Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
ETV2 is essential for the...
Target genes of ETV2 in vessel...
ETV2 in pathophysiological...
ETV2 as a novel therapeutic...
Delivery and induction of ETV2...
Conclusions
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(19):5694-5705. doi:10.7150/thno.35300 This issue Cite
Review
ETV2/ER71 Transcription Factor as a Therapeutic Vehicle for Cardiovascular Disease
Dong Hun Lee1,2*, Tae Min Kim5*, Joo Kyung Kim1, Changwon Park1,2,3,4 ![]()
1. Department of Pediatrics
2. Children's Heart Research and Outcomes Center
3. Molecular and Systems Pharmacology Program
4. Biochemistry, Cell Biology and Developmental Biology Program, Emory University School of Medicine, Atlanta, GA, USA
5. Graduate School of International Agricultural Technology and Institute of Green-Bio Science and Technology, Seoul National University, 1447 Pyeongchang-daero, Pyeongchang, Gangwon-do 25354, South Korea
*These authors equally contributed to this work.
Received 2019-3-28; Accepted 2019-6-26; Published 2019-8-9
Abstract

Cardiovascular diseases have long been the leading cause of mortality and morbidity in the United States as well as worldwide. Despite numerous efforts over the past few decades, the number of the patients with cardiovascular disease still remains high, thereby necessitating the development of novel therapeutic strategies equipped with a better understanding of the biology of the cardiovascular system. Recently, the ETS transcription factor, ETV2 (also known as ER71), has been recognized as a master regulator of the development of the cardiovascular system and plays an important role in pathophysiological angiogenesis and the endothelial cell reprogramming. Here, we discuss the detailed mechanisms underlying ETV2/ER71-regulated cardiovascular lineage development. In addition, recent reports on the novel functions of ETV2/ER71 in neovascularization and direct cell reprogramming are discussed with a focus on its therapeutic potential for cardiovascular diseases.
Keywords: ER71/ETV2, FLK1/VEGFR2, cardiovascular, angiogenesis, direct cell reprogramming
Introduction
Transcription factors perform indispensable cellular events in living organisms because they can interpret genetic information in response to biological cues. The ETS transcription factors, consisting of approximately 28 members, are involved in diverse biological processes including cell cycle control, apoptosis (programmed cell death), embryogenesis, and tumorigenesis [1]. The main distinguishing feature of ETS factors is the well conserved DNA binding domain (ETS DNA binding domain) that binds to the central sequences, 5'-GGAA/T-3', present on the promoters or enhancers of genes, leading to a wide range of biological consequences [1, 2]. Besides the ETS DNA binding domain, the pointed domain (PTN) is shared among many ETS factors and mediates protein-protein interactions. Several members of the ETS factors such as ETS1/2, ETV2 (also known as ER71), FLI1, ELK3, and ETV6 are expressed in cardiovascular lineage cells in early stage embryos [3-11]. In agreement with their expression pattern, these ETS factors have shown to be essential for the establishment and functions of the cardiovascular system constituents in the developing embryos [1, 4, 12].
Gene knockout studies have been instrumental for determining the biological consequences of genes in vivo. However, promiscuous binding specificity of the cognate binding sequences and the overlapped expression of the ETS factors often hinder the identification of the specific functions of individual ETS members. For example, Ets1 deficiency in mice is compatible with the normal vascular development, but compound knockout of Ets1 and Ets2 leads to defects in vessel formation [13, 14]. Fli1-/- mice succumb to death by embryonic day (E) 12.5 due to defective hematopoiesis and hemorrhaging in the brain [15]. Since the overall embryo morphology including the cardiovascular structure of Fli1-null embryos is normal at E8.5-E9.5, it is likely that FLI1 is important for maintaining vessel integrity at mid-gestation. Germ line deletion of Elk3 in mice develops defective vascular structures in adult retina [16]. These findings suggest overlapped or mild functions of these factors in cardiovascular development. In contrast, our studies have shown that ETV2 is indispensable for the cardiovascular development during mouse embryogenesis [5]. Our findings are consistent with the results from multiple groups working in diverse organisms such as zebrafish and Xenopus [17-21], suggesting that ETV2 is a critical regulator required for establishing the cardiovascular system in early embryogenesis. The first part of this review deals with the functions of ETV2 for cardiovascular system development. The potential applicability of ETV2 as a therapeutic agent in treating cardiovascular disease (CVD) will be discussed in the latter part.
ETV2 is essential for the establishment of the cardiovascular system
In developing mouse embryos between E8.5 and E9.5, the expression of Etv2 is preferentially detected in virtually all vascular structures including the dorsal aorta, cardinal vein, and endocardium. Etv2 expression is downregulated at E10.5 and lost completely from E11.5 onwards [5, 9, 17, 18]. In adults, the testes become the major organ for Etv2 message while its expression remains low even in the highly vascularized organs such as the heart and lung [22]. In addition, the first emerging FLK1 expressing (FLK1+) cells, which can subsequently differentiate into endothelial and hematopoietic cells [23], display enriched expression of Etv2, compared to FLK1- cells as demonstrated by the results from both in vivo mouse embryos and in vitro mouse embryonic stem cell (mESC) differentiation system [5, 24]. In agreement with the specific expression of ETV2 in hematopoietic and endothelial cell lineages in early stage embryos, gene knockdown and knockout studies also demonstrate the essential role of ETV2 in the cardiovascular system development. Such critical function of ETV2 was initially suggested from a study in zebrafish [25] by comparing the expression of genes between wild type and the zebrafish cloche mutant [26], characterized by the severe developmental defects in blood cells and endothelial lineages. The authors reported that etsrp, the zebrafish homologue of mammalian Etv2, was significantly downregulated in the mutant embryos, thereby suggesting functional significance of etsrp in blood cells and vessel development [25]. Interestingly, zebrafish embryos injected with etsrp morpholino (MO) developed severely impaired vasculatures with a significant decrease of the expression of key markers of endothelial cells, but zebrafish embryos injected with etsrp mRNA showed ectopic expression of endothelial cell markers [20]. Restored expression of etsrp in the cloche mutant embryos rescued the vascular defects. Similarly, the functional significance of etsrp in zebrafish vessel formation has also been reported by others [21]. By performing N-ethyl-N-nitrosourea (ENU)-mediated mutagenesis and positional cloning, y11 was identified as a mutant displaying defective embryonic vasculature, and etsrp was then identified as the gene responsible for these defective phenotypes. Ectopic expression of fli1 and flk1 was induced upon injection of etsrp mRNA into zebrafish embryos, but the significant reduction of endothelial and hematopoietic genes was observed in zebrafish embryos receiving etsrp MO. Subsequent studies in mouse have strengthened and expanded the findings from zebrafish. In 2008, we were the first to identify the essential function of ETV2 in cardiovascular system development in mice [5]. Etv2-deficient mouse embryos displayed a complete lack of both hematopoietic and endothelial cell lineages, resulting in embryonic lethality between E9.5 and E10.5. Overexpression of Etv2 in mESCs significantly increased the generation of cells expressing markers of hematopoietic and endothelial cells. These findings were further corroborated by the following studies from several independent groups; Etv2 mutant mouse embryos generated through the gene trap approach died in utero and showed defects in embryonic vasculature development [17]. Another report also demonstrated that Etv2-null mouse embryos failed to develop both cell lineages [18]. Studies with mESCs have provided direct evidence of the determinant role of ETV2 in this process; Etv2-/- ESCs were incapable of generating hematopoietic and endothelial cells [27]. The conserved importance of ETV2 in regulating endothelial cell development is also evident in Xenopus [19]. It is of note that hematopoiesis appears normal in Xenopus embryos treated with Etv2 MO. Collectively, these results suggest that ETV2 is essential for cardiovascular development.
Target genes of ETV2 in vessel development
In this section, we will mainly discuss the downstream targets of ETV2 that constitute a complex regulatory network for the development of embryonic vasculature. We will also discuss the mechanisms by which ETV2 activity is regulated (Figure 1A). For detailed discussion on the molecular mechanism of ETV2, the reader is directed to read the following reviews [28, 29].
Function of ETV2. (A) In early embryogenesis, ETV2 plays indispensable roles for the establishment of the cardiovascular system through the direct binding to promoters or enhancers of genes critical for the hematopoietic and endothelial cell lineages. Recent reports suggest that ETV2 can form an active transcriptional complex at least with OVOL2, GATA2, FOXC2 or TET1/2. (B) In post-natal life, ETV2 regulates the proliferation of bone marrow (BM) hematopoietic stem cells. In addition, ETV2 robustly promotes new vessel formation in pathophysiological conditions such as injury, MI, and tumorigenesis. (C) ETV2 alone or in combination with other TFs or small molecules can directly reprogram non-endothelial cells into cells displaying endothelial functionalities.
