Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Conclusions
Methods
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(15):4525-4541. doi:10.7150/thno.34676 This issue Cite
Research Paper
Functionalization of SF/HAP Scaffold with GO-PEI-miRNA inhibitor Complexes to Enhance Bone Regeneration through Activating Transcription Factor 4
Lingling Ou1, Yong Lan2, Zhiqiang Feng1, Longbao Feng2, Junjie Yang1, Yu Liu3, Liming Bian4, Jiali Tan5, Renfa Lai1, ![]() , Rui Guo6,
, Rui Guo6, ![]()
1. The First Affiliated Hospital of Jinan University, Guangzhou, 510632, China
2. Beogene Biotech (Guangzhou) CO., LTD, Guangzhou 510663, China
3. Guangzhou Chuangseed Biomedical Materials CO., LTD, Guangzhou 510663, China
4. Department of Biomedical Engineering, The Chinese University of Hong Kong, Hong Kong SAR, China
5. Department of Orthodontics, Guanghua School of Stomatology, Hospital of Stomatology, Sun Yat-sen University and Guangdong Provincial Key Laboratory of Stomatology, Guangzhou, 510055, China
6. Key Laboratory of Biomaterials of Guangdong Higher Education Institutes, Guangdong Provincial Engineering and Technological Research Center for Drug Carrier Development, Department of Biomedical Engineering, Jinan University, Guangzhou 510632, China
Received 2019-3-6; Accepted 2019-4-12; Published 2019-6-9
Abstract

Evidence indicates that microRNAs (miRNAs) play vital roles in regulating osteogenic differentiation and bone formation.
Methods: Here, we show that a polyethyleneimine (PEI)-functionalized graphene oxide (GO) complex efficiently loaded with the miR-214 inhibitor is assembled into silk fibroin/hydroxyapatite (SF/HAP) scaffolds that spatially control the release of the miR-214 inhibitor.
Results: SF/HAP/GO scaffolds with nanosized GO show high mechanical strength, and their hierarchical microporous structures promote cell adhesion and growth. The SF/HAP/GO-PEI scaffolds loaded with mir-214 inhibitor (SF/HAP/GPM) were tested for their ability to enhance osteogenic differentiation by inhibiting the expression of miR-214 while inversely increasing the expression of activating transcription factor 4 (ATF4) and activating the Akt and ERK1/2 signaling pathways in mouse osteoblastic cells (MC3T3-E1) in vitro. Similarly, the scaffolds activated the osteoblastic activity of endogenous osteoblast cells to repair critical-sized bone defects in rats without the need for loading osteoblast cells.
Conclusion: This technology is used to increase osteogenic differentiation and mineralized bone formation in bone defects, which helps to achieve cell-free scaffold-based miRNA-inhibitor therapy for bone tissue engineering.
Keywords: miRNA-214 inhibitor, GO-PEI, SF/HAP scaffold, activating transcription factor 4 (ATF4), bone regeneration
Introduction
Gene therapy offers an alternative for bone remodeling and regeneration [1, 2]. Instead of using plasmid DNA, developing microRNA (miRNA) has recently attracted much attention. miRNAs function as posttranscriptional repressors involved in the regulation of osteogenic differentiation and osteoblastic bone formation [3, 4]. miR-214 has been demonstrated to inhibit osteoblast differentiation and osteoblastic bone formation under both physiological and pathophysiological conditions [5]. Thus, miR-214 could be a therapeutic target for miRNA inhibitors to promote bone formation in the bone defect or reverse osteoporosis under pathophysiological conditions.
miRNAs cannot easily penetrate the negatively charged cell membrane with Coulomb repulsion and can rapidly degraded in vivo [6]. Therefore, vectors are required for miRNA therapy to protect and deliver miRNAs into cell types, tissues or organs [7]. Viral vectors have efficient delivery but may cause adverse immune responses, and the viral gene risks being integrated into the host genome [8, 9]. Nonviral vectors, such as Lipofectamine, cholesterol, and polymer, have poor transfection efficiencies [10-14]. For instance, frequently used DNA delivery polymer, polyethyleneimine (PEI) has been used to deliver miRNA [15, 16]. However, PEI with low molecular weight has low transfection efficiency, PEI with high molecular weight is harmful to cells [17]. Thus, developing an effective delivery platform for synthetic miRNA oligonucleotides is still ongoing.
Research has shown that graphene can load with biomolecules such as proteins, peptides and DNA for tissue regeneration and tissue engineering [18]. Negatively charged graphene oxide (GO) nanosheets bonded with cationic polymers have been used to deliver DNAs or RNAs efficiently [19]. Therefore, we designed GO-PEI complexes for a synthetic miR-inhibitor delivery system, in which a cationic PEI bound to the surface of GO and formed a shell for miR-inhibitor with controlled release. This GO-PEI-miR-inhibitor delivery method combines high transfection efficiency and controllable release duration.
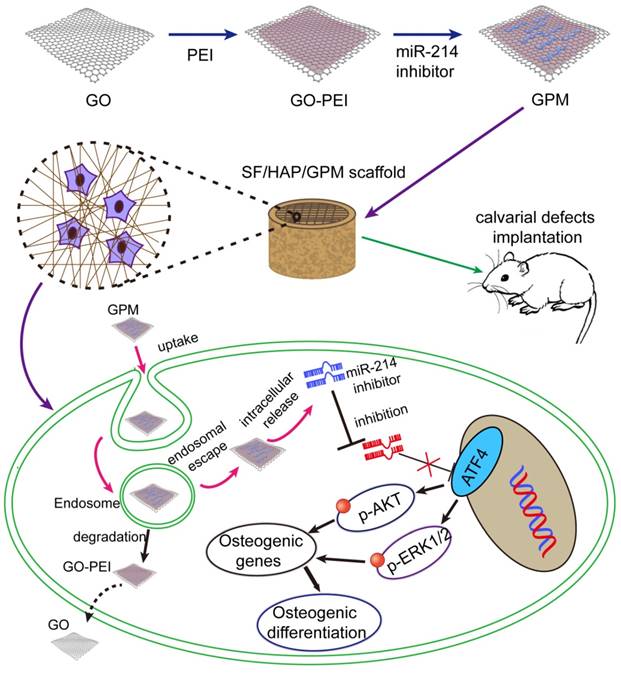
A three-dimensional microenvironment is important in tissue engineering. Available research data suggest that silk fibroin/hydroxyapatite (SF/HAP) scaffolds can be an excellent material for bone regeneration [20, 21]. GO structures also provide permissive surfaces for cell adhesion and affect cell morphology [22]. Here, we immobilize a GO-PEI-miR-inhibitor (GPM) in an SF/HAP cell-free scaffold and let miR-inhibitor release moderately, and sustained local miR-inhibitor delivery can activate osteoblast cells to repair critical-sized calvarial bone defects in mice. And schematic description of GO-PEI-miRNA inhibitor complexes immobilized on an SF/HAP scaffold was showed in Figure 1.
Schematic representation of miR-214 inhibitor delivery from GO-PEI complexes immobilized on an SF/HAP scaffold. GO and PEI formed GO-PEI complexes, and the miR-214 inhibitor was coupled to the GO-PEI complexes. The GPM complexes were then assembled into the SF/HAP scaffolds. The GPM-incorporated SF/HAP scaffolds were implanted into the calvarial bone defects of rats. GPM complexes released from the SF/HAP scaffolds could be taken up by cells through endocytosis. The intracellular release of the miR-214 inhibitor allowed its regulation of gene expression. (GPM meant GO-PEI-miR-inhibitor)
Results
1 Characterizations of GO-PEI and GO-PEI-miRNA complexes
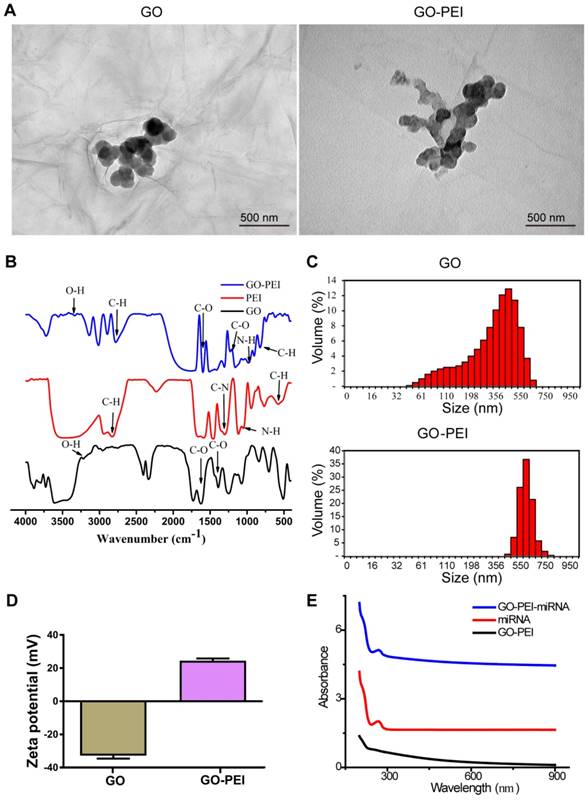
A previous report suggested that the interaction between GO and double-stranded DNA is via π-π stacking, which is relatively weak [23, 24]. Thus, we adopted GO coated with PEI, the “gold standard” cationic polymer among delivery agents, to perform the miR-214 inhibitor transfection [25, 26]. For GO-PEI complex preparation, PEI solution was slowly added into a GO solution (100 μg/mL) at ratio of 1:2 (GO:PEI). Unbound PEI was discarding by centrifuging and washing. GO-PEI complexes were stable in both phosphate-buffered solution (PBS) and culture medium without obvious aggregation as previously reported [27]. Transmission electron microscopy (TEM) analysis revealed that the functionalization of GO with PEI changed the surface slightly increased the size of GO-PEI (Figure 2A). The FT-IR was used so as to confirm the synthesis process and the results were shown in Figure 2B. The peak at 3400 cm-1 was related to the O-H, while the peaks at 1620 cm-1 and 1400 cm-1 were attributed to C=O. CO bonds existed due to the peak appearance at 1100 cm-1 and that indicated the GO had some Oxygen-containing functional groups such as O-H and C=O [27]. Furthermore, several peaks appeared in the spectrum after the functionalization procedure. The peaks at 2894 cm-1 and 2810 cm-1 attributed to the CH2 vibration of PEI, which indicated the PEI was conjugated to GO successfully. Dynamic light scattering (DLS) analysis showed the mean diameters of GO and GO-PEI were 375.51 ± 36.8 nm and 749.31 ± 35.9 nm, respectively (Figure 2C). The surface charge of GO and GO-PEI was analyzed using an electrophoretic light-scattering spectrophotometer. The zeta potential of GO was -32.73 ± 3.17 mV, whereas that of GO-PEI was 24.5 ± 2.27 mV (Figure 2D). These results demonstrated that positive charged PEI binding to negatively charged GO sheets and made the GO-PEI complexes bear positive charge [28, 29]. Moreover, positively charged PEI of GO-PEI complexes could bind and wrap negatively charged miRNA. The UV-Vis spectrum measurement was carried out for the miRNA-inhibitor-conjugated GO-PEI using a UV-Vis instrument (Shimadzu, Japan), and miRNA had an ultraviolet absorbance peak at 280 nm (Figure 2E).
