Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Conclusion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(15):4308-4323. doi:10.7150/thno.32710 This issue Cite
Research Paper
Methylation of RCAN1.4 mediated by DNMT1 and DNMT3b enhances hepatic stellate cell activation and liver fibrogenesis through Calcineurin/NFAT3 signaling
Xue-yin Pan1,2,3, Hong-mei You1,2,3, Ling Wang1,2,3, Yi-hui Bi1,2,3, Yang Yang1,2,3, Hong-wu Meng1,2,3, Xiao-ming Meng1,2,3, Tao-tao Ma1,2,3, Cheng Huang1,2,3 ![]() , Jun Li1,2,3
, Jun Li1,2,3 ![]()
1. The Key Laboratory of Major Autoimmune Diseases, Anhui Province, Anhui Institute of Innovative Drugs, School of Pharmacy, Anhui Medical University
2. The key laboratory of Anti-inflammatory of Immune medicines, Ministry of Education
3. Institute for Liver Diseases of Anhui Medical University
Received 2019-1-1; Accepted 2019-5-20; Published 2019-6-9
Abstract

Background: Liver fibrosis is characterized by extensive deposition of extracellular matrix (ECM) components in the liver. RCAN1 (regulator of calcineurin 1), an endogenous inhibitor of calcineurin (CaN), is required for ECM synthesis during hypertrophy of various organs. However, the functional role of RCAN1 in liver fibrogenesis has not yet been addressed.
Methods: We induced experimental liver fibrosis in mice by intraperitoneal injection of 10 % CCl4 twice a week. To investigate the functional role of RCAN1.4 in the progression of liver fibrosis, we specifically over-expressed RCAN1.4 in mice liver using rAAV8-packaged RCAN1.4 over-expression plasmid. Following the establishment of the fibrotic mouse model, primary hepatic stellate cells were isolated. Subsequently, we evaluated the effect of RCAN1.4 on hepatic fibrogenesis, hepatic stellate cell activation, and cell survival. The biological role and signaling events for RCAN1 were analyzed by protein-protein interaction (PPI) network. Bisulfite sequencing PCR (BSP) was used to predict the methylated CpG islands in the RCAN1.4 gene promoter. We used the chromatin immunoprecipitation (ChIP assay) to investigate DNA methyltransferases which induced decreased expression of RCAN1.4 in liver fibrosis.
Results: Two isoforms of RCAN1 protein were expressed in CCl4-induced liver fibrosis mouse model and HSC-T6 cells cultured with transforming growth factor-beta 1 (TGF-β1). RCAN1 isoform 4 (RCAN1.4) was selectively down-regulated in vivo and in vitro. The BSP analysis indicated the presence of two methylated sites in RCAN1.4 promoter and the downregulated RCAN1.4 expression levels could be restored by 5-aza-2'-deoxycytidine (5-azadC) and DNMTs-RNAi transfection in vitro. ChIP assay was used to demonstrate that the decreased RCAN1.4 expression was associated with DNMT1 and DNMT3b. Furthermore, we established a CCl4-induced liver fibrosis mouse model by injecting the recombinant adeno-associated virus-packaged RCAN1.4 (rAAV8-RCAN1.4) over-expression plasmid through the tail vein. Liver- specific-over-expression of RAN1.4 led to liver function recovery and alleviated ECM deposition. The key protein (a member of the NFAT family of proteins) identified on PPI network data was analyzed in vivo and in vitro. Our results demonstrated that RCAN1.4 over-expression alleviates, whereas its knockdown exacerbates, TGF-β1-induced liver fibrosis in vitro in a CaN/NFAT3 signaling-dependent manner.
Conclusions: RCAN1.4 could alleviate liver fibrosis through inhibition of CaN/NFAT3 signaling, and the anti-fibrosis function of RCAN1.4 could be blocked by DNA methylation mediated by DNMT1 and DNMT3b. Thus, RCAN1.4 may serve as a potential therapeutic target in the treatment of liver fibrosis.
Keywords: DNA methylation, RCAN1.4, Proliferation, Apoptosis, Calcineurin/NFAT signaling
Introduction
Liver fibrosis, characterized by extensive deposition of ECM compounds and secretion of chemokines and fibrogenic factors, is a typical scarring response to virtually all forms of chronic liver injury [1]. Although the early stages of liver fibrosis are reversible, persistent liver injury and scar formation result in end-stage cirrhosis, even hepatocellular carcinoma (HCC), ultimately requiring transplant [2]. Because the underlying cause of hepatic injury cannot be identified and eliminated, treatment of hepatic fibrosis is limited to addressing complications such as portal hypertension, progression to hepatocellular carcinoma, and liver failure [3]. Substantial improvements in the treatment of chronic liver disease have aroused interest in uncovering the mechanisms underlying hepatic fibrosis and its resolution.
Activation of resident hepatic stellate cells (HSCs) into proliferative, contractile, and fibrogenic cells in liver injury remains a dominant theme driving the field [1]. Upon liver injury, quiescent HSCs (qHSCs) are activated into proliferative, migratory myofibroblasts. It has been demonstrated that TGF-β1, an inflammatory cytokine secreted by many types of hepatic cells, is the main fibrogenic cytokine for HSC activation [4]. Activated HSCs (aHSCs) are characterized by the excessive expression of alpha-smooth muscle actin (α-SMA) and type I collagen (COL1a1), which are the main components of ECM [5]. Therefore, targeting of HSCs by inhibiting their activation and inducing apoptosis in aHSCs are proposed therapeutic approaches to reverse liver fibrosis [6].
RCAN1 gene, previously referred to as ADAPT78/DSCR1/MCIP1, is located on the q22.12 region of chromosome 21 and consists of seven exons, three of which (exons 5, 6 and 7) seem to be invariant in all RCAN1 isoforms. The first four exons are alternatively spliced/translated to produce many different mRNA isoforms [7]. So far, at least three RCAN1 mRNA and protein isoforms have been described in various organisms: RCAN1.1L (RCAN1 isoform 1 Long), RCAN1.1S (RCAN1 isoform 1 Short), and RCAN1.4 (RCAN1 isoform 4) [8]. In this study, we found that two isoforms of RCAN1 protein were expressed in a fibrotic mouse model and HSC-T6 cells cultured with TGF-β1 (10 ng/mL), and RCAN1.4 was selectively decreased during liver fibrosis progression. We, therefore, set out to uncover the functional role of RCAN1.4 in HSC activation and liver fibrosis.
Calcineurin (CaN), a calcium/calmodulin-activated serine/threonine phosphatase (PP2B), is involved in multiple physiological and pathological processes, such as regulating apoptosis [9], memory [10], and skeletal and cardiac muscle growth and differentiation [11]. It has been demonstrated that CaN can be activated by TGF-β in a time- and dose-dependent manner and is required for TGF-β-mediated ECM accumulation [12]. A previous study demonstrated that CaN is an important signaling mediator of mesangial cell hypertrophy and ECM accumulation in vitro [13]. Furthermore, inhibition of CaN with Cyclosporin A (CsA) reduced whole kidney hypertrophy and completely blocked glomerular hypertrophy and ECM accumulation in vivo [14]. CaN activation leads to dephosphorylation of NFAT (NFAT1-4) transcription factors which translocate to the nucleus and induce expression of target genes such as Insulin-like Growth Factor-1 (IGF-1) and Vascular Endothelial Growth Factor (VEGF) [15, 16]. As an endogenous inhibitor of CaN, RCAN1.4 is believed to have a functional role in regulating ECM accumulation and liver fibrogenesis. Hence, we hypothesized that RCAN1.4 might influence the survival of HSCs through CaN/NFAT signaling pathway.
Recombinant adeno-associated virus (rAAV) is a single-stranded DNA virus that can infect a broad range of cell types including dividing and non-dividing cells and is, therefore, a widely used vehicle for gene delivery [17]. Clinical trials have not only found rAAV to be consistently safe, but its vectors can instigate long-term expression. To date, rAAV vectors have been used in many clinical trials in gene therapy, and have achieved promising results from Phase 1 and Phase 2 trials [18]; 12 primate serotypes (AAV1-12) have been described [19] two of which, AAV8 and AAV9, are considered to have the highest level of hepatic transduction [20, 21]. To investigate the functional role of RCAN1.4 in the development and progression of liver fibrosis, we specifically over-expressed RCAN1.4 in mice liver through rAAV8-packed RCAN1.4 over-expression plasmid.
