Impact Factor
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials
Experimental procedures
Results
Discussion
Conclusion
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(14):4066-4083. doi:10.7150/thno.33383 This issue Cite
Research Paper
Enhanced cytotoxic T lymphocytes recruitment targeting tumor vasculatures by endoglin aptamer and IP-10 plasmid presenting liposome-based nanocarriers
Xiaomei Yang*, Jing Zhao*, Siliang Duan*, Xiaoqiong Hou, Xi Li, Zixi Hu, Zhuoran Tang, Fengzhen Mo, Xiaoling Lu ![]()
Nanobody Research Center/School of Preclinical Medicine, Guangxi Medical University, Nanning, Guangxi, 530021, China
*These authors contributed equally to this work.
Received 2019-1-21; Accepted 2019-3-24; Published 2019-5-31
Abstract

Background: Adequate recruitment of highly active tumor antigen-specific cytotoxic T lymphocytes (CTLs) remains a major challenge in cancer immunotherapy.
Objective: To construct liposome (LP)-based nanocapsules with surface endoglin aptamer (ENG-Apt) encapsulating mouse interferon-inducible protein-10 (mIP-10), with the ability to target mouse tumor vascular endothelial cells (mTECs) and enhance CTLs targeting and recruitment to the tumor vasculature.
Methods: ENG-Apt/mIP-10-LP nanocapsules were prepared by grafting DSPE-PEG2000-ENG-Apt on the surface of liposomes containing mIP-10 plasmids, characterized and assessed for the cell binding specificity in vitro. The tumor-targeting ability of ENG-Apt/mIP-10-LP nanocapsules was evaluated in vivo. The anti-tumor efficacy of ENG-Apt/mIP-10-LP nanocapsules treatment, as well as the combination treatment of ENG-Apt/mIP-10-LP nanocapsules and adoptive TRP2CD8+ T cells, were both tested in melanoma-bearing mice, by evaluation of the tumor volume and the mouse survival time. To discuss the anti-tumoral mechanism of ENG-Apt/mIP-10-LP nanocapsules-based therapies, IFN-γ secretion, proportion of TRP2CD8+ T cells among TILs, MDSCs in the tumor microenvironment and Tregs in the spleen, were determined after the treatments. Proliferation and apoptosis of tumor cells, and tumor angiogenesis were also assessed.
Results: The prepared ENG-Apt/mIP-10-LP nanocapsules possess an adequate nanometric size, good stability, high specificity to mTECs and tumor sites, along with the ability to induce mIP-10 expression in vitro and in vivo. Treatment of ENG-Apt/mIP-10-LP nanocapsules demonstrated CTLs enrichment into the tumor site, which inhibited tumor cell proliferation and angiogenesis, as well as promoted tumor-cell apoptosis, leading to a decrease in tumor progression and prolonged survival time in melanoma tumor-bearing mice. In addition, the proportion of MDSCs and Tregs was found to decrease. The combination of ENG-Apt/mIP-10-LP nanocapsules with adoptive TRP2CD8+ T cells, showed stronger abilities in inhibiting tumor growth and increasing animal survival time, thereby displayed an enhanced anti-melanoma tumor efficacy, due to the recruitment of both endogenous CD8+ T cells and exogenous TRP2CD8+ T cells in vivo.
Conclusion: ENG-Apt/mIP-10-LP nanocapsules could enhance the recruitment of both endogenous and exogenous CTLs specifically targeting melanoma tumor vasculatures and exert anti-tumoral effect, therefore provides a potentially novel strategy for tumor immunotherapy.
Keywords: Endoglin aptamer, IP-10, nanocarriers, tumor vasculatures
Introduction
The capability of tumor antigen-specific cytotoxic T lymphocytes (CTLs) in targeting and killing tumor cells, have gained much attention [1-3]. Two strategies used in anti-tumor immunotherapy, respectively based on in vivo activation of tumor-specific CTLs [4] and in vitro activation of autologous or allogeneic immune cells [5], have been proposed. In the latter approach, also known as adoptive cellular immunotherapy, activated immune cells are transfused back into the patient after in vitro induction and proliferation. However, the amount of tumor-specific CTLs that could reach the tumor site in vivo is limited. Also, the anti-tumor response of CTLs was found to be weakened, perhaps due to tumor's ability to escape the immunologic surveillance [6-7]. Efficient eradication of tumor cells remains a huge challenge in clinical application. Thus, developing a new approach to recruit sufficient tumor-specific CTLs with enhanced tumor-targeting and -killing efficacy, may provide a strategy for effective anti-tumor immunotherapy.