Characterization of GO-PEI complexes. (A) SEM images of GO and GO-PEI complexes. Scale bars: 500 nm. (B) FT-IR spectra results of GO (bottom), PEI (middle), and GO-PEI complexes (top). (C) Size distributions of GO and GO-PEI measured by DLS analysis. (D) Measured values of the zeta potential of GO and GO-PEI complexes. (E) UV-Vis spectra normalized by the extinction coefficients of the complexes at 260 nm. Black line for the GO-PEI complex, red line for the free RNA sense strand, and blue line for miRNA-conjugated GO-PEI in phosphate buffer (pH 7.4).
2 Evaluation of GO-PEI complex biocompatibility and delivery effectiveness
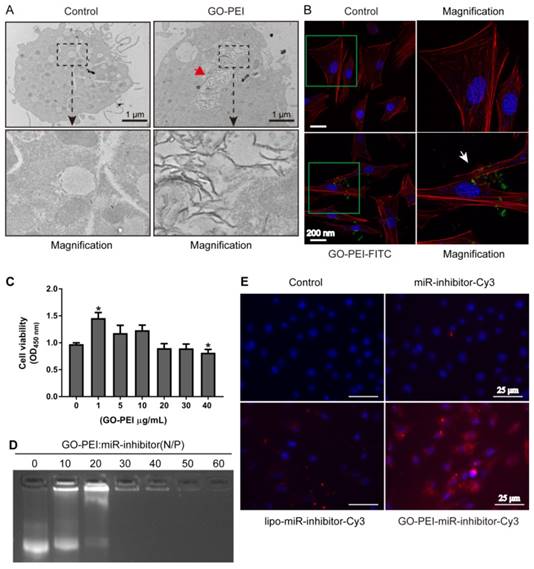
To measure the delivery efficiency of miRNA inhibitor into the cells, we first observed the uptake of GO-PEI complexes into MC3T3-E1 cells by TEM. GO-PEI was found in both the cytoplasm and vesicles inside the cells after 24 h of incubation with 3 μg/mL of GO-PEI, and no sheets were found in non-GO-PEI treated cells (named as control group) (Figure 3A). In addition, we also labeled GO-PEI with FITC to track its cellular uptake through laser scanning confocal microscopy (LSCM). FITC-labeled GO-PEI complexes (3 μg/mL) were incubated with the cells and could be detected on the cell plasma membranes after 6 h and inside the cells after 12 h. Most of GO-PEI complexes were located in the cell cytoplasm, and some were located within the nucleus (Figure 3B). To test our hypothesis that such GO-PEI complexes result in low cytotoxicity, (CCK)-8 assay was performed for MC3T3-E1 cells treated with GO-PEI [30]. There was no significant toxicity in the cells even when the concentration of GO-PEI reached 40 μg/mL. Interestingly, 1 μg/mL GO-PEI promoted cell proliferation, which is consistent with previous results showing that GO enhances the growth and differentiation potential of cells [31]. Moreover, the results demonstrated that GO-PEI-treated cells were still viable after treated for 24 h or 48 h. Thus, GO-PEI had good biocompatibility and low toxicity (Figure 3C). To further assay the affinity for miRNA inhibitors and the transfection efficiency of GO-PEI, we examined the binding capacity of GO-PEI complexes to miR-inhibitors using agarose gel electrophoresis. GO-PEI was complexed with the miR-inhibitor at various N/P ratios (molar ratio of nitrogen of PEI to phosphate groups of miRNA). The GPM complex demonstrated obvious electrophoretic retardation at the N/P ratio of 30 (Figure 3D), which meant that when GPM complex reached certain concentration, it would wrap miR-inhibitor inside and prevent miR-inhibitor being degraded. In addition, we assessed the GO-PEI complex delivery efficiency for miR-inhibitor using MC3T3-E1 cells, and the miR-inhibitor was labeled with Cy3. The fluorescence microscopy images showed that the red fluorescence was robustly increased in cells treated with GO-PEI compared to that in cells treated with the naked miR-inhibitor or in cells transfected with Lipofectamine 2000 for 24 h (Figure 3E). These results indicated that GO-PEI-miRNA had good cell uptake and transfection efficacy.
Evaluation of GO-PEI complex biocompatibility and delivery effectiveness. (A) TEM images of non-GO-PEI cells (named as control) and GO-PEI-induced cells (the concentration of GO-PEI in the culture medium was 3 μg/mL). The images below each group were the enlarged images of the black squares. The red arrowhead indicates GO-PEI inside the cells. Scale bars: 1 μm. (B) Fluorescent images of FITC-labeled GO-PEI (green) within MC3T3-E1 cells are shown. The cell cytoskeleton was stained with phalloidin (red), and the nuclei were stained with DAPI (blue). The FITC-labeled GO-PEI inside the cells was indicated by the white arrowhead. Scale bars: 200 nm. The images on the right of each group are the enlarged images of green squares. (C) The viability of MC3T3-E1 cells was measured by the CCK-8 assay after incubation with various concentrations of GO-PEI for 48 h. *p < 0.05. (D) A gel retardation assay of the mixture of GO-PEI and miRNA at different N/P ratios (0, 10, 20, 30, 40, 50 and 60). (E) Fluorescent images for GO-PEI complex delivery of Cy3-labeled miR-214 inhibitor after 24 h of incubation. miR-214 was labeled using Cy3 (orange red), and the cell nuclei were stained with DAPI (blue).
3 GO-PEI-miR-214-inhibitor promoted osteoblast activity and mineralization
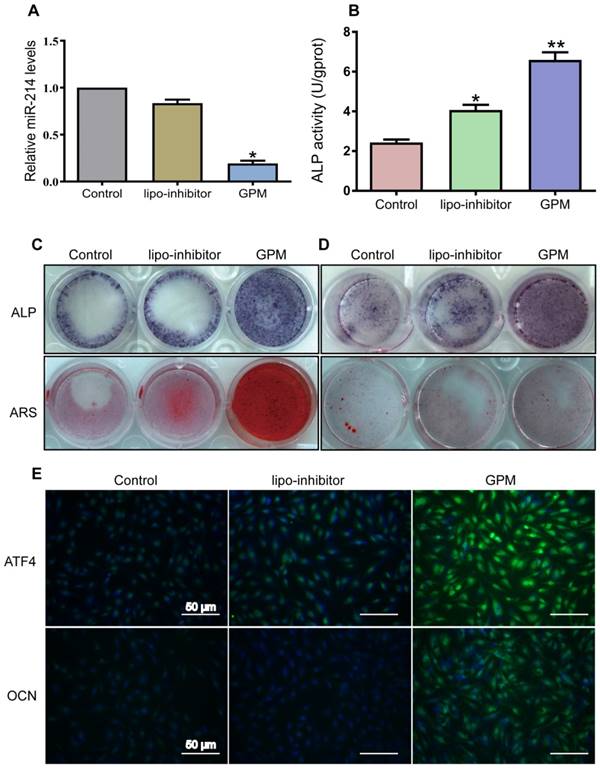
To detect whether the GO-PEI-miR-214 inhibitor exerts an effect inside the cells, we treated MC3T3-E1 cells or bone marrow mesenchymal stem cells (BMSCs) with GPM. Intracellular miR-214 levels were markedly downregulated by 24 h of GPM treatment (Figure 4A). In contrast, alkaline phosphatase (ALP) was upregulated in the cells with GPM treatment compared to that in the cells treated with lipo-transfected inhibitor and control for 72 h (Figure 4B). Consistent with these changes, GPM enhanced ALP staining and alizarin red staining (ARS) in both MC3T3-E1 cells and BMSCs, resulting in more mineral deposition in the GPM group than in the lipo-transfected inhibitor and control groups (Figure 4C and D). One of the direct targets of miR-214 is ATF4 [5], which is one of the important transcription factors required for osteoblast function [32, 33]. GMP might therapeutically inhibit miR-214 and result in the suppression of ATF4. The immunofluorescence results showed that GPM increased the protein levels of ATF4 and osteocalcin (OCN) in the MC3T3-E1 cells (Figure 4E).
