Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results
Discussion
Conclusion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(15):4391-4408. doi:10.7150/thno.32462 This issue Cite
Research Paper
ZnAs@SiO2 nanoparticles as a potential anti-tumor drug for targeting stemness and epithelial-mesenchymal transition in hepatocellular carcinoma via SHP-1/JAK2/STAT3 signaling
Yongquan Huang1,2,3#, Bin Zhou2,3#, Hui Luo3#, Junjie Mao2,3, Yin Huang2, Ke Zhang2,3, Chaoming Mei3, Yan Yan3, Hongjun Jin3, Jinhao Gao4, Zhongzhen Su1,3, Pengfei Pang2,3 ![]() , Dan Li3
, Dan Li3 ![]() , Hong Shan2,3
, Hong Shan2,3 ![]()
1. Department of Ultrasound, The Fifth Affiliated Hospital, Sun Yat-sen University, Zhuhai, Guangdong Province 519000, China
2. Center for Interventional Medicine, The Fifth Affiliated Hospital, Sun Yat-sen University, Zhuhai, Guangdong Province 519000, China
3. Guangdong Provincial Key Laboratory of Biomedical Imaging and Guangdong Provincial Engineering Research Center of Molecular Imaging, The Fifth Affiliated Hospital, Sun Yat-sen University, Zhuhai, Guangdong Province 519000, China
4. State Key Laboratory of Physical Chemistry of Solid Surfaces, The MOE Key Laboratory of Spectrochemical Analysis & Instrumentation, The Key Laboratory for Chemical Biology of Fujian Province, and Department of Chemical Biology, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen, Fujian Province 361005, China
# These authors contributed equally to this article.
Received 2018-12-21; Accepted 2019-5-15; Published 2019-6-9
Abstract

Rationale: Current therapies for hepatocellular carcinoma (HCC) are hampered by treatment failure and recurrence due to the remaining treatment-resistant liver cancer stem cells (CSCs). Stemness and epithelial-mesenchymal transition (EMT) are regarded as two fundamental characteristics of liver CSCs necessary for cancer progression; thus, drugs that simultaneously target both characteristics should prove effective in eliminating HCC and impeding recurrence. In this study, we developed new arsenic trioxide (ATO)-based nanoparticles (NPs), which are expected to be more effective than the current HCC therapy, and explored their potential mechanism.
Methods: A “one-pot” reverse emulsification approach was employed to prepare the ZnAs@SiO2 NPs. HCC cell lines, MHCC97L and Hep3b, were used to analyze the antitumor activity of ZnAs@SiO2 NPs in vitro and in vivo by quantifying cell growth and metastasis as well as to study the effect on stemness and EMT. SHP-1 siRNA was used to validate the role of the SHP-1/JAK2/STAT3 signaling pathway in mediating inhibition of stemness and EMT by ZnAs@SiO2.
Results: Compared with the current ATO treatment, ZnAs@SiO2 NPs promoted apoptosis and significantly inhibited proliferation, migration, and invasion of both MHCC97L and Hep3b cells. In the in vivo assay, ZnAs@SiO2 NPs inhibited tumor growth by 2.2-fold and metastasis by 3.5-fold as compared to ATO. The ZnAs@SiO2 NPs also inhibited tumor spheroid formation in vitro and tumor initiation in vivo and induced significant changes in the expression of stemness markers (CD133, Sox-2, and Oct-4) and EMT markers (E-cadherin, Vimentin, and Slug) both in vitro and in vivo. These effects of ZnAs@SiO2 that correlated with prognosis of HCC were mediated by the SHP-1/JAK2/STAT3 signaling.
Conclusions: ZnAs@SiO2 NPs can effectively suppress tumor initiation, growth, metastasis, and inhibit stemness and EMT through regulation of SHP-1/JAK2/STAT3 signaling pathway in liver cancer cells in vitro and in vivo. Thus, ZnAs@SiO2 NPs have immense potential for HCC treatment in the future.
Keywords: Arsenic trioxide nanoparticles, stemness, epithelial-mesenchymal transition, hepatocellular carcinoma, SHP-1
Introduction
Liver cancer was recently estimated to be the sixth most common cancer and the fourth most common cause of cancer-related deaths worldwide in 2018. Hepatocellular carcinoma (HCC) comprises 75-85% of primary liver cancer [1]. Though conventional HCC therapies, including surgical excision, ablation, chemotherapy, and liver transplantation, have been somewhat effective, cancer relapse and metastasis remain its most serious complications. Although molecular agents targeting the signaling mechanisms involved in the progression and recurrence of HCC have been explored [2], the long-term prognosis of patients with HCC is still poor. One reason for the limited efficacy of these therapeutic strategies is due to the self-renewal and drug resistance ability of a small number of cancer cells, known as cancer stem cells (CSCs). Thus, to improve outcomes in HCC therapy, it is critical to identify new methods that target both differentiated tumor cells and CSCs [3].
Despite initial successful therapy, a small population of CSCs embedded in tumors fuels tumor growth and plays a significant role in cancer relapse and metastasis [4]. CSCs, which undergo the aberrant differentiation of progeny cells, were reported to have characteristic surface markers and express stem cell genes [5]. Subsets of liver CSCs are phenotypically marked by CD133 [6]. Their elevated expression of sex-determining region Y-box 2 (Sox-2) and Octamer-binding transcription factor 4 (Oct-4) is related to stemness [7-9]. The ability of CSCs to form tumor spheroids in non-adhesive cultures in vitro, and to induce tumors on serial transplantation into immunodeficient mice, is also a manifestation of their pluripotency [10]. A further hallmark of CSCs is a high degree of epithelial-mesenchymal transition (EMT), which has been linked with cancer aggression and drug resistance. EMT activation is also closely related to stemness and therefore exacerbates tumor-initiating potential of CSCs [11, 12]. Although these two aspects of CSCs may be connected, recent studies indicate that they may occur in parallel via activation through independent pathways [13]. Thus, antitumor strategies that emphasize the dual inhibition of stemness and EMT in CSCs, as well as in the differentiated cancer cells, may enhance the ability of antitumor therapy to block tumor recurrence [5].
Arsenic trioxide (ATO) has been approved as a first-line treatment in acute promyelocytic leukemia (APL) by the U. S. Food and Drug Administration because of its remarkable ability to induce complete remission [14]. ATO has also been reported to be efficacious against other malignant tumor cell lines, such as mammary cancer, prostate carcinoma, and HCC [15]. Despite the success of ATO in treating APL, it proved unsatisfactory in treating solid tumors in vivo [16]. A multicenter phase II clinical trial also reported that ATO monotherapy did not significantly benefit patients with HCC [17]. Currently, ATO is approved by the China Food and Drug Administration only for the palliative treatment of patients with HCC. ATO inhibited markers related to stemness and EMT; it downregulated the stem cell marker Sox-2 in glioma cells, induced differentiation of CD133+ HCC cells, and inhibited the EMT process in gastric cancer cells [18-20]. However, to our knowledge, so far, no studies have focused on the ability of ATO to inhibit stemness and EMT simultaneously for cancer treatment. Furthermore, ATO has poor bioavailability and apparent adverse reactions, such as liver dysfunction and side effects on skin that restrict its clinical application in treating solid tumors. Thus, exploring novel strategies to reduce off-target effects and improve bioavailability could enhance the treatment of solid tumors.
The emergence of nanomaterials has provided a versatile platform for cancer treatment. Nanotechnology-based drug delivery systems are potent methods for improving drug efficacy and overcoming side effects and other limitations [21]. To determine whether nanoparticle-based delivery could improve the efficacy of ATO, we developed ZnAs@SiO2 nanoparticles (NPs) and tested them for their ability to inhibit cell proliferation, tumor growth, and metastasis in cultured tumor cell lines and in vivo models of tumor xenografts and metastases. We also evaluated the inhibitory effects of ZnAs@SiO2 NPs on stemness and EMT as well as explored the underlying molecular mechanisms.
Methods
Materials
Tetraethylorthosilicate (TEOS 99.9%), Zinc chloride (ZnCl2 90%), and Arsenic trioxide (As2O3 90%) were obtained from Alfa Aesar. Polyoxyethylene nonylphenyl ether (Igepal Co-520, ESI) was purchased from Sigma-Aldrich. Ethanol ammonium hydroxide, cyclohexane, and sodium metasilicate nonahydrate were purchased from Sinopharm Chemical Reagent Co. Ltd (Shanghai, China). 3-(Dimethyl(3-(trimethoxysilyl) propyl)-ammonio) propane-1-sulfonate was purchased from Meryer Chemical Technology Co., Ltd (Shanghai).
Synthesis of ZnAs@SiO2 NPs
Zinc arsenite (ZnAsOx) NPs were synthesized by a reverse microemulsion method [22]. Briefly, 1630 µL of ZnCl2 (0.1 M) was mixed with 35 mL cyclohexane (29 Vol% Igepal Co-520) to form homogeneous microemulsions. Subsequently, 0.1 M aqueous ATO (1630 µL pH ∼8) and 1.6 × 10-2 M disodium silicate were added to the solvent mixture. After 6 h of reaction at room temperature (RT), 30 µL TEOS and 500 µL ammonia were added to the silica-coated system, and ZnAsOx NPs were encapsulated in the SiO2 matrix. The ZnAs@SiO2 NPs were dispersed in phosphate buffered saline (PBS) (pH 7.4) at 37 °C and 20% v/v FBS was added to the mixture with stirring at 37 °C.
Characterization
Transmission electron microscope (TEM) images were captured by using the JEM-2100 microscope accelerating at the voltage of 200 kV. The Tecnai F20 microscope accelerating at the voltage of 300 kV was employed for the energy-dispersive X-ray (EDX) element mapping and energy dispersive X-ray spectroscopy (EDS). The inductively-coupled plasma atomic emission spectrometry (ICP-AES) was applied for determining the concentration of Zn and As. Briefly, ZnAs@SiO2 samples were determined at a specific wavelength. The concentration of the sample was compared with that of the standard sample. ICP-AES calibration was recorded using the full quantitative model (R2 =0.999). Similarly, the inductively coupled plasma mass spectroscopy (ICP-MS) was applied for determining the As concentration of tissues/organs. The Malvern Zetasizernano ZS instrument was used for the dynamic light scattering (DLS) measurements.
