Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(15):4474-4493. doi:10.7150/thno.31942 This issue Cite
Research Paper
Astrocytic connexin 43 potentiates myelin injury in ischemic white matter disease
Minghuan Wang1*, Chuan Qin1*, Xiang Luo1*, Jie Wang2, Xuxia Wang2, Minjie Xie1, Jing Hu1, Jie Cao1, Ting Hu2, Steven A. Goldman3,4,5, Maiken Nedergaard3,4 ![]() , Wei Wang1,6
, Wei Wang1,6 ![]()
1. Department of Neurology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430030, China.
2. State Key Laboratory of Magnetic Resonance and Atomic and Molecular Physics, Key Laboratory of Magnetic Resonance in Biological Systems, Wuhan Institute of Physics and Mathematics, Chinese Academy of Science, Wuhan 430030, China.
3. Center for Translational Neuromedicine, University of Rochester Medical Center, Rochester, NY 14642, USA.
4. Center for Neuroscience, University of Copenhagen Faculty of Health and Medicine, Copenhagen, Denmark.
5. Center for Neuroscience, Rigshospitalet, Copenhagen, Denmark.
6. Key Laboratory of Neurological Diseases of Chinese Ministry of Education, the School of Basic Medicine, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430030, China.
*Drs. Minghuan Wang, Chuan Qin and Xiang Luo contribute equally to this work.
Received 2018-12-2; Accepted 2019-4-19; Published 2019-6-9
Abstract

Rational: Myelin loss is a characteristic feature of both ischemic white matter disease and its associated vascular dementia, and is a hallmark of chronic cerebral hypoperfusion due to carotid artery stenosis. Yet the cellular mechanisms involved in ischemic dysmyelination are not well-understood, and no effective treatment has emerged to prevent or slow hypoperfusion-related demyelination. In a study employing the bilateral common carotid artery stenosis (BCAS) mouse model, we found reduced cerebral blood flow velocity and arteriolar pulsatility, and confirmed that prolonged BCAS provoked myelin disruption. These pathological features were associated with marked cognitive decline, in the absence of evident damage to axons.
Methods: To assess the role of astroglial communication in BCAS-associated demyelination, we investigated the effect of deleting or inhibiting connexin 43 (Cx43), a constituent of astroglial gap junctions and hemichannels.
Results: Genetic deletion and pharmacological inhibition of gap junctions both protected myelin integrity and rescued cognitive decline in the BCAS-treated mice. Gap junction inhibition also suppressed the transient increase in extracellular glutamate observed in the callosal white matter of wild-type mice exposed to BCAS.
Conclusion: These findings suggest that astrocytic Cx43 may be a viable target for attenuating the demyelination and cognitive decline associated with chronic cerebral hypoperfusion.
Keywords: connexin 43, myelin injury, ischemic white matter disease, astrocyte
Introduction
Chronic cerebral hypoperfusion arising from carotid artery stenosis is associated with disturbed cerebral hemodynamics and cognitive decline [1, 2]. Patients experiencing carotid artery stenosis often present with white matter injury and progressive cognitive impairment, even in the absence of overt symptoms or focal neurological deficits [3, 4].
Little attention has been paid to elucidating the temporal sequence of events leading to ischemic injury in white matter. Most preclinical studies have focused on hypoxic-ischemic injury to the grey matter, and a large body of literature has supported the concept that glutamate excitotoxicity is a key mediator of ischemic injury in grey matter [5, 6]. Although axons largely lack glutamate receptors [7, 8], oligodendrocytes are highly sensitive to over-activation of AMPA/kainate receptors [9, 10]. Moreover, cellular stress is linked to an increase in the number of open astrocytic Cx43 hemichannels, which release glutamate and ATP, and which have been shown to promote inflammation following ischemic insult via their activation of local astroglial and microglial cytokine release [11-13]. In a feed-forward fashion, these events may then increase the vulnerability of oligodendrocytes to glutamate excitotoxicity [14, 15].
Given this background, we asked whether chronic cerebral ischemia might preferentially target the central white matter to provoke glial loss and myelin disruption, and if so, whether that process might be attenuated by interfering with glial syncytial communication via gap junctions. To this end, we assessed the effects on the central white matter of bilateral common carotid artery stenosis (BCAS), using a murine model of BCAS that yields a white matter injury resembling that of chronic cerebral hypoperfusion in humans [16-19]. In so doing, we drew upon prior reports that white matter is affected in the BCAS hypoperfusion model [20], with disruption of myelin and nodes of Ranvier in the absence of overt axonal damage [16]. Using a combination of 2-photon imaging of blood flow, magnetic resonance imaging of myelin, and quantitative light and electron microscopy, we report herein that myelin loss precedes axonal injury in BCAS, and importantly, that the suppression of astrocytic Cx43 protects against deficits in myelin integrity and cognition otherwise resulting from chronic cerebral hypoperfusion.
Methods
Animals
All experiments were performed according to the guidelines of the Institutional Animal Care and Use Committee at Tongji Medical College, Huazhong University of Science and Technology. Male C57Bl/6 mice (age, 10-12 weeks; weight, 22-27 g) of SPF grade were obtained from Hunan SJA Laboratory Animal Co. Ltd., Hunan, China. Mice were housed in groups of 2-4 mice per cage under standard housing conditions with a 12-h/12-h dark-light cycle and provision of food and water ad libitum.
Astroglial Cx43-deficient mice
To generate a mouse line in which the gene encoding Cx43 was deleted in astrocytes only, we followed the procedure as described in a previous report [21]. We crossed female GFAP-Cre mice with floxed males, since Cre expression was in the male germline. In brief, female mGFAP-Cre transgenic mice (purchased from Jackson laboratory, line73.12, stock Number 012886) were crossbred with male Cx43fl/fl mice (purchased from Jackson laboratory, stock Number 008039). Then, the female F1 generation (GFAP-Cre+ Cx43fl/fl) were crossbred with male Cx43fl/fl mice to obtain the F2 generation: astroglial-specific Cx43-del mice (GFAP-Cre+ Cx43fl/fl) and their normal littermates (Cx43fl/fl) (Figure S1A). Rosa-LacZ reporter (Rosa) mice (kindly given by Prof. Shumin Duan) were crossbred with the mGFAP-Cre transgenic mice. Before performing further experiments, we confirmed Cre-recombinase activity in the brain of mGFAP-Cre mice [22] by gain of lacZ activity, and identified the genotypes of individual F2 mice by PCR (Figure S1B-D). Astroglial Cx43-deficiency was further confirmed by immunohistochemistry (Figure S1E-F).
Experimental design
All the experimental groups were randomized and all outcome analysis was carried out by independent investigators blinded to the treatment conditions and mouse types. Randomization was performed before the surgical procedure (BCAS or Sham-operation, Carbenoxolone (CBX)/Meclofenamic Acid (MFA), Gap19/Gap26 administration or Vehicle) using the random number generator in Graphpad. From the mean and standard deviation of outcomes for each group seen in previous studies [16, 17, 20] and from our own preliminary experiments, we calculated that five animals per group would suffice to obtain 80% power at a significance level of <0.05 with a 2-sided test. Because BCAS leads to consistent WM lesions and low mortality, we used 5-10 animals per group, since the variance depended on the particular outcome being studied. A total of 732 wild-type male mice, 171 Cx43fl/fl male mice, 12 GFAP-Cre+ Cx43fl/- male mice, 176 GFAP-Cre+ Cx43fl/fl male mice, two Rosa-LacZ mice and two GFAP-Cre+ Rosa-LacZ mice were used in the experiments. Our decision to use exclusively adult male mice was intended to minimize sex and age differences as factors in the cerebral ischemic injury. Future studies will include female mice. A flow diagram illustrates the experimental design of the study (Figure S2).
Surgical procedures
Mice were anesthetized with a mixture of ketamine (0.12 mg/g) and xylazine (0.01 mg/g) in 0.9% NaCl solution administered intraperitoneally. Chronic cerebral hypoperfusion was then induced by applying wire microcoils (Sawane Spring Co, Shizuoka, Japan) with an internal diameter of 0.18 mm around both common carotid arteries, as previously described [17, 23], to induce chronic hypoperfusion instead of complete occlusion. Sham-operated animals underwent identical surgical interventions except without application of microcoils to the common carotid arteries.
Carbenoxolone (CBX), Meclofenamic Acid (MFA), Gap26 and Gap19 treatment
ALZET osmotic mini-pumps (Cupertino, CA, model 1004; reservoir volume 100 μl, pumping rate 0.11 μl/h, with continuous application for one month) were filled with different concentrations of CBX (0.1, 0.5, or 2.5 μg/μl, Sigma-Aldrich, USA), MFA (0.1, 1, or 10 mM, Sigma-Aldrich, USA), Gap 26 (2.0 μg/μl, TOCRIS Bioscience, UK, CAS Number 197250-15-0), Gap 19 (2.0 μg/μl, TOCRIS, Bioscience, UK, CAS Number 1507930-57-5) or saline vehicle [14, 24-26]. The pumps were connected to brain infusion cannula (Brain Infusion Kit 3, ALZET, Cupertino, CA) via a catheter tube and primed by placement at 37 °C for 24 h prior to subcutaneous implantation. After creating a burr hole, the infusion cannula was directed to the right ventricle (anterior-posterior = -0.4 mm, medial-lateral = 1.0 mm, and dorsal-ventral = 3.0 mm) using the stereotaxic frame, and stabilized on the skull with dental cement. The ALZET pump was placed in a subcutaneous pocket prepared under the skin of back, and the catheter was guided under the skin to the exposed skull. The scalp wound was closed with suture, and in the same surgical session the mice underwent BCAS surgery as described above, beginning just at the start of the drug or saline delivery via minipump.
In vivo two photon imaging
Images of mouse vasculature were acquired dynamically by two-photon microscope laser scanning as described previously [27, 28]. Mice from sham operated and BCAS groups were anesthetized intraperitoneally with a mixture of ketamine (0.12 mg/g) and xylazine (0.01 mg/g) in 0.9% NaCl solution and then intubated and ventilated with a small animal ventilator (CWE). Blood gasses were monitored and body temperature was maintained at physiological parameters (~100 breaths/min, tidal volume of 0.3-0.4 ml, pO2= 80-150 mmHg, pH 7.25-7.5, body temperature of 37 °C). For vasculature imaging, we injected 0.1 ml of blood-brain barrier impermeable Texas Red-conjugated dextran-70 (MW, 70 kDa; 1% in saline; Invitrogen) to a femoral artery just before imaging. A MaiTai laser (Spectra Physics) attached to a confocal scanning system (LSM710, Carl Zeiss) and an upright microscope (20x/1.0, water immersed, WD, 1.8 mm, W-Plan-Apochromat 75 mm, DIC slide Senarmont) was used to image the cortex; the excitation wavelength was 870 nm.
