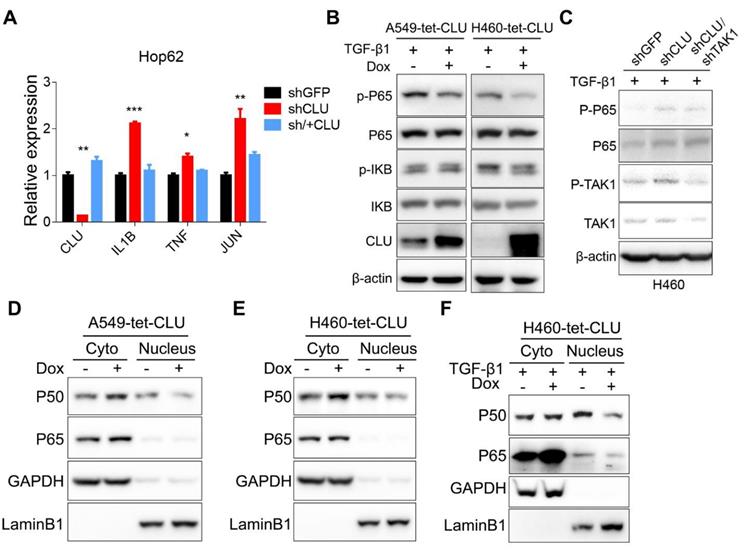
Activation of the IκB kinase (IKK) complex induces the phosphorylation and subsequent degradation of IκBs, resulting in releasing P50 and P65 complex to translocate into the nucleus to function as transcription factors. Consistently, we observed an increase of cytoplasmic and concurrent decrease of nuclear P50 and P65 in A549-tet-CLU (Figure 4D) and H460-tet-CLU cells (Figure 4E) after Dox treatment. Of note, we observed similar impact of CLU expression on NF-κB signaling in H460 cultured in the presence of TGF-β1 (Figure 4F).
Taken together, our data showed that TAK1-NF-κB axis played a critical role mediating growth-promoting effect of CLU-deficient lung cancer cells.
CLU-TAK1-NF-κB signaling axis is clinically relevant
Based on our signaling model (Figure 5A), we predicted that CLU protein level reversely correlated with NF-κB signaling activity in lung cancer cells. Interestingly, we found significant reverse correlation between CLU and TAK1 (Figure 5B). We also detected reverse correlation between CLU and IL1β or IL6, two important NF-κB target genes (Figure 5C-D).
CLU-TAK1-NF-κB signaling axis is clinically relevant. (A) Schematic model of CLU-TAK1-NF-κB signaling axis in lung cancer. (B) Correlation analysis of expression between CLU and TAK1. Expression data of lung adenocarcinoma cancer patients from TCGA data base (analyzed through UCSC Xena). (C) and (D) Correlation analysis between CLU and NF-κB target gene IL1B and IL6. (E) and (F) Correlation analysis between CLU and NF-κB target gene IL1B and IL6 in lung adenocarcinoma cancer patients positive of KRAS mutation. Pearson r and P value are shown in images. Date are presented as log2 RSEM. P value and Person r were shown on the figure.
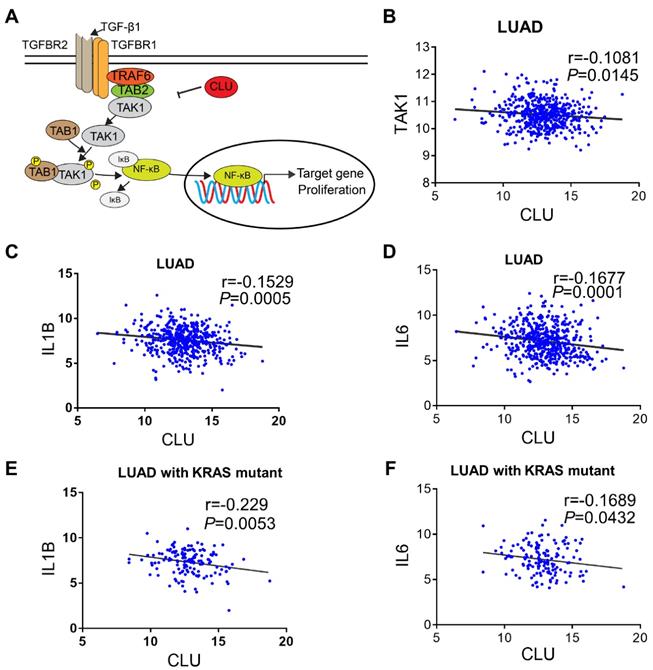
NSCLC are known to be driven by oncogenic alterations [40]. Interestingly, we found that approximately 80% of KRAS mutation positive NSCLC patient concurrently had diminished CLU expression (Table S2). Likewise, expression level of CLU was significantly reverse-correlated with that of IL1β or IL6 in these KRAS mutation positive patients (Figure 5E-F).
