Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Immune-related targets in tumor...
Small molecules as...
Small molecules for imaging...
Conclusion and perspective
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(25):7849-7871. doi:10.7150/thno.37218 This issue Cite
Review
Small molecules as theranostic agents in cancer immunology
Jindian Li1,2, Juno Van Valkenburgh1, Xingfang Hong3, Peter S. Conti1, Xianzhong Zhang2 ![]() , Kai Chen1
, Kai Chen1 ![]()
1. Molecular Imaging Center, Department of Radiology, Keck School of Medicine, University of Southern California, 2250 Alcazar Street, CSC103, Los Angeles, CA 90033, USA.
2. State Key Laboratory of Molecular Vaccinology and Molecular Diagnostics & Center for Molecular Imaging and Translational Medicine, School of Public Health, Xiamen University, Xiamen 361102, China.
3. Laboratory of Pathogen Biology, School of Basic Medical Sciences, Dali University, Dali 671000, China.
Received 2019-6-2; Accepted 2019-9-10; Published 2019-10-15
Abstract

With further research into the molecular mechanisms and roles linking immune suppression and restraint of (pre)malignancies, immunotherapies have revolutionized clinical strategies in the treatment of cancer. However, nearly 70% of patients who received immune checkpoint therapeutics showed no response. Complementary and/or synergistic effects may occur when extracellular checkpoint antibody blockades combine with small molecules targeting intracellular signal pathways up/downstream of immune checkpoints or regulating the innate and adaptive immune response. After radiolabeling with radionuclides, small molecules can also be used for estimating treatment efficacy of immune checkpoint blockades. This review not only highlights some significant intracellular pathways and immune-related targets such as the kynurenine pathway, purinergic signaling, the kinase signaling axis, chemokines, etc., but also summarizes some attractive and potentially immunosuppression-related small molecule agents, which may be synergistic with extracellular immune checkpoint blockade. In addition, opportunities for small molecule-based theranostics in cancer immunology will be discussed.
Keywords: small molecules, theranostic agents, cancer immunology, molecular imaging, targeted therapy
Introduction
Cancer is still one of the leading causes of morbidity and mortality worldwide. In 2018, 18 million new cancer cases and 9 million cancer-related deaths occurred [1]. Recent immuno-oncology therapies have seen significant success by remolding the immune system of patients to treat multiple cancers [2-6].
The mechanism of cancer immunotherapy is based on the blockade of tumor-mediated inhibition of immune responses rather than direct targeting of tumor cells. “Immune checkpoints” means the stimulation or inhibition of receptor-ligand signal axes between tumor cells and immune cells including T cells, dendritic cells (DCs), and macrophages in the tumor microenvironment [7, 8]. It wasn't until 1992 when the first immunotherapy drug - PROLEUKIN® (aldesleukin) was approved by the US Food and Drug Administration (FDA), which opened a new era of immunotherapy. Various immune checkpoint-directed antibodies such as anti-cytotoxic-T-lymphocyte-associated protein 4 (anti-CTLA-4), anti-programmed cell death 1 (anti-PD-1), anti-programmed cell death ligand 1 (anti-PD-L1), and anti-CD19 have shown to affect various cancers and are approved by the US FDA [9]. In addition, other new and promising drugs for targets such as T-cell immunoglobulin mucin 3 (TIM3), tumor necrosis factor receptor superfamily member 4 (TNFRSF4), and lymphocyte-activation gene 3 (LAG-3) are being investigated in clinical trials, such as NCT02817633 and NCT01303705 (Table 1) [10].
Representative drugs approved by the US FDA and other checkpoint inhibitors
| (Generic/Brand name) | Target | Mainly indication (Approved time) | Status |
|---|---|---|---|
| Aldesleukin | IL-2 receptor | Metastatic Melanoma (1998.1) Metastatic Renal Cell Carcinoma (mRCC) (1992.5) | Approved |
| Ipilimumab/ Yervoy | CTLA-4 | Metastatic Colorectal Cancer (mCC) (2018.7) Advanced Renal Cell Carcinoma (aRCC) (2018.4) Metastatic Melanoma (2017.7) Late-Stage Melanoma (2011.5) | Approved |
| Nivolumab/ Opdivo | PD-1 | Hepatocellular Carcinoma (HCC) (2017.9) Metastatic Urothelial Carcinoma (mUC) (2017.2) Head and Neck Cancer (HNC) (2016.11) Hodgkin Lymphoma (HL) (2016.5) mRCC (2015.11) Advanced Melanoma (2014.12) | Approved |
| Pembrolizumab/Keytruda | PD-1 | mSCLC (2019.6); Squamous Cell HNC (2019.6); aRCC (2019.4); NSCLC (2019.4); Advanced or Metastatic Merkel Cell Carcinoma (a/mMCC) (2018.12); HCC (2018.11); mNSCLC (2018.10) Primary Mediastinal Large B-Cell Lymphoma (PMBCL) (2018.6) Metastatic Cervical Cancer (mCC) (2018.6) Advanced or Metastatic Gastric Cancer (2017.9) Advanced or Metastatic UC (2017.5); HL (2017.3); mNSCLC (2016.10) Advanced Melanoma (2014.9) | Approved |
| Cemiplimab/ Libtayo | PD-1 | Squamous Cell Carcinoma (2018.9) | Approved |
| toripalimab | PD-1 | Advanced or Metastatic Melanoma | Phase 2 |
| Atezolizumab/ Tecentriq | PD-L1 | Extensive-Stage SCLC (2019.3) Metastatic Triple-Negative Breast Cancer (mTNBC) (2019.3.) Metastatic Non-Squamous NSCLC (2018.12) Advanced Bladder Cancer (2017.4) Metastatic Lung Cancer (2016.10) Urothelial Carcinoma(2016.5) | Approved |
| Avelumab/ Bavencio | PD-L1 | Advanced Renal Cell Carcinoma (2019.5) Urothelial Carcinoma (2017.5) Merkel Cell Carcinoma (2017.3) | Approved |
| Durvalumab/ Imfinzi | PD-L1 | Non-Small Cell Lung Cancer (2018.2) Urothelial Carcinoma (2017.3) | Approved |
| MEDI4736 | PD-L1 | NSCLC | Phase 3a |
| Avelumab | PD-L1 | Ovarian Cancer | Phase 2 |
| Tisagenlecleucel/Kymriah | CD19 | Large B-Cell Lymphoma (2018.5) Acute Lymphoblastic Leukemia (2017.8) | Approved |
| Axicabtagene ciloleucel/ Yescarta | CD19 | Large B-Cell Lymphoma (2017.10) | Approved |
| Cyclophosphamide | CD19 | Lymphocytic Leukemia | Phase 1 |
| Sym023 | TIM-3 | Metastatic Cancer; Solid Tumor; Lymphoma | Phase 1 |
| TSR-022 | TIM-3 | Advanced or Metastatic Solid Tumors | Phase 1 |
| MEDI6469 | TNFRSF4 | Head and Neck Cancer; Progressive Metastatic Prostate Cancer | Phase 1 |
| KHK4083 | TNFRSF4 | B-Cell Non-Hodgkin Lymphoma | Phase 2 |
| PF-04518600 | TNFRSF4 | Metastatic Renal Cell Cancer | Phase 2 |
| Sym022 | LAG-3 | Advanced Solid Tumor Malignancies or Lymphomas | Phase 1 |
| BMS 986016 | LAG-3 | Gliosarcoma and Recurrent Brain Neoplasm | Phase 1 |
However, new problems have arisen in the course of immunotherapy: 1) nearly 70% of patients who received immune checkpoint therapeutics showed no response or only showed a short-term beneficial effect with recurrence soon afterwards [8]; 2) immune checkpoint blockade increases the activity of the immune system and can result in immune-related adverse events such as myocarditis, vasculitis, heart failure, dermatitis, endocrine dysfunction, and even death [11-13]; 3) there are primary, adaptive, and acquired resistances to cancer immunotherapies [14]; and 4) the disadvantages of antibodies include their long half-life (even multiple weeks) and persistent side effects once injected into the body. Therefore, it is necessary to identify novel immune-related oncology molecular targets and small molecule drugs to expand the treatment range of tumors and/or subtypes of patients, limit adverse events and reduce resistances of immunotherapy.
Recently, combination therapies are widely considered as the most promising oncology treatment strategy. Exploiting intracellular immune-related signal pathways to improve the effect of tumor treatment is an important transition. Intracellular pathways downstream of checkpoint blockade such as the kynurenine pathway, purinergic signaling, and the kinase pathway axis have been explored, and small-molecule-mediated therapeutic agents are being developed, which may show complementary and/or synergistic effects when combined with extracellular checkpoint antibody blockades. Small-molecule drugs possess some advantages over antibodies: 1) the ability go across cellular membranes and other physiological barriers and reach intracellular targets; 2) oral bioavailability; 3) various dosage forms and excellent pharmacokinetic characteristics such as good tumor penetration, efficient delivery into brain tissues, appropriate half-life, and intracellular targets [15]; 4) lower manufacturing costs; and 5) diversified strategies for combined therapy by passing into the cytoplasm and interacting with multiple intracellular targets [16, 17]. Importantly, kinase-targeted small molecule inhibitors have been established, which are clinically effective and possess appropriate selectivity to avoid or manage clinical toxicities.
This review summarizes the recent use of small-molecule drugs in tumor immunotherapies and immunodiagnostics in (pre)clinical trials, and provides thoughts regarding their future utility both as therapeutic agents and diagnostic tracers. Several comprehensive reviews on small molecules in cancer immunotherapy have been published previously [17-19], which highlighted the small molecule-based immune mechanism, therapeutic compound structures or imaging application in immunity. This review provides a recent update on not only the immune mechanism and therapeutic compounds but also small molecule-based diagnostic radiotracers. Broad applications of small molecules as theranostic agents in cancer immunology are presented and discussed.
Immune-related targets in tumor microenvironment
Clinical investigators reported that the combination of checkpoint inhibitors with other targeted agents provide multiple points of opportunity for cancer treatments. The various cell types and targets/signal pathways involved in cancer immunity supply prosperous potential targets of intervention for small-molecule-mediated agents, including receptors, extracellular enzymes, and intracellular signal transduction pathways.
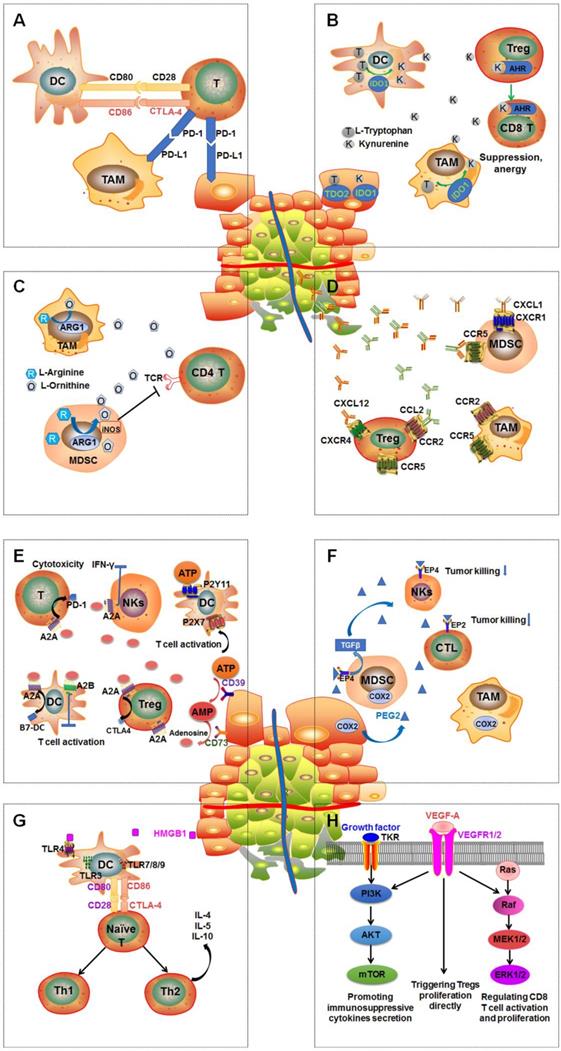
In general, the tumor microenvironment is quite complex. It is made up of tumor cells, multiple immune cells, lymphovascular cells, and extracellular matrix [20, 21]. The interactions between different tumor variants and diverse immune cells either annihilate tumors by activating immune responses or boost immune tolerance and eventually result in tumor proliferation and/or metastasis (Figure 1) [20, 22].
Multiple immunosuppressive mechanisms coexist in tumor microenvironment. A: PD-1regulates T-cell activation through binding to its ligand PD-L1. CTLA-4 can stop potentially autoreactive T cells at the initial stage of naive T-cell activation. B: Overexpression of DCs and TAMs or IDO and tryptophan 2,3‑dioxygenase (TDO2) in tumor leads to extracellular tryptophan depletion and tryptophan production, subsequently causing defective antigen presentation of DCs and Tregs activations and effector T cell function suppression. C: Upregulation of arginase 1 (ARG1) in tumor-associated macrophages (TAMs) and myeloid-derived suppressor cells (MDSCs) results in arginine the depletion, leading to MDSC-mediated immune suppression and impaired CD4+ T cell function. D: Chemokines CXCL1, CXCL12, CCL2, and CCL5 are secreted by the tumor then spread to the vasculature leading to the recruit and activity of immunosuppressive MDSCs, TAMs and Tregs through interacting with their receptors: CXCR1, CXCR4, CCR2 and CCR5. E: ATP is dephosphorylated to adenosine by CD39 and CD73. Extracellular adenosine interacts with their receptors A2AR and A2BR overexpressed on Tregs, DCs, and T cells to regulate immunosuppressive functions through boosting upregulation of CTLA-4, PD-1 and B7 proteins. F: COX2 is overexpressed on tumor cells TAMs and MDSCs to stimulate PGE2 production and then enhance the tumor proliferation and immunosuppressive function of TAMs and MDSCs. In addition, enhancing TGF-β production by MDSCs can inhibit the function of NKs. G: Secretion of the high-mobility-group box 1 (HMGB1) protein by dying tumor cells can stimulate the expression of CD80 and CD86 on DCs by binding to TLR4 overexpressed by DCs, which contributes to the differentiation if naïve and/or activated T cells into T helper 1 (Th1) and T helper 2 (Th2). H: Activation of PI3K-AKT-mTOR pathway is able to boost expression of immunosuppressive cytokines, chemokines, and checkpoint ligands and recruit regulatory immune cell subsets such as MDSCs and Tregs into tumor. RAS-RAF-MEK-ERK1/2 pathway plays a critical role in CD8 T cell activation, proliferation, and survival by regulating the production of IL-2. The activation of VEGF-A/VEGFR axis enhances PD-1 expression on CD8+ T cells leading to the exhaustion of anti-tumor immune cells. In addition, VEGF-A/VEGFR could enhance the pathway of PI3K-AKT-mTOR and RAS-RAF-MEK-ERK1/2.
