Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
1. Introduction
2. cGAS-STING signaling pathway...
3. STING-activating drugs
4. STING-activating drug...
5. Indirect STING-activating...
6. Summary and outlook
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(25):7759-7771. doi:10.7150/thno.37574 This issue Cite
Review
STING activation in cancer immunotherapy
Ting Su1,2,*, Yu Zhang1,2,*, Kristoffer Valerie3,4, Xiang-Yang Wang3,5,6, Shuibin Lin1 ![]() , Guizhi Zhu2,3,7
, Guizhi Zhu2,3,7 ![]()
1. Department of Rehabilitation Medicine, Center for Translational Medicine, Precision Medicine Institute, The First Affiliated Hospital, Sun Yat-sen University, Guangzhou, 510080, China
2. Department of Pharmaceutics and Center for Pharmaceutical Engineering and Sciences, School of Pharmacy, Richmond, VA, 23298, USA
3. Massey Cancer Center, Virginia Commonwealth University, Richmond, VA, 23298, USA
4. Department of Radiation Oncology, Virginia Commonwealth University, Richmond, VA, 23298, USA
5. Department of Human and Molecular Genetics, Virginia Commonwealth University, Richmond, VA, 23298, USA.
6. Institute of Molecular Medicine, Virginia Commonwealth University, Richmond, VA, 23298, USA.
7. Institute for Structural Biology, Drug Discovery and Development, Virginia Commonwealth University, Richmond, VA, 23219, USA
*These authors contributed equally to this work
Received 2019-6-13; Accepted 2019-9-2; Published 2019-10-15
Abstract

Cancer immunotherapy modulates and leverages the host immune system to treat cancer. The past decade has witnessed historical advancement of cancer immunotherapy. A myriad of approaches have been explored to elicit or augment anticancer innate immunity and/or adaptive immunity. Recently, activation of stimulator of interferon (IFN) genes (STING), an intracellular receptor residing in the endoplasmic reticulum, has shown great potential to enhance antitumor immunity through the induction of a variety of pro-inflammatory cytokines and chemokines, including type I IFNs. A number of natural and synthetic STING agonists have been discovered or developed, and tested in preclinical models and in the clinic for the immunotherapy of diseases such as cancer and infectious diseases. Cyclic dinucleotides (CDNs), such as cyclic dimeric guanosine monophosphate (c-di-GMP), cyclic dimeric adenosine monophosphate (c-di-AMP), and cyclic GMP-AMP (cGAMP), are a class of STING agonists that can elicit immune responses. However, natural CDNs are hydrophilic small molecules with negative charges and are susceptible to enzymatic degradation, leading to low bioavailability in target tissues yet unwanted toxicities and narrow therapeutic windows. Drug delivery systems, coupled with nucleic acid chemistry, have been exploited to address these challenges. Here, we will discuss the underlying immunological mechanisms and approaches to STING activation, with a focus on the delivery of STING agonists, for cancer immunotherapy.
Keywords: Stimulator of interferon genes (STING), cyclic dinucleotides, cyclic GMP-AMP synthase (cGAS), immunostimulatory adjuvants, drug delivery, cancer immunotherapy
1. Introduction
Stimulator of interferon genes (STING) is a signaling molecule that plays a crucial role in controlling the transcription of many host defense genes, including pro-inflammatory cytokines and chemokines, and type I interferons (IFNs) [1, 2]. STING appears to be a dimer, with 398 and 378 amino acids in humans and mice, respectively. STING is located on the membrane of endoplasmic reticulum (ER) with its C-terminal tail residing in cell cytosol [3]. In early studies, STING was observed to stimulate the transcription of innate immune genes in response to some of invading bacteria, DNA viruses or transfected DNA [1, 2, 4, 5]. Further investigation revealed that STING was strongly activated by cyclic dinucleotides (CDNs), such as cyclic di-GMP (c-di-GMP) and cyclic di-AMP (c-di-AMP), both of which can be secreted by bacteria [6, 7]. Indeed, cytosolic DNA species can also trigger STING signaling following binding to and activating cyclic GMP-AMP synthase (cGAS). Specifically, in the presence of cytosolic double-stranded DNA (dsDNA), the intracellular nucleic acid sensor cGAS uses cytosolic ATP and GTP as substrates to catalyze the production of cyclic GMP-AMP (cGAMP), which has a noncanonical 2ʹ,5ʹ-phosphodiester linkage and/or a canonical 3ʹ,5ʹ linkage (c[G(2ʹ,5ʹ)pA(3ʹ,5ʹ)p]) [8-10]. Upon binding to CDNs, STING translocates from the ER to the Golgi apparatus and further to the perinuclear microsomes or punctuate structures, which in turn recruit the downstream TANK-binding kinase 1 (TBK1) and the transcription factor interferon regulatory factor 3 (IRF3), leading to induction of type I IFNs [11]. Typically, STING is then rapidly degraded, an event that may avoid problems associated with sustained cytokine production (Figure 1A) [12]. In addition, STING is associated with the sensing of aberrant cytosolic DNA species, including self-ssDNA (single-stranded DNA) and dsDNA, to trigger host-defense-related gene expression [13]. Conversely, constitutive STING activation has been linked to autoimmune diseases [14]. For example, some gain-of-function mutations in STING result in constitutive activity and autoinflammatory diseases such as STING-associated vasculopathy [15]. In this article, we will discuss the underlying immunological mechanisms and approaches to activating STING for cancer immunotherapy, with a focus on potential drug delivery systems for STING agonists (Figure 1).
STING activation for cancer immunotherapy. (A) The cGAS-STING signaling pathway that mediate the cytosolic nucleic acid sensing and can be activated to elicit antitumor immune responses for cancer immunotherapy. Reprinted from [16], copyright (2017) Elsevier Ltd.. (B) Representative biomaterials that have been exploited to delivery STING agonists, including CDNs. (C) Schematic description of delivering STING agonists, via intratumoral vaccination or lymphoid vaccination, to elicit innate and adaptive antitumor immune responses. cGAS: cyclic GMP-AMP synthase; CDN: cyclic di-nucleotide; IFN: interferon; STING: stimulator of interferon genes; DC: dendritic cell; TCR: T cell receptor; MHC-I: major histocompatibility complex type I.
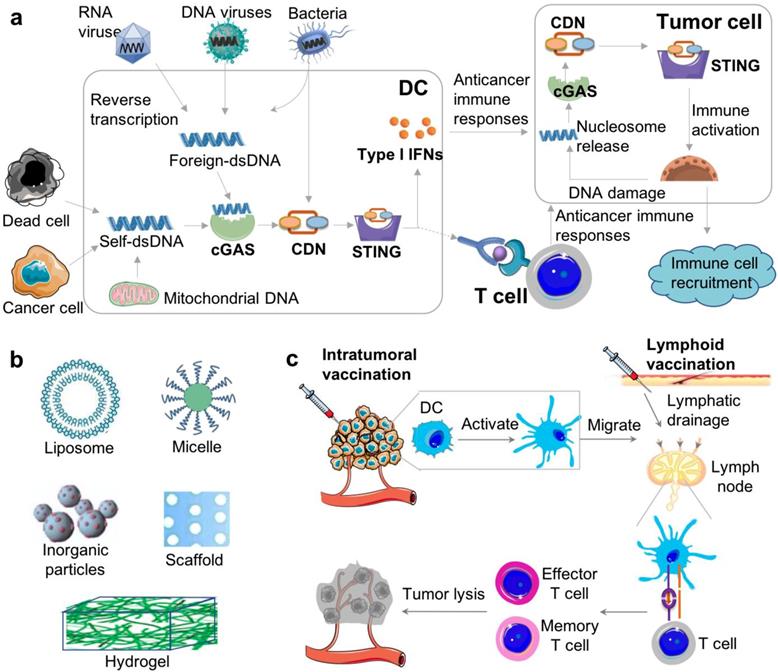
2. cGAS-STING signaling pathway in cancer and cancer immunotherapy
cGAS-STING signaling pathway has the potential to elicit or boost innate and adaptive immune responses, both of which are critical for cancer immunotherapy (Figure 1) [17]. The activation of STING drives the production of cytokines such as Type I IFNs [18]. Type I IFNs belong to a family of cytokines and consist of 16 members, including 12 IFN-α subtypes, IFN-β, IFN-ε, IFN-κ, and IFN-ω, all of which are involved in antiviral immunity [19]. Type I IFNs promote the generation of cytotoxic T cell responses as well as type 1 T helper cell (Th1)-biased responses [20]. Furthermore, type I IFNs promote the activation and functional maturation of dendritic cells (DCs), thereby facilitating antigen presentation to CD4+ T cells as well as antigen cross-presentation to CD8+ T cells [21]. STING activation triggers a multifaceted type I IFN-driven inflammatory response that stimulates DC activation and cross-presentation of tumor antigens for the subsequent T cell priming [22]. Further, recent studies have shown that the STING signaling pathway is essential for endogenous antitumor T cell responses as well as radiation-induced antitumor T cell responses [23, 24]. Consistently, STING-deficient mice have a higher susceptibility to tumor formation, diminished antitumor T cell immunity and impaired responses to immunotherapy [24]. Furthermore, the ability of immune checkpoint inhibitors to reinvigorate antitumor immune responses was also abrogated in STING-deficient mice, indicating a role of STING in the therapeutic efficacy of immune checkpoint inhibitors [25]. One hypothesis for the underlying mechanism is that DCs engulf necrotic tumor cells, and the tumor cell-derived DNA triggers STING signaling in DCs [23, 24, 26, 27]. The resulting type I IFNs, in a paracrine or autocrine manner, may elicit the production of additional cytokines in DCs that facilitate antigen presentation to CD4+ T cells and antigen cross-presentation to CD8+ T cells, thus further potentiating antitumor T cell responses (Figure 1C).