Epigenetic modification, especially DNA methylation, plays a pivotal role in silencing gene function. DNA methylation is generally catalyzed by members of the DNA methyltransferase (DNMTs) family, including DNMT1, DNMT3a and DNMT3b [22]. Recent studies by our group and also other researchers demonstrated that DNA methylation plays a pivotal role in determining liver fibrosis and HSC activation [23]. In this study, we investigated whether the decreased expression of RCAN1.4 protein in fibrotic livers was attributed to DNA methylation.
Materials and Methods
Murine model of CCl4-induced liver fibrosis
6-8-week-old male C57BL/6j mice (18-22 g weight) were purchased from the Laboratory Animal Center of Anhui Medical University. All animal procedures were reviewed and approved by the Institutional Animal Experimental Ethics Committee. Mice were housed in a temperature-controlled room (22 ℃). After a week of adaptive breeding, mice were randomly divided into vehicle-treated and CCl4-treated groups (8 mice per group). Mice were placed in a pathogen-free animal facility with access to water and food and allowed to eat and drink ad libitum. Hepatic fibrosis was generated by biweekly intraperitoneal injection of 10 % carbon tetrachloride (CCl4) in corn oil at a dose of 0.01 mL/g/mouse. Control mice were injected with the same volume of corn oil. Four weeks later, 48 h after the last injection of CCl4, mice were sacrificed under anesthesia. Blood samples and liver tissues were collected for further analysis.
Recombinant adeno-associated virus-mediated RCAN1.4 over-expression in mice
Mouse RCAN1.4 over-expression plasmid labeled with the green fluorescent protein (GFP) was obtained from Hanbio Biotechnology Co., Ltd. (Shanghai, China). RCAN1.4 plasmid was packaged with recombinant adeno-associated-virus for over-expression of RCAN1.4 in vivo. Male C57BL/6j mice (6-8 weeks of age, 18-22 g weight) were housed at the Animal Experimental Center for one week to adapt to the environment. The tails of mice were wiped with alcohol to expand the tail vein for injection. Mice were slowly injected with 100 μL recombinant adeno-associated-virus-packaged RCAN1.4 over-expression plasmid with a concentration of 1× 1011 v.g/mL/mouse through the tail vein using 0.5 mL insulin syringe. One week later, mice were intraperitoneally injected with either 10 % CCl4 or the equal volume of corn oil.
Isolation of primary hepatic stellate cells
Mice were chosen randomly from the vehicle group and CCl4-induced liver fibrosis mouse model (n=8/group). The primary hepatic stellate cells was isolated adapting the method as previously described [24]. Briefly, the mice were anesthetized, a 20-G catheter was put through the portal vein, the inferior vena cava was cut, and the liver was perfused with digestion buffer (1×PBC supplemented with type IV collagenase (Sigma-Aldrich, St. Louis, USA), pronase (Sigma-Aldrich, St. Louis, USA) and 4.76 mM CaCl2 (Sigma-Aldrich, St. Louis, USA)). After digestion, the liver was disrupted in 1 % BSA. The cell suspension was prepared by mincing, filtration, and centrifugation in the cell-Nycodenz mixture (Sigma, GER) with a density of 1.040-1.060 g/ml. To create a discontinuous gradient, the cell-Nycodenz mixture was covered with Hank's buffer (Gibco, USA). Finally, primary HSCs was collected by extracting the white cell layer from the gradient interface. The cell suspension was used in subsequent studies. The expression levels of α-SMA and COL1a1 (myofibroblast markers) were examined by Western blotting and RT-qPCR.
Chromatin immunoprecipitation (ChIP) assay
HSC-T6 cells stimulated with TGF-β1 (10 ng/mL), and corresponding control cells were cultured to ~80-90 % confluency. Subsequently, ChIP assay was performed using the SimpleChIP® Enzymatic Chromatin IP Kit (Magnetic Beads) #9003 (Cell Signaling Technology, USA) according to the manufacturer's instructions. The following antibodies were used to immuno-precipitate crosslinked protein-DNA complexes: rabbit anti-DNMT1, rabbit anti-DNMT3a, rabbit anti-DNMT3b and normal rabbit IgG. The immunoprecipitated DNA was purified for PCR analyses with primers specific for the putative binding sites within the promoter of RCAN1.4.
Statistical Analysis
Data were expressed as the mean ± s.e.m. One-way analysis of variance followed by the Newman-Keuls post hoc test (Prism 5.0 GraphPad Software, Inc, San Diego, CA, USA) was used to analyze results. p<0.05 was considered statistically significant.
Additional methods
Detailed methods are provided in Supplementary Materials.
Results
Establishment of CCl4-induced hepatic fibrosis model in C57BL/6j mice
To investigate the extent of hepatic fibrosis in CCl4-induced C57BL/6j mice, histopathological studies were performed. As shown in Figure S1A, liver tissues from CCl4-induced mice harbored fibrotic nodules and displayed hepatomegaly. Histologically, normal lobular architecture consisted of central veins and radiating hepatic cords while treatment with CCl4 resulted in prominent hepatic steatosis, necrosis, inflammatory infiltration, and formation of regenerative nodules and fibrotic septa (Figure S1B-1D and Figure S1H). Furthermore, CCl4 treatment increased hepatic expression of CD8 (a lymphocyte marker) and Ly6G (a neutrophil marker) (Figure S1F and S1G). Immunohistochemical staining showed that desmin and α-SMA, markers for HSCs and activated HSCs were extensively stained in CCl4-treated group as compared to the vehicle-treated group (Figure S1E and S1J). Serum ALT and AST levels were also elevated in CCl4-induced fibrotic mice (Figure S1I). These data indicated a successful establishment of the fibrosis model.
RCAN1.4 expression is down-regulated in vivo and in vitro
The results of double immunofluorescence showed high expression of RCAN1 (green) in α-SMA-positive cells (red) in the fibrotic area, implying that RCAN1.4 was expressed in hepatic stellate cells (Figure 1A). As shown in Figure 1B-C, the protein and mRNA levels of RCAN1.4 were significantly decreased while COL1a1 and α-SMA were elevated upon treatment with CCl4. There was no difference in RCAN1.1 expression between the CCl4-induced and vehicle-treated groups (Figure 1B-C). As displayed in Figure 1D, RCAN1 immunostaining signal was decreased in the fibrotic liver tissue from the CCl4-treated group.
Down-regulated expression of RCAN1.4 in vivo and in vitro. (A) Double immunofluorescence staining was performed to determine the colocalization of RCAN1 (green) and α-SMA (red) in the CCl4-induced liver fibrosis mouse model. DAPI (blue) images were not shown separately. (B) Protein and (C) mRNA expression of RCAN1.1, RCAN1.4, COL1a1 and α-SMA in primary hepatic stellate cells. (D) Immunohistochemistry signals of RCAN1. (E) Protein and (F) mRNA level of RCAN1.1, RCAN1.4, COL1a1 and α-SMA in HSC-T6 cells. (G) RCAN1 protein expression in HSC-T6 cells was examinated by immunofluorescence. Representative pictures are presented. Data represent the mean ± s.e.m. *p<0.05, **p<0.01, as indicated.
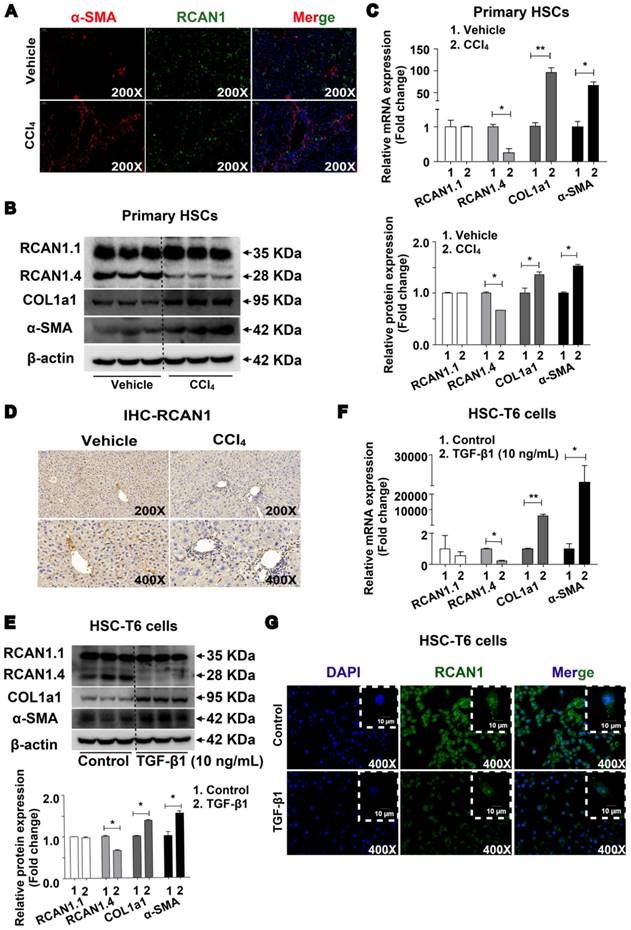
RCAN1.4 expression was also decreased in HSC-T6 cells cultured with TGF-β1 (10 ng/mL) as determined by RT-qPCR, Western blotting, and immunofluorescence staining (Figure 1E-1G). However, the protein and mRNA levels of COL1a1 and α-SMA were significantly increased in TGF-β1 (10 ng/mL)-treated HSC-T6 cells (Figure 1E-1F, S2A). The results showed that RCAN1.4 was selectively down-regulated in vivo and in vitro and may be the main member of the RCAN1 family implicated in liver fibrogenesis.