Interferon-inducible protein-10 (IP-10), also known as C-X-C motif chemokine ligand (CXCL) 10, one of the CXC chemokines, shows strong anti-tumor activity [8-10] and potent inhibition of tumor angiogenesis [11]. IP-10 plays a role by chemoattracting CXCR3+ immune cells, especially CD8+ T cells (CTLs), and promoting activation and proliferation of these cells in the tumor sites [12, 13]. Considering the strong chemotactic functions of IP-10 in gathering tumor-specific CTLs, it is important to develop a strategy to ensure the sufficient IP-10 expression in the tumor environment.
Endoglin (ENG), a homodimeric transmembrane glycoprotein, is known as a component of the transforming growth factor-β (TGF-β) receptor complex. ENG has been reported to be highly expressed on the surface of tumor neovascular endothelial cells and display a homogeneous expression in tumor tissues due to the endothelial cell-derived tumor angiogenesis [14-16], on the other hand, it is rarely expressed on normal blood vessels except for the umbilical cord of newborns [17]. By targeting the neovascular tissue of tumors, ENG avoids the usual off-target effects caused by heterogeneity of tumor-specific antigens that typically show continual genetic variations [18-20]. Therefore, ENG is projected to be a suitable marker of tumor vasculature, as well as an attractive target for anti-tumor therapy.
Among packaging materials for drug delivery, liposome has several advantages, for example, a similar structure to the cell membrane which grants them good biocompatibility, a large surface area, and lack of cytotoxicity [21], etc. Several studies have demonstrated that engineering of liposomes with systematically varied properties by multiple ligands, could thereby improve their in vivo functions [22, 23]. The known ligands used for such purposes include aptamers, proteins, peptides, antibodies, and antibody fragments [8, 24]. Aptamers are synthesized single-stranded DNA or RNA oligonucleotide molecules with distinct 3D structures in the designed purpose to mimic properties of antibodies, and have recently emerged as reliable and promising class of targeted ligands with excellent potential for diagnostic and therapeutic applications [25-27]. Aptamers have several advantages over protein antibodies, including very small molecular weight (5-40 kDa), rapid plasma clearance, non-immunogenicity, easy tissue penetration, high binding affinity and specificity, and the ease of synthesis [28]. Aptamers also display remarkable stability under various pH and temperature conditions, as well as in a wide variety of organic solvents [29, 30]. Moreover, the mass production of aptamers is simpler and cost-effective, compared to that of proteins. These advantages make aptamers more desirable for targeted drug delivery that rival antibodies in clinical therapeutics [31-33].
In the present study, we aimed to develop a new cancer immunotherapy strategy, in which ENG-Apt/mIP-10-LP nanocapsules were constructed by combining the ENG-Apt with liposomes containing the mIP-10 plasmid. We characterized the structure, and assessed their binding specificity to target cells in vitro, as well as their abilities to recruit both endogenous and exogenous CTLs targeting the tumor vasculature in vivo. To demonstrate the therapeutic efficacy for ENG-Apt/mIP-10-LP nanocapsules, a series of in vivo antitumor experiments were carried out in melanoma-bearing mice models. The mechanisms were then discussed.
Materials
POPC, cholesterol, DSPE-PEG2000, DDAB, and DSPE-PEG2000-maleimide were purchased from Avanti Polar Lipids Co., and α-tocopherol and β-cysteamine were purchased from Sigma-Aldrich Co. (St Louis, MO, USA).