GO-PEI-miR-214-inhibitor promoted osteoblast activity and mineralization. (A) Intracellular miR-214 levels in MC3T3-E1 cells after incubation with GPM for 24 h. (B) ALP activities of MC3T3-E1 cells were measured after incubation with GPM for 72 h. (C) Representative ALP and ARS staining image of MC3T3-E1 cells were shown after GPM incubation for 72 h. (D) Representative ALP and ARS staining image of mouse bone marrow stem cells were shown after GPM incubation for 72 h. (E) Fluorescent images of ATF4 and OCN expression in MC3T3-E1 cells after GPM incubation for 72 h. Scale bars: 50 μm.
4 Characterization and degradation of SF/HAP/GO-PEI-miR scaffolds
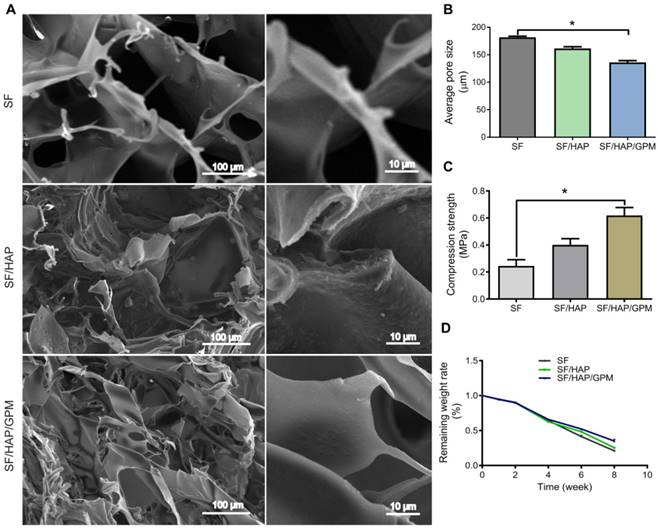
Macroscopic images of freeze-dried scaffolds (5 mm in diameter) were shown in Supplementary Figure 1A (Figure S1A). The SF content in scaffolds largely affected the mechanical properties of scaffolds because of their pore size and density, and a 5% (w/v) concentration of SF can result in porous structure applicable to tissue regeneration [34, 35]. The pore structure of scaffold was detected by scanning electron microscopy (SEM) (Figure 5A), and HAP and GO-PEI content in the scaffolds affected the pore size of scaffolds. The average pore sizes of the SF, SF/HAP scaffold and SF/HAP/GO-PEI scaffolds were 180 ± 3.5 μm, 161 ± 4.2 μm and 135 ± 5.1 μm, respectively (Figure 5B). The SF/HAP/GO-PEI scaffold had a disorganized porous structure, and the GO-PEI was firmly fixed to the walls of the SF/HAP structure. The high surface area and oxygen-containing functional groups of GO made GO-PEI and SF/HAP scaffolds to interact electrostatically [36]. Graphene can enhance the mechanical properties of scaffolds for load-bearing implant applications to tissue regenerate [37]. The compressive stress values of the SF, SF/HAP and SF/HAP/GO scaffolds were determined to be 244.9 ±79.7 kPa, 400.7 ± 80.1 kPa and 618.9 ± 101.5 kPa, respectively (Figure 5C). The degradation of the scaffolds was detected by soaking the scaffolds in PBS with lysozyme (0.5 mg/mL) to simulate a physiological environment. The degradation rate of the three SF-based scaffolds was similar, and the remaining weight was almost 90% at the first 2 weeks, 60% at 4 weeks and 30% at 8 weeks. The SF/HAP/GO-PEI scaffolds displayed better structural integrity than the other two types of scaffolds during degradation. After 8 weeks, a large amount of HAP and GO crystals still remained on the SF/HAP/GO-PEI scaffold, and the residual HAP and GO crystals provided the support for cells, which were benefit for the cell adhesion and growth, inducing matrix mineralization (Figure 5D).
Characterization and degradation of SF/HAP/GO-PEI-miR scaffolds. (A) SEM images of SF, SF/HAP, and SF/HAP/GPM scaffolds were shown, and the images on the right of each group are enlarged images. Scale bars: 100 μm and 10 μm. (B) Compression strength of the SF, SF/HAP and SF/HAP/GPM scaffolds. n = 4; *p < 0.05. (C) Degradation ratio of SF, SF/HAP and SF/HAP/GPM scaffolds changed with immersion time (2, 4, 6, 8 weeks after immersion).
5 Osteoblast cell proliferation and differentiation on SF/HAP/GPM scaffolds
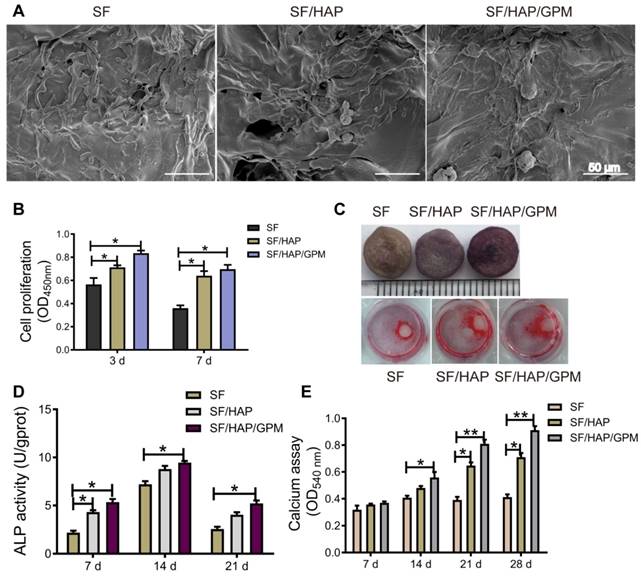
The SF/HAP/GPM scaffolds were used to promote osteoblast cell proliferation and differentiation to restore defects in bone tissue. As shown in Figure 6A, MC3T3-E1 cells grew robustly on all three types of scaffolds, suggesting that the SF-based scaffolds were suitable for cell attachment. And the high surface area of GO-PEI fixed on the walls of the SF/HAP scaffolds is beneficial for cell attachment and proliferation. Moreover, we performed CCK-8 assays to analyze the cell proliferation of SF-based scaffolds. Regardless of the substrate, SF/HAP/GPM scaffolds showed higher cell proliferation than SF and SF/HAP scaffolds. At day 3, cell proliferation reached a relatively high level; however, at day 7, the proliferation of cells decreased, and cells tended to differentiate and mineralize (Figure 6B). An ALP activity assay was performed after the cells were cultured on SF-based scaffolds for 7, 14, and 21 days. A representative image of the ALP staining of the scaffolds and ARS staining of the cells surrounding the scaffolds in the cultured plate is shown in Figure 6C. Interestingly, the cells on the plate surrounding the scaffold were also induced to osteogenic differentiation and could be dyed with ARS after 12 days of culture, and there was much more calcium accumulation surrounding the SF/HAP/GPM scaffolds (Figure 6C). The ALP activities of cells on scaffolds were analyzed and were significantly higher on SF/HAP/GPM scaffolds than on SF and SF/HAP scaffolds (Figure 6D). The intensity of ARS staining of the cells was also increased on the SF/HAP/GPM scaffolds compared to that on the SF and SF/HAP scaffolds (Figure 6E). Moreover, the results showed that the ALP activities reached a peak at 14 days, and ARS indicated obvious calcium deposition after 21 days. In conclusion, the SF/HAP/GPM scaffolds slowly released miRNA inhibitors to promote osteoblast differentiation and calcium accumulation, which are desirable for bone tissue regeneration.
Cell proliferation and osteogenic differentiation on SF/HAP/GPM scaffolds. (A) SEM images showing the morphologies of MC3T3-E1 cells on SF-based scaffolds after incubation for 3 days. Scale bars: 50 μm. (B) Proliferation of MC3T3-E1 cells on SF-based scaffolds after 3 and 7 days of incubation. (C) Image of ALP staining of SF-based scaffolds and ARS staining of cells surrounding the scaffolds after 7 days of incubation. (D) The ALP activities of MC3T3-E1 cells on SF-based scaffolds for 7, 14 or 21 days. (E) The calcium accumulation of cells on SF-based scaffolds for 7, 14, 21 or 28 days.
6 The signaling pathway by which SF/HAP/GPM scaffolds promote osteogenesis
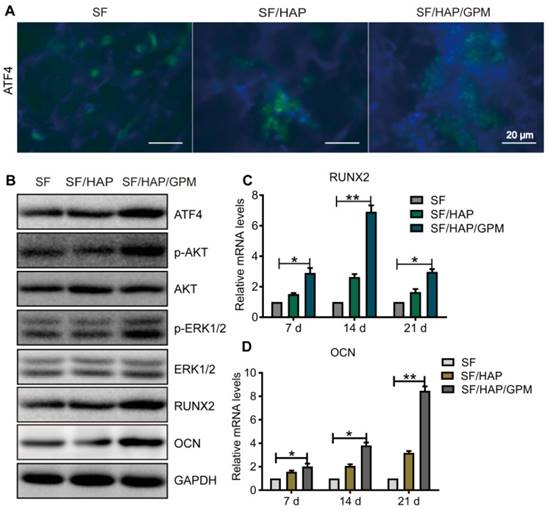
miR-214 has been reported to negatively regulate the osteoblast differentiation and bone formation by targeting osteogenic transcription factors, such as ATF4 and other osteogenic-related genes [5, 38, 39]. The above results showed that the GO-PEI-miR-214 inhibitor or SF/HAP/GPM scaffolds clearly promoted osteogenesis. To investigate the mechanism by which SF/HAP/GPM scaffolds regulate osteogenic differentiation, we performed immunofluorescence staining and western blot (WB) and qRT-PCR assays. The immunofluorescence staining results illustrated that there were many more ATF4-positive cells in SF/HAP/GPM scaffolds than in SF and SF/HAP scaffolds, which suggested that the ATF4 protein level was increased by the slowly released miR-214 inhibitor in the SF/HAP/GPM scaffolds (Figure 7A). The WB assay was performed after the cells were cultured in SF/HAP/GPM scaffolds for 14 days, and the results indicated that the protein levels of ATF4, Runx2, ALP and OCN were significantly increased in SF/HAP/GPM scaffolds compared to those in SF and SF/HAP scaffolds (Figures 7B). ATF4 is an important transcription factor involved in osteoblast differentiation and bone formation [40]. Runx2, a bone transcription factor, is necessary for ALP and OCN [41]. ALP is a differentiation marker at the initial stage of osteogenic differentiation [42], and OCN is a differentiation marker involved in the regulation of bone matrix deposition and mineralization [43]. The protein levels of phospho-ERK1/2 and phospho-Akt were also increased in SF/HAP/GPM scaffolds compared to those in SF and SF/HAP scaffolds. It has been documented that phosphoinositide 3-kinase (PI3K)/Akt and ERK1/2 signaling is involved in ATF4- or Runx2-related osteogenic differentiation of osteoblast cells [44, 45]. ATF4 can also activate Runx2 through some pathways to promote osteoblast differentiation [46]. Furthermore, the qRT-PCR results demonstrated that Runx2 was more highly expressed in SF/HAP/GPM scaffolds than in SF and SF/HAP scaffolds at 7 days, and the level of Runx2 was highest at 14 days and declined at 21 days. However, the level of OCN increased with incubation time and was highest at 21 days (Figure 7C and D).