Recombinant adeno-associated virus-mediated overexpression of RCAN1.4 protects CCl4-induced liver fibrosis in vivo
As previously described, expression of RCAN1.4, both at protein and mRNA levels, was significantly downregulated both in vivo and in vitro. We determined whether RCAN1.4-targeted therapy could prevent CCl4-induced liver fibrosis and liver injury in vivo. The eGFP signal in the liver transfected with RCAN1.4 over-expression plasmid (Figure S3A) indicated a successful infection of the virus. RCAN1.4 expression was increased in the rAAV8-RCAN1.4-transfected liver compared to rAAV8-empty vector-treated liver tissue (Figure 2A-a). As shown in Figure 2A-b and Figure S3B, overexpression of RCAN1.4 in liver tissue inhibited infiltration of inflammatory cells, hepatocyte apoptosis, and lobule destruction compared to empty-virus-treated mice in the CCl4-treated group. Figures 2A-a, d, e display overexpressed RCAN1.4 also decreased areas of blue or green matrix and α-SMA signal indicating reduced ECM deposition.
Liver-specific RCAN1.4 overexpression alleviates CCl4-induced liver fibrosis. (A) Representative RCAN1, H&E, Masson, Sirius red, and α-SMA staining of liver tissue sections. (B) Serum ALT and AST levels. (C) Protein and (D) mRNA levels of RCAN1.4, COL1a1 and α-SMA in primary hepatic stellate cells. (E) Protein levels of C-myc, Cyclin D1, Bax, Bcl2, and cleaved-caspase3. *p<0.05, **p<0.01, ***p<0.001 compared to the vehicle group, #p<0.05, ##p<0.01, ###p<0.001 compared to rAAV8-empty-treated vehicle group, $p<0.05, $$p<0.01, $$$p<0.001 compared to rAAV8-RCAN1.4-treated vehicle group, @p<0.05, @@p<0.01, as indicated. For all panels, data represent the mean ± s.e.m. for 3-4 independent experiments.
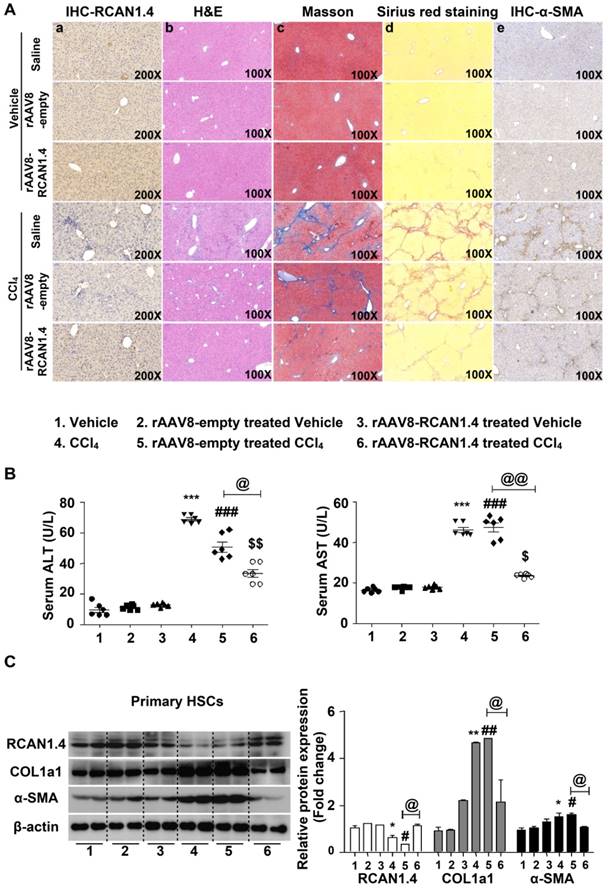
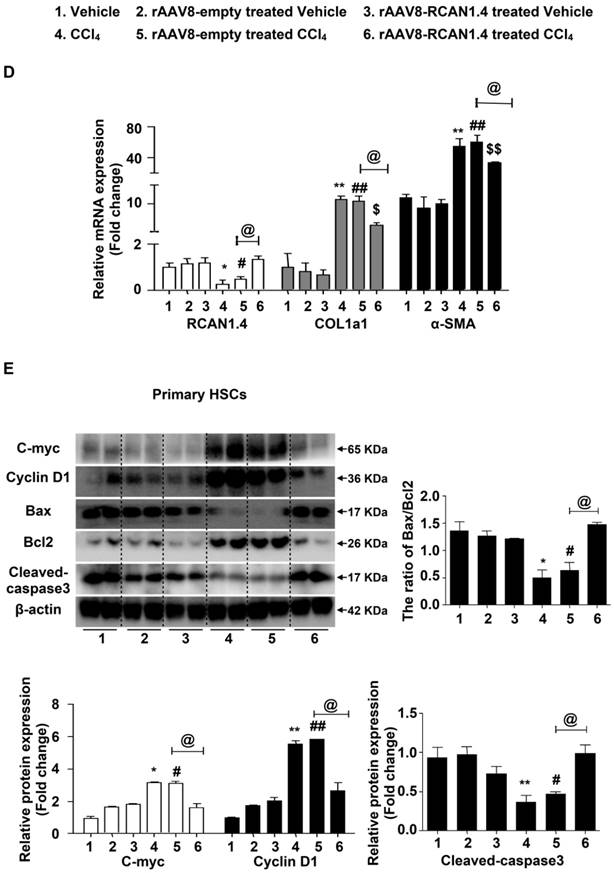
Forced expression of RCAN1.4 dramatically decreased serum ALT/AST levels in CCl4-treated mice (Figure 2B). The protein and mRNA levels of COL1a1 and α-SMA were also significantly down-regulated in primary HSCs in the RCAN1.4 overexpression fibrotic mouse model group (Figure 2C-D). Ectopic expression of RCAN1.4 in liver inhibited primary HSC proliferation and promoted primary HSC apoptosis which was indicated by reduced C-Myc and Cyclin D1 protein expression and an elevated ratio of Bax/Bcl2 and cleaved-caspase3 protein levels (Figure 2E). These results further confirmed that knock in of RCAN1.4 exerts a protective role in CCl4-induced liver fibrosis mouse model. In this setting, RCAN1.4 appears to be a potential therapeutic target for CCl4-induced liver fibrosis.
These results indicated that knocking in RCAN1.4 in vivo could alleviate liver injury and ECM accumulation. As shown in Figure S3C, the proteins interacting with RCAN1.4 were predicted by online String database. It has been reported that CaN/NFAT signaling plays a pivotal role in tissue hypertrophy and ECM accumulation [13, 25]. Therefore, we investigated the function of CaN/NFATs signaling in liver fibrosis.