In addition to T cells, the STING signaling pathway can be activated in macrophages, B cells and some other leukocytes [3, 14] to produce type I IFNs. Moreover, the STING signaling pathway can also be triggered in NK cells, which are then primed for the cytotoxic killing of tumor cells [28]. These studies provide the evidences that STING signaling pathway plays a central role in a variety of innate and adaptive immune responses that can be exploited for cancer immunotherapy.
Note that, STING can also be a double-edged sword in cancer development. Cancer cells may resist against the activation of the cGAS-STING pathway. Indeed, low STING signaling activity has been found in multiple types of cancer cells ranging from colorectal carcinoma [29], melanoma [30], to ovarian cancer [31]. STING activation can be suppressed often by genetic mutations and/or direct epigenetic silencing of either STING or cGAS. For example, Kirsten rat sarcoma gene (KRAS)- and LKB1-mutated non-small cell lung cancer cells epigenetically silenced STING and cGAS expression [32]. Consequently, loss of STING-cGAS signaling rendered these cancer cells unable to elicit antitumor immune responses. Moreover, STING activation has been found to promote the proliferation of brain metastatic cells and chemoresistance in breast cancer cells and lung cancer cells [33]. Specifically, brain metastatic cancer cells use astrocyte gap-junctional networks to transfer cGAMP to astrocytes, leading to STING activation in astrocytes and production of inflammatory cytokines. These inflammatory cytokines can activate the STAT1 and NF-κB pathways in brain metastatic cells, thereby supporting tumor growth and chemoresistance. In addition, prolonged IFN-I signaling has been shown to cause immune dysfunction [34]. Overall, the potentially opposing functions of STING activation may influence the balance between anticancer immune responses and the immune escape of cancer [35].
3. STING-activating drugs
Insight into the roles of STING in immunomodulation indicated the potential of STING agonists as cancer therapeutics to activate antitumor immune responses [22]. Small molecule STING-activating immunomodulators have been long studied for the treatment of diseases, including cancer. An early example of STING activator, 5,6‑dimethylxanthenone‑4‑acetic acid (DMXAA) (Figure 2), was investigated as an experimental anticancer immunomodulator [36]. Unfortunately, STING binding and immune activation by DMXAA was only specific for murine STING but not human STING, which is attributed to the unsuccessful clinical translation of DMXAA in human cancer patients [37]. Nonetheless, DMXAA has generated tremendous basic knowledge and highlighted the importance of species selectivity in drug development for human diseases. Indeed, small molecule STING-activating immunomodulators are still appealing for the cancer drug development. For example, Ramanjulu, et al reported the discovery of a small molecule STING agonist that is systemically efficacious to treat tumors in mice [38]. Specifically, they developed a linking strategy to synergize the effect of two symmetry-related amidobenzimidazole (ABZI)-based compounds to create linked ABZIs (diABZIs) (Figure 2), which was empowered with enhanced STING binding affinity and strong antitumor activity.
The chemical structures of representative STING agonists.
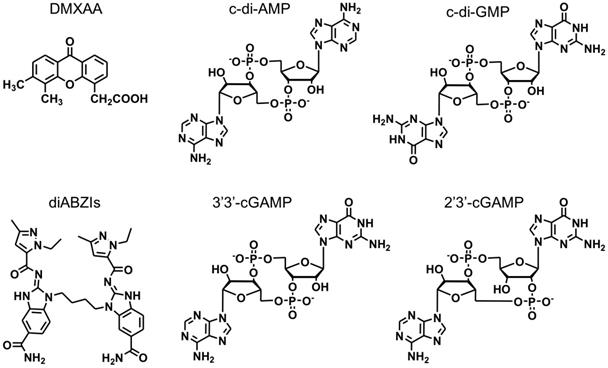
CDNs are another type of STING agonists (Figure 2). CDNs that are derived from bacteria might directly activate the STING signaling pathway. Since the late 1980s, CDNs were recognized as secondary messengers that mediate signaling transduction in prokaryotic cells. In mammalian cells, CDNs function as activators of the innate immune responses [39]. The potential anticancer activity was discovered in CDNs [40], such as c-di-GMP that inhibited the proliferation of human colon cancer cells in vitro. Later, the effect of CDNs on the host immune response was discovered [41-44]. When a model antigen β-galactosidase (β-Gal) was administered subcutaneously with c-di-GMP in vivo, a significant increase in antigen-specific IgG was observed [45]. Cellular immune responses showed that the production of not only IFN-γ, IL-10 and IL-2, but also pro-inflammatory cytokines and chemokines was greatly elevated compared with antigen β-Gal alone [45]. By intraperitoneal injection of c-di-GMP, it was found that high-dose c-di-GMP directly killed tumor cells likely via inducing immunogenic tumor cell death [46]. However, low-dose c-di-GMP improved T-cell responses and significantly reduced immune suppression by converting a subpopulation of immune-suppressing myeloid-derived suppressor cells (MDSCs) into an immune-stimulating phenotype, characterized by the production of IL-12 that can stimulate the activation of T cells [46]. One high-dose treatment followed by multiple low doses of c-di-GMP was equally effective compared with the combination of c-di-GMP and Listeria monocytogenes (LM)-based vaccine that expresses a tumor-associated antigen Mage-b (LM-Mb) [46]. Increasing evidence indicates that targeting the STING signaling pathway in the tumor can be an important approach to remodeling the tumor microenvironment for immunotherapy [22]. 3'3'-cGAMP and 2'3'-cGAMP (Figure 2) are also commonly used CDNs [47, 48]. 2'3'-cGAMP is a natural CDN. In different tumor types such as 4T1 murine breast cancer, HSC-2 squamous cell carcinomas, CT26 murine colon cancer, and B16F10 murine melanoma, intratumoral vaccination with 2'3'-cGAMP led to transient accumulation of macrophages at the tumor site and remodel the tumor immune microenvironment by, for example, repolarizing M2-like tumor-associated macrophages to antitumor M1-type macrophages [48]. In another study, intraperitoneally-injected 3'3'-cGAMP induced apoptosis in malignant B cells through STING activation [47]. Given the ability of STING agonists to elicit potent innate and adaptive immune responses, rational combination of STING agonists with immune checkpoint inhibitors have been explored for cancer immunotherapy. Intratumoral vaccination with STING agonists can potently prime innate immunity and tumor antigen-specific CD8+ T cell responses. CDNs increased tumor-specific CD8+ T cells infiltration and potentiated the therapeutic efficacy of anti-CTLA-4, anti-PD-1, and anti-4-1BB, and reprogrammed suppressive tumor-associated macrophages to a proinflammatory phenotype, namely M1 macrophage [49]. In another example, STING agonists were combined with a PD-L1 inhibitor and an OX40 agonist, resulting in not only effectively activation of innate immunity to support T cell priming, but also overcoming the antigen-enforced immune tolerance for tumor regression [50]. These results indicate the great potential of these STING agonists for versatile applications in tumor immunotherapy.
