Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Nanobody structure and...
Advantages and disadvantages of...
Nanobodies represent a versatile...
Therapeutic application of...
Theranostics in immuno-oncology...
Outlook: evolution from bench to...
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(25):7772-7791. doi:10.7150/thno.34941 This issue Cite
Review
Theranostics in immuno-oncology using nanobody derivatives
Quentin Lecocq1, Yannick De Vlaeminck1, Heleen Hanssens2, Matthias D'Huyvetter2, Geert Raes3,4, Cleo Goyvaerts1, Marleen Keyaerts2,5, Nick Devoogdt2 ![]() , Karine Breckpot1
, Karine Breckpot1 ![]()
1. Laboratory for Molecular and Cellular Therapy (LMCT), Vrije Universiteit Brussel (VUB), Laarbeeklaan 103, B-1090 Brussels
2. In Vivo Cellular and Molecular Imaging Laboratory (ICMI), VUB, Laarbeeklaan 103, B-1090 Brussels
3. Unit of Cellular and Molecular Immunology (CMIM), VUB, Pleinlaan 2, B-1050 Brussels
4. Myeloid Cell Immunology Lab, VIB Inflammation Research Center, Pleinlaan 2, B-1050 Brussels, Belgium
5. Nuclear Medicine Department, UZ Brussel, Laarbeeklaan 101, B-1090 Brussels.
* These authors share senior authorship
Received 2019-3-16; Accepted 2019-7-11; Published 2019-10-15
Abstract

Targeted therapy and immunotherapy have become mainstream in cancer treatment. However, only patient subsets benefit from these expensive therapies, and often responses are short‐lived or coincide with side effects. A growing modality in precision oncology is the development of theranostics, as this enables patient selection, treatment and monitoring. In this approach, labeled compounds and an imaging technology are used to diagnose patients and select the best treatment option, whereas for therapy, related compounds are used to target cancer cells or the tumor stroma. In this context, nanobodies and nanobody-directed therapeutics have gained interest. This interest stems from their high antigen specificity, small size, ease of labeling and engineering, allowing specific imaging and design of therapies targeting antigens on tumor cells, immune cells as well as proteins in the tumor environment. This review provides a comprehensive overview on the state-of-the-art regarding the use of nanobodies as theranostics, and their importance in the emerging field of personalized medicine.
Keywords: single domain antibody, nanobody, cancer, molecular imaging, immunotherapy
Introduction
The description of camelid single domain antibodies (sdAbs), as we know them today, was preceded by a report published by Ward S. in 1989. In this report, the binding characteristics of isolated variable domains (VH) from the heavy chain of antibodies, generated after immunizing mice with either lysozyme or keyhole-limpet hemocyanin, was described [1]. The VH genes were expressed in E. coli and the VH were characterized by nanomolar affinity for their target. However, the antigen-binding affinity, stability and solubility of the VH were lower than those of the parent antibody, posing major challenges for commercial application. It was not until 1993 that Hamers R et al. described heavy-chain-only antibodies (HCAbs) in camelids, from which high affinity, functional camelid sdAbs are derived [2].
Hamers R. and his team from the Free University Brussels (Vrije Universiteit Brussel, VUB [Dutch]) analyzed serum samples from dromedaries (Arabian camel) and discovered the presence of immunoglobulins (IgGs) lacking a light chain. These HCAbs have a molecular weight of ~90 kDa and contributed up to 75% of all serum IgGs. Also other members of the Camelidae family were shown to possess HCAbs with concentrations varying between 30-50%. Blotting experiments and radioimmunoprecipitation were used to show the high affinity of HCAbs. The antigen binding part of HCAbs was confined to one single domain, known as the variable domain of the heavy chain of the HCAb (VHH). Ghahroudi et al. were the first to show that camelid sdAbs are well expressed in E. coli, and are highly stable and soluble, making them interesting for commercial applications [3]. Thus begins the exploratory phase for the development of camelid sdAbs that resulted in the foundation of Ablynx® in 2001, as a spin-off company from the VUB and the Flemish Institute of Biotechnology (Vlaams Instituut voor Biotechnology, VIB [Dutch]), that refers to VHH and camelid sdAbs as nanobodies™.
Since their discovery, nanobodies received increasing attention, as exemplified by the growing number of studies that evaluate the application of nanobodies in the fields of biotechnology and medicine, in particular for oncology, the focus of this review (Figure 1).
Graph showing the number of publications on the description of nanobodies in oncology during the period from 2004 to 2019.
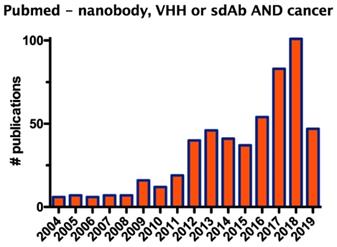
The developments in nanobody discovery together with the many advantageous properties of nanobodies led to their exploration in clinical trials conducted by large biopharmaceutical companies [4]. Meanwhile, main patent claims on nanobodies are expiring, which will fuel the growing interest in commercializing nanobodies as research, therapy and diagnostic agents. Following the description of the structure and merits of nanobodies, we will address the different diagnostic and therapeutic applications in the context of immuno-oncology in more detail.
Nanobody structure and development platforms
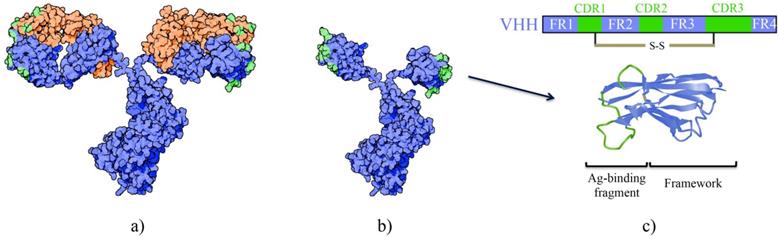
HCAbs are homodimers consisting of two heavy chains linked with a disulfide bond. These heavy chains are comprised of a variable (VHH) and a constant region however lack the CH1 domain found in conventional antibodies [5]. Three complementarity determining regions (CDRs) form the antigen binding site of HCAbs, as opposed to six CDRs found in conventional antibodies that also contain light chains (Figure 2). The solvent-exposed framework region 2 (FR2) in VHH is more hydrophilic than the corresponding VH fragment in conventional antibodies [6]. The distinctive CDR3 loop in VHH plays a crucial role in antigen binding. Especially in camels and dromedaries, the single domain can have a particularly long and diverse CDR3 that is the result of recombination of different V-D-J germline elements, junctional diversity and hypermutation, and selection during in vivo maturation of more functional and soluble nanobodies with a long CDR3 [7]. Frequently the long CDR3 extends out and allows high affinity binding to a concave epitope at active sites of proteins that are usually inaccessible to antibodies [8-10]. Moreover, besides CDR3, also CDR1 and CDR2 contribute to target binding, involving more hydrophobic amino acids in their paratope, and a surprisingly high amount of residues in framework regions make contacts with the antigen. It is suggested that the interaction of nanobodies to their targets are more similar to general protein-to-protein interactions instead of antibody-to-antigen interactions [10]. Other differences to conventional antibodies have evolved to ensure large repertoire diversity and high binding capacity in the absence of light chains and include (1) an extended CDR1 region towards the N-terminal end, (2) involvement of FR2 in shaping the CDR3 loop and (3) extensive somatic hypermutation [11]. Finally, disulfide bonds present in the VHH, especially those derived from camel and dromedary, confer extra stability [12].
A schematic representation of the differences between a conventional antibody (a) and a HCAb (b). The antigen-binding domain from the HCAb is referred to as a VHH, nanobody or sdAb (c).
The generation of a VHH library against an antigen of interest has already been described in numerous publications. The vast majority of isolated nanobodies described to date are isolated using the same procedure, namely selections of phage libraries displaying VHH retrieved from immunized camelids [13]. In short, an animal from the Camelidae family like an alpaca or a dromedary is immunized with a source of antigen (frequently recombinant protein). Approximately 40 days later, peripheral blood lymphocytes are isolated and subsequent isolation of RNA is performed. The VHH gene fragments are amplified using a PCR and cloned in a phagemid vector to an in vivo matured VHH library. The library is phage-displayed and subjected to several consecutive rounds of biopanning on solid phase coated recombinant target protein or on cells, enriching antigen-specific phages with each round. Recently, newer techniques have been reported that allow improved screening of nanobody immune libraries using yeast surface display platforms or genetically encoded barcoding peptides [14-16]. Finally, positive clones are cloned in an appropriate expression vector allowing nanobody production in microbial hosts like E. coli, S. cerevisiae or P. pastoris [17-19], in mammalian cells and plants [20]. Bacteria produce the nanobodies in their cytosol or in periplasm. Nanobody extraction from the periplasm is performed through osmotic shock, while lysis using sonication or by freeze-thaw cycles is necessary for nanobody extraction from the cytosol. Yeast and mammalian cells secrete nanobodies at high yields in the culture supernatant, guaranteeing correct post-translational modifications like the formation of disulfide bonds, but care should be taken to avoid nanobody glycosylation as this can have a negative impact on its functionality and in vivo behavior [20].
The classic acquisition of a VHH library by immunizing camelidae is straightforward but inconvenient from the point of view of animal protection and costs to maintain these large animals. Transgenic mice that express HCAbs by their B cells were generated by Janssens et al. and could serve as an alternative host for immunization [21]. This transgenic mouse was realized by recombining two llama variable V regions and the human D, J, Cμ and/or Cγ constant regions to generate a hybrid llama/human antibody locus. Recently, efforts has been made to develop in vitro selection of nanobodies against a target protein by for example cDNA display methods with synthetic or semi-synthetic libraries [22-25]. Although some studies reported this method for selection of nanobodies against their target of interest, it must be mentioned that affinities were usually lower, in the range of 10-7 to 10-8 M, and that this approach demands large library sizes and sophisticated selection procedures, which currently is prohibitive for widespread implementation [26-28].
Advantages and disadvantages of nanobodies
A key characteristic of nanobodies is their small size. Nanobodies are 2.5 nm in diameter, 4 nm in height with a molecular mass of around 12-15 kDa. Other advantageous traits include (1) high affinity, (2) high specificity, (3) low off-target accumulation, (4) high (thermo)stability and (5) good solubility [29]. Due to their small size, nanobodies have the distinct feature of penetrating dense tissues like tumors very well [30]. Additionally, some nanobodies have been described to even transmigrate through the brain endothelial cell layer [31-33]. Moreover, nanobodies allow targeting hidden epitopes of certain proteins and at locations difficult to approach by conventional antibodies, which include ion channels [34], G protein-coupled receptors [35,36] and immune synapses [37]. Ease of molecular engineering, low immunogenicity, and ease and relative low cost of production are other advantages that make nanobodies interesting compounds for various immuno-oncology applications [38].
For certain therapeutic applications, maximal delivery of the nanobody in the cancer lesion is desired, which is a disadvantage of these fast-clearing proteins. Several strategies can be explored to enhance the delivery of nanobodies to the tumor site. One of these is extending the serum half-life of the nanobody using different formatting options. Examples hereof are fusion of the nanobody directly with albumin, polyethylene glycol (PEG; PEGylation) or IgG-Fc (allowing recycling through the neonatal Fc receptor [FcR]), or indirectly with a second nanobody that binds albumin or the neonatal FcR, thereby generating bispecific constructs [39-42]. Another option to obtain maximal delivery in the tumor site is to use a gene therapy approach, which could be highly feasible for nanobodies as these are simple proteins expressed from one single gene [43,44]. A continuous, local secretion of nanobodies by genetically engineered cells in the tumor microenvironment (TME) will maximize their delivery to the target of interest and will moreover minimize the risks associated with systemic distribution. Additionally, nanobodies are well suited for cytosolic expression due to their ability to fold in reducing environments like the cytosol, a feature that has been widely exploited for in cellulo tracking of intracellular proteins via microscopic imaging [45,46].
As a result of their easy cloning, nanobodies have also been explored for other gene-based immuno-oncology approaches, as exemplified by the use of nanobodies to design (1) vaccines targeted to antigen-presenting cell (APC) subsets or (2) chimeric antigen receptors (CARs) to direct the specificity of cancer killing cells [47-57]. The ease of molecular engineering is also exploited to generate multi-specific nanobody formats purposed to engage cancer killing immune cells, in particular cytotoxic T lymphocytes (CTLs) and natural killer (NK) cells. An example hereof is the development of so-called bispecific light T cell engagers (LiTEs) in which scFvs targeting the CD3 molecule on T cells are coupled to nanobodies targeting EGFR tumor on cancer cells. Consequently a bridge is formed between cancer cells and T cells and these T cells are activated through cross-linking of the CD3 molecule [58,59]. The ability to design multi-specific and chimeric nanobody formats is furthermore exploited to generate so-called bispecific killer cell engagers (BiKEs), nanobody-based cancer therapy agents that engage NK cells to exert antibody-dependent cell-mediated cytotoxicity (ADCC) [60,61]. ADCC is triggered when Fc is bound by FcRs on NK cells. The lack of an Fc in nanobodies implies that nanobodies in some cases might exhibit lower therapeutic potency compared to antibodies. To circumvent this, nanobodies targeting the FcγRIII have been developed [62]. Coupling of this nanobody to an anti-carcinoembryonic antigen (CEA) specific nanobody resulted in potent killing of CEA+ cells [63]. Consequently, combining an FcR-specific nanobody with a nanobody binding to a target of interest could be conceived when Fc effector functions are required. As these multi-specific nanobodies are still relatively small in size, they retain a good tumor penetration capacity.
Another potential disadvantage of nanobodies is that the human immune system can potentially perceive them as foreign, as they are derived from camelid HCAbs. Nonetheless, nanobodies are considered to be weakly immunogenic due to their high similarity with human VH fragments and their properties, including size, monomeric form, solubility and lack of an Fc fragment [64]. Furthermore, it is suggested that the immunogenicity of nanobodies could be minimized by humanization [65,66]. Nanobody humanization is generally performed by surface veneering into residues that are encoded in the human germline. The change of each individual residue replacement, in particular in FR2, should be carefully monitored as this can influence both nanobody functionality and, importantly, its solubility which is an important trigger of immunogenicity [65,67]. The necessity to humanize the nanobody sequence is further questioned by the observation that anti-drug immune responses against a humanized tetravalent nanobody targeting death receptor (DR) 5 on cancer cells, were observed and were the main reason to stop clinical evaluation [68]. Some studies support the notion that nanobodies and even VH fragments can be immunogenic [68-70], while other studies with other nanobody constructs showed no signs of immunogenicity [71-73]. To date, there is a lack of understanding on the immunogenicity of non-humanized and humanized nanobodies due to insufficient data. Therefore, studies on this subject are warranted to demonstrate the possible limitations on the extent of manipulating nanobodies. As nanobodies are increasingly used in clinical studies, data will become available to address this question. Meanwhile, several companies focus on development of nanobodies and their use for diagnosis and/or therapy. Examples hereof are Ablynx®, Novamab, NanoMab, Orionis Biosciences, Helix Biopharma, Nanjing Legend Biotech and Camel-IDS, which have partnered with leading pharmaceutical companies to develop nanobodies among others to tackle cancer. Of these companies, Ablynx® recently brought an anti-von Willebrand factor-specific nanobody to the market for the treatment of thrombotic thrombocytopenic purpura, highlighting the potential of nanobodies as innovative medicines [74].