CTLA-4, PD-1 and PD-L1 are considered the most prominent immune pathway checkpoints [23]. CTLA-4 and PD-1 are mainly overexpressed by T cells and increase the tolerance of immune cells. PD-L1 is mainly expressed on tumor cells, which binds to PD-1 on immune cells to induce immune suppression. The activation of the PD-1/PD-L1 signal axis also can inhibit proliferation and survival of effector T cells, and secretions of interferon gamma (IFN-γ), interleukin-2 (IL-2), and tumor necrosis factor alpha (TNF-α) [24, 25]. CTLA-4 is upregulated on activated T cells. CTLA-4 can resist CD28 activity via binding to CD80 and CD86 with a much higher affinity than that of CD28, subsequently inducing inhibition of T-cell activation. In addition, activated CD8+ T cells also overexpress CTLA-4, which counteracts the activity of helper T cells downstream and boosts the regulatory T cells (Tregs) immunosuppressive activity. Therefore, CTLA-4 plays a significant role in the early development of immune tolerance. CTLA-4 inhibitors are able to stimulate activated T-cell activation, subsequently showing antitumor immune response [26].
Tumor-associated macrophages (TAMs) and myeloid-derived suppressor cells (MDSCs) are also able to cause the suppression of immune effectors [27]. M2 macrophages present anti-inflammatory and pro-tumorigenic effects such as promoting tumor neovasculature formation, invasion and metastasis. In addition, MDSCs can secrete transforming growth factor beta (TGF-β) and IL-10 to produce direct immunosuppressive effects on T effector cells or induce Tregs generation. Spleen MDSCs are able to downregulate the cell adhesion molecule L-selectin on CD4+ and CD8+ T cells, resulting in a decrease in the activation and homing of CD8+ cells in lymph nodes [28]. Cancer-associated fibroblasts (CAFs) can be stimulated by TGF-β and fibroblast growth factor (FGF), thereby promoting tumorigenesis, lymphatic vascularization, and metastasis [29]. The function of MDSCs, DCs, macrophages and TAMs can be regulated by indoleamine 2,3-dioxygenase 1 (IDO1), chemokines (CXCRs), arginase 1 (ARG1), or toll-like receptors (TLRs) [17].
Reprogramming of energy metabolism of cancer cells is very important in immunosuppression. Intratumoral hypoxia induces upregulation of hypoxia inducible factor-1α (HIF-1α) through regulating ATP-binding cassette (ABC) transporters. On one hand, accumulated ATP stimulates antitumor immune response via the P2 purinergic receptors (P2XRs or P2YRs) mainly expressed on macrophages, DCs, CD4+ T cells, and CD8+ T cells. On the other hand, accumulated ATP can be further degraded to adenosine by the catalysis of ectonucleotidases CD39 and CD73 mainly overexpressed on tumor cells, B cells and Tregs [30]. Extracellular adenosine mediates immunosuppression by interacting with four subtypes of adenosine receptors, A1R (presented on neutrophils and immature DCs), A2AR (presented on most immune cells and platelets), A2BR (presented on tumor cell macrophages, DCs, and mast cells), and A3R (presented on neutrophils and mast cells) [31]. In addition, intracellular cyclic AMP (one downstream signaling molecule of ATP) of MDSCs, TAMs, Tregs and tumor cells, is also associated with immunosuppression by COX2 (overexpressed on tumor cells, MDSCs, TAMs, and Tregs), EP2 receptor (presented on cytotoxic T lymphocytes (CTLs) and Tregs), and EP4 receptor (mainly expressed on DCs, natural killer cells (NKs), TH1, and TH17 cells) to regulate MDSCs, Tregs, NKs, and tumor cells [17].
Intracellular signal transduction pathways which are involved in immune resistance have been thoroughly explored. Various kinase signal pathways are the key regulatory factors in the immune system [32]. Activin-like kinase 5 (ALK5) can diminish TGF-β signaling leading to activation of CD8+ T cells, stimulation of NKs, and generation of CTLs [33]. Phosphatidylinositol-3-OH kinase (PI3Kδ) is able to attenuate Tregs function, causing activation of effector T cells [34]. Colony stimulating factor 1 (CSF1) stimulates M1 to M2 polarization, and then boosts tumor proliferation and survival [35]. Small-molecule drugs, such as vemurafenib and dabrafenib (v-Raf murine sarcoma viral oncogene homolog B1 [BRAF] inhibitors), cobimetinib and trametinib (mitogen-activated extracellular signal-regulated kinase [MEK] inhibitors), and sorafenib, and pazopanib (vascular endothelial growth factor [VEGF] inhibitors) have been approved by the US FDA for the treatment of multiple cancers [36].
Small molecules as immunotheranostics
Current strategies of immunotherapy aim to reverse immune resistance either by promoting the recognition of tumor-associated antigens or by modulating signals of T cell co-receptors through biological modalities. Multiple clinical trials suggested that small molecule-based approaches of targeted multiple immune-related targets mentioned above show complementary and/or synergistic effect with immune checkpoint inhibitors, which can further promote the response rates of patients and improve survival rates. Nearly 25% of immunotherapy clinical trials combine small molecules as partners for immune checkpoint blockades [37]. Therefore, it is necessary to summarize the latest developments of small-molecule-mediated targeting agents as immunotherapies in cancer, and to offer considerations about their utility both as mono-agents and/or in combination with other anti-cancer drugs.
PD-1/PD-L1 immune checkpoint
PD-1 is overexpressed by multiple immune cells such as activated T cells, B cells, NKs, DCs, and TAMs, and it is a critical regulator protein for immune inhibition in the innate and adaptive immune systems [38]. PD-L1 is overexpressed on various solid tumors and hematological malignancies [39, 40]. The PD-1/PD-L1 signal axis inhibits the T cell functions by creating a “molecular shield” in the tumor microenvironment. The interactions of PD-1 and PD-L1 may promote T cell exhaustion, or induce the CD4+ and CD8+ T cell apoptosis, or enhance immunosuppression of intratumoral Tregs. To date, multiple PD-1/PD-L1 inhibitor antibodies have been effective in some advanced cancer types; however, a remarkable proportion of patients remain resistant to these antibody-based immunotherapies. In order to further expand the response rates of patients to immunotherapies, various small molecule drugs are being explored (Figure 2).
Small-molecule inhibitors of PD-1/PD-L1.
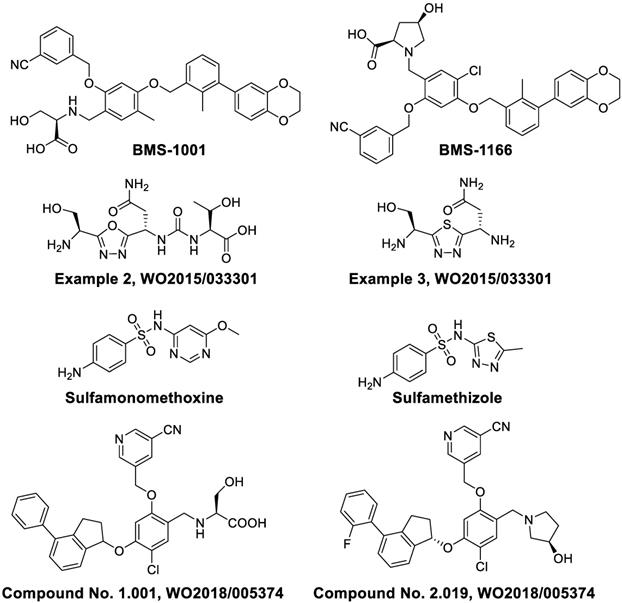
Researchers at Bristol-Myers Squibb (BMS) first synthesized multiple biaryl drugs as PD-1/PD-L1 and CD80/PD-L1 interaction small molecule inhibitors (WO2015/034820). The interaction mechanism may induce the dimerization of PD-L1, subsequently occluding the PD-1 interaction surface [41]. The best lead compounds are BMS-1001 and BMS-1166 which can induce dimerization of PD-L1 to exert therapeutic activities [42]. BMS-1001 and BMS-1166 can completely restore anti-CD3-mediated T cell activation in nuclear factor of activated T cells (NFAT) luciferase reporter-transfected Jurkat T cells [43]. In addition, scientists from Aurigene Discovery Technologies Limited synthesized two compounds with 1,3,4-oxadiazole and 1,3,4-thiadiazole scaffolds (Figure 2, Examples 2 and 3, respectively), which are able to inhibit the PD-1 signaling pathway (WO2011/082400 A3). Sharpe and colleagues synthesized and tested small-molecule PD-1 modulators sulfamonomethoxine and sulfamethizole and their derivatives, which both inhibited the expression of PD-1 in transgenic mouse T cells (WO2011/082400 A3). Most recently, the company Curis Inc. reported compounds CA-170 and CA-327, which not only bind to PD-L1 but also antagonize VISTA or TIM3 binding respectively [44]. They can boost T cell proliferation and cytokine secretion. CA-170 is being evaluated in a trial of clinical phase I in humans with advanced solid tumors or lymphomas (NCT02812875). However, their structures have not been disclosed yet.
In 2018, a patent (WO2018/005374 A1) reported by ChemoCentryx Inc. described immunomodulatory inhibitors (Compound No. 1.001 and Compound No. 2.019) that are able to inhibit the PD-1 pathway as shown by ELISA platform-based biochemical interaction assay using human PD-L1.
Amino acid catabolism
The metabolism of amino acids plays an important role in regulating the innate immune response when diseases occur. Particularly the catabolism of tryptophan and arginine can regulate the immune responses to T cell proliferation and activation. The metabolic pathway of tryptophan catabolized to kynurenine is an essential regulator in maintaining the immunosuppressive microenvironment in many types of cancers. Indoleamine-2,3-dioxygenase (IDO) and TRP-2,3-dioxygenase (TDO) are the key and rate-limiting enzymes in establishing and maintaining immune privilege in tumor immune escape [45]. Recruitment of ARG1-expressing MDSCs at a tumor site results in the depletion of L-arginine, which causes reduced proliferation of T-cells and NKs and inhibition of the antitumor immune response. Recruitment of ARG1-overexpressed MDSCs at a tumor site causes L-arginine depletion, which decreases the proliferation of T-cells and NKs and inhibition of the antitumor immune response.
Indoleamine-pyrrole 2,3-dioxygenase (IDO)
The IDO family consists of IDO isozymes (IDO1 and IDO2) and tryptophan 2,3‑dioxygenase (TDO2), catalyzing tryptophan to N-formylkynurenine and subsequently to kynurenine and other metabolites. IDOs are overexpressed in macrophages, DCs and various tumor types and contribute to immunosuppression, leading to poor prognosis. The IDO pathway can diminish immune antigen recognition by inducing differentiation and hyper-activation of Tregs and inhibiting immune responses of effector T cells and decreasing DC function. The interaction of kynurenine with aryl hydrocarbon receptor has been verified as a pivotal pathway in immunosuppression functions.
D-1MT, or indoximod (Figure 3), the D-enantiomer of 1-methyl tryptophan, has been investigated in combination with pembrolizumab in a clinical phase II trial for patients with metastatic melanoma (NCT02073123). Indoximod in combination with a taxane compound is also being investigated in a phase II clinical study for treating patients with metastatic breast cancer (NCT01792050). INCB024360 (Figure 3), or epacadostat, a high selective human IDO1 antagonist, boosts effector T cells and NKs growth, enhances IFN-γ production, increases the amounts of CD86high DCs and diminishes Tregs conversion [46]. Although phase III clinical trials (NCT02752074) showed that epacadostat in combination with pembrolizumab for the treatment of melanoma did not reveal superior outcome compared to pembrolizumab alone, the failed trial may have several caveats: 1) it is uncertain as to whether the target was adequately inhibited; 2) a mechanistic rationale for the combination needs to be tested further. Nevertheless, regarding immune-related toxic effects, epacadostat in combination with pembrolizumab therapy seemed to be well tolerated compared with other immunotherapy combinations such as ipilimumab plus pembrolizumab [47]. Better rationalized compounds and trial designs will be significant in the future to accurately evaluate medical impact. IDO1 antagonist navoximod (NLG-919/GDC-0919) has also entered a clinical phase I trial for the therapy of advanced solid tumors (NCT02471846). The combination of navoximod and atezolizumab showed acceptable tolerability, safety, and pharmacokinetics for patients with advanced cancer. However, there was no clear evidence of benefit from adding navoximod to atezolizumab although activity was observed. In a CT26 colon carcinoma model with high IDO1 activity, PF-06840003 can reduce over 80% intratumoral kynurenine levels and inhibited tumor growth both in monotherapy and, with an increased efficacy, in combination with a humanized anti-PD-L1 antibody avelumab [48]. AMG-1 [49], miconazole [50], imidazothiazole derivative BITMC [51], and a 2-aminophenylurea derivatives DFPTA and CUPCA (Figure 3) are some promising lead compounds [52].
Chemical structures of IDO, TDO, and arginase inhibitors.