4. STING-activating drug delivery systems in cancer immunotherapy
As discussed above, small molecule STING agonists as well as cytosolic DNA species can stimulate the STING signaling pathway to promote antigen presentation and T cell priming for tumor eradication [51-54]. However, the intrinsic negative charges, susceptibility to enzymatic degradation, hydrophilicity, as well as small sizes of CDNs pose challenges to the biostability, bioavailability, delivery, and retention of CDNs in target tissues and cells. Drug delivery systems involving biomaterials at a variety of scales (from nanocarriers, microcarriers, to macromaterials) have been engineered to overcome tissue and cell barriers to improve the therapeutic efficacy while ameliorating adverse side effects (Table 1). In general, these delivery systems can be applied under different contexts. Typically, the smaller the drug carriers, the easier for them to be transported via lymphatic drainage which is often need in local vaccination; by comparison, relatively large drug carriers such as large microparticles and hydrogels tend to be retained locally, which may be great for in situ vaccination or intratumoral implantation. Worth noting that, macromaterials such as hydrogels may involve invasive procedure, with the exception of injectable macromaterials. At the tissue level, drug delivery systems have been developed to transport and retain STING agonists in the tissues, such as lymph nodes for lymphoid vaccination, or tumors for intratumoral vaccination. At the cell level, since STING is located in the ER, drug delivery systems are expected to deliver STING agonists across cell membrane and even escape from the endosomal compartment if endocytosis is involved. A variety of such drug delivery systems have been engineered based on nanoparticles [55-58], microparticles [59, 60], as well as macromaterials such as hydrogels [61, 62] (Table 1). In this section, we will discuss the application of STING agonists for cancer immunotherapy, with a focus on drug delivery systems for CDN-based STING agonists.
STING-activating delivery systems for cancer immunotherapies.
| Nanocarriers | Payload CDNs | Tumor models | Administration routes | References | |
|---|---|---|---|---|---|
| Liposome | PEG-containing lipids | 2'3'-cGAMP | Melanoma | Intratumoral | [63] |
| A pH-sensitive cationic lipid (YSK05) | c-di-GMP | Lung metastatic melanoma | Intravenous | [64] | |
| A pH-sensitive cationic lipid (YSK05) | c-di-GMP | T cell lymphoma | Subcutaneous | [65] | |
| PEGylated lipid | c-di-GMP | Lymphoma; Melanoma | Subcutaneous | [58] | |
| Soy-PC-DOTAP liposome | 3'3'-cGAMP | Basal-like triple-negative breast cancers; melanoma | Intravenous | [57] | |
| Polymeric nanoparticles | Poly(beta-amino ester) (PBAE) | ML-RR-CDA | Melanoma | Intratumoral | [66] |
| In situ crosslinked PEG- DBP polymersomes | 2'3'-cGAMP | Melanoma | Intratumoral; intravenous | [56] | |
| Acetalated dextran (Ace-DEX) microparticles | 3'3'-cGAMP | Melanoma | Intraperitoneal; intramuscular; intravenous; intratumoral | [59,67,68] | |
| Ultra-pH-sensitive copolymers | -- | Melanoma | Subcutaneous | [69,70] | |
| Others | Cationic silica nanoparticles (CSiNPs) | c-di-GMP | Melanoma | Intratumoral | [85] |
| Irradiated GM-CSF-secreting whole-cell vaccine | CDN derivative | Melanoma | Subcutaneous | [71] | |
| Lipid-coated silica microsphere | c-di-GMP | Pancreatic cancer | Implants | [72] | |
| LPEI/HA hydrogels | cGAMP | -- | Intratumoral | [62] | |
| Peptide STINGel | ML RR-S2 CDA | Oral cancer cell | Intratumoral | [61] | |
GM-CSF: granulocyte-macrophage colony-stimulating factor; PEG-DBP: poly(ethylene glycol)-block-[(2-(diethylamino)ethyl methacrylate)-co-(butyl methacrylate)-co-(pyridyl disulfide ethyl methacrylate)] copolymers. LPEI: linear poly-ethyleneimine; HA: hyaluronic acid.
4.1. Nanocarrier-based STING-activating delivery
STING-activating drugs can induce profound antitumor immune responses. However, the clinical translation of CDN-based STING agonists can be confronted by drawbacks of CDNs. First, CDNs are susceptible to enzymatic degradation by phosphodiesterases [73]. Second, the hydrophilicity and small sizes (molecular weights lower than 1 kDa) of CDNs facilitate random diffusion and clearance upon typical administration into the body. Third, the negative charges of CDNs refrains CDNs from cell membrane penetration and cell uptake [57, 74]. These drawbacks render CDNs to have poor pharmacokinetics and pharmacodynamics, short half-lives, unwanted systemic dissemination that may further cause toxic cytokine storms, and limited bioavailability. Nucleic acid chemistry has been employed to chemically modify CDNs to improve the biostability of CDNs. For instance, one STING agonist called ADU-S100, also known as ML RR-S2 CDA (dithio-(RP,RP)-[cyclic[A(2',5')pA(3',5')p]), is a phosphodiesterase-resistant CDN that has been tested in the clinic. By chemical modifications, its biostability has been improved which further promotes their antitumor efficacy in a series of cancer cells [75]. To address the complications associated with the hydrophilicity and negative charges of CDNs, cationic and/or encapsulating drug carriers have been exploited to improve the tissue and cell delivery of CDNs, while minimizing systemic toxicity [58, 76]. Injection of unformulated “free” STING agonists may lead to rapid dissemination into the blood and subsequently cause systemic cytokine storm that can be harmful [77, 78]. In one example, c-di-GMP-incorporated nanoparticles elicited 8.2-fold more antigen-specific IgG titers than the “free” c-di-GMP counterpart at the same dose. While elevating the dose of c-di-GMP promoted the production of antibody titer, this is accompanied by the elevated production of systemic inflammatory cytokines such as IL-6, TNF-α, and IFN-β [58]. Nanoparticles smaller than 200 nm in diameters can typically be taken up by the peripheral APCs or drained from interstitial spaces to the lymphatic lumen and then transported to draining lymph nodes [79, 80], which host a variety of immune cells and orchestrate immune responses that are critical for cancer immunotherapy. Typically, nanoparticles with diameters of approximately 50 nm have been found to be especially efficient at uptake and retention in lymph nodes [81, 82]. Thus, rationally-designed nanoparticulate delivery systems hold tremendous potential to promote CDN delivery and advance the application of CDNs as potent immunotherapeutics [58]. A series of CDN delivery systems have been developed using nanocarriers such as liposomes [57, 58, 65, 74, 83], polymeric nanoparticles [55, 59, 68, 84], as well as inorganic materials [72, 85].
Liposomes, which can have positive charges and aqueous cores, are great candidates for encapsulating STING agonists. The positive charge on liposomes can not only promote the encapsulation of negatively-charged CDNs, but can also facilitate intracellular liposome delivery by electrostatically interacting with negatively-charged cell membrane [57, 58, 65, 74, 83]. For example, the encapsulation of c-di-GMP in PEGylated lipid nanoparticles concurrently minimized systemic dissemination and markedly enhanced lymph nodes accumulation compared with free c-di-GMP [58]. When co-delivered with a peptide antigen, the c-di-GMP-delivering nanoparticles increased antigen-specific CD8+ T cell responses. Moreover, the durable antibody titers were substantially higher than those promoted by a TLR4 agonist monophosphoryl lipid A, indicating that the nanoformulation improved the delivery of c-di-GMP and promoted the immune responses of c-di-GMP. This approach implies that the delivery and the cancer therapeutic efficacy of STING agonists such as CDNs can be effectively improved via drug delivery systems based on rationally-designed nanocarriers. Besides functioning as vaccine adjuvants, nanoparticulate STING agonists such as PEGylated liposomes loaded with cGAMP, can also be used to overcome the immunosuppressive tumor microenvironment [74]. When intratumorally administered, this liposomal formulation of cGAMP significantly enhanced the tumor retention of cGAMP and the colocalization of cGAMP with tumor-associated APCs, which may explain the superior type I IFN induction and adaptive immune responses to clear established melanoma and to resist a second tumor challenge. Even in PD-L1-insensitive models of triple-negative breast (TNBC) cancer which has poor prognosis and few effective treatment options, STING agonist-loaded liposomes effectively repolarized M2-like macrophages into M1-type macrophages and elicited STING-dependent antitumor immunity [57]. Note that, these CDN nanoparticles can elicit a potent and durable immune response that prevents relapse [57, 74].