TAK1 inhibitor synergizes with existing therapeutics to treat CLU deficient lung cancer
Our above data have shown that a portion of lung cancer patients are concurrently Kras mutation positive and CLU-deficient (designated K+/C- patients). We then went on to test whether TAK1 inhibitor may synergize with existing drugs for treating this portion of lung cancers. We chose H358 cells (KrasG12C) to model K+/C- lung cancer patients (Figure S1F).
Our results have shown that TAK1 inhibition may synergize with existing drug to treat CLU deficient lung cancer patients. Indeed, correlation between lower CLU expression and stronger sensitivity of NSCLC cells to TAK1 inhibition was confirmed when comparing NSCLC cell lines against their corresponding CLU-knockdown derivatives (Figure S6A-F). Earlier, we reported that MEK1/2 inhibitor partially regressed mutant Kras driven lung cancers [41]. In our pilot CCK8 assay experiment, we found that while singlet drug treatment with TAK1 inhibitor (NG25) or MEK1/2 inhibitor (Trametinib) partially inhibited the growth rate of H358, combinational treatment almost completely blocked the cell growth (Figure 6A). Likewise, we also observed similar effect on colony formation in 2-D plates (Figure 6B -C).
TAK1 inhibitor synergizes with existing therapeutics to treat CLU deficient lung cancer. (A) NG25 synergizes with Trametinib to inhibit growth of H358 cells. 200 cells/well were seeded in 96 wells plate and cultured for 5 days with indicated treatment. Viability of cells analyzed with CCK8 assay. Statistics done on day 5 with two-tailed t-test. (B) NG25 synergizes with Trametinib to inhibit 2-D colony formation of H358 cells. Cells were culture for 7 days with indicated treatment. (C) Quantification of B, one-way ANOVA test. (D) NG25 synergizes with Trametinib to inhibit growth of Hop62-shCLU derived xenograft tumor. Hop62-shCLU cells (3 million) were s.c. implanted in the flanks of nude mice. 1-week post implantation, mice were treated with NG25 (4 mg/kg/Day, intravenous injection), Trametinib (Tram, 1 mg/kg/Day, gavage), or combination for 15 days. n > 4 in each group. two-tailed t-test. (E) Image of tumors harvested in D. (F) Weight of tumors harvested in D. one-way ANOVA test. (G) NG25 synergizes with Trametinib to shrink CLU deficient KrasG12D driven lung cancer in transgenic mouse model. Lsl-KrasG12D/+ mice were treated with pSECC-sgCLU lentivirus by nasal inhalation. Tumor burdens were documented with CT. Mice were treated as indicated. NG25 (4 mg/kg/Day, intravenous injection), Trametinib (Tram, 1 mg/kg/Day, gavage), or combination for 12 days. n > 4 in each group. (H) relative tumor volume of mice of G. (I) Representative pathology images of mice of G. (J) Quantification of tumor numbers of mice of G. (K) Quantification of size of tumor in G. one-way ANOVA test on comparison of singlet treatment with the combo treatment. All the data plotted are mean ± s.e.m., ***P < 0.005, ****P < 0.0001, n = 3
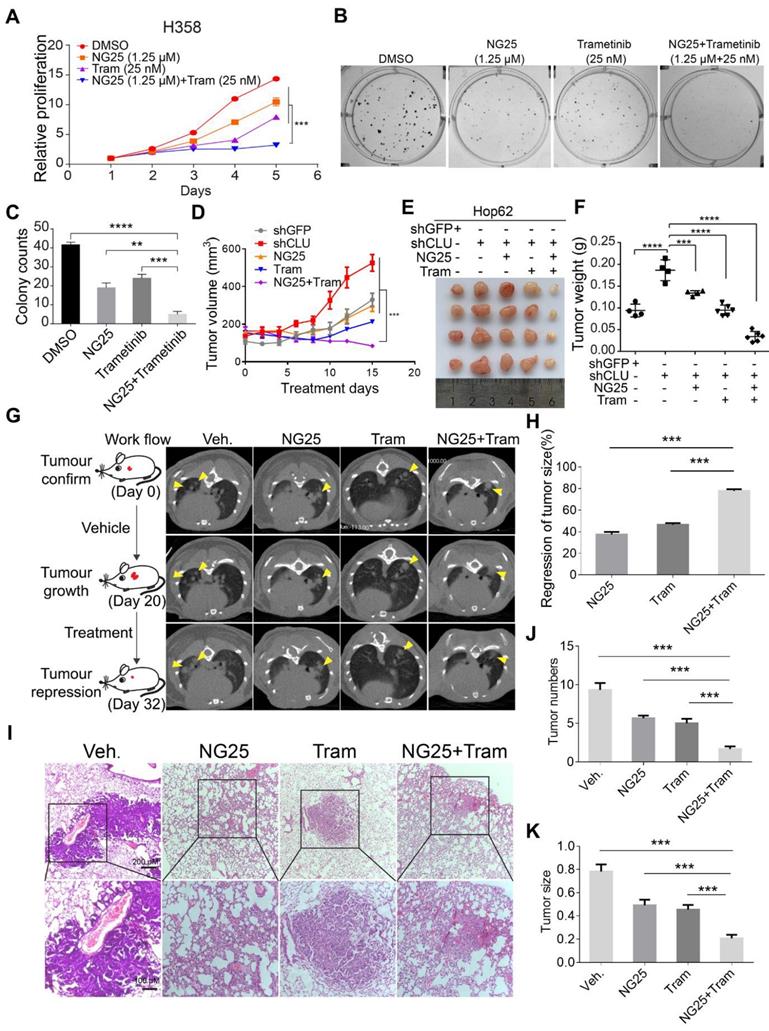