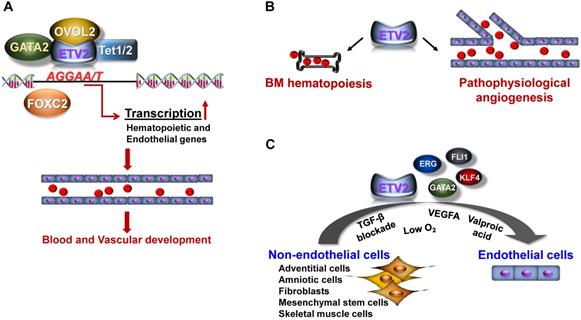
As discussed, ETV2 is an ETS factor which can target genes containing the consensus sequences (5'-GGAA/T-3') present on enhancers or promoter regions [2]. We were the first to report that ETV2 can directly bind to the Flk1 promoter through the ETS binding elements and activate the expression of Flk1 by performing the luciferase-based promoter assay and chromatin immunoprecipitation (ChIP)-PCR analysis [5]. In subsequent studies, including ours, several key genes for vessel development and angiogenesis including Tie2, Cdh5, Vegfr3, Robo4, and Notch4 have been identified as direct targets of ETV2 [27, 30-32]. Recently, a genome-wide analysis using ChIP-sequencing revealed that ETV2 has a wide range of direct downstream genes, further providing important insights into the molecular interaction between ETV2 and other signaling pathways [33]. In this study, we found that ETV2 can directly activate genes closely associated with VEGF, Rho-GTPase and MAPK signaling as well as the aforementioned genes, all of which have been implicated in vessel development or hematopoiesis. Interestingly, ETV2 appears to activate its own expression and induces other ETS factors such as FLI1 and ERG [33]. Considering the sequence specificity among the ETS factors, it is expected that other ETS factors can also bind some, if not all, of the ETV2 target genes and replace ETV2 function when ETV2 expression becomes silent [5, 9, 17, 18]. Interestingly, the message of Fli1 is rapidly induced by overexpression of ETV2, whereas Fli1 deficiency does not affect the expression of ETV2 and the emergence of FLK1+ cells [33]. A ChIP-PCR analysis revealed that day 4-5 embryoid bodies (EBs, cell aggregates of differentiating mESCs) expressing diminished level of Etv2 showed enhanced in vivo binding of FLI1 on Tie2, Cdh5 and Gata2, compared to day 3 EBs with high level expression of Etv2 [33]. Fli1-/- mESCs displayed a significantly reduced expression level of these genes, suggesting that FLI1 can, at least partly, inherit ETV2 function when ETV2 expression is depleted. This observation was further supported by another study [34] in which FLI1 failed to bind promoters of Tie2 and Cdh5, when ETV2 is highly expressed. Interestingly, in the absence of Etv2, FLI1 is able to occupy these genes in mouse embryos. Moreover, Tie2 and Cdh5 expressions were reduced in Fli1 knockout mice embryos, compared to control embryos. A similar relationship between ETV2 and FLI1 was also reported in zebrafish. Data from another group showed that inhibition of etsrp leads to severe vascular defects in zebrafish embryos, but a lack of both fli1a and fli1b is compatible with embryonic vessel development [35]. Additionally, etsrp MO;fli1b-deficient embryos, not etsrp MO;fli1a- deficient embryos, displayed severe vascular defects, compared to etsrp MO embryos. The authors also demonstrated that etsrp and fli1b can redundantly induce the expression of the same target genes. Taken together, these results suggest that ETV2 initiates the cardiovascular developmental cascade by directly activating a wide range of genes in early embryos and later becomes silent after embryonic vessels have been established. In turn, other ETV2-induced ETS factors, such as FLI1, could partly replenish the vascular function of ETV2 by regulating subsets of the targets of ETV2 to mainly ensure the subsequent maturation and maintenance of vasculatures for successful embryogenesis. Sustained expression of ETV2 in mouse embryos leads to acquisition of endothelial character in non-endothelial organs [36]. This supports the notion that ETV2 is short-lived, but critical for embryonic vessel formation [37].
Apart from blood vessel formation, it has also been suggested that ETV2 is involved in the development of lymphatic vasculature. In zebrafish, etsrp expression can be detected in cells fated to lymphatic vessels at 24-56 hours post fertilization (hpf) [38]. Inhibition of etsrp by photoactivatable etsrp MO leads to defects in lymphatic vessel formation with impaired expression of lymphatic markers such as vegfr3 (flt4) and lyve1b [38]. In agreement with these findings, the ChIP-sequencing assay and the promoter-based luciferase experiments reveal both vegfr3 and lyve1b as direct downstream targets of etsrp [33, 38]. Considering the evolutionarily conserved function of ETV2 and the increasing importance of the lymphatic vessels in pathophysiological events including CVD and cancer [39-43], a further detailed investigation on the role of ETV2 in lymphatic compartment in mammals would be warranted.
As other transcription factors, studies have shown that the activity of ETV2 can be regulated via its interacting proteins. Several endothelial genes including FOXC2 [30], OVOL2 [24], or GATA2 [44] have been reported to interact with ETV2 in the generation of cardiovascular lineages (Figure 1A). Although detailed mechanisms of how these interactions control the functional activity of ETV2 need further investigations, the enhanced stability of ETV2 has been suggested as one of the mechanisms as demonstrated by the high level of ETV2 protein upon the interaction with OVOL2 [24, 45], a zinc finger transcription factor known to play a critical role in angiogenesis [24, 45]. Interestingly, a recent study found that ETV2 can also regulate the expression of its target genes through DNA methylation/demethylation by interacting with TET1/2 [32], epigenetic modifiers responsible for DNA demethylation [46]. The authors showed that ETV2 can directly bind the proximal promoter of Robo4, an important endothelial gene [47], through the ETS binding elements. Importantly, the interaction between ETV2 and TET proteins cooperatively potentiates DNA demethylation on Robo4 proximal promoter, leading to the expression of Robo4 in non-endothelial cells. Whether the expression of other endothelial genes can also be regulated by the interaction of ETV2 and TET1/2 is unclear. Nonetheless, this report suggests a novel insight into the mechanisms of ETV2 function in regulating its downstream target genes. Therefore, a comprehensive investigation to uncover the protein components of the ETV2 transcriptional complex would aid our current understanding of the functions of ETV2.
ETV2 in pathophysiological angiogenesis and cell fate reprogramming
The potent role of ETV2 in vascular formation during embryo development led the investigators to explore the unknown functions of ETV2 in post-natal angiogenesis under the pathophysiological conditions. In the following sections, we will discuss recent findings from our laboratory and other groups regarding the therapeutic potential of ETV2 in mediating neovascularization and direct cell reprogramming (Figure 1B-C).
Endothelial ETV2 is required for angiogenesis in response to pathophysiological stimuli
As discussed, ETV2 is specifically expressed in the vasculatures within a narrow developmental window between E8.5 and E10.5. In accordance with the transient expression pattern of ETV2, mice deficient in endothelial Etv2 (i.e., Tie2-Cre or Cdh5-Cre;Etv2floxed/floxed mice) are born alive and develop normal vascular structures [48, 49], suggesting that ETV2 is dispensable for steady-state vessel formation. There is a prevailing notion that embryonic events or signaling pathways become critical for the development of diseases or pathophysiological events in adults. This prompted us to investigate the function of ETV2 in post-natal life. Interestingly, we found rapid upregulation of Etv2 in endothelial cells of mouse hindlimbs in response to ischemic injury [49], suggesting an important function of reactivated endothelial Etv2 in injury-induced angiogenesis. By employing conditional knockout mice, we showed that the absence of Etv2 in endothelial cells led to a significantly compromised vascular regeneration in response to injury such as eye injury, skin wounding, or hindlimb ischemic injury. Inversely, the overexpression of Etv2 by injecting lentiviral Etv2 into mice with ischemic injury facilitated recovery from the impaired blood perfusion and augmented the new vessel formation. Furthermore, the damage to tissues caused by the ischemic insult was repaired upon the injection of lentiviral Etv2. Mechanistically, ETV2 was able to activate critical genes for vessel development and angiogenesis, such as Flk1, Cdh5 and Vegfa/b/c, as seen in developing embryos. Similarly, upregulated level of Etv2 in tumor associated endothelial cells (TAECs) has also been reported in a previous study [48]. The authors demonstrated that the deletion of Etv2 in endothelial cells or systemic delivery of siEtv2 into tumor bearing mice impairs tumorigenesis and angiogenesis. As ETV2 function is conserved in other vertebrates, etsrp expression in zebrafish, which is normally diminished by 2-4 dpf (days post fertilization) in developing embryos, is significantly induced not only in embryonic vasculatures, but also in tumor-associated vessels upon the transplantation of tumor cells [37]. While wild type zebrafish injected with the tumor cells induced tumor-associated angiogenesis, blocking the function of etsrp using etsrp null mutant or photoactivatable etsrp MO led to severe impairment of the angiogenesis. Although beyond the vascular system itself, hematopoietic Etv2 is also reactivated upon injury and plays an important function in bone marrow (BM) hematopoiesis (Figure 1B and also see next pages for further discussion) [50]. Taken together, these results clearly suggest that reactivation of endothelial Etv2 in response to pathophysiological stimuli is a central step for neovascularization and tissue repair (Figure 1B). In addition, the profound vascular regulatory functions of ETV2 in vivo upon delivery into mice in the form of lentiviral Etv2 or siEtv2-nanoparticle complex raise the potential applicability of ETV2 as a therapeutic agent for diseases related to the dysfunctional vessel formation.