The signaling pathway by which SF/HAP/GPM scaffolds promote osteogenesis. (A) Immunofluorescence staining of ATF4 in MC3T3-E1 cells on SF, SF/HAP and SF/HAP/GPM scaffolds. Scale bars: 20 μm. (B) The expression levels of ATF4, p-Akt, Akt, p-ERK1/2, ERK1/2, RUNX2 and OCN in MC3T3-E1 cells incubated on SF, SF/HAP and SF/HAP/GPM scaffolds for 14 days. (C and D) The expression levels of RUNX2 and OCN on SF, SF/HAP and SF/HAP/GPM scaffolds were evaluated by qRT-PCR.
7 SF/HAP/GPM scaffolds enhanced bone regeneration in calvarial defects
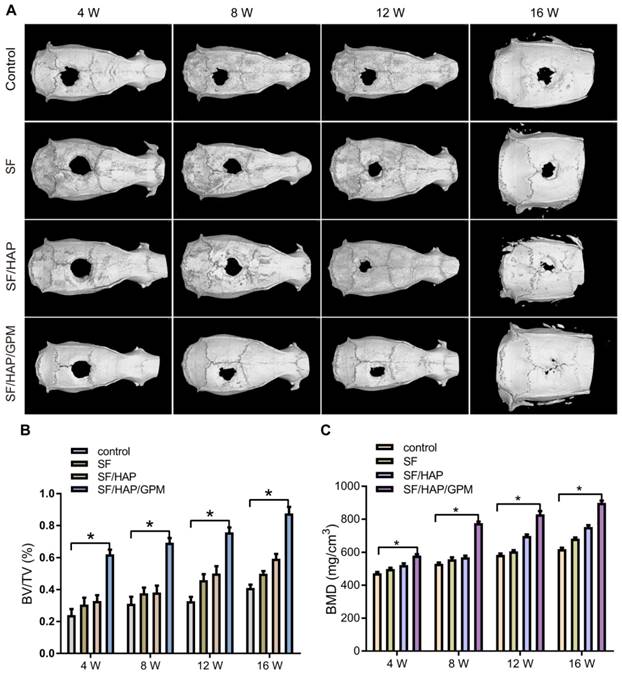
The osteogenic capability of SF/HAP/GPM scaffolds was observed by detecting bone regeneration after transplanting miR-214 inhibitor-releasing scaffolds in a calvarial bone-defect model. The SF-based scaffolds were cut into 2 mm thick sections of 5 mm in diameter, and the calvarial defect in rats could be completely filled with the scaffolds (Figure S1B). The new bone formation in the defects was detected by micro-CT after 4, 8, 12 and 16 weeks of implantation, and three-dimensional representative images are shown in Figure 8A. SF/HAP/GPM scaffolds appeared to have a weak effect on mineralization at week 4; however, by week 12, the SF/HAP/GPM groups demonstrated enhanced bony ingrowth into the defect area; by week 16, the defect was totally filled with mineralized (mature) bone tissue. The bone formation of the SF/HAP scaffold was not as good as that of the SF/HAP/GPM scaffold, and there was only some new bone generation at the periphery of the defect in the SF/HAP scaffold groups at week 16. SF scaffolds had worse osteogenic function than SF/HAP scaffolds. The untreated group showed minimal new bone generation and mineralization at the defect site at week 16 (Figure 8A). Moreover, the regenerated bone volume/total volume (BV/TV) ratio in the SF/HAP/GPM scaffold group (0.87 ± 0.07) was much greater than in the SF (0.5 ± 0.02) and SF/HAP scaffold groups (0.59 ± 0.05) at week 16 (Figure 8B). Consistently, the bone mineral density (BMD) of new bone in the SF/HAP/GPM scaffolds (899.77 ± 22.83 mg/m3) was significantly higher than that in either the SF/HAP (752.9 ± 19.13 mg/m3) or SF scaffolds (682.73 ± 13.44 mg/m3) at week 16 (Figure 8C). Overall, the data indicated the excellent osteoconductive capacity of the SF/HAP/GPM scaffolds in vivo.
Increased bone regeneration in calvarial defects after SF/HAP/GPM scaffold implantation. (A) Representative images of the reconstructed micro-CT of calvarial defects after implanting SF, SF/HAP and SF/HAP/GPM scaffolds for 4, 8, 12 and 16 weeks. (B) The bone volume/total volume (BV/TV) ratio was quantified from micro-CT analysis. (C) The BMD was measured by micro-CT.
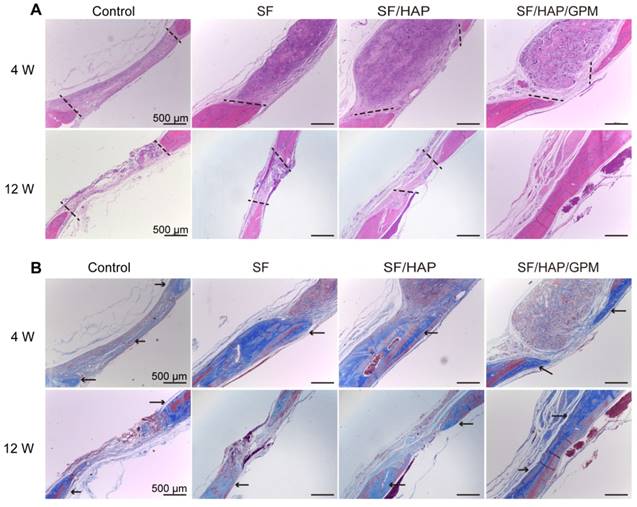
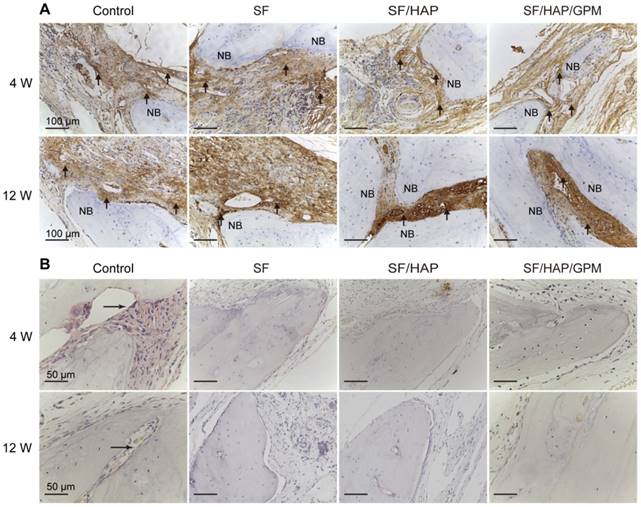
The SF/HAP/GPM scaffolds were also implanted subcutaneously on the back of nude mice and showed good biocompatibility. There were no obvious inflammatory reactions in the SF/HAP/GPM scaffolds after 4 weeks of implantation. Moreover, the scaffolds maintained physical integrity until week 4 while allowing fibrous tissues and capillary vessels to grow into the scaffolds (Figure S1C). Moreover, the hematoxylin and eosin (H&E) staining of scaffold-implanted calvarial defects presented in Figure 9A shows that the scaffold materials partly degraded, and fibroblasts were observed around the residual material after 4 weeks; after 8 weeks, most of the residual material was eliminated, and new bone ingrowth was observed. H&E staining primarily shows cell migration and proliferation, vessel formation and osteoid formation in defects. There was more new bone formation in the defects in SF/HAP/GPM scaffolds than in SF and SF/HAP scaffolds after 4 or 8 weeks. The enhanced new bone generation by SF/HAP/GPM scaffolds was also detected by conducting Masson's trichrome staining, which revealed new bone collagen or osteoid as blue staining in the sections. Blue-colored bone collagen almost filled the defect area in the SF/HAP/GPM group after 8 weeks, which was in contrast with the observations in the other groups. In contrast, there were loose collagen and little osteoid tissue in the SF and SF/HAP scaffold groups (Figure 9B). Therefore, the SF/HAP/GPM scaffolds induced more bone collagen regeneration in the defects than the other three types of SF scaffolds. The results were ascribed to miR-214 inhibitor release from SF/HAP/GPM scaffolds to inhibit miR-214 function. The protein level of the late differentiation marker, OCN, was further confirmed by immunohistochemistry staining and the highly intense brown staining in the SF/HAP/GPM scaffold-treated defects (Figure 10A). The OCN positive staining in the SF/HAP/GPM group was the highest along the four groups at 12 W. Moreover, tartrate-resistant acidic phosphatase (TRAP) (osteoclast marker) staining showed that osteoclast cells were not present in any of the SF-based scaffolds except the control group (Figure 10B). Collectively, these findings demonstrated that the SF/HAP/GPM scaffold had high osteoinductivity in vivo.