We then went on to evaluate the treatment effect of combination of NG25 and Trametinib in vivo. To this end, we subcutaneously (s.c.) inoculated Hop62-shCLU cell in nude mice and randomized them for treatment with vehicle, NG25, Trametinib or combination of NG25 and Trametinib (designated combo) when tumors reached a volume of around 150 mm3. Interestingly, we found that while NG25 or Trametinib singlet treatment slowed down the growth rate of Hop62-shCLU xenograft tumors, combo treatment shrank the tumor (Figure 6D-E). Consistently, we observed that the weight of tumors in combo treated cohort was significantly lower than that of vehicle, NG25 or Trametinib treated cohort by the end of experiment (Figure 6F). Furthermore, combo treatment significantly inhibited proliferation and increased the apoptosis of the Hop62-shCLU tumors as revealed by Ki67 and Caspase3 immunochemistry (Figure S6G-I).
Mouse models of autochthonous lung cancer faithfully recapitulate the clinical course of lung cancer development and microenvironment of lung cancer patients. We then tested combination treatment on CLU knockout mouse model. For this purpose, we generated a cohort of K-CLU mice (see Figure 1 for methods and tumor pathology) and randomized them for treatment with NG25, Trametinib or combo after documenting tumor burden through CT imaging. As expected, we saw significant tumor growth in all groups of mice during the initial 20 days of vehicle treatment. Interestingly, CT imaging revealed dramatic tumor shrinkage in combo treated group in comparison to partial regression of tumor in NG25 or Trametinib treated group (Figure 6G-H). Consistently, pathological analysis revealed significantly lower tumor numbers and smaller tumor nodules in the cohort of combo treatment (Figure 6I-K). In line with this, we found occasional foci of tumor nodules with intra-tumoral spaces, thickened alveolar wall and slight fibrosis in NG25 or trametinib treated mice, all of which are much heavier in combo treated mice (Figure 6I), indicative of drastic remodeling process to replace previous tumor area. To test the translational significance of our combinational treatment in clinic, we assayed toxicity through blood biochemical assay to analyze the damage of liver (ALT and AST), kidney (CREA and UREA) and heart (LDH) of healthy C57J/B6 mice treated with the above dosing scheme. Our results revealed no significant toxicity in singlet- or combo- treated mice (Figure S6J-N). Taken together, our data showed unique sensitivity of CLU deficient lung cancer cells to TAK1 inhibitors.
Discussion
Our current work highlighted an interesting concept: loss of function of a TSG rewired the signaling network and created an Achilles' heel in cancer cells, which could be exploited in cancer precision medicine. In case of CLU, its inactivation resulted in hyperactivation of TAK1- NF-κB signaling axis and TAK1 inhibitor exhibited exquisite cytotoxicity to CLU deficient lung cancer cells.
CLU protein is reported to undergo an intricate process of post-translational maturation. The N-terminal secretory signal peptide of 22 aa is cleaved off from CLU precursor, which is subsequently cleaved between residues Arg227-Ser228 to generate an α-chain and a β-chain. These 2 halves are assembled in antiparallel fashion to generate a heterodimeric molecule with five disulfide bridges [42]. Consistent with previous report, we found that lung cancer cells harbored intracellular full-length and cleaved Clusterin and secreted cleaved protein [43].