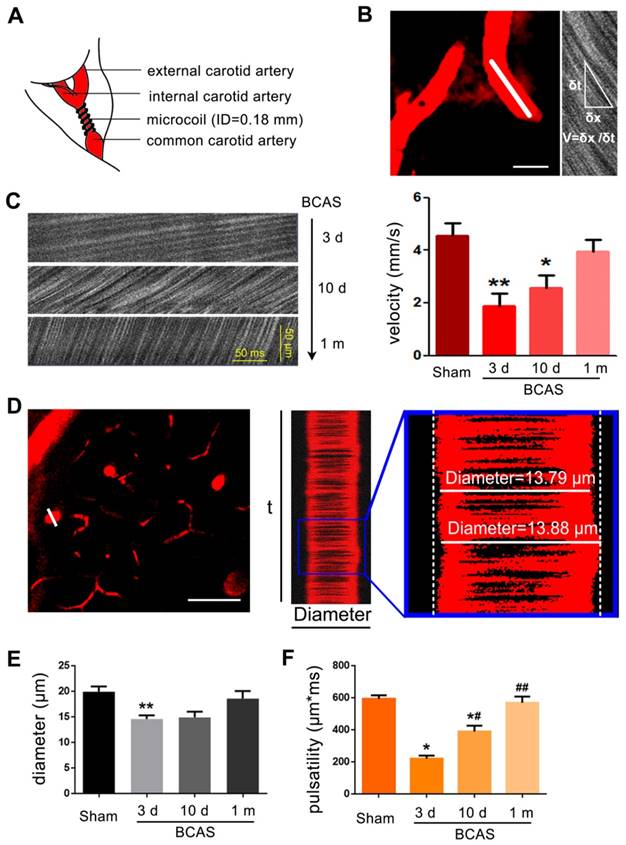
To measure vessel diameters, we acquired 3000 ms X-T line scans orthogonal to the vessel axis in penetrating arteries 50-150 μm below the cortical surface. The line-scans were collected along the centerline of each vessel at a scan rate of 1.6 kHz/line. RBC velocity was determined from the slope of the line-scan. Planar image stacks (256 by 256 pixels) were acquired to establish the vessel diameter. The mean (SD) diameters of (N=3) penetrating arteries measured per mouse was 10.0 ± 5.1 μm (n=6 mice for each group). The vessel wall pulsatility (derived units m*ms) was calculated as the absolute value of area under the diameter-time plot, integrated about the running average over the 3000 ms epoch (Figure 1).
Reduction of blood flow velocity and arteriole pulsatility after BCAS. (A) Schematic of BCAS model. (B) Representative images showed the penetrating artery at a depth of 60 μm below cerebral cortex surface in BCAS mice at three days post-surgery. Red blood cell (RBC) velocity was detected by two photon line scanning from the penetrating arterioles centerline axis (white line). The streaks within the line scan images represent non-fluorescent RBCs moving across a fluorescent background, where δx represents the distance traveled by RBCs during the time interval, δt. The RBC velocity is then calculated from the regression slope as shown in the equation. Scale bar, 2 µm. (C) Representative images of RBC movement in penetrating vessels and comparison of RBC velocity along a penetrating arteriole at three days, ten days, and one month post BCAS. **p˂0.01 **p˂0.01 versus Sham, one-way ANOVA with Dunnett's post-hoc test, n=6 mice for each group and sampling of three vessels for each mouse. (D) Cortical penetrating arteries were selected and the X-T line scans (white lines) were generated orthogonal to the vessel axis. Vascular pulsatility was then calculated as the absolute value of the integral of vascular diameter during a three second recording. The penetrating arteriole diameters and pulsatility were evaluated by two photon imaging at three days, ten days, and one month post-BCAS. Scale bar, 20 µm. (E-F) A bar histogram comparison of penetrating arteriole diameters and pulsatility at three days, ten days and one month post BCAS compared to sham operated mice *p˂0.05 **p˂0.01 versus Sham, #p˂0.05 ##p˂0.01 versus three days post BCAS, one-way ANOVA with Dunnett's post-hoc test, n=6 mice in each group and sampling of three vessels in each mouse.
Immunohistochemistry
At three days, ten days, one month, or three months after surgery, mice were perfused with 20 ml of 0.9% heparinized phosphate-buffered saline (PBS) and then 20 ml 4 % paraformaldehyde (PFA) while deeply anesthetized. After brain removal, coronal slices (10 μm thick) were prepared at -20 °C using a constant temperature freezing microtome (CM 1900, Leica, Germany). The primary antibodies used in this study were: myelin-associated glycoprotein (MAG, 1:100, Santa Cruz Biotechnology), myelin basic protein (MBP, 1:100, Millipore), neurofilament-Light (NF-L), neurofilament-Medium (NF-M), and neurofilament-Heavy (NF-H) (1:1000, Cell Signaling Technology), Caspr (1:100, UC Davies NIH NeuroMab), panNfasc (kindly provided by Prof. Peter Brophy), type Ⅵ sodium channel (Nav1.6, 1:200, Millipore), connexin43 (Cx43, 1:200-1:250, Abcam) , glial fibrillary acidic protein (GFAP, 1:200, Sigma), microtubule-associated protein2 (Map2, 1:200-1:400, Abcam), neuronal nuclei (NeuN, 1:200, Chemicon), ionized calcium binding adapter molecule 1 (Iba-1, 1:500, Wako Pure Chemical Industries), macrosialin (CD68, 1:200, BIO-RAD), Neurofilament H Non-Phosphorylated (SMI32, 1:200, Cell Signaling Technology) and glutathione-s-transferase π (GSTπ, 1:200, Abcam). Secondary antibodies, which included FITC, Cy3, AlexaFluor488, and AlexaFluor647, purchased from Jackson ImmunoResearch Laboratories. Sections were incubated with primary antibodies overnight at 4 °C in PBS (pH 7.4), along with 0.1% Triton X-100. Images were acquired by confocal microscopy (FV1200, Olympus, Japan). Confocal settings were kept the same during every single immunofluorescence staining experiment.
Nodes of Ranvier Imaging
For visualizing the nodes of Ranvier, images were acquired with a 100× oil immersion objective. Length measurements and co-localization assessment were performed using Imaris (Bitplane AG, Switzerland) and ImageJ (NIH, USA) software as previously described [16].
Sodium channel cluster (Nav1.6) length was assessed after one month of hypoperfusion. The identity of the nodes of Ranvier was confirmed by Caspr/Neurofascin double labeling. All length measurements were performed using ImageJ [16]. For Caspr and Neurofascin, common epitope labeling co-localization analysis [29] was performed using Imaris software (Bitplane AG, Switzerland), and Pearson coefficients were determined for the correlations.
Luxol fast blue staining and X-gal staining, Electron Microscopy, Western Blots and Eight-arm radial maze test
A detailed description of these experimental procedures is provided in the online-only Data Supplement. The methodology for scoring the severity of WM lesions was as previously reported. [17, 30] In brief, to gauge the severity of white matter lesions, the tissue was graded by two independent investigators as follows: normal (grade 0), disarrangement of nerve fibers (grade 1), formation of marked vacuoles (grade 2), or disappearance of myelinated fibers (grade 3).
Magnetic resonance imaging
At one month post-surgery, mice were re-anesthetized and placed in a magnetic resonance imaging (MRI)-compatible holder. Structural MRI data were collected using a Bruker 7 T preclinical scanner with a 72 mm volume coil and a phased array mouse brain coil, as previously described [31]. In brief, axial diffusivity (AD), mean diffusivity (MD), fractional anisotropy (FA), and radial diffusivity (RD) maps were generated using the Bruker software PARAVISION 5.0 and processed for region of interest (ROI) analysis (Figure S2A). White matter ROIs, including corpus callosum, fimbria, internal capsule, anterior commissure, and optic tract, were selected from within T2-weighted structural volumes by an observer blinded to intervention, and then transferred onto the parametric maps for measurements.
Microdialysis
Cerebral microdialysis was used to determine the interstitial concentrations of glutamate, adenosine, and lactate in living mice. All experiments were performed in non-anesthetized freely moving mice at day zero, three days or one month after surgery. Microdialysis probes (MD-2211 for probes, MD-2255 for cannula, BASi, USA) in the white matter (anterior-posterior = -1.0 mm, medial-lateral = 1.0 mm, and dorsal-ventral = 1.8 mm) were continuously perfused with artificial cerebrospinal fluid (aCSF) at flow-rates of 0.1, 0.5, 1.0, and 1.5 μl/min (BASi microdialysis pump, USA). Four dialysate samples (5 μl) were collected at 30 min intervals to determine the baseline concentrations. Perfusion of the probes at 1 μl/min was maintained two hours before the actual sampling to attain the steady-state concentrations of the analytes. The prepared samples were stored at -80 °C for further analysis.
Chromatography
The microdialysate samples were analyzed for glutamate using a Dionex Ultimate 3000 HPLC system as previously described [32]. In brief, the calibration standards of glutamate prepared at five different concentration levels (0.10, 0.50, 1.0, 5.0, and 15.0 μM) were derivatized with o-phthalaldehyde in 5 mM sodium sulfite and detected with colormetric detection for the calibration curve generation. The calibration curves were constructed by plotting the peak areas of compounds (y, nA*min) against their concentration (x, μM), and used to calculate by interpolation the glutamate concentrations in both aCSF and real microdialysate samples from the mouse brain.
The microdialysate samples from white matter were run under the same conditions as the standards [32]. The intra-day and inter-day variabilities for the compounds were assessed by obtaining triplicate samples on the same day and on three consecutive days, respectively.
Enzymatic colorimetric assays
Portions of microdialysate samples were analyzed for lactate and adenosine content using enzymatic lactate and adenosine assay kits (Biovision, Mountainview, CA, USA) according to the manufacturers' guidelines. Fluorescence was measured at 590 nm (lactate) and 587 nm (adenosine) using a microplate reader (Model 680 Bio-Rad, Hercules, CA, USA) at an excitation wavelength of 535 nm.
1H NMR analysis
The dissected white matter samples were collected in Eppendorf tubes and frozen by immersion in liquid nitrogen as soon as possible post mortem. The white matter samples were later homogenized in 0.1 M HCl/methanol at room temperature using a Qiagen Tissue-Lyzer (RetschGmBH, Germany) operating at 20 Hz for 90 s. Then the homogenized mixtures were decanted and centrifuged three times (16000 g, for 10 min at 4 °C), with pooling of the supernatants. The collected liquid was lyophilized and dissolved in 540 μl 12.5 mMPi (90 mM K2HPO4 and 35 mM KH2PO4) and 60 μl D2O for 1H NMR analysis.
The 1H NMR spectra were acquired and data analysis performed as previously described [33, 34]. In brief, all 1H-NMR spectra were acquired at 298 K on a Bruker Avance III 600 MHz NMR spectrometer (operating at 600.13 MHz for 1H) equipped with an inverse cryogenic probe using a standard solvent-suppressed one-dimensional pulse sequence (the standard WATERGATE pulse sequence). A total of 32 k data points and 64 transients were collected for each spectrum with a spectral width of 20 ppm. The 1H-NMR spectra were analyzed with the NMRSpec software [35] for measurement of metabolite concentrations. We have successfully used the NMRSpec software for metabolomics in several previous studies [33].
Statistical analysis
Statistical analyses were performed using SPSS 19.0. All data were expressed as the mean ± SEM (except for in vivo two photo imaging mentioned above). Data were first tested for normality using the Kolmogorov-Smirnov test. Single comparisons to control results were made using 2-tailed Student's t-test and multiple comparisons within the same data set were analyzed by one-way analysis of variance (ANOVA) with Dunnett's post-hoc test. Mann-Whitney U test was used to compare the severity of WM lesions between the groups. One-way ANOVA with repeated analysis was conducted in the eight-arm maze test. Differences were considered statistically significant at a P value of <0.05.