Ectopic expression of RCAN1.4 inhibits liver fibrosis and promotes aHSC apoptosis in vivo
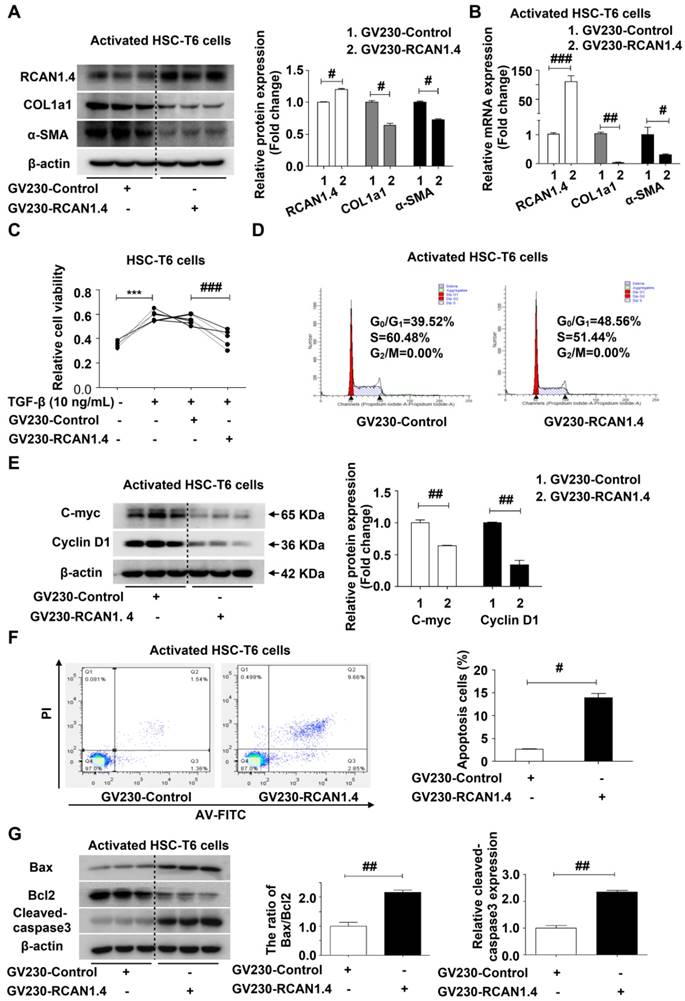
The aberrant expression of RCAN1.4 in activated HSCs (aHSCs), both in vivo and in vitro, and the liver-protective role of RCAN1.4 over-expression in vivo prompted us to investigate the underlying molecular mechanisms of RCAN1.4 in HSC activation and liver fibrosis. We used GV230-RCAN1.4 plasmid to over-express RCAN1.4 in activated HSC-T6 cells (aHSCs). Forced ectopic expression of RCAN1.4 could down-regulate COL1a1 and α-SMA protein and mRNA levels in aHSCs (Figure 3A-3B) and decrease their viability (Figure 3C). The mRNA expression of vimentin, S100A4, and fibronectin was also attenuated in aHSCs transfected with GV230-RCAN1.4 plasmid (Figure S4A). Furthermore, over-expression of RCAN1.4 could induce cell cycle arrest in G0/G1 phase in aHSCs (Figure 3D) and the expression of cell-cycle-associated proteins, C-Myc and Cyclin D1 were notably down-regulated in GV230-RCAN1.4 plasmid-transfected group compared to GV230-control plasmid-transfected group (Figure 3E). These results implied that increased expression of RCAN1.4 in aHSCs could inhibit their activation. We further investigated the influence of forced expression of RCAN1.4 on the survival of aHSCs by flow cytometry. Forced RCAN1.4 expression increased the percentage of apoptotic cells (Figure 3F), and the apoptosis-related proteins (the ratio of Bax/Bcl-2 and cleaved-caspase3) were elevated in GV230-RCAN1.4 plasmid transfected group compared to GV230-control plasmid transfected group (Figure 3G). Thus, these data suggested that ectopic expression of RCAN1.4 could promote apoptosis of aHSCs in vitro.
Effect of RCAN1.4 expression on activated HSC-T6 cells. (A) Protein and (B) mRNA levels of RCAN1.4, COL1a1, and α-SMA. (C) Relative cell viability of HSC-T6 cells was measured by CCK-8 assay. (D) Cell cycle of HSC-T6 cells were analyzed by flow cytometry. (E) Expression of cell-cycle-related protein C-Myc and Cyclin D1. (F) Effect of RCAN1.4 on apoptosis of TGF-β1-activated HSC-T6 cells was determined by flow cytometry. Representative images of three independent experiments are shown. (G) Expression levels of apoptosis-associated proteins (Bax, Bcl2 and cleaved-caspase3 protein). ***p<0.001, #p<0.05, ##p<0.01, ###p<0.001, as indicated. Representative images of three independent experiments are shown. For all panels, data represent the mean ± s.e.m. for 3-4 independent experiments in vitro.
RCAN1.4 silencing enhances the activation of HSC-T6 cells in vitro
We further confirmed the ability of RCAN1.4 to inhibit HSC activation using RCAN1.4-RNAi transfection. Figure S5A shows that RCAN1.4 mRNA expression in RCAN1.4-RNAi transfected group decreased significantly by approximately 80 % compared to the scrambled-RNAi transfected group. RCAN1.4 knockdown increased COL1a1 and α-SMA protein expression in aHSCs. RT-qPCR results were similar to Western blotting analysis (Figure 4B), indicating that RCAN1.4 silencing could enhance the activation of HSCs. Cell viability was increased in the RCAN1.4-RNAi-transfected group compared with the scrambled-RNAi-transfected group (Figure 4C). Flow cytometry analysis showed that RCAN1.4 silencing could advance HSCs into G2/M phase (Figure 4D). Furthermore, cell-cycle-related proteins (C-Myc and Cyclin D1) were elevated in RCAN1.4-RNAi-transfected cells (Figure 4E). These data indicated that knockdown of RCAN1.4 expression enhanced the activation of HSCs.
Effect of RCAN1.4 silencing on activated HSC-T6 cells. (A) Protein and (B) mRNA expression of RCAN1.4, COL1a1, and α-SMA. (C) Relative cell viability of HSC-T6 cells was measured by CCK-8 analysis. (D) Cell cycle was determined by flow cytometry. (E) The cell-cycle-related proteins C-Myc and Cyclin D1 levels were analyzed by western blotting analysis. (F) Effect of RCAN1.4 silencing on apoptosis of activated HSC-T6 cells was detected by flow cytometry. (G) The expression levels of apoptosis-associated proteins (Bax, Bcl2 and cleaved-caspase3 protein) were detected by western blotting assay. Representative images of three independent experiments are shown. ***p<0.001, #p<0.05, ###p<0.001, as indicated. For all panels, data represent the mean ± s.e.m. for 3-4 independent experiments in vitro.
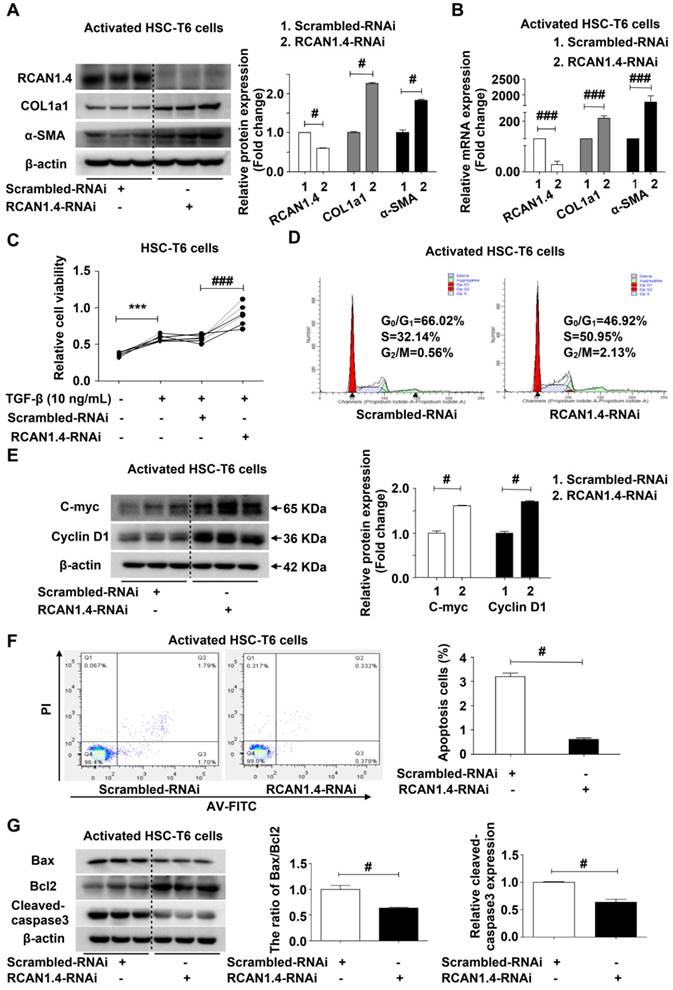
We further examined the influence of RCAN1.4 silencing on apoptosis of activated HSC-T6 cells by flow cytometry and observed a decrease in apoptotic cell proportion in the RCAN1.4-RNAi transfected group (Figure 4F). Moreover, the apoptosis-associated proteins (the ratio of Bax/Bcl-2, cleaved-caspase3) were down-regulated in the RCAN1.4-RNAi-transfected group (Figure 4G). The migration of HSCs, as detected by scratch wound healing experiment and Transwell assay, was significantly increased by depletion of RCAN1.4 (Figure S5B and S5C). Taken together, these results indicated that depletion of RCAN1.4 could aggravate liver fibrosis.