Cell fate reprogramming by ETV2
ETV2 has also additional therapeutic potential owing to its critical role in the direct reprogramming of non-endothelial cells into endothelial cells (Figure 1C). Along with the groundbreaking iPSC generation from somatic cells [51, 52] with diverse methods, a significant effort has been made to convert one cell type into a distinct one by bypassing the pluripotent stage through the overexpression of cell lineage specific transcription factors [53-56] or miRNAs [57-60]. The first successful demonstration of conversion of non-endothelial cells into functional endothelial cells was achieved in human amniotic cells (ACs) with a transient overexpression of ETV2, followed by the prolonged duration of other ETS factors, ERG and FLI1 in conjunction with TGF-β signaling blockade [61]. Transplantation of the converted endothelial cells into mouse models of neovascularization proves the functionality of the converted cells in vivo. It is important to note that ETV2 appears dispensable once ACs start expressing endothelial genes and that further maturation steps depend on ERG and FLI1, which is reminiscent of the sequence of events occurring in the developing embryos. In addition, it was shown that the battery of key endothelial transcription factors, ETV2, FLI1, GATA2, and KLF4, were able to generate cells with an endothelial phenotype directly from the human fibroblast [62]. These results strongly suggest the feasibility of direct cell fate conversion for endothelial cell generation using the endothelial transcription factors. However, potential genetic burden caused by using a combination of multiple transcription factors and viral vector-mediated delivery is one of the major obstacles that needs to be overcome for clinical use of the reprogrammed cells. As a first step towards tackling the issue, Morita et al. reported direct conversion of the human dermal fibroblasts (HDFs) into endothelial-like cells using ETV2 alone [63]. Delivery of the reprogrammed endothelial cells into mouse ischemic hindlimbs results in augmented perfusion with concomitant enhancement of neovascularization. The authors claimed that a transient expression of ETV2 was sufficient for direct reprogramming. A modified method similar to this study demonstrated that culturing ETV2-transduced HDFs in hypoxic condition in the presence of VEGFA increased the efficiency of direct reprogramming of HDFs into cells expressing endothelial markers [64]. We have also demonstrated a potent role for ETV2 in mediating the direct cell conversion process [65]. In our study, we found two waves of the reprogramming process; a week-long overexpression of ETV2 in HDFs induces so called early reprogrammed endothelial cells (rEC) characterized by substantial expression of key endothelial surface molecules including KDR and CDH5 with insignificant expression of mature endothelial markers such as CD31 and VWF. Despite the immature nature of early rECs, they are functional as evidenced by a significant enhancement of perfusion recovery from ischemic injury upon injection of the cells in a mouse model of hindlimb ischemia. The generation of endothelial cells displaying mature phenotype (i.e., late rECs) requires transient re-expression of ETV2 together with the treatment of an epigenetic modifier, valproic acid (VPA) during the cultivation of early rECs. Such treatments confer mature endothelial cell characteristics to the early rECs, such as high expression of CD31 and production of nitric oxide. In addition, genome wide RNA sequencing results show a close relation of late rECs to HUVECs or HUMVECs. Thus, our reprogramming protocol could be a novel system to study endothelial cell generation and maturation. Further, we believe that the early rECs could be useful as potential therapies and the late rECs would be a reliable source for drug testing. The function of ETV2 in mediating direct cell reprogramming into endothelial cells is further supported by studies in other species. For example, mouse adult skin fibroblasts can be directly reprogrammed into endothelial cells using a combination of ETV2 and other transcription factors including FOXO1, KLF2, TAL1, and LMO2 [66]. Mouse adventitial SCA1+ cells transduced with the adenoviral Etv2 become cells displaying the endothelial functionalities in vivo and in vitro [67]. In both studies, implantation of the directly converted cells in mouse vascular injury models leads to enhanced recovery from the injury by facilitating revascularization. In addition, a heat shock-mediated induction of etsrp in zebrafish embryos prior to 30 hpf can convert skeletal muscles into endothelial cells [68]. Taken together, these results strongly suggest that ETV2 plays a critical function in direct cell conversion of non-endothelial somatic cells into endothelial cells, which could be used as cell therapy for CVD.
ETV2 as a novel therapeutic agent for diseases with vascular defects
Dysfunctional blood vessel or uncontrolled vasculature formation is one of the leading factors that cause devastating diseases such as cardiovascular, cerebrovascular disease, age-related macular degeneration, post-operative complications after vascular surgeries, chronic wounds, and cancer. CVD ranks as the number one disease in the United States as well as worldwide [69], and treating patients with myocardial infarction (MI) and peripheral artery disease (PAD) remains one of the most profound challenges due to the diverse causes of these diseases. PAD can progress to critical limb ischemia, which causes a high incidence of amputation and mortality, necessitating efficient ways to treat the disease. Since the main cause of CVD is dysfunctional blood vessels resulting in loss or dysregulation of vasculatures, approaches for enhancing functional re-vascularization from the damaged tissues has gained extensive interest. For example, delivery of pro-angiogenic factors such as VEGFA, HGF, and FGF has been tested and found to have therapeutic benefits in animal systems [70-75]. Mice undergoing ischemic MI show a long-term survival rate accompanying promoted myocardial function and enhanced angiogenesis upon reception of modified RNA form of VEGFA [76]. Despite promising outcomes from animal or preclinical trials, the beneficial effects of pro-angiogenic approach are minimal at best in clinical trials with patients [77]. This might be at least partly due to the intrinsic nature of the factors tested, most of which signal through their cognate receptor, limiting the width of angiogenic repertoires in patients who mostly have other comorbidities. Based on the basic developmental biology and recent progress in cell reprogramming, we understand that some lineage specific transcription factors dictate the expression of a wide range of downstream target genes, reshaping genetic and epigenetic status of cells. Studies have revealed that ETV2 can induce diverse endothelial genes through its DNA binding ability and its interacting proteins [5, 24, 30, 32, 33, 44], leading to strong vasculogenic and angiogenic events as well as cell fate reprogramming, acting as a master regulator of the vasculature formation and function. Hence, delivery of ETV2 alone or in combination with other pro-angiogenic factors would be a novel and efficient strategy for patients with MI or PAD (Figure 2). As a proof of principle, our group has already provided a promising data showing that ETV2 is involved in promoting vascular regeneration and tissue repair in a mouse model of hindlimb ischemia [49]. Furthermore, injection of lentiviral or adeno-associated viral Etv2 into mouse MI hearts enhances the recovery of heart function and myocardial angiogenesis [78]. It is worthwhile to mention that ischemic hindlimbs that received lentiviral Etv2 express reduced levels of inflammatory genes and develop a significantly lower degree of fibrosis [49]. The same findings are also observed in mouse MI hearts upon injection of the lentiviral Etv2 [78]. Since unresolved or sustained inflammation may be detrimental and lead to chronic inflammatory diseases [79], these results further suggest the therapeutic advantages of ETV2 in reparative angiogenesis for various inflammatory vascular diseases.
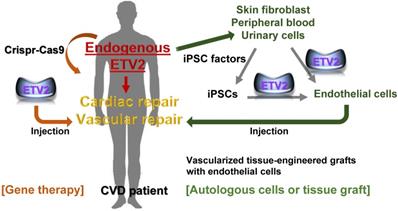
The potential of therapeutic use of ETV2 in cardiovascular diseases. Cardiac or vascular lesions caused by MI or PAD may be repaired by injecting ETV2, preferably in a non-genetic or non-viral form. In autologous cell transplantation model, reprogrammed endothelial cells can be obtained from non-endothelial cells including skin fibroblasts, peripheral blood or urinary cells via transient expression of ETV2 in these non-endothelial cells. Alternatively, iPSC-derived endothelial cells can be directly generated upon the overexpression of ETV2. Finally, vascular graft containing a patient's own reprogrammed endothelial cells would be an ideal option for repairing large vessels. Although far from being currently feasible, other approaches such as inducing endogenous ETV2 transcription machinery using the Crispr-Cas9 would provide an innovative strategy for vascular repair.