Nanobodies represent a versatile platform for imaging of cancer cells and their environment
The role of immune cells in shaping a tumor and in influencing the outcome of cancer therapy is generally recognized [75,76]. As a consequence when diagnosing cancer, one would like to know as much as possible about the tumor, such as the presence of targetable tumor antigens and the immune contexture, to plan and monitor the most effective treatment. This can be achieved via molecular imaging, the use of labeled indicator molecules that can be imaged in a noninvasive manner. Nanobodies are attractive tools for this purpose [77].
Modalities for nanobody-based imaging
For noninvasive imaging, nanobodies need to be labeled with an imaging probe that can consist of a radioisotope, fluorescent dye, microbubble or a chemical like gadolinium, allowing imaging via technologies such as single photon emission computed tomography (SPECT), positron emission tomography (PET), optical imaging (OI), ultrasound (US) and magnetic resonance imaging (MRI) [30]. While these different imaging modalities exist, the majority of nanobody-mediated imaging studies use SPECT and PET, because these radioisotope-based techniques have a high sensitivity and offer quantitative information [78-80]. In preclinical studies, nanobodies often contain a genetically inserted C-terminal hexahistidine tag for purification purposes, which can be complexed with 99mTc(CO3), a γ-emitting radionuclide that is easily detectable using SPECT [81]. For PET, which is clinically more relevant, nanobodies are labeled with positron-emitting radionuclides, exemplified by 18F, 64Cu, 68Ga and 89Zr. Frequently used radioisotopes are 18F and 68Ga because of their short half-life, 68 and 110 minutes respectively, matching the biological half-life of nanobodies when injected intravenously [82]. For coupling of these radioisotopes to nanobodies site-specific labeling is desired to obtain homogenous and consistent tracers. A good example of site-specific conjugation is the transpeptidase sortase A-mediated ligation. The latter catalyzes the formation of a peptide bond between the C-terminally expressed LPXTG peptide motif of the nanobody and the N-terminal oligo-glycine motif on the label [83,84]. Another example is the conjugation of maleimide-functionalized labels on an unpaired cysteine on the nanobody [85-87]. An alternative to radiolabeling of nanobodies is the use of fluorescent dyes that can be combined with OI. For in vivo imaging, near-infrared (NIR) emitting fluorophores are the label of choice, as these provide strong contrast and resolution combined with signal detection in depths ranging from several hundred µm to one cm [88]. Examples of NIR fluorophores include IRDye-680RD or -800CW, Cy5 and AlexaFluor 680 [77]. Advantages of OI are its flexibility, simplicity and cost-effective character, as in contrast to radioisotope-mediated imaging, it does not require dedicated facilities. This imaging modality is often used to study surface lesions during surgical or endoscopic procedures, as OI dyes have limited tissue penetrating capacity compared to radioisotope-based imaging. The use of US has been studied as an alternative to radiolabeled nanobodies while retaining the ability for high-resolution images [89]. This type of imaging requires conjugation of nanobodies to US contrast agents, microbubbles or nanobubbles that after intravenous administration allow the molecular characterization of the vascular wall [90,91]. Finally, Prantner et al. evaluated cross-reactive nanobodies targeting mesothelin-expressing ovarian cancer for MRI imaging [92]. Their nanobody-coated superparamagnetic nanoparticles allowed mesothelin-detection in xenografted tumors using MRI.
Imaging of cancer cells
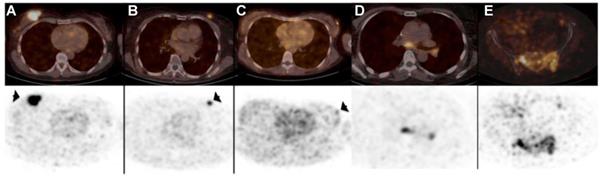
Nanobody-based imaging has been extensively studied for detection of cancer cells themselves, mostly in preclinical studies. Antigens that have been targeted include but are not limited to CEA, epidermal growth factor receptor (EGFR), HER2, prostate-specific membrane antigen (PSMA), CD20 and CD38, as reviewed by Debie et al. [77]. With regard to clinical testing the most advanced nanobody-based imaging agent is the 68Ga-coupled anti-HER2 nanobody 2Rs15d for PET imaging of breast cancer patients [93]. Results of the first clinical trial with nanobody 2Rs15d were published in 2016 and showed detection and imaging of HER2 in the primary tumor as well as local or distant metastases as soon as 60 minutes post-injection without any adverse effects, such as renal toxicity and tracer-induced antibodies [73]. The detection of HER2 using this nanobody was shown to be highly specific reaching SUV values of 11.8 for the primary lesions and 6.0 for metastases (Figure 3). Moreover, background uptake was very low with the exception of signals observed in the kidneys, intestines and liver. Recently, a phase II clinical trial evaluating the potential of 68Ga-NOTA-2Rs15d to detect brain metastasis has been initiated (NCT03924466). Implementation of nanobody-based imaging of cancer markers can be a guide for therapy selection, in particular as targeted therapies have been developed for many of these cancer markers, some of which are based on the use of nanobodies, as exemplified by the use of anti-CD20 and anti-HER2 nanobodies for targeted therapy [94-96].
PET/CT scans (top) and PET scans (bottom) after injection of 68Ga-HER2-Nanobody showing uptake in primary breast carcinoma lesions (arrows) (A-C) and metastatic lesions in lymph nodes in mediastinum and left hilar region (D) and bone metastasis in pelvis (E). Adapted with permission from [73], copyright 2016.
Imaging the tumor stroma
Solid tumors can be considered as abnormal organs comprising multiple immune cell types, new blood vessels and extracellular matrix (ECM). As cancer cells intimately interact with their surroundings and as this crosstalk often promotes tumor progression and even therapy resistance, it is of interest to visualize (and target) these components as well. Recently, the generation of diverse nanobody libraries against ECM proteins associated with cancer was described [97]. From these libraries, a nanobody that targets the alternatively spliced EIIIB domain of fibronectin, an important component of the ECM and neovasculature, was used to show the broad applicability of ECM-targeted nanobodies to image tumors and metastases using two-photon immuno-fluorescence and noninvasive immuno-PET/CT imaging [98]. In line with this topic is the visualization of newly forming blood vessels. As endothelial cells that line blood vessels are genetically more stable than cancer cells, they have been proposed as targets for anti-angiogenic therapy [99]. Visualization of tumor endothelial cells that express vascular cell adhesion molecule-1 (VCAM-1) has been described using anti-VCAM-1 nanobody decorated microbubbles [91], and also radiotracer variants of this nanobody are available [100,101]. This is important as a correlation between VCAM-1 expression on endothelial cells and vessel density as well as metastasis was reported [102,103]. Moreover, VCAM-1 expression on cancer cells has been linked to immune evasion [104]. It was shown using the TC-1 lung epithelial cancer model that tumors with high VCAM-1 expression were devoid of CD8+ tumor-specific T cells [105]. These studies suggest that VCAM-1 nanobodies could provide valuable information on the resistance of tumors to immune-mediated killing when combined with nanobodies generated to image CD8+ T cells. Such nanobodies have been developed by Rashidian et al. and were studied as a means to predict response to anti-cytotoxic T lymphocyte antigen-4 (CTLA-4) immune checkpoint (ICP) therapy in a mouse melanoma model [106].
As blockade of inhibitory ICPs, such as CTLA-4 and programmed death-1 (PD-1): programmed death-ligand 1 (PD-L1), receives increasing attention in immuno-oncology, nanobody-based probes to image the expression of these ICPs have been developed. PET/CT imaging of naive and tumor bearing mice was performed using the 18F or 89Zr radiolabeled anti-CTLA-4 nanobody H11 [107]. Nanobodies to image the expression of PD-L1 in the TME have also been developed [108]. High contrast images showing mouse PD-L1 expression in syngeneic mouse tumors were obtained 1 hour after inoculation of a 99mTc-labeled nanobody using SPECT [109]. Moreover, human PD-L1 specific nanobodies were generated for SPECT [110,111] and PET imaging [112]. Regarding SPECT imaging, the nanobody K2 demonstrated several interesting properties, among which high specificity and affinity for human PD-L1, low kidney retention and competition with the FDA approved anti-PD-L1 antibody, avelumab [111]. For PET imaging, 89Zr labeled KN035, a bivalent nanobody containing an Fc tail, was able to monitor PD-L1 levels using PET in nude mice bearing human xenografts [112]. Additionally, a first-in-human study demonstrated that SPECT imaging with nanobody 99mTc-NM-01 is safe and suited to evaluate PD-L1 levels at the tumor site as soon as 2 hours after injection [110].
Imaging of proteins such as CD8, VCAM-1, CTLA-4 and PD-L1 provide a first glance on the immune contexture and possibly immune resistance of tumors. However, certain immune cell types like macrophages have been designated as culprits in the development of tumors and in the failure of many immunotherapies [75]. Macrophages in the TME can present themselves in many forms, and efforts have been placed in identifying markers that can discriminate pro- from antitumor macrophages [113]. It was reported that macrophage mannose receptor (MMR, CD206) is highly expressed on tumor-promoting macrophages. Nanobodies to detect MMR have been developed and were shown to bind MMR+ macrophages in hypoxic regions. These 99mTc-labeled anti-MMR nanobodies were also used to image MMR levels in tumors using SPECT/CT [114].
A comparison of this nanobody labeled with 99mTc versus 18F showed that labeling with 18F reduced both liver and kidney uptake [115]. The first steps towards clinical translation of this approach using a 68Ga-NOTA-anti-MMR nanobody for PET/CT imaging were recently taken [116]. In contrast to MMR, expression of MHC-II on macrophages (and dendritic cells [DCs]) is considered a good prognostic marker, as it is associated with antigen presentation to CD4+ T cells. An MHC-II targeting nanobody that was labeled with 64Cu was able to image MHC-II+ cells in the spleen and bones of NOD/SCID humanized mice using PET/CT [117]. Further, detection of 1 mm-sized tumors with a high affinity 18F-labeled anti-MHC-II nanobody was shown to be more accurate than with 18F-FDG [118]. Other probes that were developed to image (and target) APCs, include the probes DC1.8 and DC2.1 [119]. DC2.1 was shown to recognize a wide range of myeloid cells, while DC1.8 only bound immature DCs. This binding pattern was reflected in their biodistribution when labeled with 99mTc, with DC2.1 showing accumulation in liver, spleen and lungs, and DC1.8 primarily showing signals in skin. With the growing interest in targeting tumor-associated DCs to unleash an antitumor immune response, further studies on the potential of DC-targeting nanobodies to image specific DC subsets is warranted [120].
Therapeutic application of nanobodies and nanobody derivatives in immuno-oncology
Surgery, chemotherapy and radiotherapy have long been considered the best options for cancer treatment. These therapies are an indiscriminate warfare, coinciding with damaging side effects and failing to protect against recurring cancer cells. Our growing understanding on how cancer develops has opened new avenues to treat cancer. Cancer is driven by corrupted messages from our own genes, caused and influenced by a range of factors, not in the least our own immune system. The identification of molecular accelerators of cancer cells, such as HER2, led to the development of molecularly targeted treatments, designed to bind and override faulty molecules in cancer cells. Moreover, the identification of immune cells and immune pathways acting as a brake or accelerator on cancer cells, led to the idea of manipulating the immune system to fight cancer. In particular the latter has gained increasing interest, as with immunotherapy comes the promise of a systemic treatment leading to a long-lasting immunological memory capable of tackling recurrent cancer cells.
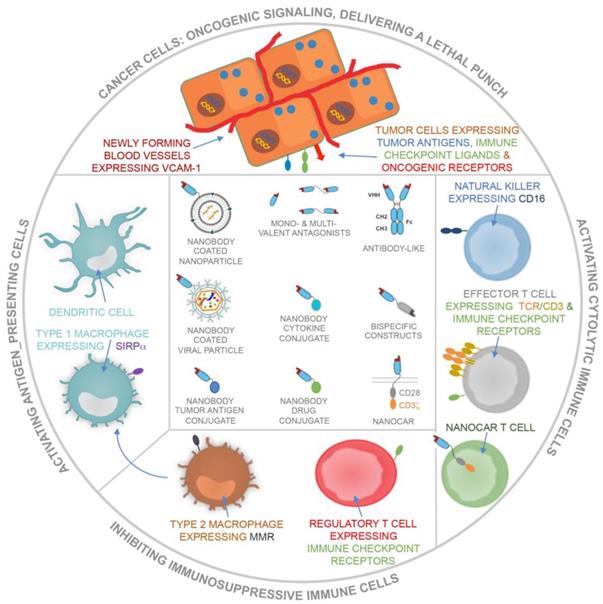
Nanobodies have been studied extensively in the context of targeted cancer therapy and immunotherapy. Strategies embracing nanobodies in the fight against cancer can be categorized into the following types: the use of nanobodies to (1) dampen oncogenic signals, (2) deliver a lethal punch to cancer cells, (3) design cancer vaccines, (4) engage cytolytic cells and (5) prevent immunosuppressive events (Figure 4). Below we discuss the use of nanobodies according to these categories.
Schematic representation of the use of nanobodies and nanobody-derivatives for targeting of cancer cells and blood endothelial cells or for modulation of immune cells that can either activate (APCs, including DCs and type 1 macrophages), exert (cytolytic immune cells, including NK cells and CTLs) or suppress (type 2 macrophages and Tregs) antitumor immune responses.
Exploiting nanobodies to dampen oncogenic signals
Several types and subtypes of cancer are characterized by dysregulation of receptor tyrosine kinase (RTK) signaling, leading to an imbalance between cell growth, proliferation and cell death. Examples of RTKs that frequently have alterations in cancer cells, and therefore are constitutively providing oncogenic signals, are VEGFR, EGFR, HER2 and c-MET (hepatocyte growth factor receptor). Nanobodies that antagonize these RTKs have been developed. Additionally, nanobodies that bind VEGF and hepatocyte growth factor, the unique ligands of VEGFR and c-MET have been developed as well [121].
The versatility of nanobodies in the context of targeted therapy is exemplified by studies on targeting of EGFR and VEGF/VEGFR signaling. In the context of EGFR targeting, a biparatopic anti-EGFR nanobody fused to an anti-albumin nanobody, referred to as CONAN-1 was developed [122]. CONAN-1 was reported to have a serum half-life of 2-3 days and to inhibit tumor outgrowth albeit at a lower potency as the EGFR targeting antibody cetuximab (Erbitux®). This lower potency may be related to a lack of Fc on the nanobody construct and suggests additional involvement of immune cells in eradication of EGFR-expressing tumors. Multi-specific nanobody formats have also been generated to target signaling mediated by VEGF and its receptor. A tri-specific nanobody targeting VEGF, angiopoietin-2 and serum albumin, has been co-developed by Ablynx® and Boehringer Ingelheim. This construct, referred to as BI1836880, inhibits signaling mediated by VEGF and angiopoietin-2 and was shown in different in vivo models to be superior in efficacy in comparison to the antibody bevacizumab (Avastin®) [123]. Inhibition of endothelial cell proliferation has also been shown with various monovalent nanobodies against different isoforms of VEGF, showing that the necessity to generate multivalent or multi-specific nanobody formats should be evaluated on a per case basis [124-126]. A further improvement of efficacy of serum half-life extended antagonistic nanobodies can perhaps be augmented by arming these compounds with cytotoxic effectors, as discussed in the following paragraph.