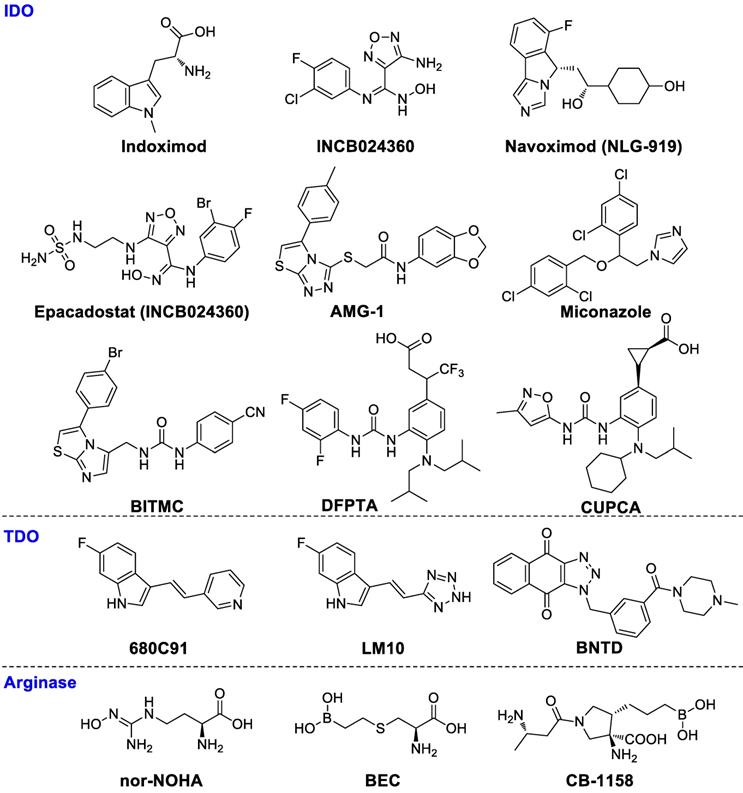
Tryptophan-2,3-dioxygenase (TDO)
TDO is significantly overexpressed in glioma, lung, breast and colorectal cancer, which is closely related to malignant progression and poor survival [53-58]. In glioma, the higher tumor TDO expression is negatively correlated with CD8+ immune cell infiltration [53]. Salter et al. first reported a new TDO inhibitor 680C91 (Figure 3), which could effectively inhibit TDO activity, but the solubility and bioavailability were poor [59]. LM10 is a potent TDO inhibitor with higher solubility and better bioavailability as compared to 680C91. The plasma concentration of LM10 is about 330 times over the 680C91 after seven days of oral administration of 160 mg/kg/day. LM10 demonstrated convincing anti-tumor activity in a preclinical assay showing approximately 57% inhibition of mice with tumor progression compared to normal drinking water (P < 0.001) [54]. Wu and coworkers synthesized a highly potent TDO inhibitor BNTD, but further evaluations should be done to verify its therapeutic effects in vivo [60]. In a word, TDO inhibitors as novel cancer treatments need further investigations.
Arginase
The arginine catabolism pathway is a promising approach to reversing immune suppression in the tumor microenvironment [61]. MDSC and TAMs both express ARG1, which can decompose the amino acid arginine into ornithine and urea. The consumption of extracellular arginine leads to the CD3ζ chain depletion of T-cell receptors (TCRs), subsequently causing tolerance of T cell responses to antigens. The concentration of ARG1 in MDSCs is elevated in breast cancer and renal cell carcinoma [62, 63]. ARG1 inhibition has been shown to prevent lung carcinoma proliferation in mice [64]. Small molecule arginase inhibitors including nor-NOHA [65, 66], BEC [67] and CB-1158 are under evaluation.
Chemokines and chemokine receptors
Chemokines are the pivotal mediators of cancer related chronic inflammation, which modify expression in malignancies, and mediate leukocyte activation and recruitment, angiogenesis, and the proliferation and metastasis of cancer cells. More importantly, the appropriate recruitment of immune cells is orchestrated by the temporal and spatial expression of chemokines and chemokine receptors [68].
CXCR family
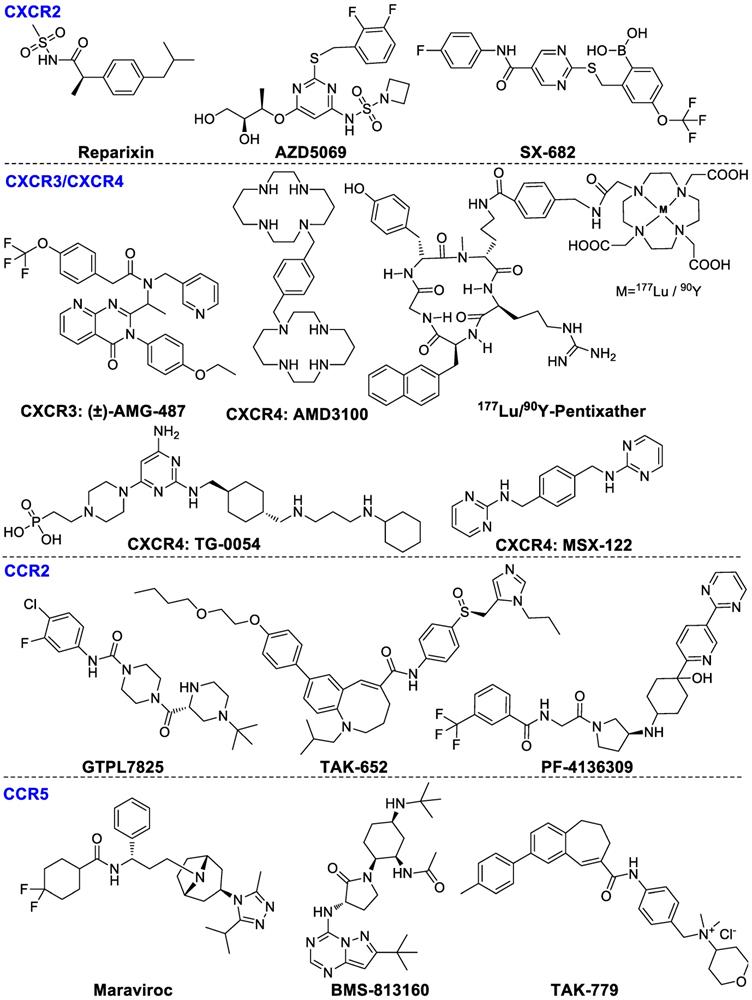
Chemokines, secreted proteins, are critical for lymphoid system development and homing, retention, infiltration and activation of T cells to tumors [69]. Chemokines are divided into four main subgroups: CC, XC, CXC, and CX3C. Chemokines are mainly found on the surface of immunocytes, tumor cells and stromal cells. The main immunological function of CXCR2 is to regulate trafficking of neutrophils from the bone marrow to inflammation sites [70]. In addition, CXCR2 also modulates MDSCs migrations and mediates local immunosuppression [71]. CXCR2-targeted reparixin and PF-04136309 have been investigated in clinical studies for the therapy of breast cancer and pancreatic neoplasms patients respectively [17]. The combination AZD5069 (CXCR2 antagonist) with durvalumab (ant-PD-L1) is being investigated in phase Ib/II trials in patients with advanced solid malignancies and metastatic pancreatic ductal adenocarcinoma [72]. SX-682 [73], a dual CXCR1/2 antagonist, in combination with pembrolizumab is also being investigated in clinical phase I for metastatic melanoma therapy (NCT03161431). CXCR3 is mainly overexpressed by effector CD8+ T cells, NKs and TH1 cells. The ligands of CXCR3 are CXC-chemokine ligand 9 (CXCL9) and CXCL10, whose elevated levels are related with enhanced amounts of tumor-infiltrating CD8+ T cells, subsequently decreased cancer metastasis and improved survival rates of colon cancer and ovarian cancer patients. CXCR3-targeted AMG487 remarkably decreased metastasis and enhanced host anti-tumor immunity in a 4T1 mammary tumor model [74]. The CXCR4-CXCL12 signaling pathway mediates Tregs homing to the bone marrow [75] and plasmacytoid precursor dendritic cells transitioning into tumors [76], regulating metastasis and vascularization of the tumor [77]. CXCR4 antagonist plerixafor (AMD3100) combining with anti-PD-1 induced T-cell rapid accumulation among cancer cells and acted synergistically with α-PD-L1 to significantly decrease tumor volume [78]. In addition, CXCR4-targeted endoradiotherapy with 177Lu- or 90Y-pentixather were well-tolerated and exerted anti-myeloma activity even at patients with advanced stage multiple myeloma. Nevertheless, further assessment of toxicity studies and prospectively designed clinical trials is highly warranted [79]. Pentixather also can be developed as different diagnostic agents when it is radiolabeled with shorter half-life radionuclides such as 68Ga [80, 81]. Other CXCR4 inhibitors such as TG‑0054 and MSX-122 are being investigated in clinical studies currently [82, 83]. The ligand structures of CXCR family are summarized in Figure 4.
Ligands of chemokine receptors CXCR2, CXCR3, CXCR4, CCR2, and CCR5.
CCR family
The CCR2-CCL2 pathway axis is able to induce macrophage migration into the tumor microenvironment and stimulate tumor proliferation and invasion [84]. Various CCR2 antagonists have been investigated in clinical studies including GTPL7825, TAK-652 and PF-04136309. PF-4136309 can enhance antitumor effects of the immune system and inhibit tumor proliferation and invasion in patients with pancreatic cancer [85].
CCR5 is primarily expressed by lymphocytes, macrophages and metastatic tumor cells. The upregulation of CCR5 on CD8+ T cells, TH1 cells, monocytes and macrophages promotes Tregs infiltration and stimulates progenitor cells differentiation into TAMs and MDSCs [86]. Maraviroc was shown to block CCR5 and inhibit tumor metastases in colorectal cancer patients in a clinical phase I evaluation (NCT01736813). In addition, BMS-813160, a dual CCR2/5 antagonist, in combination with nivolumab has been developed for the therapy of patients with colorectal and pancreatic cancers (NCT03184870). TAK-779 is able to block migration of tumor-associated Tregs consequently inhibiting tumor growth specifically in pancreatic adenocarcinoma [87]. The ligand structures of CCR family are summarized in Figure 4.
Purinergic signaling
Hypoxia activates tumor cells to release the pro-inflammatory adenosine triphosphate (ATP), which is subsequently dephosphorylated to immunosuppressive adenosine by CD39 and CD73. The biological actions of adenosine and ATP depend on the activation of purinergic receptors such as P2Xs, P2Ys, CD39, CD73 and adenosine receptors, which are significantly overexpressed on tumor cells and infiltrating immune cells [88].
P2 family
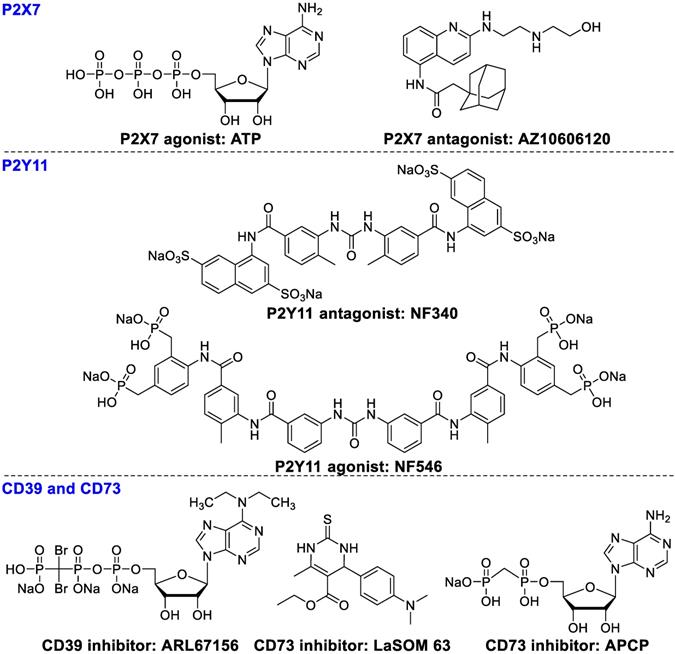
P2Xs (ion channel receptors) and P2Ys (G protein-coupled receptors) are overexpressed on various immune cells in the tumor microenvironment. High levels of extracellular ATP can stimulate P2X expression in macrophages and DCs, inducing IL-1β secretion, subsequently enhancing the cytotoxicity of CD8+ T cells. Extracellular ATP can also induce apoptosis of Tregs and diminish immunosuppressive activity [89]. However, some studies have shown the opposite results, where overexpression of P2Xs promotes tumor growth and survival in vivo . AZ10606120 (Figure 5), an antagonist of P2X7, significantly inhibited tumor growth. These contradictory data may result from a slow upgrading of extracellular ATP levels, which causes differences in acute apoptotic response. P2Y11 receptors moderate ATP-induced semi-maturation of monocyte-derived dendritic cells and mediate dendritic cell-based immunotherapy [90]. P2Y11 antagonist NF546 stimulated thrombospondin-1 and interleukin 8 (IL-8) release and inhibited lipopolysaccharide-stimulated IL-12 secretion, whereas agonist NF340 reversed these effects [91].
Ligands of P2X7. P2Y11, CD39, and CD73.
CD39 and CD73
CD39 and CD73 play a pivotal role in tumor immunosuppression through converting ATP and ADP to AMP and then to adenosine, resulting in immunosuppression and subsequently the onset and progression of tumor growth [92-94]. CD39 is overexpressed on endothelial cells, leukocytes, and B cells [95]. CD39 modulates immune and tumor cells to promote tumor growth by catalyzing extracellular ATP or ADP to AMP [96, 97]. Subsequently, AMP is hydrolyzed by CD73 into adenosine, which is responsible for immunosuppressive and anti-inflammatory functions of Tregs [93, 98]. In addition, T-cell subsets Thpp cells also overexpress CD73 and suppress the CD4+ or CD8+ T cell proliferation in the presence of exogenous AMP [99]. ARL67156 (Figure 5) inhibits the activity of CD39 and partially overwhelms hyporesponsiveness of T cell in some patients with follicular lymphoma [100]. LaSOM 63 is able to inhibit the activity of Ecto-5' Nucleotidase/CD73 subsequently causing glioma cell apoptosis [101]. APCP, a selective CD73 inhibitor, inhibited tumor proliferation and enhanced efficacy of adoptive T cell therapy [102].
Adenosine A2A receptor (A2AR) and adenosine A2B receptor (A2BR)
After ATP is dephosphorylated to adenosine by CD39 and CD73, the accumulated extracellular adenosine interacts with receptors A1R, A2AR, A2BR and A3R which regulate immunosuppressive functions [31, 103]. Cyclic AMP (cAMP) is a downstream signaling molecule of adenosine receptors, which is stimulated by A2AR and A2BR, thereby enhancing immunosuppressive functions [104-106].