Enhanced collagen deposition in calvarial bone defects after SF/HAP/GPM scaffolds implantation. (A) Histological sections of control, SF, SF/HAP and SF/HAP/ GPM scaffolds were stained with H&E after 4 and 12 weeks of implantation. (B) Histological sections of control, SF, SF/HAP and SF/HAP/ GPM scaffolds were stained with Masson's trichrome after implantation for 4 and 12 weeks. The area of two black dotted lines mean the defect area and the arrowheads point the new bone collagen. Scale bars: 500 μm.
Enhanced osteogenesis in calvarial bone defects after SF/HAP/GPM scaffolds implantation. (A) Osteocalcin (OCN) immunohistochemical staining of the calvarial bones implanted with SF, SF/HAP and SF/HAP/ GPM scaffolds for 4 and 12 weeks. NB, new bone; the arrowheads point the OCN positive staining. (B) TRAP staining of the calvarial bones implanted with SF, SF/HAP and SF/HAP/ GPM scaffolds for 4 and 12 weeks. NB, new bone; the arrowheads point the TRAP-stained cells.
Discussion
Many miRNAs are negative regulator involved in osteogenic differentiation [4, 47]. Elevated miR-214 was reported to be correlated with less bone formation in bone specimen of aged patients with osteoporosis [5]. It was suggested that therapeutic inhibition of miR-214 in osteoblasts might block the negative effect on bone formation and promote osteoblastic calcification. Recently, miRNAs have been utilized as novel therapeutic agents for several advantages, such as small size and having a completely conserved sequence among species [48]. However, to be successful therapeutic agents, miRNAs need to overcome many challenges, including efficient delivery, biological stability, short in vivo half-life, and targeting [49].
In our work, a nonviral GO-PEI vector with high miR-214 inhibitor-binding affinity and an SF/HAP/GPM scaffold with slow miR-214 inhibitor release were developed with negligible cytotoxicity. To synthesize the GO-PEI complex, GO was mixed with PEI at a ratio of 1:2, and the positively charged PEI was bound to the negatively charged GO via electrostatic interactions and form stable GO-PEI complexes. Positive charged PEI polymers of GO-PEI complexes can efficiently load genes via the layer-by-layer assembly process. Moreover, GO-PEI complex can protect miRNAs from RNase-mediated degradation [26]. The size of the GO-PEI complexes was approximately 600 nm, which facilitated cell uptake [50]. Moreover, a large amount of bioactive compounds could be bind to GO due to its large surface area (2630 m2/g), and the release could be controlled by chemical-physical modifications [51, 52].
When the GO-PEI and miR-214 inhibitor were complexed, the negatively charged miR-214 inhibitor was wrapped into PEI and loaded onto the GO-PEI complexes. We hypothesized that such a GO-PEI-miR-214-inhibitor complex decreased PEI toxicity and increased the miRNAs accessibility and the interaction with the cell, thus GO-PEI complex were served as an excellent miRNA-inhibitor vector. GO-PEI can also protect miRNA inhibitors from extracellular degradation [53]. After the GO-PEI-miR-214 inhibitor enwrapped in endosomes, the cationic PEI component can preserve the biological activity of the miRNAs inhibitor and averted endosomal swelling and lysis by buffering the acidic environment in the endosomes [54]. miRNA inhibitors release controllably in GO-PEI complexes and GO-PEI degrades gradually in the cytosol.
In addition, graphene-based materials are widely used to enhance bone regeneration alone or incorporated into bone implants and scaffolds [37]. GO-PEI-miR-214 inhibitors were assembled into SF/HAP scaffolds, where the released miR-214 inhibitors locally transfected osteoblast cells to repair calvarial defect without the need to externally seed stem cells. SF/HAP composites have been used for bone constructs and have certain properties of bone regeneration [55]. GO-PEI assembled into SF/HAP scaffolds increased compression stress and fracture toughness, showed a strong porous network, and were electrically conductive and cytocompatible with MC3T3-E1 cells. This in situ 3D scaffold could direct cells to grow into the defect. Our animal data showed that the long-term (longer than 4 weeks) delivery of miR-214 inhibitor remained highly efficient and functional, and the healing speed of calvarial defects increased after 4 weeks.
With multiple functions, miR-214 was reported to inhibit osteoblast differentiation and bone formation through the ATF4 pathway [5]. Wang et al. showed that antagomiR-214 promoted osteoblast activity and mineralization in vitro and increased bone mass in aged ovariectomy-induced osteoporotic mice, but the dose of antagomiR-214 used in the experiment was up to 200 μM in vitro and 10 mg/kg in vivo (extremely high use dose). In this work, we demonstrated that SF/HAP/GPM-delivered miR-214 inhibitors effectively enhanced osteogenic gene expression in vitro compared to the Lipofectamine 2000 vector and promoted the regeneration of calvarial bone defects without seeding stem cells in vivo.
From a mechanistic perspective, miR-214 restrains the osteogenic capability of osteoblasts by functionally targeting ATF4, and miR-214 promotes osteoclast differentiation through the PI3k/Akt pathway [56]. Therefore, we examined this mechanism and found that the miR-214 inhibitor could be effectively transfected into cells by GO-PEI and could decrease the expression levels of miR-214 in osteoblasts. Furthermore, we revealed that the miR-214 inhibitor in the scaffolds not only enhanced ATF4 expression level and osteoblast activity but also suppressed osteoclast activity. ATF4 is crucial in the late stage of osteoblast differentiation [40], and the activation of ATF4 is accompanied by activated ERK1/2 and Akt signaling [57]. The upregulation of ERK1/2 and Akt phosphorylation substantially enhanced the protein level of Runx2 and stimulated ALP activity. The increase in ALP activity was followed by the enhanced synthesis of OCN, reflecting calcium mineral deposition [45].
It has been demonstrated to the use of growth factors [58], advanced scaffolds [59, 60] or advanced pore/surface structures [61, 62] have facilitated bone repair in tissue engineering. In this work, the novel SF/HAP/GO-PEI scaffolds with the controlled miRNA-inhibitor delivery prolonged the expression of multiple osteogenic genes and proteins, resulting in good bone regeneration in the defects without seeding stem cells.
Conclusions
A high miR-214 expression level correlates with reduced osteoblast differentiation and osteoblastic bone formation [63], which is related to skeletal disorders [5, 64]. In this work, miR-214 inhibitor delivered by SF/HAP/GPM scaffolds efficiently and released controllably showed good osteogenic capacity for bone defects. This technology could also deliver other nucleic acids or inhibitors to repair other tissues or to treat other diseases. Taken together, the results of this study show that the SF/HAP/GPM scaffold is a potential candidate for bone repair and regeneration in vivo.
Methods
Preparation of GO-PEI
GO was purchased (Aladdin, Shanghai, China) and ultrasonicated into a size of ~300 nm, and GO-PEI complexes were prepared as previously [27]. Briefly, we slowly added PEI (25-kDa) solution (1 mg/mL, Sigma-Aldrich, Missouri, USA) into a GO solution (1 mg/mL) in 10 min. The GO-PEI complexes were obtained by mixing PEI solutions with GO solutions at a GO:PEI weight ratio of 1:2. After ultrasonic treatment for 15 min, the mixed solution was stirred overnight, and then washed four times and resuspended in deionized water. The size of GO and GO-PEI was measured by a DLS spectrophotometer (Otsuka Electronics, Osaka, Japan). GO and GO-PEI were also detected by TEM (HT7700, Hitachi, Japan). FT-IR spectroscopy was utilized to measure the functional groups on the surface of GO, PEI, and GO-PEI complexes. The spectra were taken from 4000 to 400 cm-1 on an FT-IR instrument (Vertex 70, Bruker, Germany) at room temperature. The UV-Vis absorption spectra from 190 to 800 nm for the GO, GO-PEI and GO-PEI-miRNA samples (10 μg/mL) were obtained by an UV-Vis spectrophotometer (Shimadzu, Japan). The surface charge of the samples in deionized water or cell culture media was measured by a Zetasizer instrument (Malvern Nano ZS, Malvern, UK).
Gel retardation assay and cellular uptake of GO-PEI
The miR-214 inhibitor was bought from Ribobio (Ribobio, Guangzhou, China), and miRBase accession number is MIMAT0000661. To determine the appropriate method of GO-PEI to carry miRNA inhibitors, we used different N/P ratios for the GO-PEI-miRNA inhibitor complexes. The solution of GO-PEI and miRNA inhibitor were slowly mixed and put on ice for 30 min, and then were heated at 65 °C for 10 min. The GO-PEI-miRNA inhibitor solution (9 μL) were added with 3 μL of 4× loading buffer, loaded on a 1% (w/v) agarose gel and then electrophoresed at 100 V for 25 min. Cellular uptake of GO-PEI was measured by fluorescence analysis. Briefly, GO-PEI (1 mg/mL) was mixed for 2 h with FITC dye (Bioss Inc., Beijing, China) or miRNA-Cy3 solution (Ribobio, Guangzhou, China) at an N/P ratio of 1:30. The GO-PEI complexes were centrifuged and washed prior to incubating with MC3T3-E1 cells for 24 h. Then, the GO-PEI-treated cells were observed under a confocal microscope (Carl Zeiss, Inc., Jena, Germany) or a fluorescence microscope (Olympus Co., Ltd., Japan). Cellular uptake of GO-PEI was also measured by TEM (HT7700).