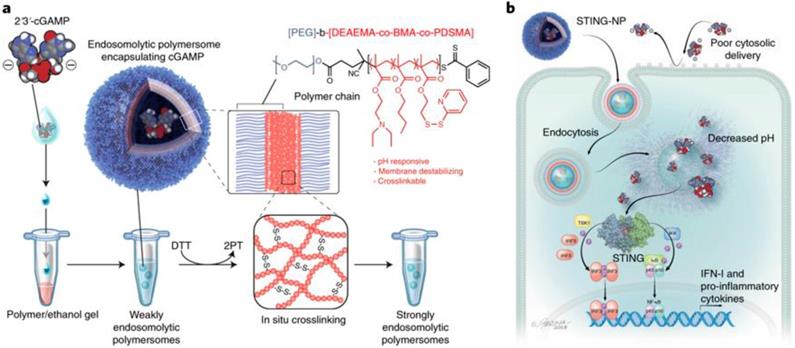
Polymeric nanoparticles represent another promising class of nanocarriers for the delivery of STING agonists such as CDNs for caner immunotherapy [55, 56, 59, 68]. Polymeric nanoparticles can be tailor-designed with defined topological structures, functional modifications, controlled drug loading and release kinetics, as well as good biodegradability and good safety [81, 84, 86-88]. These characteristic features have empowered polymeric nanocarriers to be one of the most successful class of drug nanocarriers. For example, a biodegradable poly(beta-amino ester) (PBAE) cationic polymer was developed to form PBAE/CDN polymeric nanoparticles through electrostatic interaction between positively charged PBAE and negatively charged CDNs [55]. The resulting nanoparticles can be effectively and selectively taken up by monocytes and macrophages, indicating the potential of these CDN nanoparticles as immune adjuvants. This selectivity of uptake might be attributed to the end-capping group in the polymers. When combined with an immune checkpoint inhibitor, these CDN nanoparticles showed an order of magnitude reduction of the dose needed to eliminate established poorly immunogenic melanoma. In another example of polymeric nanoparticles for CDN delivery, pH-responsive polymersomes were designed to load cGAMP [69, 70]. The pH-responsive polymersomes can disassemble in the acidic endolysosome upon endocytosis-mediated cell uptake, allowing conditional cGAMP release from the polymersome carriers (Figure 3) [56]. This polymersome delivery system potentiated the immunostimulatory activity of cGAMP by two to three orders of magnitudes in multiple immune cells in vitro. A single-dose intratumoral administration of such CDN-loaded polymersomes in a mouse melanoma model remodeled the tumor immune milieu, as characterized by increased populations of tumor-infiltrating neutrophils as well as CD8+ and CD4+ T cells, activated DCs indicated by CD86 expression, which altogether reprogram the tumor microenvironment to be 'hot' or T cell-inflamed for efficacious immunotherapy. As a result, when administered intratumorally or intravenously, these cGAMP-loaded polymersomes increased the therapeutic efficacy.
Polymersome-based CDN delivery for cancer immunotherapy. a) Schematic illustration of using pH-responsive diblock copolymers to formulate 2′3′-cGAMP-loaded endosomolytic polymersomes. b) Schematic description of intracellular uptake of the intracellular delivery of 2′3′-cGAMP via polymersomes (STING-NPs), the endosomal release of 2′3′-cGAMP from STING-NPs, and the endosomal escape of 2′3′-cGAMP to the cytosol for STING activation. Reprinted from [56], copyright (2019) Nature Publishing Group.
In addition to liposomes and polymeric nanocarriers, some other types of nanocarriers have been investigated to deliver CDNs for cancer immunotherapy. In one example, cationic silica nanoparticles (CSiNPs), which can induce necrotic tumor cell death, were used to further deliver c-di-GMP and elicit strong antitumor immune responses upon intratumoral vaccination [85].In a melanoma mouse model, it was shown that the STING agonists cooperate with the release of tumor-associated antigens and local inflammation in tumor microenvironment induced by the CSiNPs to enhance the immunotherapeutic efficacy.
4.2. Micromaterial- or macromaterial-based STING-activating drug delivery
Drug delivery systems based on micromaterials, such as microparticles, have also been developed for STING agonists in cancer immunotherapy. For example, acetylated dextran (Ace-DEX) were developed to synthesize polymeric microparticles for cGAMP loading by electrospray [60]. Through a one-step synthesis, the pendant hydroxyl groups of water soluble dextran were converted into acid-sensitive acetal groups. These microparticles demonstrated the potential as a potent vaccine adjuvant to elicit or augment humoral and cellular immune responses including type-I IFN production, antibody production, as well as germinal center B cell and memory T cell responses. Further, the therapeutic efficacy of cGAMP-loaded microparticles was investigated in two murine tumor models. Compared with three clinically-relevant immune-activating drugs (imiquimod, murabutide, and poly(I:C)), intratumorally-administered cGAMP-loaded microparticles generated robust innate and adaptive anti-cancer immune responses and enhanced type-I IFN responses by up to 50 times [59]. In another example, the Ace-DEX microparticles were studied for the co-delivery of cGAMP and R848 (a TLR7/8 agonist), and the resulting Ace-DEX microparticles co-loaded with cGAMP and R848 were found to elicit strong immune responses when administered at extremely low doses [68].
Hydrogel-based micromaterials or macromaterials are another interesting class of delivery platform for STING-activating immunomodulators. For example, submicron-sized microparticulate hydrogels were fabricated from linear poly-ethyleneimine (LPEI)/hyaluronic acid (HA), and were loaded with cGAMPs as vaccine adjuvants [62]. The resulting microgels mediated efficient intracellular delivery via uptake by phagocytic macrophages, leading to enhanced cytokine induction compared with conventional cationic Lipofectamine. In another example, a peptide hydrogel, called STINGel, was developed as an injectable peptide hydrogel that controllably delivered CDNs. STINGel was formulated through the electrostatic interactions between negatively charged CDNs and the positively charged peptide [61]. The controlled release of CDNs from STINGel created a high local CDN concentration that lasted for at least seven days. Such local STING agonist depots around the STINGel can efficiently remodel the tumor immune microenvironment to improve the immunotherapeutic efficacy.
STING-activating drug delivery systems have also been developed to boost the anticancer immune responses in adoptive cell transfer therapy. In one example, a biopolymer scaffold was developed to deliver STING agonists (c-di-GMP) along with chimeric antigen receptor T (CAR-T) cells, and such STING agonist-delivering scaffold was found to prime robust tumor-specific host lymphocyte responses to eliminate local and distant (metastatic) tumors (Figure 4) [72]. Specifically, the implantable scaffold of porous alginate matrices were loaded with mesoporous silica microparticles that were loaded with c-di-GMP, and those silica microparticles were further modified with stimulatory anti-CD3/CD28/CD137 antibodies on their phospholipid membrane to facilitate their interaction with CAR-T cells.
Schematic illustration of an implantable biomaterial scaffold that co-delivered CAR-T cells and CDNs for synergistic tumor immunotherapy. a) The scaffold that was loaded with CAR-T cells and microspheres of STING agonists interact with the tumor bed. b) The Macro- and microscope image of the porous alginate matrices that are functionalized with c-di-GMP-loading mesoporous silica microparticles. Reprinted from [72], copyright (2017) American Society for Clinical Investigation.