The oligonucleotides of ENG-Apt was synthesized by Shanghai Sangon Ltd., Co. (Sangon, Shanghai, China) according to the gene sequence of ENG-Apt and sequence of T6 Tag (5'CCCCCGATGCTTTCGCTTTTCCTTTCGCTTTTGTTCGCTTCGTCCCTGCTTCCTTTCTTG-3'-T6-HS) sequence with FITC-labeled or unlabeled at the 5' end. The plasmid GV 143 mIP-10 containing an enhanced green fluorescent protein (EGFP) reporter was constructed by ChemGene Co. (Shanghai, China).
The 180-188 residues of H-2Kb restricted tyrosinase-related protein 2 (TRP2, peptide sequence: SVYDFFVWL) were synthesized by GenScript Inc. (GenScript, Nanjing, China). Kb-TRP2 monomers and TRP2 tetramer-PE were synthesized by Beijing KuangBo Biological Technology Co. (KuangBo, Beijing, China). The ENG Apt was synthesized by Shanghai Sangon Ltd., Co. (Sangon, Shanghai, China). CD8+ T magnetic bead sorting kit (Invitrogen, Carlsbad, CA, USA) and CD105 MultiSort Kit(PE), mouse (Miltenyi Biotec, Germany) were used.
Cells and cell culture
C57BL/6 mouse-derived melanoma cell line B16 and 293T cells were purchased from the Shanghai Institute of Cell Biology, Chinese Academy of Sciences. 293T-mE cells (293T cells stably expressing CD105) were generated by Bioladder Co. (Shanghai, China). B16 cells were cultured in RPMI 1640 supplemented with 10% bovine fetal serum, 1% streptomycin, and 1% penicillin at 37 °C in an atmosphere of 5% CO2. 293T and 293T-mE cells were cultured in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 μg/mL streptomycin for screening 293T-mE cells after the addition of 500 μg/mL G418.
To isolate CD105+ mouse tumor vascular endothelial cells (mTECs), tumor tissue sections (about 1 cm in diameter) were excised from C57BL/6 mice and digested by collagenase I solution (1 mg/mL) for 0.5 h at 37 °C. Then, the suspensions were passed through a cell strainer and washed with PBS. Cells were enriched by centrifugation. To obtain CD105+ T cells, individual cells were enriched and isolated using a mouse CD105 MultiSort Kit (PE) (Miltenyi Biotec, Germany). The cells were then resuspended in EGM-2MV (Clonetics,Walkersville, MD, USA) and the concentration was adjusted into 1×105/mL upon seeding into 6-well plates.
To prepare artificial antigen-presenting cells (aAPCs), 100 μg/mL of TRP2 antigen was firstly incubated with 20 μg/mL of recombinant soluble Dimeric mouse H-2Kb:Ig Fusion protein (BD Bioscience), at 37 oC overnight to generate H-2Kb:Ig-TRP2 complexes and H-2Kb :Ig-Mut1 complexes. Artificial beads (Dynabeads® M-450 Epoxy, Invitrogen, Norway) were incubated with H-2Kb:Ig-TRP2 complexes and anti-mouse CD28 monoclonal antibody (2 μg/mL) at 4 oC for 1 h to make H-2 Kb:Ig:TRP2-aAPCs.
To obtain CD8+ T cells, individual cells from C57BL/6 (H-2Kb) mice spleens were enriched and isolated using mouse CD8 Dynabeads® Flow CompTM (Invitrogen, Carlsbad, CA, USA). The cells were then resuspended in RPMI 1640 supplemented with 10% fetal bovine serum, 1% streptomycin, and 1% penicillin at 37 °C in an atmosphere of 5% CO2, and the concentration was adjusted to 2×106/mL upon seeding into 12-well plates.
To prepare TRP2-specific T cells, CD8+ T cells were co-stimulated with H-2Kb:Ig-TRP2-aAPCs (a CD8+ T cells to aAPCs ratio of 1:3 ), recombinant mouse interleukin-21 (rmIL-21, 25 ng/mL, R&D Systems, Minneapolis, MN, USA), and rmIL-15 (10 ng/mL, R&D Systems) for 2 weeks and then isolated by sorting with magnetic beads coated with TRP2 specific tetramer to obtain TRP2-positive cells.