Results
Hypoperfusion leads to reduced RBC velocity, arteriolar pulsatility, and diameter
Several studies have reported that the white matter is particularly vulnerable to chronic hypoperfusion [13, 16, 17]. To assess the evolution of hypoperfusion-associated white matter pathology, we adopted the BCAS model of chronic hypoperfusion, in which coils are placed around both common carotid arteries permanently (Figure 1A). The RBC velocity was evaluated by two photon in vivo imaging with line scanning at three days, ten days and one month post BCAS (Figure 1B-C). The reduction of RBC velocity along arterioles was most striking at three days post-BCAS, when there was an approximately 55% reduction relative to the velocity in sham controls. RBC velocity along the arterioles recovered gradually, but plateaued at 80% of baseline ten days post-BCAS. No statistically significant reduction of cerebral RBC velocity was noted at one month following BCAS (Figure 1C). We next assessed the penetrating arteriole diameter and pulsatility (Figure 1D). The mean diameter of penetrating arterioles was 25% lower at three days post-BCAS, but there was no discernible reduction at ten days or one month post-BCAS (Figure 1E). We also analyzed the rapid pulsatile movements of the vascular wall, taking advantage of the high temporal resolution of two photon imaging in vivo [27, 36]. Pulsatility analysis showed a transient initial reduction by as much as 60%, which had recovered at one month post-surgery to levels close to those observed in sham operated mice (Figure 1F). Overall, these observations show that bilateral common artery stenosis significantly reduced arteriole pulsatility and diameter and likewise reduced cerebral blood flow for at least ten days (Figure 1).
Chronic hypoperfusion leads to loss of white matter integrity
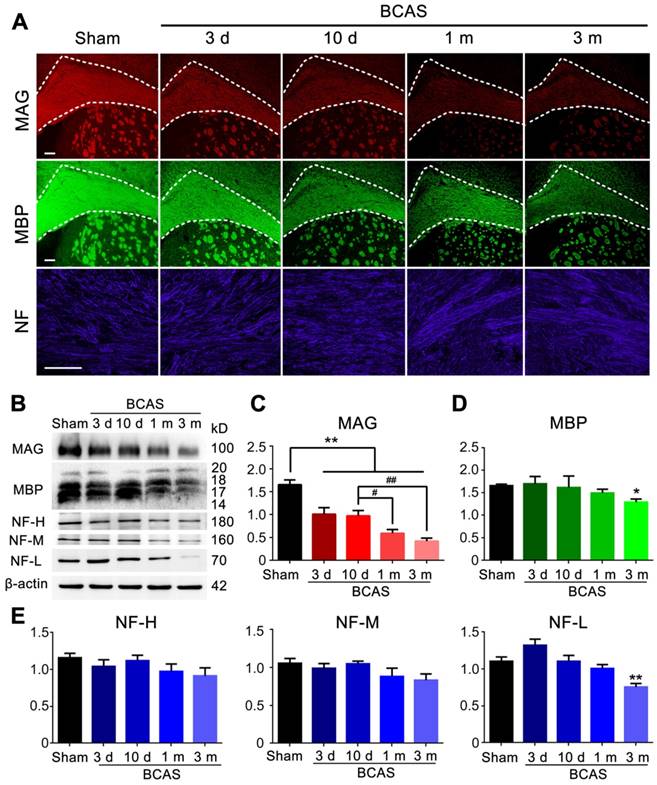
We next asked if reduced cerebral blood flow and penetrating arteriole pulsatility would affect myelin integrity. The expression of myelin associated glycoprotein (MAG), a marker for myelin-axon integrity [37], as well as the expression of myelin basic protein (MBP) and the axonal marker neurofilament (NF)[38, 39], respectively, were quantified at three days, ten days, one month, and three months post-injury in the BCAS group using immunochemistry and western blots; all results were compared to those obtained from the sham-operated group.
A sharp drop in MAG expression was noted at day three, which progressed throughout the three months observation period. The MAG expression was significantly decreased at one month compared with ten days after BCAS, but there was no significant change of MAG expression between one and three months post-BCAS (Figure 2A-C). In contrast, the expression of MBP showed an overall decreasing trend and a significant decrease lasting until three months post-BCAS. The neurofilament-heavy (NF-H), neurofilament-medium (NF-M), neurofilament-light (NF-L) all remained stable until three months post-BCAS, at which point NF-L displayed significant reductions (Figure 2A-E). Thus, compromised myelin-axon integrity was already evident by three days after hypoperfusion injury, even though the myelin still appeared intact, as defined by MBP immunofluorescence and western blots analysis. Concurrently, the expression of the neurofilament proteins NF-H, NF-M, NF-L, comprising a canonical set of markers of axonal integrity, exhibited no detectable changes. Thus, we confirmed previous observations [16] that mice with chronic hypoperfusion exhibited a significant disruption of myelin-axon integrity at early time-points following BCAS, prior to any histological evidence of axonal damage.
Myelin disruption was observed in chronic hypoperfusion mice. (A) Representative images depicting immunofluorescent labeling of myelin-associated glycoprotein (MAG), myelin basic protein (MBP), and neurofilament (NF) in coronal slices from sham-operated mice and bilateral common carotid artery stenosis (BCAS) mice at different time points post-surgery (three days, ten days, one month and three months). Scale bar, 100 µm. (B) The expression of MAG, MBP and NF (three major NF subunits based upon their molecular mass: lowest (NF-L), the middle (NF-M) and the highest (NF-H)) was determined by western blots at three days, ten days, one month, and three months post-BCAS and quantitative analysis of results in (B) was performed (C-E). *p˂0.05, **p˂0.01 versus Sham, #p˂0.05 ##p˂0.01 versus ten days post-BCAS, one-way ANOVA with Dunnett's post-hoc test, n=6 mice for each group.
To critically extend this observation and assess the sequence of injury in the BCAS model of chronic hypoperfusion, we next utilized MR diffusion tensor imaging (DTI) to measure the integrity of the myelin sheath and axonal structures in the medial part of corpus callosum (CCm), peripheral part of corpus callosum (CCp), anterior commissure (AC), fimbria of the hippocampus (F), internal capsule (IC), and optic tract (OT) (Figure S3A). We found that the fractional anisotropy (FA) decreased in the IC and OT at day three, in CCm at day ten, and in F at one month; these FA decreases progressed with hypoperfusion post-BCAS, indicating progressive loss of white matter integrity (Figure 3A-B, Figure S3C). The radial diffusivity (RD) was slightly different between groups, and the increase in RD was significant in the CCm, F, and IC at one month post-BCAS, demonstrating that the disruption of myelin had occurred by this time (Figure 3C-D, Figure S3D). The FA and RD values also differed at one month post-hypoperfusion, consistent with the time course of white matter pathology. There was no significant change in mean diffusivity (MD) in any region except for the OT, where no changes presented until three months post-hypoperfusion (Figure S3E), while axial diffusivity (AD) remained roughly constant until three months in F, IC, and OT (Figure S3F). These data suggested that there was no loss to axonal integrity until three months post-hypoperfusion. The T2-weighted imaging data revealed no overt structural changes in any of the mice studied (Figure S3B), which was consistent with the histological observations.
White matter disruption following BCAS detected by MRI and electron microscopy. (A, C) Selected regions of 7.0 Tesla (T) MRI for inspecting the integrity of myelin sheath and axonal structures in different regions of white matter (the medial part of corpus callosum (CCm), peripheral part of corpus callosum (CCp), anterior commissure (AC), fimbria of hippocampus (F), the internal capsule (IC) and the optic tract (OT)) at different time points (three days, ten days, one month, and three months) post BCAS. (B, D) Quantitative analysis of fractional anisotropy (FA) and radial diffusivity (RD) at different time points (three days, ten days, one month and three months). Arrowheads indicate vacuoles and delamination, which are classical signs of disruption of myelinated structures. *p˂0.05, **p˂0.01 versus Sham, one-way ANOVA with Dunnett's post-hoc test, n=6 mice for sham and three months, n=5 for three and ten days, and n=7 for one month. (E) Representative electron microscopy images of axons in corpus callosum of sham-operated and BCAS mice at three days, ten days, one month and three months post-BCAS. Representative electron microscopy images are from sham-operated and BCAS mice at one month post-BCAS. Scale bar, 2 µm. (F) Scatter plot diagram of the g ratios (the inner axonal diameter relative to the total outer diameter) and axon diameter was performed at one month post-injury. (G) Quantitative analysis of g ratios at different time points (three days, ten days, one month and three months) post injury. **p˂0.01, p>0.05 versus Sham, one-way ANOVA with Dunnett's post-hoc test, n=200 myelinated axons (40 axons per mouse, 5 mice per group) for each group.
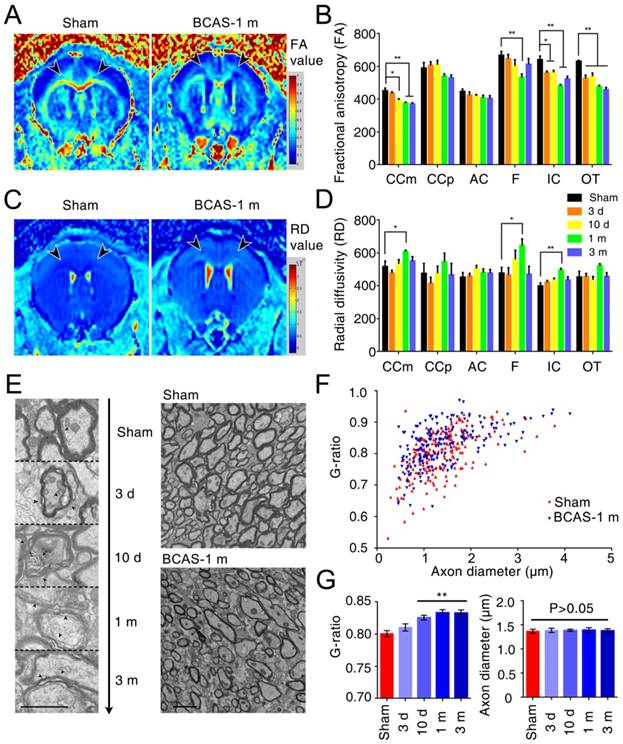
We also detected ultrastructural damage of the CCm in the hypoperfused mice, finding vacuoles and delamination in BCAS mice from three days post-injury and persisting throughout the three months observation period, whereas well-defined lamellar myelin sheaths remained intact in sham-operated mice (Figure 3E). Quantitative analysis of g ratios confirmed the ultrastructural disruption of myelin first presenting at ten days post-hypoperfusion in BCAS mice, while the axons remained intact (Figure 3F-G).
In the next phase we applied luxol fast blue (LFB) staining as a highly sensitive marker of myelin loss. There was decreased LFB staining in the CCm first appearing at three days, and with progression thereafter. The caudate-putamen (CPu), AC, IC exhibited significant loss in staining at one and three months, whereas F displayed a tendency of myelin loss, but was not significantly altered. The most severe rarefaction of myelin was evident in the CCm (Figure 4A-B). Consistent with the preservation of axons and NF, the density of Map2- and NeuN-immunoreactive neuronal cell bodies remained normal throughout the observation period, as previously reported [16] (Figure S4C-E). Together, these multiple approaches including quantitative immunohistochemistry, electron microscopy, and MRI, all indicated that white matter injury induced by chronic hypoperfusion results in early disruption of the relationship of axons with their myelin sheaths, followed by late demyelination. These changes were most notable in the CCm. In contrast, the number of NeuN- and Map2-defined neurons and their axons remained intact during the three months observation period. A schematic diagram compares the temporo-spatial pattern of myelin injury in the BCAS model detected by multiple alternative approaches (Figure S5).