Scaffold preparation
Nanosized HAP particles were prepared as described [65]. Equal volumes of a 0.3 M ammonium hydrogen phosphate solution and a 0.5 M calcium chloride solution were mixed and stirred for 2 h. The mixture was aged at room temperature for 24 h, the precipitates were centrifuged and freeze-dried for 24 h. The SF solution was also prepared as previously described [66]. Briefly, silk fibroin extracted from cocoons was dried and dissolved in a solution of 9.3 M LiBr for 5 h at 60 °C to get the SF solution. The SF solution was dialyzed for 3 days and centrifuged at 5,000 rpm for 10 (25 °C). 1 mL of SF solution (4%, w/v) was mixed with HAP nanoparticles (10 mg) in a well (a 48-well plate) to form the SF/HAP scaffold after freeze-drying for 24 h. The SF/HAP/GO-PEI scaffold or SF/HAP/GO-PEI-miR-214 inhibitor scaffold was fabricated by mixing 0.5 mL of the SF solution (4%, w/v), 10 mg of HAP nanoparticles and 0.5 mL of GO-PEI or GO-PEI-miRNA inhibitor (1 mg/mL) in a well and freeze-drying for 24 h. All SF-based scaffolds were soaked in a 90% (v/v) methanol solution for 30 min to transform into the water-insoluble scaffolds by inducing the structural transition [67].
Scaffold characterization
The morphologies of the different SF-based scaffolds were observed by SEM (LEO1530 VP, Philips, Netherlands). The pore sizes of the scaffolds were measured by observing 40 random pores in the SEM images using ImageJ software. The compressive load and extension of the SF-based scaffolds were measured by a Bose ElectroForce-3220 instrument (TA Instruments, Eden Prairie, MN, USA), and 5 samples of each scaffold group were analyzed [20].
Scaffold degradation
The degradation of the SF-based scaffolds was examined recording the weight change of the scaffold after infusion in PBS with lysozyme (0.5 mg/mL, Amresco) which was similar to circulating blood. The degradation buffer was changed every 2 days. The scaffolds were freeze-dried and weighed at time intervals of 1, 2, 4, 6 and 8 weeks. The percentage of scaffold degradation was determined by the following formula [68]:
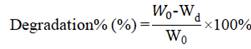
W0 is the original weight of the scaffold and Wd is the weight after degradation.
Cell culture
The purchased MC3T3-E1 cells (Geneseed, Guangzhou, China) were cultured in α-MEM medium (containing 10% FBS and 1% penicillin/streptomycin, PS) at 37 °C in a 5% CO2 incubator. The BMSCs were harvested from the bone marrow of BABL/c mice and cultured in Dulbecco's modified Eagle's medium (DMEM, Gibco, USA). To induce the BMSCs differentiation, the cells were cultured in osteogenic medium supplemented with 10% FBS, 1% antibiotics, 50 μM ascorbic acid (Sigma, Missouri, USA), 10 mM β-glycerol phosphate (Sigma), and 0.1 μM dexamethasone (Sigma). The scaffolds were immersed in complete medium (DMEM medium, 10% FBS, 1% antibiotics) overnight, the MC3T3-E1 cells and BMSCs were then seeded (1×106 cells/scaffold) on the SF, SF/HAP, and SF/HAP/GO-PEI-miR-214 inhibitor scaffolds. The scaffolds were kept in the complete medium for 1 day to allow cell adhesion and growth, and then changed with osteogenic medium to induce osteogenic differentiation.
Cell proliferation
Cell proliferation was examined by the CCK-8 assay kit as described [69]. Briefly, the cells were seeded on a 96-well plate or SF-based scaffolds and cultured for 3 or 7 days. 10 μL of CCK-8 solution was added to each well and incubated for 3 h at 37 °C in the incubator. The absorbance was tested by a microplate absorbance reader (Bio-Rad, CA, USA) at 450 nm. All experiments were repeated 3 times.
ALP activity and ARS staining
The cells on scaffolds were fixed in 4% paraformaldehyde on days 7 14, 21 and 28. ALP staining (Rainbow, Shanghai, China) and ARS staining (Sigma) were performed as described [70, 71]. Briefly, the cells were lysed with 0.1% Triton X-100, and the protein concentration was measured by a BCA protein assay kit. ALP activity was determined at 405 nm. The scaffolds were stained with ARS solution (40 mM) for 30 min and washed with deionized water 5 times, and then the staining on scaffolds was eluted by the 10% cetylpyridinium chloride for 1 h. The eluted dye solution was collected and read at the absorbance of 540 nm in a spectrophotometer (Thermo Spectronics, Waltham, MA, USA).
Real-time RT-PCR analysis
The cells on the scaffolds were detached using a 0.25% trypsin and collected after centrifugation at 250×g. Total RNA was isolated using an RNeasy Mini Kit (Qiagen, USA). cDNAs were synthesized by a cDNA synthesis kit (#K1622, Fermentas, EU). And then cDNAs and forward (1 μL) and reverse primers (1 μL) were added into a SYBR reaction mixture (20 μL). The qPCR cycles were set as follow: 2 mins at 50 °C, 10 min at 95 °C, 50 cycles; 15 s at 95 °C, and 1 min at 60 °C, then 4 °C to cool down. miRNA qRT-PCR was performed using the Bulge-LoopTM miRNA qRT-PCR kit (Ribobio, Guangzhou, China). Each sample was performed 3 times. The primers used for qPCR were: RUNX2, forward, AGTCCCAACTTCCTGTGCTC, reverse, TCGGCGGAGTAGTTCTCATC; OCN, forward, CGGCCCTGAGTCTGACAAA, reverse, TAGCGCCGGAGTCTGTTC; mmu-miR-214-3p (MIMAT0000661), ACAGCAGGCACAGACAGGCAGU.
Immunofluorescence staining and western blotting assay
The cells were fixed by 4% (w/v) paraformaldehyde and permeabilized with 0.1% (v/v) Triton X-100. The permeable cells were incubated with 3% (v/v) normal goat serum (Sigma-Aldrich) for 1 h to block nonspecific binding of the primary antibodies and incubated with mouse monoclonal anti-ATF4, anti-OCN antibodies (1:300; Santa Cruz Biotechnology, Santa Cruz, CA, USA) and rhodamine-phalloidin (1:40, Invitrogen, CA, USA) at 4 °C overnight. And then the cells were incubated with secondary antibodies (Alexa Fluor-488 goat anti-mouse IgG; Invitrogen) and incubated for 1 h at room temperature. Nuclei were stained with DAPI (1:500, Sigma) at room temperature for 20 min. Fluorescence images were observed by CLSM and fluorescence microscopy (Olympus IX71; Olympus). The fluorescence intensity of images from 3 independent cultures was quantitatively analyzed using ImageJ software. For Western blot, the cells were lysed and the same amount of proteins was loaded onto a 12% SDS-PAGE gel (Beyotime, Nanjing, China), and then transferred onto PVDF membranes. The membranes were blocked by 5% BSA solution and incubated with monoclonal antibodies of ATF4, p-Akt, Akt, p-ERK1/2, ERK1/2, RUNX2, and OCN (CST, MA, USA), followed by HRP-conjugated secondary antibodies (CST). GAPDH was used as an internal control. The membranes were exposed using an ECL WB substrate kit (Beyotime) and analyzed by Image J software (NIH, MD, USA).
Subcutaneous implantation
The animal procedure was approved by the Institutional Animal Care and Use Committee of Jinan University. The GPM scaffolds were implanted in subcutaneous on the backs of nude mice as previously described [72]. Briefly, the female nude mice (6∼8-week old) were anesthetized with pentobarbital sodium (Sigma-Aldrich) at 15 mg/kg. Two subcutaneous pockets were created on the disinfected dorsum by blunt dissection, and one scaffold was implanted in one pocket. To determine in vivo degradation of scaffolds, the implants were harvested at 2, 4, and 6 weeks after the mice were sacrificed. Four scaffolds were implanted randomly in subcutaneous for each group (n = 4).
Calvarial bone-defect model construction
SD rats (200-250 g) were divided randomly into four groups. The surgical procedures of the calvarial bone-defect model were performed as described [73, 74]. Rats were anesthetized, shaved and sterilized in the skull. A linear scalp incision was made and full-thickness flaps were elevated through the skin along the sagittal suture of the skull. A 5-mm craniotomy defect was made in the parietal bone by a slow-speed dental drill (Dentium, Korea), and the wounds were copiously irrigated with physiological saline (0.9%) while drilling. The 5-mm calvarial bone was carefully removed to avoid injuring the underlying dura or brain. The scaffolds were placed in the defects after hemostasis; defects without scaffolds served as controls. The sagittal incisions were closed by suturing. When the rats recovered from anesthesia, gave buprenorphine (0.08 mg/kg) every 12 h for 48 h to control pain. The cranial bones of mice were harvested at 4, 8 and 12 weeks postoperative.
Microcomputed tomography (micro-CT)
To examine the repair of calvarial bone defect using micro-CT (mCT100, Scanco Medical, Basserdorf, Switzerland), the calvarial bone were fitted in a cylindrical sample holder and scanned with direction parallel to the coronal aspect (with a voxel size of 12 mm). The trabecular bones were measured and segmented for 3D reconstruction, and BMD and relative bone volume (BV/TV) were calculated.
Histology and immunohistochemistry
The rats were sacrificed and the skulls were harvested at 4, 8 or 12 weeks after implantation. The calvarial bone samples were fixed in 4% paraformaldehyde for 48 h at 4 °C and decalcified in 10% EDTA for 4 weeks. Soft tissue and decalcified bone samples were embedded in paraffin and sectioned into slices (10 μm thick). The sections were stained with H&E or Masson's trichrome (Beyotime). For immunohistochemistry, the sections were incubated with the anti-OCN primary antibody (1:200, Santa Cruz, CA, USA) overnight at 4 °C after blocking in horse serum (Solarbio, Beijing, China). And then the sections were incubated with a secondary antibody (1:500, Santa Cruz, CA, USA) for 2 h at room temperature. Three samples for each group were examined by light microscopy (Olympus IX71).