Exploiting cancer cell-specific nanobodies to deliver a lethal punch to cancer cells
Nanobodies that do not have antagonistic traits, yet target cancer cells, have been coupled to other technology platforms to deliver a targeted, lethal punch to cancer cells. Nanobodies have been coupled to (1) tumor necrosis factor-related apoptosis inducing ligand (TRAIL), a death inducing ligand [44,127], (2) a truncated form of the Pseudomonas exotoxin A [127-130], (3) various drugs and drug-loaded nanoparticles [131-138], (4) photosensitizers [139-143] and (5) therapeutic radionuclides [95,96,144-149]. The reason to couple nanobodies to these toxic moieties is to bring them close to cancer cells, while minimizing toxic effects to healthy tissues, hence reducing potential adverse effects. Another strategy with a similar purpose is coupling of cancer cell-targeted nanobodies to enzymes for prodrug activation, ensuring drug activity only in the vicinity of cancer cells [150,151].
Targeting of DRs on cancer cells using TRAIL is an interesting strategy that was shown to act in concert with EGFR-targeting [152]. Keeping this in mind, a bi-functional molecule consisting of an anti-EGFR nanobody coupled to TRAIL was generated [44,153]. It was shown in vitro that the bi-functional molecule inhibited growth of several cancer cell types that did not respond well to EGFR blockade or DR engagement as a stand-alone treatment. Binding of EGFR by the nanobody induced DR clustering at the cancer cell membrane, thereby sensitizing these cells to TRAIL and downstream caspase-mediated apoptosis. Stem cells engineered to express the anti-EGFR nanobody-TRAIL (ENb-TRAIL) fusion protein were used in vivo in an orthotopic resection model of primary glioblastoma as a continuous source of the bi-functional molecule [44,153]. ENb-TRAIL simultaneously binds to EGFR and DR5 receptor, present on tumor cells, leading to receptor clustering and the induction of apoptotic signals, which resulted in a significant increase in survival [44,153].
The truncated form of the Pseudomonas exotoxin (PE38) has been studied in conjunction with anti-VEGFR [128], anti-CD7 [127,129] and anti-CD38 [130] nanobodies. To generate so-called targeted toxins the nanobody in its monovalent or bivalent format is genetically coupled via a linker to the PE38 toxin. Both the VEGFR and anti-CD38 targeted immunotoxins were shown to compromise antigen-specific tumor cells in vitro, an effect that was also observed in vivo using CD7-targeted immunotoxins.
Among the drugs that are frequently used to treat various cancer types are cisplatin and its analogues, carboplatin and oxaliplatin as well as doxorubicin, RTK inhibitors and death effector molecules. As these drugs lack selectivity, nanobodies have been used to target them to cancer cells. In the case of delivery of platin-based chemotherapy two fusion proteins, termed NGC and NGCA, with different functional modules, were developed by Huang et al [131]. The NGC fusion protein is composed of a biparatopic anti-EGFR nanobody for cancer cell targeting, a high-affinity gadolinium-binding domain for MRI imaging, and a C3-tag for drug conjugation. In addition, the NGCA fusion protein further contains an anti-albumin nanobody at the C-terminus of NGC, as this prolongs the in vivo circulation time. These nanobody containing fusion proteins were coupled to a maleimide-functionalized Pt(IV) prodrug (Mal-Pt), which was synthesized from cisplatin and coupled to the C3-tag. This theranostic drug delivery system enabled drug accumulation in EGFR+ tumors, which was most pronounced using NGCA, and delayed tumor growth with little toxicity when compared to classical treatment with cisplatin. Doxorubicin is another small molecule chemotherapy agent for which targeted delivery is of interest. This can be achieved by site-selective cysteine bioconjugation of a thioether propargyl carbamate linker bearing the anti-cancer drug to a nanobody against the HER2 antigen [132]. Release of doxorubicin was achieved with palladium, resulting in decaging of propargyl carbamate protected lysine or tyrosine residues. It was shown that this reaction is suitable for drug delivery to cells. However, for in vivo applications the development of new palladium compounds is still needed. Another means of delivering doxorubicin is after its encapsulation in nanoparticles [154] such as liposomes [138], polymer-based nanoparticles and micelles [155], overall nanoparticles that could be coupled to nanobodies. Coupling of nanobodies to these nanoparticles often makes use of chemical reactive moieties that are present on the nanoparticle as a consequence of its PEGylation. The nanobody is coupled via cysteine chemistry or modified with N-succinimidyl-S-acetylthioacetate, as this does not affect the nanobody's binding capacity [85,156]. Because this results in the presence of multiple nanobodies on the nanoparticle, a high avidity is obtained [157]. The anti-EGFR nanobody has been used in view of nanoparticle targeting, guiding both the particle and exerting its antagonistic function. Talleli et al. studied targeted delivery of micelles [155] and in a follow-up study doxorubicin-loaded micelles [133]. It was shown that these thermosensitive, biodegradable polymeric micelles themselves inhibit tumor growth in vivo, and that encapsulation of doxorubicin further increased the tumor inhibiting effects. Liposomes coated with anti-EGFR nanobodies, and loaded with the anti-IGFR-1R kinase inhibitor AG538 have been studied by van der Meel et al. to inhibit tumor growth [134]. This is a particularly interesting approach as it has been shown that there is a crosstalk between EGFR and IGF-1R, which can lead to acquired resistance against EGFR targeted drugs [158]. The latter is explained by the interaction of EGFR and IGF-1R, which can be directly through the association between their respective receptors and by inducing the availability of each other's ligands, or indirectly through the interaction of common binding partners or downstream signaling molecules. Subsequently, this provides a rationale to dual targeted therapies [158]. Without AG538, the anti-EGFR coated liposomes induced downregulation of EGFR. Loading the liposomes with AG538 additionally affected IGF-1R signaling and further increased the inhibitory effect on cancer cell growth in vitro [134]. In vivo, growth inhibition was observed in one of the two cell lines examined in a mouse xenograft model [135]. Also nanobody-coated polymers have been studied for delivery of drugs to cancer cells. Polymers decorated with anti-HER2 [136] or anti-MUC1 nanobodies [137] were shown to enable selective cancer cell targeting. Encapsulation of plasmid DNA and hence delivery of genetic material was achieved in this way. More specifically, MUC1-targeted delivery of plasmid DNA encoding for truncated-Bid (under the control of a MUC1 promotor) was shown to result in expression of the transgene, resulting in considerable cell death [137].
Instead of delivering a drug, cancer cell-specific nanobodies have also been studied to deliver enzymes that cleave prodrugs into their active form, as such ensuring minimal off-target drug activity. An example hereof is the fusion of the enzyme β-lactamase from E. cloacae P99 to a high-affinity anti-CEA nanobody. The chosen β-lactamase enzyme efficiently converted the prodrug 7-(4-carboxybutanamido) cephalosporin mustard in phenyl-enediamine mustard at the surface of CEA-expressing colon cancer cells in vitro, thereby inducing cytotoxicity. This nanobody-enzyme conjugate induced tumor regression and in some cases even cured mice of established tumor xenografts [150]. Another example is the immunoconjugate designated L-DOS47, which is comprised of a nanobody targeting CEACAM6 coupled to an urease enzyme that converts urea into ammonia in situ and as such induces toxicity. The specificity and activity of L-DOS47 was shown and this nanobody-conjugate is being explored in a phase I/II clinical trial for non-small cell lung cancer [151].
Hitting a photosensitizer with light of a particular wavelength in an oxygenated environment results in formation of reactive oxygen species (ROS). These ROS in turn damage proteins, lipids and/or nucleic acids, and as such induce cell death. Monovalent (7D12) and biparatopic (7D12-9G8) nanobodies targeting EGFR have been used to selectively deliver the photosensitizer IRDye700DX, a fluorescent dye that also allows OI. In vitro studies showed that these anti-EGFR-IRDye700DX conjugates specifically induced cell death of EGFR overexpressing cells in low nanomolar concentrations, while IRDye700DX alone or anti-EGFR-IRDye700DX conjugates in the absence of light did not induce cell death [140]. Internalization of IRDye700DX was observed when the photosensitizer was coupled to the biparatopic format of the anti-EGFR nanobody, which resulted in a more pronounced induction of cell death [140]. However, enhanced cytotoxicity upon internalization of nanobody photosensitizer conjugates was contradicted in a study by van Lith et al. [141]. Herein, the anti-EGFR nanobody was conjugated to a cell-penetrating peptide (CPP) to enhance internalization of the IRDye700DX photosensitizer. This discrepancy is likely explained by the biparatopic format of the anti-EGFR nanobody used by Heukers et al., which might induce EGFR downregulation and as a consequence reduced oncogenic signaling [140], much in the same way as observed when anti-EGFR nanobodies are coupled to nanoparticles [134,155]. Induction of selective tumor cell death was also studied in vivo in immunodeficient mice growing orthotopic tongue tumors, showing that both 7D12 and 7D12-9G8 anti-EGFR-IRDye700DX conjugates allowed selective OI of the tumors, and that in particular the 7D12 anti-EGFR-IRDye700DX conjugate consistently induced near-complete tumor eradication with little to no toxicity in healthy tissues [139]. The potency of this strategy warrants further studies to translate this technology from bench to bedside.
Similar as photosensitizers, branched gold nanoparticles kill cancer cells when excited by NIR-light, but by generating heat instead of ROS. Van de Broek et al. and d'Hollander et al. site-directionally conjugated such gold nanostars to anti-HER2 nanobodies and showed in vitro that they precisely killed HER2-expressing cancer cells when triggered by laser-light [142]. Moreover, in vivo these HER2 nanobodies coupled to gold nanostars were able to reach HER2+ xenografted tumors [143].
Targeted radionuclide therapy (TRT) is a systemic treatment with radioactively labeled cancer-specific probes purposed to selectively hit diseased cells. As radioactive labels α- or β--emitters are used, which release their energy in the proximity of the cancer cells, thereby causing irreparable DNA damage. The potential of nanobodies as targeting moieties for TRT in preclinical cancer models has been extensively investigated using the therapeutic β--particle emitting radionuclides Lutetium-177 and Iodine-131, and more recently the α-emitting radionuclides Astatine-211, Actinium-225 and Bismuth-213 [95,96,144,145]. Early proof-of-concept studies using β--particle emitting radionuclides coupled to nanobodies showed delivery of lethal radiation doses to developing tumors with a negligible level of irradiation to healthy tissues, except for kidneys, which were the dose-limiting organs. However, a generic method to reduce kidney retention of radiolabeled nanobodies was described, in particular the use of untagged nanobodies with co-infusion of the plasma expander Gelofusin [95]. Preclinical studies on β--TRT have been performed in several cancer models, including multiple myeloma [144], breast and ovarian cancer [95,145], and non-Hodgkin lymphoma [96], using nanobodies targeting the 5T2MM paraprotein, HER2 and CD20, respectively. In these studies, nanobody-based β--TRT led to a significant blockade of tumor growth and as a consequence a significant difference in event-free survival between the treated and the control groups. However, repeated dosing was required to achieve an effective therapeutic dose in tumors, which is in part explained by the low linear energy transfer (LET) of β--particles (LET: 0.1-1 keV/µm) and the rapid clearance of nanobodies. Because α-particles have a higher LET (50-100 keV/µm) with a short penetrating range (40-80 µm), α-TRT has been proposed as an attractive strategy to eradicate residual cancer cells [146,147]. Several research groups have reported on the characterization of HER2- and PSMA-targeting nanobodies labeled with the α-emitting radionuclides Astatine-211, Actinium-225 and Bismuth-213 [147-149]. In these studies specific uptake of the radiolabeled nanobodies was shown with high tumor-to-normal organ ratios (except for kidneys). Moreover, it was shown using Bismuth-213 coupled to anti-PSMA nanobodies that DNA double-strand breaks were induced in vivo in tumor cells [149]. Taken together, these studies support the further development of nanobody-mediated TRT.
Exploiting nanobodies to design cancer vaccines targeted to antigen-presenting cells
The goal of cancer vaccination is to activate CTLs. These can selectively recognize and kill cancer cells irrespective of their location, and can form an immunological memory, ready to act when cancer cells with the same properties pop up. Activation of CTLs requires tumor antigen presentation in MHC-I molecules and co-stimulation by professional APCs of which DCs have been extensively studied for cancer vaccination [56,159].
Adoptive transfer of autologous DCs manipulated in vitro to present tumor antigens has proven successful in several clinical trials [160]. However, to circumvent the laborious and expensive procedure of generating ex vivo DCs, strategies have been developed to manipulate DCs in situ. Nanobodies have been coupled to several technology platforms designed to deliver tumor antigens or potentiate the APC's antigen-presenting capacity. It is contended that targeting of APCs will enhance the vaccine efficacy and will reduce potential adverse effects.
Nanobodies have been generated against multiple proteins that are expressed on the surface of APCs, including CD11b, CD36, MHC-II [161], CD1d [162], PD-L2 [163] and Clec9a [164]. Moreover several nanobodies that target APCs, however for which the antigen has not yet been identified, have been described, including nanobody R3_13 [47], DC1.8 and DC2.1 [119]. In vivo biodistribution studies suggest that DC1.8 targets conventional DCs (cDCs) of murine origin, while DC2.1 targets myeloid APCs, including cDCs, plasmacytoid DCs and macrophages. These nanobodies were studied in the context of virus-based cancer vaccines, based on adenoviruses [165] and lentiviruses [47,49,55,57,166]. Incorporation of the nanobody into lentiviral vectors is achieved using the nanobody display technology, which exploits the budding mechanism of lentiviral vectors to incorporate the nanobody together with a binding-defective, fusion-competent glycoprotein in the viral envelope [49,167]. This strategy allows high titer production of lentiviral vectors coated with nanobodies. When using DC1.8 and DC2.1, selective, nanobody-dependent infection of mouse DCs and macrophages both in vitro and in situ was shown. Moreover, this strategy was translated to a human model, showing selective infection of in vitro generated or lymph node-derived DCs and macrophages [47]. An adenovirus-based vaccine was also successfully redirected to DCs by replacement of the adenoviral fiber knob with fiber-fibritin chimeras fused to DC1.8 [165]. Against the expectation, the APC-targeted lentiviral cancer vaccine was less potent in activation of CTLs than non-targeted lentiviral cancer vaccines [57]. This was explained by (1) a strong induction of type I interferon (IFN), which is a result of recognition of viral components by the APCs and hampered the translation of the delivered tumor antigen; and (2) lack of stromal cell transduction with reduced MHC-I mediated antigen presentation [168]. In contrast, enhanced immunogenicity was observed for peptide-based vaccines when these were coupled to nanobodies specific for MHC-II, CD11b or CD36 [161]. It was observed that coupling of peptides to the nanobody targeted to MHC-II elicited strong activation of CD4+ T cells, which support APCs and CD8+ T cells in their function. The strongest activation of CD8+ T cells was observed when using the anti-CD11b nanobody for targeting [161]. These studies suggest that nanobodies are indeed suited to target tumor antigens to APCs, however point out that a smart choice of a technology platform is required to ensure that the APC reaches its full potential to present the tumor antigens.