Animals
Female C57BL/6 mice (4-6 weeks, H-2Kb, SPF grade) were purchased from Beijing Vital River Laboratory Animal Technology Co. (Vital River, Beijing, China). Female nude BALB/c mice (4-6 weeks old and weighing approximately 20 g) were purchased from the Experimental Animal Center of Guangxi Medical University. All animal experiments were performed in compliance with Guangxi Medical University animal protection and regulations.
Experimental procedures
Nanocapsules preparation and characterization
Assembly of ENG-Apt/mIP-10-LP
To obtain ENG-Apt-PEG2000-DSPE, ENG-Apt oligonucleotides and maleimide-PEG2000-DSPE were mixed under a molar ratio of 1:5, and incubated at 4 °C under nitrogen protection for 24 h in dark. PEGylated liposomes were prepared from a mixture of POPC, Chol, DSPE-PEG-2000, DDAB, and α-tocopherol at a molar ratio of 52:40:5:3:0.2. Then 200 μg of mIP-10 plasmid was added to the liposome suspension 0.4 mL by gradual dropping with 35% ethanol and 4 nmol CaCl2, the suspension was ultra-filtered in a centrifuge tube (MWCO = 7-10K), and dialyzed (50 mM Tris-HCl, 140 mM NaCl, pH = 7) to obtain IP-10 gene-carrying liposomes after five repeated cycles of freezing, thawing, and homogenization. The ENG-Apt-PEG2000-DSPE solution 6 μL was added to 200 μL liposome suspension to form ENG-Apt-decorated liposomes. The resultant solution was dialyzed in an ultrafiltration tube (MWCO=3500-5000) to remove unreacted ENG-Apt. The preparation scheme for ENG-Apt-PEG2000-DSPE is shown in Figure S1.
DiR-PEGylated liposomes were prepared from mixing POPC, Chol, DSPE-PEG-2000, DDAB, α-tocopherol, and DiR at a molar ratio of 52:40:5:3:0.2:0.5. DiR-labeled LP-mIP-10 and ENG-Apt/mIP-10-LP were then prepared following the steps for LP-mIP-10 synthesis described as above.
Morphology, size, and zeta potential measurements
The morphology of ENG-Apt/mIP-10-LP nanocapsules was examined using transmission electron microscopy (TEM) at 100 kV. Samples were suspended in deionized water droplets and embedded in a carbon-coated copper carrier and then negatively stained with 1% phosphotungstic acid for 2 min.
To measure nanocapsules size and zeta potential, samples were suspended in deionized water in 1:100 (v/v). Upon obtaining a suspension with an appropriate particle concentration and viscosity, the particle size and zeta potential were measured using a nanometer laser particle size analyzer (Malvern Instruments, Worcestershire, UK).
Electrophoretic mobility shift assay (EMSA)
Encapsulation of IP-10 phosphorylated DNA within liposomes was verified using agarose gel electrophoresis. The mIP-10-LP complex (containing 75 ng IP-10 pDNA) was purified by 1% (w/v) agarose gel electrophoresis with GoldView staining (Dongsheng Biotech, China). Band shifts upon electrophoresis in Tris-acetate-EDTA (TAE) buffer were evaluated after recording using a chemiluminescence imaging system (Bio-Rad, USA).
N/P Ratio optimization
To optimize the nitrogen/phosphate (N/P) ratio, 80 μg, 40 μg, 20 μg, 10 μg, and 5 μg of plasmids were respectively set into five tubes loaded with 100 μL of empty LP, to achieve ENG-Apt/mIP-10-LP with N/P ratio of 1:4, 1:2, 1:1, 2:1 and 4:1.
Stability analysis
ENG-Apt/mIP-10-LP (20 μL) was respectively added into high-glucose DMEM medium alone, 100% human serum alone, or high glucose DMEM containing 20% fetal bovine serum (FBS), with an incubation time frame of 10 min, 0.5 h, 2 h, 4 h, 6 h or 8 h. As the controls, mIP-10 plasmid aliquots were also respectively incubated in above three liquid conditions for 10 min, 0.5 h, 2 h, 4 h, 6 h or 8 h, then analyzed in 1% agarose gel at 100 mA electrophoresis for 30 min. The results were visualized by a chemiluminescence imager system.