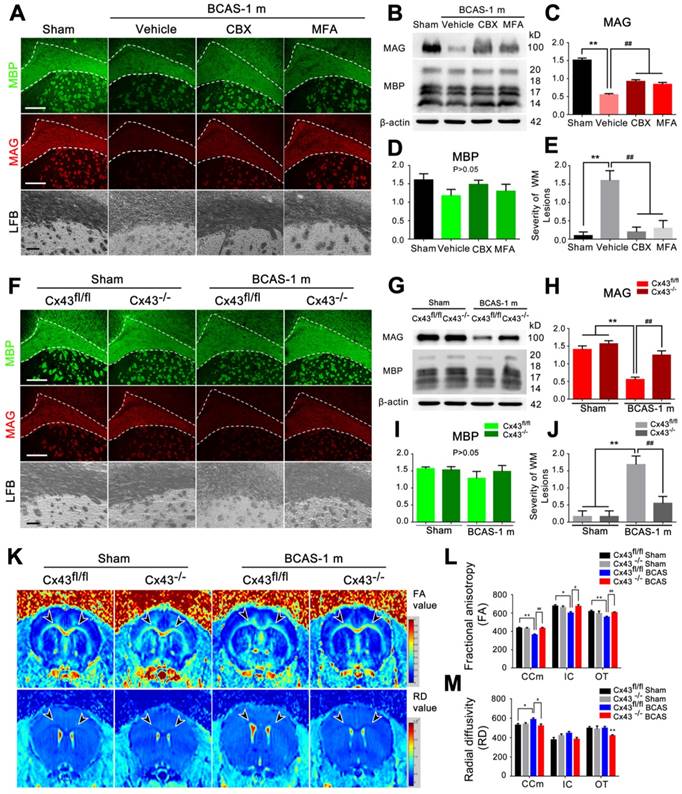
BCAS-associated white matter disruption was protected by gap junction inhibition. (A) Representative confocal images of coronal sections labeled with MBP and MAG from sham-operated and carbenoxolone (CBX) and meclofenamic acid (MFA) treated groups. The severity of white matter lesions was also assessed by Luxol fast blue (LFB) staining. Quantitative analysis was performed in (E). **p˂0.01 versus Sham, ##p˂0.01 versus Vehicle, one-way ANOVA with Dunnett's post-hoc test, n=10 mice per group. Scale bars, 300 µm and 50 µm. (B) Expression of MAG and MBP was determined by western blots in mice from sham-operated and vehicle, CBX and MFA treated group. (C-D) Quantitative analysis of (B). **p˂0.01 versus Sham, ##p˂0.01 versus Vehicle, one-way ANOVA with Dunnett's post-hoc test, n=6 mice per group. (F) Representative confocal images of coronal sections stained for MBP and MAG from Cx43fl/fl BCAS mice, GFAP-Cre+ Cx43fl/fl (Cx43-/-) mice and sham-operated littermate controls at one month post-surgery. Expression of MAG and MBP was determined by western blots (G-I) and severity of white matter lesion was assessed by LFB staining. Quantitative analysis was performed in (J). **p˂0.01 versus Sham, ##p˂0.01 versus Cx43fl/fl BCAS mice, one-way ANOVA with Dunnett's post-hoc test, n=6 mice per group. Scale bars, 300 µm, 50 µm. (K) 7 T MRI scan detecting the integrity of myelin sheath and axonal structures in corpus callosum, internal capsule and optic tract in Cx43fl/fl BCAS mice, Cx43-/- BCAS mice and sham-operated control at one month post-surgery. (L-M) Quantitative analysis of fractional anisotropy value (FA) and axial diffusivity (RD) value in (K). *p<0.05, **p<0.01 versus Cx43fl/fl Sham, #p<0.05, ## p<0.01 versus Cx43fl/fl BCAS mice, one-way ANOVA with Dunnett's post-hoc test, n=4 for Cx43-/- sham-operated control, n=5 for Cx43fl/fl BCAS mice, Cx43-/- BCAS mice and Cx43fl/fl sham-operated control.
White matter damage was reduced by gap junction inhibition
We previously showed that pharmacological inhibition of gap junctions or deletion of astrocytic connexin 43 (Cx43) can ameliorate delayed neuronal death in a mouse model of middle cerebral artery occlusion [40]. On that basis, we now asked whether the white matter impairment in BCAS mice is also modulated by Cx43. To this end, we analyzed the expression of Cx43 at three days, ten days, one month, and three months post-BCAS. We found that Cx43 expression increased at three days, peaking at one month post-BCAS, which was accompanied by astrocytic activation, detected as an upregulation of GFAP (Figure S6A-C).
Carbenoxolone (CBX) and meclofenamic acid (MFA) are two commonly used inhibitors of gap junction proteins [14, 41], including Cx43 [24, 25]. We treated BCAS mice by administering increasing concentrations of CBX (0.1, 0.5 and 2.5 μg/μl) and MFA (0.1, 1 and 10 mM) with an osmotic minipump (Figure S6D-E). The minimum effective concentration to ameliorate myelin loss was 0.5 μg/μl for CBX and 1 mM for MFA (Figure S6D-E), which proved sufficient to block the BCAS-associated upregulation of Cx43 and activation of astrocytes (Figure S6F-H). To assess potential harmful effects of long time intraventricular infusion of the Cx inhibitors to mice, wild-type control mice were next treated with CBX (0.5 μg/μl), MFA (1 mM), or vehicle (saline, same volume) continuously for one month (Figure S7). An eight-arm maze test was performed on these mice at the end of the one month treatment. No difference in performance was observed between the three groups, i.e. CBX, MFA, or vehicle (Figure S7A). LFB staining revealed no structural damage to myelin after intraventricular infusion of CBX or MFA for one month (Figure S7C). Next, we tested the expression of MAG and MBP in CBX-treated and MFA-treated mice at one month post-BCAS. The reduction in MAG expression in BCAS mice one month post-injury was partly reversed by treatment with the connexin inhibitors CBX and MFA, suggesting partial pharmacological rescue of the insult to myelin sheaths (Figure 4A-D). Myelin integrity was preserved by these treatments, as assessed by LFB staining (Figure 4A and 4E). Thus, two different gap junction blockers substantially rescued the late white matter damage caused by hypoperfusion (Figure 4A-E).
To specifically test for a critical role of astrocytic Cx43 in white matter damage induced by BCAS, we next exposed astrocyte-specific Cx43 conditional knockout mice (Figure S1A-F) to BCAS. We first compared Cx30 expression in the conditional knockout mice and wild-type mice using western blots (Figure S8). We saw a minor increase of Cx30 expression in Cx43 conditional knockout mice compare to wild-type, while Cx30 expression did not change upon chronic hypoperfusion in the conditional knockout mice (Figure S8A-B). The expression of Cx43 to western blots of white matter did not differ between Cx43fl/fl and wild-type control mice (Figure S8C-D). Both the reduction in MAG expression and loss of white matter integrity were less pronounced at one month post-BCAS in mice with deletion of Cx43, as assessed by immunofluorescence, western blots and LFB staining (Figure 4F-J). On that basis, we next analyzed using structural MRI the effect of Cx43 deletion on BCAS-associated loss of myelin integrity. The decrease in FA of the white matter was significantly lower in mice with deletion of Cx43 (Figure 4K and 4L). A similar protective change in the RD value was detected in the CCm (Figure 4K and 4M), consistent with the attenuation of white matter pathology in the Cx43 nulls. Moreover, we evaluated the protective effect of genetic ablation of Cx43 on oligodendrocytes, finding that the number of GSTπ-positive cells was decreased in Cx43fl/fl mice at one month post BCAS, whereas GSTπ expression was unaffected in Cx43 knockout mice. (Figure S9) We also examined the axonal damage was evaluated by immunostaining of Neurofilament H Non-Phosphorylated (SMI32) at three days, ten days and one month post-injury in the BCAS group. Elevated expression of SMI32 was evident in BCAS mice only at one month post injury, and this increase was rescued by either pharmacological blockade by treated with Gap26 or Gap19 or genetic ablation of Cx43 (Figure S10).
Thus, using several different complementary approaches, we found that the loss of white matter integrity following BCAS was reversed by gap junction inhibition or astroglial Cx43 deletion. Overall, these results point to astrocytic communication via Cx43 (channels or hemichannels) as a contributor to myelin loss in the setting of BCAS.
Structural disruption of the nodes of Ranvier was retained if BCAS was combined with inhibition of gap junctions or deletion of Cx43
The nodes of Ranvier, which are flanked by myelinated axon segments, are important sites for action potential propagation based on the clustering of voltage-gated sodium channels (Nav1.6). Immunohistochemistry for Caspr and panNfasc reveals two key proteins that are essential for the integrity of the paranodal junction and the nodes of Ranvier [42, 43].
We first tested whether chronic cerebral hypoperfusion would cause damage to paranodal septate-like junctions. A significant reduction in the co-localization of Caspr and panNfasc within the paranodes was observed at one month post-hypoperfusion, suggesting a breakdown in the paranodal structure (Figure 5A and 5C). On that basis, we next asked whether disruption of paranodal structure would result in the breakdown of the nodes of Ranvier. We immunostained coronal brain sections for Caspr, and also for Nav1.6 (which is usually confined to the nodes and is sequestered by Caspr). However, in response to hypoperfusion, Nav1.6 expression extended beyond the territory of Caspr immunoreactivity, as shown by the superimposition of Nav1.6 and Caspr staining (Figure 5A). In addition, we saw a significant increase in the length of the Nav1.6-positive clusters in fibres within the corpus callosum in BCAS mice at one month post-hypoperfusion (Figure 5B and 5D).
Disruption of axon-glial interaction was reversed by gap junction blocking. (A) Representative confocal images immunostained for Caspr/panNfasc and Caspr/Nav1.6 in response to hypoperfusion in CBX, MFA, and vehicle-treated mice as well as sham-operated controls at one month post-surgery. Scale bar, 10 µm. (B) A higher magnification of (A) is shown. (C) Quantitative analysis of the Caspr/panNfasc co-localization. **p˂0.01 versus Sham, #p˂0.05, ##p˂0.01 versus Vehicle, one-way ANOVA with Dunnett's post-hoc test, n=6 mice per group. (D) Summary of the length of Nav1.6 domain in the corpus callosum is shown in the curve. p˂0.001 CBX-, MFA-treated versus Vehicle, Vehicle versus Sham, one-way ANOVA with Dunnett's post-hoc test, n=6 mice for each group and 100 Nav1.6 domains for each mouse. (E) Representative confocal images labeled with Caspr/panNfasc and Caspr/Nav1.6 in Cx43fl/fl, Cx43-/- mice and sham-operated controls at one month post BCAS. Scale bar, 10 µm. (F) A higher magnification of (E) is shown. (G) Quantitative analysis of Caspr/panNfasc colocalization is shown in histogram. n=6 mice per group. **p˂0.01 versus Sham, #p˂0.05 versus Cx43lox/lox (Cx43fl/fl) BCAS mice, one-way ANOVA with Dunnett's post-hoc test. (H) Summary of the length of the Nav1.6 clusters in the corpus callosum is shown in the curve. p˂0.001 Cx43fl/fl BCAS mice versus Sham, Cx43-/- BCAS mice versus Cx43fl/fl BCAS mice, one-way ANOVA with Dunnett's post-hoc test, n=6 mice for each group and 100 Nav1.6 domains for each mouse.