DCs can acquire tumor antigens in tumors. However tumor-derived factors render these DCs unable to activate CD4+ and CD8+ T cells. Therefore, strategies have been explored to activate these DCs, of which delivery of cytokines is an example [166]. As systemic delivery of cytokines is often correlated to toxicity, nanobodies have been used to engineer 'Activity-on-Target cytokines' or AcTakines. These mutant cytokines display a reduced receptor-binding affinity, however upon fusion to nanobodies their activity is restored on the nanobody bound cell populations. In the case of IFN or 'AcTaferon', this hypothesis was confirmed with a nanobody specific for programmed death-ligand 2 (PD-L2) on APCs as it appeared up to 1,000-fold more potent on target cells, allowing specific signaling in selected cell types only [163]. A strong antitumor activity was also observed when a nanobody against the cDC1 marker Clec9A was used as adapter in murine melanoma, breast carcinoma and lymphoma models and against human lymphoma in humanized mice without any detectable toxic side effects [164,169].
While the strategies above use nanobodies to deliver tumor antigens or cytokines to APCs, agonistic nanobodies that target CD1d have been used 'as such' to potentiate DCs [162]. CD1d is an antigen-presenting molecule involved in the presentation of glycolipids to NKT cells, which are recognized as major contributors to antitumor immunity [170]. Similar to CD4+ T cells, NKT cells support APCs and CTLs in their activity, and can exert direct cytolytic effects on cancer cells. Lameris et al. generated anti-CD1d nanobodies and showed that 2 out of 22 nanobodies had the capacity to induce NKT cell-independent production of interleukin (IL) 12 by monocyte-derived DCs, thereby supporting the DC's ability to activate antitumor immunity. This study shows that nanobodies can be used as such to potentiate the APC's stimulatory capacity, provided that the target is well chosen. Other potential targets for agonistic nanobodies include co-stimulatory molecules that signal towards APCs, such as CD40 [171] and CD83 [172].
Exploiting nanobodies to engage cytolytic cells
Cancer vaccination is one approach in which nanobodies are studied to activate cytolytic immune cells to recognize and kill cancer cells. This strategy however requires targeting of APCs. Alternative approaches using nanobodies have been designed that act directly on cytolytic immune cells, thereby bypassing the targeting of APCs.
A multitude of cytokines acts as signals for T cell activation, proliferation and survival. To target these signals to T cells within the TME, and therefore avoid overwhelming systemic T cell activation, cytokines have been coupled to nanobodies targeting antigens highly expressed in the TME. An anti-CEA nanobody-Fc fusion was further coupled to IL-15 linked to its soluble receptor IL15-R to increase its function, and this construct was used in a xenograft model, showing strong antitumor effects associated with CD8+ T cell recruitment [173]. Also IL-2 and IFN-γ have been coupled to nanobodies, in this case targeting PD-L1. It was shown that the anti-PD-L1 nanobodies delivered the cytokines in the TME using a melanoma as well as pancreatic cancer model. Combining the nanobody-mediated delivery of IL-2 and IFN-γ with tumor-specific antibody therapy resulted in tumor growth inhibition, which coincided with increased CD8+ T cell numbers. Delivery of IL-2 however also increased regulatory T cell numbers, warranting the use of a mutated form of IL-2 with affinity for binding to the IL-2 receptor on CD8+ T cells. Next to increasing CD8+ T cell numbers, delivery of IFN-γ acted on myeloid cells, redirecting them towards an MHC-II+ phenotype, which is correlated to an antitumor function [174].
While cytokines provide important signals during the activation of CD8+ T cells, they only act as co-stimuli. Without engagement of the TCR and subsequent CD3ζ signaling, CD8+ T cells cannot differentiate into CTLs. Therefore, strategies have been developed to mimic CD3ζ signaling in the absence of MHC-I:peptide mediated TCR triggering. These rely on binding of CD3 by agonistic antibodies or antibody fragments that are cross-linked. Nanobodies that bind EGFR on cancer cells have been coupled in their monovalent format or as a trimer to anti-CD3 scFvs to ensure cross-linking and as such T cell activation [58,59]. Such constructs are referred to as LiTEs or bispecific light T cell engagers, and were shown to retain high tissue penetration capacity and to enable strong levels of T cell activation. To further increase the T cell activating capacity of LiTEs, nanobodies binding 4-1BB, a co-stimulatory receptor on T cells, have been incorporated in the construct, showing accumulation in the TME and efficient activation of antitumor responses [175]. Activation of γδ T cells, in particular Vγ9Vδ2 T cells that exert antitumor activity, has been achieved using a similar principle. Activation of TCR signaling without functional recognition of MHC-peptide complexes was achieved using agonistic nanobodies that bind the Vγ9Vδ2 TCR and that were coupled to anti-EGFR nanobodies to ensure cross-linking. When combined with Vγ9Vδ2 T cells this construct enhanced the cytolytic response of the Vγ9Vδ2 T cells against EGFR+ cancer cell lines in vitro as well as in xenografted mice [176].
To ensure on-target cytolytic activity of CD8+ T cells, CAR T cells have been developed. These are patient-derived T cells that are genetically modified to express a CAR that combines T cell cytotoxicity with the MHC-independent antigen recognition of antibodies. The concept was first introduced by Gross et al., who cloned VH and VL parts of an antibody to the intracellular parts of TCRα and TCRβ in two different genes [177]. This design was then further adjusted by the same group, when they assembled one single genetic construct, consisting of a scFv, coupled to intracellular FcRγ or CD3ζ in 1993 [178]. This set the basis for further CAR design and adjustment over the years, which is nowadays conventionally build-up of a single gene encoding an antigen recognizing part (conventionally a scFv), coupled to a spacer (derived from CD8α, CD28, IgG1 or IgG4), a transmembrane domain (derived from CD3ζ, CD4, CD8α or CD28) and to intracellular co-stimulatory (CD28, 4-1BB, OX40, ICOS and/or CD27 derived) and activation (usually CD3ζ) domains [179]. While most research has been focused on the optimization of the intracellular signaling domain, fewer efforts have been made in the optimization of the format of the extracellular, antigen binding part of the CAR [180]. ScFvs derived from clinically approved or tested antibodies are used most commonly because their behavior and target-specificity in patients is well characterized. However, some drawbacks are attributed to this antigen-binding format in a CAR context. For instance, scFvs can be immunogenic and instable. This has been proven to limit clinical efficacy because of an anti-CAR immune response, and to lead to premature T cell exhaustion [181-183]. The latter was found to be due to the vulnerability of scFv framework regions to aggregation, which leads to antigen-independent T cell activation, shortly followed by premature exhaustion, limiting CAR T cell persistence in vivo [184,185]. Consequently, several groups have successfully replaced the scFv with a nanobody in a CAR context, to create nanoCARs. These nanoCARs target different tumor cell-associated markers, such as PSMA [52] and MUC1 [51] for solid tumors and CD38 for myeloma [53]. Other groups have developed nanoCARs directed against markers of tumor stroma, such as PD-L1, EIIIB fibronectin splice variant and VEGFR2 [54,186]. Although difficult to compare directly with scFv-based CARs because of differences in protocols and constructs, these studies show that nanoCARs make valid alternatives for classical CARs and might open the door to a more efficient CAR design.
The concept of universal CARs (uniCARs), which are CAR T cells recognizing a molecule or peptide that is coupled to a cancer-targeting module (TM), administered separately, has also been introduced with nanobodies [187]. In this pre-targeting approach the TM will guide uniCARs to the tumor target in a concentration-dependent manner. In the absence of a TM, the uniCAR T cells are inactive. This allows the repeated stop-and-go retargeting of tumor cells [188]. Recently also, nanobody-based CAR NK cells have been validated in xenografts of T cell acute lymphoblastic leukemia [189], opening doors to off-the shelf products.
Altogether, several recent studies show how nanobodies are slowly finding their way in CAR cell therapy, with different side tracks being explored. Though most studies using nanoCARs are still in the early stages, few examples prove that nanoCARs are paving the way to clinical applications. The CAR T cell therapy from Nanjing Legend Biotech® uses two BCMA-specific nanobodies in tandem, targeting myeloma cells and is proving to be effective in phase I/II clinical trials (NCT03758417) [190,191]. More recently, a phase I trial with CD19/CD20 bispecific nanoCAR T cell therapy for B cell lymphoma was initiated (NCT03881761). Results from these trials will prove whether nanobody-based CAR T cell therapy can add value in the field of adoptive T cell transfer, at points where scFvs fall short.
Next to CD8+ T cells, NK cells, which are innate cytolytic immune cells, can be harnessed in the fight against cancer. They can exert direct cytolytic functions or can recruit DCs and aid T cell activation through cytokine and chemokine production. Modification of NK cells with nanoCARs is one way to exploit the antitumor properties of NK cells [189]. Another strategy that has been explored is the development of BiKEs or bispecific killer cell engagers. Herein the ability of NK cells to exert cytolytic functions when the receptor CD16 (FcRIII) is bound is used. As coupling of cancer-specific nanobodies to Fc fragments can impact on the nanobodies' properties, as excellently reviewed by Saunders KO [192] describing the multifaceted functions of Fc, a nanobody that binds CD16 with high affinity has been generated, designated "C21" [62]. Multimerization of this nanobody allowed activation of NK cells, as shown by the production of IL-2 and IFN-γ [62]. Coupling of this nanobody to an anti-MUC-1 [61] or an anti-CEA [193,194] nanobody was shown to mediate tumor growth inhibition when injected on a daily basis in mice receiving peripheral blood mononuclear cells and xenografted with MUC-1+ or CEA+ colon carcinoma tumors. Comparison of trastuzumab (Herceptin®) to a construct in which nanobody "C21" was coupled to a bsFab build from anti-HER2 nanobodies showed comparable efficacy in vivo on cells that are highly HER2+, such as SK-BR-3 and BT474, however superior activity of the nanobody-based construct when HER2 expression was low (MCF-7 model) [195].
Exploiting nanobodies to revert immunosuppressive events
Tumor cells exploit different suppressive pathways and cells to avoid antitumor immune responses, allowing them to continue their growth undisturbed and eventually to metastasize. In this paragraph we address the following immunosuppressive mechanisms, (1) inhibitory ICPs, (2) immunosuppressive cytokines and (3) myeloid cells like M2-macrophages, against which nanobodies have been developed.
The discovery of the ICPs CTLA-4 and PD-L1 led to the award of the Nobel prize for medicine to James Allison and Tasuku Honjo in 2018 and impacted the current therapeutic landscape for patients with solid and hematological tumor malignancies [196]. Regarding the ICP CTLA-4, efforts have been taken to develop nanobodies for therapeutic applications. In the study of Wan R et al., the treatment of melanoma (B16) bearing mice with mouse CTLA-4 targeting nanobody "NB16" significantly delayed their tumor growth and survival time [197]. In contrast, Ingram JR et al. demonstrated that nanobody-mediated blocking of mouse CTLA-4 only induced minimal effects on antitumor responses [107]. Interestingly, the lack in therapeutic efficacy was related to the lack of an Fc portion on nanobodies. Fusion of nanobody "H11" to a murine IgG2a constant region could dramatically enhance its antitumor effect. These studies again highlight that the necessity to modulate the nanobody format should be evaluated on a per case basis. Therapeutic nanobodies have also been developed against the ICP PD-L1 in order to counteract immune inhibition through the ligation with its receptor PD-1 that is highly expressed on activated T cells [111,198-200]. Nanobody mediated blockade of PD-L1, present on DCs, could significantly increase the activity of T cells upon antigen presentation, an effect that was not seen with the FDA-approved anti-PD-L1 antibody Avelumab [198]. Moreover, the study of Zhang et al. demonstrated in vivo therapeutic effects of their PD-L1 targeting nanobody when fused to a human IgG1 Fc [199]. This nanobody, designated as KN035, is currently being evaluated in 2 different clinical trials for the treatment of patients with advanced solid tumors (NCT03101488, NCT03248843).
Another ICP ligand is CD47. CD47 is expressed in a wide range of malignancies and negatively regulates phagocytosis through the interaction with its receptor SIRP1α, expressed on macrophages. A study using a mouse melanoma model has shown that blockade of the CD47-SIRP1α interaction using nanobody "A4" was only able to induce tumor rejection responses when locally delivered and combined with antitumor antigen antibodies, PD-L1 blockade and/or GVAX vaccination [201]. In contrast with the Fc fused CTLA-4 blocking nanobodies mentioned above, fusion of "A4" to a constant region did not improve its therapeutic efficacy and even induced anemia since CD47 is also expressed by red blood cells.
The cytokine tumor necrosis factor alpha (TNF-α) works immunosuppressive by increasing the expansion of immunosuppressive myeloid cells and regulatory T cells at the tumor site [202,203]. Nanobodies targeting TNF-α were able to reduce lung metastasis in vivo and worked synergic with the antimitotic agent paclitaxel [204].
Several strategies have been explored to directly target immunosuppressive myeloid cells like for example macrophages. The drugs gefitinib and simvastatin are known to have an effect on the activation of tumor-associated macrophages [205,206]. Liposome coupled, PD-L1 targeting nanobodies were loaded with these drugs and showed a potent antitumor effect when administered in vivo [207]. A switch in M1/M2 ratio in the treated mice in favor of M1-macrophages was observed. Moreover, nanobodies targeting MMR, present on pro-tumor M2-macrophages, have been generated and used in therapeutic settings when fused to other proteins and nanoparticles. Coupling of the MMR specific nanobody to pro-apoptotic proteins like second-mitochondria derived activator of caspases showed specific targeting to and modulation of M2-macrophages [208]. Moreover, Nuhn et al. conjugated anti-MMR nanobodies to polymeric maleimide-functionalized nanogels and showed effective nanoparticle delivery to MMR-expressing macrophages in vitro and in vivo when administered in tumor bearing mice [209]. Consequently, they observed a significant reduction of M2-macrophages in the TME, which could pave the road for targeted eradication, or modulation of immunosuppressive cells.
Theranostics in immuno-oncology using nanobodies and nanobody derivatives
The focus for the treatment of most cancers is evolving to more expensive therapies where predictive markers are not only clinically relevant but also an economic requirement. Diagnostic tests like immunohistochemistry are current practice but are unable to portray whole tumor expression levels and this is even worse for metastatic lesions. This could explain the failure to accurately predict outcome responses in all patients. Whole body, noninvasive imaging modalities such as PET, SPECT, MRI and OI, using nanobody-based tracers, could fulfill these shortcomings and could be implemented repetitively without the need of collecting invasive biopsies.
Until now, we described the use of nanobodies in the context of immuno-oncology for either imaging or therapeutic applications. We are convinced that many of the described nanobodies hold the potential to be used as molecular imaging probes as well as therapeutic agents. The term “theranostic” was initially put forward to describe the development of diagnostic tests alongside the application of a therapy targeted towards a specific molecular feature, as exemplified by Herceptin® and HercepTest®, which were simultaneously approved by the FDA in 1998 for the treatment and diagnosis of Her2 expressing breast cancers [210]. Hence, historically, the theranostics paradigm refers to different methodologies and compounds that unite diagnosis and therapy of the same molecular pathways and disease indication in a practice that is also called precision medicine. Currently, the term theranostics is used in a much stricter sense and rather refers to agents that are identical or closely related and that harbor the potential to be used both for diagnostic as well as for therapeutic purposes. In this paragraph we will elaborate on nanobody-based compounds for use as true theranostics. A summary of published examples is made in Table 1.