Cell culture
Human HCC cell lines Hep3b, HepG2, and Bel7402 were acquired from the American Type Culture Collection (ATCC, Manassas, VA). MHCC97L cell line were obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). All cells were maintained in Dulbecco's Modified Eagle Medium (DMEM, Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS, Invitrogen, Carlsbad, CA) and penicillin (100 IU/mL)/streptomycin (100 µg/mL) in a humidified atmosphere with 5% CO2 and 95% air at 37 °C.
Cell viability assay
The cell viability was detected using the MTS assay (Promega, WI). 5×103 cells per well were seeded in 96-well plates and cultured for 24h. Subsequently, the cells were incubated with indicated concentrations of PBS, ATO, or ZnAs@SiO2 NPs. A spectrophotometer at 490nm wavelength was used to determine the absorbance. The cell viability was presented as a percentage OD value of the treated cells versus that of the control group. The half-maximal inhibitory concentration (IC50) was calculated to quantify the 50% inhibitory effect versus the PBS-treated control. Cell growth curves were depicted according to the OD value at indicated time points.
Colony formation assay
After treatment with PBS, ATO, or ZnAs@SiO2 NPs for 24 h, cells were seeded in 6-well plates at a density of 5×102 cells per well. After culturing for 14 days, cells were washed twice with PBS. Paraformaldehyde was used to fix the cell colonies for 30 minutes. Subsequently, crystal violet was used for staining for another 30 minutes. All assays were performed in triplicates.
Wound healing assay
6×105 cells per well were cultured in 6-well plates until the cells reached 90-100% confluency. Wounds were made using a 200 µL pipette tip and cells were rinsed 3 times with PBS. Cells were then incubated with PBS, 5 µM of ATO or ZnAs@SiO2 NPs for 24 h. Wound healing area was photographed and calculated.
Transwell migration and invasion assays
Transwell migration and invasion assays were carried out using a 24-well cell culture insert and BD Transwell chambers (BD Biosciences). After 24h incubation with PBS, ATO, or ZnAs@SiO2 NPs, 2×104 suspended in 200 µL DMEM without FBS were plated in the top chamber, and 600 µL DMEM with 10% FBS was added to the lower chamber. After culturing for 24 h, cells of the bottom surface were fixed using paraformaldehyde for 30 min and subsequently stained by crystal violet while cells adhered to the upper surface were removed using cotton swabs. Subsequently, the chambers were rinsed with water and the cells of the undersurface of the chamber were counted in five random fields under the microscope. For Transwell invasion assay, a similar protocol was adopted except that the top Transwell chambers were precoated with diluted Matrigel (BD Biocoat, Bedford, MA) and 5×104 cells suspended in 200 µl DMEM without FBS were plated in the top chamber.
Apoptosis assessment
Flow cytometry was used to assess apoptosis of HCC cells after treatment with PBS, ATO, or ZnAs@SiO2 NPs at indicated concentrations for 24 h based on staining with the Annexin V/PI Kit (KeyGEN, Nanjing, China). Cells were digested using 0.25% trypsin without EDTA (Gibco BRL, Grand Island, NY), then washed with PBS twice. After staining with Annexin V and propidium iodide, apoptosis assessment was performed by flow cytometry (Beckman Coulter, CA).
Real-time quantitative polymerase chain reaction (RT-qPCR)
An RNeasy Mini Kit (Qiagen Germany) was used to isolate total RNA from HCC cells, and cDNA Reverse Transcription Kit (Applied Biosystems) was used for reverse transcription. RT-qPCR was carried out on a BIO-RAD Real-time PCR system with SYBR green mix (Applied Biosystems). Primers were designed and synthesized by Sangon Biotech (Shanghai, China). The primers sequences are listed in Table S1.
Western blot analysis
Total lysates of HCC cells were prepared, and proteins were extracted after centrifugation. 30 µg proteins of each sample were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotted with indicated antibodies. The visualization of protein signals was performed using a chemiluminescence reaction system following the manufacturer's recommenddations. Antibody for cleaved PARP, cleaved Caspase-3, CD133, Sox-2, Oct-4, Vimentin, SHP-1, JAK2, STAT3, p-STAT3, and β-Actin were purchased from Cell Signaling Technology (Danvers, MA). Slug, Snail, p-JAK2, and the second antibodies were acquired from Abcam (Cambridge, UK).
Use of IncuCyte ZOOM
MHCC97L and Hep3b cells treated with PBS, ATO, or ZnAs@SiO2 NPs were incubated at 37 °C and monitored in the presence of cleaved Caspase-3 dye for 24 h using the IncuCyte ZOOM (Essen BioScience). The fluorescent pictures were automatically obtained across the indicated periods.
Spheroid formation
2×103 HCC cells treated with PBS, ATO, or ZnAs@SiO2 NPs were cultured for 7 days in low attachment 6-well plates (Corning, NY) with serum-free F12 medium (SFM) containing appropriate amounts of EGF (Sigma-Aldrich; 10 ng/mL), B27 supplement (Invitrogen, Life Technologies, Carlsbad, CA), and insulin (Sigma-Aldrich; 10 mg/mL). Subsequently, a phase contrast microscope was used to record photos of the spheroids, and their number and size were analyzed.
siRNA transfection
MHCC97L or Hep3b cells were transfected with control or targeted siRNAs (#1 or #2) using Lipofectamine 2000 (Invitrogen). Briefly, a mixture of siRNA (100 nM final concentration) and transfection reagents were incubated for 20 min at RT, added to the cells, and incubated for 48 h. Subsequently, cells were harvested and analyzed for RNA and protein expression. The siRNA sequences are listed in Table S2.
Hemolysis rate test
Hemolysis was tested to determine the blood compatibility of ZnAs@SiO2. Fresh anticoagulated blood from human volunteers (2 mL) was diluted with 2.5 mL of saline. The ZnAs@SiO2 was prepared in saline and kept at 37 °C for 30 min. The diluted blood (0.2 mL) was added to ZnAs@SiO2. The mixture was kept at 37 °C for 60 min and then centrifuged at 1500 rpm for 10 min. The supernatant was transferred to a 96-well plate where the absorbance was measured at 545 nm. Positive controls consisted of 0.2 mL diluted blood in 10 mL deionized water while negative controls consisted of 0.2 mL diluted blood in 10 mL of saline. Hemolysis rate was calculated as follows: Hemolysis rate = [(Dt-Dnc) / (Dpc-Dnc)] × 100%, where Dt, Dnc, and Dpc are the absorbance values of the sample, negative, and positive controls, respectively.
Animal study
All animal experiments were carried according to the Institutional Ethical Guidelines for Animal Experiments of the fifth affiliated hospital of Sun Yat-sen University. In vivo tumorigenicity assay was performed by subcutaneous inoculation into the flanks of 4-week old female nonobese diabetic/severe combined immunodeficiency (NOD-SCID) mice.
To investigate the inhibitory effect of ZnAs@SiO2 NPs on HCC cell growth in vivo, xenograft tumor model was created in 6 week-old female BALB/c nude mice following the previously described method [23, 24]. Briefly, 5×106 MHCC97L cells in 100 µL cold PBS were injected subcutaneously into the flanks of nude mice. When the tumors developed to 100 mm3, PBS, ATO, or ZnAs@SiO2 NPs were injected into the mice through the tail vein every 3 days. To monitor tumor growth in real-time, bioluminescence images were taken every 3 days as previously described [25].
In vivo bio-distribution and whole blood circulation analysis
The isolated tissues/organs were excised from mice 24 h post injection, weighed, lysed, and the concentration of As element was analyzed by ICP-MS. For pharmacokinetic analysis, 20 µL blood was sampled at retroorbital locations at the indicated time points post-injection. Subsequently, the blood samples were lysed at room temperature overnight in 500 mL nitrolysis solution (a mixture of concentrated nitric acid and hydrogen peroxide) and the concentration of As element was analyzed by ICP-MS. After comparing to a fully quantitative model, the results were calculated and recorded as the percentage of injected dose per gram (%ID/g) of tissue. The Winnonlin software was used for calculating the half-life and two-compartment was used for fitting.
Immunohistochemistry (IHC) staining
The tumor tissue stemness and EMT expression pattern were determined using IHC staining. Sections of the tumor specimens were subjected to rehydration and antigen retrieval followed by incubation with the first antibodies overnight in a humidified atmosphere at 4°C. After washing with PBS 3 times, horseradish peroxidase-conjugated second antibodies (ZSGB, China) were applied for 30 min at RT. The patterns were then detected by staining with 3, 5-diaminobenzidine (DAB) and hematoxylin.
Statistical analysis
The data from at least three independent experiments were expressed as n ± SE. Differences between two groups were analyzed by Student's t-test while one-way ANOVA was applied for comparison of more than two subgroups. P < 0.05 was adopted as the criterion for statistical significance. All statistical analyses were performed by SPSS 13.0 software.