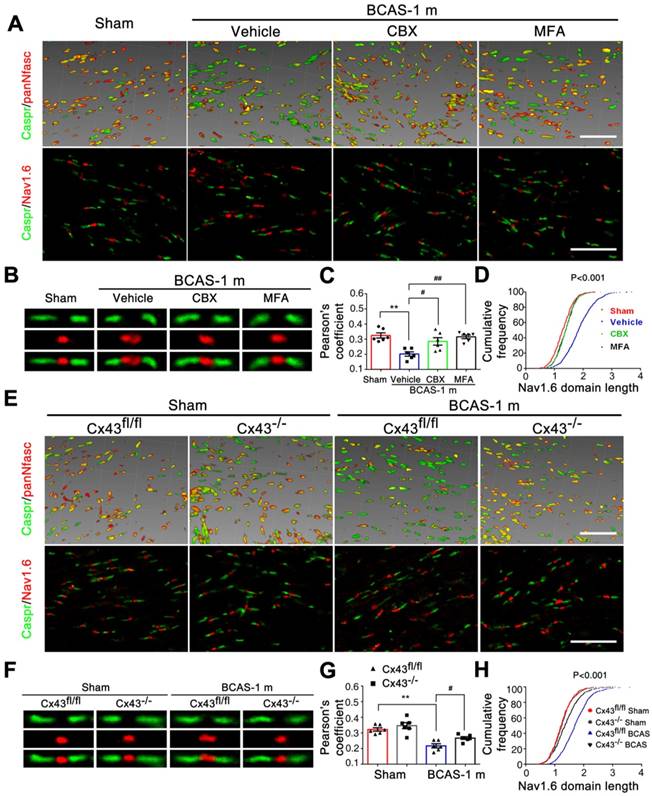
We next tested the protective effects of CBX and MFA treatment on the nodes of Ranvier after hypoperfusion injury. There was no alteration in either the length of Nav1.6 domains or the co-localization of pan-Neurofascin with Caspr in CCm of mice treated with either Cx43 inhibitor (Figure 5A-D). To further investigate the protective effect of astrocytic gap junction blockade on myelin integrity, we analyzed the length of Nav1.6 channels and neurofascin and Caspr localization in the astrocyte-specific Cx43 conditional knockout mice. There was no evident difference in these markers between the GFAP-Cre+ Cx43fl/fl mice and their littermates under physiological conditions, whereas the length increase in the Nav1.6 channels and selective delocalization of neurofascin and Caspr immunostaining in CCm in response to hypoperfusion injury were both retained by the knockout mice (Figure 5E-H). This finding confirmed that selective deletion of astrocytic Cx43 rescued the damage to myelin caused by hypoperfusionWe also assessed whether the ultrastructural damage to paranodes was ameliorated by pharmacological inhibition of gap junctions. We found that the g ratio was significantly decreased in mice treated with CBX and MFA (Figure 6A-D). In addition, there was no significant change in the g ratio in astrocytic Cx43 knockout mice following BCAS, further confirming the myelin protective effect of gap junction inhibition and deletion of Cx43 (Figure 6E-H).
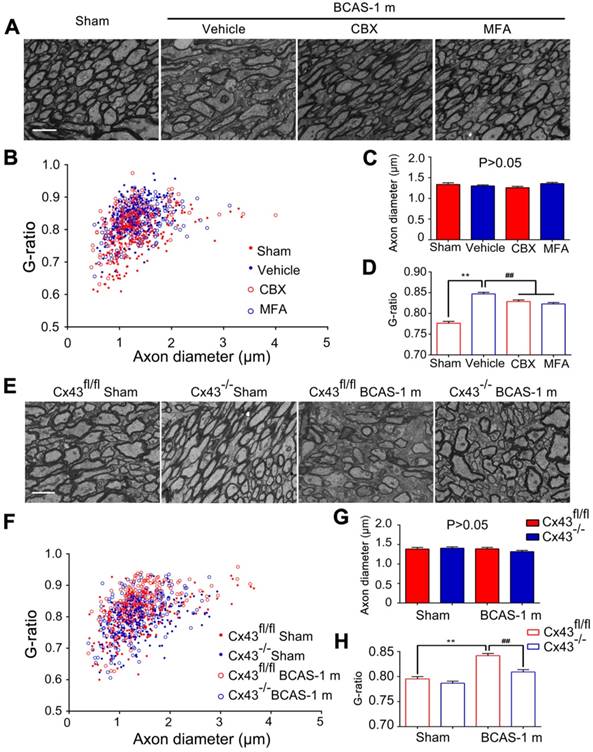
Ultrastructural myelin injury was attenuated by inhibition of connexin 43. (A) Representative electron microscopy images from brains collected at one month post BCAS in CBX, MFA, and vehicle-treated mice, as well as their sham-operated controls. Scale bar, 2.5 µm. (B-D) Quantitative analysis of the g ratios and axon diameters was performed. **p˂0.01 versus Sham, ##p˂0.01 versus Vehicle, one-way ANOVA with Dunnett's post-hoc test, n=200 myelinated axons (40 axons per mouse, five mice per group) for each group. (E) Representative electron microscopy images in Cx43fl/fl mice and GFAP-Cre+ Cx43fl/fl (Cx43-/-) mice at one month post BCAS as well as their sham-operated controls. Scale bar, 2.5 µm. (F-H) Quantitative analysis of the g ratios and axon diameters was performed. **p˂0.01 versus Sham, ##p˂0.01 versus Cx43fl/fl BCAS mice, one-way ANOVA with Dunnett's post-hoc test, n=200 myelinated axons (40 axons per mouse, five mice per group) for each group.
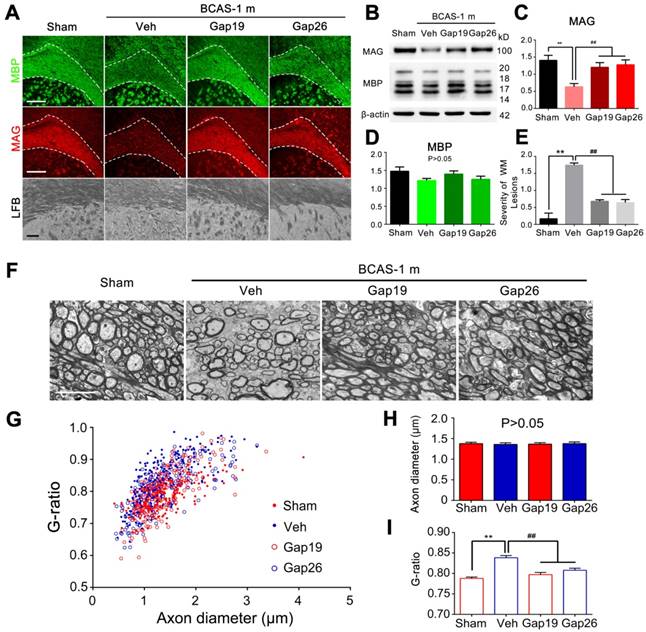
As a more specific approach to assess the role of Cx43 hemichannels in myelin injury in BCAS mice, we next administrated the two Cx43 mimetic peptides Gap19 and Gap26 separately by intraventricular administrated for one month. Prior studies have shown that Gap19 and Gap26 each selectively inhibit Cx43 hemichannels [14, 26]. We sought to determine if Gap19 and Gap26 could attenuate the myelin disruption in BCAS mice, based on immunofluorescence staining with MAG and MBP. Our results indicated that the reduction in MAG expression in BCAS mice one month post-injury was partly reversed by treatment with Gap19 or Gap26 (Figure 7A). Using western blots, we found that the decrease in MAG and MBP expression in BACS mice was attenuated when treated with either Gap19 or Gap26 (Figure 7B-D). Also, we saw no structural damage to myelin in BACS mice treated with either Gap19 or Gap26 when detected by LFB staining (Figure 7A, E). Besides, we assessed the effect of Cx43 deletion on ischemic white matter injury at three months post BCAS through LFB staining. Our results showed that Cx43 knockout also protected the myelin from injury at three months post hypoperfusion (Figure S11). Moreover, the g ratio was significantly lower in BCAS mice treated with Gap19 or Gap26 than in vehicle controls, further confirming that the ultrastructural damage to paranodes was ameliorated by specific pharmacological inhibition of Cx43 (Figure 7F-I).
Protection from BCAS-associated white matter disruption by selective inhibition of Cx43 by two mimetic peptides, Gap19 and Gap26. (A) Representative confocal images of coronal sections labeled with MBP and MAG from sham-operated and Gap19 or Gap26-treated groups. The severity of white matter lesions was also assessed by Luxol fast blue (LFB) staining. Quantitative analysis was performed in (E). **p˂0.01 versus Sham, ##p˂0.01 versus Vehicle, one-way ANOVA with Dunnett's post-hoc test, n=6 mice per group. Scale bars, 300 µm and 50 µm. (B) Expression of MAG and MBP was determined by western blots in mice from sham-operated and vehicle, Gap19, and Gap26-treated groups. (C-D) Quantitative analysis of (B). **p˂0.01 versus Sham, ##p˂0.01 versus Vehicle, one-way ANOVA with Dunnett's post-hoc test, n=6 mice per group. (F) Representative electron microscopy images from brains collected at one month post BCAS in CBX, MFA and vehicle treated mice as well as their sham-operated controls. Scale bar, 2.5 µm. (G-I) Quantitative analysis of the g ratios and axon diameters was performed. **p˂0.01 versus Sham, ##p˂0.01 versus Vehicle, one-way ANOVA with Dunnett's post-hoc test, n=200 myelinated axons (40 axons per mouse, five mice per group) for each group.
BCAS-associated deficits in working memory were reversed by inhibition of astroglial connexin43
We next asked whether pharmacological inhibition of gap junctions would attenuate the cognitive impairment induced by BCAS [2]. Working memory was assessed using an eight-arm maze test. The sham-operated mice made a declining number of revisiting errors over the training period, while the BCAS mice showed impaired working memory, with no improvement in revisiting errors (Figure 8A). The number of different arm choices in the first eight entries (ranging from four for chance performance to eight for a perfect performance) is another measure of working memory. The BCAS mice performed much worse than the sham-operated mice in each successive training session (Figure 8B), yet displayed no impairment in spatial reference memory (Figure 8C).
Working memory impairment was improved by inhibition of astroglial Cx43. The working memory of mice from each group was assessed by the eight-arm maze test at one month post-injury. One-way ANOVA with repeated analysis. (A-C) At one month post injury the BCAS mice made many more revisiting errors (p<0.001) and fewer different arm choices in the first eight entries (p=0.002) compared to the sham-operated mice. No impairment of spatial reference memory was revealed for BCAS mice in the eight-arm maze test (p=0.776). n=8 mice for each group. (D-F) There were far fewer revisiting errors in MFA- and CBX-treated groups (p<0.001) and far more different arm choices made in the first eight entries (p<0.05) compared to vehicle-treated mice, while no obvious difference of reference memory errors was observed (p>0.05). n=9 for Sham, n=10 for Vehicle and MFA- and CBX-treated groups. (G-I) At one month after BCAS, GFAP-Cre+ Cx43fl/fl (Cx43-/-) mice made fewer revisiting errors (p<0.001) and more different arm choices in the first eight entries (p=0.002) compared to their littermate controls, while no obvious difference in reference memory errors was evident (p>0.05). n=8 mice for each group. (J-L) There were far fewer revisiting errors in the Gap26- and Gap19-treated groups (p<0.01), and far more different arm choices made in the first eight entries (p<0.05) comparing to vehicle-treated mice, while no obvious difference of reference memory errors was observed (p>0.05). n=8 mice for each group.