In addition to inducing angiogenesis, inhibition of pathological angiogenesis or uncontrolled vessel formation can be a constructive means for preventing vascular-related diseases including tumorigenesis. As discussed, reactivation of Etv2 occurs in TAECs, while interfering the function of ETV2 leads to decreased tumor angiogenesis in a mouse tumor model of xenotransplantation [48]. Interestingly, a recent study demonstrates an augmented expression of ETV2 in the patients of high grade of glioblastoma and reveals an inverse correlation between the expression level of ETV2 and survival rate of patients of high grade of glioblastoma [80]. Further, ETV2 can stimulate the reprogramming of glioblastoma neural stem-like cells into cells with an endothelial phenotype. Additionally, deletion of ETV2 in glioblastoma inhibits this process, suggesting novel functions of ETV2 in tumor-derived endothelial cell generation (i.e., vasculogenesis). Therefore, these findings suggest potent and broad spectrum of therapeutic potential of ETV2 in treating patients suffering from a diverse range of diseases including CAD, PAD and cancer.
Cell-based therapy is an additional method for treating ischemic vascular diseases (Figure 2). As discussed, upon overexpression of ETV2 in HDFs, reprogrammed endothelial cells exhibit an endothelial phenotype and show augmented recovery of perfusion accompanying enhanced neovascularization in a mouse model of hindlimb ischemia. This is probably due to the paracrine effect and direct incorporation of the reprogrammed cells into the vessels of the ischemic tissues [65]. In line with these findings, a recent study [81] reported that ETV2 can induce direct reprogramming of different somatic cells including human skeletal muscle cells (hSkMCs), adipose-derived mesenchymal stem cells, umbilical cord-derived mesenchymal stem cells, human embryonic lung fibroblast cells, and human skin fibroblast cells into endothelial like cells with hSkMCs being the best in terms of efficiency. In this study, the authors showed that co-culture of CDH5+ cells generated from ETV2-infected hSkMCs and skeletal muscle cells on PLGA/PLLA scaffolds or decellularized scaffolds leads to the formation of vascular-like structures in the engineered muscle tissues. By transplanting the engineered muscle tissue into athymic nude mice, they also demonstrated that the injected tissues survived and formed functional vascular network within the host as evidenced by the presence of red blood cells in the lumen. An additional study further expanded the applicability of ETV2 in direct cell reprogramming with human cell sources by demonstrating that a short period of ETV2 expression with inhibition of TGF-β signaling was able to generate cells with endothelial functionalities from human adipose-derived stem cells and human umbilical cord-derived mesenchymal stem cells [82]. To make the current findings applicable to cell replacement therapy, direct reprogramming with patient-derived somatic cells would be an important prerequisite, taking into consideration immune compatibility. An additional obstacle that must be overcome is making the procedures for obtaining the cells as minimally invasive as possible. Peripheral blood and urinary cells would fit best as sources for reprogrammed endothelial cell generation (Figure 2).
ETV2 could also be exploited as a potential therapy by harnessing the biological traits of exosomes, extracellular vesicles ranging between 30-100 nm in diameter which contain proteins, mRNAs and miRNAs [83]. The components of exosomes often reflect the pathophysiological status and the contents in exosomes are usually being transported to other cells. This biological function can be utilized for developing novel clinical therapies. Indeed, recent research in exosome biology has shown that exosomes derived from diverse sources can be used as biomarkers for certain diseases including CVDs and as therapeutics [84, 85]. Regarding ETV2-exosomes, a recent study reported that CD31+ cells generated from lentiviral ETV2 infected HDFs were able to produce exosomes [86]. In a mouse model of hindlimb ischemia, the authors claimed that mice injected with the exosomes recovered efficiently from the ischemic damage, compared to mice injected with vehicles. However, further detailed analysis on the functions and mechanisms of the exosomes should be followed to firmly establish the potential role of the ETV2-exosomes in mediating neovascularization.
Delivery and induction of ETV2 for therapeutic approaches
An issue that should be addressed is the fact that the observed beneficial effects of ETV2 were made possible with the viral particle delivery system, challenging the applicability of ETV2 for gene therapy or cell-based therapy for clinical use. Thus, a non-viral or even non-genetic delivery method for ETV2 in vitro and in vivo should be devised to eliminate the potential risk of the insertional mutagenesis caused by viral form of ETV2, resulting in chromosome instability. Success in cell reprogramming with modified mRNAs or chemical/small molecules [87, 88] has already been achieved. Therefore, a modified mRNA form of ETV2 or chemical/small molecules that can replace the lentiviral or adenoviral ETV2 would be of particular interest from a clinical perspective. Generation of hematopoietic cells from human pluripotent stem cells (hPSC) by transfecting modified mRNA of ETV2 and GATA2 has been reported [89]. The same group has also shown that modified mRNA of ETV2 alone is capable of inducing cells displaying an endothelial phenotype from multiple lines of hPSCs and non-human primate iPSCs [90], raising the feasibility of ETV2 as a therapeutic agent. Specific induction of endogenous ETV2 with a Crispr-Cas9 approach (e.g., delivery of sgRNA specific to endogenous ETV2 promoter/ enhancer and dCas9-VP64) would be another option to pursue [91-93]. In addition, efforts should be made for the development of targeted delivery methods of modified RNA form of ETV2, siRNAs or small molecules to allow the clinical potential of ETV2 to be realized.
It would also be important to identify upstream signaling that can induce the expression of ETV2 in the vascularization as this may be of significance for therapeutic purposes. In this regard, we have previously reported that ETV2 functions downstream of BMP, WNT and NOTCH signaling in generating cardiovascular lineages from mESCs [5]. Additionally, NKX2-5, foxc1a/fox1b, MESP1-CREB, and IP3Rs-Ca2+-CALCINEURIN-NFATc3 [17, 94-97] have been identified as direct upstream regulators of ETV2. Among them, we will focus on two important signaling events, calcium signaling and reactive oxygen species (ROS). Recent studies have shown that calcium (Ca2+) signaling plays a critical role in Etv2 expression. Sequence analysis on the Etv2 gene locus revealed two conserved cAMP response element (CRE) in the promoter and 5'-untranslated region (UTR). Mechanistically, the cAMP/protein kinase A (PKA) pathway can induce Etv2 expression via direct binding of CRE binding protein (CREB) to the CRE located in the 5'-UTR of Etv2 [97]. Since Ca2+ can activate adenylyl cyclases which are responsible for the conversion of ATP into cAMP [98], these findings suggest that intracellular Ca2+ influx can act as an important upstream inducer of ETV2 expression via cAMP-PKA-CREB signaling. Another study demonstrated that Mesp1, a bHLH family transcription factor for cardiac mesoderm specification [99], can activate transcription of Etv2 by the MESP1-CREB interaction in CRE within the Etv2 5'-UTR [95]. More recently, it was shown that NFATc3, an effector DNA-binding protein downstream of the Ca2+/ CALCINEURIN pathway, occupies an important position in promoting hematopoietic lineage commitment from FLK1+ mesoderm by direct binding to the Etv2 promoter [96]. Blocking Ca2+/CALCINEURIN signaling in differentiating mESCs by deleting inositol 1,4,5-trisphosphate receptors (IP3Rs), which are the major calcium releasing channels in endoplasmic reticulum, or by treating CALCINEURIN inhibitors, led to a decrease in Etv2 expression. However, treatment of ionomycin, or overexpression of a constitutively active form of CALCINEURIN or NFATc3 was able to induce the expression of Etv2 in wild type and IP3Rs deficient mESCs. Further, it was shown that NFATc3 can directly occupy the NFAT and NFκB binding sites in the Etv2 upstream region in vivo [96]. Taken together, these findings suggest that identifying bioactive molecules that can regulate Ca2+ related signaling seems to be an effective option that can induce the expression of ETV2 in mediating vascularization in adults. One candidate would be adrenomedullin, a vasodilatory hormone peptide, which has been shown to exhibit various effects on vessels such as inhibiting endothelial cell apoptosis and promoting vessel growth through several important signaling pathways including Ca2+ and cAMP [100, 101]. Importantly, administration or overexpression of adrenomedullin promoted (lymph)angiogenesis, while reduced expression of adrenomedullin led to impairment of vessel recovery in murine models of vascular injury [102-106]. At least in differentiating mESC, adrenomedullin was able to increase the generation of ETV2+ cells and the expression of Etv2 [97]. The functional relation between adrenomedullin and ETV2 in pathophysiological angiogenesis in adults needs further investigations.