Results
Synthesis and characterization of ZnAs@SiO2 NPs
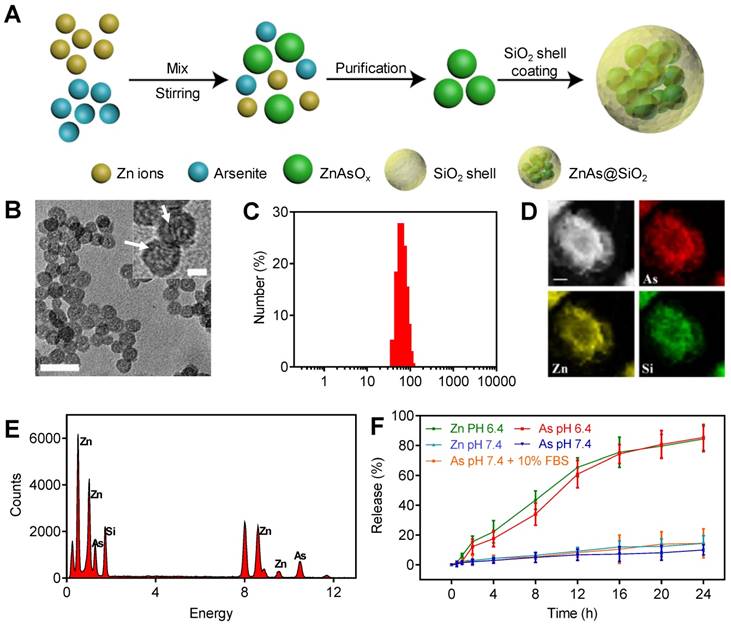
The ZnAs@SiO2 NPs were prepared and functionalized by a reverse microemulsion method, as illustrated in Figure 1A. Briefly, we first synthesized ZnAsOx nanocomplexes by precipitating zinc chloride and aqueous ATO in water nanodroplets. Subsequently, we added TEOS to the microemulsion and coated the hollow silicon shell (HSS) on the ZnAsOx NPs in situ to form ZnAs@SiO2 core-shell nanostructures. TEM images showed that ZnAs@SiO2 NPs were highly uniform, with a diameter of 35 ± 2 nm (Figure 1B). We could observe the ZnAsOx nanocomplexes embedded in the silica shells. DLS analysis indicated that the ZnAsOx@SiO2 NPs were monodisperse in water with a hydrodynamic diameter of 51 ± 3 nm (Figure 1C). The increase in hydrodynamic diameter could be attributed to the hydration layer on the surface of the ZnAs@SiO2 NPs. Scanning TEM and EDX element mapping showed that the Zn and As elements were located in the core of the HSS (Figure 1D). EDS analysis further confirmed the successful fabrication of ZnAs@SiO2 (Figure 1E). To study the ion release efficiency of ZnAs@SiO2 NPs, they were incubated with PBS at pH values of 7.4 (with or without 10% FBS) and 6.4, chosen to represent the physiological pH of the blood and the acidic tumor microenvironment, respectively. Our results showed that the ion release efficiency of the ZnAs@SiO2 NPs at pH 6.4 was much higher than that at pH 7.4 (Figure 1F). Also, ZnAs@SiO2 NPs were stable when 10% FBS was added to PBS mimicking the blood environment. These observations indicated that ZnAs@SiO2 NPs could remain stable in the circulation, whereas the weakly acidic tumor microenvironment facilitated their release suggesting that the ZnAs@SiO2 NPs have enhanced antitumor effects while their toxic side effects may be reduced.
Synthesis and characteristics of ZnAs@SiO2 NPs. (A) Depiction of ZnAs@SiO2 NPs synthesis. (B) TEM image of ZnAs@SiO2 NPs. Scale bar, 50 nm. Insert: the high-magnification TEM image of ZnAs@SiO2 NPs indicating the formation of zinc arsenite nano-complexes (white arrows) in the silica shell. Scale bar, 10 nm. (C) DLS profile of ZnAs@SiO2 NPs. (D) EDX mapping images of the ZnAs@SiO2 NPs. Scale bar, 1 µm. (E) EDS of accumulative ZnAs@SiO2 NPs in the copper mesh. (F) Zn and As ions release from HSS at different pH values of 7.4 (with or without 10% FBS) and 6.4 (n = 3).
ZnAs@SiO2 NPs inhibit proliferation and promote apoptosis of HCC cells in vitro
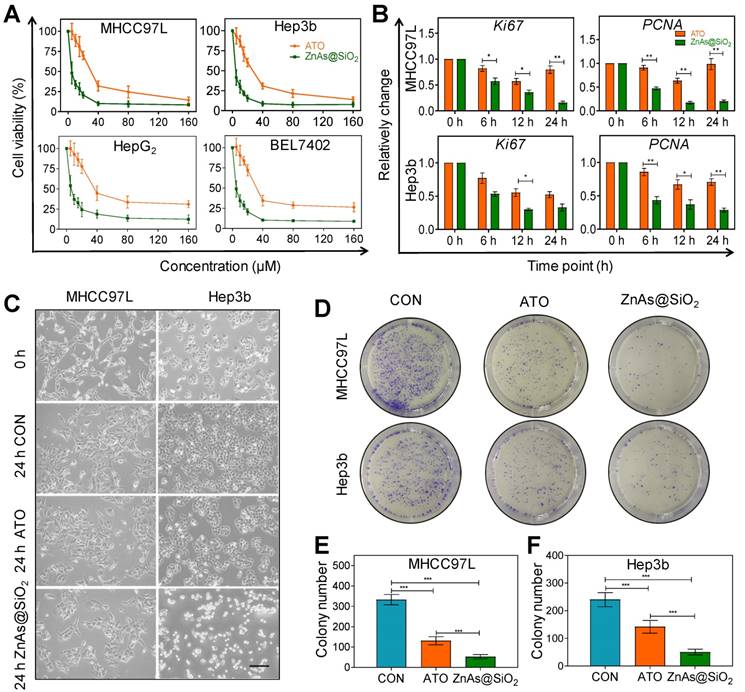
We tested the cytotoxicity of HSS and Zn ions toward HCC cells prior to investigating the effect of ZnAs@SiO2 NPs. Neither element alone affected the proliferation of MHCC97L and Hep3b cells after incubation for 24 h, even at concentrations up to 160 µM (Figure S1). Multiple HCC cell lines were then exposed to ATO or ZnAs@SiO2 NPs for 24 h at various concentrations to compare their antitumor efficacies. The results showed that ZnAs@SiO2 NPs inhibited proliferation to a significantly greater extent than ATO in all cell lines (Figure 2A). The half-maximal inhibitory concentrations (IC50) for both hepatocyte and HCC cell lines were determined. The IC50 values (48 h) showed that ZnAs@SiO2 NPs exhibited more than 2-fold greater inhibitory efficacy than ATO in all four HCC cell lines (Table 1, Figure S2). Also, the cytotoxicity of ZnAs@SiO2 for hepatocyte cell lines MIHA and L02 was lower than that for HCC cell lines (Table 1). Patient-derived primary hepatocytes and primary HCC cells were also exposed to ATO or ZnAs@SiO2 NPs for 24 h at indicated concentrations. ZnAs@SiO2 NPs were more cytotoxic to primary HCC cells than ATO, while both ATO and ZnAs@SiO2 showed no obvious cytotoxicity to primary hepatocytes (Figure S3). These results collectively suggest that ZnAs@SiO2 NPs specifically kill HCC cells sparing normal liver cells. Cell viability assays at various concentrations for up to 72 h of incubation also showed that ZnAs@SiO2 NPs had a higher growth-inhibitory effect on HCC cells than ATO. A trend toward growth inhibition was noted in a time-dependent manner even when cells were incubated with as little as 5 µM of ZnAs@SiO2 NPs, whereas 20 µM of ATO showed a slight growth trend (Figure S4). No cytotoxicity was observed at a concentration of 1 µM. Concentrations of ZnAs@SiO2 NPs greater than 5 µM resulted in a high rate of cell death; therefore 5 µM was adopted as the optimal concentration for further experiments. Changes in mRNA levels of Ki67 and PCNA, indicators of proliferation, also confirmed significantly less proliferation after treatment with 5 µM of ZnAs@SiO2 NPs than with ATO (Figure 2B) (P<0.05). Morphological changes were more obvious after ZnAs@SiO2 NPs treatment with changes in both cell density and the spindle shape of the cells (Figure 2C). Furthermore, the colony formation assay showed that ATO decreased the colony-forming ability of MHCC97L and Hep3b cells by 60.66 ± 10.26% and 40.83 ± 4.08%, respectively, while ZnAs@SiO2 NPs reduced colony numbers by 84.18 ± 7.36% and 79.17 ± 3.75%, respectively (Figure 2D-F).
ZnAs@SiO2 NPs inhibit proliferation of HCC cells in vitro. (A) Cell viability of MHCC97L, Hep3b, HepG2, and Bel7402 cells treated with different concentrations of ATO or ZnAs@SiO2 NPs for 24 h (n = 3). (B) mRNA levels of proliferation indicators Ki67 and PCNA in MHCC97L and Hep3b cells after treatment with ATO or ZnAs@SiO2 NPs for 24 h. (C) Morphology of MHCC97L and Hep3b cells before and after treatment with PBS, ATO or ZnAs@SiO2 NPs for 24 h. Scale bar, 200 µm. (D) Colony forming ability of MHCC97L and Hep3b cells after treatment with PBS, ATO, or ZnAs@SiO2 NPs for 24 h. (E, F) Quantification of colony formation assay in MHCC97L and Hep3b cells. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
IC50 values of hepatocellular cells and HCC cell lines after treatment with ATO or ZnAs@SiO2 NPs for 48 h. Data are expressed as mean ± standard deviation (n=3).