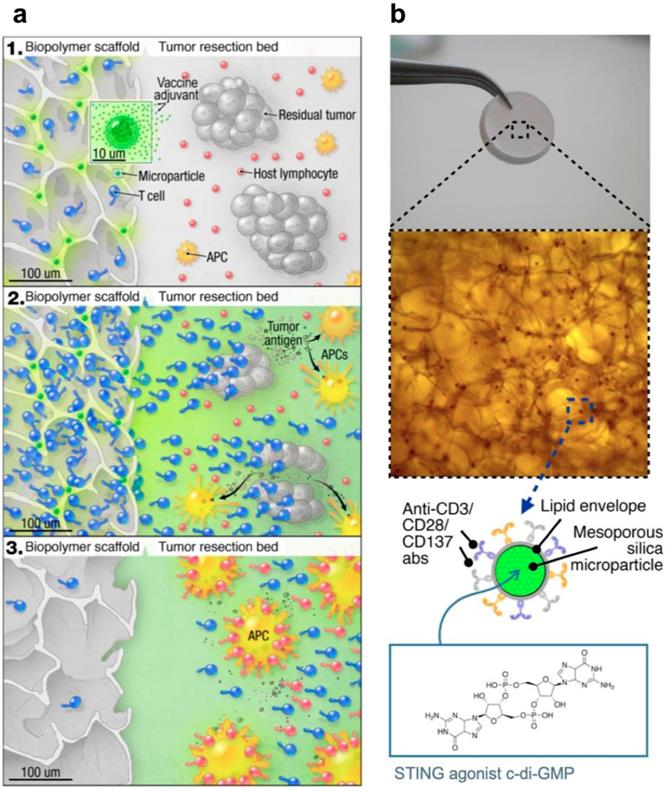
In orthotopic tumor models of inoperable pancreatic cancer and incompletely resected melanoma, this scaffold was directly implanted in the tumor tissues, and the scaffold-mediated CAR-T cell delivery induced tumor regression more effectively when compared to systemic CAR-T cell injection. Armed with STING agonists to remodel the tumor immune milieu, this strategy may provide an effective treatment for solid heterogeneous tumors that have poor responses to conventional T cell therapies. Collectively, these results indicate that rationally-designed micromaterials and macromaterials can be developed for efficient delivery of STING agonists as immunostimulatory adjuvants for versatile combination immunotherapy of cancer.
4.3. STING-activating nanoparticles
In addition to serving as carriers for STING agonists, synthetic materials per se have also been developed to activate the STING signaling pathway [69, 70]. In a recent study, a library of ultra-pH-sensitive copolymers consisting of different tertiary amines was found to activate STING for tumor immunotherapy (Figure 5) [69]. The polymers per se could activate antigen-presenting cells (APCs), especially DCs in draining lymph nodes and stimulate type I IFN production in a STING-dependent manner. When tumor antigens were delivered via these STING-activating nanovaccines, potent and durable antigen-specific T cell responses were elicited, which resulted in robust immunotherapeutic efficacy in multiple murine cancer models. The unique STING activation characteristics of these polymers indicate their potential for application in cancer immunotherapy. In a follow-up study, STING-activating nanoparticles were combined with ionizing radiation [70]. This combination of STING-activating nanovaccine with local radiotherapy reverted the immunosuppressive environment in a STING-dependent manner, leading to synergistic radioimmunotherapy in both primary and metastatic tumors.
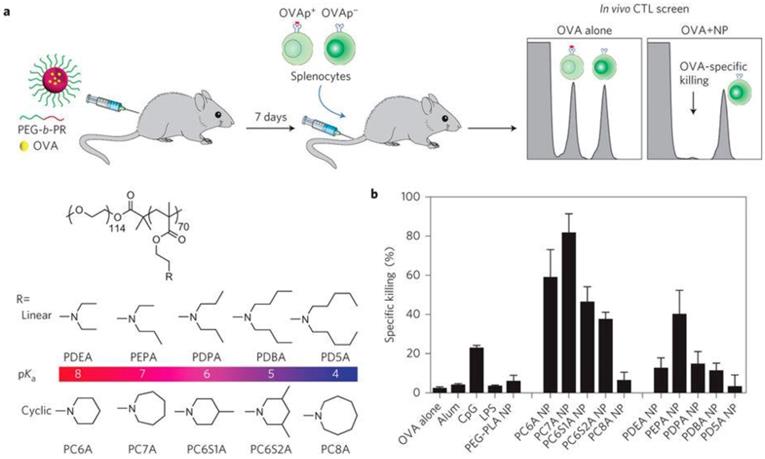
Intrinsically STING-activating nanoparticles for tumor immunotherapy. a) Schematic illustration of a series of polymer nanoparticles that were screened for immunostimulation and the generation of strong antigen specific CTL responses when loaded with a model antigen ovalbumin (OVA). b) Quantitative comparison of antigen specific CTL responses elicited by different polymer nanoparticles. Reprinted from [69] copyright (2017) Nature Publishing Group.
5. Indirect STING-activating therapy
Radiation or some chemotherapeutic drugs can induce immunogenic cell death (ICD). Tumor cell ICD may further induce innate and adaptive antitumor immune responses [89, 90]. For example, irradiation was found to induce the production of type I IFNs and elicit adaptive immune responses to support tumor regression [91]. Among a variety of mechanisms that could induce type I IFN production, Weichselbaum and co-workers found that STING, but not myeloid differentiation primary-response protein 88 (MyD88) or TIR-domain-containing adapter-inducing interferon-β (TRIF), is indispensable for the induction of type I IFNs and promotion of the antitumor effect of radiation [52]. The mechanism of radiation-induced immunostimulation is dose-dependent. When radiation was delivered at a high dose, the induction of three prime repair exonuclease 1 (Trex1) in irradiated cancer cells can degrade the DNA accumulating in the cytosol, which precluded the activation of cGAS-STING-IFN-I pathway and dampens the radiation-induced immunostimulation. By contrast, when radiation was given below the threshold dose for Trex1 induction, cancers cells can be optimally stimulated to produce IFNβ and activate specific DCs, which was essential for the priming of tumor-specific CD8+ T cells [92]. These studies have provided new insight as to the design of radiotherapy or radioimmunotherapy for the optimal treatment outcome.
In addition to radiotherapy, some chemotherapeutic antitumor drugs that interfere with genomic DNA synthesis or induce genomic DNA damage may induce the production of cytosolic DNA, which trigger cGAS-STING signaling pathway and subsequently elicit antitumor immune responses [93, 94]. For example, Topotecan (TPT) can inhibit topoisomerases and trigger DNA double-strand breaks to cause cell death.[68] This process induces the generation of danger-associated molecules, triggers DC activation, and activates a STING-dependent pathway for antitumor cytokine production. Notably, the antitumor effects of TPT decreased in STING-deficient mice, suggesting that type I IFN production was induced through the cGAS-STING axis and that cGAS-STING axis may play important roles in TPT-induced therapeutic efficacy [95]. In another example, hydroxyurea and cisplatin were shown to cause DNA damage in BRCA1-deficient breast tumors, which upregulated the secretion of C-X-C motif chemokine 10 (CXCL10) and chemokine (C-C motif) ligand 5 (CCL5) chemokine in a DNA damage-associated manner involving a STING-TBK1-IRF3 signaling pathway [96]. Recently, a poly(ADP-ribose) polymerase (PARP) inhibitor Olaparib was shown to trigger robust STING-dependent antitumor immune responses in breast cancer type 1 (BRCA1)-deficient ovarian cancer, which induces robust adaptive and innate antitumor immune responses. These results shed lights on the mechanisms of the therapeutic effects of PARP inhibitors in BRCA1-deficient tumors [97]. With accumulating evidence supporting that the effects of chemotherapeutic drugs involve immunostimulatory pathways such as cGAS-STING, it is expected that the delineation of the immune-related signaling pathways will help map out the comprehensive mechanisms of action for these drugs, and guide the rational drug combinations for cancer therapy.