RCAN1.4 functions as an endogenous inhibitor of calcineurin-NFAT3 signaling in liver fibrosis
It has been demonstrated that both RCAN1.1 and RCAN1.4 inhibit the activity of CaN, an intracellular phosphatase involved in many biological processes [26]. Furthermore, RCAN1 has been demonstrated to especially inhibit nuclear translocation of NFAT mediated by CaN[27]. Therefore, we assessed the protein expression levels of CaN in aHSCs after knock-in and knockdown of RCAN1.4. The CaN expression was significantly reduced when RCAN1.4 was over-expressed (Figure 5A), while its expression increased when RCAN1.4 was knocked down in activated HSC-T6 cells (Figure 5B). The nuclear distribution of NFAT1-4 was assessed by Western blot analysis. We observed that over-expression of RCAN1.4 inhibited the nuclear accumulation of NFAT3, while knockdown of RCAN1.4 promoted NFAT3 nuclear translocation (Figure 5C, S6A). However, the total protein level of NFAT3 was not affected by altered RCAN1.4 expression (Figure S6B). Thus, these results indicated that altered nuclear level of NFAT3 was not attributed to a change in total NFAT3 expression but the translocation of NFAT3 to the nucleus. Interestingly, the expression of NFAT1, NFAT2, and NFAT4 was not affected by altered RCAN1.4 expression (Figure 5C and S6A). More importantly, the expression of CaN and nuclear accumulation of NFAT3 could be inhibited by elevated RCAN1.4 expression in vivo (Figure S6C).
RCAN1.4 inhibits the nuclear translocation of NFAT3 in liver fibrosis. (A)(B) Western blot analysis of CaN protein level. (C) The nuclear proteins were extracted 24 h after transfection of GV230-RCAN1.4 plasmid or RCAN1.4-RNAi in activated HSC-T6 cells. The NFAT1, NFAT2, NFAT3, and NFAT4 protein levels were measured by Western blotting analysis. Representative images of three independent experiments are shown. (D) Protein level of NFAT3 in the nucleus. #p<0.05, $p<0.05, @p<0.05, %p<0.05, ^^p<0.01, &&p<0.01, as indicated. Representative images of three independent experiments are shown. For all panels, data represent the mean ± s.e.m. for 3-4 independent experiments in vitro.
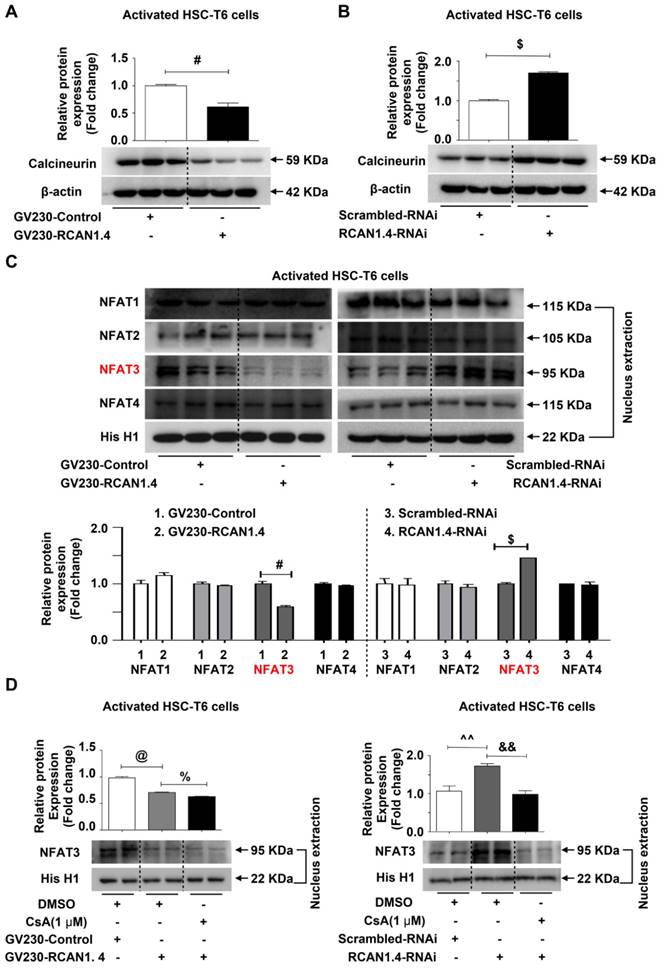
We treated HSC-T6 cells with CsA [28], an inhibitor of CaN, and examined NFAT3 nuclear distribution levels. CsA treatment reversed NFAT3 nuclear distribution in RCAN1.4 over-expression and silencing groups separately (Figure 5D). It has been reported that inhibition of CaN/NFAT3 pathway could attenuate cardiac hypertrophy [29]. To investigate whether heterologous expression of NFAT3 could reverse myofibroblast phenotype, we performed NFAT3 overexpression experiments. The transfection efficiency of NFAT3 overexpression plasmid was analyzed by Western blot analysis (Figure S6D). Forced expression of NFAT3 aggravated fibrosis as indicated by elevated COL1a1 and α-SMA expression (Figure S6E). However, simultaneous overexpression of RCAN1.4 and NFAT3 could reverse the influence of NFAT3 on myofibroblast marker expression (Figure S6F). These data indicated that RCAN1.4 negatively regulated NFAT3 nuclear accumulation through inhibiting CaN activity in HSC-T6 cells and primary hepatic stellate cells.
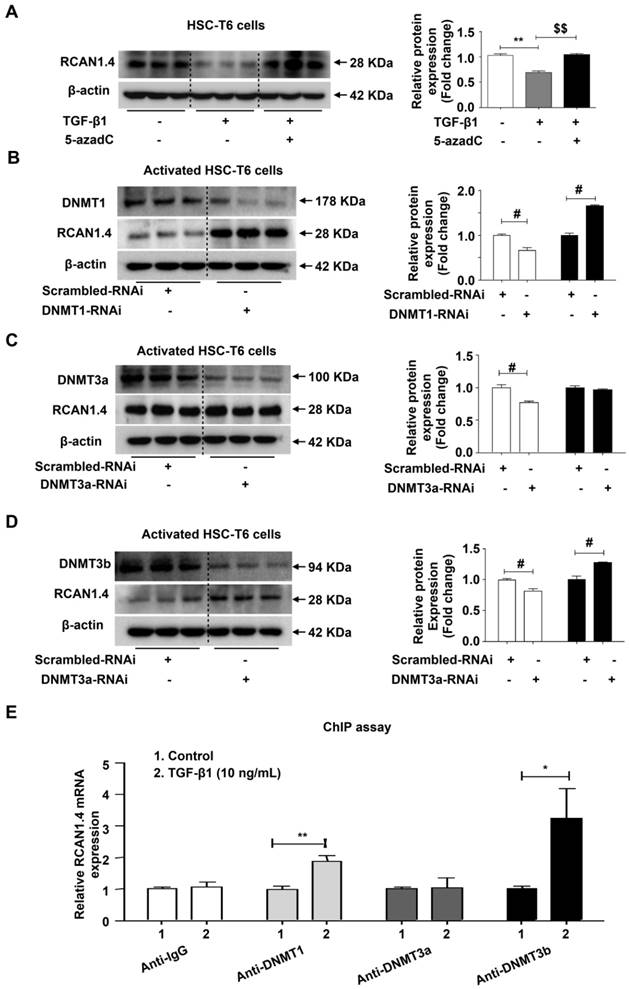
Down-regulated RCAN1.4 expression is mediated by DNMT1 and DNMT3b
Studies performed by our laboratory and other groups have demonstrated that epigenetic modifications, especially DNA methylation, play an important role in the regulation of HSC activation and liver fibrosis [30]. DNA methylation is mainly catalyzed by three DNA methyltransferases: DNMT1, DNMT3a and DNMT3b [22]. It has been demonstrated that methyltransferases are upregulated in activated HSCs [23] and that elevated DNMTs lead to aberrant DNA methylation patterns [31]. Consistent with the previous studies, we observed distinct upregulation of DNMT1, DNMT3a and DNMT3b protein expression both in vivo and in vitro (Figure S7A).