| Cell lines | IC50 ATO (µM) | IC50 ZnAs@SiO2 (µM) | IC50 ATO/IC50 ZnAs@SiO2 |
|---|---|---|---|
| MIHA | 65.15 ± 5.07 | 29.71 ± 4.18 | 2.19 |
| L02 | 68.52 ± 6.32 | 32.43 ± 4.33 | 2.11 |
| MHCC97L | 9.95 ± 1.01 | 3.91 ± 0.85 | 2.54 |
| Hep3b | 10.45 ± 2.16 | 3.58 ± 0.97 | 2.92 |
| HepG2 | 20.13 ± 2.84 | 7.91 ± 1.20 | 2.54 |
| Bel7402 | 10.15 ± 1.72 | 4.34 ± 0.76 | 2.34 |
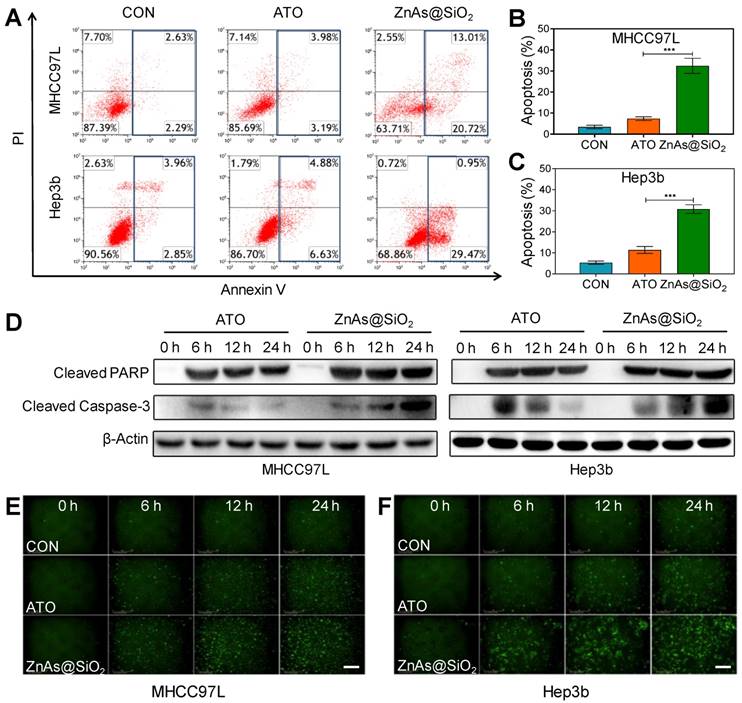
We examined whether ZnAs@SiO2 NPs induced apoptosis in HCC cells. Flow cytometry analysis demonstrated higher levels of apoptosis after treatment with ZnAs@SiO2 than with ATO (Figures 3A-C, S5). Also, compared to ATO, ZnAs@SiO2 treatment significantly elevated the levels of cleaved PARP and cleaved caspase-3 at the indicated time points (Figure 3D). Immunofluorescent staining of cleaved caspase-3 was comparable to the results of Western blots showing a significantly higher level of cleaved caspase-3 in both MHCC97L and Hep3b cells after treatment with ZnAs@SiO2 NPs than with ATO (Figure 3E, F). Collectively, these results suggest that ZnAs@SiO2 NPs are more efficacious than ATO against HCC cells in vitro.
ZnAs@SiO2 NPs promote apoptosis in HCC cells in vitro. (A) Apoptosis analysis of MHCC97L and Hep3b cells after treatment with PBS, ATO or ZnAs@SiO2 NPs for 24 h. (B, C) Quantification of apoptosis rate of MHCC97L and Hep3b cells after treatment with PBS, ATO, or ZnAs@SiO2 NPs for 24 h. (D) Western blots of cleaved PARP and cleaved Caspase-3 after treatment with ATO or ZnAs@SiO2 NPs at indicated time points. (E, F) Cleaved Casepase3 immunofluorescent staining after treatment with PBS, ATO, or ZnAs@SiO2 NPs at indicated time points in MHCC97L and Hep3b cells. Scale bar, 200 µm. ***, P < 0.001.
ZnAs@SiO2 NPs inhibit migration and invasion of HCC cells in vitro
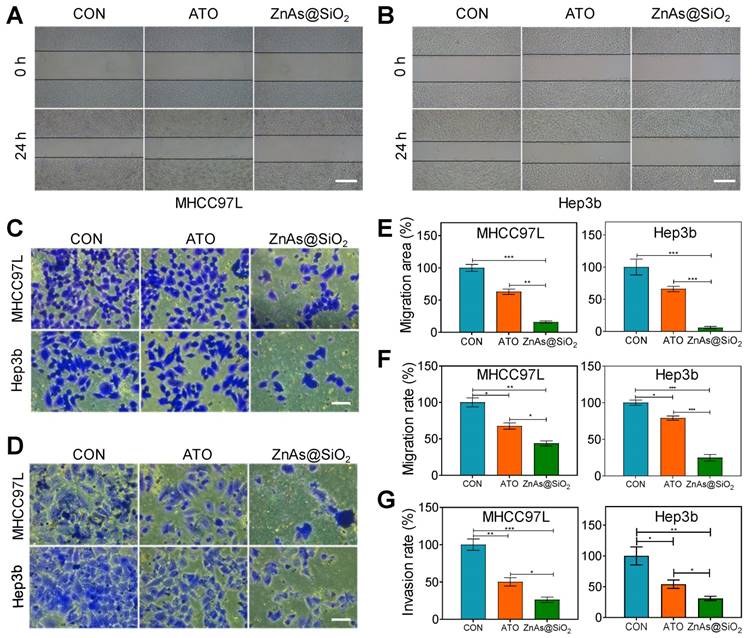
Because the primary cause of treatment failure or recurrence in cancer is metastasis, in which the cells migrate to and invade other tissues, drugs with the ability to inhibit tumor metastasis are highly desirable in treating cancer. Using wound-healing (Figure 4A, B) and Transwell assays (Figure 4C, D), we compared the effects of ATO and ZnAs@SiO2 NPs at equal concentrations of As (5 µM) on the migration and invasion of HCC cells. ATO decreased the relative healing area of MHCC97L and Hep3b cells by 37.06 ± 7.19% and 34.02 ± 7.18%, respectively, whereas ZnAs@SiO2 NPs reduced the relative healing area by 84.04 ± 2.95% and 94.02 ± 3.26%, respectively (Figure 4A, B, E). The Transwell migration assay indicated reduced migration after ATO treatment in both MHCC97L and Hep3b cells by 32.51 ± 7.46% and 21.09 ± 4.92%, respectively whereas ZnAs@SiO2 NPs reduced migration by 56.32 ± 6.06% and 75.08 ± 7.85%, respectively (Figure 4C, F). Furthermore, the Transwell invasion assay revealed that in both cell lines, ATO decreased the invasion ratio (49.79 ± 9.72% and 46.18 ± 11.75%, respectively), although ZnAs@SiO2 NPs reduced it more potently (73.82 ± 6.35% and 69.10 ± 6.06%, respectively; Figure 4D, G). These observations indicate that ZnAs@SiO2 NPs are better able to inhibit the migration- and invasion-like activities, which are markers of metastatic capacity.
ZnAs@SiO2 NPs inhibit the migration and invasion of HCC cells in vitro. (A, B) Wound-healing ability of HCC97L and Hep3b cells after treatment with PBS, ATO, or ZnAs@SiO2 NPs for 24 h. Scale bar, 500 µm. (C) Migration and (D) invasion of HCC97L and Hep3b cells after treatment with PBS, ATO, or ZnAs@SiO2 NPs for 24 h. Scale bar, 50 µm. Quantification of (E) wound-healing, (F) migration, and (G) invasion of HCC97L and Hep3b cells after treatment with PBS, ATO, or ZnAs@SiO2 NPs for 24 h. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
ZnAs@SiO2 NPs inhibit the stemness and EMT of HCC cells in vitro
To determine whether ZnAs@SiO2 NPs affect stemness in HCC cells, a tumor spheroid-formation assay was conducted. The results showed that ZnAs@SiO2 NPs led to a more significant decrease in the ability of HCC cells to initiate tumor spheroid formation in a serial passage in secondary transplantations than ATO (P<0.05) (Figure 5A-D). RT-qPCR and Western blot assays were used to examine the effect of ZnAs@SiO2 NPs on the expression of stemness markers in HCC cells. Results showed that stemness markers, including CD133, Sox-2, and Oct-4, were downregulated at both the mRNA and protein levels significantly more after incubation with ZnAs@SiO2 NPs than with ATO (Figures S6, 5E, 5F). Collectively, these data suggest that ZnAs@SiO2 NPs inhibit stemness in HCC cells.
ZnAs@SiO2 NPs inhibit stemness and EMT in HCC cells in vitro. (A,B) Typical images of tumor spheroids forming ability of MHCC97L and Hep3b cells after treatment with PBS, ATO, or ZnAs@SiO2 NPs for 24h. Scale bar, 50 µm. (C, D) Quantification of tumor spheroids forming ability of MHCC97L and Hep3b cells after treatment with PBS, ATO, or ZnAs@SiO2 NPs for 24 h. (E, F) Protein levels of stemness markers, CD133, Sox-2, and Oct-4, in MHCC97L and Hep3b cells after treatment with ATO or ZnAs@SiO2 NPs at indicated time points. (G, H) Protein levels of EMT markers, E-cadherin, Snail, Slug, and Vimentin in MHCC97L and Hep3b cells after treatment with ATO or ZnAs@SiO2 NPs at indicated time points. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
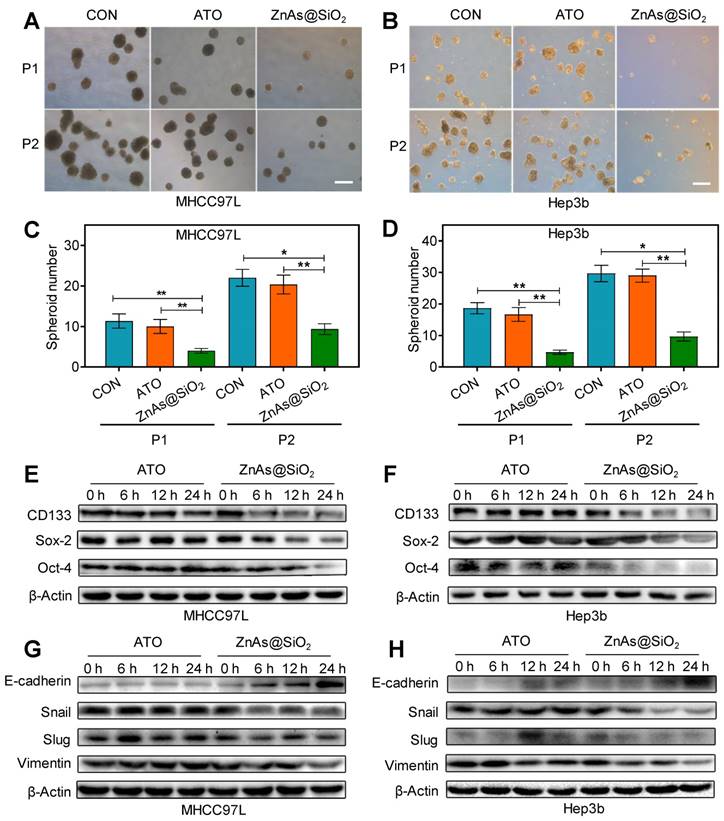
EMT is a highly dynamic process implicated in various biological processes during which cells do not necessarily exist in 'pure' epithelial or mesenchymal states. There are cells with mixed features of the two termed as the intermediate cell states (ICSs), which play an important role in EMT, stemness, and tumor metastasis [26]. The potency of both ATO and ZnAs@SiO2 may be measured by the dynamic processes rather than one pure state. Therefore, we examined both epithelial and mesenchymal markers to monitor the existence of ICSs. Treatment with ZnAs@SiO2 NPs resulted in reduced mesenchymal and increased epithelial marker expression, as demonstrated by the elevated expression of E-cadherin and decreased expression of Vimentin, Slug, and Snail. In contrast, cells incubated with ATO did not increase the E-cadherin level and seemingly decreased the expression of mesenchymal markers less potently than ZnAs@SiO2 NPs at 24 h (Figures S7, 5G, 5H, S8). These findings suggest that ZnAs@SiO2 NPs inhibit EMT more effectively than ATO by modulating the expression of transition-related genes. However, the inhibitory effect of ATO on Snail and Vimentin at the protein level was not as apparent as that at the mRNA level. The protein levels in cells are balanced by two processes: de novo synthesis and protein degradation. Decreased levels of Snail and Vimentin RNAs were inconsistent with their protein expression and may be the result of increased protein stability. Moreover, protein expression may lag behind the mRNA for a short period and lead to the inconsistency between mRNA and protein level at a given time point.