Another fundamental approach on Etv2 induction have focused on ROS, whose function is also involved in promoting physiological angiogenesis if properly controlled [107]. Upon hematopoietic cell injury by 5-FU injection, the expression of hematopoietic Etv2 and the ROS production increases in hematopoietic stem and progenitor cells (HSPCs) as defined by c-KIT+Sca1+Lin- [50]. Remarkably, the expression of Etv2 was augmented in HPSCs after being treated with an ROS activator, whereas 5-FU induced augmentation of Etv2 expression was abrogated in the presence of a ROS scavenger and an NADPH oxidase inhibitor, suggesting that ROS function as a critical player in Etv2 activation [50]. More direct evidence on ROS-mediated Etv2 induction was shown in TAECs from lung carcinoma in a tumor graft mice model. A higher level of ROS was detected in TAECs than in endothelial cells from the normal lung or hindlimb tissues. Treatment of an ROS scavenger or siRNA against Etv2 in vivo resulted in down-regulation of the expression of Etv2 in TAEC, and led to reduction of the tumor volume [48]. These results indicate that ROS functions as an important inducer of Etv2. Collectively, these studies present a novel direction for the development of therapeutic strategies by bringing Ca2+-related signaling, ROS, other intermediate signaling molecules to the territory of ETV2 in vascular disease.
Conclusions
ETV2 has increasingly gained attention due to its potent vasculo-angiogenic properties. Over the past decade, studies have revealed the important functions of ETV2 and its underlying mechanisms. However, our current knowledge about ETV2 is still too limited to make this molecule therapeutically feasible. The advent of new research tools such as the next generation sequencing, Crispr-Cas9, epitranscriptomics and biomaterials will facilitate understanding of the mechanisms and functions of ETV2. This, in turn, could aid the development of safe and novel ETV2 therapeutic options to treat diseases related to vessel dysfunction. As discussed above, preclinical testing of ETV2 in inflammatory vascular diseases (e.g., atherosclerosis) would also be important since vascular diseases are often accompanied with chronic inflammation. Lastly, it is important to note that the therapeutic applicability of ETV2 can be expanded to the lymphatic vascular system. Experimental results from several animal models including rabbit and dog suggest a close association of impaired lymphatic flow and CVD [39, 108-110]. Moreover, etsrp plays an important function in regulating lymphatic vessel formation in zebrafish [38]. Thus, further detailed studies regarding ETV2-lymphatic vessel/disease would be an interesting area to pursue for basic research and translational purposes.
Although rodent models have significantly contributed to the understanding of early vessel development and pathophysiology of various CVDs, it should also be noted that their cardiovascular physiology and inflammatory profiles are different from humans [111, 112]. Accordingly, large animal models are often preferred because they are accepted as more clinically relevant models of human physiology, despite the limited number of available reports [113]. In a sheep model of MI, when injected directly into the myocardium one hour after coronary artery ligation with a plasmid DNA expressing human VEGFA165 under CMV promoter/enhancer, the resting myocardial perfusion was increased while the infarct area was reduced [114]. Additionally, the capillary density was higher in the VEGFA165-treated animals than in placebo group. More recently, another study in a swine model of MI reported that adenovirus carrying human SCF enhanced cardiac functions, with an increase of c-KIT+ cells, vessel density, and a decrease in apoptosis [115]. In a dog model of hindlimb ischemia, human bone marrow- derived mesenchymal stem cells together with the sustained release of bFGF via heparin-conjugated fibrin promoted the angiogenic effect, as indicated by the increased arterioles and capillaries, as well as PDGF- and VEGFA-positive cells 6 months after transplantation [116]. Large animal models are essential, not only for their usefulness in translating preclinical results into clinical research, but also for determining optimal dosage and delivery route for human use [117]. Together with well-developed interventional approaches, the use of large animal models with a special focus on ETV2 will be instrumental for confirming the initial findings from rodent studies, and finally will significantly contribute to the development of novel gene- or cell-based therapeutics for CVDs.
Abbreviations
ACs: amniotic cells; BMP: bone Morphogenetic Protein; CAD: coronary artery disease; c-AMP: cyclic adenosine monophosphate; CAS9: CRISPR associated protein 9; CD31: cluster of differentiation 31 (also known as PECAM1); CDH5: cadherin 5 (also known as VEcadherin); c-KIT: receptor tyrosine kinase (also known as stem cell factor receptor, CD117); CREB: c-AMP response element binding protein; CLI: critical limb ischemia; CMV: cytomegalovirus; Crispr: clustered regularly interspaced short palindromic repeats; CVD: cardiovascular disease; E: embryonic day; EB: embryoid body; ELK3: ETS domain-containing protein; ERG: ETS-related gene; ESCs: embryonic stem cells; ETS: E26-AMV virus oncogene cellular homolog, a transcription factor; ETV2: ETS Variant 2 (also known as ER71 or etsrp); ETV6: ETS Variant 6; FGF: fibroblast growth factor; FLI1: friend leukemia virus integration 1; FOXO1: forkhead box O1; 5-FU: 5-fluorouracil; GATA2: GATA-binding factor 2; HDF: human dermal fibroblast; HGF: hepatocyte growth factor; HSPCs: hematopoietic stem and progenitor cells; HSFs: human skin fibroblast cells; hSkMCs: human skeletal muscle cells; IP3R: inositol trisphosphate receptor; iPSCs: induced pluripotent stem cells; KDR: kinase insert domain receptor (also known as FLK1 or VEGFR2); KLF2/4: kruppel like factor 2/4; LMO2: lim domain only 2; MAPK: mitogen activated protein kinase; MESP1: mesoderm posterior protein 1; MI: myocardial infarction; MO: morpholino; NFATc3: nuclear factor of activated T cells 3; NKX2-5: NK2 homeobox 5; NO: nitric oxide; NOTCH4: neurogenic locus notch homolog protein 4; PAD: peripheral artery disease; PDGF: platelet-derived growth factor; PI3K: Phosphoinositide 3-kinase; PKB: Protein kinase B; PLLA: polylactic acid; PLGA: poly lactic-co-glycolic acid; PSC: pluripotent stem cell; Rho-GTPase: ras homolog gene family GTPase activating protein; ROS: reactive oxygen species; SCA1: stem cells antigen1; SCF: stem cell factor; sgRNA: single guide RNA; TAL1: T-Cell acute lymphocytic leukemia protein 1 (also known as SCL); TET1/2: ten-eleven translocation ½; TGF: transforming growth factor; TIE2: TEK tyrosine kinase; VEGF: vascular endothelial growth factor; VEGFR3: vascular endothelial growth factor receptor 3; VWF: von willebrand factor; VPA: valproic acid; WNT: wingless-related integration site
Acknowledgements
This work was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2018R1D1A1A02085481) (T.M.K), and National Institutes of Health (NIH) R01 HL119291 (to C.P.), Children's Miracle Network 660085-1116 (to C.P.) and Children's Heart Research and Outcomes Center and Children's Healthcare of Atlanta 00060337 (to C.P.).