We predicted the existence of two methylated CpG sites in the RCAN1.4 gene promoter (Figure S7B). It has been shown that 5-azadC, an inhibitor of DNA methyltransferase, can block the enzymatic activity of all three methyltransferases [32]. We have previously demonstrated that 5-azadC could reduce liver injury and inhibit the expression of COL1a1 and α-SMA in primary HSCs [33]. To investigate whether the down-regulated expression of RCAN1.4 in activated HSCs was attributed to DNA methylation, 5-azadC [34] and DNMTs-RNAi were employed. As shown in Figure 6A, the decreased RCAN1.4 protein level in TGF-β1-treated group could be restored by culturing activated HSC-T6 cells with 5-azadC. RCAN1.4 expression was elevated in DNMT1-RNAi- and DNMT3b-RNAi-transfected groups but not in DNMT3a-RNAi-transfected group (Figure 6B-D). The results of RT-qPCR were similar to Western blot analysis (Figure S7C). Furthermore, ChIP assay showed that RCAN1.4 gene could be pulled down by anti-DNMT1 and anti-DNMT3b antibodies, but not by anti-DNMT3a and negative control anti-IgG antibodies (Figure 6E). These results indicated direct binding of DNMT1 and DNMT3b with the RCAN1.4 promoter in HSC-T6 cells. Hence, these data demonstrated that decreased RCAN1.4 expression was associated with elevated expression of DNMT1 and DNMT3b methyltransferases.
Decreased expression of RCAN1.4 was mediated by DNMT1 and DNMT3b. (A) Restoration of down-regulated RCAN1.4 protein level in TGF-β1-treated HSC-T6 cells by 5-azadC (1 mM). (B)-(D) Protein levels of DNMT1, DNMT3a, DNMT3b, and RCAN1.4. (E) ChIP assay showing the interaction of DNMT1, DNMT3a, and DNMT3b with the potential binding sites on the RCAN1.4 promoter. (F) Prediction of methylated CpG sites in the RCAN1.4 gene promoter. *p<0.05, **p<0.01, $$p<0.01, #p<0.05, ##p<0.01, as indicated. Values represent the mean ±s.e.m.
Discussion
In the current study, we observed that RCAN1.4 expression was selectively down-regulated in hepatic fibrosis and was associated with pathological processes in vivo and in vitro. We detected two methylated CpG islands in the RCAN1.4 gene promoter. Furthermore, the expression of RCAN1.4 could be restored by 5-azadC and DNMTs-RNAi which indicated that the reduced RCAN1.4 expression was attributed to DNA methylation. Using the ChIP assay, we confirmed that methylation of RCAN1.4 promoter was induced by DNMT1 and DNMT3b. We demonstrated that accelerated liver fibrosis, caused by decreased expression of RCAN1.4, could be alleviated by forced RCAN1.4 expression. We then set out to elucidate the functional role of RCAN1.4 in liver fibrosis.
In previous studies, overexpression of RCAN1.4 has been shown to block VEGF-calcineurin-NFAT-mediated endothelial cell activation in vitro [35, 36]. A microarray study analyzing gene transcription changes following VEGF treatment of endothelial cells reported that RCAN1.4 is the most obviously changed transcript, consistent with its important role in VEGF-mediated angiogenesis [37]. Recently, a role for CaN/NFAT signaling in cancer progression has been identified. Most notably, NFAT mediated breast cancer cell invasion and metastasis as a consequence of downregulated AKT expression [38]. RCAN1.4 has been shown to inhibit cancer cell proliferation, migration, and invasion in hepatocellular carcinoma by preventing nuclear translocation of NFAT1 [15]. Sun et al. demonstrated that overexpression of RCAN1.4 exacerbated calcium overloading-induced neuronal apoptosis through the caspase3 apoptotic pathway [39]. However, other groups demonstrated that RCAN1.1L prevented hypoxia-induced cell apoptosis through the induction of mitophagy [40]. Similarly, targeted deletion of both RCAN1.1 and RCAN1.4 promoted apoptosis, rather than proliferation, of endothelial cells by stimulating VEGF [41]. Interestingly, Ryeom et al. predicted that there is a delicate equilibrium between RCAN1 and CaN/NFAT suggesting that the CaN/NFAT-RCAN1 pathway can be a susceptible target for antiangiogenic cancer therapies [41]. Taken together, these conflicting reports suggest a complex role for the RCAN1 gene in cell survival or cell death under stress conditions.
In this study, we demonstrated that knocking in of RCAN1.4 alleviated liver fibrosis and promoted apoptosis of activated HSCs in vivo and in vitro. Furthermore, forced RCAN1.4 expression could inhibit the protein expression and activity of CaN, subsequently preventing the nuclear translocation of NFAT3 and could also promote apoptosis of activated HSCs with elevated cleaved-caspase3 protein levels. However, it needs to be further investigated whether the role of RCAN1.4 in regulating HSC proliferation can also be attributed to IGF-1 and VEGF as suggested in a previous study [15].
Fibrosis in the liver, as with fibrosis in other organs (such as lung, heart, kidney, or skin) is characterized by excessive deposition of ECM [42] which leads to the destruction of organ structure and impairment of organ function. Activated fibroblasts are key mediators of organ fibrosis [42]. Diabetic nephropathy is characterized by excessive accumulation of ECM proteins in the glomerulainterstitium [43, 44]. It has been reported that elevated level of Insulin-like growth factor-I (IGF-1) in the urine was associated with hypertrophy and progression of kidney disease in patients with type I diabetes [45]. IGF-1 could activate Erk1/Erk2, MAPK, and PI3K signaling in many different cell types [46], but neither of them was required for hypertrophy or accumulation of ECM proteins [13]. In animal models, inhibitors of CaN could prevent hypertrophy successfully [13, 25, 47]. As mentioned earlier, RCAN1 negatively regulates the dephosphorylation activity of CaN. Therefore, elevated RCAN1.4 expression is believed to decrease ECM accumulation. However, it has been reported that over-expression of RCAN1.4 increased the production of collagens in Mes-13 cells [48]. The reasonable explanation for this paradox is provided by several studies showing that RCAN1 is not only an inhibitor of CaN, but can also interact with several proteins, including Raf-1 [49], NIK [50], CREB [51], SCFβ-TrCP ubiquitin ligase [52], and Tollip[53]. RCAN1 has also been shown to modulate the activity of nuclear factor κB (NF-κB) [54] and ERK signaling [55]. Further studies are required to address the mechanism underlying RCAN1 function in regulating ECM deposition in liver fibrosis.
It has been demonstrated that RCAN1 plays a role in Alzheimer's disease (AD) pathogenesis [56]. Aging is the most notable risk factor for AD as well as many other age-related human diseases [57, 58]. Fibrosis is a major cause of morbidity and mortality in the aging process of humans worldwide. Treating fibrosis is a potential strategy for slowing the aging process and prolonging lifespan [59, 60].
In the current study, we demonstrated that over-expression of RCAN1.4 could alleviate liver fibrosis, decrease ECM accumulation in vivo, and inhibit activation of HSCs in vivo and in vitro. While studying the underlying mechanisms for the reversal of liver fibrosis, we demonstrated that RCAN1.4 could inhibit CaN/NFAT3 signaling pathway and subsequently reduce ECM protein accumulation. Thus our data indicate a crucial role of RCAN1.4 via CaN/NFAT3 signaling in regulating hypertrophy and ECM accumulation and provide a potential therapeutic strategy for fibrotic diseases.
Conclusion
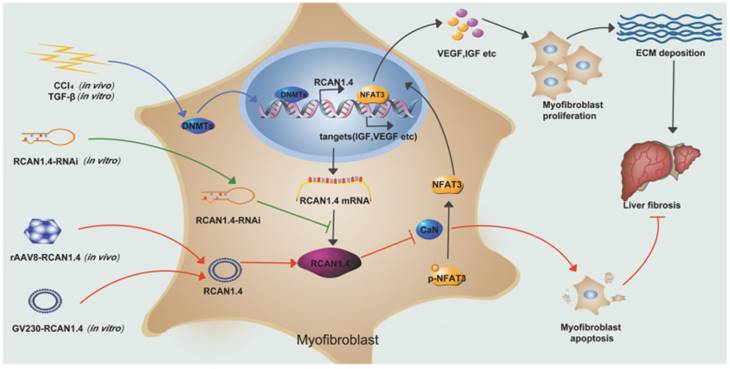
Our study has illustrated the central role of RCAN1.4 in liver fibrosis (Figure 7). The expression of RCAN1.4 was downregulated both in vivo and in vitro by methyltransferases DNMT1 and DNMT3b. Furthermore, CCl4-induced liver fibrosis and ECM deposition could be reversed by forced expression of RCAN1.4. Ectopic expression of RCAN1.4 in vivo effectively attenuated liver injury, inhibited activation of qHSCs, and promoted apoptosis of aHSCs through CaN/NFAT3 signaling pathway. The results illustrated in Figure 7 suggest that RCAN1.4 can serve as a therapeutic target in the treatment of liver fibrosis.