Published theranostic applications (pre-clinical and clinical) of nanobodies in immuno-oncology. Diagnostic technologies are labeled blue, whereas therapeutic approaches are depicted in orange. Examples and references are shown per targeted antigen.
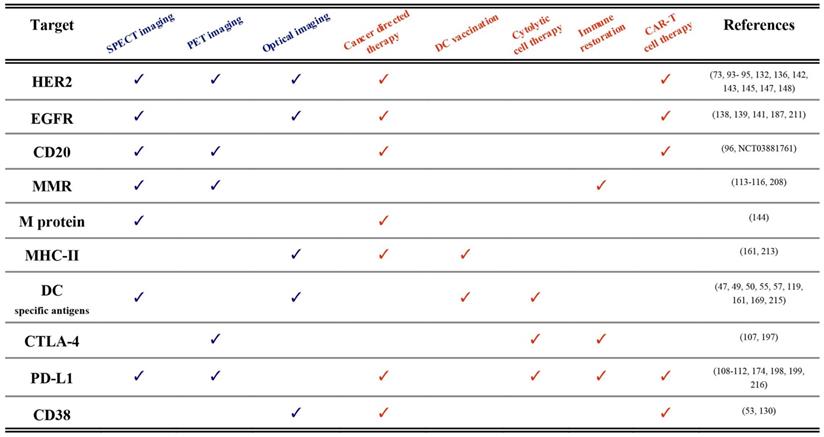
First of all, nanobodies targeting cancer-specific membrane proteins (e.g. HER2, EGFR, M-protein, CD20 and CD38) have been evaluated for both imaging and therapeutic applications [73,95,96,122,130,144,147,211]. The clearest example of nanobody theranostics is where both diagnostic tracers and therapeutic compounds are radiolabeled, in a TRT approach [96,144]. The radiolabel can be different, e.g. Gallium-68 or Fluor-18 for PET imaging and Actinium-225 for α-TRT. But sometimes the radiolabel is the same such as Iodine-131 labeled nanobodies that are first used at low doses in SPECT imaging for diagnosis and dose estimations, and then at higher doses for TRT [145]. Of importance is that the diagnostic and therapeutic nanobody-radiopharmaceuticals have similar pharmacokinetics and biodistribution profile.
Nuclear imaging with nanobodies could be a guide for other nanobody-based therapeutic modalities as well. Examples of these are nanobody-toxin fusions [18,127,128] or nanoCAR T cells [50,52,53]. In these settings PET imaging is the dominant diagnostic modality due to its superior sensitivity, resolution and quantifiability. In the nanoCAR T application PET could be useful to predict homing of the re-targeted T cells to the tumor.
Another clear example of nanobody-theranostics is OI and photodynamic therapy [121]. Here the cancer-specificity of NIR-labeled nanobodies is first exploited to visualize the tumors (diagnosis) and help the surgeon to better delineate tumor boundaries (therapy) [212]. After the image-guided surgery, residual cells can then be killed by activating the nanobody-photosensitizer-conjugates on the membrane of the targeted cells by light.
Next, nanobodies targeting specific immune cells subsets like DCs can be used to detect their presence in mm-sized tumors using different imaging modalities [119]. The same nanobodies can then be used to target peptide vaccines or cytokines to these cells and elicit immunogenicity responses [49,213-215].
Finally, nanobodies have been generated against ICPs due to their important role in dampening antitumor immune responses. For example, CTLA-4 or PD-L1 targeting nanobodies, when labeled with radioisotopes, can be used to noninvasively detect using PET or SPECT imaging which type of immunosuppressive pathway dominates the TME [109,111,112,197]. Then, these nanobodies when administered as such, or combined with chemokines/cytokines or implemented in nanoCARs can restore the antitumor immune responses in vivo [54,107,174,198,199,216].
Outlook: evolution from bench to bedside
Since their first observation, over a quarter century ago, many nanobody-based pharmaceuticals, after a preclinical phase, are now in or on the path towards clinical testing in the area of oncology. Here the nanobodies block essential protein-protein interactions, re-target effector cells or act as nanobody-drug conjugates as well as tracers for molecular imaging. The extraordinary features of nanobodies - small size, stability, affinity, easy of engineering in multifunctional constructs - will continue to fulfill a central role in future scientific research in the field of antibody engineering. Preclinical and clinical data such as those with HER2 targeting nanobodies support the claims of the great potential of labeled nanobodies as diagnostic agents. Moreover, increasing pre-clinical studies have evaluated the use of nanobodies to successfully combat a variety of different tumor models in vivo. This report contains a state-of-the-art overview of the use of nanobodies as therapeutic and diagnostic agents in the field of immuno-oncology and their promise as theranostics in the context of cancer. Their small size makes them ideally suited for noninvasive imaging of malignant cells or processes linked to malignancy such as angiogenesis and immunosuppression. Consequently, the potential of visualizing these markers will act as a modality to guide and monitor treatments of patients. Furthermore, nanobodies are more and more studied in therapy applications. Despite the lack of any Fc-mediated effector functions, pre-clinical studies hint towards a substantial value in applications such as the blockade of receptor signaling present on tumor cells and/or immune cells and targeted delivery of various agents ranging from toxins, pro-apoptotic proteins, radionuclides to cytokines. Preclinical studies on the applications of nanobodies in immuno-oncology are vast, and translation of some of these methodologies in a clinical setting is either already happening or a possibility in the near future.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ward ES, Gussow D, Griffiths AD, Jones PT, Winter G. Binding activities of a repertoire of single immunoglobulin variable domains secreted from Escherichia coli. Nature. 1989;341(6242):544-6
2. Hamers-Casterman C, Atarhouch T, Muyldermans S, Robinson G, Hamers C, Songa EB. et al. Naturally occurring antibodies devoid of light chains. Nature. 1993;363(6428):446-8
3. Arbabi Ghahroudi M, Desmyter A, Wyns L, Hamers R, Muyldermans S. Selection and identification of single domain antibody fragments from camel heavy-chain antibodies. FEBS Lett. 1997;414(3):521-6
4. Steeland S, Vandenbroucke RE, Libert C. Nanobodies as therapeutics: big opportunities for small antibodies. Drug Discov Today. 2016;21(7):1076-113
5. Vu KB, Ghahroudi MA, Wyns L, Muyldermans S. Comparison of llama VH sequences from conventional and heavy chain antibodies. Mol Immunol. 1997;34(16-17):1121-31
6. Muyldermans S, Atarhouch T, Saldanha J, Barbosa JA, Hamers R. Sequence and structure of VH domain from naturally occurring camel heavy chain immunoglobulins lacking light chains. Protein Eng. 1994;7(9):1129-35
7. Henry KA, MacKenzie CR. Antigen recognition by single-domain antibodies: structural latitudes and constraints. MAbs. 2018;10(6):815-26
8. De Genst E, Silence K, Decanniere K, Conrath K, Loris R, Kinne J. et al. Molecular basis for the preferential cleft recognition by dromedary heavy-chain antibodies. Proc Natl Acad Sci U S A. 2006;103(12):4586-91
9. Al Qaraghuli MM, Ferro VA. Analysis of the binding loops configuration and surface adaptation of different crystallized single-domain antibodies in response to various antigens. J Mol Recognit. 2017:30 (4). doi: 10.1002/jmr.2592
10. Zavrtanik U, Lukan J, Loris R, Lah J, Hadzi S. Structural Basis of Epitope Recognition by Heavy-Chain Camelid Antibodies. J Mol Biol. 2018;430(21):4369-86
11. Nguyen VK, Su C, Muyldermans S, van der Loo W. Heavy-chain antibodies in Camelidae; a case of evolutionary innovation. Immunogenetics. 2002;54(1):39-47
12. Govaert J, Pellis M, Deschacht N, Vincke C, Conrath K, Muyldermans S. et al. Dual beneficial effect of interloop disulfide bond for single domain antibody fragments. J Biol Chem. 2012;287(3):1970-9
13. Pardon E, Laeremans T, Triest S, Rasmussen SGF, Wohlkonig A, Ruf A. et al. A general protocol for the generation of Nanobodies for structural biology. Nat Protoc. 2014;9(3):674-93
14. Liu W, Song H, Chen Q, Yu J, Xian M, Nian R. et al. Recent advances in the selection and identification of antigen-specific nanobodies. Mol Immunol. 2018;96:37-47
15. Uchanski T, Zogg T, Yin J, Yuan D, Wohlkonig A, Fischer B. et al. An improved yeast surface display platform for the screening of nanobody immune libraries. Sci Rep. 2019;9(1):382. doi: 10.1038/s41598-018-37212-3
16. Egloff P, Zimmermann I, Arnold FM, Hutter CAJ, Morger D, Opitz L. et al. Engineered peptide barcodes for in-depth analyses of binding protein libraries. Nat Methods. 2019;16(5):421-8
17. Makvandi-Nejad S, Fjallman T, Arbabi-Ghahroudi M, MacKenzie CR, Hall JC. Selection and expression of recombinant single domain antibodies from a hyper-immunized library against the hapten azoxystrobin. J Immunol Methods. 2011;373(1-2):8-18
18. Ezzine A, M'hirsi El Adab S, Bouhaouala-Zahar B, Hmila I, Baciou L, Marzouki MN. Efficient expression of the anti-AahI' scorpion toxin nanobody under a new functional form in a Pichia pastoris system. Biotechnol Appl Biochem. 2012;59(1):15-21
19. Gorlani A, de Haard H, Verrips T. Expression of VHHs in Saccharomyces cerevisiae. Methods Mol Biol. 2012;911:277-86
20. Liu Y, Huang H. Expression of single-domain antibody in different systems. Appl Microbiol Biotechnol. 2018;102(2):539-51
21. Janssens R, Dekker S, Hendriks RW, Panayotou G, van Remoortere A, San JK. et al. Generation of heavy-chain-only antibodies in mice. Proc Natl Acad Sci U S A. 2006;103(41):15130-5
22. Moutel S, Bery N, Bernard V, Keller L, Lemesre E, de Marco A. et al. NaLi-H1: A universal synthetic library of humanized nanobodies providing highly functional antibodies and intrabodies. Elife. 2016:5 doi: 10.7554/eLife.16228
23. Monegal A, Ami D, Martinelli C, Huang H, Aliprandi M, Capasso P. et al. Immunological applications of single-domain llama recombinant antibodies isolated from a naive library. Protein Eng Des Sel. 2009;22(4):273-80
24. Goldman ER, Anderson GP, Liu JL, Delehanty JB, Sherwood LJ, Osborn LE. et al. Facile generation of heat-stable antiviral and antitoxin single domain antibodies from a semisynthetic llama library. Anal Chem. 2006;78(24):8245-55
25. Yan J, Li G, Hu Y, Ou W, Wan Y. Construction of a synthetic phage-displayed Nanobody library with CDR3 regions randomized by trinucleotide cassettes for diagnostic applications. J Transl Med. 2014;12:343. doi: 10.1186/s12967-014-0343-6
26. Comor L, Dolinska S, Bhide K, Pulzova L, Jimenez-Munguia I, Bencurova E. et al. Joining the in vitro immunization of alpaca lymphocytes and phage display: rapid and cost effective pipeline for sdAb synthesis. Microb Cell Fact. 2017;16(1):13. doi: 10.1186/s12934-017-0630-z
27. Suzuki T, Mochizuki Y, Kimura S, Akazawa-Ogawa Y, Hagihara Y, Nemoto N. Anti-survivin single-domain antibodies derived from an artificial library including three synthetic random regions by in vitro selection using cDNA display. Biochem Biophys Res Commun. 2018;503(3):2054-60
28. Itoh K, Reis AH, Hayhurst A, Sokol SY. Isolation of nanobodies against Xenopus embryonic antigens using immune and non-immune phage display libraries. PLoS One. 2019;14(5):e0216083
29. van der Linden RH, Frenken LG, de Geus B, Harmsen MM, Ruuls RC, Stok W. et al. Comparison of physical chemical properties of llama VHH antibody fragments and mouse monoclonal antibodies. Biochim Biophys Acta. 1999;1431(1):37-46
30. Chakravarty R, Goel S, Cai W. Nanobody: the “magic bullet” for molecular imaging? Theranostics. 2014;4(4):386-98
31. Muruganandam A, Tanha J, Narang S, Stanimirovic D. Selection of phage-displayed llama single-domain antibodies that transmigrate across human blood-brain barrier endothelium. FASEB J Off Publ Fed Am Soc Exp Biol. 2002;16(2):240-2
32. Abulrob A, Sprong H, Van Bergen en Henegouwen P, Stanimirovic D. The blood-brain barrier transmigrating single domain antibody: mechanisms of transport and antigenic epitopes in human brain endothelial cells. J Neurochem. 2005;95(4):1201-14
33. Li T, Bourgeois J-P, Celli S, Glacial F, Le Sourd A-M, Mecheri S. et al. Cell-penetrating anti-GFAP VHH and corresponding fluorescent fusion protein VHH-GFP spontaneously cross the blood-brain barrier and specifically recognize astrocytes: application to brain imaging. FASEB J Off Publ Fed Am Soc Exp Biol. 2012;26(10):3969-79
34. Danquah W, Meyer-Schwesinger C, Rissiek B, Pinto C, Serracant-Prat A, Amadi M. et al. Nanobodies that block gating of the P2X7 ion channel ameliorate inflammation. Sci Transl Med. 2016;8(366):366ra162
35. Bradley ME, Dombrecht B, Manini J, Willis J, Vlerick D, De Taeye S. et al. Potent and efficacious inhibition of CXCR2 signaling by biparatopic nanobodies combining two distinct modes of action. Mol Pharmacol. 2015;87(2):251-62
36. Manglik A, Kobilka BK, Steyaert J. Nanobodies to Study G Protein-Coupled Receptor Structure and Function. Annu Rev Pharmacol Toxicol. 2017;57:19-37
37. Cartwright ANR, Griggs J, Davis DM. The immune synapse clears and excludes molecules above a size threshold. Nat Commun. 2014;5:5479. doi: 10.1038/ncomms6479
38. Chanier T, Chames P. Nanobody Engineering: Toward Next Generation Immunotherapies and Immunoimaging of Cancer. Antibodies. 2019;8(1):13. doi: 10.3390/antib8010013