Cytotoxicity detection by MTT assay
293T, 293T-mE, mTEC, and B16 cells were resuspended into 5×104/mL in appropriate medium after digestion. Then 100 μL aliquots of cell suspensions were transferred to 96-well plates and incubated in a humidified incubator at 37 °C with 5% CO2 overnight. In addition to the blank group (medium alone) and control group (cells alone), plasmid-containing nanocapsules were resuspended in 100 μL of serum-free medium. The nanocapsules-containing suspensions then were added to each experimental group (5 wells/group), according to an equal-amount elemental ratio of ENG-Apt/mIP-10-LP with different N/P ratio [1:4 (2.5 μL), 1:2 (5 μL), 1:1 (10 μL), 2:1 (20 μL), and 4:1 (40 μL)], followed by incubation for 48 h. MTT solution (5 mg/mL, 20 μL/well) was added to each well and incubated at 37 °C for 4 h in dark. The supernatant was then discarded and 150 μL/well of DMSO was added and mixed prior to absorbance measurement at 490 nm in a microplate reader.
Binding specificity of ENG-Apt/mIP-10-LP nanocapsules
The binding specificity of fluorescent labeled ENG-Apt/mIP-10-LP nanocapsules to mIP-10-LPs with 293T, 293T-mE, B16, and mTEC cells were observed under fluorescence microscopy and quantitated by flow cytometry. For morphological observation, cells were placed onto cover slips in 6-well plates at 2×105 cells/well. Fluorescein isothiocyanate (FITC) and DilC18(3) (DiI)-labeled ENG-Apt/mIP-10-LP nanocapsules, DiI-labeled mIP-10-LPs, or unlabeled ENG-Apt/mIP-10-LP nanocapsules were respectively added to the cells before incubation for 2 h at 37 °C in 5% CO2 with 95% humidity. The binding specificity of ENG-Apt/mIP-10-LP nanocapsules to mTECs, 293T-mECs, and mTECs, was also determined by ENG-Apt blocking test. Cells were additionally incubated with excess amount of unlabeled ENG-Apt for 30 min in prior, then incubated with an equimolar amount of fluorescent ENG-Apt/mIP-10-LP nanocapsules for another 2 h of incubation as above. Cell nuclei were stained with 4, 6-diamidino-2-phenylindole (DAPI) for 5 min, and the cells were then fixed with 4% paraformaldehyde for 30 min at room temperature. The fluorescence intensity within cells was observed under a fluorescence microscope (Nikon, Japan). All experiments were repeated three times.
To evaluate the expression of IP-10-EGFP after ENG-Apt/mIP-10-LP nanocapsules uptake by the cells, 293T, B16, 293T-mE and mTEC cells were cultured with FITC-labeled ENG-Apt/mIP-10-LP nanocapsules for 2 h and resuspended in 200 μL phosphate-buffered solution (PBS) after washing. Fluorescence intensity of cells in different groups was quantitatively analyzed by a fluorescence spectrophotometer. Furthermore, 293T-mEs and mTECs were cultured with DiI-labeled mIP-10-LPs or ENG-Apt/mIP-10-LP nanocapsules for 4 h, and washed with PBS to remove non-phagocytic fluorescent materials. 100 μL of 0.1% Triton X-100 was added, and then the cells were centrifuged. The cell pellet was resuspended in 200 μL of PBS, and the fluorescence intensity was detected using a fluorescence spectrophotometer which evaluated the cellular uptake of DiI dye.
mIP-10 expression in mTECs
Totally 4×105 mTECs/well were seeded in 6-well plates and allowed to proliferate overnight to reach 70% confluence. The culture medium was replaced with 1 mL medium containing ENG-Apt/mIP-10-LP nanocapsules or mIP-10-LPs for 6 h. Next, the cells were cultured for 24 h at 37 °C in 5% CO2 with 95% humidity. Treatment with GV 143 mIP-10 plasmid served as a control. EGFP expression was observed by fluorescence microscopy, and EGFP expression efficiency in each group was analyzed by fluorescence-activated cell sorting (FACSCaliburTM, BD, CA, USA) after trypsinization for 24 h.