Authors' contributions
DHL and TMK: wrote the manuscript, CP: conceptualized, designed, wrote the manuscript, prepared figures with a help of DH, TMK and JKK.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Hollenhorst PC, McIntosh LP, Graves BJ. Genomic and biochemical insights into the specificity of ETS transcription factors. Annu Rev Biochem. 2011;80:437-471
2. Hollenhorst PC, Jones DA, Graves BJ. Expression profiles frame the promoter specificity dilemma of the ETS family of transcription factors. Nucleic Acids Res. 2004;32:5693-5702
3. Ayadi A, Suelves M, Dolle P, Wasylyk B. Net, an Ets ternary complex transcription factor, is expressed in sites of vasculogenesis, angiogenesis, and chondrogenesis during mouse development. Mech Dev. 2001;102:205-208
4. Craig MP, Sumanas S. ETS transcription factors in embryonic vascular development. Angiogenesis. 2016;19:275-285
5. Lee D, Park C, Lee H, Lugus JJ, Kim SH, Arentson E. et al. ER71 acts downstream of BMP, Notch, and Wnt signaling in blood and vessel progenitor specification. Cell Stem Cell. 2008;2:497-507
6. Liu F, Patient R. Genome-wide analysis of the zebrafish ETS family identifies three genes required for hemangioblast differentiation or angiogenesis. Circ Res. 2008;103:1147-1154
7. Maroulakou IG, Papas TS, Green JE. Differential expression of ets-1 and ets-2 proto-oncogenes during murine embryogenesis. Oncogene. 1994;9:1551-1565
8. Melet F, Motro B, Rossi DJ, Zhang L, Bernstein A. Generation of a novel Fli-1 protein by gene targeting leads to a defect in thymus development and a delay in Friend virus-induced erythroleukemia. Mol Cell Biol. 1996;16:2708-2718
9. Rasmussen TL, Kweon J, Diekmann MA, Belema-Bedada F, Song Q, Bowlin K. et al. ER71 directs mesodermal fate decisions during embryogenesis. Development. 2011;138:4801-4812
10. Stiegler P, Wolff CM, Meyer D, Senan F, Durliat M, Hourdry J. et al. The c-ets-1 proto-oncogenes in Xenopus laevis: expression during oogenesis and embryogenesis. Mech Dev. 1993;41:163-174
11. Wang LC, Kuo F, Fujiwara Y, Gilliland DG, Golub TR, Orkin SH. Yolk sac angiogenic defect and intra-embryonic apoptosis in mice lacking the Ets-related factor TEL. EMBO J. 1997;16:4374-4383
12. Park C, Kim TM, Malik AB. Transcriptional regulation of endothelial cell and vascular development. Circ Res. 2013;112:1380-1400
13. Barton K, Muthusamy N, Fischer C, Ting CN, Walunas TL, Lanier LL. et al. The Ets-1 transcription factor is required for the development of natural killer cells in mice. Immunity. 1998;9:555-563
14. Wei G, Srinivasan R, Cantemir-Stone CZ, Sharma SM, Santhanam R, Weinstein M. et al. Ets1 and Ets2 are required for endothelial cell survival during embryonic angiogenesis. Blood. 2009;114:1123-1130
15. Spyropoulos DD, Pharr PN, Lavenburg KR, Jackers P, Papas TS, Ogawa M. et al. Hemorrhage, impaired hematopoiesis, and lethality in mouse embryos carrying a targeted disruption of the Fli1 transcription factor. Mol Cell Biol. 2000;20:5643-5652
16. Weinl C, Wasylyk C, Garcia Garrido M, Sothilingam V, Beck SC, Riehle H. et al. Elk3 deficiency causes transient impairment in post-natal retinal vascular development and formation of tortuous arteries in adult murine retinae. PLoS One. 2014;9:e107048
17. Ferdous A, Caprioli A, Iacovino M, Martin CM, Morris J, Richardson JA. et al. Nkx2-5 transactivates the Ets-related protein 71 gene and specifies an endothelial/endocardial fate in the developing embryo. Proc Natl Acad Sci U S A. 2009;106:814-819
18. Kataoka H, Hayashi M, Nakagawa R, Tanaka Y, Izumi N, Nishikawa S. et al. Etv2/ER71 induces vascular mesoderm from Flk1+PDGFRalpha+ primitive mesoderm. Blood. 2011;118:6975-6986
19. Neuhaus H, Muller F, Hollemann T. Xenopus er71 is involved in vascular development. Dev Dyn. 2010;239:3436-3445
20. Sumanas S, Lin S. Ets1-related protein is a key regulator of vasculogenesis in zebrafish. PLoS Biol. 2006;4:e10
21. Pham VN, Lawson ND, Mugford JW, Dye L, Castranova D, Lo B. et al. Combinatorial function of ETS transcription factors in the developing vasculature. Dev Biol. 2007;303:772-783
22. De Haro L, Janknecht R. Cloning of the murine ER71 gene (Etsrp71) and initial characterization of its promoter. Genomics. 2005;85:493-502
23. Choi K, Kennedy M, Kazarov A, Papadimitriou JC, Keller G. A common precursor for hematopoietic and endothelial cells. Development. 1998;125:725-732
24. Kim JY, Lee RH, Kim TM, Kim DW, Jeon YJ, Huh SH. et al. OVOL2 is a critical regulator of ER71/ETV2 in generating FLK1+, hematopoietic, and endothelial cells from embryonic stem cells. Blood. 2014;124:2948-2952
25. Sumanas S, Jorniak T, Lin S. Identification of novel vascular endothelial-specific genes by the microarray analysis of the zebrafish cloche mutants. Blood. 2005;106:534-541
26. Stainier DY, Weinstein BM, Detrich HW 3rd, Zon LI, Fishman MC. Cloche, an early acting zebrafish gene, is required by both the endothelial and hematopoietic lineages. Development. 1995;121:3141-3150
27. Liu F, Kang I, Park C, Chang LW, Wang W, Lee D. et al. ER71 specifies Flk-1+ hemangiogenic mesoderm by inhibiting cardiac mesoderm and Wnt signaling. Blood. 2012;119:3295-3305
28. Oh SY, Kim JY, Park C. The ETS Factor, ETV2: a Master Regulator for Vascular Endothelial Cell Development. Mol Cells. 2015;38:1029-1036
29. Sumanas S, Choi K. ETS Transcription Factor ETV2/ER71/Etsrp in Hematopoietic and Vascular Development. Curr Top Dev Biol. 2016;118:77-111
30. De Val S, Chi NC, Meadows SM, Minovitsky S, Anderson JP, Harris IS. et al. Combinatorial regulation of endothelial gene expression by ets and forkhead transcription factors. Cell. 2008;135:1053-1064
31. Lee D, Kim T, Lim DS. The Er71 is an important regulator of hematopoietic stem cells in adult mice. Stem Cells. 2011;29:539-548
32. Tanaka T, Izawa K, Maniwa Y, Okamura M, Okada A, Yamaguchi T. et al. ETV2-TET1/TET2 Complexes Induce Endothelial Cell-Specific Robo4 Expression via Promoter Demethylation. Sci Rep. 2018;8:5653
33. Liu F, Li D, Yu YY, Kang I, Cha MJ, Kim JY. et al. Induction of hematopoietic and endothelial cell program orchestrated by ETS transcription factor ER71/ETV2. EMBO Rep. 2015;16:654-669
34. Abedin MJ, Nguyen A, Jiang N, Perry CE, Shelton JM, Watson DK. et al. Fli1 acts downstream of Etv2 to govern cell survival and vascular homeostasis via positive autoregulation. Circ Res. 2014;114:1690-1699
35. Craig MP, Grajevskaja V, Liao HK, Balciuniene J, Ekker SC, Park JS. et al. Etv2 and fli1b function together as key regulators of vasculogenesis and angiogenesis. Arterioscler Thromb Vasc Biol. 2015;35:865-876
36. Hayashi M, Pluchinotta M, Momiyama A, Tanaka Y, Nishikawa S, Kataoka H. Endothelialization and altered hematopoiesis by persistent Etv2 expression in mice. Exp Hematol. 2012;40:738-750
37. Baltrunaite K, Craig MP, Palencia Desai S, Chaturvedi P, Pandey RN, Hegde RS. et al. ETS transcription factors Etv2 and Fli1b are required for tumor angiogenesis. Angiogenesis. 2017;20:307-323
38. Davis JA, Koenig AL, Lubert A, Chestnut B, Liu F, Palencia Desai S. et al. ETS transcription factor Etsrp / Etv2 is required for lymphangiogenesis and directly regulates vegfr3 / flt4 expression. Dev Biol. 2018;440:40-52
39. Cui Y. Pulmonary hemodynamic effects resulting from mediastinal lymphatic obstruction in anesthetized rabbits. Med Sci Res. 1999;27:345-348
40. Ludwig LL, Schertel ER, Pratt JW, McClure DE, Ying AJ, Heck CF. et al. Impairment of left ventricular function by acute cardiac lymphatic obstruction. Cardiovasc Res. 1997;33:164-171