Statistical analysis
The experimental data are presented as the mean ± SD. All experiments were performed in triplicate (n ≥ 3). Statistical differences among groups were analyzed by one-way ANOVA or between two groups were analyzed using Student's t-test. A P-value < 0.05 was considered statistical significant.
Abbreviations
GO, graphene oxide; GO-PEI, graphene oxide-polyethyleneimine; GPM, GO-PEI-mir-214 inhibitor; SF/HAP, silk fibroin/hydroxyapatite; SF/HAP/GO, silk fibroin/hydroxyapatite/ graphene oxide; ATF4, activating transcription factor 4; Runx2, Runt-related transcription factor 2; ALP, alkaline phosphatase; OCN, osteocalcin; TRAP, tartrate-resistant acidic phosphatase.
Supplementary Material
Supplementary figure.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 81703120, 31271019), the Science and Technology Program of Guangzhou (201508020115, 201601010270, 2017010160489, 201704030083), Natural Science Foundation of Guangdong Province (2017A030310365), and Guangdong Provincial Medical Research Foundation (A2016360), the Pearl River S&T Nova Program of Guangzhou (201710010155, 201806010072), and the Science and Technology Project of Guangdong Province (2015A010101313, 2017A050506011, 2017B090911012, 2018A050506040, 2018A050506019, 2018A050506021).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Giatsidis G, Dalla Venezia E, Bassetto F. The role of gene therapy in regenerative surgery: updated insights. Plast Reconstr Surg. 2013;131:1425-35
2. Bez M, Sheyn D, Tawackoli W. In situ bone tissue engineering via ultrasound-mediated gene delivery to endogenous progenitor cells in mini-pigs. Sci Transl Med. 2017:9
3. Zhang J, Tu Q, Bonewald LF, He X, Stein G, Lian J. et al. Effects of miR-335-5p in modulating osteogenic differentiation by specifically downregulating Wnt antagonist DKK1. J Bone Miner Res. 2011;26:1953-63
4. Eskildsen T, Taipaleenmaki H, Stenvang J, Abdallah BM, Ditzel N, Nossent AY. et al. MicroRNA-138 regulates osteogenic differentiation of human stromal (mesenchymal) stem cells in vivo. Proc Natl Acad Sci U S A. 2011;108:6139-44
5. Wang X, Guo B, Li Q, Peng J, Yang Z, Wang A. et al. miR-214 targets ATF4 to inhibit bone formation. Nat Med. 2013;19:93-100
6. Verma IM. Medicine. Gene therapy that works. Science. 2013;341:853-5
7. Guzman-Villanueva D, El-Sherbiny IM, Herrera-Ruiz D, Vlassov AV, Smyth HD. Formulation approaches to short interfering RNA and MicroRNA: challenges and implications. J Pharm Sci. 2012;101:4046-66
8. Miyazaki Y, Adachi H, Katsuno M, Minamiyama M, Jiang YM, Huang Z. et al. Viral delivery of miR-196a ameliorates the SBMA phenotype via the silencing of CELF2. Nat Med. 2012;18:1136-41
9. Nayak S, Herzog RW. Progress and prospects: immune responses to viral vectors. Gene Ther. 2010;17:295-304
10. Yin L, Song Z, Kim KH, Zheng N, Gabrielson NP, Cheng J. Non-viral gene delivery via membrane-penetrating, mannose-targeting supramolecular self-assembled nanocomplexes. Adv Mater. 2013;25:3063-70
11. Dalby B, Cates S, Harris A, Ohki EC, Tilkins ML, Price PJ. et al. Advanced transfection with Lipofectamine 2000 reagent: primary neurons, siRNA, and high-throughput applications. Methods. 2004;33:95-103
12. Li J, Tan S, Kooger R, Zhang C, Zhang Y. MicroRNAs as novel biological targets for detection and regulation. Chem Soc Rev. 2014;43:506-17
13. Yin H, Kanasty RL, Eltoukhy AA, Vegas AJ, Dorkin JR, Anderson DG. Non-viral vectors for gene-based therapy. Nat Rev Genet. 2014;15:541-55
14. Whitehead KA, Dorkin JR, Vegas AJ, Chang PH, Veiseh O, Matthews J. et al. Degradable lipid nanoparticles with predictable in vivo siRNA delivery activity. Nat Commun. 2014;5:4277
15. Parayath NN, Parikh A, Amiji MM. Repolarization of Tumor-Associated Macrophages in a Genetically Engineered Nonsmall Cell Lung Cancer Model by Intraperitoneal Administration of Hyaluronic Acid-Based Nanoparticles Encapsulating MicroRNA-125b. Nano Lett. 2018;18:3571-3579
16. Ibrahim AF, Weirauch U, Thomas M, Grunweller A, Hartmann RK, Aigner A. MicroRNA replacement therapy for miR-145 and miR-33a is efficacious in a model of colon carcinoma. Cancer Res. 2011;71:5214-24
17. Kirtane AR, Panyam J. Polymer nanoparticles: Weighing up gene delivery. Nat Nanotechnol. 2013;8:805-6
18. Chung C, Kim YK, Shin D, Ryoo SR, Hong BH, Min DH. Biomedical applications of graphene and graphene oxide. Acc Chem Res. 2013;46:2211-24
19. Yin D, Li Y, Lin H, Guo B, Du Y, Li X. et al. Functional graphene oxide as a plasmid-based Stat3 siRNA carrier inhibits mouse malignant melanoma growth in vivo. Nanotechnology. 2013;24:105102
20. Liu H, Xu GW, Wang YF, Zhao HS, Xiong S, Wu Y. et al. Composite scaffolds of nano-hydroxyapatite and silk fibroin enhance mesenchymal stem cell-based bone regeneration via the interleukin 1 alpha autocrine/paracrine signaling loop. Biomaterials. 2015;49:103-12
21. Shen X, Zhang Y, Gu Y, Xu Y, Liu Y, Li B. et al. Sequential and sustained release of SDF-1 and BMP-2 from silk fibroin-nanohydroxyapatite scaffold for the enhancement of bone regeneration. Biomaterials. 2016;106:205-16
22. Sun Y, Zhang K, Deng R, Ren X, Wu C, Li J. Tunable stiffness of graphene oxide/polyacrylamide composite scaffolds regulates cytoskeleton assembly. Chem Sci. 2018;9:6516-6522
23. Tang Z, Wu H, Cort JR, Buchko GW, Zhang Y, Shao Y. et al. Constraint of DNA on functionalized graphene improves its biostability and specificity. Small. 2010;6:1205-9
24. Tang LA, Wang J, Loh KP. Graphene-based SELDI probe with ultrahigh extraction and sensitivity for DNA oligomer. J Am Chem Soc. 2010;132:10976-7
25. Hobel S, Koburger I, John M, Czubayko F, Hadwiger P, Vornlocher HP. et al. Polyethylenimine/small interfering RNA-mediated knockdown of vascular endothelial growth factor in vivo exerts anti-tumor effects synergistically with Bevacizumab. J Gene Med. 2010;12:287-300
26. Choi HY, Lee TJ, Yang GM, Oh J, Won J, Han J. et al. Efficient mRNA delivery with graphene oxide-polyethylenimine for generation of footprint-free human induced pluripotent stem cells. J Control Release. 2016;235:222-235
27. Feng L, Zhang S, Liu Z. Graphene based gene transfection. Nanoscale. 2011;3:1252-7
28. Li MJ, Liu CM, Xie YB, Cao HB, Zhao H, Zhang Y. The evolution of surface charge on graphene oxide during the reduction and its application in electroanalysis. Carbon. 2014;66:302-311
29. Tenzer S, Docter D, Kuharev J, Musyanovych A, Fetz V, Hecht R. et al. Rapid formation of plasma protein corona critically affects nanoparticle pathophysiology. Nat Nanotechnol. 2013;8:772-81
30. Liao KH, Lin YS, Macosko CW, Haynes CL. Cytotoxicity of graphene oxide and graphene in human erythrocytes and skin fibroblasts. ACS Appl Mater Interfaces. 2011;3:2607-15
31. Halim A, Luo Q, Ju Y, Song G. A Mini Review Focused on the Recent Applications of Graphene Oxide in Stem Cell Growth and Differentiation. Nanomaterials (Basel). 2018;8:736
32. Yu S, Zhu K, Lai Y, Zhao Z, Fan J, Im HJ. et al. ATF4 promotes beta-catenin expression and osteoblastic differentiation of bone marrow mesenchymal stem cells. Int J Biol Sci. 2013;9:256-66
33. Wang W, Lian N, Ma Y, Li L, Gallant RC, Elefteriou F. et al. Chondrocytic Atf4 regulates osteoblast differentiation and function via Ihh. Development. 2012;139:601-11
34. Davis NE, Beenken-Rothkopf LN, Mirsoian A, Kojic N, Kaplan DL, Barron AE. et al. Enhanced function of pancreatic islets co-encapsulated with ECM proteins and mesenchymal stromal cells in a silk hydrogel. Biomaterials. 2012;33:6691-7
35. Kim HH, Park JB, Kang MJ, Park YH. Surface-modified silk hydrogel containing hydroxyapatite nanoparticle with hyaluronic acid-dopamine conjugate. Int J Biol Macromol. 2014;70:516-22
36. Nayak TR, Andersen H, Makam VS, Khaw C, Bae S, Xu X. et al. Graphene for controlled and accelerated osteogenic differentiation of human mesenchymal stem cells. ACS Nano. 2011;5:4670-8