The STING-dependent antitumor immune responses may mediate the therapeutic activity of oncolytic viruses. As biological nanoparticles that have tumor tropism, oncolytic viruses can target multiple steps in the cancer-immunity cycle [98]. These viruses can lyse tumor cells, release tumor antigens (e.g., neoantigens) and danger signals as well as proinflammatory factors such as type I INFs, all of which drive antitumor immune responses. Engineered oncolytic viruses may additionally express cancer therapeutics of interest to drive antitumor immune responses and remodel the tumor immune microenvironment. Following viral infection, viruses can be recognized by pattern recognition receptors (PRRs), such as cGAS and STING. cGAS-STING signaling pathway can sense the genomic elements of viruses, thereby triggering the expression of type I IFNs, the release of chemokines to recruit lymphoid cells that can be leveraged for tumor therapy [98-100].
6. Summary and outlook
The cGAS-STING signaling pathway is a critical process in immune sensing that results in the production of type I IFNs, pro-inflammatory cytokines, and chemokines. The characteristic features of STING activation enable STING to be a potential target for cancer immunotherapy, and STING agonists have been investigated for cancer immunotherapy. Optimal cancer immunotherapeutic efficacy is hinged on the effective delivery of such STING agonists to the desired tissue and cell populations. Specifically, a variety of cell populations in tumor and/or immune tissues such as lymph nodes and spleens have shown the potential as targets for STING activation in cancer immunotherapy. Therefore, at the tissue level, drug delivery systems that enable efficient delivery of STING agonists to tumor and/or lymph nodes have been enthusiastically explored. Given the intracellular location of STING in the ER, STING agonists are expected to penetrate cell membrane to interact with STING. CDNs and CDN derivatives are a representative class of small-molecule STING agonists. Natural CDNs are hydrophilic, negatively charged, and are susceptible to enzymatic degradation, all of which present challenges for the tissue and cell delivery of CDNs in cancer immunotherapy. To address these complications, a variety of CDN delivery systems have been developed to improve their efficacy of cancer immunotherapy. In this article, we have summarized recent progress in the development of biomaterial-based STING agonist delivery systems using nanoparticles or microparticles as well as hydrogels. The therapeutic benefit of these delivery systems in preclinical tumor models has been thus far encouraging and insightful for their clinical translation. Worth noting, STING activation appears to be amenable for versatile evidence-based combination with synergistic therapeutics to further improve cancer therapeutic efficacy. Combinations of STING agonists with immune checkpoint blockade has been under clinical investigation for cancer immunotherapy. Moreover, accumulating evidence suggests that STING activation may be involved, largely via intracellular nucleic acid sensing, in the process of apoptosis, pyroptosis, necroptosis, and autophagy [101]. Given the complexity of the immune modulation network, caution has to be taken in the design of STING-activation-based combination therapy to improve therapeutic outcome while improving or at least not compromising the safety profiles of treatment. Comprehensive delineation of the underlying mechanisms and systematic optimization of immunotherapy involving STING-activating modalities will be intriguing for the design and development of rational combination treatment. Overall, STING activation has shown tremendous potential for cancer immunotherapy, and drug delivery systems can further promote the efficacy of combination cancer immunotherapy.
Acknowledgements
G.Z. acknowledges partial support from the Center for Pharmaceutical Engineering and Sciences - VCU School of Pharmacy, National Institutes of Health (NIH) Clinical and Translational Science Award KL2 Scholarship and Endowment Fund from VCU C. Kenneth and Dianne Wright Center for Clinical and Translational Research (UL1TR002649), American Cancer Society Institutional Research Grants (IRG-18-159-43), Pilot Research Grant from Massey Cancer Center (P30 CA106059), and VCU Presidential Research Quest Fund. X.W. acknowledges support from NIH (CA099326). S.L. was supported by grants from National Natural Science Foundation of China (81772999) and Guangzhou People's Livelihood Science and Technology Project (201903010006). Y.Z. was supported by grant from National Science Foundation for Young Scientists of China (201804038).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455:674-678
2. Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461:788-792
3. Barber GN. STING-dependent cytosolic DNA sensing pathways. Trends Immunol. 2014;35:88-93
4. Jin L, Waterman PM, Jonscher KR, Short CM, Reisdorph NA, Cambier JC. MPYS, a novel membrane tetraspanner, is associated with major histocompatibility complex class II and mediates transduction of apoptotic signals. Mol Cell Biol. 2008;28:5014-5026
5. Zhong B, Yang Y, Li S, Wang YY, Li Y, Diao F. et al. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity. 2008;29:538-550
6. Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M. et al. STING is a direct innate immune sensor of cyclic di-GMP. Nature. 2011;478:515-518
7. Woodward JJ, Iavarone AT, Portnoy DA. c-di-AMP Secreted by Intracellular Listeria monocytogenes Activates a Host Type I Interferon Response. Science. 2010;328:1703-1705
8. Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP Synthase Is a Cytosolic DNA Sensor That Activates the Type I Interferon Pathway. Science. 2013;339:786-791
9. Ablasser A, Goldeck M, Cavlar T, Deimling T, Witte G, Rohl I. et al. cGAS produces a 2'-5'-linked cyclic dinucleotide second messenger that activates STING. Nature. 2013;498:380-384
10. Diner EJ, Burdette DL, Wilson SC, Monroe KM, Kellenberger CA, Hyodo M. et al. The innate immune DNA sensor cGAS produces a noncanonical cyclic dinucleotide that activates human STING. Cell Rep. 2013;3:1355-1361
11. Shu HB, Wang YY. Adding to the STING. Immunity. 2014;41:871-873
12. Konno H, Konno K, Barber GN. Cyclic dinucleotides trigger ULK1 (ATG1) phosphorylation of STING to prevent sustained innate immune signaling. Cell. 2013;155:688-698
13. Abe T, Harashima A, Xia T, Konno H, Konno K, Morales A. et al. STING recognition of cytoplasmic DNA instigates cellular defense. Mol Cell. 2013;50:5-15
14. Barber GN. STING: infection, inflammation and cancer. Nat Rev Immunol. 2015;15:760-770
15. Liu Y, Jesus AA, Marrero B, Yang D, Ramsey SE, Sanchez GAM. et al. Activated STING in a vascular and pulmonary syndrome. N Engl J Med. 2014;371:507-518
16. Ng KW, Marshall EA, Bell JC, Lam WL. cGAS-STING and Cancer: Dichotomous Roles in Tumor Immunity and Development. Trends Immunol. 2018;39:44-54
17. Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. 2013;14:1014-1022
18. Zitvogel L, Galluzzi L, Kepp O, Smyth MJ, Kroemer G. Type I interferons in anticancer immunity. Nat Rev Immunol. 2015;15:405-414
19. Gonzalez-Navajas JM, Lee J, David M, Raz E. Immunomodulatory functions of type I interferons. Nat Rev Immunol. 2012;12:125-135