Earlier report has shown that CLU expression promotes migration ability of NSCLC cells [44]. However, clinical evidence for EMT/metastasis promoting activity of CLU is lacking in literature. Careful comparison between primary and metastatic NSCLC nodules are needed before clinical relevance of EMT/metastasis promoting activity of CLU can be solidified. Moreover, clinical evidence of significant association between higher CLU expression level and recurrence-free survival [45] suggests EMT/metastasis promoting activity of CLU detected in vitro to be an artifact. In contrast, clinical data, cell line data, and in vivo data in our current work strongly argue that CLU is a tumor suppressor in lung cancer. Likewise, significant correlation between higher CLU expression in clinic has been reported [45]. In the same study, authors showed that higher CLU level significantly correlated with longer overall survival [45]. Taken these evidences together, we conclude that CLU is a NSCLC tumor suppressor per se in lung cancer. Our work revealed that CLU negatively regulated TAK1 activity and the downstream NK-κB signaling. Of note, Bonacini et. al. have previously reported that NK-κB signaling pathway was negatively regulated by CLU in prostate cancer [46].
Considering facts that TGFBR1 directly recruits TRAF6[37], that TAB2 mediates TAK1-TRAF6 interaction [38] and our observation that there exists a complex of TGFBR1/TRAF6/TAB2/TAK1 in lung cancer cells and 293T cells (Figures 3C-F & Figure S4A-E), our data argue the following working model for CLU's tumor suppressive role in NSCLC cells: CLU binds TGFBR1 and competes against TRAF6 for binding TGFBR1. CLU thus interferes recruitment of complex of TRAF6/TAB2/TAK1 by activated TGFBR1. We also envisioned that CLU binds TGFBR1 on a motif different from that bound by TRAF6.
Although clinical trials are been conducted targeting G12C mutant Kras (AMG 510 (NCT03600883) and MRTX849 (NCT03785249)) along with active research on siRNA delivery [47], KRAS mutation positive NSCLC remains the most difficult subtype of lung cancers to treat. Our current work suggested another strategy: concurrent dysfunction of TSGs in KRAS mutation positive tumors may create a new Achilles' heel. In case of CLU inactivation, lung cancer cells become uniquely sensitive to TAK1/NF-κB inhibition. Most strikingly, treatment data on cell line, xenograft tumor, and autochthonous lung cancer in transgenic mouse models showed that combinational inhibition of TAK1 and MEK1/2 effectively shrank lung cancer concurrently positive for KRAS mutation and CLU deficiency. Interestingly, combinational treatment with MEK/TAK1 inhibition was earlier found to be effective in eliciting apoptosis of Kras signaling dependent colon cancer cells through inhibition of mTOR, Wnt and NF-κB signaling [48].
TAK1 is an important mediator for TGFBRs' signaling. Of note, TGF-β signaling is involved in organizing an immunosuppressive micro-environment in tumor foci by inducing Tregs [49, 50]. It is expected that TAK1 inhibition not only sensitizes NSCLC cells to targeting therapies, but normalizes immunity against tumors.
In short, here we identified CLU as a potent TSG in lung cancer. In activation of CLU leads to hyperactivation of TGFBR/TAK1 signaling axis. We show that this event is relevant to KRAS mutation positive NSCLC and that TAK1 inhibition synergizes with existing drugs to treat a portion of KRAS mutation positive NSCLC patients.
Materials and methods
Animal care and use
All mice were housed in a pathogen-free environment in Jinan University. Experimental protocols were approved by the Institutional Committee for Animal Care and Use at Jinan University and all animal work was performed in accordance with the approved protocol.
Data base analysis of CLU expression in human lung cancer and correlation between expression of CLU and TAK1-NF-κB target genes
For comparison the CLU level between lung adenocarcinoma against para-tumoral tissues with GEO database. We downloaded the data from the NCBI GEO data sets (GSE10072 and GSE7670 with probe set 208792_s_at). In addition, we also compared CLU expression between normal tissue, para-tumoral and primary tumor tissues was performed with “Compare tumor vs normal within or across tissue types” function of UCSC Xena (http://xena.ucsc.edu/compare-tissue/) [51]. The data base includes 1410 lung cancer samples from UCSC RNA-seq Compendium, where The Cancer Genome Atlas (TCGA) and the Genotype-Tissue Expression (GTEx, normal tissue of individuals without cancer) were re-aligned to hg38 genome by the same RNA-seq pipeline. The 'RSEM norm_count' dataset which was normalized by the upper quartile method was been chosen for the gene expression comparison. For correlation analysis between CLU and TAK1, IL1b, IL6 in lung adenocarcinoma patients, we acquired the gene expression data from TCGA database by searching UCSC Xena and the gene expression correlation was analyzed by GraphPad 6.0. For the expression specificity analysis of the CLU with Kras mutant in lung adenocarcinoma patients, patients were divided into CLU high and low expression groups according to the median of CLU expression. The expression specificity between the patients with Kras mutation with high or low CLU expression was analysis with Fisher' exact test, P < 0.05 is considered as significant correlation. Expression correlation analysis between CLU and IL1B or IL6 in these Kras mutant patient was analyzed by performed GraphPad 6.0. All the original data could be found in Table S1.