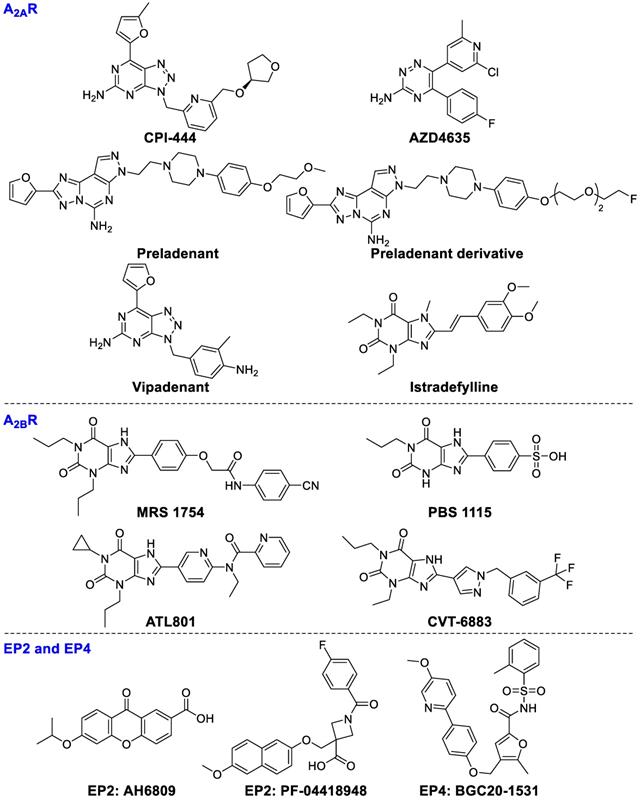
A2AR is mainly expressed on lymphocytes, NKs, DCs, and T cells. Activation of A2AR on T cells markedly inhibits TCR-mediated cytotoxicity and cytokine production, and restrains proliferation of T cells [107]. On the other hand, A2AR activation can boost Tregs expansion which ultimately enhances immunosuppressive activity [108]. CPI-444, a selective A2AR inhibitor, was used as a mono-drug or combined with atezolizumab (anti-PD-L1 antibody) for the therapies of patients with advanced non-small cell lung cancer (NSCLC), renal cell carcinoma (RCC), melanoma, and triple negative breast cancer (TNBC) (NCT02655822). The combination of CPI-444 and anti-PD-1 led to a synergistic inhibition of tumor growth (eliminating tumors in 90% of treated mice) and prolonged survival time compared to either agent alone [109]. Based on the promising results, Phase 1b clinical study has been initiated (NCT02655822). Co-targeting A2AR (PBF‑509, structure not disclosed) with durvalumab is being evaluated in patients with NSCLC (NCT02403193). AZD4635 as a mono-agent or combined with durvalumab (ant-PD-L1) is being investigated for the therapy of patients with advanced solid malignancies, NSCLC, metastatic castrate-resistant prostate carcinoma (mCRPC), and colorectal carcinoma (CC) (NCT02740985), but it has not been completed until now. A2AR antagonist preladenant (SCH58261) could enhance NKs activity in mice with B16 melanoma metastasis [110]. A fluorinated polyethylene glycol (PEG) derivative of preladenant is confirmed as a promising immunotherapeutic agent [111]. Vipadenant and istradefylline are being evaluated in phase II and III studies in Parkinson's disease, which may be promising for treating cancer patients [112].
A2BR receptor is the least sensitive of the four adenosine receptors for the requirement of adenosine concentrations to achieve physiological functions. Expression of A2BR is enormously increased under hypoxic conditions. Activation of A2BR mainly promotes M1 macrophage to M2 macrophage switching, subsequently inhibiting the antitumor T cell activities and promoting angiogenesis in tumors. Under hypoxia, the A2BR overexpression on mature DCs can polarize DCs to a Th2-stimulating phenotype. MRS1754, an A2BR inhibitor, can enhance the secretion of IL-12p70 and TNF-α and increase the production of Th1 cytokine IFN-c in an mDCs-T-cell co-culture system [113]. PBS1115, an A2BR-selective antagonist, increases the accumulation of tumor-infiltrating MDSCs in vivo [114]. ATL801 not only inhibited the growth of 4T1 breast and MB49 bladder tumors but also reduced the metastasis of breast cancer cells, though it significantly increased the concentration of IFN-γ and chemokine CXCL10 [115]. CVT-6883, a potent selective A2BR antagonist, has entered into clinical trials to treat pulmonary inflammation and injury [116]. Therefore, CVT-6883 is a very promising candidate drug for tumor immunotherapy.
Elevation of cyclic AMP (EP2 and EP4)
The expression of inducible cyclooxygenase (COX2) is correlated with lower survival rates of patients. The metabolites of COX2 are associated with immune tolerance of tumors through stimulating prostaglandin E2 (PGE2) generation to boost tumor proliferation and migration. COX2 upregulation leads to sustained high concentrations of PGE2 [117]. PGE2 activates its receptors EP2 and EP4 leading to elevation of cAMP levels [118], consequently promoting various immune-suppressive cells activity including Tregs, TAMs, and MDSCs [119-121]. AH6809, an EP2 receptor antagonist, can diminish Tregs-mediated immune tolerance [122]. PF‑04418948 (EP2 antagonist) [123] and BGC20‑1531 (EP4 antagonist) [124] have been studied in clinical trials for various non-oncology candidates, which may soon expand to anticancer therapy. The chemical structures of ligands are presented in Figure 6.
Ligands of A2AR, A2BR, EP2, and EP4.
Toll-like receptor (TLR) and stimulator of interferon genes protein (STING)
TLRs and STING are regarded as crucial components of the innate immune sensing of tumors. The activation of TLRs and STING in the innate immune system can enhance the secretion of pro-inflammatory cytokines and T-cell recruitment factors subsequently modulate innate immunity, which is able to resist tumor-induced immunosuppression and shows a synergistic effect with present cancer therapies [18].
TLR
The TLR family is a critical member of the innate immune system [125]. TLRs are primarily expressed by DCs, B cells, neutrophils, monocytes and macrophages, along with the gastrointestinal tract and lungs which are exposed to the external environment. TLRs take part in recognizing pathogen-associated and damage-associated molecular patterns [126]. The TLR superfamily contains 13 members; TLR3, TLR7, TLR8 and TLR9 are distributed in the endosomal compartment and others in the cytoplasm. TLR agonists are being evaluated in (pre)clinical studies.
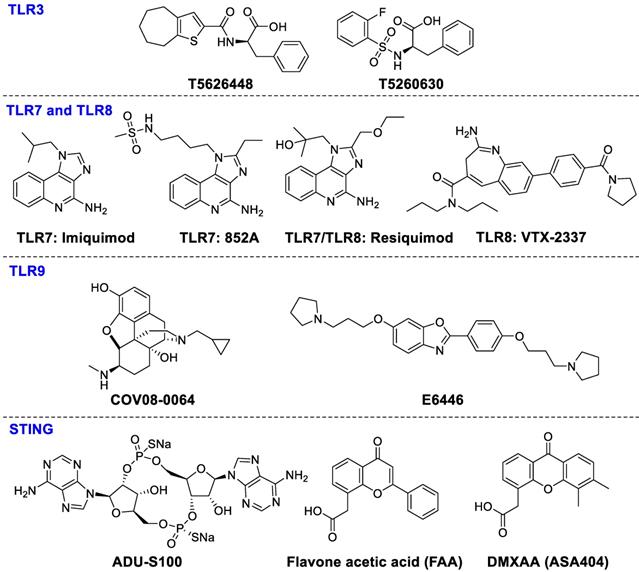
The agonists of TLR3, TLR7, TLR8 and TLR9 make up the majority of preclinical and clinical trials of TLRs. TLR3 signaling is stimulated by dsRNA subsequently causing secretion of pro-inflammatory cytokines and type I interferons such as IL-1, and IL-6, TNF-R to stimulate immune cell activation and recruitment during inflammation or viral infection [127]. A series of TLR3-targeted inhibitors, such as T5626448 and T5260630, were evaluated in vitro [128]. TLR7 and TLR8 are the key targets in the recognition of single-stranded RNA in certain cell types, such as pDC [129]. Imiquimod significantly enhanced CD8+ T cell accumulation in spleen and draining lymph nodes after administration of DC vaccination [130, 131]. Resiquimod, stimulating TLR7 and TLR8, is able to activate immune responses effectively against viral infections and tumors. Resiquimod is in clinical phase II studies for the therapies of viral skin lesions and skin cancer [132]. 852A (TLR7 agonist) and VTX-2337 (TLR8 agonist) have been investigated in phase I for treating subjects with advanced solid tumors and lymphoma [133, 134]. TLR9 has high affinity for unmethylated and CpG-rich DNA, which is an endosomal receptor for dsDNA in the extracellular compartment. TLR9 agonists COV08-0064 and E6446, can inhibit TLR9-mediated sterile inflammation in acute liver injury and acute pancreatitis models and restrain responses of deleterious inflammation in rodent malaria, respectively [135, 136]. Although several trials with TLR modulators are underway, more investigation should be done to achieve more clinical benefits. The chemical structures of TLR ligands are summarized in Figure 7.
Ligands of TLR3, TLR7, TLR8, TLR9, and STING.
STING
Transmembrane protein 173 (TMEM173), is expressed in T cells, DCs, and macrophages as well as in various epithelial and endothelial cells. STING activation results in the secretion of cytokines, interferons, and T-cell recruitment factors subsequently modulating innate immunity [137, 138].
STING signals can be activated by cyclic dinucleotides such as cyclic di-GMP and cGAMP, which can induce the expression of interferon-β [139]. Recently, ADU-S100 ((R, R)-S2-CDA) is being evaluated in a clinical phase I study for treating advanced/metastatic solid tumors and lymphomas (NCT02675439). MK-1454 (structure undisclosed) is being studied for treating advanced/metastatic solid tumors or lymphoma in a clinical phase I study (NCT03010176). Flavone acetic acid (FAA) and DMXAA (ASA404) both failed in clinical trials for the therapy of advanced cancer [140]. Despite some compounds being in clinical trials for antitumor therapy, administration methods and combinations with other drugs need to be further investigated. The chemical structures of STING ligands are presented in Figure 7.
Kinase
Kinase signaling pathways can drive many hallmark phenotypes of tumor biology such as metabolism, proliferation, and metastasis. Tumor cells can exert a considerable impact on the microenvironment to inhibit anti-tumor immune responses and escape the pivotal phylactic mechanism. The application of kinase inhibitors can directly inhibit tumor cells, reduce their immunosuppressive influences, and shift the local immunosuppression toward a proinflammatory state, subsequently boosting the activity of the immune activators. Therefore, the combination of kinase inhibitors such as PI3K, MAPK, BRAF, and MEK1/2 inhibitors with immune checkpoint inhibitors is a significant opportunistic proposition to increase the utility of immune modulation in oncology. Various pathways involving kinase inhibitors need to be elucidated to optimize their application in this setting [141].
PI3K-AKT-mTOR
Recently, inhibiting the PI3K-AKT-mTOR signal pathway has been evaluated to promote the production of immunosuppressive cytokines [142, 143], the tumor infiltration of MDSCs and Tregs, thereby inhibiting proliferation, migration and survival of tumor cells [144, 145]. Thus, it is understandable that PI3K-AKT-mTOR inhibitors plus checkpoint blockade would be effective [146]. PI3Kα, PI3Kβ, PI3Kγ and PI3Kδ from the PI3K family are well studied in anti-tumor immunotherapy. PI3Kγ and PI3Kδ are primarily expressed by B and T cells and myeloid lineage cells [17].
PI3Kγ
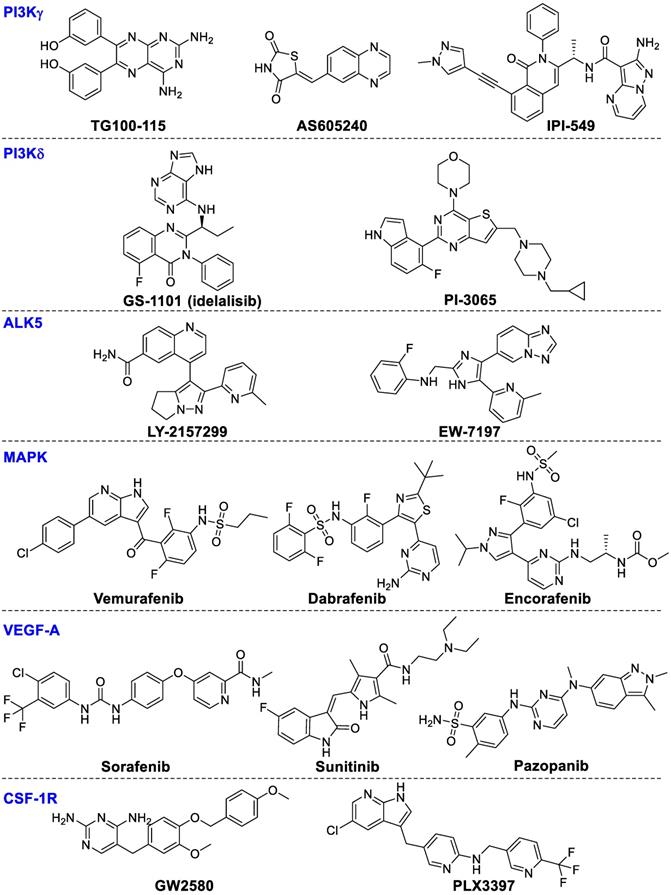
PI3Kγ mainly regulates the innate immune response of myeloid cells by regulating integrin α4β1-dependent macrophage chemotaxis into tumors and suppressing the proliferation and metastasis of tumors [147]. TG100-115 and AS605240 could inhibit inflammation, angiogenesis and tumor proliferation in lung carcinoma but without directly affecting tumor cells [147]. Infinity (IPI-549) could powerfully inhibit PI3K-γ-mediated neutrophil migration and is presently in clinical phase I studies for the therapy of advanced solid tumors [148]. In addition, the combination of IPI-549 with anti-PD-1 treatment enhanced gene expression of anti-tumor immunity and inhibited gene expression of immune-suppressors, thereby hampering tumor growth in primary tumors from human papilloma virus (HPV)+ head and neck squamous cell carcinoma [149]. These results suggest that PI3Kγ inhibition can synergize with T-cell-targeted immunotherapy to promote anti-tumor immune response.
PI3Kδ
PI3Kδ is mainly involved in modulating B-cell proliferation and differentiation. GS‑1101 (idelalisib), a PI3Kδ selective inhibitor, has been approved for treating chronic lymphocytic leukemia. Recently, preclinical results suggest PI3Kδ inhibition in Tregs results in boosting anti-tumor T-cell function and restricting tumor proliferation [34]. Idelalisib in combination with pembrolizumab is being investigated for the therapy of chronic lymphocytic leukemia and non-Hodgkin lymphomas (NCT02332980). PI‑3065, a selective PI3Kδ inhibitor, can inhibit tumor proliferation and metastasis in 4T1 breast cancer models. The likely mechanism of PI‑3065's immune regulatory effect may result from enhancing the anti-tumor immune effect though inhibiting Tregs and MDSC function. These results showed PI3K-AKT-mTOR signaling pathways are important targets for the regulation of innate immunity. The chemical structures of the PI3Kγ or PI3Kδ inhibitors are summarized in Figure 8.