41. Ullal SR, Kluge TH, Gerbode F. Functional and pathologic changes in the heart following chronic cardiac lymphatic obstruction. Surgery. 1972;71:328-334
42. Lund AW, Duraes FV, Hirosue S, Raghavan VR, Nembrini C, Thomas SN. et al. VEGF-C promotes immune tolerance in B16 melanomas and cross-presentation of tumor antigen by lymph node lymphatics. Cell Rep. 2012;1:191-199
43. Hajrasouliha AR, Funaki T, Sadrai Z, Hattori T, Chauhan SK, Dana R. Vascular endothelial growth factor-C promotes alloimmunity by amplifying antigen-presenting cell maturation and lymphangiogenesis. Invest Ophthalmol Vis Sci. 2012;53:1244-1250
44. Shi X, Richard J, Zirbes KM, Gong W, Lin G, Kyba M. et al. Cooperative interaction of Etv2 and Gata2 regulates the development of endothelial and hematopoietic lineages. Dev Biol. 2014;389:208-218
45. Unezaki S, Horai R, Sudo K, Iwakura Y, Ito S. Ovol2/Movo, a homologue of Drosophila ovo, is required for angiogenesis, heart formation and placental development in mice. Genes Cells. 2007;12:773-785
46. Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA. et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333:1300-1303
47. Bedell VM, Yeo SY, Park KW, Chung J, Seth P, Shivalingappa V. et al. Roundabout4 is essential for angiogenesis in vivo. Proc Natl Acad Sci U S A. 2005;102:6373-6378
48. Kabir AU, Lee TJ, Pan H, Berry JC, Krchma K, Wu J. et al. Requisite endothelial reactivation and effective siRNA nanoparticle targeting of Etv2/Er71 in tumor angiogenesis. JCI Insight. 2018;3(8):e97349
49. Park C, Lee TJ, Bhang SH, Liu F, Nakamura R, Oladipupo SS. et al. Injury-Mediated Vascular Regeneration Requires Endothelial ER71/ETV2. Arterioscler Thromb Vasc Biol. 2016;36:86-96
50. Xu C, Lee TJ, Sakurai T, Krchma K, Liu F, Li D. et al. ETV2/ER71 regulates hematopoietic regeneration by promoting hematopoietic stem cell proliferation. J Exp Med. 2017;214:1643-1653
51. Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K. et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861-872
52. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663-676
53. Huang P, He Z, Ji S, Sun H, Xiang D, Liu C. et al. Induction of functional hepatocyte-like cells from mouse fibroblasts by defined factors. Nature. 2011;475:386-389
54. Ieda M, Fu JD, Delgado-Olguin P, Vedantham V, Hayashi Y, Bruneau BG. et al. Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell. 2010;142:375-386
55. Song K, Nam YJ, Luo X, Qi X, Tan W, Huang GN. et al. Heart repair by reprogramming non-myocytes with cardiac transcription factors. Nature. 2012;485:599-604
56. Vierbuchen T, Ostermeier A, Pang ZP, Kokubu Y, Sudhof TC, Wernig M. Direct conversion of fibroblasts to functional neurons by defined factors. Nature. 2010;463:1035-1041
57. Ambasudhan R, Talantova M, Coleman R, Yuan X, Zhu S, Lipton SA. et al. Direct reprogramming of adult human fibroblasts to functional neurons under defined conditions. Cell Stem Cell. 2011;9:113-118
58. Jayawardena TM, Egemnazarov B, Finch EA, Zhang L, Payne JA, Pandya K. et al. MicroRNA-mediated in vitro and in vivo direct reprogramming of cardiac fibroblasts to cardiomyocytes. Circ Res. 2012;110:1465-1473
59. Jayawardena TM, Finch EA, Zhang L, Zhang H, Hodgkinson CP, Pratt RE. et al. MicroRNA induced cardiac reprogramming in vivo: evidence for mature cardiac myocytes and improved cardiac function. Circ Res. 2015;116:418-424
60. Yoo AS, Sun AX, Li L, Shcheglovitov A, Portmann T, Li Y. et al. MicroRNA-mediated conversion of human fibroblasts to neurons. Nature. 2011;476:228-231
61. Ginsberg M, James D, Ding BS, Nolan D, Geng F, Butler JM. et al. Efficient direct reprogramming of mature amniotic cells into endothelial cells by ETS factors and TGFbeta suppression. Cell. 2012;151:559-575
62. Wong WT, Cooke JP. Therapeutic transdifferentiation of human fibroblasts into endothelial cells using forced expression of lineage-specific transcription factors. J Tissue Eng. 2016;7:1-10
63. Morita R, Suzuki M, Kasahara H, Shimizu N, Shichita T, Sekiya T. et al. ETS transcription factor ETV2 directly converts human fibroblasts into functional endothelial cells. Proc Natl Acad Sci U S A. 2015;112:160-165
64. Van Pham P, Vu NB, Nguyen HT, Huynh OT, Truong MT. Significant improvement of direct reprogramming efficacy of fibroblasts into progenitor endothelial cells by ETV2 and hypoxia. Stem Cell Res Ther. 2016;7:104
65. Lee S, Park C, Han JW, Kim JY, Cho K, Kim EJ. et al. Direct Reprogramming of Human Dermal Fibroblasts Into Endothelial Cells Using ER71/ETV2. Circ Res. 2017;120:848-861
66. Han JK, Chang SH, Cho HJ, Choi SB, Ahn HS, Lee J. et al. Direct conversion of adult skin fibroblasts to endothelial cells by defined factors. Circulation. 2014;130:1168-1178
67. Le Bras A, Yu B, Issa Bhaloo S, Hong X, Zhang Z, Hu Y. et al. Adventitial Sca1+ Cells Transduced With ETV2 Are Committed to the Endothelial Fate and Improve Vascular Remodeling After Injury. Arterioscler Thromb Vasc Biol. 2018;38:232-244
68. Veldman MB, Zhao C, Gomez GA, Lindgren AG, Huang H, Yang H. et al. Transdifferentiation of fast skeletal muscle into functional endothelium in vivo by transcription factor Etv2. PLoS Biol. 2013;11:e1001590
69. Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M. et al. Heart disease and stroke statistics-2015 update: a report from the American Heart Association. Circulation. 2015;131:e29-e322
70. Choi JS, Kim KB, Han W, Kim DS, Park JS, Lee JJ. et al. Efficacy of therapeutic angiogenesis by intramyocardial injection of pCK-VEGF165 in pigs. Ann Thorac Surg. 2006;82:679-686
71. Gao MH, Lai NC, McKirnan MD, Roth DA, Rubanyi GM, Dalton N. et al. Increased regional function and perfusion after intracoronary delivery of adenovirus encoding fibroblast growth factor 4: report of preclinical data. Hum Gene Ther. 2004;15:574-587
72. Hao X, Mansson-Broberg A, Grinnemo KH, Siddiqui AJ, Dellgren G, Brodin LA. et al. Myocardial angiogenesis after plasmid or adenoviral VEGF-A(165) gene transfer in rat myocardial infarction model. Cardiovasc Res. 2007;73:481-487
73. Horvath KA, Doukas J, Lu CY, Belkind N, Greene R, Pierce GF. et al. Myocardial functional recovery after fibroblast growth factor 2 gene therapy as assessed by echocardiography and magnetic resonance imaging. Ann Thorac Surg. 2002;74:481-486
74. Lahteenvuo JE, Lahteenvuo MT, Kivela A, Rosenlew C, Falkevall A, Klar J. et al. Vascular endothelial growth factor-B induces myocardium-specific angiogenesis and arteriogenesis via vascular endothelial growth factor receptor-1- and neuropilin receptor-1-dependent mechanisms. Circulation. 2009;119:845-856
75. Nakamura T, Mizuno S, Matsumoto K, Sawa Y, Matsuda H, Nakamura T. Myocardial protection from ischemia/reperfusion injury by endogenous and exogenous HGF. J Clin Invest. 2000;106:1511-1519
76. Zangi L, Lui KO, von Gise A, Ma Q, Ebina W, Ptaszek LM. et al. Modified mRNA directs the fate of heart progenitor cells and induces vascular regeneration after myocardial infarction. Nat Biotechnol. 2013;31:898-907
77. Ishikawa K, Weber T, Hajjar RJ. Human Cardiac Gene Therapy. Circ Res. 2018;123:601-613
78. Lee S, Lee DH, Park BW, Kim R, Hoang AD, Woo SK. et al. In vivo transduction of ETV2 improves cardiac function and induces vascular regeneration following myocardial infarction. Exp Mol Med. 2019;51:13
79. Serhan CN. Treating inflammation and infection in the 21st century: new hints from decoding resolution mediators and mechanisms. FASEB J. 2017;31:1273-1288