Survival curve analysis
The survival curve analysis of CLU expression in lung cancer patients was using Kaplan-Meier-Plotter (http://kmplot.com/analysis/index.php?cancer=lung&p=service) by searching the TGCA database. Total and Stage I lung cancer patient were analyzed respectively in this study.
Cell culture and cell engineering
Hop62 (KRAS G12C), NCI-H460 (KRAS G61H), A549 (KRAS G12S), H358 (KRAS G12C), HCC827 (EGFR G746-A750 deletion), H1975 (EGFR T790M and L858R), EKVX (non-KRAS), Hop92 (non-KRAS), H3255 (non-KRAS), H446 (non-KRAS) and H322 (non-KRAS) were purchased from ATCC (American Typical Culture Collection, Manassas, VA, USA). PC9 (EGFR G746-A750 deletion) and HEK293T were kindly provided by Dr. Kwok-Kin Wong (The Helen and Martin Kimmel Center for Stem Cell Biology, NYU). All the NSCLC cell lines were cultured in RPMI-1640 with 10% fetal bovine serum (FBS, Gibco, Life Technologies, Carlsbad, CA, USA). HEK293T cell was cultured in DMEM with 10% FBS. To generate the CLU knockdown NSCLC cell lines (shCLU), Hop62, H460, A549 and EKVX cell lines were infected with lentivirus packaged from pLKO-puro vector harboring shRNA sequence targeting CLU gene. To generate cell lines for Dox-inducible over-expression of CLU, H460, A549, EKVX and HEK293T cell were infected with lentivirus coding CLU (packaged from pLVX-tetone-CLU-puro vector). For CLU re-expression, Hop62 cell lines were infected with lentivirus encoding shRNA-resistant CLU gene (packaged from pLVX-tetone-zeo vector). To generate cell lines for Dox-inducible TAK1 knockdown, Hop62-shCLU cells were infected with lentivirus packaged from pLKO-tet-zeo vector harboring shRNA targeting TAK1 mRNA. Cells were selected with puromycin (2 μg/mL) for one week or zeomycin (300 μg/mL) for one week in appropriate cases.
Bimolecular fluorescence complementation (BiFC) assay
293T-tet-CLU cells were co-transferred with 250 ng each of pCAG-TGFBR1-C-luc and pCAG-TRAF6-Nluc; or pCAG-TAB2-Nluc and pCAG-TAK1-Luc; pCAG-TAK1-CLUc and pCAG-TRAF6-Nluc; or pCAG-TAK1-CLUc and pCAG-TAB2-Nluc respectively in 24 well plate. 50 ng Renilla vector was also co-transferred in all the above experiments as the transfection efficiency control. 1 μg/mL of Dox was added to induce CLU expression in 293T-tet-CLU cell. Fluorescence detection was performed with the Dual Luminescent Kit and the Microplate Reader.
Mouse treatment
All the transgenic mice were C57BL6 background about 6-8 weeks old with no restrictions on sex. To generate KrasG12D/CLU-/- (designated K-CLU) transgenic mice model, CLU sgRNAs were cloned into pSECC vector which co-expressing Cre and CRISPR/CAS9 (generously provided by F.J. Sanchez-Rivera and T. Jacks, Koch Institute for Integrative Cancer Research at MIT). Lentivirus of pSECC-sgCLU was packaged in 293T, validated by infecting NIH-3T3 cells and administered into lsl-KrasG12D mice through nasal instillation. pSECC-TdTomato recombinant lentivirus was used as control (designated K-Ctl). Lung tumor formation in K-CLU mice were compared to K-Ctl mice 13 weeks post-infection. To generate lung cancer transgenic mice model for Dox induced CLU expression, lentivirus of pLVX-tet-CLU was packaged in 293T, validated by infecting NIH-3T3 cells and administered nasally into Tet-KrasG12D/CC10rtTA mice, Tet-EGFR L858R/CC10rtTA mice or Tet-EML4-ALK/CC10rtTA mice, respectively. pLVX-tet-mCherry virus was used as control. The lung tumor burdens were recorded through computed tomography scan (CT, PINGSENG Healthcare) after 2 around mouths of Dox diet feeding. Mice were treated with NG25 (intraperitoneal injection, i.p, 4 mg/kg/Day), Trametinib (gavaged, 1 mg/kg/Day), NG25 and Trametinib combination or vehicle. The tumor size of the mice was analysis with ImageJ. Tumor burden comparison analysis followed our previous published study [29].
For xenograft tumor model, 2 × 106 engineered cancer cells were subcutaneously implanted into the two flanks of the nude mice. Tumors were left to fix for 1 week. Tumor-bearing mice were treated with NG25 (intraperitoneal injection, i.p, 4 mg/kg/Day), Trametinib (gavaged, 1mg/kg/Day), NG25 and Trametinib combination or vehicle. 15 days after treatment, the mice were sacrificed and the tumors burden was dissected. The tumor volume is calculated by the formula: volume = (width)2 × length × 0.5.