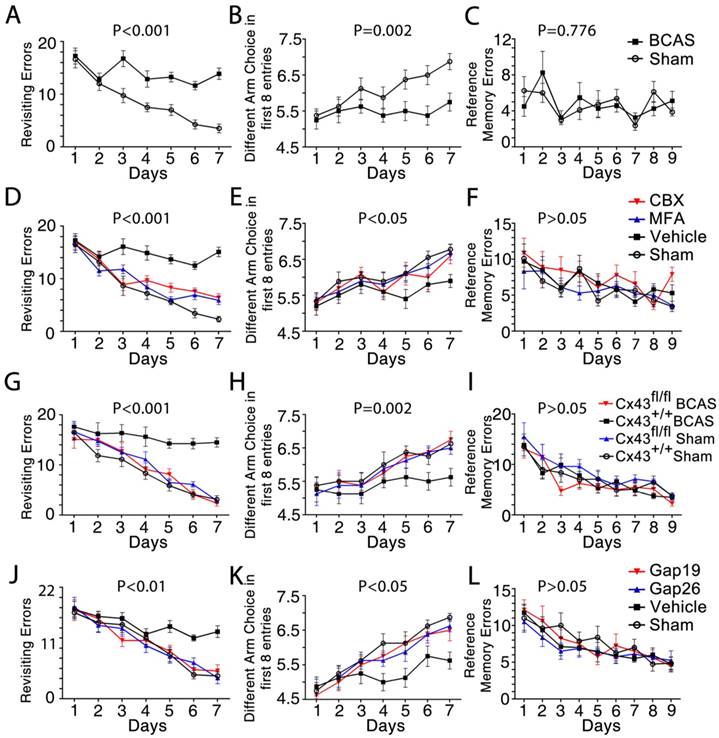
This cognitive decline in BCAS mice was reversed by treatment with the gap junction blockers MFA, CBX, or the Cx43 inhibitors Gap19 and Gap26, an effect apparent at one month post-BCAS. Particularly, the MFA-, CBX-, Gap19- and Gap26- treated mouse groups all made fewer revisiting errors (Figure 8D, 8J) and more distinct arm choices in the first eight entries compared to vehicle-treated mice (Figure 8E, 8K). Similarly, genetic deletion of astrocytic Cx43 ameliorated cognitive function at one month post-BCAS. Knockout of astrocytic Cx43 resulted in fewer revisiting errors (Figure 8G), and a greater number of different arm choices in the first eight entries compared to their littermate controls (Figure 8H). However, we saw no obvious differences in spatial reference memory between the groups (Figure 8F, 8I and 8L).
Overall, these results showed that BCAS mice displayed impaired working memory. These impairments were reversed in mice treated with any of four Cx43 blockers, as well as in Cx43 knockout mice, indicating a role of Cx43 in both mediating and sustaining BCAS-triggered deficits in working memory.
Interstitial glutamate increases were abolished by ablating astrocytic connexin 43
Astrocytes control and promote the homeostatic regulation of extracellular glutamate and potassium concentrations, as well as that of other neuroactive agents [12]. Astrocytic Cx43 can, however, act as a conduit for the release of neurotransmitters, including glutamate and ATP in pathological conditions [44, 45]. We here used microdialysis to evaluate the effect of Cx43 knock-out on the interstitial concentrations of glutamate, adenosine and lactate in BCAS mice. We saw a >50% increase in extracellular glutamate (Figure 9A-C) and a nearly 30% increase in the extracellular lactate concentration at three days post-hypoperfusion injury, relative to the levels in sham-operated mice (Figure 9D). These differences subsequently resolved, such that there were no remaining significant differences in interstitial glutamate or lactate concentrations in BCAS mice compared to sham-operated mice at one month post-injury. Deletion of astrocytic Cx43 abolished the increases in interstitial glutamate and lactate of BCAS mice at three days post-hypoperfusion (Figure 9C-D). We did not observe any changes in extracellular adenosine concentration in either the acute (three days) or chronic (one month) phases after BCAS injury in Cx43 knockout or control mice (Figure 9E). The total brain glutamate concentration was also assessed by 1H NMR analysis. The total concentration of glutamate in extracts of the corpus callosum was significantly increased at three days following hypoperfusion, whereas there was no such change in Cx43 knockout mice (Figure 9F-G).
Interstitial glutamate levels post-BCAS were normalized by ablation of astroglial connexin43. (A) Representative chromatograms of microdialysis results depicting the elution profiles of standard amino acid (AA) solutions and microdialysate samples after derivatization with OPA-sulfite for the detection of glutamate. The dashed square inset indicated the profile of the extracellular glutamate concentration. Black curve: standard AA solution (10 µM); Green curve: microdialysate from white matter. (B) A higher magnification image of a representative microdialysis chromatogram. The black curve depicts results for Cx43fl/fl mice and the red curve GFAP-Cre+ Cx43fl/fl (Cx43-/-) mice. (C-E) The interstitial concentrations of glutamate (C), lactate (D) and adenosine (E) in corpus callosum in Cx43fl/fl and Cx43-/- BCAS mice as well as sham-operated controls at three days and one month post-surgery. *p˂0.05 versus Sham, #p˂0.05, ##p˂0.01 versus Cx43fl/fl mice at three days post-BCAS, one-way ANOVA with Dunnett's post-hoc test, n=6 mice for each group. (F) The normalized 1H-NMR spectra of white matter tissue extracts, with the dashed square insets, indicating the 13C-NMR spectra profile of Glu4 in white matter tissue extracts. This figure was derived from the average values of Glu4 concentration in white matter tissue extract and standard error (SE), where SE was calculated based on the standard deviation and the number of replicates. (G) The proton signals connected with the C4 in Glutamate in the NMR spectra, which was the same as the dash square in Fig. 9G. The black curve depicts the results for Cx43fl/fl mice and the red curve for GFAP-Cre+ Cx43fl/fl (Cx43-/-) mice. The green band shows the range of standard error for Cx43fl/fl mice and the blue band shows the corresponding range for Cx43-/- mice. Glu4: sum of all resonances of Glu labeled at the carbon 4 position. p˂0.01 versus Cx43fl/fl mice three days post-BCAS, one-way ANOVA with Dunnett's post-hoc test, n=8 for Cx43-/- sham-operated mice and Cx43-/- mice at three days post BCAS, n=9 for Cx43-/- mice at one month post BCAS, n=7 for all the littermate control groups.
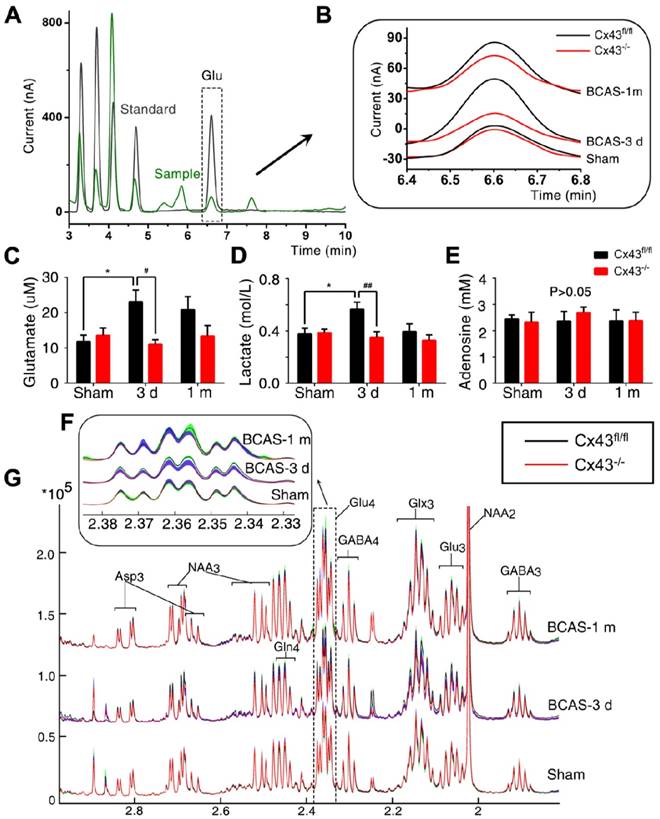
Administering the Cx43 mimetics (gap26 and gap19) to BCAS mice for only three days lowered the extracellular glutamate content, although glutamate content was slightly higher than in BCAS mice treated for one month. (Figure S12A-C) Administering the Cx43 mimetic peptides for three days by osmotic minipumps rescued disruption of axon-myelin integrity in the CCm (Figure S12D-G).
Together, these results suggested that the contribution of Cx43 to ischemic white matter injury involved increased concentrations of total brain and extracellular glutamate levels and possibly also lactate in the early post-injury phase.
Microglial activation was attenuated by knockout of astroglial connexin43
Astrocytic Cx43 hemichannels aggravate the inflammatory response to injury and cellular stress by releasing glutamate and ATP, which subsequently activate microglia, triggering the additional release of proinflammatory cytokines and thus increasing the susceptibility of oligodendrocytes to excitotoxicity [14, 15, 46]. We therefore asked whether microglial activation post-BCAS was affected by the deletion of Cx43. Chronic hypoperfusion triggered a significant increase in Iba-1 and CD68 positive microglial cells, which peaked at ten days post-BCAS injury. Genetic deletion of Cx43 significantly reduced the activation of microglial cells(Figure S13A-C). Moreover, increased expressions of IL1-β, TNF-α and NOX2 was detected in the Cx43-/- BCAS mice as well as the sham controls, but their expression was lower in Cx43-/- mice compared with Cx43fl/fl mice at one month post BCAS, which suggests lower neuroinflammation in Cx43-/- mice post hypoperfusion injury. Thus, this observation suggested that the presence of astrocytic Cx43 contributed to the activation of microglia and additional release of proinflammatory cytokines after hypoperfusion injury in mice (Figure S14).
Discussion
This study assessed the cellular mechanisms involved in white matter involution and degeneration in a murine model of chronic brain hypoperfusion. In our model of chronic carotid hypoperfusion, we place obstructive coils around the common carotid arteries to suppress both cerebral blood flow and arterial pulsatility, with these effects peaking at three days, followed by a slow recovery over the month thereafter. Blood flow is reduced in this model of chronic hypoperfusion [47, 48], and is linked to a progressive atrophy of white matter, in the absence of overt pathology in either cortical or subcortical gray matter regions [13, 16, 17]. The white matter loss suffered by these mice was characterized by early oligodendrocytic loss and myelin atrophy, which occurred prior to any detectable axonal degeneration. Remarkably, we noted that deletion of astrocytic Cx43, as well as pharmacological blockade of gap junctions, substantially protected against this white matter injury, and attenuated the loss of myelin induced by carotid stenosis. The cognitive performance of hypoperfused mice was also better preserved in those test animals subjected to genetic deletion of Cx43, to gap junction inhibitors, or peptides blocking Cx43 hemichannels.
Together, these results indicate that chronic hypoperfusion primarily targets the white matter glial syncytium, which constitutes a network of functionally interconnected astrocytes and oligodendrocytes. As such, the delayed axonal injury may represent a bystander effect of a dysfunctional glial syncytium. These observations are intriguing in the light of the well-established sensitivity of neurons to ischemic injury. Unlike glia, neurons die within minutes of ischemia due to Ca2+ overload and caspase activation initiated by excessive and uncontrolled NMDA receptor activation [49].
Given the high energy demands placed upon myelinated neurons by saltatory conduction, it may be surprising that oligodendrocytes should prove more vulnerable than their associated axons to chronic hypoperfusion injury. Yet oligodendrocytes are strictly dependent upon the local supply of glucose and oxygen, whereas axons traverse long distances, and may be robust to local pockets of hypoperfusion, if perfusion is preserved in adjacent regions and elsewhere along their path. In addition, axons in the adult brain express low levels of NMDA and AMPA receptors, while oligodendrocytes express moderate amounts of both excitatory receptors [7, 8]; axons may thus be relatively tolerant to increases in extracellular glutamate. In particular, oligodendrocytes are highly sensitive to AMPA/kainate receptor activation [7, 50, 51] and consequent Ca2+ and Na+ influx, which can respectively mediate oligodendrocyte death following hypoxia in both young and adult mice [9]. Thus, multiple mechanisms may contribute to the relative resistance of white matter axons and vulnerability of oligodendrocytes following bilateral carotid stenosis.