Chemical structures of kinase inhibitors.
Activin receptor-like kinase 5 (ALK5)
TGF-β binds to ALK5 and TGF-β receptor type 2 to mediate phosphorylation of SMAD2 and SMAD3. Recently, LY-2157299, a selective ALK5 inhibitor, was able to block TGF-β signaling and inhibit tumor progression in preclinical models. LY-2157299 has entered a Phase I clinical study to evaluate antitumor activity in glioma patients [150]. EW-7197 was reported to enhance activation of cytotoxic T lymphocytes thereby inhibiting tumor growth in melanoma-bearing mice [33].
Mitogen-activated protein kinase (MAPK)
The MAPK signal axis is activated by various mechanisms, and is a very important target for pathway targeting therapies especially for melanoma metastasis treatment [151, 152]. Among the MAPK signaling cascades, the RAS-RAF-MEK-ERK1/2 pathway is very important for CD8 T cell activation, proliferation, and survival, subsequently regulating tumor proliferation and survival [153, 154]. Inhibiting the MAPK signaling axis by MEK and B-Raf inhibitors has been an effective therapy for patients with metastatic tumors bearing B-Raf mutations [155]. The approved B-Raf inhibitors include vemurafenib and dabrafenib, while encorafenib is being evaluated in multiple phase III trials. In addition, various MEK inhibitors also have been approved such as cobimetinib and trametinib, while binimetinib is presently being studied in various clinical phase III studies. Combinations of MEK inhibitor trametinib with checkpoint inhibitors were more effective than any single drug [156]. Clinical evaluation of such combination strategies is underway. It is possible to expand these combinational therapeutic strategies towards other cancer types beyond melanoma.
Vascular endothelial growth factor A (VEGF-A)
VEGF-A, a proangiogenic factor produced by malignancies, can enhance the expression of PD-1 on CD8+ T cells through an overexpressed VEGF receptor causing the exhaustion of cytotoxic immune cells, which could be reversed by anti-angiogenic agents targeting VEGF-A-VEGFR [157]. Some small-molecule VEGF inhibitors including sorafenib, sunitinib and pazopanib have been approved for renal cell cancer. VEGF inhibition enhanced the amounts of tumor-infiltrated effector T-cells and reduced Tregs accumulation in the tumor microenvironment in patients with primary and metastatic renal cell carcinoma [158]. VEGF inhibitors in combination with anti-PD-1 or anti-PD-L1 showed positive treatment benefits in patients with renal cell carcinoma or clear-cell metastatic renal cell carcinoma (NCT01472081).
CSF-1 receptor (CSF-1R)
CSF-1R signaling is important for recruitment and function of distinct tumor-infiltrating myeloid cells subsets, including TAMs and MDSCs [159]. CSF-1R inhibitor GW2580 combined with an anti-VEGFR-2 antibody synergistically inhibits tumor proliferation and hampers tumor angiogenesis [159]. PLX3397, a selective CSF-1R inhibitor, is being investigated in clinical trials alone or combined with paclitaxel or checkpoint immunotherapies, or radiation therapy for treating patients with breast cancer, metastatic pancreatic cancer, glioblastoma, and other cancers [17, 160].
Small molecules for imaging cancer immunotherapy
With the increase in development of personalized-medicine approaches, discoveries of novel immune mechanisms and more selective-targeted drugs approvals, clinicians and researchers need novel methods for exploring the interaction and relationships between tumor cells, the immune system, and immunotherapy agents. It is essential to diagnose whether the patient expresses the related targets before drug administration [161]. In addition, it is critical to track the dynamic changes of the targets in immune cells and in tumor cells, to guide clinicians when to switch one drug to another or to determine if a patient no longer needs to receive costly therapy [19]. In this way, imaging will provide guidance for physicians to make better decisions on therapeutic regimens and patient follow-up. Several comprehensive reviews on molecular imaging in immunotherapy have been published previously [19, 161-169].
Antibodies can be quickly radiolabeled for imaging via conjugation to a contrast agent or radionuclide. In addition, radiolabeled antibodies keep their naturally high specificity and binding affinity toward their cognate antigens. However, the slow clearance rate and relatively poor penetration into target tissues are drawbacks of radiolabeled-antibody tracers [19]. Clinicians must often wait several days before the background signal clears from blood circulation and various non-target tissues. Conversely, radiolabeled small molecules tracers not only possess excellent pharmacokinetic characteristics but also can go across cellular membranes and other physiological barriers and reach intracellular targets. In addition, the manufacturing costs of small molecules tracers are lower than radiolabeled antibodies.
PD-1/PD-L1 immune checkpoint
Early detection of therapy response is very pivotal for patients undergoing anti-cancer immunotherapy. 18F-labeled fluorodeoxyglucose (18F-FDG) (Figure 9) is the most widely used PET probe in nuclear medicine, which is being tested in immunotherapy settings. Chen and co-workers reported that the uptake of 18F-FDG has a positive correlation with the expression of both PD-1 and PD-L1 in patients with bladder tumors [170]. In addition, Ferdinand and colleagues showed that 18F-FDG PET can reliably identify cancer patients who will most benefit from PD-1-therapy as early as two weeks after therapy initiation in stage IV melanoma (Figure 10A) [171]. However, 18F-FDG used as an immuno-imaging PET tracer needs to be further evaluated in additional clinical trials. It is also expected that the results from 18F-FDG can be compared with other specific PD-L1 binding peptide probes, such as 64Cu-WL12 [172].
Small molecule-based PET probes for immuno-oncology imaging
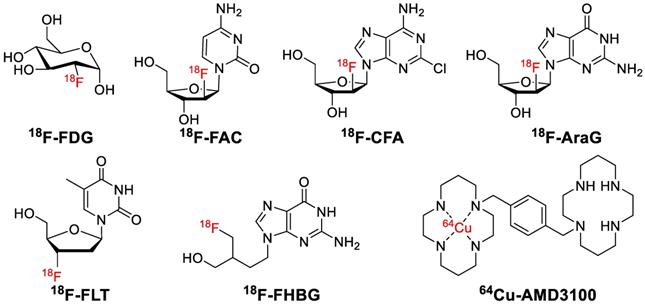
A: Whole-body 18F-FDG-PET examination at two time points: before PD-1-therapy start (t0, base-line) (A1) and two weeks (t1, study examination) (A2). B: 18F-FAC was used to evaluate lymphoid organs and immune activation, tu: tumor; ln: lymph node; sp: spleen; gi: gastrointestinal tract; b: bone. C: Detection of immune responses after immunotherapy in glioblastoma using 18F-CFA. D: Pharmacokinetics of 18F-AraG in a healthy human volunteer to prepare to visualize activated T cells in acute graft-versus-host disease. E: Early identification of antigen-specific immune responses in vivo by18F-FLT. F: Reporter gene imaging of targeted T-cell immunotherapy in recurrent glioma by 18F-FHBG, F1: Pre-immunotherapy using CD8+ cytotoxic T lymphocytes (CTLs), F2: Post-immunotherapy using CD8+ CTLs. G: 64Cu-AMD3100-PET/CT for imaging of CXCR4 expression in subcutaneous brain tumor xenografts.
Metabolism of T Cell
PET probes targeting vital metabolic pathways, such as glucose metabolism and nucleotide synthesis and metabolism can potentially be used to monitor the efficacy of immunotherapy involved in innate and adaptive immunity [165].
18F-FAC and 18F-CFA
In 2008, Radu and co-workers synthesized 1-(2'-deoxy-2'-[18F]fluoroarabinofuranosyl) cytosine (18F-FAC) to map the deoxyribonucleotide salvage pathway [173]. 18F-FAC was able to visualize lymphoid organs and was adequately sensitive to localize immune activation in an antitumor immunity mouse model. Additionally, early changes of lymphoid mass in systemic autoimmunity was detected by 18F-FAC (Figure 10B), which allowed for the real-time evaluation of immunosuppressive therapy. All the results confirmed 18F-FAC can be used for monitoring the process of immune response. However, the clinical application was limited by its rapid catabolism. Kim and colleagues developed an analogue of 18F-FAC, 18F-Clofarabine (18F-CFA), which accumulates in tissues with high dCK expression such as hematopoietic bone marrow and secondary lymphoid organs (Figure 10C) [174]. Further studies proved that 18F-CFA might be a promising tracer to image the host antitumor immune response against intracranial tumors [175].
18F-AraG
9-(β-D-Arabinofuranosyl)guanine (AraG) is an analog of guanosine that has a demonstrable effectiveness for the therapy of T-cell lymphoblastic disease such as recurrent T-cell lymphoblastic leukemia and T-cell lymphoblastic lymphoma. AraG is triphosphorylated by multiple kinases to AraGTP, which preferentially distributes in malignant T-cells. 18F-AraG was synthesized and radiolabeled by Ronald et al in 2011, which showed favorable pharmacokinetic properties in healthy humans. PET imaging of 18F-AraG may also provide essential information for the early diagnosis of activated T cells in acute graft-versus-host disease (Figure 10D) [176].
18F-FLT
Fluorothymidine (FLT), a nucleoside analog, can be quickly absorbed by nucleoside transporters expressed on proliferating cells and then is phosphorylated by the S-phase specific thymidine kinase 1 (TK1), subsequently trapping it within the cells [177]. The uptake level of 18F-FLT in lymph nodes correlates to the level of antigen-specific IgG antibodies and antigen-specific proliferation of T cells in the blood of patients with metastatic melanoma who received dendritic cell vaccine therapy (Figure 10E) [178].
Reporter genes of targeted T cells
Reporter gene imaging of engineered T cells is possible when the T cells are transfected with a PET reporter gene which encodes a protein. Reporter gene imaging plays an important role in visualizing their targeting/trafficking, proliferation/expansion, and retention/death using highly sensitive reporter systems, which would provide useful theranostic information [179].
Herpes simplex virus type 1 thymidine kinase (HSV-TK) is a viral nucleoside kinase which is coded by the HSV-TK gene (HSV-tk), whose substrates are thymidine or non-natural nucleosides [180]. Preclinical results showed that 18F-labeled 9-[4-fluoro-3-(hydroxymethyl)butyl]guanine (18F-FHBG) was more sensitive than 14C-labeled 1-(2'-deoxy-2'-fluoro-β-D-arabinofuranosyl)-5-iodouracil (FIAU) (14C-FIAU) in the HSV1-sr39tk system [181]. In 2017, Khun and co-workers reported immunotherapy using CD8+ cytotoxic T lymphocytes engineered to express both HSV1-TK and IL-13 zetakine chimeric antigen receptor (CAR), which is a promising therapy strategy for patients with recurrent glioma (Figure 10F) [179]. Although 18F-FHBG PET imaging was safe and facilitated the longitudinal imaging of T cells stably transfected with a PET reporter gene in patients, problems with specificity and viral gene editing in humans may limit their primary application to ex vivo immune cell manipulation.
CXCR4-based immune cells
CXCR4 plays a pivotal role in recruiting immune cells and homing stem cells and progenitor cells [182-184]. CXCR4 is overexpressed on multiple human tumor types including esophageal, prostate, ovarian, and renal cell carcinoma, boosting tumor proliferation and metastasis [185, 186]. Jacobson and co-workers synthesized CXCR4-specific tracer 64Cu-AMD3100, which showed accumulation in CXCR4-expressing organs and tissues [187]. Then Nimmagadda and colleagues reported that 64Cu-AMD3100 possessed optimized pharmacokinetics and can be applied to decipher graded levels of CXCR4 expression in subcutaneous brain tumor xenografts (Figure 10G) [188]. 64Cu-AMD3100 is a promising PET tracer for diagnosis of CXCR4 expression.
Conclusion and perspective
Despite cancer immunotherapy having achieved clinical successes in the past decade, only about 30% of patients have benefited from immunotherapies. There are still many challenges for immuno-theranostics in cancer: 1) additional immune regulatory mechanisms to expand patients' response rates to immunotherapy need to be explored; 2) special immune-competent animal models are required, including transplantable, spontaneous, carcinogen-induced, or genetically engineered humanized malignancies [189]; 3) immunotherapeutic effects may also be obstructed by external conditions such as certain bacterial or viral infections, which will modify the immune system [190, 191]; 4) reducing or avoiding toxicities caused by general systemic immune activation and developing safer and more effective drugs is essential [192]; and 5) more advanced imaging techniques and better characteristic probes need to be developed in order to achieve earlier diagnosis and offer more biological information about multiple cancers [19].
The combination of small-molecule drugs with biologic checkpoint inhibitors is an effective strategy to increase response rates of patients and the efficacy of immunotherapy. It is critical to develop promising imaging technologies and probes to monitor target expression, estimate therapeutic efficacy and potential toxic reactions, and identify who will benefit from immunotherapies. In order to achieve better cancer immune theranostic effect, 1) both intracellular signal pathways down/upstream of immune checkpoints and related therapeutic agents need to be explored; 2) new imaging techniques such as quantum-inspired imaging need to be developed to provide clearer images; 3) mathematic modeling to increasingly derive guiding principles for imaging design and application needs to be optimized; 4) radiomics enabling data to be extracted and applied to improve cancer diagnostic, prognostic, and predictive accuracy should be employed; and 5) more immunoimaging agents need to be developed to keep pace with drug development [19, 193, 194]. In addition, immunotherapies can be extended to autoimmune diseases such as graft-versus-host disease [176] and rheumatoid arthritis [195], multiple sclerosis [196] and neurodegenerative diseases [197, 198]. Furthermore, artificial intelligence and machine learning will likely improve the efficiency of drug screening and help physicians make better treatment plans.