20. Tough DF. Modulation of T-cell function by type I interferon. Immunol Cell Biol. 2012;90:492-497
21. Longhi MP, Trumpfheller C, Idoyaga J, Caskey M, Matos I, Kluger C. et al. Dendritic cells require a systemic type I interferon response to mature and induce CD4+ Th1 immunity with poly IC as adjuvant. J Exp Med. 2009;206:1589-1602
22. Corrales L, McWhirter SM, Dubensky TW Jr, Gajewski TF. The host STING pathway at the interface of cancer and immunity. J Clin Invest. 2016;126:2404-2411
23. Woo SR, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MY. et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity. 2014;41:830-842
24. Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A. et al. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity. 2014;41:843-852
25. Wang H, Hu S, Chen X, Shi H, Chen C, Sun L. et al. cGAS is essential for the antitumor effect of immune checkpoint blockade. Proc Natl Acad Sci USA. 2017;114:1637-1642
26. Ahn J, Gutman D, Saijo S, Barber GN. STING manifests self DNA-dependent inflammatory disease. Proc Natl Acad Sci USA. 2012;109:19386-19391
27. Klarquist J, Hennies CM, Lehn MA, Reboulet RA, Feau S, Janssen EM. STING-mediated DNA sensing promotes antitumor and autoimmune responses to dying cells. J Immunol. 2014;193:6124-6134
28. Sundararaman SK, Barbie DA. Tumor cGAMP Awakens the Natural Killers. Immunity. 2018;49:585-587
29. Xia T, Konno H, Ahn J, Barber GN. Deregulation of STING Signaling in Colorectal Carcinoma Constrains DNA Damage Responses and Correlates With Tumorigenesis. Cell Rep. 2016;14:282-297
30. Xia T, Konno H, Barber GN. Recurrent Loss of STING Signaling in Melanoma Correlates with Susceptibility to Viral Oncolysis. Cancer Res. 2016;76:6747-6759
31. Queiroz NMGPd, Xia T, Konno H, Barber GN. Ovarian Cancer Cells Commonly Exhibit Defective STING Signaling Which Affects Sensitivity to Viral Oncolysis. Mol Cancer Res. 2019;17:974-986
32. Kitajima S, Ivanova E, Guo S, Yoshida R, Campisi M, Sundararaman SK. et al. Suppression of STING Associated with LKB1 Loss in KRAS-Driven Lung Cancer. Cancer Discov. 2019;9:34-45
33. Chen Q, Boire A, Jin X, Valiente M, Er EE, Lopez-Soto A. et al. Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature. 2016;533:493-498
34. Snell LM, McGaha TL, Brooks DG. Type I Interferon in Chronic Virus Infection and Cancer. Trends Immunol. 2017;38:542-557
35. Benci JL, Xu B, Qiu Y, Wu TJ, Dada H, Twyman-Saint Victor C. et al. Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell. 2016;167:1540-1554.e12
36. Baguley BC, Ching L-M. Immunomodulatory Actions of Xanthenone Anticancer Agents. BioDrugs. 1997;8:119-127
37. Roberts ZJ, Goutagny N, Perera PY, Kato H, Kumar H, Kawai T. et al. The chemotherapeutic agent DMXAA potently and specifically activates the TBK1-IRF-3 signaling axis. J Exp Med. 2007;204:1559-1569
38. Ramanjulu JM, Pesiridis GS, Yang J, Concha N, Singhaus R, Zhang SY. et al. Design of amidobenzimidazole STING receptor agonists with systemic activity. Nature. 2018;564:439-443
39. Danilchanka O, Mekalanos JJ. Cyclic dinucleotides and the innate immune response. Cell. 2013;154:962-970
40. Karaolis DK, Cheng K, Lipsky M, Elnabawi A, Catalano J, Hyodo M. et al. 3',5'-Cyclic diguanylic acid (c-di-GMP) inhibits basal and growth factor-stimulated human colon cancer cell proliferation. Biochem Biophys Res Commun. 2005;329:40-45
41. Karaolis DK, Means TK, Yang D, Takahashi M, Yoshimura T, Muraille E. et al. Bacterial c-di-GMP is an immunostimulatory molecule. J Immunol. 2007;178:2171-2181
42. Ogunniyi AD, Paton JC, Kirby AC, McCullers JA, Cook J, Hyodo M. et al. c-di-GMP is an effective immunomodulator and vaccine adjuvant against pneumococcal infection. Vaccine. 2008;26:4676-4685
43. Karaolis DK, Newstead MW, Zeng X, Hyodo M, Hayakawa Y, Bhan U. et al. Cyclic di-GMP stimulates protective innate immunity in bacterial pneumonia. Infect Immun. 2007;75:4942-4950
44. Dubensky TW Jr, Kanne DB, Leong ML. Rationale, progress and development of vaccines utilizing STING-activating cyclic dinucleotide adjuvants. Ther Adv Vaccines. 2013;1:131-143
45. Ebensen T, Schulze K, Riese P, Link C, Morr M, Guzman CA. The bacterial second messenger cyclic diGMP exhibits potent adjuvant properties. Vaccine. 2007;25:1464-1469
46. Chandra D, Quispe-Tintaya W, Jahangir A, Asafu-Adjei D, Ramos I, Sintim HO. et al. STING ligand c-di-GMP improves cancer vaccination against metastatic breast cancer. Cancer Immunol Res. 2014;2:901-910
47. Tang CH, Zundell JA, Ranatunga S, Lin C, Nefedova Y, Del Valle JR. et al. Agonist-Mediated Activation of STING Induces Apoptosis in Malignant B Cells. Cancer Res. 2016;76:2137-2152
48. Ohkuri T, Kosaka A, Ishibashi K, Kumai T, Hirata Y, Ohara K. et al. Intratumoral administration of cGAMP transiently accumulates potent macrophages for anti-tumor immunity at a mouse tumor site. Cancer Immunol Immunother. 2017;66:705-716
49. Ager CR, Reilley MJ, Nicholas C, Bartkowiak T, Jaiswal AR, Curran MA. Intratumoral STING Activation with T-cell Checkpoint Modulation Generates Systemic Antitumor Immunity. Cancer Immunol Res. 2017;5:676-684
50. Foote JB, Kok M, Leatherman JM, Armstrong TD, Marcinkowski BC, Ojalvo LS. et al. A STING Agonist Given with OX40 Receptor and PD-L1 Modulators Primes Immunity and Reduces Tumor Growth in Tolerized Mice. Cancer Immunol Res. 2017;5:468-479
51. Ahn J, Xia T, Rabasa Capote A, Betancourt D, Barber GN. Extrinsic Phagocyte-Dependent STING Signaling Dictates the Immunogenicity of Dying Cells. Cancer Cell. 2018;33:862-873.e5
52. Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A. et al. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity. 2014;41:843-852
53. Wang H, Hu S, Chen X, Shi H, Chen C, Sun L. et al. cGAS is essential for the antitumor effect of immune checkpoint blockade. Proc Natl Acad Sci USA. 2017;114:1637-1642
54. Woo S-R, Fuertes Mercedes B, Corrales L, Spranger S, Furdyna Michael J, Leung Michael YK. et al. STING-Dependent Cytosolic DNA Sensing Mediates Innate Immune Recognition of Immunogenic Tumors. Immunity. 2014;41:830-842
55. Wilson DR, Sen R, Sunshine JC, Pardoll DM, Green JJ, Kim YJ. Biodegradable STING agonist nanoparticles for enhanced cancer immunotherapy. Nanomed - Nanotechnol. 2018;14:237-246
56. Shae D, Becker KW, Christov P, Yun DS, Lytton-Jean AKR, Sevimli S. et al. Endosomolytic polymersomes increase the activity of cyclic dinucleotide STING agonists to enhance cancer immunotherapy. Nat Nanotechnol. 2019;14:269-278
57. Cheng N, Watkins-Schulz R, Junkins RD, David CN, Johnson BM, Montgomery SA. et al. A nanoparticle-incorporated STING activator enhances antitumor immunity in PD-L1-insensitive models of triple-negative breast cancer. JCI Insight. 2018;3(22):e120638
58. Hanson MC, Crespo MP, Abraham W, Moynihan KD, Szeto GL, Chen SH. et al. Nanoparticulate STING agonists are potent lymph node-targeted vaccine adjuvants. J Clin Invest. 2015;125:2532-2546
59. Watkins-Schulz R, Tiet P, Gallovic MD, Junkins RD, Batty C, Bachelder EM. et al. A microparticle platform for STING-targeted immunotherapy enhances natural killer cell- and CD8+ T cell-mediated anti-tumor immunity. Biomater. 2019;205:94-105
60. Junkins RD, Gallovic MD, Johnson BM, Collier MA, Watkins-Schulz R, Cheng N. et al. A robust microparticle platform for a STING-targeted adjuvant that enhances both humoral and cellular immunity during vaccination. J Control Release. 2018;270:1-13