39. Vosjan MJWD, Vercammen J, Kolkman JA, Stigter-van Walsum M, Revets H, van Dongen GAMS. Nanobodies targeting the hepatocyte growth factor: potential new drugs for molecular cancer therapy. Mol Cancer Ther. 2012;11(4):1017-25
40. Krasniqi A, D'Huyvetter M, Devoogdt N, Frejd FY, Sorensen J, Orlova A. et al. Same-Day Imaging Using Small Proteins: Clinical Experience and Translational Prospects in Oncology. J Nucl Med. 2018;59(6):885-91
41. Van Roy M, Ververken C, Beirnaert E, Hoefman S, Kolkman J, Vierboom M. et al. The preclinical pharmacology of the high affinity anti-IL-6R Nanobody(R) ALX-0061 supports its clinical development in rheumatoid arthritis. Arthritis Res Ther. 2015;17:135. doi: 10.1186/s13075-015-0651-0
42. Pan H, Liu J, Deng W, Xing J, Li Q, Wang Z. Site-specific PEGylation of an anti-CEA/CD3 bispecific antibody improves its antitumor efficacy. Int J Nanomedicine. 2018;13:3189-201
43. Verhelle A, Nair N, Everaert I, Van Overbeke W, Supply L, Zwaenepoel O. et al. AAV9 delivered bispecific nanobody attenuates amyloid burden in the gelsolin amyloidosis mouse model. Hum Mol Genet. 2017;26(7):1353-64
44. van de Water JAJM, Bagci-Onder T, Agarwal AS, Wakimoto H, Roovers RC, Zhu Y. et al. Therapeutic stem cells expressing variants of EGFR-specific nanobodies have antitumor effects. Proc Natl Acad Sci U S A. 2012;109(41):16642-7
45. Rothbauer U, Zolghadr K, Tillib S, Nowak D, Schermelleh L, Gahl A. et al. Targeting and tracing antigens in live cells with fluorescent nanobodies. Nat Methods. 2006;3(11):887-9
46. Beghein E, Gettemans J. Nanobody Technology: A Versatile Toolkit for Microscopic Imaging, Protein-Protein Interaction Analysis, and Protein Function Exploration. Front Immunol. 2017;8:771. doi: 10.3389/fimmu.2017.00771
47. Goyvaerts C, Dingemans J, De Groeve K, Heirman C, Van Gulck E, Vanham G. et al. Targeting of human antigen-presenting cell subsets. J Virol. 2013;87(20):11304-8
48. Cire S, Da Rocha S, Yao R, Fisson S, Buchholz CJ, Collins MK. et al. Immunization of mice with lentiviral vectors targeted to MHC class II+ cells is due to preferential transduction of dendritic cells in vivo. PLoS One. 2014;9(7):e101644
49. Goyvaerts C, De Groeve K, Dingemans J, Van Lint S, Robays L, Heirman C. et al. Development of the Nanobody display technology to target lentiviral vectors to antigen-presenting cells. Gene Ther. 2012;19(12):1133-40
50. Fang T, Van Elssen CHMJ, Duarte JN, Guzman JS, Chahal JS, Ling J. et al. Targeted antigen delivery by an anti-class II MHC VHH elicits focused alphaMUC1(Tn) immunity. Chem Sci. 2017;8(8):5591-7
51. Bakhtiari SHA, Rahbarizadeh F, Hasannia S, Ahmadvand D, Iri-Sofla FJ, Rasaee MJ. Anti-MUC1 nanobody can redirect T-body cytotoxic effector function. Hybridoma (Larchmt). 2009;28(2):85-92
52. Hassani M, Hajari Taheri F, Sharifzadeh Z, Arashkia A, Hadjati J, van Weerden WM. et al. Construction of a chimeric antigen receptor bearing a nanobody against prostate a specific membrane antigen in prostate cancer. J Cell Biochem. 2019;120(6):10787-95
53. An N, Hou YN, Zhang QX, Li T, Zhang QL, Fang C. et al. Anti-Multiple Myeloma Activity of Nanobody-Based Anti-CD38 Chimeric Antigen Receptor T Cells. Mol Pharm. 2018;15(10):4577-88
54. Xie YJ, Dougan M, Jailkhani N, Ingram J, Fang T, Kummer L. et al. Nanobody-based CAR T cells that target the tumor microenvironment inhibit the growth of solid tumors in immunocompetent mice. Proc Natl Acad Sci U S A. 2019;116(16):7624-31
55. Goyvaerts C, De Vlaeminck Y, Escors D, Lienenklaus S, Keyaerts M, Raes G. et al. Antigen-presenting cell-targeted lentiviral vectors do not support the development of productive T-cell effector responses: implications for in vivo targeted vaccine delivery. Gene Ther. 2017;24(6):370-5
56. Goyvaerts C, Breckpot K. Pros and Cons of Antigen-Presenting Cell Targeted Tumor Vaccines. J Immunol Res. 2015;2015:785634. doi: 10.1155/2015/785634
57. Goyvaerts C, Kurt DG, Van Lint S, Heirman C, Van Ginderachter JA, De Baetselier P. et al. Immunogenicity of targeted lentivectors. Oncotarget. 2014;5(3):704-15
58. Molgaard K, Harwood SL, Compte M, Merino N, Bonet J, Alvarez-Cienfuegos A. et al. Bispecific light T-cell engagers for gene-based immunotherapy of epidermal growth factor receptor (EGFR)-positive malignancies. Cancer Immunol Immunother. 2018;67(8):1251-60
59. Harwood SL, Alvarez-Cienfuegos A, Nunez-Prado N, Compte M, Hernandez-Perez S, Merino N. et al. ATTACK, a novel bispecific T cell-recruiting antibody with trivalent EGFR binding and monovalent CD3 binding for cancer immunotherapy. Oncoimmunology. 2017;7(1):e1377874
60. Gonzalez C, Chames P, Kerfelec B, Baty D, Robert P, Limozin L. Nanobody-CD16 Catch Bond Reveals NK Cell Mechanosensitivity. Biophys J. 2019;116(8):1516-26
61. Li Y, Zhou C, Li J, Liu J, Lin L, Li L. et al. Single domain based bispecific antibody, Muc1-Bi-1, and its humanized form, Muc1-Bi-2, induce potent cancer cell killing in muc1 positive tumor cells. PLoS One. 2018;13(1):e0191024
62. Behar G, Siberil S, Groulet A, Chames P, Pugniere M, Boix C. et al. Isolation and characterization of anti-FcgammaRIII (CD16) llama single-domain antibodies that activate natural killer cells. Protein Eng Des Sel. 2008;21(1):1-10
63. Li J, Zhou C, Dong B, Zhong H, Chen S, Li Q. et al. Single domain antibody-based bispecific antibody induces potent specific anti-tumor activity. Cancer Biol Ther. 2016;17(12):1231-9
64. Muyldermans S. Nanobodies: natural single-domain antibodies. Annu Rev Biochem. 2013;82:775-97
65. Vincke C, Loris R, Saerens D, Martinez-Rodriguez S, Muyldermans S, Conrath K. General strategy to humanize a camelid single-domain antibody and identification of a universal humanized nanobody scaffold. J Biol Chem. 2009;284(5):3273-84
66. Arbabi-Ghahroudi M. Camelid Single-Domain Antibodies: Historical Perspective and Future Outlook. Front Immunol. 2017;8:1589. doi: 10.3389/fimmu.2017.01589
67. Conrath K, Vincke C, Stijlemans B, Schymkowitz J, Decanniere K, Wyns L. et al. Antigen binding and solubility effects upon the veneering of a camel VHH in framework-2 to mimic a VH. J Mol Biol. 2005;350(1):112-25
68. Papadopoulos KP, Isaacs R, Bilic S, Kentsch K, Huet HA, Hofmann M. et al. Unexpected hepatotoxicity in a phase I study of TAS266, a novel tetravalent agonistic Nanobody(R) targeting the DR5 receptor. Cancer Chemother Pharmacol. 2015;75(5):887-95
69. Holland MC, Wurthner JU, Morley PJ, Birchler MA, Lambert J, Albayaty M. et al. Autoantibodies to variable heavy (VH) chain Ig sequences in humans impact the safety and clinical pharmacology of a VH domain antibody antagonist of TNF-alpha receptor 1. J Clin Immunol. 2013;33(7):1192-203
70. Peyvandi F, Scully M, Kremer Hovinga JA, Cataland S, Knobl P, Wu H. et al. Caplacizumab for Acquired Thrombotic Thrombocytopenic Purpura. N Engl J Med. 2016;374(6):511-22
71. Bartunek J, Barbato E, Heyndrickx G, Vanderheyden M, Wijns W, Holz J-B. Novel antiplatelet agents: ALX-0081, a Nanobody directed towards von Willebrand factor. J Cardiovasc Transl Res. 2013;6(3):355-63
72. Detalle L, Stohr T, Palomo C, Piedra PA, Gilbert BE, Mas V. et al. Generation and Characterization of ALX-0171, a Potent Novel Therapeutic Nanobody for the Treatment of Respiratory Syncytial Virus Infection. Antimicrob Agents Chemother. 2016;60(1):6-13
73. Keyaerts M, Xavier C, Heemskerk J, Devoogdt N, Everaert H, Ackaert C. et al. Phase I Study of 68Ga-HER2-Nanobody for PET/CT Assessment of HER2 Expression in Breast Carcinoma. J Nucl Med. 2016;57(1):27-33
74. Picod A, Coppo P. When targeted therapies alleviate the burden of TPE: The example of immune-mediated TTP. Transfus Apher Sci. 2019;58(3):273-7
75. Awad RM, De Vlaeminck Y, Maebe J, Goyvaerts C, Breckpot K. Turn Back the TIMe: Targeting Tumor Infiltrating Myeloid Cells to Revert Cancer Progression. Front Immunol. 2018;9:1977. doi: 10.3389/fimmu.2018.01977
76. Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M. et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. 2018;24(5):541-50
77. Debie P, Devoogdt N, Hernot S. Targeted Nanobody-Based Molecular Tracers for Nuclear Imaging and Image-Guided Surgery. Antibodies. 2019:8 (1). doi:10.3390/antib8010012
78. Gomes CM, Abrunhosa AJ, Ramos P, Pauwels EKJ. Molecular imaging with SPECT as a tool for drug development. Adv Drug Deliv Rev. 2011;63(7):547-54
79. Ametamey SM, Honer M, Schubiger PA. Molecular imaging with PET. Chem Rev. 2008;108(5):1501-16
80. Hicks RJ, Hofman MS. Is there still a role for SPECT-CT in oncology in the PET-CT era? Nature reviews Clinical oncology. 2012;9:712-20
81. Xavier C, Devoogdt N, Hernot S, Vaneycken I, D'Huyvetter M, De Vos J. et al. Site-specific labeling of his-tagged Nanobodies with (9)(9)mTc: a practical guide. Methods Mol Biol. 2012;911:485-90
82. Vaneycken I, D'huyvetter M, Hernot S, De Vos J, Xavier C, Devoogdt N. et al. Immuno-imaging using nanobodies. Curr Opin Biotechnol. 2011;22(6):877-81
83. Massa S, Vikani N, Betti C, Ballet S, Vanderhaegen S, Steyaert J. et al. Sortase A-mediated site-specific labeling of camelid single-domain antibody-fragments: a versatile strategy for multiple molecular imaging modalities. Contrast Media Mol Imaging. 2016;11(5):328-39
84. Rashidian M, Wang L, Edens JG, Jacobsen JT, Hossain I, Wang Q. et al. Enzyme-Mediated Modification of Single-Domain Antibodies for Imaging Modalities with Different Characteristics. Angew Chem Int Ed Engl. 2016;55(2):528-33
85. Massa S, Xavier C, De Vos J, Caveliers V, Lahoutte T, Muyldermans S. et al. Site-Specific Labeling of Cysteine-Tagged Camelid Single-Domain Antibody-Fragments for Use in Molecular Imaging. Bioconjug Chem. 2014;25(5):979-88
86. Bernardim B, Cal PMSD, Matos MJ, Oliveira BL, Martinez-Saez N, Albuquerque IS. et al. Stoichiometric and irreversible cysteine-selective protein modification using carbonylacrylic reagents. Nat Commun. 2016;7:13128. doi: 10.1038/ncomms13128
87. Vandesquille M, Li T, Po C, Ganneau C, Lenormand P, Dudeffant C. et al. Chemically-defined camelid antibody bioconjugate for the magnetic resonance imaging of Alzheimer's disease. MAbs. 2017;9(6):1016-27
88. Hong G, Antaris AL, Dai H. Near-infrared fluorophores for biomedical imaging. Nat Biomed Eng. 2017;1:10. doi: 10.1038/s41551-016-0010
89. Ntziachristos V, Razansky D. Optical and opto-acoustic imaging. Recent Results Cancer Res. 2013;187:133-50
90. Fan X, Wang L, Guo Y, Tu Z, Li L, Tong H. et al. Ultrasonic Nanobubbles Carrying Anti-PSMA Nanobody: Construction and Application in Prostate Cancer-Targeted Imaging. PLoS One. 2015;10(6):e0127419
91. Hernot S, Unnikrishnan S, Du Z, Shevchenko T, Cosyns B, Broisat A. et al. Nanobody-coupled microbubbles as novel molecular tracer. J Control Release. 2012;158(2):346-53
92. Prantner AM, Yin C, Kamat K, Sharma K, Lowenthal AC, Madrid PB. et al. Molecular Imaging of Mesothelin-Expressing Ovarian Cancer with a Human and Mouse Cross-Reactive Nanobody. Mol Pharm. 2018;15(4):1403-11
93. Xavier C, Vaneycken I, D'huyvetter M, Heemskerk J, Keyaerts M, Vincke C. et al. Synthesis, preclinical validation, dosimetry, and toxicity of 68Ga-NOTA-anti-HER2 Nanobodies for iPET imaging of HER2 receptor expression in cancer. J Nucl Med. 2013;54(5):776-84
94. Pruszynski M, Koumarianou E, Vaidyanathan G, Revets H, Devoogdt N, Lahoutte T. et al. Targeting breast carcinoma with radioiodinated anti-HER2 Nanobody. Nucl Med Biol. 2013;40(1):52-9
95. D'Huyvetter M, Vincke C, Xavier C, Aerts A, Impens N, Baatout S. et al. Targeted radionuclide therapy with A 177Lu-labeled anti-HER2 nanobody. Theranostics. 2014;4(7):708-20
96. Krasniqi A, D'Huyvetter M, Xavier C, Van der Jeught K, Muyldermans S, Van Der Heyden J. et al. Theranostic Radiolabeled Anti-CD20 sdAb for Targeted Radionuclide Therapy of Non-Hodgkin Lymphoma. Mol Cancer Ther. 2017;16(12):2828-39
97. Jailkhani N, Ingram JR, Rashidian M, Rickelt S, Tian C, Mak H. et al. Noninvasive imaging of tumor progression, metastasis, and fibrosis using a nanobody targeting the extracellular matrix. Proc Natl Acad Sci U S A. 2019 doi:10.1073/pnas.1817442116
98. Astrof S, Crowley D, George EL, Fukuda T, Sekiguchi K, Hanahan D. et al. Direct test of potential roles of EIIIA and EIIIB alternatively spliced segments of fibronectin in physiological and tumor angiogenesis. Mol Cell Biol. 2004;24(19):8662-70
99. Uldry E, Faes S, Demartines N, Dormond O. Fine-Tuning Tumor Endothelial Cells to Selectively Kill Cancer. Int J Mol Sci. 2017;18(7):1401. doi::10.3390/ijms18071401