Acknowledgements
This work was supported by the USC Research Center for Liver Diseases Pilot Funding (NIH Grant No. P30 DK048522), the Whittier Foundation for Translational Research, the USC Department of Radiology, and the China Scholarship Council (CSC) program (No. 201806310056).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394-424
2. Forde PM, Chaft JE, Smith KN, Anagnostou V, Cottrell TR, Hellmann MD. et al. Neoadjuvant PD-1 blockade in resectable lung cancer. N Engl J Med. 2018;378:1976-86
3. Leighl NB, Hellmann MD, Hui R, Carcereny E, Felip E, Ahn M-J. et al. Pembrolizumab in patients with advanced non-small-cell lung cancer (KEYNOTE-001): 3-year results from an open-label, phase 1 study. Lancet Respir Med. 2019;7:347-57
4. Rini BI, Powles T, Atkins MB, Escudier B, McDermott DF, Suarez C. et al. Atezolizumab plus bevacizumab versus sunitinib in patients with previously untreated metastatic renal cell carcinoma (IMmotion151): a multicentre, open-label, phase 3, randomised controlled trial. Lancet. 2019;393:2404-15
5. Mok TSK, Wu YL, Kudaba I, Kowalski DM, Cho BC, Turna HZ. et al. Pembrolizumab versus chemotherapy for previously untreated, PD-L1-expressing, locally advanced or metastatic non-small-cell lung cancer (KEYNOTE-042): a randomised, open-label, controlled, phase 3 trial. Lancet. 2019;393:1819-30
6. Friedrich MJ. Immunotherapy 2.0: Improving the response to checkpoint inhibitors. JAMA. 2019;321:131-3
7. Allison JP, Krummel M. The Yin and Yang of T cell costimulation. Science. 1995;270:932
8. Nishino M, Ramaiya NH, Hatabu H, Hodi FS. Monitoring immune-checkpoint blockade: response evaluation and biomarker development. Nat Rev Clin Oncol. 2017;14:655-68
9. Hargadon KM, Johnson CE, Williams CJ. Immune checkpoint blockade therapy for cancer: an overview of FDA-approved immune checkpoint inhibitors. Int Immunopharmacol. 2018;62:29-39
10. Yan H, Qiu W, Koehne de Gonzalez AK, Wei JS, Tu M, Xi CH. et al. HHLA2 is a novel immune checkpoint protein in pancreatic ductal adenocarcinoma and predicts post-surgical survival. Cancer Lett. 2019;442:333-40
11. Hu JR, Florido R, Lipson EJ, Naidoo J, Ardehali R, Tocchetti CG. et al. Cardiovascular toxicities associated with immune checkpoint inhibitors. Cardiovas Res. 2019;115:854-68
12. Johnson DB, Chandra S, Sosman JA. Immune checkpoint inhibitor toxicity in 2018. JAMA. 2018;320:1702-3
13. Wang DY, Salem JE, Cohen JV, Chandra S, Menzer C, Ye F. et al. Fatal toxic effects associated with immune checkpoint inhibitors: a systematic review and meta-analysis. JAMA Oncol. 2018;4:1721-8
14. Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. 2017;168:707-23
15. Cheng B, Yuan WE, Su J, Liu Y, Chen J. Recent advances in small molecule based cancer immunotherapy. Eur J Med Chem. 2018;157:582-98
16. Imai K, Takaoka A. Comparing antibody and small-molecule therapies for cancer. Nat Rev Cancer. 2006;6:714-27
17. Adams JL, Smothers J, Srinivasan R, Hoos A. Big opportunities for small molecules in immuno-oncology. Nat Rev Drug Discov. 2015;14:603-22
18. Bayard R. Huck LK, and Klaus Urbahns. Small molecules drive big improvements in immuno-oncology therapies. Angew Chem Int Ed. 2018;57:4412-28
19. Mayer AT, Gambhir SS. The immunoimaging toolbox. J Nucl Med. 2018;59:1174-82
20. Vinay DS, Ryan EP, Pawelec G, Talib WH, Stagg J, Elkord E. et al. Immune evasion in cancer: mechanistic basis and therapeutic strategies. Semin Cancer Biol. 2015;35(Suppl):S185-98
21. Ozpiskin OM, Zhang L, Li JJ. Immune targets in the tumor microenvironment treated by radiotherapy. Theranostics. 2019;9:1215-31
22. Spano D, Zollo M. Tumor microenvironment: a main actor in the metastasis process. Clin Exp Metastasis. 2012;29:381-95
23. Hoos A. Development of immuno-oncology drugs - from CTLA4 to PD1 to the next generations. Nat Rev Drug Discov. 2016;15:235-47
24. Buchbinder EI, Desai A. CTLA-4 and PD-1 pathways: similarities, differences, and implications of their inhibition. Am J Clin Oncol. 2016;39:98-106
25. Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677-704
26. Boutros C, Tarhini A, Routier E, Lambotte O, Ladurie FL, Carbonnel F. et al. Safety profiles of anti-CTLA-4 and anti-PD-1 antibodies alone and in combination. Nat Rev Clin Oncol. 2016;13:473-86
27. Sica A, Bronte V. Altered macrophage differentiation and immune dysfunction in tumor development. J Clin Invest. 2007;117:1155-66
28. Groth C, Hu X, Weber R, Fleming V, Altevogt P, Utikal J. et al. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br J Cancer. 2019;120:16-25
29. Madar S, Goldstein I, Rotter V. 'Cancer associated fibroblasts' - more than meets the eye. Trends Mol Med. 2013;19:447-53
30. Vijayan D, Young A, Teng MWL, Smyth MJ. Targeting immunosuppressive adenosine in cancer. Nat Rev Cancer. 2017;17:709-24
31. Cekic C, Linden J. Purinergic regulation of the immune system. Nat Rev Immunol. 2016;16:177-92
32. Ferguson FM, Gray NS. Kinase inhibitors: the road ahead. Nat Rev Drug Discov. 2018;17:353-77
33. Yoon JH, Jung SM, Park SH, Kato M, Yamashita T, Lee IK. et al. Activin receptor-like kinase5 inhibition suppresses mouse melanoma by ubiquitin degradation of Smad4, thereby derepressing eomesodermin in cytotoxic T lymphocytes. EMBO Mol Med. 2013;5:1720-39
34. Ali K, Soond DR, Pineiro R, Hagemann T, Pearce W, Lim EL. et al. Inactivation of PI(3)K p110δ breaks regulatory T-cell-mediated immune tolerance to cancer. Nature. 2014;510:407-11
35. Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF. et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. 2013;19:1264-72
36. Wu P, Nielsen TE, Clausen MH. FDA-approved small-molecule kinase inhibitors. Trends Pharmacol Sci. 2015;36:422-39
37. Plieth J, Elmhirst E. PD-1/PD-L1 combination therapies. EvaluatePharma. 2017; May, 2017. https://info.evaluategroup.com/PD1-EPV.html
38. Zou W, Wolchok JD, Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: mechanisms, response biomarkers, and combinations. Sci Transl Med. 2016;8:328rv4
39. Shi L, Chen S, Yang L, Y L. The role of PD-1 and PD-L1 in T-cell immune suppression in patients with hematological malignancies. J Hematol Oncol. 2013;6:1-6
40. Lu JC, Zeng HY, Sun QM, Meng QN, Huang XY, Zhang PF. et al. Distinct PD-L1/PD1 profiles and clinical implications in intrahepatic cholangiocarcinoma patients with different risk factors. Theranostics. 2019;9:4678-87
41. Zak KM, Grudnik P, Guzik K, Zieba BJ, Musielak B, Dömling A. et al. Structural basis for small molecule targeting of the programmed death ligand 1 (PD-L1). Oncotarget. 2016;7:30323-35
42. Soremekun OS, Olotu FA, Agoni C, Soliman MES. Recruiting monomer for dimer formation: resolving the antagonistic mechanisms of novel immune check point inhibitors against Programmed Death Ligand-1 in cancer immunotherapy. Mol Simulat. 2019;45:777-89
43. Skalniak L, Zak KM, Guzik K, Magiera K, Musielak B, Pachota M. et al. Small-molecule inhibitors of PD-1/PD-L1 immune checkpoint alleviate the PD-L1-induced exhaustion of T-cells. Oncotarget. 2017;8:72167-81
44. Kerr WG, Chisholm JD. The next generation of immunotherapy for cancer: small molecules could make big waves. J Immunol. 2019;202:11-9
45. Platten M, Doeberitz N, Oezen I, Wick W, Ochs K. Cancer immunotherapy by targeting IDO1/TDO and their downstream effectors. Front Immunol. 2014;5:1-7
46. Liu X, Shin N, Koblish HK, Yang G, Wang Q, Wang K. et al. Selective inhibition of IDO1 effectively regulates mediators of antitumor immunity. Blood. 2010;115:3520-30
47. Muller AJ, Manfredi MG, Zakharia Y, Prendergast GC. Inhibiting IDO pathways to treat cancer: lessons from the ECHO-301 trial and beyond. Semin Immunopathol. 2019;41:41-8
48. Gomes B, Driessens G, Bartlett D, Cai D, Cauwenberghs S, Crosignani S. et al. Characterization of the selective indoleamine 2,3-dioxygenase-1 (IDO1) catalytic inhibitor EOS200271/PF-06840003 supports IDO1 as a critical resistance mechanism to PD-L1 blockade therapy. Mol Cancer Ther. 2018;17:2530-42
49. Meininger D, Zalameda L, Liu Y, Stepan LP, Borges L, McCarter JD. et al. Purification and kinetic characterization of human indoleamine 2,3-dioxygenases 1 and 2 (IDO1 and IDO2) and discovery of selective IDO1 inhibitors. Biochim Biophys Acta. 2011;1814:1947-54
50. Rohrig UF, Majjigapu SR, Chambon M, Bron S, Pilotte L, Colau D. et al. Detailed analysis and follow-up studies of a high-throughput screening for indoleamine 2,3-dioxygenase 1 (IDO1) inhibitors. Eur J Med Chem. 2014;84:284-301
51. Tojo S, Kohno T, Tanaka T, Kamioka S, Ota Y, Ishii T. et al. Crystal structures and structure-activity relationships of imidazothiazole derivatives as IDO1 inhibitors. ACS Med Chem Lett. 2014;5:1119-23
52. Weinmann H. Cancer immunotherapy: selected targets and small-molecule modulators. ChemMedChem. 2016;11:450-66
53. Opitz CA, Litzenburger UM, Sahm F, Ott M, Tritschler I, Trump S. et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature. 2011;478:197-203
54. Pilotte L, Larrieu P, Stroobant V, Colau D, Dolusic E, Frederick R. et al. Reversal of tumoral immune resistance by inhibition of tryptophan 2,3-dioxygenase. Proc Natl Acad Sci U S A. 2012;109:2497-502
55. D'Amato NC, Rogers TJ, Gordon MA, Greene LI, Cochrane DR, Spoelstra NS. et al. A TDO2-AhR signaling axis facilitates anoikis resistance and metastasis in triple-negative breast cancer. Cancer Res. 2015;75:4651-64
56. Hsu YL, Hung JY, Chiang SY, Jian SF, Wu CY, Lin YS. et al. Lung cancer-derived galectin-1 contributes to cancer associated fibroblast-mediated cancer progression and immune suppression through TDO2/kynurenine axis. Oncotarget. 2016;7:27584-98
57. Chen IC, Lee KH, Hsu YH, Wang WR, Chen CM, Cheng YW. Expression pattern and clinicopathological relevance of the indoleamine 2,3-Dioxygenase 1/tryptophan 2,3-dioxygenase protein in colorectal cancer. Dis Markers. 2016;2016:8169724
58. Yu CP, Song YL, Zhu ZM, Huang B, Xiao YQ, Luo DY. Targeting TDO in cancer immunotherapy. Med Oncol. 2017;34:73
59. Salter M, Hazelwood R, Pogson CI. et al. The effects of a novel and selective inhibitor of tryptophan 2,3-dioxygenase on tryptophan and serotonin metabolism in the rat. Biochem Pharmacol. 1995;49:1435-42
60. Wu JS, Lin SY, Liao FY, Hsiao WC, Lee LC, Peng YH. et al. Identification of substituted naphthotriazolediones as novel tryptophan 2,3-dioxygenase (TDO) inhibitors through structure-based virtual screening. J Med Chem. 2015;58:7807-19
61. Arlauckas SP, Garren SB, Garris CS, Kohler RH, Oh J, Pittet MJ. et al. Arg1 expression defines immunosuppressive subsets of tumor-associated macrophages. Theranostics. 2018;8:5842-54
62. Boniface J, Mao Y, Schmidt-Mende J, Kiessling R, Poschke I. Expression patterns of the immunomodulatory enzyme arginase 1 in blood, lymph nodes and tumor tissue of early-stage breast cancer patients. Oncoimmunology. 2012;1:1305-12
63. Rodriguez PC, Ernstoff MS, Hernandez C, Atkins M, Zabaleta J, Sierra R. et al. Arginase I-producing myeloid-derived suppressor cells in renal cell carcinoma are a subpopulation of activated granulocytes. Cancer Res. 2009;69:1553-60
64. Paulo C. Rodriguez, David G. Quiceno, Jovanny Zabaleta, Blair Ortiz, Arnold H. Zea, Maria B. Piazuelo, et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res. 2004;64:5839-49
65. Havlinova Z, Hroch M, Nagy A, Sispera L, Holecek M, Chladek J. Single- and multiple-dose pharmacokinetics of arginase inhibitor Nω-hydroxy-nor-L-arginine, and its effect on plasma amino acids concentrations in Wistar rats. Gen Physiol Biophys. 2014;33:189-98
66. Havlinova Z, Babicova A, Hroch M, Chladek J. Comparative pharmacokinetics of Nω-hydroxy-nor-L-arginine, an arginase inhibitor, after single-dose intravenous, intraperitoneal and intratracheal administration to brown Norway rats. Xenobiotica. 2013;43:886-94
67. Noel N. Kim, J. David Cox, Ricky F. Baggio, Frances A. Emig, Sanjay K. Mistry, Sandy L. Harper, et al. Probing erectile function: S-(2-boronoethyl)-L-cysteine binds to arginase as a transition state analogue and enhances smooth muscle relaxation in human penile corpus cavernosum. Biochemistry. 2001;40:2678-88
68. Poeta V, Massara M, Capucetti A, Bonecchi R. Chemokines and chemokine receptors: new targets for cancer immunotherapy. Front Immunol. 2019;10:1-10
69. Nagarsheth N, Wicha MS, Zou W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat Rev Immunol. 2017;17:559-72
70. Eash KJ, Greenbaum AM, Gopalan PK, Link DC. CXCR2 and CXCR4 antagonistically regulate neutrophil trafficking from murine bone marrow. J Clin Invest. 2010;120:2423-31