Statistical analysis
All the data were presented as mean values ± s.e.m. Differences between two groups compared with unpaired two-tailed t-test, while multiple comparisons was used one-way ANOVA with Bonferroni post hoc test. Statistical analyses were performed with GraphPad Prism 6.0, P < 0.05 was deemed to be statistically significant.
Supplementary Material
Supplementary figures.
Supplementary table S1.
Supplementary table S2.
Acknowledgements
This work was financially supported by NSFC (81972778, 81672309, 31701188, 31800723), Guangzhou Research Project of Science and Technology for Citizen Health (201803010124), the Fundamental Research Funds for the Central Universities (21619101), and the Science and Technology Program of Guangzhou (201604020162), Science and Technology Program of Guangdong (2017B020227001).
Author contributions
L.C. and Q.H. conceived the idea, supervised the study, and wrote the paper. Z.C. performed most of the experiments, analyzed the data and contributed to the manuscript composition. Z.F., X.D., G.Z., L.L, R.L., G.R., L.Y., and W.C. performed experiments. Q.Z. contribute to data analysis. W.L. contribute the computer analysis. Q.H. provided helpful comments.
Clinical relevance
Clinical success of precision medicine for non-small cell lung cancer (NSCLC) is severely limited by de novo as well as acquired drug resistance. Loss of function of tumor suppressor genes (TSGs) has been reported to confer drug resistance. Here we show Clusterin (CLU) as a potent and clinically relevant TSG in lung cancer. CLU effectively inhibits TGFBR1 to recruit TRAF6/TAB2/TAK1 complex and inhibits activation of TAK1-NF-κB axis to curb cell growth. A significant portion of Kras mutation positive NSCLC patients concurrently harbor low CLU protein level. We further show that combinational treatment with inhibitors targeting TAK1 and MEK1/2 effectively shrank Kras mutation positive/CLU deficient NSCLC. We thus put forward a concept that loss of function of a TSG rewires signaling network and thereby creates an Achilles' heel in tumor cells which could be exploited in precision medicine. Meanwhile, our strategy for treating a portion of Kras mutation positive NSCLC patients deserves further study.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Lai D, Visser-Grieve S, Yang X. Tumour suppressor genes in chemotherapeutic drug response. Biosci Rep. 2012;32:361-74
2. Huang S, Benavente S, Armstrong EA, Li C, Wheeler DL, Harari PM. p53 modulates acquired resistance to EGFR inhibitors and radiation. Cancer Res. 2011;71:7071-9
3. Skoulidis F, Goldberg ME, Greenawalt DM, Hellmann MD, Awad MM, Gainor JF. et al. STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer Discov. 2018;8:822-35
4. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394-424
5. Fong KM, Sekido Y, Gazdar AF, Minna JD. Lung cancer. 9: Molecular biology of lung cancer: clinical implications. Thorax. 2003;58:892-900
6. Larsen JE, Minna JD. Molecular biology of lung cancer: clinical implications. Clin Chest Med. 2011;32:703-40
7. Yoda S, Dagogo-Jack I, Hata AN. Targeting oncogenic drivers in lung cancer: Recent progress, current challenges and future opportunities. Pharmacol Ther. 2019;193:20-30
8. Meliala ITS, Hosea R, Kasim V, Wu S. The biological implications of Yin Yang 1 in the hallmarks of cancer. Theranostics. 2020;10:4183-200
9. Blaschuk O, Burdzy K, Fritz IB. Purification and characterization of a cell-aggregating factor (clusterin), the major glycoprotein in ram rete testis fluid. J Biol Chem. 1983;258:7714-20
10. Nelson AR, Sagare AP, Zlokovic BV. Role of clusterin in the brain vascular clearance of amyloid-beta. Proc Natl Acad Sci U S A. 2017;114:8681-2
11. Lin CC, Tsai P, Sun HY, Hsu MC, Lee JC, Wu IC. et al. Apolipoprotein J, a glucose-upregulated molecular chaperone, stabilizes core and NS5A to promote infectious hepatitis C virus virion production. J Hepatol. 2014;61:984-93
12. Ischia J, So AI. The role of heat shock proteins in bladder cancer. Nat Rev Urol. 2013;10:386-95