61. Leach DG, Dharmaraj N, Piotrowski SL, Lopez-Silva TL, Lei YL, Sikora AG. et al. STINGel: Controlled release of a cyclic dinucleotide for enhanced cancer immunotherapy. Biomater. 2018;163:67-75
62. Lee E, Jang H-E, Kang YY, Kim J, Ahn J-H, Mok H. Submicron-sized hydrogels incorporating cyclic dinucleotides for selective delivery and elevated cytokine release in macrophages. Acta Biomater. 2016;29:271-281
63. Koshy ST, Cheung AS, Gu L, Graveline AR, Mooney DJ. Liposomal Delivery Enhances Immune Activation by STING Agonists for Cancer Immunotherapy. Adv Biosyst. 2017:1 1600013
64. Nakamura T, Miyabe H, Hyodo M, Sato Y, Hayakawa Y, Harashima H. Liposomes loaded with a STING pathway ligand, cyclic di-GMP, enhance cancer immunotherapy against metastatic melanoma. J Control Release. 2015;216:149-157
65. Miyabe H, Hyodo M, Nakamura T, Sato Y, Hayakawa Y, Harashima H. A new adjuvant delivery system 'cyclic di-GMP/YSK05 liposome' for cancer immunotherapy. J Control Release. 2014;184:20-27
66. Wilson DR, Sen R, Sunshine JC, Pardoll DM, Green JJ, Kim YJ. Biodegradable STING agonist nanoparticles for enhanced cancer immunotherapy. Nanomedicine. 2018;14:237-246
67. Junkins RD, Gallovic MD, Johnson BM, Collier MA, Watkins-Schulz R, Cheng N. et al. A robust microparticle platform for a STING-targeted adjuvant that enhances both humoral and cellular immunity during vaccination. J Control Release. 2018;270:1-13
68. Collier MA, Junkins RD, Gallovic MD, Johnson BM, Johnson MM, Macintyre AN. et al. Acetalated Dextran Microparticles for Codelivery of STING and TLR7/8 Agonists. Mol Pharm. 2018;15:4933-4946
69. Luo M, Wang H, Wang Z, Cai H, Lu Z, Li Y. et al. A STING-activating nanovaccine for cancer immunotherapy. Nat Nanotechnol. 2017;12:648-654
70. Luo M, Liu Z, Zhang X, Han C, Samandi LZ, Dong C. et al. Synergistic STING activation by PC7A nanovaccine and ionizing radiation improves cancer immunotherapy. J Control Release. 2019;300:154-160
71. Fu J, Kanne DB, Leong M, Glickman LH, McWhirter SM, Lemmens E. et al. STING agonist formulated cancer vaccines can cure established tumors resistant to PD-1 blockade. Sci Transl Med. 2015;7:283ra52
72. Smith TT, Moffett HF, Stephan SB, Opel CF, Dumigan AG, Jiang X. et al. Biopolymers codelivering engineered T cells and STING agonists can eliminate heterogeneous tumors. J Clin Invest. 2017;127:2176-2191
73. Li L, Yin Q, Kuss P, Maliga Z, Millán JL, Wu H. et al. Hydrolysis of 2′3′-cGAMP by ENPP1 and design of nonhydrolyzable analogs. Nat Chem Biol. 2014;10:1043-1048
74. Koshy ST, Cheung AS, Gu L, Graveline AR, Mooney DJ. Liposomal Delivery Enhances Immune Activation by STING Agonists for Cancer Immunotherapy. Adv Biosys. 2017;1:1600013
75. Corrales L, Glickman Laura H, McWhirter Sarah M, Kanne David B, Sivick Kelsey E, Katibah George E. et al. Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Rep. 2015;11:1018-1030
76. Wang C, Ye Y, Hu Q, Bellotti A, Gu Z. Tailoring Biomaterials for Cancer Immunotherapy: Emerging Trends and Future Outlook. Adv Mater. 2017;29:1606036
77. Milling L, Zhang Y, Irvine DJ. Delivering safer immunotherapies for cancer. Adv Drug Deliv Rev. 2017;114:79-101
78. Hu Q, Ren H, Li G, Wang D, Zhou Q, Wu J. et al. STING-mediated intestinal barrier dysfunction contributes to lethal sepsis. EBioMedicine. 2019;41:497-508
79. Luo M, Samandi LZ, Wang Z, Chen ZJ, Gao J. Synthetic nanovaccines for immunotherapy. J Control Release. 2017;263:200-210
80. Song W, Musetti SN, Huang L. Nanomaterials for cancer immunotherapy. Biomater. 2017;148:16-30
81. Senapati S, Mahanta AK, Kumar S, Maiti P. Controlled drug delivery vehicles for cancer treatment and their performance. Signal Transduct Targeted Ther. 2018;3:7
82. Schudel A, Francis DM, Thomas SN. Material design for lymph node drug delivery. Nat Rev Mater. 2019;4:415-428
83. Nakamura T, Miyabe H, Hyodo M, Sato Y, Hayakawa Y, Harashima H. Liposomes loaded with a STING pathway ligand, cyclic di-GMP, enhance cancer immunotherapy against metastatic melanoma. J Control Release. 2015;216:149-157
84. Soppimath KS, Aminabhavi TM, Kulkarni AR, Rudzinski WE. Biodegradable polymeric nanoparticles as drug delivery devices. J Control Release. 2001;70:1-20
85. An M, Yu C, Xi J, Reyes J, Mao G, Wei W-Z. et al. Induction of necrotic cell death and activation of STING in the tumor microenvironment via cationic silica nanoparticles leading to enhanced antitumor immunity. Nanoscale. 2018;10:9311-9319
86. Hawker CJ, Wooley KL. The Convergence of Synthetic Organic and Polymer Chemistries. Science. 2005;309:1200-1205
87. Kamaly N, Yameen B, Wu J, Farokhzad OC. Degradable Controlled-Release Polymers and Polymeric Nanoparticles: Mechanisms of Controlling Drug Release. Chem Rev. 2016;116:2602-2663
88. Schaffert D, Wagner E. Gene therapy progress and prospects: synthetic polymer-based systems. Gene Ther. 2008;15:1131-1138
89. Green DR, Ferguson T, Zitvogel L, Kroemer G. Immunogenic and tolerogenic cell death. Nat Rev Immunol. 2009;9:353-363
90. Pépin G, Gantier MP. cGAS-STING Activation in the Tumor Microenvironment and Its Role in Cancer Immunity. Xu D, editor. Regulation of Inflammatory Signaling in Health and Disease. 2017 p. 175-194
91. Burnette BC, Liang H, Lee Y, Chlewicki L, Khodarev NN, Weichselbaum RR. et al. The Efficacy of Radiotherapy Relies upon Induction of Type I Interferon-Dependent Innate and Adaptive Immunity. Cancer Res. 2011;71:2488-2496
92. Vanpouille-Box C, Alard A, Aryankalayil MJ, Sarfraz Y, Diamond JM, Schneider RJ. et al. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat Commun. 2017;8:15618
93. Yum S, Li M, Frankel AE, Chen ZJ. Roles of the cGAS-STING Pathway in Cancer Immunosurveillance and Immunotherapy. Annu Rev Cancer Biol. 2019;3:323-344
94. Corrales L, McWhirter SM, Dubensky TW Jr, Gajewski TF. The host STING pathway at the interface of cancer and immunity. J Clin Invest. 2016;126:2404-2411
95. Kitai Y, Kawasaki T, Sueyoshi T, Kobiyama K, Ishii KJ, Zou J. et al. DNA-Containing Exosomes Derived from Cancer Cells Treated with Topotecan Activate a STING-Dependent Pathway and Reinforce Antitumor Immunity. J Immunol. 2017;198:1649-1659
96. Parkes EE, Walker SM, Taggart LE, McCabe N, Knight LA, Wilkinson R. et al. Activation of STING-Dependent Innate Immune Signaling By S-Phase-Specific DNA Damage in Breast Cancer. J Natl Cancer Inst. 2016:109
97. Ding L, Kim H-J, Wang Q, Kearns M, Jiang T, Ohlson CE. et al. PARP Inhibition Elicits STING-Dependent Antitumor Immunity in Brca1-Deficient Ovarian Cancer. Cell Rep. 2018;25:2972-2980.e5
98. Bommareddy PK, Shettigar M, Kaufman HL. Integrating oncolytic viruses in combination cancer immunotherapy. Nat Rev Immunol. 2018;18:498-513
99. Cai X, Chiu Y-H, Chen Zhijian J. The cGAS-cGAMP-STING Pathway of Cytosolic DNA Sensing and Signaling. Mol Cell. 2014;54:289-296
100. Kell AM, Gale M. RIG-I in RNA virus recognition. Virol. 2015;479-480:110-121
101. Sun F, Liu Z, Yang Z, Liu S, Guan W. The emerging role of STING-dependent signaling on cell death. Immunol Res. 2019;67:290-296
Author contact
![]() Corresponding author: Shuibin Lin (linshb6sysu.edu.cn), Guizhi Zhu (gzhu2edu)
Corresponding author: Shuibin Lin (linshb6sysu.edu.cn), Guizhi Zhu (gzhu2edu)