37. Shin SR, Li YC, Jang HL, Khoshakhlagh P, Akbari M, Nasajpour A. et al. Graphene-based materials for tissue engineering. Adv Drug Deliv Rev. 2016;105:255-274
38. Yao S, Zhao W, Ou Q, Liang L, Lin X. MicroRNA-214 Suppresses Osteogenic Differentiation of Human Periodontal Ligament Stem Cells by Targeting ATF4. 2017. 2017:3028647
39. Shi K, Lu J, Zhao Y, Wang L, Li J, Qi B. et al. MicroRNA-214 suppresses osteogenic differentiation of C2C12 myoblast cells by targeting Osterix. Bone. 2013;55:487-94
40. Yang X, Matsuda K, Bialek P, Jacquot S, Masuoka HC, Schinke T. et al. ATF4 is a substrate of RSK2 and an essential regulator of osteoblast biology; implication for Coffin-Lowry Syndrome. Cell. 2004;117:387-98
41. Duan S, Yang X, Mei F, Tang Y, Li X, Shi Y. et al. Enhanced osteogenic differentiation of mesenchymal stem cells on poly(L-lactide) nanofibrous scaffolds containing carbon nanomaterials. J Biomed Mater Res A. 2015;103:1424-35
42. Lai GJ, Shalumon KT, Chen JP. Response of human mesenchymal stem cells to intrafibrillar nanohydroxyapatite content and extrafibrillar nanohydroxyapatite in biomimetic chitosan/silk fibroin/nanohydroxyapatite nanofibrous membrane scaffolds. Int J Nanomedicine. 2015;10:567-84
43. Jiang X, Zhao J, Wang S, Sun X, Zhang X, Chen J. et al. Mandibular repair in rats with premineralized silk scaffolds and BMP-2-modified bMSCs. Biomaterials. 2009;30:4522-32
44. Ren D, Wei F, Hu L, Yang S, Wang C, Yuan X. Phosphorylation of Runx2, induced by cyclic mechanical tension via ERK1/2 pathway, contributes to osteodifferentiation of human periodontal ligament fibroblasts. J Cell Physiol. 2015;230:2426-36
45. Baker N, Sohn J, Tuan RS. Promotion of human mesenchymal stem cell osteogenesis by PI3-kinase/Akt signaling, and the influence of caveolin-1/cholesterol homeostasis. Stem Cell Res Ther. 2015;6:238
46. Lin KL, Chou CH, Hsieh SC, Hwa SY, Lee MT, Wang FF. Transcriptional upregulation of DDR2 by ATF4 facilitates osteoblastic differentiation through p38 MAPK-mediated Runx2 activation. J Bone Miner Res. 2010;25:2489-503
47. Chen L, Holmstrom K, Qiu W, Ditzel N, Shi K, Hokland L. et al. MicroRNA-34a inhibits osteoblast differentiation and in vivo bone formation of human stromal stem cells. Stem Cells. 2014;32:902-12
48. van Rooij E, Purcell AL, Levin AA. Developing microRNA therapeutics. Circ Res. 2012;110:496-507
49. Burnett JC, Rossi JJ. RNA-based therapeutics: current progress and future prospects. Chem Biol. 2012;19:60-71
50. Mu Q, Su G, Li L, Gilbertson BO, Yu LH, Zhang Q. et al. Size-dependent cell uptake of protein-coated graphene oxide nanosheets. ACS Appl Mater Interfaces. 2012;4:2259-66
51. Liu Z, Liu B, Ding J, Liu J. Fluorescent sensors using DNA-functionalized graphene oxide. Anal Bioanal Chem. 2014;406:6885-902
52. Weaver CL, Cui XT. Directed Neural Stem Cell Differentiation with a Functionalized Graphene Oxide Nanocomposite. Adv Healthc Mater. 2015;4:1408-16
53. Xiang S, Tong H, Shi Q, Fernandes JC, Jin T, Dai K. et al. Uptake mechanisms of non-viral gene delivery. J Control Release. 2012;158:371-8
54. Mintzer MA, Simanek EE. Nonviral vectors for gene delivery. Chem Rev. 2009;109:259-302
55. Farokhi M, Mottaghitalab F, Samani S, Shokrgozar MA, Kundu SC, Reis RL. et al. Silk fibroin/hydroxyapatite composites for bone tissue engineering. Biotechnol Adv. 2018;36:68-91
56. Zhao C, Sun W, Zhang P, Ling S, Li Y, Zhao D. et al. miR-214 promotes osteoclastogenesis by targeting Pten/PI3k/Akt pathway. RNA Biol. 2015;12:343-53
57. Wang C, Lin K, Chang J, Sun J. Osteogenesis and angiogenesis induced by porous beta-CaSiO(3)/PDLGA composite scaffold via activation of AMPK/ERK1/2 and PI3K/Akt pathways. Biomaterials. 2013;34:64-77
58. Wei G, Jin Q, Giannobile WV, Ma PX. The enhancement of osteogenesis by nano-fibrous scaffolds incorporating rhBMP-7 nanospheres. Biomaterials. 2007;28:2087-96
59. Wubneh A, Tsekoura EK, Ayranci C, Uludag H. Current state of fabrication technologies and materials for bone tissue engineering. Acta Biomater. 2018;80:1-30
60. Hasan A, Morshed M, Memic A, Hassan S, Webster TJ, Marei HE. Nanoparticles in tissue engineering: applications, challenges and prospects. Int J Nanomedicine. 2018;13:5637-5655
61. Neffe AT, Pierce BF, Tronci G, Ma N, Pittermann E, Gebauer T. et al. One step creation of multifunctional 3D architectured hydrogels inducing bone regeneration. Adv Mater. 2015;27:1738-44
62. Kim HD, Amirthalingam S, Kim SL, Lee SS, Rangasamy J, Hwang NS. Biomimetic Materials and Fabrication Approaches for Bone Tissue Engineering. Adv Healthc Mater. 2017:6
63. Li D, Liu J, Guo B, Liang C, Dang L, Lu C. et al. Osteoclast-derived exosomal miR-214-3p inhibits osteoblastic bone formation. Nat Commun. 2016;7:10872
64. Sun Y, Kuek V, Liu Y, Tickner J, Yuan Y. MiR-214 is an important regulator of the musculoskeletal metabolism and disease. J Cell Physiol. 2018;234:231-45
65. Pang YX, Bao X. Influence of temperature, ripening time and calcination on the morphology and crystallinity of hydroxyapatite nanoparticles. Journal of the European Ceramic Society. 2003;23:1697-1704
66. Kundu J, Dewan M, Ghoshal S, Kundu SC. Mulberry non-engineered silk gland protein vis-a-vis silk cocoon protein engineered by silkworms as biomaterial matrices. J Mater Sci Mater Med. 2008;19:2679-89
67. Wongpanit P, Ueda H, Tabata Y, Rujiravanit R. In vitro and in vivo release of basic fibroblast growth factor using a silk fibroin scaffold as delivery carrier. J Biomater Sci Polym Ed. 2010;21:1403-19
68. Xie C, Lu X, Han L, Xu J, Wang Z, Jiang L. et al. Biomimetic Mineralized Hierarchical Graphene Oxide/Chitosan Scaffolds with Adsorbability for Immobilization of Nanoparticles for Biomedical Applications. ACS Appl Mater Interfaces. 2016;8:1707-17
69. Yue H, Wei W, Yue Z, Wang B, Luo N, Gao Y. et al. The role of the lateral dimension of graphene oxide in the regulation of cellular responses. Biomaterials. 2012;33:4013-21
70. Zou D, Zhang Z, Ye D, Tang A, Deng L, Han W. et al. Repair of critical-sized rat calvarial defects using genetically engineered bone marrow-derived mesenchymal stem cells overexpressing hypoxia-inducible factor-1alpha. Stem Cells. 2011;29:1380-90
71. Deng Y, Zhou H, Zou D, Xie Q, Bi X, Gu P. et al. The role of miR-31-modified adipose tissue-derived stem cells in repairing rat critical-sized calvarial defects. Biomaterials. 2013;34:6717-28
72. Zhang X, Li Y, Chen YE, Chen J, Ma PX. Cell-free 3D scaffold with two-stage delivery of miRNA-26a to regenerate critical-sized bone defects. Nat Commun. 2016;7:10376
73. Bi L, Rahaman MN, Day DE, Brown Z, Samujh C, Liu X. et al. Effect of bioactive borate glass microstructure on bone regeneration, angiogenesis, and hydroxyapatite conversion in a rat calvarial defect model. Acta Biomater. 2013;9:8015-26
74. Fu H, Rahaman MN, Brown RF, Day DE. Evaluation of bone regeneration in implants composed of hollow HA microspheres loaded with transforming growth factor beta1 in a rat calvarial defect model. Acta Biomater. 2013;9:5718-27
Author contact
![]() Corresponding authors: Renfa Lai, the first affiliated hospital of Jinan University, No.613 West Huangpu Avenue, 510632, Guangzhou, China. E-mail: Prof.Dr.Laicom. Rui Guo, Key Laboratory of Biomaterials of Guangdong Higher Education Institutes, Guangdong Provincial Engineering and Technological Research Center for Drug Carrier Development, Department of Biomedical Engineering, Jinan University, Guangzhou 510632, China. E-mail: guoruiedu.cn.
Corresponding authors: Renfa Lai, the first affiliated hospital of Jinan University, No.613 West Huangpu Avenue, 510632, Guangzhou, China. E-mail: Prof.Dr.Laicom. Rui Guo, Key Laboratory of Biomaterials of Guangdong Higher Education Institutes, Guangdong Provincial Engineering and Technological Research Center for Drug Carrier Development, Department of Biomedical Engineering, Jinan University, Guangzhou 510632, China. E-mail: guoruiedu.cn.