71. Highfill S, Cui Y, Giles A, Smith J, Zhang H, Morse E. et al. Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Sci Transl Med. 2014;6:237ra67
72. Nicholls DJ, Wiley K, Dainty I, MacIntosh F, Phillips C, Gaw A. et al. Pharmacological characterization of AZD5069, a slowly reversible CXC chemokine receptor 2 antagonist. J Pharmacol Exp Ther. 2015;353:340-50
73. Lu X, Horner JW, Paul E, Shang X, Troncoso P, Deng P. et al. Effective combinatorial immunotherapy for castration-resistant prostate cancer. Nature. 2017;543:728-32
74. Lacotte S, Brun S, Muller S, Dumortier H. CXCR3, inflammation, and autoimmune diseases. Ann N Y Acad Sci. 2009;1173:310-7
75. Zou L, Barnett B, Safah H, LaRussa V, Hogan M, Mottram P. et al. Bone marrow is a reservoir for CD4+CD25+ regulatory T cells that traffic through CXCL12/CXCR4 signals. Cancer Res. 2004;64:8451-5
76. Zou W, Machelon V, Hermin A, Borvak J, Nome F, Isaeva T. et al. Stromal-derived factor-1 in human tumors recruits and alters the function of plasmacytoid precursor dendritic cells. Nature. 2001;7:1339-46
77. Müller A, Homey B, Soto H, Ge N, Catron D, Buchanan M. et al. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410:50-6
78. Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS. et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci U S A. 2013;110:20212-7
79. Lapa C, Herrmann K, Schirbel A, Hanscheid H, Luckerath K, Schottelius M. et al. CXCR4-directed endoradiotherapy induces high response rates in extramedullary relapsed multiple myeloma. Theranostics. 2017;7:1589-97
80. Haug AR, Leisser A, Wadsak W, Mitterhauser M, Pfaff S, Kropf S. et al. Prospective non-invasive evaluation of CXCR4 expression for the diagnosis of MALT lymphoma using [68Ga]Ga-Pentixafor-PET/MRI. Theranostics. 2019;9:3653-8
81. Lapa C, Schreder M, Schirbel A, Samnick S, Kortum KM, Herrmann K. et al. [68Ga]Pentixafor-PET/CT for imaging of chemokine receptor CXCR4 expression in multiple myeloma - Comparison to [18F]FDG and laboratory values. Theranostics. 2017;7:205-12
82. Debnath B, Xu S, Grande F, Garofalo A, Neamati N. Small molecule inhibitors of CXCR4. Theranostics. 2013;3:47-75
83. Peled A, Abraham M, Avivi I, Rowe JM, Beider K, Wald H. et al. The high-affinity CXCR4 antagonist BKT140 is safe and induces a robust mobilization of human CD34+ cells in patients with multiple myeloma. Clin Cancer Res. 2014;20:469-79
84. Stewart TJ, Smyth MJ. Improving cancer immunotherapy by targeting tumor-induced immune suppression. Cancer Metastasis Rev. 2011;30:125-40
85. Ziranu P, Atzori F, Puzzoni M, Demurtas L, Astara G, Scartozzi M. Effective combinatorial immunotherapy for castration-resistant prostate cancer: new future chance? Transl Cancer Res. 2017;6:S1014-7
86. Weitzenfeld P, Ben-Baruch A. The chemokine system, and its CCR5 and CXCR4 receptors, as potential targets for personalized therapy in cancer. Cancer Lett. 2014;352:36-53
87. Tan M, Goedegebuure PS, Belt BA, Flaherty B, Sankpal N, Gillanders WE. et al. Disruption of CCR5-dependent homing of regulatory T cells inhibits tumor growth in a murine model of pancreatic cancer. J Immunol. 2009;182:1746-55
88. Leone RD, Emens LA. Targeting adenosine for cancer immunotherapy. J Immunother Cancer. 2018;6:1-9
89. Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C. et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1β-dependent adaptive immunity against tumors. Nature Med. 2009;15:1170-8
90. Wilkin F, Duhant X, Bruyns C, Suarez-Huerta N, Boeynaems JM, Robaye B. The P2Y11 receptor mediates the ATP-induced maturation of human monocyte-derived dendritic dells. J Immunol. 2001;166:7172-7
91. Meis S, Hamacher A, Hongwiset D, Marzian C, Wiese M, Eckstein N. et al. NF546 [4,4'-(carbonylbis(imino-3,1-phenylene-carbonylimino-3,1-(4-methyl-phenylene)-carbonylimino))-bis(1,3-xylene-α,α'-diphosphonic acid) tetrasodium salt] is a non-nucleotide P2Y11 agonist and stimulates release of interleukin-8 from human monocyte-derived dendritic cells. J Pharmacol Exp Ther. 2009;332:238-47
92. Beavis PA, Stagg J, Darcy PK, Smyth MJ. CD73: a potent suppressor of antitumor immune responses. Trends Immunol. 2012;33:231-7
93. Antonioli L, Pacher P, Vizi ES, Hasko G. CD39 and CD73 in immunity and inflammation. Trends Mol Med. 2013;19:355-67
94. Allard B, Longhi MS, Robson SC, Stagg J. The ectonucleotidases CD39 and CD73: novel checkpoint inhibitor targets. Immunol Rev. 2017;276:121-44
95. Kansas G, Wood G, Tedder T. Expression, distribution, and biochemistry of human CD39. Role in activation-associated homotypic adhesion of lymphocytes. J Immunol. 1991;146:2235-44
96. Bono MR, Fernandez D, Flores-Santibanez F, Rosemblatt M, Sauma D. CD73 and CD39 ectonucleotidases in T cell differentiation: beyond immunosuppression. FEBS Lett. 2015;589:3454-60
97. Takenaka MC, Robson S, Quintana FJ. Regulation of the T cell response by CD39. Trends Immunol. 2016;37:427-39
98. Schuler PJ, Saze Z, Hong CS, Muller L, Gillespie DG, Cheng D. et al. Human CD4+ CD39+ regulatory T cells produce adenosine upon co-expression of surface CD73 or contact with CD73+ exosomes or CD73+ cells. Clin Exp Immunol. 2014;177:531-43
99. Kobie JJ, Shah PR, Yang L, Rebhahn JA, Fowell DJ, Mosmann TR. T regulatory and primed uncommitted CD4 T cells express CD73, which suppresses effector CD4 T cells by converting 5'-ddenosine monophosphate to adenosine. J Immunol. 2006;177:6780-6
100. Hilchey SP, Kobie JJ, Cochran MR, Secor-Socha S, Wang JC, Hyrien O. et al. Human follicular lymphoma CD39+-infiltrating T cells contribute to adenosine-mediated T cell hyporesponsiveness. J Immunol. 2009;183:6157-66
101. Figueiró F, Mendes FB, Corbelini PF, Janarelli F, Jandrey EH, Russowsky D. et al. A monastrol-derived compound, LaSOM 63, inhibits ecto-5'Nucleotidase/CD73 activity and induces apoptotic cell death of glioma cell lines. Anticancer Res. 2014;34:1837-42
102. Wang L, Fan J, Thompson LF, Zhang Y, Shin T, Curiel TJ. et al. CD73 has distinct roles in nonhematopoietic and hematopoietic cells to promote tumor growth in mice. J Clin Invest. 2011;121:2371-82
103. Vijayan D, Young A, Teng MWL, Smyth MJ. Targeting immunosuppressive adenosine in cancer. Nat Rev Cancer. 2017;17:709-24
104. A Ohta, Sitkovsky M. Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature. 2001;41:916-20
105. Ohta A, Gorelik E, Prasad SJ, Ronchese F, Lukashev D, Wong MK. et al. A2A adenosine receptor protects tumors from antitumor T cells. Proc Natl Acad Sci U S A. 2006;103:13132-7
106. Vecchio EA, Tan CY, Gregory KJ, Christopoulos A, White PJ, May LT. Ligand-independent adenosine A2B receptor constitutive activity as a promoter of prostate cancer cell proliferation. J Pharmacol Exp Ther. 2016;357:36-44
107. Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A. et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med. 2007;204:1257-65
108. Ohta A, Kini R, Ohta A, Subramanian M, Madasu M, Sitkovsky M. The development and immunosuppressive functions of CD4+ CD25+ FoxP3+ regulatory T cells are under influence of the adenosine-A2A adenosine receptor pathway. Front Immunol. 2012;3:1-12
109. Willingham S, Ho P, Leone R, Choy C, Powell J, McCaffery I. et al. Adenosine A2A receptor antagonist, CPI-444, blocks adenosine-mediated T cell suppression and exhibits anti-tumor activity alone and in combination with anti-PD-1 and anti-PD-L1. Ann Oncol. 2016;27:vi359-78
110. Beavis PA, Divisekera U, Paget C, Chow MT, John LB, Devaud C. et al. Blockade of A2A receptors potently suppresses the metastasis of CD73+ tumors. Proc Natl Acad Sci U S A. 2013;110:14711-6
111. Yuan G, Jankins TC, Patrick CG Jr, Philbrook P, Sears O, Hatfield S. et al. Fluorinated adenosine A2A receptor antagonists inspired by preladenant as potential cancer immunotherapeutics. Int J Med Chem. 2017;2017:4852537
112. Allard D, Turcotte M, Stagg J. Targeting A2 adenosine receptors in cancer. Immunol Cell Biol. 2017;95:333-9
113. Yang M, Ma C, Liu S, Shao Q, Gao W, Song B. et al. HIF-dependent induction of adenosine receptor A2b skews human dendritic cells to a Th2-stimulating phenotype under hypoxia. Immunol Cell Biol. 2010;88:165-71
114. Chen JF, Eltzschig HK, Fredholm BB. Adenosine receptors as drug targets ─ what are the challenges? Nat Rev Drug Discov. 2013;12:265-86
115. Cekic C, Sag D, Li Y, Theodorescu D, Strieter RM, Linden J. Adenosine A2B receptor blockade slows growth of bladder and breast tumors. J Immunol. 2012;188:198-205
116. Kalla RV, Zablocki J. Progress in the discovery of selective, high affinity A2B adenosine receptor antagonists as clinical candidates. Purinerg Signal. 2009;5:21-9
117. Kaidi A, Qualtrough D, Williams AC, Paraskeva C. Direct transcriptional up-regulation of cyclooxygenase-2 by hypoxia-inducible factor (HIF)-1 promotes colorectal tumor cell survival and enhances HIF-1 transcriptional activity during hypoxia. Cancer Res. 2006;66:6683-91
118. Duan B, Davis R, Sadat EL, Collins J, Sternweis PC, Yuan D. et al. Distinct roles of adenylyl cyclase VII in regulating the immune responses in mice. J Immunol. 2010;185:335-44
119. Nakanishi Y, Nakatsuji M, Seno H, Ishizu S, Akitake-Kawano R, Kanda K. et al. COX-2 inhibition alters the phenotype of tumor-associated macrophages from M2 to M1 in ApcMin/+ mouse polyps. Carcinogenesis. 2011;32:1333-9
120. Mahic M, Yaqub S, Johansson CC, Tasken K, Aandahl EM. FOXP3+CD4+CD25+ adaptive regulatory T cells express cyclooxygenase-2 and suppress effector T cells by a prostaglandin E2-dependent mechanism. J Immunol. 2006;177:246-54
121. Obermajer N, Muthuswamy R, Lesnock J, Edwards RP, Kalinski P. Positive feedback between PGE2 and COX2 redirects the differentiation of human dendritic cells toward stable myeloid-derived suppressor cells. Blood. 2011;118:5498-505
122. Mandapathil M, Szczepanski MJ, Szajnik M, Ren J, Jackson EK, Johnson JT. et al. Adenosine and prostaglandin E2 cooperate in the suppression of immune responses mediated by adaptive regulatory T cells. J Biol Chem. 2010;285:27571-80
123. Birrell MA, Nials AT. At last, a truly selective EP2 receptor antagonist. Br J Pharmacol. 2011;164:1845-6
124. Antonova M, Wienecke T, Maubach K, Thomas E, Olesen J, Ashina M. The pharmacological effect of BGC20-1531, a novel prostanoid EP4 receptor antagonist, in the prostaglandin E2 human model of headache. J Headache Pain. 2011;12:551-9
125. Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11:373-84
126. Junt T, Barchet W. Translating nucleic acid-sensing pathways into therapies. Nature Rev Immunol. 2015;15:529-44
127. Barton G, Medzhitov R. Toll-like receptor signaling pathways. Science. 2003;300:1524-5
128. Cheng K, Wang X, Yin H. Small-molecule inhibitors of the TLR3/dsRNA complex. J Am Chem Soc. 2011;133:3764-7
129. Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S. et al. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science. 2004;303:1526-9
130. Guha M. Anticancer TLR agonists on the ropes. Nat Rev Drug Discov. 2012;11:503-5
131. Geisse J, Caro I, Lindholm J, Golitz L, Stampone P, Owens M. Imiquimod 5% cream for the treatment of superficial basal cell carcinoma: results from two phase III, randomized, vehicle-controlled studies. J Am Acad Dermatol. 2004;50:722-33
132. Meyer T, Surber C, French L, Stockfleth E. Resiquimod, a topical drug for viral skin lesions and skin cancer. Expert Opin Inv drugs. 2013;22:149-59
133. Dudek AZ, Yunis C, Harrison LI, Kumar S, Hawkinson R, Cooley S. et al. First in human phase I trial of 852A, a novel systemic toll-like receptor 7 agonist, to activate innate immune responses in patients with advanced cancer. Clin Cancer Res. 2007;13:7119-25
134. Northfelt DW, Ramanathan RK, Cohen PA, Von Hoff DD, Weiss GJ, Dietsch GN. et al. A phase I dose-finding study of the novel Toll-like receptor 8 agonist VTX-2337 in adult subjects with advanced solid tumors or lymphoma. Clin Cancer Res. 2014;20:3683-91
135. Hoque R, Farooq A, Malik A, Trawick BN, Berberich DW, McClurg JP. et al. A novel small-molecule enantiomeric analogue of traditional (-)-morphinans has specific TLR9 antagonist properties and reduces sterile inflammation-induced organ damage. J Immunol. 2013;190:4297-304
136. Franklin BS, Ishizaka ST, Lamphier M, Gusovsky F, Hansen H, Rose J. et al. Therapeutical targeting of nucleic acid-sensing Toll-like receptors prevents experimental cerebral malaria. Proc Natl Acad Sci U S A. 2011;108:3689-94