Treatment of melanoma-bearing mice with ENG-Apt/mIP-10-LP nanocapsules
To study distribution of ENG-Apt/mIP-10-LP nanocapsules in melanoma-bearing mice, 4-week-old female inbred BALB/c athymic nude mice (SPF, 18-22 g) were randomized into 3 groups (n = 5). B16 cells (5×105 cells/mouse) were implanted via subcutaneous injection into the left armpit of mouse. To evaluate tumor growth, the volume of the resulting tumors was recorded using vernier calipers every 3 d. Tumor volume (V) was calculated according to the formula: V=0.5ab2, where “a” representing the largest diameter and “b” representing the perpendicular diameter. Once the tumors reached a volume of 500 mm3, tumor-bearing mice received one of three treatments via intravenous tail injections: 0.6 mg DilC18(7) (DiR)/kg body weight (DiR control), DiR-labeled mIP-10-LPs, and ENG-Apt/mIP-10-LP nanocapsules. The distribution of the fluorescent particles in each group was detected using an in vivo imaging system (In Vivo FX PRO, Bruker, Billerica, MA, USA) (excitation at 720 nm, emission at 790 nm) at 2 h, 6 h, 24 h, 48 h and 72 h after injection.
Treatment of melanoma-bearing mice with ENG-Apt/mIP-10-LP nanocapsules
Four- to six-week-old female C57BL/6 mice (H-2Kb, SPF level, 20-25 g) were used for the following experiment. B16 cells (5×105 cells/mouse) were inoculated into the right groin area of each mouse via subcutaneous injection. On the fifth day after the tumor cell inoculation, tiny black patches were seen around the injection sites subcutaneously. These tumor-bearing mice were then randomized into four groups (n = 5), and respectively treated with PBS, mIP-10 plasmid, mIP-10-LPs, or ENG-Apt/mIP-10-LP nanocapsules at 5 d, 8 d, 11 d, and 14 d after tumor cells inoculation via tail vein intravenous injections. The size of tumors of all animals were measured with vernier caliper every 3 d after tumor cell inoculation, followed by tumor volume calculating and animal growth curve plotting as described above. Mice were monitored daily for survival, and corresponding plots were prepared. Another 32 mice subjected to the same treatments (n = 8) were monitored to generate survival curves.
Treatment of melanoma-bearing mice with ENG-Apt/mIP-10-LP nanocapsules combined with adoptive TRP2-specific CD8+ T cells
To test the therapeutic efficacy for combination treatment of ENG-Apt/mIP-10-LP nanocapsules and TRP2-specific CD8+ T cells, additional mice were randomized into five groups (n=5). The different treatments for the groups were as follows: (1) PBS, (2) ENG-Apt/mIP-10-LP nanocapsules, (3) ENG-Apt/mIP-10-LP nanocapsules combined with CD8+ T cells, (4) TRP2-specific CD8+ T cells, and (5) ENG-Apt/mIP-10-LP nanocapsules combined with TRP2-specific CD8+ T cells. Four days after subcutaneous inoculation of tumor cells, CD8+ T cells or TRP2-specific CD8+ T cells were resuspended in PBS at 1.5×107 cells/mL (200 µL/mouse) via the tail vein injection in vivo. The next day, PBS and ENG-Apt/mIP-10-LP nanocapsules (mIP-10 plasmid, 50 µg/mouse) were also injected intravenously in the corresponding groups. Mice of different groups were treated at 5 d, 8 d, 11 d, and 14 d after tumor cells inoculation via tail vein intravenous injections, and tumor volume was measured with vernier calipers during the treatment (every 3 d as described above) to generate tumor growth curves. Another 40 mice subjected to the same treatments (n = 8) were monitored to generate survival curves.