Schematic depicting the mechanisms of RCAN1.4 to prevent liver fibrosis. RCAN1.4 was selectively downregulated in liver fibrosis mediated by DNMT1 and DNMT3b. Forced RCAN1.4 expression could alleviate liver injury, inhibit HSC activation, proliferation and promote activated HSC apoptosis through CaN/NFAT3 signaling pathway in vivo and in vitro. However, silencing of RCAN1.4 exacerbates liver fibrosis in vitro. In briefly, RCAN1.4 may serve as a potential therapeutic target in the treatment of liver fibrosis.
Abbreviations
RCAN1: regulator of calcineurin 1; RCAN1.4: RCAN1 isoform 4; RCAN1.1L: RCAN1 isoform 1 Long; RCAN1.1S: RCAN1 isoform 1 Short; ECM: extracellular matrix; CaN: calcineurin; TGF-β1: transforming growth factor-beta 1; 5-azadC: 5-aza-2'-deoxycytidin; DNMT: DNA methyltransferase; NFAT: the nuclear factor of activated T cells; HCC: hepatocellular carcinoma; HSCs: hepatic stellate cells; qHSCs: quiescent HSCs; aHSCs: activated HSCs; α-SMA: alpha smooth muscle actin; COL1a1: type I collagen; CsA: cyclosporin A; IGF-1: insulin-like growth factor-1; VEGF: vascular endothelial growth factor; rAAV: recombinant adeno-associated virus; CCl4: carbon tetrachloride; DMEM: Dulbecco's modified Eagle's medium; FBS: fetal bovine serum; BSA: bovine serum albumin; ALT: alanine aminotransferase; AST: aspartate aminotransferase; H&E: hematoxylin and eosin; IHC: immunohistochemistry; DAB: 3, 30-diaminobenzidine tetrahydrochloride; RT-q-PCR: real-time quantitative PCR; PI: propidium iodide; ChIP: chromatin immunoprecipitation; AD: Alzheimer's disease; BSP: bisulfite sequencing.
Supplementary Material
Supplementary methods, figures and tables.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (No. 81770609) and Anhui Medical University of Science and Technology (No. 1704a0802161). We are grateful to Dr. Iqbal Ali for helps in polishing my manuscript.
Author's contributions
X.Y.P conceived and carried out experiments, analyzed the data, and wrote the manuscript. H.M.Y, L.W, and H.W.M isolated primary HSCs and performed Western blotting analysis. Y.Y analyzed the data. Y.H.B modified the pictures. C.H., X.M.M. and T.T.M participated in the design of the study. J.L. conceived the study and revised the manuscript. All authors contributed to the writing of the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209-18
2. Mokdad AA, Lopez AD, Shahraz S, Lozano R, Mokdad AH, Stanaway J. et al. Liver cirrhosis mortality in 187 countries between 1980 and 2010: a systematic analysis. BMC Med. 2014;12:145
3. Troeger JS, Mederacke I, Gwak GY, Dapito DH, Mu X, Hsu CC. et al. Deactivation of hepatic stellate cells during liver fibrosis resolution in mice. Gastroenterology. 2012;143:1073-83 e22
4. Friedman SL. Cytokines and fibrogenesis. Semin Liver Dis. 1999;19:129-40
5. Mederacke I, Hsu CC, Troeger JS, Huebener P, Mu X, Dapito DH. et al. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat Commun. 2013;4:2823
6. Kisseleva T, Brenner DA. Role of hepatic stellate cells in fibrogenesis and the reversal of fibrosis. J Gastroenterol Hepatol. 2007;22(Suppl 1):S73-8
7. Ermak G, Harris CD, Davies KJ. The DSCR1 (Adapt78) isoform 1 protein calcipressin 1 inhibits calcineurin and protects against acute calcium-mediated stress damage, including transient oxidative stress. FASEB J. 2002;16:814-24
8. Genesca L, Aubareda A, Fuentes JJ, Estivill X, De La Luna S, Perez-Riba M. Phosphorylation of calcipressin 1 increases its ability to inhibit calcineurin and decreases calcipressin half-life. Biochem J. 2003;374:567-75
9. Shibasaki F, McKeon F. Calcineurin functions in Ca(2+)-activated cell death in mammalian cells. J Cell Biol. 1995;131:735-43
10. Liu J, Wang W, Wang F, Cai F, Hu ZL, Yang YJ. et al. Phosphatidylinositol-linked novel D(1) dopamine receptor facilitates long-term depression in rat hippocampal CA1 synapses. Neuropharmacology. 2009;57:164-71
11. Chin ER, Olson EN, Richardson JA, Yang Q, Humphries C, Shelton JM. et al. A calcineurin-dependent transcriptional pathway controls skeletal muscle fiber type. Genes Dev. 1998;12:2499-509
12. Gooch JL, Gorin Y, Zhang BX, Abboud HE. Involvement of calcineurin in transforming growth factor-beta-mediated regulation of extracellular matrix accumulation. J Biol Chem. 2004;279:15561-70
13. Gooch JL, Tang Y, Ricono JM, Abboud HE. Insulin-like growth factor-I induces renal cell hypertrophy via a calcineurin-dependent mechanism. J Biol Chem. 2001;276:42492-500
14. Gooch JL, Barnes JL, Garcia S, Abboud HE. Calcineurin is activated in diabetes and is required for glomerular hypertrophy and ECM accumulation. Am J Physiol Renal Physiol. 2003;284:F144-54
15. Jin H, Wang C, Jin G, Ruan H, Gu D, Wei L. et al. Regulator of Calcineurin 1 Gene Isoform 4, Down-regulated in Hepatocellular Carcinoma, Prevents Proliferation, Migration, and Invasive Activity of Cancer Cells and Metastasis of Orthotopic Tumors by Inhibiting Nuclear Translocation of NFAT1. Gastroenterology. 2017;153:799-811 e33
16. Macian F. NFAT proteins: key regulators of T-cell development and function. Nat Rev Immunol. 2005;5:472-84
17. Alexander IE, Russell DW, Spence AM, Miller AD. Effects of gamma irradiation on the transduction of dividing and nondividing cells in brain and muscle of rats by adeno-associated virus vectors. Hum Gene Ther. 1996;7:841-50
18. Nam HJ, Lane MD, Padron E, Gurda B, McKenna R, Kohlbrenner E. et al. Structure of adeno-associated virus serotype 8, a gene therapy vector. J Virol. 2007;81:12260-71
19. Gao GP, Alvira MR, Wang L, Calcedo R, Johnston J, Wilson JM. Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. Proc Natl Acad Sci U S A. 2002;99:11854-9
20. Nakai H, Fuess S, Storm TA, Muramatsu S, Nara Y, Kay MA. Unrestricted hepatocyte transduction with adeno-associated virus serotype 8 vectors in mice. J Virol. 2005;79:214-24
21. Inagaki K, Fuess S, Storm TA, Gibson GA, McTiernan CF, Kay MA. et al. Robust systemic transduction with AAV9 vectors in mice: efficient global cardiac gene transfer superior to that of AAV8. Mol Ther. 2006;14:45-53
22. Valente S, Liu Y, Schnekenburger M, Zwergel C, Cosconati S, Gros C. et al. Selective non-nucleoside inhibitors of human DNA methyltransferases active in cancer including in cancer stem cells. J Med Chem. 2014;57:701-13
23. Cai SP, Cheng XY, Chen PJ, Pan XY, Xu T, Huang C. et al. Transmembrane protein 88 attenuates liver fibrosis by promoting apoptosis and reversion of activated hepatic stellate cells. Mol Immunol. 2016;80:58-67
24. Pan XY, Yang Y, Meng HW, Li HD, Chen X, Huang HM. et al. DNA Methylation of PTGIS Enhances Hepatic Stellate Cells Activation and Liver Fibrogenesis. Front Pharmacol. 2018;9:553