We also noted that conditional glial deletion of Cx43 under the regulatory control of the GFAP promotor, or long-term administrations of MFA, CBX, Gap19, or Gap26, all markedly reduced the severity of myelin pathology in BCAS mice. Oligodendrocytes and astrocytes are extensively coupled by gap junctions, forming a functionally intercommunicative glial syncytium. Among the various glial gap junction connexins, oligodendrocytes largely express Cx29, Cx32 and Cx47 [52], whereas astrocytes express Cx30 and Cx43 [12, 53]. The two cell types are thus coupled by heterotypic gap junctions, typically composed of either Cx32 docked with Cx30, or Cx47 docked with Cx43 [54]. Mice lacking either Cx32 or Cx47 are viable through adulthood, although histological examination has revealed that they are prone to vacuolization in myelin sheaths [31, 55]. In contrast, mice with deletion of both Cx32 and Cx47 die at two months of age, due to severe myelin pathology [55, 56]. Similarly, while deletions of either Cx30 or Cx43 alone have not been reported to affect myelin integrity, mice lacking both Cx30 and Cx43 exhibit extensive degeneration of myelin and vacuole formation [54, 57]. These findings suggest that oligodendrocytes depend vitally upon their heteromeric gap junction coupling with astrocytes. At first glance, it might seem paradoxical then that deletion of Cx43 should reduce the severity of white matter injury after hypoperfusion. Yet the number of functional gap junctions has been reported to fall sharply in the setting of cellular stress, as induced by ischemia or inflammation, and this decline is accompanied by an increased abundance of gap junction hemichannels, which connect the cytosol directly with the extracellular space [11, 44, 58]. As noted above, Cx43 hemichannels exert a key role in neuroinflammation, by acting as a conduit for the release of glutamate and ATP, which it turn promote release of purinergic transmitters and cytokines [59-61]. As such, mice with deletion of Cx43 or Cx30 exhibited less injury, neuroinflammation, and neuropathic pain than did wild-type controls in a spinal cord injury model [62]. Thus, BCAS may have induced less severe injury in Cx43 knockout mice than in wild-types because of the contribution of Cx43 hemichannels (as distinct from functional gap junctions) to the propagation of ischemic injury in white matter. We tested this hypothesis by administering the Cx43 mimetic peptides Gap19 and Gap26. In previous publications, Gap 19 blocked Cx43 hemichannels in a dose-dependent manner, without affecting Cx43 gap junction function [26]. Gap26 was also suggested to mainly inhibit Cx43 hemichannels.[59] The protective effect of these two peptides against ischemia myelin injury in BCAS mice suggests that the Cx43 hemichannels mediate deterioration after chronic hypoperfusion injury in BCAS mice. In fact, we observed reduced microgliosis, as well as astrogliosis in the Cx43 knockout mice, in which the post-injury extracellular glutamate concentrations were lower as well. Inflammatory cytokines have been shown to increase the calcium-dependent glutamate release in astrocytes, and impaired glutamate buffering has been implicated in inflammatory conditions such as multiple sclerosis [63]. In this regard, Saggu et al. reported reduced white matter injury in BCAS mice expressing a dominant negative inhibitor of the inflammatory signal mediator nuclear factor kappa B (NF-kB) when its expression was placed under the control of the GFAP promoter [13]. Thus, delayed Cx43-mediated enhancement of local inflammation might play a novel role in the myelin damage observed after BCAS. It is important to note that MFA and CBX inhibit gap junctions more than Cx43 [24, 25]. To overcome this technical limitation, we also tested the protective effects of Cx43 mimetic peptides Gap19 and Gap26, as well as the effect of genetic deletion of Cx43 specifically in astrocytes. Of note, we first determined the minimal effective doses of MFA and CBX in initial experiments and then showed that prolonged administrations of CBX, MFA, Gap19 and Gap26 were without detectable toxicity.
Although white matter occupies almost half of the volume of the adult human brain [64], few studies have focused on the molecular events leading to white matter atrophy in vascular dementia, the most common type of dementia after Alzheimer's disease - which is itself associated with vascular pathology [3]. The relatively sparse capillary density of the white matter may represent a contributing factor in the selective white matter loss that characterizes the chronic hypoperfusion model [9, 65]. Furthermore, many of the white matter arterioles are secondary or tertiary branches, which are inherently more prone to hypoperfusion following proximal occlusion [66].
The murine BCAS model utilized in this study replicates some of the key features of vascular dementia, including white matter pathology and cognitive decline, whereas other typically co-morbid features, such as the lipohyalinosis and small vessel pathology associated with hypertension and diabetes, are lacking [67]. Nonetheless, the post-mortem brain pathology of vascular dementia patients shares a number of features with the anatomic pathology noted in our post-BCAS mice, which included both reactive micro- and astrogliosis [68], as well as frank demyelination. As such, our data suggest that the Cx43 gap junctions may comprise a therapeutic target in vascular dementia, particularly in those patients with risk factors for chronic carotid artery or carotid-distribution diseases, such as smoking, hyperlipidemia and hypertension. More broadly, the relative protection of the post-ischemic central white matter by gap junction inhibition suggests the interdependency of astrocytes and oligodendrocytes in the maintenance and health of forebrain myelin.
Abbreviations
Bilateral common carotid artery stenosis (BCAS), connexin 43 (Cx43), carbenoxolone (CBX), meclofenamic Acid (MFA), phosphate-buffered saline (PBS), paraformaldehyde (PFA), neurofilament-Medium (NF-M), type Ⅵ sodium channel (Nav1.6), glial fibrillary acidic protein (GFAP), microtubule-associated protein2 (Map2), neuronal nuclei (NeuN), ionized calcium binding adapter molecule 1 (Iba-1), macrosialin (CD68), axial diffusivity (AD), mean diffusivity (MD), fractional anisotropy (FA), and radial diffusivity (RD), myelin associated glycoprotein (MAG), myelin basic protein (MBP), diffusion tensor imaging (DTI), medial part of corpus callosum (CCm), peripheral part of corpus callosum (CCp), anterior commissure (AC), fimbria of the hippocampus (F), internal capsule (IC), and optic tract (OT), caudate-putamen (CPu), luxol fast blue (LFB).
Supplementary Material
Supplementary methods and figures.
Acknowledgements
The study was supported by the National Natural Science Foundation of China (61327902, 91332108, 81571113, 81873749 and 81301000) and the Program for Changjiang Scholars and Innovative Research Team in University (IRT_14R20). We thank Prof. Peter J. Brophy (University of Edinburgh, Centre for Neuroregeneration, EdInburgh, United Kingdom) for kindly providing the antibody against panNeurofascin and technical assistance. We thank Prof. Shumin Duan (Department of Neurobiology, Key Laboratory of Medical Neurobiology of the Ministry of Health of China, Key Laboratory of Neurobiology, Zhejiang University School of Medicine) for kindly providing Rosa-LacZ reporter mice. We thank Dan Xue (University of Rochester) for help with graphical design and Professor Paul Cumming for comments on the manuscript. Drs. Nedergaard and Goldman are supported by the Adelson Medical Research Foundation, Novo Nordisk Foundation, Lundbeck Foundation, and NINDS; Dr. Goldman is additionally supported by CHDI and NIMH, and Dr. Nedergaard by the Leducq Foundation and Horizon 2020 SVDs@target.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Johnston SC, O'Meara ES, Manolio TA, Lefkowitz D, O'Leary DH, Goldstein S. et al. Cognitive impairment and decline are associated with carotid artery disease in patients without clinically evident cerebrovascular disease. Ann Intern Med. 2004;140:237-47
2. Buratti L, Balucani C, Viticchi G, Falsetti L, Altamura C, Avitabile E. et al. Cognitive deterioration in bilateral asymptomatic severe carotid stenosis. Stroke. 2014;45:2072-7
3. Iadecola C. The pathobiology of vascular dementia. Neuron. 2013;80:844-66
4. Mathiesen EB, Waterloo K, Joakimsen O, Bakke SJ, Jacobsen EA, Bonaa KH. Reduced neuropsychological test performance in asymptomatic carotid stenosis: The Tromso Study. Neurology. 2004;62:695-701
5. Andrabi SA, Kang HC, Haince JF, Lee YI, Zhang J, Chi ZK. et al. Iduna protects the brain from glutamate excitotoxicity and stroke by interfering with poly(ADP-ribose) polymer-induced cell death. Nat Med. 2011;17:692-U82
6. Borsello T, Clarke PG, Hirt L, Vercelli A, Repici M, Schorderet DF. et al. A peptide inhibitor of c-Jun N-terminal kinase protects against excitotoxicity and cerebral ischemia. Nat Med. 2003;9:1180-6
7. Tekkok SB, Goldberg MP. Ampa/kainate receptor activation mediates hypoxic oligodendrocyte death and axonal injury in cerebral white matter. J Neurosci. 2001;21:4237-48
8. Petralia RS, Wang YX, Hua F, Yi Z, Zhou A, Ge L. et al. Organization of NMDA receptors at extrasynaptic locations. Neuroscience. 2010;167:68-87
9. Baltan S, Besancon EF, Mbow B, Ye Z, Hamner MA, Ransom BR. White matter vulnerability to ischemic injury increases with age because of enhanced excitotoxicity. J Neurosci. 2008;28:1479-89
10. McDonald JW, Althomsons SP, Hyrc KL, Choi DW, Goldberg MP. Oligodendrocytes from forebrain are highly vulnerable to AMPA/kainate receptor-mediated excitotoxicity. Nat Med. 1998;4:291-7
11. Lin JH, Weigel H, Cotrina ML, Liu S, Bueno E, Hansen AJ. et al. Gap-junction-mediated propagation and amplification of cell injury. Nat Neurosci. 1998;1:494-500
12. Bennett MV, Contreras JE, Bukauskas FF, Saez JC. New roles for astrocytes: gap junction hemichannels have something to communicate. Trends Neurosci. 2003;26:610-7
13. Saggu R, Schumacher T, Gerich F, Rakers C, Tai K, Delekate A. et al. Astroglial NF-kB contributes to white matter damage and cognitive impairment in a mouse model of vascular dementia. Acta Neuropathol Commun. 2016;4:76
14. Chen G, Park CK, Xie RG, Berta T, Nedergaard M, Ji RR. Connexin-43 induces chemokine release from spinal cord astrocytes to maintain late-phase neuropathic pain in mice. Brain. 2014;137:2193-209