100. Broisat A, Toczek J, Dumas LS, Ahmadi M, Bacot S, Perret P. et al. 99mTc-cAbVCAM1-5 imaging is a sensitive and reproducible tool for the detection of inflamed atherosclerotic lesions in mice. J Nucl Med. 2014;55(10):1678-84
101. Bala G, Blykers A, Xavier C, Descamps B, Broisat A, Ghezzi C. et al. Targeting of vascular cell adhesion molecule-1 by 18F-labelled nanobodies for PET/CT imaging of inflamed atherosclerotic plaques. Eur Heart J Cardiovasc Imaging. 2016;17(9):1001-8
102. Ding Y-B, Chen G-Y, Xia J-G, Zang X-W, Yang H-Y, Yang L. Association of VCAM-1 overexpression with oncogenesis, tumor angiogenesis and metastasis of gastric carcinoma. World J Gastroenterol. 2003;9(7):1409-14
103. Scalici JM, Arapovic S, Saks EJ, Atkins KA, Petroni G, Duska LR. et al. Mesothelium expression of vascular cell adhesion molecule-1 (VCAM-1) is associated with an unfavorable prognosis in epithelial ovarian cancer (EOC). Cancer. 2017;123(6):977-84
104. Lin K-Y, Lu D, Hung C-F, Peng S, Huang L, Jie C. et al. Ectopic expression of vascular cell adhesion molecule-1 as a new mechanism for tumor immune evasion. Cancer Res. 2007;67(4):1832-41
105. Wu T-C. The role of vascular cell adhesion molecule-1 in tumor immune evasion. Cancer Res. 2007;67(13):6003-6
106. Rashidian M, Ingram JR, Dougan M, Dongre A, Whang KA, LeGall C. et al. Predicting the response to CTLA-4 blockade by longitudinal noninvasive monitoring of CD8 T cells. J Exp Med. 2017;214(8):2243-55
107. Ingram JR, Blomberg OS, Rashidian M, Ali L, Garforth S, Fedorov E. et al. Anti-CTLA-4 therapy requires an Fc domain for efficacy. Proc Natl Acad Sci U S A. 2018;115(15):3912-7
108. Broos K, Lecocq Q, Raes G, Devoogdt N, Keyaerts M, Breckpot K. Noninvasive imaging of the PD-1:PD-L1 immune checkpoint: Embracing nuclear medicine for the benefit of personalized immunotherapy. Theranostics. 2018;8(13):3559-70
109. Broos K, Keyaerts M, Lecocq Q, Renmans D, Nguyen T, Escors D. et al. Non-invasive assessment of murine PD-L1 levels in syngeneic tumor models by nuclear imaging with nanobody tracers. Oncotarget. 2017;8(26):41932-46
110. Xing Y, Chand G, Liu C, Cook GJR, O' Doherty J, Zhao L. et al. Early phase I study of a (99m)Tc labeled anti-PD-L1 single domain antibody in SPECT/CT assessment of programmed death ligand-1 expression in non-small cell lung cancer. J Nucl Med. 2019 doi: 10.2967/jnumed.118.224170
111. Broos K, Lecocq Q, Xavier C, Bridoux J, Nguyen TT, Corthals J. et al. Evaluating a Single Domain Antibody Targeting Human PD-L1 as a Nuclear Imaging and Therapeutic Agent. Cancers (Basel). 2019:11 (6). doi: 10.3390/cancers11060872
112. Li D, Cheng S, Zou S, Zhu D, Zhu T, Wang P. et al. Immuno-PET Imaging of (89)Zr Labeled Anti-PD-L1 Domain Antibody. Mol Pharm. 2018;15(4):1674-81
113. Bolli E, Movahedi K, Laoui D, Van Ginderachter JA. Novel insights in the regulation and function of macrophages in the tumor microenvironment. Curr Opin Oncol. 2017;29(1):55-61
114. Movahedi K, Schoonooghe S, Laoui D, Houbracken I, Waelput W, Breckpot K. et al. Nanobody-based targeting of the macrophage mannose receptor for effective in vivo imaging of tumor-associated macrophages. Cancer Res. 2012;72(16):4165-77
115. Blykers A, Schoonooghe S, Xavier C, D'hoe K, Laoui D, D'Huyvetter M. et al. PET Imaging of Macrophage Mannose Receptor-Expressing Macrophages in Tumor Stroma Using 18F-Radiolabeled Camelid Single-Domain Antibody Fragments. J Nucl Med. 2015;56(8):1265-71
116. Xavier C, Blykers A, Laoui D, Bolli E, Vaneyken I, Bridoux J. et al. Clinical Translation of [(68)Ga]Ga-NOTA-anti-MMR-sdAb for PET/CT Imaging of Protumorigenic Macrophages. Mol imaging Biol MIB Off Publ Acad Mol Imaging. 2019 doi: 10.1007/s11307-018-01302-5
117. Van Elssen CHMJ, Rashidian M, Vrbanac V, Wucherpfennig KW, Habre Z El, Sticht J. et al. Noninvasive Imaging of Human Immune Responses in a Human Xenograft Model of Graft-Versus-Host Disease. J Nucl Med. 2017;58(6):1003-8
118. Rashidian M, Keliher E, Dougan M, Juras PK, Cavallari M, Wojtkiewicz GR. et al. The use of (18)F-2-fluorodeoxyglucose (FDG) to label antibody fragments for immuno-PET of pancreatic cancer. ACS Cent Sci. 2015;1(3):142-7
119. De Groeve K, Deschacht N, De Koninck C, Caveliers V, Lahoutte T, Devoogdt N. et al. Nanobodies as tools for in vivo imaging of specific immune cell types. J Nucl Med. 2010;51(5):782-9
120. Murgaski A, Bardet PMR, Arnouk SM, Clappaert EJ, Laoui D. Unleashing Tumour-Dendritic Cells to Fight Cancer by Tackling Their Three A's: Abundance, Activation and Antigen-Delivery. Cancers (Basel). 2019:11 (5). doi: 10.3390/cancers11050670
121. Bannas P, Hambach J, Koch-Nolte F. Nanobodies and Nanobody-Based Human Heavy Chain Antibodies As Antitumor Therapeutics. Front Immunol. 2017;8:1603. doi: 10.3389/fimmu.2017.01603
122. Roovers RC, Vosjan MJWD, Laeremans T, el Khoulati R, de Bruin RCG, Ferguson KM. et al. A biparatopic anti-EGFR nanobody efficiently inhibits solid tumour growth. Int J cancer. 2011;129(8):2013-24
123. Gschwind A, Ott RG, Boucneau J, Buyse M-A, Depla E USAP. Bispecific Binding Molecules Binding to VEGF and Ang2. US Patents US 20170247475 A1. 2017
124. Kazemi-Lomedasht F, Behdani M, Bagheri KP, Habibi-Anbouhi M, Abolhassani M, Arezumand R. et al. Inhibition of angiogenesis in human endothelial cell using VEGF specific nanobody. Mol Immunol. 2015;65(1):58-67
125. Farajpour Z, Rahbarizadeh F, Kazemi B, Ahmadvand D. A nanobody directed to a functional epitope on VEGF, as a novel strategy for cancer treatment. Biochem Biophys Res Commun. 2014;446(1):132-6
126. Ebrahimizadeh W, Mousavi Gargari SLM, Javidan Z, Rajabibazl M. Production of Novel VHH Nanobody Inhibiting Angiogenesis by Targeting Binding Site of VEGF. Appl Biochem Biotechnol. 2015;176(7):1985-95
127. Yu Y, Li J, Zhu X, Tang X, Bao Y, Sun X. et al. Humanized CD7 nanobody-based immunotoxins exhibit promising anti-T-cell acute lymphoblastic leukemia potential. Int J Nanomedicine. 2017;12:1969-83
128. Behdani M, Zeinali S, Karimipour M, Khanahmad H, Schoonooghe S, Aslemarz A. et al. Development of VEGFR2-specific Nanobody Pseudomonas exotoxin A conjugated to provide efficient inhibition of tumor cell growth. N Biotechnol. 2013;30(2):205-9
129. Tang J, Li J, Zhu X, Yu Y, Chen D, Yuan L. et al. Novel CD7-specific nanobody-based immunotoxins potently enhanced apoptosis of CD7-positive malignant cells. Oncotarget. 2016;7(23):34070-83
130. Li T, Qi S, Unger M, Hou YN, Deng QW, Liu J. et al. Immuno-targeting the multifunctional CD38 using nanobody. Sci Rep. 2016;6:27055. doi: 10.1038/srep27055
131. Huang H, Wu T, Shi H, Wu Y, Yang H, Zhong K. et al. Modular design of nanobody-drug conjugates for targeted-delivery of platinum anticancer drugs with an MRI contrast agent. Chem Commun (Camb). 2019;55(35):5175-8
132. Stenton BJ, Oliveira BL, Matos MJ, Sinatra L, Bernardes GJL. A thioether-directed palladium-cleavable linker for targeted bioorthogonal drug decaging. Chem Sci. 2018;9(17):4185-9
133. Talelli M, Oliveira S, Rijcken CJF, Pieters EHE, Etrych T, Ulbrich K. et al. Intrinsically active nanobody-modified polymeric micelles for tumor-targeted combination therapy. Biomaterials. 2013;34(4):1255-60
134. van der Meel R, Oliveira S, Altintas I, Haselberg R, van der Veeken J, Roovers RC. et al. Tumor-targeted Nanobullets: Anti-EGFR nanobody-liposomes loaded with anti-IGF-1R kinase inhibitor for cancer treatment. J Control Release. 2012;159(2):281-9
135. van der Meel R, Oliveira S, Altintas I, Heukers R, Pieters EHE, van Bergen en Henegouwen PMP. et al. Inhibition of tumor growth by targeted anti-EGFR/IGF-1R nanobullets depends on efficient blocking of cell survival pathways. Mol Pharm. 2013;10(10):3717-27
136. Zou T, Dembele F, Beugnet A, Sengmanivong L, Trepout S, Marco S. et al. Nanobody-functionalized PEG-b-PCL polymersomes and their targeting study. J Biotechnol. 2015;214:147-55
137. Sadeqzadeh E, Rahbarizadeh F, Ahmadvand D, Rasaee MJ, Parhamifar L, Moghimi SM. Combined MUC1-specific nanobody-tagged PEG-polyethylenimine polyplex targeting and transcriptional targeting of tBid transgene for directed killing of MUC1 over-expressing tumour cells. J Control Release. 2011;156(1):85-91
138. Oliveira S, Schiffelers RM, van der Veeken J, van der Meel R, Vongpromek R, van Bergen En Henegouwen PMP. et al. Downregulation of EGFR by a novel multivalent nanobody-liposome platform. J Control Release. 2010;145(2):165-75
139. van Driel PBAA, Boonstra MC, Slooter MD, Heukers R, Stammes MA, Snoeks TJA. et al. EGFR targeted nanobody-photosensitizer conjugates for photodynamic therapy in a pre-clinical model of head and neck cancer. J Control Release. 2016;229:93-105
140. Heukers R, van Bergen en Henegouwen PMP, Oliveira S. Nanobody-photosensitizer conjugates for targeted photodynamic therapy. Nanomedicine. 2014;10(7):1441-51
141. van Lith SAM, van den Brand D, Wallbrecher R, Wubbeke L, van Duijnhoven SMJ, Makinen PI. et al. The effect of subcellular localization on the efficiency of EGFR-targeted VHH photosensitizer conjugates. Eur J Pharm Biopharm. 2018;124:63-72
142. Van de Broek B, Devoogdt N, D'Hollander A, Gijs H-L, Jans K, Lagae L. et al. Specific cell targeting with nanobody conjugated branched gold nanoparticles for photothermal therapy. ACS Nano. 2011;5(6):4319-28
143. D'Hollander A, Jans H, Velde G Vande, Verstraete C, Massa S, Devoogdt N. et al. Limiting the protein corona: A successful strategy for in vivo active targeting of anti-HER2 nanobody-functionalized nanostars. Biomaterials. 2017;123:15-23
144. Lemaire M, D'Huyvetter M, Lahoutte T, Van Valckenborgh E, Menu E, De Bruyne E. et al. Imaging and radioimmunotherapy of multiple myeloma with anti-idiotypic Nanobodies. Leukemia. England. 2014;28:444-7
145. D'Huyvetter M, De Vos J, Xavier C, Pruszynski M, Sterckx YGJ, Massa S. et al. (131)I-labeled Anti-HER2 Camelid sdAb as a Theranostic Tool in Cancer Treatment. Clin Cancer Res. 2017;23(21):6616-28
146. Dekempeneer Y, Keyaerts M, Krasniqi A, Puttemans J, Muyldermans S, Lahoutte T. et al. Targeted alpha therapy using short-lived alpha-particles and the promise of nanobodies as targeting vehicle. Expert Opin Biol Ther. 2016;16(8):1035-47
147. Pruszynski M, D'Huyvetter M, Bruchertseifer F, Morgenstern A, Lahoutte T. Evaluation of an Anti-HER2 Nanobody Labeled with (225)Ac for Targeted alpha-Particle Therapy of Cancer. Mol Pharm. 2018;15(4):1457-66
148. Choi J, Vaidyanathan G, Koumarianou E, Kang CM, Zalutsky MR. Astatine-211 labeled anti-HER2 5F7 single domain antibody fragment conjugates: radiolabeling and preliminary evaluation. Nucl Med Biol. 2018;56:10-20
149. Nonnekens J, Chatalic KLS, Molkenboer-Kuenen JDM, Beerens CEMT, Bruchertseifer F, Morgenstern A. et al. (213)Bi-Labeled Prostate-Specific Membrane Antigen-Targeting Agents Induce DNA Double-Strand Breaks in Prostate Cancer Xenografts. Cancer Biother Radiopharm. 2017;32(2):67-73
150. Cortez-Retamozo V, Backmann N, Senter PD, Wernery U, De Baetselier P, Muyldermans S. et al. Efficient cancer therapy with a nanobody-based conjugate. Cancer Res. 2004;64(8):2853-7
151. Tian B, Wong WY, Hegmann E, Gaspar K, Kumar P, Chao H. Production and characterization of a camelid single domain antibody-urease enzyme conjugate for the treatment of cancer. Bioconjug Chem. 2015;26(6):1144-55
152. Shrader M, Pino MS, Lashinger L, Bar-Eli M, Adam L, Dinney CPN. et al. Gefitinib reverses TRAIL resistance in human bladder cancer cell lines via inhibition of AKT-mediated X-linked inhibitor of apoptosis protein expression. Cancer Res. 2007;67(4):1430-5
153. Zhu Y, Bassoff N, Reinshagen C, Bhere D, Nowicki MO, Lawler SE. et al. Bi-specific molecule against EGFR and death receptors simultaneously targets proliferation and death pathways in tumors. Sci Rep. 2017;7(1):2602. doi: 10.1038/s41598-017-02483-9
154. Deepa K, Singha S, Panda T. Doxorubicin nanoconjugates. J Nanosci Nanotechnol. 2014;14(1):892-904
155. Talelli M, Rijcken CJF, Oliveira S, van der Meel R, van Bergen En Henegouwen PMP, Lammers T. et al. Nanobody-shell functionalized thermosensitive core-crosslinked polymeric micelles for active drug targeting. J Control Release. 2011;151(2):183-92
156. Heukers R, Altintas I, Raghoenath S, De Zan E, Pepermans R, Roovers RC. et al. Targeting hepatocyte growth factor receptor (Met) positive tumor cells using internalizing nanobody-decorated albumin nanoparticles. Biomaterials. 2014;35(1):601-10