80. Zhao C, Gomez GA, Zhao Y, Yang Y, Cao D, Lu J. et al. ETV2 mediates endothelial transdifferentiation of glioblastoma. Signal Transduct Target Ther. 2018;3:4
81. Yan G, Yan R, Chen C, Chen C, Zhao Y, Qin W. et al. Engineering vascularized skeletal muscle tissue with transcriptional factor ETV2-induced autologous endothelial cells. Protein Cell. 2019;10:217-222
82. Cheng F, Zhang Y, Wang Y, Jiang Q, Zhao CJ, Deng J. et al. Conversion of human adipose-derived stem cells into functional and expandable endothelial-like cells for cell-based therapies. Stem Cell Res Ther. 2018;9:350
83. Bjorge IM, Kim SY, Mano JF, Kalionis B, Chrzanowski W. Extracellular vesicles, exosomes and shedding vesicles in regenerative medicine - a new paradigm for tissue repair. Biomater Sci. 2017;6:60-78
84. Dickhout A, Koenen RR. Extracellular Vesicles as Biomarkers in Cardiovascular Disease; Chances and Risks. Front Cardiovasc Med. 2018;5:113
85. Boulanger CM, Loyer X, Rautou PE, Amabile N. Extracellular vesicles in coronary artery disease. Nat Rev Cardiol. 2017;14:259-272
86. Van Pham P, Vu NB, Dao TT, Le HT, Phi LT, Huynh OT. et al. Extracellular vesicles of ETV2 transfected fibroblasts stimulate endothelial cells and improve neovascularization in a murine model of hindlimb ischemia. Cytotechnology. 2017;69:801-814
87. Hodgkinson CP, Kang MH, Dal-Pra S, Mirotsou M, Dzau VJ. MicroRNAs and Cardiac Regeneration. Circ Res. 2015;116:1700-1711
88. Xie X, Fu Y, Liu J. Chemical reprogramming and transdifferentiation. Curr Opin Genet Dev. 2017;46:104-113
89. Elcheva I, Brok-Volchanskaya V, Kumar A, Liu P, Lee JH, Tong L. et al. Direct induction of haematoendothelial programs in human pluripotent stem cells by transcriptional regulators. Nat Commun. 2014;5:4372
90. Suknuntha K, Tao L, Brok-Volchanskaya V, D'Souza SS, Kumar A, Slukvin I. Optimization of Synthetic mRNA for Highly Efficient Translation and its Application in the Generation of Endothelial and Hematopoietic Cells from Human and Primate Pluripotent Stem Cells. Stem Cell Rev. 2018;14:525-534
91. Black JB, Adler AF, Wang HG, D'Ippolito AM, Hutchinson HA, Reddy TE. et al. Targeted Epigenetic Remodeling of Endogenous Loci by CRISPR/Cas9-Based Transcriptional Activators Directly Converts Fibroblasts to Neuronal Cells. Cell Stem Cell. 2016;19:406-414
92. Weltner J, Balboa D, Katayama S, Bespalov M, Krjutskov K, Jouhilahti EM. et al. Human pluripotent reprogramming with CRISPR activators. Nat Commun. 2018;9:2643
93. Xiong K, Zhou Y, Blichfeld KA, Hyttel P, Bolund L, Freude KK. et al. RNA-Guided Activation of Pluripotency Genes in Human Fibroblasts. Cell Reprogram. 2017;19:189-198
94. Veldman MB, Lin S. Etsrp/Etv2 is directly regulated by Foxc1a/b in the zebrafish angioblast. Circ Res. 2012;110:220-229
95. Shi X, Zirbes KM, Rasmussen TL, Ferdous A, Garry MG, Koyano-Nakagawa N. et al. The transcription factor Mesp1 interacts with cAMP-responsive element binding protein 1 (Creb1) and coactivates Ets variant 2 (Etv2) gene expression. J Biol Chem. 2015;290:9614-9625
96. Wang YJ, Huang J, Liu W, Kou X, Tang H, Wang H. et al. IP3R-mediated Ca2+ signals govern hematopoietic and cardiac divergence of Flk1+ cells via the calcineurin-NFATc3-Etv2 pathway. J Mol Cell Biol. 2017;9:274-288
97. Yamamizu K, Matsunaga T, Katayama S, Kataoka H, Takayama N, Eto K. et al. PKA/CREB signaling triggers initiation of endothelial and hematopoietic cell differentiation via Etv2 induction. Stem Cells. 2012;30:687-696
98. Hanoune J, Defer N. Regulation and role of adenylyl cyclase isoforms. Annu Rev Pharmacol Toxicol. 2001;41:145-174
99. Bondue A, Lapouge G, Paulissen C, Semeraro C, Iacovino M, Kyba M. et al. Mesp1 acts as a master regulator of multipotent cardiovascular progenitor specification. Cell Stem Cell. 2008;3:69-84
100. Shimekake Y, Nagata K, Ohta S, Kambayashi Y, Teraoka H, Kitamura K. et al. Adrenomedullin Stimulates Two Signal Transduction Pathways, cAMP Accumulation and Ca2+ Mobilization, in Bovine Aortic Endothelial Cells. J Biol Chem. 1995;9:4412-4417
101. Yuan M, Wang Q, Li C, Tao L, Zhang H, Wang H. et al. Adrenomedullin in vascular endothelial injury and combination therapy: Time for a new paradigm. Curr Vasc Pharmacol. 2015;13:459-466
102. Trincot CE, Xu W, Zhang H, Kulikauskas MR, Caranasos TG, Jensen BC. et al. Adrenomedullin Induces Cardiac Lymphangiogenesis After Myocardial Infarction and Regulates Cardiac Edema Via Connexin 43. Circ Res. 2019;124:101-113
103. Iwase T, Nagaya N, Fujii T, Itoh T, Ishibashi-Ueda H, Yamagishi M. et al. Adrenomedullin enhances angiogenic potency of bone marrow transplantation in a rat model of hindlimb ischemia. Circulation. 2005;111:356-362
104. Tokunaga N, Nagaya N, Shirai M, Tanaka E, Ishibashi-Ueda H, Harada-Shiba M. et al. Adrenomedullin gene transfer induces therapeutic angiogenesis in a rabbit model of chronic hind limb ischemia: benefits of a novel nonviral vector, gelatin. Circulation. 2004;109:526-531
105. Iimuro S, Shindo T, Moriyama N, Amaki T, Niu P, Takeda N. et al. Angiogenic effects of adrenomedullin in ischemia and tumor growth. Circ Res. 2004;95:415-423
106. Imai Y, Shiindo T, Maemura K, Kurihara Y, Nagai R, Kurihara H. Evidence for the physiological and pathological roles of adrenomedullin from genetic engineering in mice. Ann N Y Acad Sci. 2001;947:26-33
107. Kim YW, Byzova TV. Oxidative stress in angiogenesis and vascular disease. Blood. 2014;123:625-631
108. Miller AJ, Pick R, Katz LN. Ventricular endomyocardial pathology produced by chronic cardiac lymphatic obstruction in the dog. Circ Res. 1960;8:941-947
109. Solti F, Lengyel E, Jellinek H, Schneider F, Juhasz-Nagy A, Kekesi V. Coronary arteriopathy after lymphatic blockade: an experimental study in dogs. Lymphology. 1994;27:173-180
110. Sun SC, Lie JT. Cardiac lymphatic obstruction: ultrastructure of acute-phase myocardial injury in dogs. Mayo Clin Proc. 1977;52:785-792
111. Dixon JA, Spinale FG. Large animal models of heart failure: a critical link in the translation of basic science to clinical practice. Circ Heart Fail. 2009;2:262-271
112. Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W. et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci U S A. 2013;110:3507-3512
113. Savoji H, Mohammadi MH, Rafatian N, Toroghi MK, Wang EY, Zhao Y. et al. Cardiovascular disease models: A game changing paradigm in drug discovery and screening. Biomaterials. 2019;198:3-26
114. Vera Janavel G, Crottogini A, Cabeza Meckert P, Cuniberti L, Mele A, Papouchado M. et al. Plasmid-mediated VEGF gene transfer induces cardiomyogenesis and reduces myocardial infarct size in sheep. Gene Ther. 2006;13:1133-1142
115. Ishikawa K, Fish K, Aguero J, Yaniz-Galende E, Jeong D, Kho C. et al. Stem cell factor gene transfer improves cardiac function after myocardial infarction in swine. Circ Heart Fail. 2015;8:167-174
116. Kim AK, Kim MH, Kim BS, Kim DI. Long-term Angiogenesis Efficacy Using a Heparin-Conjugated Fibrin (HCF) Delivery System with HBM-MSCs. Int J Stem Cells. 2012;5:23-30
117. Harding J, Roberts RM, Mirochnitchenko O. Large animal models for stem cell therapy. Stem Cell Res Ther. 2013;4:23
Author contact
![]() Corresponding author: Changwon Park, Department of Pediatrics, Emory University School of Medicine, 2015 Uppergate Dr. Atlanta GA, Tel:404-727-7143, Fax: 404-727-5737, Email:cpark23edu
Corresponding author: Changwon Park, Department of Pediatrics, Emory University School of Medicine, 2015 Uppergate Dr. Atlanta GA, Tel:404-727-7143, Fax: 404-727-5737, Email:cpark23edu