25. Lim HW, De Windt LJ, Steinberg L, Taigen T, Witt SA, Kimball TR. et al. Calcineurin expression, activation, and function in cardiac pressure-overload hypertrophy. Circulation. 2000;101:2431-7
26. Rothermel B, Vega RB, Yang J, Wu H, Bassel-Duby R, Williams RS. A protein encoded within the Down syndrome critical region is enriched in striated muscles and inhibits calcineurin signaling. J Biol Chem. 2000;275:8719-25
27. Crabtree GR, Schreiber SL. SnapShot: Ca2+-calcineurin-NFAT signaling. Cell. 2009;138:210 e1
28. Rusnak F, Mertz P. Calcineurin: form and function. Physiol Rev. 2000;80:1483-521
29. Lu Y, Zhao M, Liu JJ, He X, Yu XJ, Liu LZ. et al. Long-term administration of pyridostigmine attenuates pressure overload-induced cardiac hypertrophy by inhibiting calcineurin signalling. J Cell Mol Med. 2017;21:2106-16
30. Wu Y, Bu F, Yu H, Li W, Huang C, Meng X. et al. Methylation of Septin9 mediated by DNMT3a enhances hepatic stellate cells activation and liver fibrogenesis. Toxicol Appl Pharmacol. 2017;315:35-49
31. Chen T, Li E. Structure and function of eukaryotic DNA methyltransferases. Curr Top Dev Biol. 2004;60:55-89
32. Wang X, Chen E, Yang X, Wang Y, Quan Z, Wu X. et al. 5-azacytidine inhibits the proliferation of bladder cancer cells via reversal of the aberrant hypermethylation of the hepaCAM gene. Oncol Rep. 2016;35:1375-84
33. Chen X, Li WX, Chen Y, Li XF, Li HD, Huang HM. et al. Suppression of SUN2 by DNA methylation is associated with HSCs activation and hepatic fibrosis. Cell Death Dis. 2018;9:1021
34. Lindner DJ, Wu Y, Haney R, Jacobs BS, Fruehauf JP, Tuthill R. et al. Thrombospondin-1 expression in melanoma is blocked by methylation and targeted reversal by 5-Aza-deoxycytidine suppresses angiogenesis. Matrix Biol. 2013;32:123-32
35. Hesser BA, Liang XH, Camenisch G, Yang S, Lewin DA, Scheller R. et al. Down syndrome critical region protein 1 (DSCR1), a novel VEGF target gene that regulates expression of inflammatory markers on activated endothelial cells. Blood. 2004;104:149-58
36. Minami T, Horiuchi K, Miura M, Abid MR, Takabe W, Noguchi N. et al. Vascular endothelial growth factor- and thrombin-induced termination factor, Down syndrome critical region-1, attenuates endothelial cell proliferation and angiogenesis. J Biol Chem. 2004;279:50537-54
37. Abe M, Sato Y. cDNA microarray analysis of the gene expression profile of VEGF-activated human umbilical vein endothelial cells. Angiogenesis. 2001;4:289-98
38. Jauliac S, Lopez-Rodriguez C, Shaw LM, Brown LF, Rao A, Toker A. The role of NFAT transcription factors in integrin-mediated carcinoma invasion. Nat Cell Biol. 2002;4:540-4
39. Sun X, Wu Y, Herculano B, Song W. RCAN1 overexpression exacerbates calcium overloading-induced neuronal apoptosis. PloS one. 2014;9:e95471
40. Sun L, Hao Y, An R, Li H, Xi C, Shen G. Overexpression of Rcan1-1L inhibits hypoxia-induced cell apoptosis through induction of mitophagy. Mol Cells. 2014;37:785-94
41. Ryeom S, Baek KH, Rioth MJ, Lynch RC, Zaslavsky A, Birsner A. et al. Targeted deletion of the calcineurin inhibitor DSCR1 suppresses tumor growth. Cancer cell. 2008;13:420-31
42. Brenner DA, Waterboer T, Choi SK, Lindquist JN, Stefanovic B, Burchardt E. et al. New aspects of hepatic fibrosis. J Hepatol. 2000;32:32-8
43. Iidaka K, McCoy J, Kimmelsteil P. The glomerular mesangium. A quantitative analysis. Lab Invest. 1968;19:573-9
44. Kawano K, Arakawa M, McCoy J, Porch J, Kimmelstiel P. Quantitative study of glomeruli. Focal glomerulonephritis and diabetic glomerulosclerosis. Lab Invest. 1969;21:269-75
45. Verrotti A, Cieri F, Petitti MT, Morgese G, Chiarelli F. Growth hormone and IGF-I in diabetic children with and without microalbuminuria. Diabetes Nutr Metab. 1999;12:271-6
46. Yenush L, White MF. The IRS-signalling system during insulin and cytokine action. Bioessays. 1997;19:491-500
47. Murat A, Pellieux C, Brunner HR, Pedrazzini T. Calcineurin blockade prevents cardiac mitogen-activated protein kinase activation and hypertrophy in renovascular hypertension. J Biol Chem. 2000;275:40867-73
48. Jang C, Lim JH, Park CW, Cho YJ. Regulator of Calcineurin 1 Isoform 4 (RCAN1.4) Is Overexpressed in the Glomeruli of Diabetic Mice. Korean J Physiol Pharmacol. 2011;15:299-305
49. Cho YJ, Abe M, Kim SY, Sato Y. Raf-1 is a binding partner of DSCR1. Arch Biochem Biophys. 2005;439:121-8
50. Lee EJ, Seo SR, Um JW, Park J, Oh Y, Chung KC. NF-kappaB-inducing kinase phosphorylates and blocks the degradation of Down syndrome candidate region 1. J Biol Chem. 2008;283:3392-400
51. Seo SR, Chung KC. CREB activates proteasomal degradation of DSCR1/RCAN1. FEBS letters. 2008;582:1889-93
52. Asada S, Ikeda A, Nagao R, Hama H, Sudo T, Fukamizu A. et al. Oxidative stress-induced ubiquitination of RCAN1 mediated by SCFbeta-TrCP ubiquitin ligase. Int J Mol Med. 2008;22:95-104
53. Lee JY, Lee HJ, Lee EJ, Jang SH, Kim H, Yoon JH. et al. Down syndrome candidate region-1 protein interacts with Tollip and positively modulates interleukin-1 receptor-mediated signaling. Biochim Biophys Acta. 2009;1790:1673-80
54. Kim YS, Cho KO, Lee HJ, Kim SY, Sato Y, Cho YJ. Down syndrome candidate region 1 increases the stability of the IkappaBalpha protein: implications for its anti-inflammatory effects. J Biol Chem. 2006;281:39051-61
55. Lee HJ, Kim YS, Sato Y, Cho YJ. RCAN1-4 knockdown attenuates cell growth through the inhibition of Ras signaling. FEBS letters. 2009;583:2557-64
56. Wong H, Levenga J, Cain P, Rothermel B, Klann E, Hoeffer C. RCAN1 overexpression promotes age-dependent mitochondrial dysregulation related to neurodegeneration in Alzheimer's disease. Acta Neuropathol. 2015;130:829-43
57. Jackaman C, Tomay F, Duong L, Abdol Razak NB, Pixley FJ, Metharom P. et al. Aging and cancer: The role of macrophages and neutrophils. Ageing Res Rev. 2017;36:105-16
58. Proshkina EN, Shaposhnikov MV, Sadritdinova AF, Kudryavtseva AV, Moskalev AA. Basic mechanisms of longevity: A case study of Drosophila pro-longevity genes. Ageing Res Rev. 2015;24:218-31
59. Kahn BB, Alquier T, Carling D, Hardie DG. AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 2005;1:15-25
60. Minokoshi Y, Kim YB, Peroni OD, Fryer LG, Muller C, Carling D. et al. Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature. 2002;415:339-43
Author contact
![]() Corresponding authors: Li Jun, Professor of Pharmacology, Huang Cheng, Professor of Pharmacology, School of Pharmacy, Anhui Medical University, Hefei, Anhui, China. Tel: +86-551-65161217; Fax: +86-551-65161217; E-mail address: ljedu.cn, huangchengedu.cn
Corresponding authors: Li Jun, Professor of Pharmacology, Huang Cheng, Professor of Pharmacology, School of Pharmacy, Anhui Medical University, Hefei, Anhui, China. Tel: +86-551-65161217; Fax: +86-551-65161217; E-mail address: ljedu.cn, huangchengedu.cn