ZnAs@SiO2 NPs inhibit stemness and EMT through regulating SHP-1/JAK2/STAT3 signaling pathway
Recent studies showed that Janus kinase 2 (JAK2) signal transducer and activator of transcription 3 (STAT3) play a pivotal role in stemness [27] and EMT [28]. Dephosphorylation of p-JAK2 and p-STAT3 mediated by SH2-containing protein tyrosine phosphatase 1 (SHP-1) can prevent tumor formation and invasion [29]. Here we explored whether ZnAs@SiO2 NPs inhibit stemness and EMT through regulation of SHP-1/JAK2/STAT3 signaling pathway. Western blot analysis showed that, compared to ATO, treatment with ZnAs@SiO2 upregulated SHP-1 and downregulated p-JAK2 and p-STAT3 levels, while the expression of total JAK or STAT3 showed no apparent variations between the two treatments (Figure 6A). To validate the role of SHP-1/JAK2/STAT3 axis in stemness and EMT, negative control or SHP-1 siRNAs were used. Following transfection, expression of SHP-1 both at mRNA and protein levels showed that siRNA #1 was more efficient than siRNA #2 (Figure. S9). We then transfected both MHCC97L and Hep3b cells with SHP-1 siRNA #1 before treatment with or without 5 µM ZnAs@SiO2 for 24h. The results showed that treatment with si-SHP-1 suppressed SHP-1 and resulted in upregulation of p-JAK and p-STAT3, their inhibition by ZnAs@SiO2 was reversed, and the inhibition of stemness (CD133, Sox-2, and Oct-4) and EMT (E-cadherin, Vimentin, and Snail) was reduced (Figure 6B, C). Furthermore, colony formation assay showed the si-SHP-1 reversal of ZnAs@SiO2 effect which was manifested in reduced colony numbers. This effect was also observed in the sphere formation assay in which si-SHP-1 reversed the ZnAs@SiO2-mediated decrease in both the sphere numbers and size (Figure 6D-F). Transwell migration and invasion assay also revealed the decreased migration and invasion rate of HCC cells by ZnAs@SiO2 were restored by si-SHP-1 (Figure 6G-I). These data suggest that ZnAs@SiO2 NPs inhibit the stemness and EMT of HCC cells through regulation of the SHP-1/JAK2/STAT3 signaling pathway.
ZnAs@SiO2 NPs inhibit stemness and EMT via SHP-1/JAK2/STAT3 signaling pathway. (A) Protein levels of SHP-1, p-JAK2, JAK2, p-STAT3 and STAT3 in MHCC97L and Hep3b cells after treatment with PBS, ATO or ZnAs@SiO2 NPs for 24 h. (B) ZnAs@SiO2 NPs inhibit JAK2/STAT3 mediated by SHP-1. (C) ZnAs@SiO2 NPs inhibit stemness and EMT mediated by SHP-1. (D-I) Colony formation assay (D), Sphere formation assay (E), Transwell migration assay (G) and invasion assay (H) of MHCC97L and Hep3b cells after treatment with ZnAs@SiO2 NPs with or without si-SHP-1 and quantification (F, I), Scale bar, 50 µm. (J) Data from TCGA public database showing the negative correlation between mRNA levels of SHP-1 and tumor TNM staging and Child-Pugh grading. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
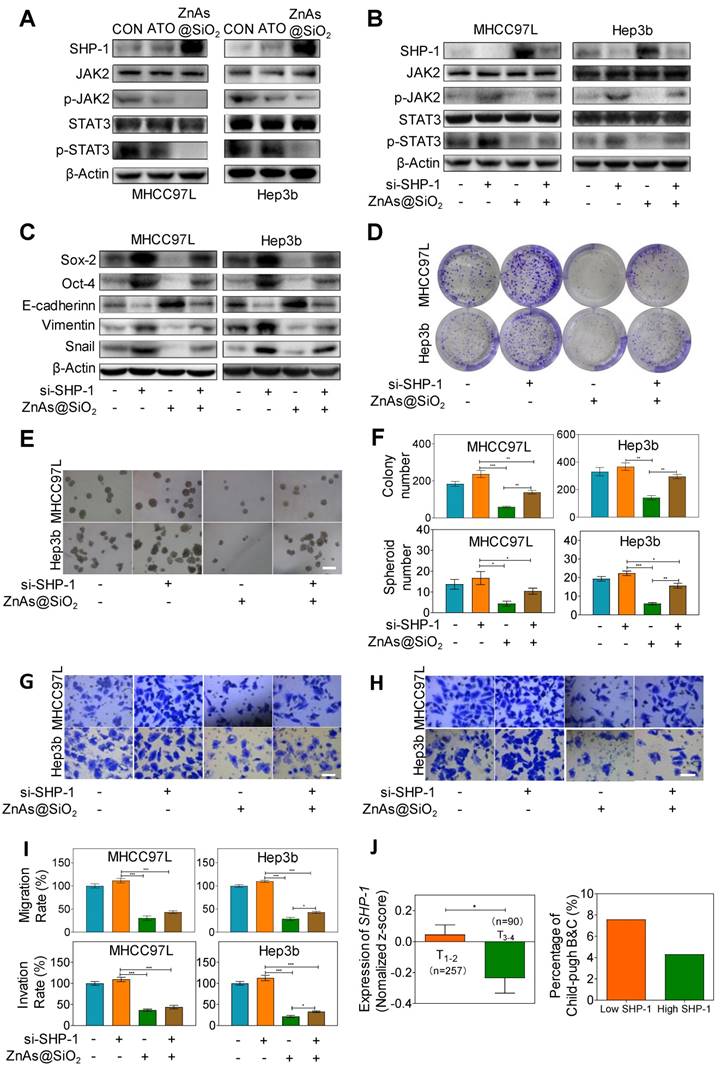
We also assessed the clinical significance of SHP-1. The mRNA reads per kilobase of exon model per million of HCC samples from a TCGA public database were selected and analyzed to compare abundances by GraphPad Prism 8 software. The results demonstrated that the level of SHP-1 in phase T1-2 patients was higher than that of phase T3-4 patients, and there was a higher Child-Pugh grading B&C in high SHP-1 group than low SHP-1 group (Figure. 6J). These observations suggested that SHP-1 might be a tumor suppressor of HCC. Regulation of SHP-1 by ZnAs@SiO2 may, therefore, provide a better strategy for treating liver cancer through inhibiting stemness and EMT.
Pharmacokinetics, bio-distribution and blood compatibility of ZnAs@SiO2 NPs in vivo
To study the pharmacokinetic behavior of ZnAs@SiO2 NPs, blood from tumor-bearing mice was sampled at selected time points post-injection and the concentration of arsenic was measured by ICP-MS. The results showed that the blood circulation half-life of ZnAs@SiO2 NPs was significantly increased (28.81 h), more than 2-fold as that of ATO (14.30 h). ZnAs@SiO2 NPs could be retained in the blood at a concentration of 11.05 %ID/g 6 h after injection (Figure 7A) which was much higher than that of 6.30 %ID/g for ATO. The significantly higher blood arsenic concentration of ZnAs@SiO2 NPs is likely to improve drug retention in the systemic circulation and facilitate time-dependent drug accumulation at tumor sites through the EPR effect. The bio-distribution of ZnAs@SiO2 in MHCC97L-tumor-bearing mice was measured 24 h after the treatment and showed high accumulation of ZnAs@SiO2 NPs of 6.27 ± 3.25% and 3.05 ± 1.47% in liver and spleen, respectively, which may be due to the phagocytosis of the reticuloendothelial system. Tumor uptake of ZnAs@SiO2 was 14.54 ± 5.27%ID/g compared to free ATO which was 5.92 ± 1.47%ID/g (Figure 7B). These results clearly showed that ZnAs@SiO2 could more efficiently accumulate at tumor sites resulting in a higher concentration of ATO-based Nano drugs.
Pharmacokinetics, bio-distribution, and blood compatibility in vivo. (A) Circulation curves of ATO and ZnAs@SiO2 NPs in mice by the measurement of As ions. Concentration of As was determined by ICP-MS. (n = 3) (B) Bio-distribution of As ions in mice at 24 h after intravenous injection of ATO and ZnAs@SiO2 NPs (n = 3). (C) HE-stained images of tissue sections from main organs: liver, lung, heart, kidney, and spleen of the mice after injections of PBS, ATO, or ZnAs@SiO2 NPs for 6 times in 21 days. Scale bar, 50 µm. (D-F) Blood analysis of ALT (D), Cr (E), APTT (F) after injections of PBS, ATO, or ZnAs@SiO2 NPs for 6 times in 21 days. (G) Hemolysis rate of ATO and ZnAs@SiO2 NPs. *, P < 0.05; ***, P < 0.001.