Flow cytometric analysis
Tumors were separated from mice, washed and incubated in a solution containing collagenase type-I (200 U/mL) and DNase I (200 μg/mL) for 1 h at 37 °C. Single cell suspension of the bone marrow was prepared by blowing the morrow fluids out from the cavity by 1 mL of sterile PBS and pipetting. Splenocytes were isolated from the mouse spleen by grinding using disposal pestles over copper mesh. Red blood cells were removed from the cell suspensions using Erythrocyte Lysate buffer (Solarbio, Beijing, China) according to the manufacture's instruction. Then, the cell suspension from either resource above were passed through a cell strainer and washed with sterile PBS. Cell density was adjusted into 1 × 106 cells/100 μL for fluorescence-activated cell sorting (FACS) analysis.
All fluorescently labeled antibodies were purchased from eBioscience. The following antibodies were used for phenotypic analysis: CD11b-FITC, Ly-6 (Gr-1)-PE, CD4-FITC, CD25-PECy5, FoxP3-PE, CD8α-FITC/PE, CD8β-PerCPCy5.5. Cells isolated from bone marrow, spleen, and tumor tissues of mice were adjusted to a concentration of 1×107 cells/mL. Percentages of CD11b+Gr1+ cells in the bone marrow, spleen, and tumor tissues, along, CD4+CD25+FoxP3+ T regulatory cells (Tregs) in the spleen and tumor tissues, as well as, TRP2CD8+ T cells in tumor-infiltrating lymphocytes were analyzed via flow cytometry. All cells were incubated at 4 °C in the dark for 30 min.
Anti-mouse CD4-FITC (0.5 μL) and anti-mouse CD25-PE-Cy5 (0.25 μL) antibodies were added to spleen cells and tumor-infiltrating lymphocytes (TILs) followed by incubation in the dark for 30 min. Once the cells were ruptured, anti-FoxP3-PE (2 μL) was added and incubated at 4 °C for an extra 30 min. Subsequently, cells were washed twice for flow cytometric analysis.
Detection of IFN-γ secretion by ELISPOT assay
TILs were collected for ELISPOT analysis after the previously described treatments. Briefly, 96-well plates containing polyvinylidene difluoride (PVDF) films were coated with mouse anti-IFN-γ antibody, and incubated with RPMI 1640 medium (Gibco, USA) for 10 min at room temperature. The supernatant was discarded, and 2×105 of collected cells were seeded into each well and incubated with TRP2 peptide-plus B16 cells for 24 h at 37 °C. Subsequently, the samples were washed and 100 μL of biotinylated antibodies were added to each well followed by 1 h of incubation at 37°C. Then, 100 μL (2.5 μg) horseradish peroxidase (HRP)-avidin was added to each well and incubated for another 1 h at 37°C. In the last step, 100 μL of dyes (ELISPOT kit, Dakewe Biotech Company, China) were added to each well and incubated for 25 min in the dark. The plates were then read by a CTL-Immunospot® Analyzer (CTL, Shaker Heights, OH, USA), and the data was analyzed using Immunospot 5.0 Pro software.
Immuno-histochemical staining
Excised mouse tumor tissues were fixed in paraffin and prepared for histological examination. After dewaxing, 4 μm paraffin-embedded sections were incubated with 3% H2O2 for 10 min at room temperature and then with 5% bovine serum albumin for 30 min at 37 °C. The sections were then stained with anti-mouse proliferating cell nuclear antigen expression (PCNA) monoclonal antibody (1:200 dilution, Boster, Wuhan, China), rat anti-mouse CD31 antibody (1:50 dilution, Abcam), and rabbit anti-mouse CXCL10/IP-10 antibody (1:100 dilution, Biosynthesis) and kept at 4 °C overnight. After three washes with PBS, the sections were stained with HRP-labeled goat anti-mouse, goat anti-rat, or goat anti-rabbit IgG secondary antibodies (Zhongshan Golden Bridge, China) and incubated for 30 min at 37 °C. Sections treated as described without the primary antibody were used as negative controls. The sections were counterstained with DAB, examined by microscopy (400×), and analyzed using Image-Pro Plus® software (Media Cybernetics).