13. Oh SB, Kim MS, Park S, Son H, Kim SY, Kim MS. et al. Clusterin contributes to early stage of Alzheimer's disease pathogenesis. Brain Pathol. 2019;29:217-31
14. Trougakos IP, So A, Jansen B, Gleave ME, Gonos ES. Silencing expression of the clusterin/apolipoprotein j gene in human cancer cells using small interfering RNA induces spontaneous apoptosis, reduced growth ability, and cell sensitization to genotoxic and oxidative stress. Cancer Res. 2004;64:1834-42
15. Pucci S, Bonanno E, Pichiorri F, Angeloni C, Spagnoli LG. Modulation of different clusterin isoforms in human colon tumorigenesis. Oncogene. 2004;23:2298-304
16. July LV, Beraldi E, So A, Fazli L, Evans K, English JC. et al. Nucleotide-based therapies targeting clusterin chemosensitize human lung adenocarcinoma cells both in vitro and in vivo. Molecular cancer therapeutics. 2004;3:223-32
17. Guitart K, Loers G, Buck F, Bork U, Schachner M, Kleene R. Improvement of neuronal cell survival by astrocyte-derived exosomes under hypoxic and ischemic conditions depends on prion protein. Glia. 2016;64:896-910
18. Makridakis M, Roubelakis MG, Bitsika V, Dimuccio V, Samiotaki M, Kossida S. et al. Analysis of secreted proteins for the study of bladder cancer cell aggressiveness. J Proteome Res. 2010;9:3243-59
19. Lau SH, Sham JS, Xie D, Tzang CH, Tang D, Ma N. et al. Clusterin plays an important role in hepatocellular carcinoma metastasis. Oncogene. 2006;25:1242-50
20. Wang C, Jiang K, Kang X, Gao D, Sun C, Li Y. et al. Tumor-derived secretory clusterin induces epithelial-mesenchymal transition and facilitates hepatocellular carcinoma metastasis. Int J Biochem Cell Biol. 2012;44:2308-20
21. Miyake H, Gleave ME, Arakawa S, Kamidono S, Hara I. Introducing the clusterin gene into human renal cell carcinoma cells enhances their metastatic potential. J Urol. 2002;167:2203-8
22. Shiota M, Zardan A, Takeuchi A, Kumano M, Beraldi E, Naito S. et al. Clusterin mediates TGF-beta-induced epithelial-mesenchymal transition and metastasis via Twist1 in prostate cancer cells. Cancer Res. 2012;72:5261-72
23. Chayka O, Corvetta D, Dews M, Caccamo AE, Piotrowska I, Santilli G. et al. Clusterin, a haploinsufficient tumor suppressor gene in neuroblastomas. J Natl Cancer Inst. 2009;101:663-77
24. Bettuzzi S, Davalli P, Davoli S, Chayka O, Rizzi F, Belloni L. et al. Genetic inactivation of ApoJ/clusterin: effects on prostate tumourigenesis and metastatic spread. Oncogene. 2009;28:4344-52
25. Thomas-Tikhonenko A, Viard-Leveugle I, Dews M, Wehrli P, Sevignani C, Yu D. et al. Myc-transformed epithelial cells down-regulate clusterin, which inhibits their growth in vitro and carcinogenesis in vivo. Cancer Res. 2004;64:3126-36
26. Yan Y, Luo K, Zhang H, Chai W. RNA interference-mediated secretory clusterin gene silencing inhibits proliferation and promotes apoptosis of human non-small cell lung cancer cells. Hepatogastroenterology. 2013;60:70-5
27. Albert JM, Gonzalez A, Massion PP, Chen H, Olson SJ, Shyr Y. et al. Cytoplasmic clusterin expression is associated with longer survival in patients with resected non-small cell lung cancer. Cancer epidemiology, biomarkers & prevention: a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2007;16:1845-51
28. Panico F, Casali C, Rossi G, Rizzi F, Morandi U, Bettuzzi S. et al. Prognostic role of clusterin in resected adenocarcinomas of the lung. Lung Cancer. 2013;79:294-9
29. Wu Q, Tian Y, Zhang J, Tong X, Huang H, Li S. et al. In vivo CRISPR screening unveils histone demethylase UTX as an important epigenetic regulator in lung tumorigenesis. Proc Natl Acad Sci U S A. 2018;115:E3978-E86
30. Sanchez-Rivera FJ, Papagiannakopoulos T, Romero R, Tammela T, Bauer MR, Bhutkar A. et al. Rapid modelling of cooperating genetic events in cancer through somatic genome editing. Nature. 2014;516:428-31
31. Zhang H, Cao J, Li L, Liu Y, Zhao H, Li N. et al. Identification of urine protein biomarkers with the potential for early detection of lung cancer. Sci Rep. 2015;5:11805