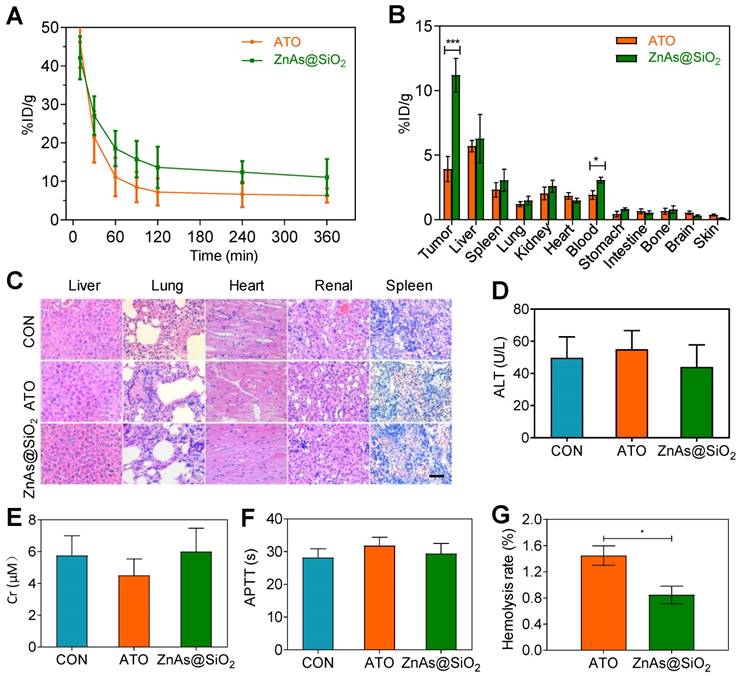
We also assessed systemic toxicity using hematoxylin-eosin (HE) staining (Figure 7C), blood index analyses of both biochemical (Figures 7D, 7E, S10) and coagulation properties (Figures 7F, S11), and M-mode echocardiography for evaluating the left ventricular eject function (LVEF) (Figure S12). The results indicated minimal side effects and acceptable biocompatibility of ZnAs@SiO2 NPs. Hemolysis rate of ATO and ZnAs@SiO2 NPs was also tested and found to be below the threshold value of 5% of the positive control even at the concentration of 0.4 mM (Figure 7G) indicating good biocompatibility of ZnAs@SiO2 NPs.
ZnAs@SiO2 NPs inhibit tumor initiation, growth, stemness, EMT, and lung metastasis of HCC cells in vivo
Given the in vitro effects of ZnAs@SiO2 NPs, we sought to determine whether these results would translate to in vivo conditions using a subcutaneous xenograft mouse model. Implanted MHCC97L cells treated with ZnAs@SiO2 NPs for 24 h failed to initiate tumor formation after 6 weeks, whereas implants of 5×104 and 1×105 MHCC97L cells incubated with ATO generated tumors at rates of 12.5% and 37.5%, respectively (Table 2).
In vivo tumor development experiments of HCC cells after treatment with PBS, WATO or ZnAs@SiO2 NPs for 24 h in NOD-SCID mice.
| Cells | Incidence | Incidence |
|---|---|---|
| Cell number | 100000 | 50000 |
| CON | 6/8 (75%) | 3/8 (37.5%) |
| ATO | 3/8 (37.5%) | 1/8 (12.5%) |
| ZnAs@SiO2 | 0/8 (0%) | 0/8 (0%) |
In vivo evaluation of the therapeutic effect of ZnAs@SiO2 NPs were performed in a nude mouse xenograft model bearing human HCC tumors. When the tumors reached ∼100 mm3, PBS, ATO, or ZnAs@SiO2 NPs were injected intravenously into the mice (2.0 mg As/kg each [22, 30]) every 3 days from day 0 to day 18 and tumor volume was measured at each time point (Figure 8A). Both ATO and ZnAs@SiO2 NPs inhibited tumor growth, whereas tumors in the mice treated with PBS grew rapidly (Figures 8B-D, S13). ZnAs@SiO2 NPs delayed tumor growth significantly and more efficiently than ATO by 2.2-fold. This result could be attributed to the prolonged circulation time of the nanodrug ZnAs@SiO2, its slow release in the neutral environment, and increased release at the tumor site which, together with the EPR effect, resulted in increased drug accumulation at the tumor site. Importantly, the treated mice maintained a consistent weight (Figure S14) over the course of the treatment, indicating that the ZnAs@SiO2 NPs had minimal side effects.
ZnAs@SiO2 NPs inhibit tumor growth and metastasis in vivo. (A) Schematic diagram of the subcutaneous xenograft model experimental setups. (B) Photograph of tumor grafts excised 21 days after injection. (C) Tumor growth curves in mice after treatment with PBS, ATO and ZnAsO@SiO2. (D) Tumor weight of mice after treatment by PBS, ATO, and ZnAsO@SiO2. (E, F) Photographs and histology images of the lung metastasis model after injections of PBS, ATO, or ZnAs@SiO2 NPs for 21 days and quantification. Scale bar, 200 µm. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
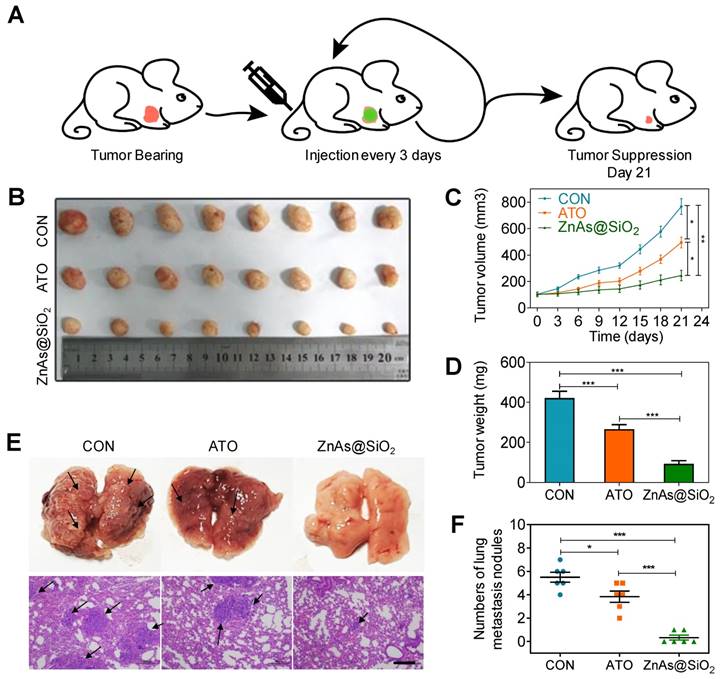
To confirm that ZnAs@SiO2 NPs could inhibit metastasis of HCC cells in vivo, a lung metastasis animal model was established through tail vein injection. HE staining was used to confirm metastasis to the lungs, and the numbers of metastatic tumors was counted. The results showed that ZnAs@SiO2 NPs decreased the formation of metastatic tumors in the lungs more significantly than ATO (Figure 8E, F). These observations indicate that ZnAs@SiO2 NPs have better ability to inhibit metastasis in vivo.
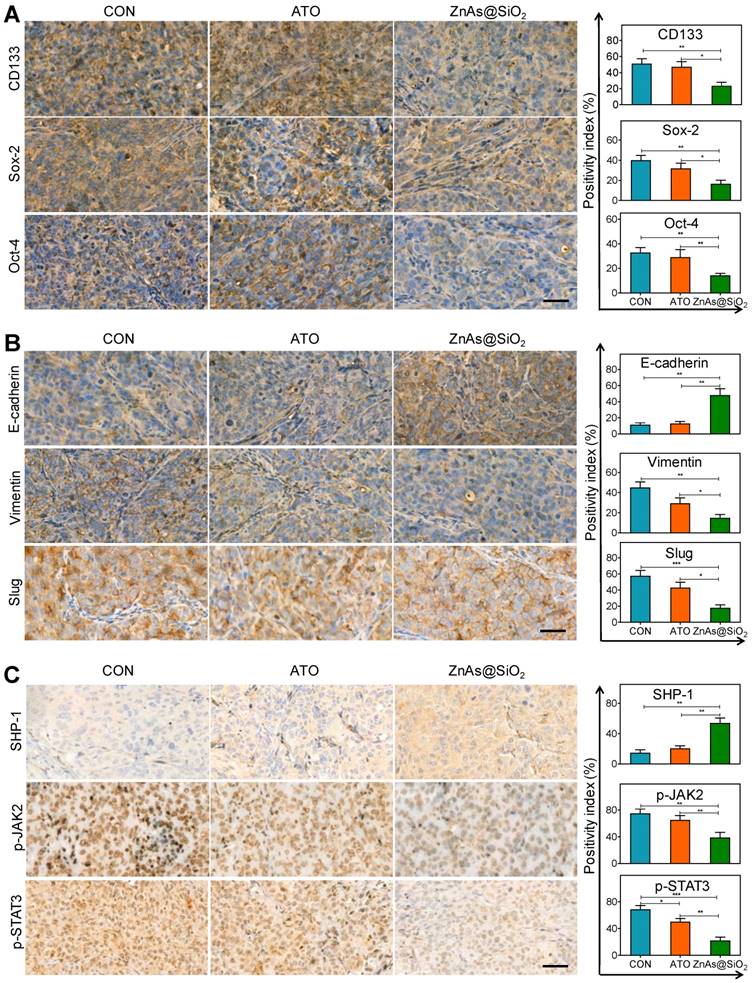
We also analyzed the tumor tissues for the expression of stemness and EMT markers. The results showed that ZnAs@SiO2 NPs significantly reduced the expression of CD133, Sox-2, and Oct-4 (Figure 9A). Also, following ZnAs@SiO2 NPs treatment, Slug and Vimentin levels were significantly decreased, and E-cadherin was significantly increased (Figure 9B). However, ATO treatment failed to elicit such changes. These results suggest that the in vitro effects of ZnAs@SiO2 NPs will translate to the in vivo situation. Given that SHP-1/JAK2/STAT3 signaling pathway mediates the inhibition of stemness and EMT of ZnAs@SiO2 NPs in vitro, we explored the effect of NPs on this pathway in vivo. The IHC results of tumor sections confirmed the superior effect of ZnAs@SiO2 NPs on the upregulation of SHP-1 and inhibition of phosphorylation of JAK/STAT3 compared with ATO (Figure 9C), which was consistent with their effect on stemness and EMT (Figure 9A, B). This cascade effect leads to suppression of tumor initiation, growth, and metastasis.