157. Huet HA, Growney JD, Johnson JA, Li J, Bilic S, Ostrom L. et al. Multivalent nanobodies targeting death receptor 5 elicit superior tumor cell killing through efficient caspase induction. MAbs. 2014;6(6):1560-70
158. van der Veeken J, Oliveira S, Schiffelers RM, Storm G, van Bergen En Henegouwen PMP, Roovers RC. Crosstalk between epidermal growth factor receptor- and insulin-like growth factor-1 receptor signaling: implications for cancer therapy. Curr Cancer Drug Targets. 2009;9(6):748-60
159. Goyvaerts C, Breckpot K. The Journey of in vivo Virus Engineered Dendritic Cells From Bench to Bedside: A Bumpy Road. Front Immunol. 2018;9:2052. doi: 10.3389/fimmu.2018.02052
160. Garg AD, Vara Perez M, Schaaf M, Agostinis P, Zitvogel L, Kroemer G. et al. Trial watch: Dendritic cell-based anticancer immunotherapy. Oncoimmunology. 2017;6(7):e1328341
161. Duarte JN, Cragnolini JJ, Swee LK, Bilate AM, Bader J, Ingram JR. et al. Generation of Immunity against Pathogens via Single-Domain Antibody-Antigen Constructs. J Immunol. 2016;197(12):4838-47
162. Lameris R, de Bruin RCG, van Bergen En Henegouwen PMP, Verheul HM, Zweegman S, de Gruijl TD. et al. Generation and characterization of CD1d-specific single-domain antibodies with distinct functional features. Immunology. 2016;149(1):111-21
163. Garcin G, Paul F, Staufenbiel M, Bordat Y, Van der Heyden J, Wilmes S. et al. High efficiency cell-specific targeting of cytokine activity. Nat Commun. 2014;5:3016. doi: 10.1038/ncomms4016
164. Tullett KM, Lahoud MH, Radford KJ. Harnessing Human Cross-Presenting CLEC9A(+)XCR1(+) Dendritic Cells for Immunotherapy. Front Immunol. 2014;5:239. doi: 10.3389/fimmu.2014.00239
165. Sharma PK, Dmitriev IP, Kashentseva EA, Raes G, Li L, Kim SW. et al. Development of an adenovirus vector vaccine platform for targeting dendritic cells. Cancer Gene Ther. 2018;25(1-2):27-38
166. Van der Jeught K, Bialkowski L, Daszkiewicz L, Broos K, Goyvaerts C, Renmans D. et al. Targeting the tumor microenvironment to enhance antitumor immune responses. Oncotarget. 2015;6(3):1359-81
167. Cronin J, Zhang X-Y, Reiser J. Altering the tropism of lentiviral vectors through pseudotyping. Curr Gene Ther. 2005;5(4):387-98
168. Breckpot K, Escors D, Arce F, Lopes L, Karwacz K, Van Lint S. et al. HIV-1 lentiviral vector immunogenicity is mediated by Toll-like receptor 3 (TLR3) and TLR7. J Virol. 2010;84(11):5627-36
169. Cauwels A, Van Lint S, Paul F, Garcin G, De Koker S, Van Parys A. et al. Delivering Type I Interferon to Dendritic Cells Empowers Tumor Eradication and Immune Combination Treatments. Cancer Res. 2018;78(2):463-74
170. Bae E-A, Seo H, Kim I-K, Jeon I, Kang C-Y. Roles of NKT cells in cancer immunotherapy. Arch Pharm Res. 2019;42(7):543-8
171. McLellan AD, Sorg R V, Williams LA, Hart DN. Human dendritic cells activate T lymphocytes via a CD40: CD40 ligand-dependent pathway. Eur J Immunol. 1996;26(6):1204-10
172. Aerts-Toegaert C, Heirman C, Tuyaerts S, Corthals J, Aerts JL, Bonehill A. et al. CD83 expression on dendritic cells and T cells: correlation with effective immune responses. Eur J Immunol. 2007;37(3):686-95
173. Liu Y, Wang Y, Xing J, Li Y, Liu J, Wang Z. A novel multifunctional anti-CEA-IL15 molecule displays potent antitumor activities. Drug Des Devel Ther. 2018;12:2645-54
174. Dougan M, Ingram JR, Jeong H-J, Mosaheb MM, Bruck PT, Ali L. et al. Targeting Cytokine Therapy to the Pancreatic Tumor Microenvironment Using PD-L1-Specific VHHs. Cancer Immunol Res. 2018;6(4):389-401
175. Compte M, Harwood SL, Munoz IG, Navarro R, Zonca M, Perez-Chacon G. et al. A tumor-targeted trimeric 4-1BB-agonistic antibody induces potent anti-tumor immunity without systemic toxicity. Nat Commun. 2018;9(1):4809. doi: 10.1038/s41467-018-07195-w
176. de Bruin RCG, Veluchamy JP, Lougheed SM, Schneiders FL, Lopez-Lastra S, Lameris R. et al. A bispecific nanobody approach to leverage the potent and widely applicable tumor cytolytic capacity of Vgamma9Vdelta2-T cells. Oncoimmunology. 2017;7(1):e1375641
177. Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci U S A. 1989;86(24):10024-8
178. Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci U S A. 1993;90(2):720-4
179. Lee Y-H, Kim CH. Evolution of chimeric antigen receptor (CAR) T cell therapy: current status and future perspectives. Arch Pharm Res. 2019;42(7):607-16
180. van der Stegen SJC, Hamieh M, Sadelain M. The pharmacology of second-generation chimeric antigen receptors. Nat Rev Drug Discov. 2015;14(7):499-509
181. Hege KM, Bergsland EK, Fisher GA, Nemunaitis JJ, Warren RS, McArthur JG. et al. Safety, tumor trafficking and immunogenicity of chimeric antigen receptor (CAR)-T cells specific for TAG-72 in colorectal cancer. J Immunother cancer. 2017;5:22. doi: 10.1186/s40425-017-0222-9
182. Teng MWL, Kershaw MH, Jackson JT, Smyth MJ, Darcy PK. Adoptive transfer of chimeric FcepsilonRI gene-modified human T cells for cancer immunotherapy. Hum Gene Ther. 2006;17(11):1134-43
183. Lamers CHJ, Willemsen R, van Elzakker P, van Steenbergen-Langeveld S, Broertjes M, Oosterwijk-Wakka J. et al. Immune responses to transgene and retroviral vector in patients treated with ex vivo-engineered T cells. Blood. 2011;117(1):72-82
184. Nieba L, Honegger A, Krebber C, Pluckthun A. Disrupting the hydrophobic patches at the antibody variable/constant domain interface: improved in vivo folding and physical characterization of an engineered scFv fragment. Protein Eng. 1997;10(4):435-44
185. Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M. et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med. 2015;21(6):581-90
186. Hajari Taheri F, Hassani M, Sharifzadeh Z, Behdani M, Arashkia A, Abolhassani M. T cell engineered with a novel nanobody-based chimeric antigen receptor against VEGFR2 as a candidate for tumor immunotherapy. IUBMB Life. 2019 doi: 10.1002/iub.2019
187. Albert S, Arndt C, Feldmann A, Bergmann R, Bachmann D, Koristka S. et al. A novel nanobody-based target module for retargeting of T lymphocytes to EGFR-expressing cancer cells via the modular UniCAR platform. Oncoimmunology. 2017;6(4):e1287246
188. Bachmann M. The UniCAR system: A modular CAR T cell approach to improve the safety of CAR T cells. Immunol Lett. 2019;211:13-22
189. You F, Wang Y, Jiang L, Zhu X, Chen D, Yuan L. et al. A novel CD7 chimeric antigen receptor-modified NK-92MI cell line targeting T-cell acute lymphoblastic leukemia. Am J Cancer Res. 2019;9(1):64-78
190. Zhao W-H, Liu J, Wang B-Y, Chen Y-X, Cao X-M, Yang Y. et al. A phase 1, open-label study of LCAR-B38M, a chimeric antigen receptor T cell therapy directed against B cell maturation antigen, in patients with relapsed or refractory multiple myeloma. J Hematol Oncol. 2018;11(1):141
191. Xu J, Chen L-J, Yang S-S, Sun Y, Wu W, Liu Y-F. et al. Exploratory trial of a biepitopic CAR T-targeting B cell maturation antigen in relapsed/refractory multiple myeloma. Proc Natl Acad Sci U S A. 2019;116(19):9543-51
192. Saunders KO. Conceptual Approaches to Modulating Antibody Effector Functions and Circulation Half-Life. Front Immunol. 2019;10:1296. doi: 10.3389/fimmu.2019.01296
193. Dong B, Zhou C, He P, Li J, Chen S, Miao J. et al. A novel bispecific antibody, BiSS, with potent anti-cancer activities. Cancer Biol Ther. 2016;17(4):364-70
194. Rozan C, Cornillon A, Petiard C, Chartier M, Behar G, Boix C. et al. Single-domain antibody-based and linker-free bispecific antibodies targeting FcgammaRIII induce potent antitumor activity without recruiting regulatory T cells. Mol Cancer Ther. 2013;12(8):1481-91
195. Turini M, Chames P, Bruhns P, Baty D, Kerfelec B. A FcgammaRIII-engaging bispecific antibody expands the range of HER2-expressing breast tumors eligible to antibody therapy. Oncotarget. 2014;5(14):5304-19
196. Wei SC, Duffy CR, Allison JP. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018;8(9):1069-86
197. Wan R, Liu A, Hou X, Lai Z, Li J, Yang N. et al. Screening and antitumor effect of an antiCTLA4 nanobody. Oncol Rep. 2018;39(2):511-8
198. Broos K, Lecocq Q, De Keersmaecker B, Raes G, Corthals J, Lion E, Thielemans K, Devoogdt N, Keyaerts M, Breckpot K. et al. Single domain antibody-mediated blockade of programmed death-ligand 1 on dendritic cells enhances CD8 T-cell activation and cytokine production. Vaccines. 2019:10 3390/vaccines7030085
199. Zhang F, Wei H, Wang X, Bai Y, Wang P, Wu J. et al. Structural basis of a novel PD-L1 nanobody for immune checkpoint blockade. Cell Discov. 2017;3:17004. doi: 10.1038/celldisc.2017.4
200. Sun X, Yan X, Zhuo W, Gu J, Zuo K, Liu W. et al. PD-L1 Nanobody Competitively Inhibits the Formation of the PD-1/PD-L1 Complex: Comparative Molecular Dynamics Simulations. Int J Mol Sci. 2018;19(7):1984. doi: 10.3390/ijms19071984
201. Ingram JR, Blomberg OS, Sockolosky JT, Ali L, Schmidt FI, Pishesha N. et al. Localized CD47 blockade enhances immunotherapy for murine melanoma. Proc Natl Acad Sci U S A. 2017;114(38):10184-9
202. Zhao X, Rong L, Zhao X, Li X, Liu X, Deng J. et al. TNF signaling drives myeloid-derived suppressor cell accumulation. J Clin Invest. 2012;122(11):4094-104
203. Okubo Y, Mera T, Wang L, Faustman DL. Homogeneous expansion of human T-regulatory cells via tumor necrosis factor receptor 2. Sci Rep. 2013;3:3153. doi: 10.1038/srep03153
204. Ji X, Peng Z, Li X, Yan Z, Yang Y, Qiao Z. et al. Neutralization of TNFalpha in tumor with a novel nanobody potentiates paclitaxel-therapy and inhibits metastasis in breast cancer. Cancer Lett. 2017;386:24-34
205. Alupei MC, Licarete E, Patras L, Banciu M. Liposomal simvastatin inhibits tumor growth via targeting tumor-associated macrophages-mediated oxidative stress. Cancer Lett. 2015;356(2 Pt B):946-52
206. Tariq M, Zhang J-Q, Liang G-K, He Q-J, Ding L, Yang B. Gefitinib inhibits M2-like polarization of tumor-associated macrophages in Lewis lung cancer by targeting the STAT6 signaling pathway. Acta Pharmacol Sin. 2017;38(11):1501-11
207. Yin W, Yu X, Kang X, Zhao Y, Zhao P, Jin H. et al. Remodeling Tumor-Associated Macrophages and Neovascularization Overcomes EGFR(T790M) -Associated Drug Resistance by PD-L1 Nanobody-Mediated Codelivery. Small. 2018;14(47):e1802372
208. De Vlaeminck Y, Lecocq Q, Giron P, Heirman C, Geeraerts X, Bolli E. et al. Single-domain antibody fusion proteins can target and shuttle functional proteins into macrophage mannose receptor expressing macrophages. J Control Release. 2019;299:107-20
209. Nuhn L, Bolli E, Massa S, Vandenberghe I, Movahedi K, Devreese B. et al. Targeting Protumoral Tumor-Associated Macrophages with Nanobody-Functionalized Nanogels through Strain Promoted Azide Alkyne Cycloaddition Ligation. Bioconjug Chem. 2018;29(7):2394-405
210. Gilham I. Theranostics - An emerging tool in drug discovery and commercialisation. Available from: https://www.ddw-online.com/precision-medicine/p148484-theranostics-an-emerging-tool-in-drug-discovery-and-commercialisation.html
211. Huang L, Gainkam LOT, Caveliers V, Vanhove C, Keyaerts M, De Baetselier P. et al. SPECT imaging with 99mTc-labeled EGFR-specific nanobody for in vivo monitoring of EGFR expression. Mol imaging Biol MIB Off Publ Acad Mol Imaging. 2008;10(3):167-75
212. Hernot S, van Manen L, Debie P, Mieog JSD, Vahrmeijer AL. Latest developments in molecular tracers for fluorescence image-guided cancer surgery. Lancet Oncol. 2019;20(7):e354-67
213. Fang T, Duarte JN, Ling J, Li Z, Guzman JS, Ploegh HL. Structurally Defined alphaMHC-II Nanobody-Drug Conjugates: A Therapeutic and Imaging System for B-Cell Lymphoma. Angew Chem Int Ed Engl. 2016;55(7):2416-20
214. Cauwels A, Van Lint S, Garcin G, Bultinck J, Paul F, Gerlo S. et al. A safe and highly efficient tumor-targeted type I interferon immunotherapy depends on the tumor microenvironment. Oncoimmunology. 2018;7(3):e1398876
215. Peyrassol X, Laeremans T, Gouwy M, Lahura V, Debulpaep M, Van Damme J. et al. Development by Genetic Immunization of Monovalent Antibodies (Nanobodies) Behaving as Antagonists of the Human ChemR23 Receptor. J Immunol. 2016;196(6):2893-901
216. Fang T, Li R, Li Z, Cho J, Guzman JS, Kamm RD. et al. Remodeling of the Tumor Microenvironment by a Chemokine/Anti-PD-L1 Nanobody Fusion Protein. Mol Pharm. 2019;16(6):2838-44
Author contact
![]() Corresponding authors: Karine Breckpot: Tel: 0032-2-477.45.65, Mail: karine.breckpotbe; Nick Devoogdt: Tel: 0032-2-477.49.91, Mail: ndevoogdbe
Corresponding authors: Karine Breckpot: Tel: 0032-2-477.45.65, Mail: karine.breckpotbe; Nick Devoogdt: Tel: 0032-2-477.49.91, Mail: ndevoogdbe