137. Woo SR, Corrales L, Gajewski TF. The STING pathway and the T cell-inflamed tumor microenvironment. Trends Immunol. 2015;36:250-6
138. Barber GN. STING: infection, inflammation and cancer. Nat Rev Immunol. 2015;15:760-70
139. Fu J, Kanne DB, Leong M, Glickman LH, McWhirter SM, Lemmens E. et al. STING agonist formulated cancer vaccines can cure established tumors resistant to PD-1 blockade. Sci Transl Med. 2015;7:283ra52
140. Vargas R, Lizon B, Apetoh L. Rationale for stimulator of interferon genes-targeted cancer immunotherapy. Eur J Cancer. 2017;75:86-97
141. Gross S, Rahal R, Stransky N, Lengauer C, Hoeflich KP. Targeting cancer with kinase inhibitors. J Clin Invest. 2015;125:1780-9
142. Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT. et al. Loss of PTEN promotes resistance to T cell-mediated immunotherapy. Cancer Discov. 2016;6:202-16
143. Ying H, Elpek KG, Vinjamoori A, Zimmerman SM, Chu GC, Yan H. et al. PTEN is a major tumor suppressor in pancreatic ductal adenocarcinoma and regulates an NF-kappaB-cytokine network. Cancer Discov. 2011;1:158-69
144. O'Donnell JS, Massi D, Teng MWL, Mandala M. PI3K-AKT-mTOR inhibition in cancer immunotherapy, redux. Semin Cancer Biol. 2018;48:91-103
145. Xue G, Zippelius A, Wicki A, Mandala M, Tang F, Massi D. et al. Integrated Akt/PKB signaling in immunomodulation and its potential role in cancer immunotherapy. J Natl Cancer Inst. 2015:107
146. Liu J, Blake SJ, Yong MC, Harjunpaa H, Ngiow SF, Takeda K. et al. Improved efficacy of neoadjuvant compared to adjuvant immunotherapy to eradicate metastatic disease. Cancer Discov. 2016;6:1382-99
147. Schmid MC, Avraamides CJ, Dippold HC, Franco I, Foubert P, Ellies LG. et al. Receptor tyrosine kinases and TLR/IL1Rs unexpectedly activate myeloid cell PI3kgamma, a single convergent point promoting tumor inflammation and progression. Cancer Cell. 2011;19:715-27
148. Evans CA, Liu T, Lescarbeau A, Nair SJ, Grenier L, Pradeilles JA. et al. Discovery of a selective phosphoinositide-3-Kinase (PI3K)-gamma inhibitor (IPI-549) as an immuno-oncology clinical candidate. ACS Med Chem Lett. 2016;7:862-7
149. Kaneda MM, Messer KS, Ralainirina N, Li H, Leem CJ, Gorjestani S. et al. PI3Kgamma is a molecular switch that controls immune suppression. Nature. 2016;539:437-42
150. Rodon J, Carducci MA, Sepulveda-Sanchez JM, Azaro A, Calvo E, Seoane J. et al. First-in-human dose study of the novel transforming growth factor-beta receptor I kinase inhibitor LY2157299 monohydrate in patients with advanced cancer and glioma. Clin Cancer Res. 2015;21:553-60
151. Sharma A, Tran MA, Liang S, Sharma AK, Amin S, Smith CD. et al. Targeting mitogen-activated protein kinase/extracellular signal-regulated kinase kinase in the mutant (V600E) B-Raf signaling cascade effectively inhibits melanoma lung metastases. Cancer Res. 2006;66:8200-9
152. Panka DJ, Atkins MB, Mier JW. Targeting the mitogen-activated protein kinase pathway in the treatment of malignant melanoma. Clin Cancer Res. 2006;12:2371s-5s
153. Cheng Y, Zhang G, Li G. Targeting MAPK pathway in melanoma therapy. Cancer Metastasis Rev. 2013;32:567-84
154. D'Souza WN, Chang CF, Fischer AM, Li M, Hedrick SM. The Erk2 MAPK regulates CD8 T cell proliferation and survival. J Immunol. 2008;181:7617-29
155. Menzies AM, Long GV. Dabrafenib and trametinib, alone and in combination for BRAF-mutant metastatic melanoma. Clin Cancer Res. 2014;20:2035-43
156. Liu L, Mayes PA, Eastman S, Shi H, Yadavilli S, Zhang T. et al. The BRAF and MEK inhibitors Dabrafenib and Trametinib: effects on immune function and in combination with immunomodulatory antibodies targeting PD-1, PD-L1, and CTLA-4. Clin Cancer Res. 2015;21:1639-51
157. Voron T, Colussi O, Marcheteau E, Pernot S, Nizard M, Pointet AL. et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J Exp Med. 2015;212:139-48
158. Desar IM, Jacobs JH, Hulsbergen-vandeKaa CA, Oyen WJ, Mulders PF, van der Graaf WT. et al. Sorafenib reduces the percentage of tumour infiltrating regulatory T cells in renal cell carcinoma patients. Int J Cancer. 2011;129:507-12
159. Priceman S, Sung J, Shaposhnik Z, Burton J, Collado A, Moughon D. et al. Targeting distinct tumor-infiltrating myeloid cells by inhibiting CSF-1 receptor: combating tumor evasion of antiangiogenic therapy. Blood. 2010;115:1461-71
160. Mok S, Koya RC, Tsui C, Xu J, Robert L, Wu L. et al. Inhibition of CSF-1 receptor improves the antitumor efficacy of adoptive cell transfer immunotherapy. Cancer Res. 2013;74:153-61
161. Ehlerding EB, England CG, McNeel DG, Cai W. Molecular imaging of immunotherapy targets in cancer. J Nucl Med. 2016;57:1487-92
162. Weissleder R, Schwaiger M, Gambhir S, Hricak H. Imaging approaches to optimize molecular therapies. Sci Transl Med. 2016;8:355ps16
163. Liu Z, Li Z. Molecular imaging in tracking tumor-specific cytotoxic T lymphocytes (CTLs). Theranostics. 2014;4:990-1001
164. Vaz SC, Capacho AS, Oliveira FP, Gil N, Teixeira Barros C, Parreira A. et al. Radiopharmacology and molecular imaging of PD-L1 expression in cancer. Clin Trans Imag. 2018;6:429-39
165. Wei W, Jiang D, Ehlerding EB, Luo Q, Cai W. Noninvasive PET imaging of T cells. Trends in Cancer. 2018;4:359-73
166. Du Y, Jin Y, Sun W, Fang J, Zheng J, Tian J. Advances in molecular imaging of immune checkpoint targets in malignancies: current and future prospect. Eur Radiol. 2019;29:4294-302
167. Nishino M, Hatabu H, Hodi FS. Imaging of cancer immunotherapy: current approaches and future directions. Radiology. 2019;290:9-22
168. Helfen A, Roth J, Ng T, Eisenblaetter M. In vivo imaging of pro- and antitumoral cellular components of the tumor microenvironment. J Nucl Med. 2018;59:183-8
169. Broos K, Lecocq Q, Raes G, Devoogdt N, Keyaerts M, Breckpot K. Noninvasive imaging of the PD-1:PD-L1 immune checkpoint: Embracing nuclear medicine for the benefit of personalized immunotherapy. Theranostics. 2018;8:3559-70
170. Chen R, Zhou X, Liu J, Huang G. Relationship between the expression of PD-1/PD-L1 and 18F-FDG uptake in bladder cancer. Eur J Nucl Med Mol Imaging. 2019;46:848-54
171. Seith F, Forschner A, Schmidt H, Pfannenberg C, Guckel B, Nikolaou K. et al. 18F-FDG-PET detects complete response to PD1-therapy in melanoma patients two weeks after therapy start. Eur J Nucl Med Mol Imaging. 2018;45:95-101
172. Chatterjee S, Lesniak WG, Miller MS, Lisok A, Sikorska E, Wharram B. et al. Rapid PD-L1 detection in tumors with PET using a highly specific peptide. Biochem Biophys Res Commun. 2017;483:258-63
173. Radu CG, Shu CJ, Nair-Gill E, Shelly SM, Barrio JR, Satyamurthy N. et al. Molecular imaging of lymphoid organs and immune activation by positron emission tomography with a new [18F]-labeled 2'-deoxycytidine analog. Nat Med. 2008;14:783-8
174. Kim W, Le TM, Wei L, Poddar S, Bazzy J, Wang X. et al. [18F]CFA as a clinically translatable probe for PET imaging of deoxycytidine kinase activity. Proc Natl Acad Sci U S A. 2016;113:4027-32
175. Antonios J, Soto H, Everson R, Moughon D, Wang A, Orpilla J. et al. Detection of immune responses after immunotherapy in glioblastoma using PET and MRI. Proc Natl Acad Sci USA. 2017;114:10220-5
176. Ronald JA, Kim BS, Gowrishankar G, Namavari M, Alam IS, D'Souza A. et al. A PET imaging strategy to visualize activated T cells in acute graft-versus-host disease elicited by allogenic hematopoietic cell transplant. Cancer Res. 2017;77:2893-902
177. Shields AF, Grierson JR, Dohmen BM, Machulla HJ, Stayanoff JC, Lawhorn-Crews JM. et al. Imaging proliferation in vivo with [F-18]FLT and positron emission tomography. Nat Med. 1998;4:1334-6
178. Vanderhoek M, Juckett MB, Perlman SB, Nickles RJ, Jeraj R. Early assessment of treatment response in patients with AML using [18F]FLT PET imaging. Leuk Res. 2011;35:310-6
179. Keu KV, Witney TH, Yaghoubi S, Rosenberg J, Kurien A, Magnusson R. et al. Reporter gene imaging of targeted T cell immunotherapy in recurrent glioma. Sci Transl Med. 2017;9:eaag2196
180. Gambhir S, Herschman H, Cherry S, Barrio J, Satyamurthy N, Toyokuni T. et al. Imaging transgene expression with radionuclide imaging technologies. Neoplasia. 2000;2:118-38
181. Min JJ, Iyer M, Gambhir SS. Comparison of [18F]FHBG and [14C]FIAU for imaging of HSV1-tk reporter gene expression: adenoviral infection vs stable transfection. Eur J Nucl Med Mol Imaging. 2003;30:1547-60
182. Zlotnik A, Burkhardt AM, Homey B. Homeostatic chemokine receptors and organ-specific metastasis. Nat Rev Immunol. 2011;11:597-606
183. Jacobson O, Weiss ID. CXCR4 chemokine receptor overview: biology, pathology and applications in imaging and therapy. Theranostics. 2013;3:1-2
184. Li J, Peng C, Guo Z, Shi C, Zhuang R, Hong X. et al. Radioiodinated pentixather for SPECT imaging of expression of the chemokine receptor CXCR4 in rat myocardial-infarction-reperfusion models. Anal Chem. 2018;90:9614-20
185. Zhao H, Guo L, Zhao H, Zhao J, Weng H, Zhao B. CXCR4 over-expression and survival in cancer: a system review and meta-analysis. Oncotarget. 2014;6:5022-40
186. Domanska UM, Kruizinga RC, Nagengast WB, Timmer-Bosscha H, Huls G, de Vries EG. et al. A review on CXCR4/CXCL12 axis in oncology: no place to hide. Eur J Cancer. 2013;49:219-30
187. Jacobson O, Weiss ID, Szajek L, Farber JM, Kiesewetter DO. 64Cu-AMD3100 - a novel imaging agent for targeting chemokine receptor CXCR4. Bioorg Med Chem. 2009;17:1486-93
188. Nimmagadda S, Pullambhatla M, Stone K, Green G, Bhujwalla ZM, Pomper MG. Molecular imaging of CXCR4 receptor expression in human cancer xenografts with [64Cu]AMD3100 positron emission tomography. Cancer Research. 2010;70:3935-44
189. Zitvogel L, Pitt JM, Daillere R, Smyth MJ, Kroemer G. Mouse models in oncoimmunology. Nat Rev Cancer. 2016;16:759-73
190. Das R, Verma R, Sznol M, Boddupalli CS, Gettinger SN, Kluger H. et al. Combination therapy with anti-CTLA-4 and anti-PD-1 leads to distinct immunologic changes in vivo. J Immunol. 2015;194:950-9
191. Kokolus KM, Capitano ML, Lee CT, Eng JW, Waight JD, Hylander BL. et al. Baseline tumor growth and immune control in laboratory mice are significantly influenced by subthermoneutral housing temperature. Proc Natl Acad Sci U S A. 2013;110:20176-81
192. Weber JS, Yang JC, Atkins MB, Disis ML. Toxicities of immunotherapy for the practitioner. J Clin Oncol. 2015;33:2092-9
193. Altmann Y, McLaughlin S, Padgett MJ, Goyal VK, Hero AO, Faccio D. Quantum-inspired computational imaging. Science. 2018;361:eaat2298
194. Lambin P, Leijenaar RTH, Deist TM, Peerlings J, de Jong EEC, van Timmeren J. et al. Radiomics: the bridge between medical imaging and personalized medicine. Nature Rev Clin Oncol. 2017;14:749-62
195. Semerano L, Minichiello E, Bessis N, Boissier MC. Novel immunotherapeutic avenues for rheumatoid arthritis. Trends Mol Med. 2016;22:214-29
196. James ML, Hoehne A, Mayer AT, Lechtenberg K, Moreno M, Gowrishankar G. et al. Imaging B cells in a mouse model of multiple sclerosis using 64Cu-Rituximab PET. J Nucl Med. 2017;58:1845-51
197. Liu Y, Aguzzi A. Immunotherapy for neurodegeneration? Science. 2019;364:130-1
198. Herline K, Prelli F, Mehta P, MacMurray C, Goni F, Wisniewski T. Immunotherapy to improve cognition and reduce pathological species in an Alzheimer's disease mouse model. Alzheimers Res Ther. 2018;10:54-72
Author contact
![]() Corresponding author: Dr. Xianzhong Zhang at the State Key Laboratory of Molecular Vaccinology and Molecular Diagnostics & Center for Molecular Imaging and Translational Medicine, School of Public Health, Xiamen University, Xiamen 361102, China; Tel and Fax: +86-592-2880645; E-mail: zhangxzhedu.cn; ORCID: 0000-0001-8591-8301. Dr. Kai Chen at the Molecular Imaging Center, Department of Radiology, Keck School of Medicine, University of Southern California, 2250 Alcazar Street, CSC103, Los Angeles, CA 90033, USA; Tel: +1 (323) 442-3858; Fax: +1 (323) 442-3253; E-mail: chenkaiusc.edu; ORCID: 0000-0002-8647-1182.
Corresponding author: Dr. Xianzhong Zhang at the State Key Laboratory of Molecular Vaccinology and Molecular Diagnostics & Center for Molecular Imaging and Translational Medicine, School of Public Health, Xiamen University, Xiamen 361102, China; Tel and Fax: +86-592-2880645; E-mail: zhangxzhedu.cn; ORCID: 0000-0001-8591-8301. Dr. Kai Chen at the Molecular Imaging Center, Department of Radiology, Keck School of Medicine, University of Southern California, 2250 Alcazar Street, CSC103, Los Angeles, CA 90033, USA; Tel: +1 (323) 442-3858; Fax: +1 (323) 442-3253; E-mail: chenkaiusc.edu; ORCID: 0000-0002-8647-1182.