ZnAs@SiO2 NPs inhibit EMT, stemness and regulate SHP-1/JAK2/STAT3 signaling pathway in vivo. (A) Immunohistochemical staining and quantification of stemness markers, CD133, Sox-2, and Oct-4, in tumor sections after treatment with PBS, ATO, or ZnAs@SiO2 NPs for 21 days. Scale bar, 50 µm. (B) Immunohistochemical staining and quantification of EMT markers, E-cadherin, Slug, and Vimentin, in tumor sections after treatment with PBS, ATO, or ZnAs@SiO2 NPs for 21 days. Scale bar, 50 µm. (C) Immunohistochemical staining and quantification of SHP-1, p-JAK2, and p-STAT3 in tumor sections after treatment with PBS, ATO, or ZnAs@SiO2 NPs for 21 days. Scale bar, 50 µm. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Discussion
CSCs are believed to be the root cause of recurrence and metastasis [31, 32]. Stemness and EMT endow CSCs with the ability to disseminate and initiate tumors at secondary sites. Therefore, strategies inhibiting both stemness and EMT may significantly improve treatment outcomes. ATO was used previously as a radical drug for acute promyelocytic leukemia. The inhibition of stemness or EMT by ATO has been reported [18-20], and although its effects on solid tumors were dissatisfactory, it was still recommended as palliative care for HCC treatment.
Nanoparticle delivery systems can reduce side effects and improve efficacy and biocompatibility compared with conventional chemotherapeutic drugs [33]. In this study, we successfully synthesized an HSS-coated nanodrug with high uniformity to deliver As to HCC cells. Due to its intrinsic properties and other advantages, including biodegradability, inorganic components, attractive stability, excellent biocompatibility, and a high surface-to-volume ratio, HSS has been employed as a carrier and has achieved high therapeutic efficacy without adverse reactions [22, 30]. The biodegradation products of silicon NPs are also extremely biocompatible with various tissues [34]; furthermore, through renal clearance, they were readily cleared from a mouse model and produced little toxicity in vivo. The microemulsion method used in this study involves uniform-size nanoreactors with aqueous core resulting from a specific ratio of the aqueous phase to the surfactant. The confinement of reactants by micelles facilitates the nucleation and growth of inorganic NPs [22]. Within this reverse micelle system, it is also feasible to provide an ideal template in situ silica coating leading to more uniform sizes, better appearance, and higher drug loading of the ZnAs@SiO2 (Figure 1A-C).
Recently, various nanomaterials have been explored to provide new delivery systems to improve the therapeutic effect of cancer treatment. In our study, ZnAs@SiO2 NPs exhibited superior efficacy in inhibiting proliferation (Figures 2, S2-4), inducing apoptosis (Figures 3, S5), and preventing migration and invasion (Figure 4) of HCC cells which may be related to the higher cellular uptake of ZnAs@SiO2 NPs, a function of their nanosize, compared with ATO. NPs exhibit enhanced cancer cell adherence because of their high surface-to-volume ratio and small size.
Stemness and EMT are two fundamental characteristics of CSCs and account for tumor metastasis and recurrence [35, 36]. Previous experimental studies have shown that stemness and EMT are interconnected via underlying gene regulatory networks [37]. However, recent studies suggest that these two traits can be uncoupled [13]. Mesenchymal cancer cells can exhibit a low degree of stemness [38] and cancer cells with high stemness can also exhibit a low grade of EMT [39]. The reduction in the ability of cells to form spheroids (Figure 5A-D) and initiate tumors in vivo (Table 2), together with the decrease in stemness markers (Figure S6, 5E, 5F), demonstrated that ZnAs@SiO2 NPs inhibited the self-renewal of HCC cells more effectively than ATO. The nanodrug also inhibited the expression of EMT-related markers in the HCC cells more effectively than ATO (Figure S7, 5G, 5H, S8). These observations may represent the mechanism underlying the greater efficacy of ZnAs@SiO2 NPs, especially about inhibiting metastatic behavior in vitro (Figure 4). These results indicate that ZnAs@SiO2 NPs may very effectively kill CSCs in HCC tumors by simultaneously modulating stemness and EMT.
JAK2/STAT3 signaling axis was reported to be closely associated with maintaining stemness of cancer cells [40] and regulation of EMT [41]. SHP-1 inhibits this signaling pathway through dephosphorylation of JAK2/STAT3 [18]. In our study, we explored whether ZnAs@SiO2 NPs inhibit stemness and EMT by regulating SHP-1/JAK2/STAT3 signaling pathway. The results showed that ZnAs@SiO2 promoted SHP-1 and inactivated JAK2/STAT3 more potently than ATO (Figures 6A, 9C). Knockdown of SHP-1 reversed the anti-stemness and EMT effects of ZnAs@SiO2 (Figure 6B-I). These results suggested that the inhibitory effect of ZnAs@SiO2 on stemness and EMT was mediated by SHP-1/JAK2/STAT3 signaling pathway. The simultaneous modulation of stemness and EMT through SHP-1/JAK2/STAT3 signaling pathway makes ZnAs@SiO2 a potent drug for inhibiting malignant properties of HCC cells. Furthermore, from the analysis of the TCGA database, we found that high levels of SHP-1 transcripts correlated with better TNM staging and Child-Pugh grading in patients with HCC (Figure 6J). SHP-1 has also been demonstrated as a tumor suppressor of hepatocarcinogenesis and HCC progression and was proposed as a novel prognostic biomarker and therapeutic target for HCC [29]. Therefore, ZnAs@SiO2 may improve the prognosis of HCC by enhancing the expression of SHP-1.
Notably, ZnAs@SiO2 led to the augmented therapeutic efficacy of solid tumors (Figure 8B-D), suppressed metastasis nodules (Figure 8E, F) as well as inhibited stemness and EMT (Figures 5, 9A, 9B). The acidic microenvironment is a feature of most solid tumors. ZnAs@SiO2 NPs were better able to release As at the lower pH value of 6.4 (Figure 1F), which mimics the tumor extracellular pH and lysosomal environment. This pH-triggered release behavior of ZnAs@SiO2 was due to the degradation of the core shell of the ZnAsOx complex resulting in accelerated drug release. EPR effect would work concomitantly with this pH-dependent release resulting in considerable enrichment and delivery of ZnAs@SiO2 at the tumor site (Figure 7B). This suggests that the therapeutic efficacy of ZnAs@SiO2 NPs would be augmented once it is within the acidic tumor environment. Another reason for the enhanced efficacy of ZnAs@SiO2 NPs was their prolonged circulation time and higher blood concentration (Figure 7A) resulting from its slow release at the neutral pH, which may also be responsible for its minimal side effects in vivo (Figures 7C-G, S10-12) [42].
Previous reports suggested that CSCs differentiate only into non-CSCs (NCSCs). However, a recent study showed that NCSCs in the tumor tissue could de-differentiate into new CSCs, thereby regenerating the tumorigenic potential [43, 44]. CSCs and NCSCs appear to be able to interconvert and maintain a dynamic equilibrium. To date, various CSCs have been selectively targeted with NPs for cancer therapy. However, selectively eliminating CSCs is insufficient in preventing relapse because of the existence of residual NCSCs. Therefore, an effective therapeutic strategy requires not only depleting CSCs but also eradicating NCSCs [31]. Targeting CSCs by inhibiting stemness and EMT, as well as NCSCs by ZnAs@SiO2 NPs, may deplete both cell types in HCC effectively, and thus be a significantly more effective treatment for solid tumors and metastasis.
Our study has identified areas that should be pursued in the future to further improve the efficacy of ZnAs@SiO2 NPs. Since NPs can actively target tumor cells, surface modifications of HSS by chemical manipulations may further improve drug delivery and cellular uptake. Despite current advances, drug resistance remains problematic [45] requiring the use of combination therapies with synergistic therapeutic efficacy to suppress resistance [46]. However, uneven bio-distribution can impair this synergistic interaction. Nanotechnology enables co-delivery of two synergistic drugs by encapsulating them in the same vehicle and resulting in better pharmacokinetics [47]. ZnAs@SiO2 NPs can be further developed to co-deliver As with another HCC drug, such as sorafenib, to improve its therapeutic efficacy.
Conclusion
In this study, we synthesized ZnAs@SiO2 NPs using a “one-pot” reverse microemulsion method for As delivery. ZnAs@SiO2 NPs significantly inhibited tumor growth and metastasis of HCC cells. More importantly, by activating the SHP-1/JAK2/STAT3 signaling pathway, ZnAs@SiO2 NPs inhibited stemness and EMT more efficiently than ATO. Thus, this new drug delivery system may hold tremendous potential to improve HCC treatment.
Abbreviations
ATO: arsenic trioxide; CSCs: cancer stem cells; DAB: diaminobenzidine; DLS: dynamic light scattering; EMT: epithelial-mesenchymal transition; EDS: energy dispersive X-ray spectroscopy; EDX: energy-dispersive X-ray; HCC: hepatocellular carcinoma; HE: hematoxylin-eosin; ICP-MS: inductively coupled plasma mass spectroscopy; ICSs: intermediate cell states; IC50: half-maximal inhibitory concentration; IHC: Immunohistochemistry; JAK2: Janus kinase 2; NCSCs: non-cancer stem cells; NOD-SCID: nonobese diabetic/severe combined immunodeficiency; NPs: nanoparticles; Oct-4: octamer-binding transcription factor 4; RT: room temperature; SHP-1: SH2-containing protein tyrosine phosphatase 1; Sox-2: sex-determining region Y-box 2; STAT3: signal transducer and activator of transcription 3; TEM: transmission electron microscope.