15. Orellana JA, Froger N, Ezan P, Jiang JX, Bennett MV, Naus CC. et al. ATP and glutamate released via astroglial connexin 43 hemichannels mediate neuronal death through activation of pannexin 1 hemichannels. J Neurochem. 2011;118:826-40
16. Reimer MM, McQueen J, Searcy L, Scullion G, Zonta B, Desmazieres A. et al. Rapid disruption of axon-glial integrity in response to mild cerebral hypoperfusion. J Neurosci. 2011;31:18185-94
17. Shibata M, Ohtani R, Ihara M, Tomimoto H. White matter lesions and glial activation in a novel mouse model of chronic cerebral hypoperfusion. Stroke. 2004;35:2598-603
18. Miyamoto N, Maki T, Pham LD, Hayakawa K, Seo JH, Mandeville ET. et al. Oxidative stress interferes with white matter renewal after prolonged cerebral hypoperfusion in mice. Stroke. 2013;44:3516-21
19. Srinivasan VJ, Yu E, Radhakrishnan H, Can A, Climov M, Leahy C. et al. Micro-heterogeneity of flow in a mouse model of chronic cerebral hypoperfusion revealed by longitudinal Doppler optical coherence tomography and angiography. J Cereb Blood Flow Metab. 2015;35:1552-60
20. Shibata M, Yamasaki N, Miyakawa T, Kalaria RN, Fujita Y, Ohtani R. et al. Selective impairment of working memory in a mouse model of chronic cerebral hypoperfusion. Stroke. 2007;38:2826-32
21. Garcia AD, Doan NB, Imura T, Bush TG, Sofroniew MV. GFAP-expressing progenitors are the principal source of constitutive neurogenesis in adult mouse forebrain. Nat Neurosci. 2004;7:1233-41
22. Theis M, de Wit C, Schlaeger TM, Eckardt D, Kruger O, Doring B. et al. Endothelium-specific replacement of the connexin43 coding region by a lacZ reporter gene. Genesis. 2001;29:1-13
23. Coltman R, Spain A, Tsenkina Y, Fowler JH, Smith J, Scullion G. et al. Selective white matter pathology induces a specific impairment in spatial working memory. Neurobiol Aging. 2011;32:2324 e7-12
24. Chen Q, Boire A, Jin X, Valiente M, Er EE, Lopez-Soto A. et al. Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature. 2016;533:493-8
25. Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S. et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. 2005;8:752-8
26. Abudara V, Bechberger J, Freitas-Andrade M, De Bock M, Wang N, Bultynck G. et al. The connexin43 mimetic peptide Gap19 inhibits hemichannels without altering gap junctional communication in astrocytes. Front Cell Neurosci. 2014;8:306
27. Iliff JJ, Wang M, Zeppenfeld DM, Venkataraman A, Plog BA, Liao Y. et al. Cerebral arterial pulsation drives paravascular CSF-interstitial fluid exchange in the murine brain. J Neurosci. 2013;33:18190-9
28. Nishimura N, Schaffer CB, Friedman B, Lyden PD, Kleinfeld D. Penetrating arterioles are a bottleneck in the perfusion of neocortex. Proc Natl Acad Sci U S A. 2007;104:365-70
29. Rickman C, Medine CN, Bergmann A, Duncan RR. Functionally and spatially distinct modes of munc18-syntaxin 1 interaction. J Biol Chem. 2007;282:12097-103
30. Wakita H, Tomimoto H, Akiguchi I, Kimura J. Glial activation and white matter changes in the rat brain induced by chronic cerebral hypoperfusion: an immunohistochemical study. Acta Neuropathol. 1994;87:484-92
31. Holland PR, Bastin ME, Jansen MA, Merrifield GD, Coltman RB, Scott F. et al. MRI is a sensitive marker of subtle white matter pathology in hypoperfused mice. Neurobiol Aging. 2011;32:2325 e1-6
32. Chen S, Yuan J, Yao S, Jin Y, Chen G, Tian W. et al. Lipopolysaccharides may aggravate apoptosis through accumulation of autophagosomes in alveolar macrophages of human silicosis. Autophagy. 2015;11:2346-57
33. Kamal GM, Wang X, Bin Y, Wang J, Sun P, Zhang X. et al. Compositional differences among Chinese soy sauce types studied by (13)C NMR spectroscopy coupled with multivariate statistical analysis. Talanta. 2016;158:89-99
34. Kamal GM, Yuan B, Hussain AI, Wang J, Jiang B, Zhang X. et al. (13)C-NMR-Based Metabolomic Profiling of Typical Asian Soy Sauces. Molecules. 2016:21
35. Liu Y, Cheng J, Liu H, Deng Y, Wang J, Xu F. NMRSpec: An integrated software package for processing and analyzing one dimensional nuclear magnetic resonance spectra. Chemometr Intell Lab Syst. 2017;162:142-8
36. Kress BT, Iliff JJ, Xia M, Wang M, Wei HS, Zeppenfeld D. et al. Impairment of paravascular clearance pathways in the aging brain. Ann Neurol. 2014;76:845-61
37. Schnaar RL, Lopez PH. Myelin-associated glycoprotein and its axonal receptors. J Neurosci Res. 2009;87:3267-76
38. Singh S, Metz I, Amor S, van der Valk P, Stadelmann C, Bruck W. Microglial nodules in early multiple sclerosis white matter are associated with degenerating axons. Acta neuropathologica. 2013;125:595-608
39. Vassall KA, Bamm VV, Harauz G. MyelStones: the executive roles of myelin basic protein in myelin assembly and destabilization in multiple sclerosis. Biochem J. 2015;472:17-32
40. Xie M, Yi C, Luo X, Xu S, Yu Z, Tang Y. et al. Glial gap junctional communication involvement in hippocampal damage after middle cerebral artery occlusion. Ann Neurol. 2011;70:121-32
41. Xu G, Wang W, Kimelberg HK, Zhou M. Electrical coupling of astrocytes in rat hippocampal slices under physiological and simulated ischemic conditions. Glia. 2010;58:481-93
42. Sherman DL, Tait S, Melrose S, Johnson R, Zonta B, Court FA. et al. Neurofascins are required to establish axonal domains for saltatory conduction. Neuron. 2005;48:737-42
43. Thaxton C, Pillai AM, Pribisko AL, Dupree JL, Bhat MA. Nodes of Ranvier act as barriers to restrict invasion of flanking paranodal domains in myelinated axons. Neuron. 2011;69:244-57
44. Cotrina ML, Lin JH, Alves-Rodrigues A, Liu S, Li J, Azmi-Ghadimi H. et al. Connexins regulate calcium signaling by controlling ATP release. Proc Natl Acad Sci U S A. 1998;95:15735-40
45. Ye ZC, Wyeth MS, Baltan-Tekkok S, Ransom BR. Functional hemichannels in astrocytes: a novel mechanism of glutamate release. J Neurosci. 2003;23:3588-96
46. Matute C, Alberdi E, Domercq M, Sanchez-Gomez MV, Perez-Samartin A, Rodriguez-Antiguedad A. et al. Excitotoxic damage to white matter. J Anat. 2007;210:693-702
47. Shi Y, Thrippleton MJ, Makin SD, Marshall I, Geerlings MI, de Craen AJ. et al. Cerebral blood flow in small vessel disease: A systematic review and meta-analysis. J Cereb Blood Flow Metab. 2016;36:1653-67
48. Brown R, Benveniste H, Black SE, Charpak S, Dichgans M, Joutel A. et al. Understanding the role of the perivascular space in cerebral small vessel disease. Cardiovasc Res. 2018
49. George PM, Steinberg GK. Novel Stroke Therapeutics: Unraveling Stroke Pathophysiology and Its Impact on Clinical Treatments. Neuron. 2015;87:297-309
50. Agrawal SK, Fehlings MG. Role of NMDA and non-NMDA ionotropic glutamate receptors in traumatic spinal cord axonal injury. J Neurosci. 1997;17:1055-63
51. Li S, Stys PK. Mechanisms of ionotropic glutamate receptor-mediated excitotoxicity in isolated spinal cord white matter. J Neurosci. 2000;20:1190-8
52. May D, Tress O, Seifert G, Willecke K. Connexin47 protein phosphorylation and stability in oligodendrocytes depend on expression of Connexin43 protein in astrocytes. J Neurosci. 2013;33:7985-96
53. Magnotti LM, Goodenough DA, Paul DL. Functional heterotypic interactions between astrocyte and oligodendrocyte connexins. Glia. 2011;59:26-34
54. Cotrina ML, Nedergaard M. Brain connexins in demyelinating diseases: therapeutic potential of glial targets. Brain Res. 2012;1487:61-8
55. Odermatt B, Wellershaus K, Wallraff A, Seifert G, Degen J, Euwens C. et al. Connexin 47 (Cx47)-deficient mice with enhanced green fluorescent protein reporter gene reveal predominant oligodendrocytic expression of Cx47 and display vacuolized myelin in the CNS. J Neurosci. 2003;23:4549-59
56. Menichella DM, Goodenough DA, Sirkowski E, Scherer SS, Paul DL. Connexins are critical for normal myelination in the CNS. J Neurosci. 2003;23:5963-73
57. Lutz SE, Zhao Y, Gulinello M, Lee SC, Raine CS, Brosnan CF. Deletion of astrocyte connexins 43 and 30 leads to a dysmyelinating phenotype and hippocampal CA1 vacuolation. J Neurosci. 2009;29:7743-52
58. Arumugam H, Liu X, Colombo PJ, Corriveau RA, Belousov AB. NMDA receptors regulate developmental gap junction uncoupling via CREB signaling. Nat Neurosci. 2005;8:1720-6
59. Jiang S, Yuan H, Duan L, Cao R, Gao B, Xiong YF. et al. Glutamate release through connexin 43 by cultured astrocytes in a stimulated hypertonicity model. Brain Res. 2011;1392:8-15
60. Brancaccio M, Edwards MD, Patton AP, Smyllie NJ, Chesham JE, Maywood ES. et al. Cell-autonomous clock of astrocytes drives circadian behavior in mammals. Science. 2019;363:187-92
61. Jiang S, Wang YQ, Xu CF, Li YN, Guo R, Li L. Involvement of connexin43 in the infrasonic noise-induced glutamate release by cultured astrocytes. Neurochem Res. 2014;39:833-42
62. Chen MJ, Kress B, Han X, Moll K, Peng W, Ji RR. et al. Astrocytic CX43 hemichannels and gap junctions play a crucial role in development of chronic neuropathic pain following spinal cord injury. Glia. 2012;60:1660-70
63. Macrez R, Stys PK, Vivien D, Lipton SA, Docagne F. Mechanisms of glutamate toxicity in multiple sclerosis: biomarker and therapeutic opportunities. Lancet Neurol. 2016;15:1089-102
64. Zhang K, Sejnowski TJ. A universal scaling law between gray matter and white matter of cerebral cortex. Proc Natl Acad Sci U S A. 2000;97:5621-6
65. Ballabh P, Braun A, Nedergaard M. Anatomic analysis of blood vessels in germinal matrix, cerebral cortex, and white matter in developing infants. Pediatr Res. 2004;56:117-24
66. Brown WR, Thore CR. Review: cerebral microvascular pathology in ageing and neurodegeneration. Neuropathol Appl Neurobiol. 2011;37:56-74
67. Hainsworth AH, Allan SM, Boltze J, Cunningham C, Farris C, Head E. et al. Translational models for vascular cognitive impairment: a review including larger species. BMC Med. 2017;15:16
68. Snyder HM, Corriveau RA, Craft S, Faber JE, Greenberg SM, Knopman D. et al. Vascular contributions to cognitive impairment and dementia including Alzheimer's disease. Alzheimers Dement. 2015;11:710-7
Author contact
![]() Corresponding authors: Wei Wang, Department of Neurology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430030, China. wwang126.com or Maiken Nedergaard, Center for Translational Neuromedicine, University of Rochester Medical Center, Rochester, NY 14642, USA. nedergaardrochester.edu.
Corresponding authors: Wei Wang, Department of Neurology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430030, China. wwang126.com or Maiken Nedergaard, Center for Translational Neuromedicine, University of Rochester Medical Center, Rochester, NY 14642, USA. nedergaardrochester.edu.