32. Gao L, Hu Y, Tian Y, Fan Z, Wang K, Li H. et al. Lung cancer deficient in the tumor suppressor GATA4 is sensitive to TGFBR1 inhibition. Nat Commun. 2019;10:1665
33. Ninomiya-Tsuji J, Kishimoto K, Hiyama A, Inoue J, Cao Z, Matsumoto K. The kinase TAK1 can activate the NIK-I kappaB as well as the MAP kinase cascade in the IL-1 signalling pathway. Nature. 1999;398:252-6
34. Kim SI, Choi ME. TGF-beta-activated kinase-1: New insights into the mechanism of TGF-beta signaling and kidney disease. Kidney Res Clin Pract. 2012;31:94-105
35. Ma FY, Tesch GH, Ozols E, Xie M, Schneider MD, Nikolic-Paterson DJ. TGF-beta1-activated kinase-1 regulates inflammation and fibrosis in the obstructed kidney. Am J Physiol Renal Physiol. 2011;300:F1410-21
36. Reddy KB, Karode MC, Harmony AK, Howe PH. Interaction of transforming growth factor beta receptors with apolipoprotein J/clusterin. Biochemistry. 1996;35:309-14
37. Sorrentino A, Thakur N, Grimsby S, Marcusson A, von Bulow V, Schuster N. et al. The type I TGF-beta receptor engages TRAF6 to activate TAK1 in a receptor kinase-independent manner. Nat Cell Biol. 2008;10:1199-207
38. Takaesu G, Kishida S, Hiyama A, Yamaguchi K, Shibuya H, Irie K. et al. TAB2, a novel adaptor protein, mediates activation of TAK1 MAPKKK by linking TAK1 to TRAF6 in the IL-1 signal transduction pathway. Mol Cell. 2000;5:649-58
39. Shyu YJ, Hu CD. Fluorescence complementation: an emerging tool for biological research. Trends Biotechnol. 2008;26:622-30
40. Stella GM, Scabini R, Inghilleri S, Cemmi F, Corso S, Pozzi E. et al. EGFR and KRAS mutational profiling in fresh non-small cell lung cancer (NSCLC) cells. J Cancer Res Clin Oncol. 2013;139:1327-35
41. Ji H, Wang Z, Perera SA, Li D, Liang MC, Zaghlul S. et al. Mutations in BRAF and KRAS converge on activation of the mitogen-activated protein kinase pathway in lung cancer mouse models. Cancer Res. 2007;67:4933-9
42. Fini ME, Bauskar A, Jeong S, Wilson MR. Clusterin in the eye: An old dog with new tricks at the ocular surface. Exp Eye Res. 2016;147:57-71
43. Chaiwatanasirikul KA, Sala A. The tumour-suppressive function of CLU is explained by its localisation and interaction with HSP60. Cell Death Dis. 2011;2:e219
44. Chou TY, Chen WC, Lee AC, Hung SM, Shih NY, Chen MY. Clusterin silencing in human lung adenocarcinoma cells induces a mesenchymal-to-epithelial transition through modulating the ERK/Slug pathway. Cell Signal. 2009;21:704-11
45. Panico F, Rizzi F, Fabbri LM, Bettuzzi S, Luppi F. Clusterin (CLU) and lung cancer. Adv Cancer Res. 2009;105:63-76
46. Bonacini M, Negri A, Davalli P, Naponelli V, Ramazzina I, Lenzi C. et al. Clusterin Silencing in Prostate Cancer Induces Matrix Metalloproteinases by an NF-kappaB-Dependent Mechanism. J Oncol. 2019;2019:4081624
47. Yin F, Hu K, Chen Y, Yu M, Wang D, Wang Q. et al. SiRNA Delivery with PEGylated Graphene Oxide Nanosheets for Combined Photothermal and Genetherapy for Pancreatic Cancer. Theranostics. 2017;7:1133-48
48. McNew KL, Whipple WJ, Mehta AK, Grant TJ, Ray L, Kenny C. et al. MEK and TAK1 Regulate Apoptosis in Colon Cancer Cells with KRAS-Dependent Activation of Proinflammatory Signaling. Mol Cancer Res. 2016;14:1204-16
49. Fu S, Zhang N, Yopp AC, Chen D, Mao M, Chen D. et al. TGF-beta induces Foxp3 + T-regulatory cells from CD4 + CD25 - precursors. Am J Transplant. 2004;4:1614-27
50. Panagi M, Voutouri C, Mpekris F, Papageorgis P, Martin MR, Martin JD. et al. TGF-beta inhibition combined with cytotoxic nanomedicine normalizes triple negative breast cancer microenvironment towards anti-tumor immunity. Theranostics. 2020;10:1910-22
51. Goldman MJ, Craft B, Hastie M, Repecka K, McDade F, Kamath A. et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat Biotechnol. 2020;38:675-8
Author contact
![]() Corresponding author: L.C. (email: chenliangedu.cn), Q.H. (email: tqyhejnu.edu.cn), L.Y. (yulei118com) or G.R. (15868197173com).
Corresponding author: L.C. (email: chenliangedu.cn), Q.H. (email: tqyhejnu.edu.cn), L.Y. (yulei118com) or G.R. (15868197173com).