Supplementary Material
Supplementary methods, figures and table.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81430041, 81620108017, 81771879, 81871462), Science and Technology Planning Project of Guangzhou (201604020098, 201610010006), and Natural Science Foundation of Guangdong Province (2018A0303130070).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global Cancer Statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;6:394-424
2. Hernandez-Gea V, Toffanin S, Friedman SL, Llovet JM. Role of the microenvironment in the pathogenesis and treatment of hepatocellular carcinoma. Gastroenterology. 2013;144:512-27
3. Dragu DL, Necula LG, Bleotu C, Diaconu CC, Chivu-Economescu M. Therapies targeting cancer stem cells: Current trends and future challenges. World J Stem Cells. 2015;7:1185-201
4. Batlle E, Clevers H. Cancer stem cells revisited. Nat Med. 2017;23:1124-34
5. Garg M. Epithelial plasticity and cancer stem cells: Major mechanisms of cancer pathogenesis and therapy resistance. World J Stem Cells. 2017;9:118-26
6. Chai S, Ng KY, Tong M, Lau EY, Lee TK, Chan KW. et al. Octamer 4/microRNA-1246 signaling axis drives Wnt/beta-catenin activation in liver cancer stem cells. Hepatology. 2016;64:2062-76
7. Di J, Duiveman-de Boer T, Zusterzeel PL, Figdor CG, Massuger LF, Torensma R. The stem cell markers Oct4A, Nanog and c-Myc are expressed in ascites cells and tumor tissue of ovarian cancer patients. Cell Oncol (Dordr). 2013;36:363-74
8. Sainz B Jr, Alcala S, Garcia E, Sanchez-Ripoll Y, Azevedo MM, Cioffi M. et al. Microenvironmental hCAP-18/LL-37 promotes pancreatic ductal adenocarcinoma by activating its cancer stem cell compartment. Gut. 2015;64:1921-35
9. Hermann PC, Sancho P, Canamero M, Martinelli P, Madriles F, Michl P. et al. Nicotine promotes initiation and progression of KRAS-induced pancreatic cancer via Gata6-dependent dedifferentiation of acinar cells in mice. Gastroenterology. 2014;147:1119-33 e4
10. O'Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445:106-10
11. Puisieux A, Brabletz T, Caramel J. Oncogenic roles of EMT-inducing transcription factors. Nat Cell Biol. 2014;16:488-94
12. Scheel C, Weinberg RA. Cancer stem cells and epithelial-mesenchymal transition: concepts and molecular links. Semin Cancer Biol. 2012;22:396-403
13. Nieto MA, Huang RY, Jackson RA, Thiery JP. Emt: 2016. Cell. 2016;166:21-45
14. Wang ZY, Chen Z. Acute promyelocytic leukemia: from highly fatal to highly curable. Blood. 2008;111:2505-15
15. Miller WH Jr, Schipper HM, Lee JS, Singer J, Waxman S. Mechanisms of action of arsenic trioxide. Cancer Res. 2002;62:3893-903
16. Subbarayan PR, Ardalan B. In the war against solid tumors arsenic trioxide needs partners. J Gastrointest Cancer. 2014;45:363-71
17. Lin CC, Hsu C, Hsu CH, Hsu WL, Cheng AL, Yang CH. Arsenic trioxide in patients with hepatocellular carcinoma: a phase II trial. Invest New Drugs. 2007;25:77-84
18. Kim SH, Yoo HS, Joo MK, Kim T, Park JJ, Lee BJ. et al. Arsenic trioxide attenuates STAT-3 activity and epithelial-mesenchymal transition through induction of SHP-1 in gastric cancer cells. BMC Cancer. 2018;18:150
19. Sun H, Zhang S. Arsenic trioxide regulates the apoptosis of glioma cell and glioma stem cell via down-regulation of stem cell marker Sox2. Biochem Biophys Res Commun. 2011;410:692-7
20. Zhang KZ, Zhang QB, Zhang QB, Sun HC, Ao JY, Chai ZT. et al. Arsenic trioxide induces differentiation of CD133+ hepatocellular carcinoma cells and prolongs posthepatectomy survival by targeting GLI1 expression in a mouse model. J Hematol Oncol. 2014;7:28
21. Zhang RX, Cai P, Zhang T, Chen K, Li J, Cheng J. et al. Polymer-lipid hybrid nanoparticles synchronize pharmacokinetics of co-encapsulated doxorubicin-mitomycin C and enable their spatiotemporal co-delivery and local bioavailability in breast tumor. Nanomedicine. 2016;12:1279-90
22. Zhang Z, Liu H, Zhou H, Zhu X, Zhao Z, Chi X. et al. A facile route to core-shell nanoparticulate formation of arsenic trioxide for effective solid tumor treatment. Nanoscale. 2016;8:4373-80
23. Li D, Liu S, Liu R, Zhou Y, Park R, Naga K. et al. EphB4-targeted imaging with antibody h131, h131-F(ab')2 and h131-Fab. Mol Pharm. 2013;10:4527-33
24. Li D, Liu S, Liu R, Park R, Hughes L, Krasnoperov V. et al. Targeting the EphB4 receptor for cancer diagnosis and therapy monitoring. Mol Pharm. 2013;10:329-36
25. Wu C, Li J, Pang P, Liu J, Zhu K, Li D. et al. Polymeric vector-mediated gene transfection of MSCs for dual bioluminescent and MRI tracking in vivo. Biomaterials. 2014;35:8249-60
26. Sha Y, Haensel D, Gutierrez G, Du H, Dai X, Nie Q. Intermediate cell states in epithelial-to-mesenchymal transition. Phys Biol. 2019;16:021001
27. Abubaker K, Luwor RB, Zhu H, McNally O, Quinn MA, Burns CJ. et al. Inhibition of the JAK2/STAT3 pathway in ovarian cancer results in the loss of cancer stem cell-like characteristics and a reduced tumor burden. BMC Cancer. 2014;14:317
28. Colomiere M, Ward AC, Riley C, Trenerry MK, Cameron-Smith D, Findlay J. et al. Cross talk of signals between EGFR and IL-6R through JAK2/STAT3 mediate epithelial-mesenchymal transition in ovarian carcinomas. Br J Cancer. 2009;100:134-44
29. Wen LZ, Ding K, Wang ZR, Ding CH, Lei SJ, Liu JP. et al. SHP-1 Acts as a Tumor suppressor in hepatocarcinogenesis and HCC progression. Cancer Res. 2018;78:4680-91
30. Zhao Z, Wang X, Zhang Z, Zhang H, Liu H, Zhu X. et al. Real-time monitoring of arsenic trioxide release and delivery by activatable T(1) imaging. ACS Nano. 2015;9:2749-59
31. Zhou G, Latchoumanin O, Bagdesar M, Hebbard L, Duan W, Liddle C. et al. Aptamer-based therapeutic approaches to target cancer stem cells. Theranostics. 2017;7:3948-61
32. Lin X, Chen W, Wei F, Zhou BP, Hung MC, Xie X. Nanoparticle delivery of miR-34a eradicates long-term-cultured breast cancer stem cells via targeting C22ORF28 directly. Theranostics. 2017;7:4805-24
33. Cho K, Wang X, Nie S, Chen ZG, Shin DM. Therapeutic nanoparticles for drug delivery in cancer. Clin Cancer Res. 2008;14:1310-6
34. Peng F, Su Y, Zhong Y, Fan C, Lee ST, He Y. Silicon nanomaterials platform for bioimaging, biosensing, and cancer therapy. Acc Chem Res. 2014;47:612-23
35. Yeh HW, Hsu EC, Lee SS, Lang YD, Lin YC, Chang CY. et al. PSPC1 mediates TGF-beta1 autocrine signalling and Smad2/3 target switching to promote EMT, stemness and metastasis. Nat Cell Biol. 2018;20:479-91
36. Lupia M, Angiolini F, Bertalot G, Freddi S, Sachsenmeier KF, Chisci E. et al. CD73 Regulates stemness and epithelial-mesenchymal transition in ovarian cancer-initiating cells. Stem Cell Reports. 2018;10:1412-25
37. May CD, Sphyris N, Evans KW, Werden SJ, Guo W, Mani SA. Epithelial-mesenchymal transition and cancer stem cells: a dangerously dynamic duo in breast cancer progression. Breast Cancer Res. 2011;13:202
38. Drasin DJ, Guarnieri AL, Neelakantan D, Kim J, Cabrera JH, Wang CA. et al. TWIST1-Induced miR-424 reversibly drives mesenchymal programming while inhibiting tumor initiation. Cancer Res. 2015;75:1908-21
39. Chang CC, Hsu WH, Wang CC, Chou CH, Kuo MY, Lin BR. et al. Connective tissue growth factor activates pluripotency genes and mesenchymal-epithelial transition in head and neck cancer cells. Cancer Res. 2013;73:4147-57
40. Zhu L, Cheng X, Shi J, Jiacheng L, Chen G, Jin H. et al. Crosstalk between bone marrow-derived myofibroblasts and gastric cancer cells regulates cancer stemness and promotes tumorigenesis. Oncogene. 2016;35:5388-99
41. Chang JC. Cancer stem cells: Role in tumor growth, recurrence, metastasis, and treatment resistance. Medicine (Baltimore). 2016;95:S20-5
42. Tong R, Chiang HH, Kohane DS. Photoswitchable nanoparticles for in vivo cancer chemotherapy. Proc Natl Acad Sci U S A. 2013;110:19048-53
43. Shibue T, Weinberg RA. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol. 2017;14:611-29
44. Marjanovic ND, Weinberg RA, Chaffer CL. Cell plasticity and heterogeneity in cancer. Clin Chem. 2013;59:168-79
45. Konieczkowski DJ, Johannessen CM, Garraway LA. A convergence-based framework for cancer drug resistance. Cancer Cell. 2018;33:801-15
46. Saputra EC, Huang L, Chen Y, Tucker-Kellogg L. Combination therapy and the evolution of resistance: the theoretical merits of synergism and antagonism in cancer. Cancer Res. 2018;78:2419-31
47. Liu Y, Fang J, Kim YJ, Wong MK, Wang P. Codelivery of doxorubicin and paclitaxel by cross-linked multilamellar liposome enables synergistic antitumor activity. Mol Pharm. 2014;11:1651-61
Author contact
![]() Corresponding authors: shanhongsysu.edu.cn, lidan25sysu.edu.cn, pangpfsysu.edu.cn
Corresponding authors: shanhongsysu.edu.cn, lidan25sysu.edu.cn, pangpfsysu.edu.cn