Detection of tumor cell apoptosis by TUNEL assay
TUNEL assay was performed to detect apoptosis in paraffin-embedded sections of harvested tumor tissues. In situ cell death detection kit, Fluorescein (Roche Diagnostics, Hoffman-La Roche, Basel, Switzerland) was used, and 50 μL reaction mixture was applied to each slide according to the kit instructions before incubation at 37 °C for 60 min. Nuclei were counterstained with DAPI (0.5 μg/mL) at room temperature in the dark for 30 min. Five different regions on each slide were selected for photographing (n = 5 slides per group), and automatic analysis software (Image-Pro Plus 5.0) was used to count stained cells.
In situ tetramer staining (ISTS)
Fresh tumor tissues were embedded in optimal cutting temperature (OCT) medium and fixed in ice-cold acetone for 15 min; frozen sections were cut at a thickness of 6 μm. After three washes with PBS, the sections were incubated with a mixture of TRP2 tetramer-PE (5 μL), CD8-FITC (3 μL), fetal bovine serum (2 μL), and PBS (90 μL) at 37 °C for 30 min. Posterior to three additional washes with PBS, 0.5 μg/mL DAPI was applied for 30 min at room temperature to stain the nuclei. The slides were mounted with an antifade agent and observed under fluorescence microscope.
Statistical analysis
All data was analyzed using SPSS 16.0 statistical software (SPSS Inc, Chicago, IL, USA) and expressed as mean values. Differences between data from the experimental and control groups were analyzed using one-way analysis of variance (ANOVA) to determine statistical significance. Survival rates were calculated using the Kaplan-Meier method and analyzed using the log-rank test. A p value≤ 0.05 was considered statistically significant.
Results
Characterization of ENG-Apt/mIP-10-LP nanocapsules
ENG-Apt-PEG2000-DSPE was firstly synthesized via thiol-maleimide “click” reaction according to the schematic shown in Figure S1. TEM images reveal that the prepared nanocapsules possessed a uniform particle size as well as high dispersion characteristics. The cationic liposome presented a smooth surface and spherical appearance. The size of ENG-Apt/mIP-10-LP nanocapsules, as analyzed by dynamic light scattering, was uniform at a diameter of 145 nm approximately, which was slightly larger than the diameter of LP (shown as Figure 1 A, B). Figure 1C shows the average particle size of the nanocapsules decreased as the ratio of N/P was adjusted from 1:4 to 1:1 and the zeta potential gradually increased. With an N/P ratio of 1:1, the particle size of ENG-Apt/mIP-10-LP nanocapsules was 145 nm and the zeta potential was approximately -17 mV.
Characterization of ENG-Apt/mIP-10-LP nanocapsules: Representative TEM images and size distributions of (A) LPs and (B) ENG-Apt/mIP-10-LP nanocapsules. Scale bar on TEM images is 200 nm (upper) and 100 nm (blow). N/P ratio of LPs and ENG-Apt/mIP-10-LP nanocapsules is 1:1.(C) shows the particle sizes and zeta potentials of different types of nanoparticles and ENG-Apt/mIP-10-LP nanocapsules prepared with different ratios of N/P. (D) Detection of mIP-10 expression after treatment with mIP-10-LPs or ENG-Apt/mIP-10-LP nanocapsules in 1% agarose gel electrophoresis (Lane 1: mIP-10, Lane 2: mIP-10-LPs, Lane 3: ENG-Apt/mIP-10-LP, Lane 4: mIP-10-LPs + Triton X-100, Lane 5: ENG-Apt/mIP-10-LP + Triton X-100, Lane 6: LP + Triton X-100, Lane 7: mIP-10 + Triton X-100). (E) Identification of ENG-Apt binding to LP in 3% gel retardation assay analysis of ENG-Aptamer binding to LP surface (Lane 1: ENG-Aptamer, Lane 2: LP, Lane 3: LP + ENG-Aptamer, Lane 4: unpurified ENG-Apt-LP, Lane 5: purified ENG-Apt-LP).
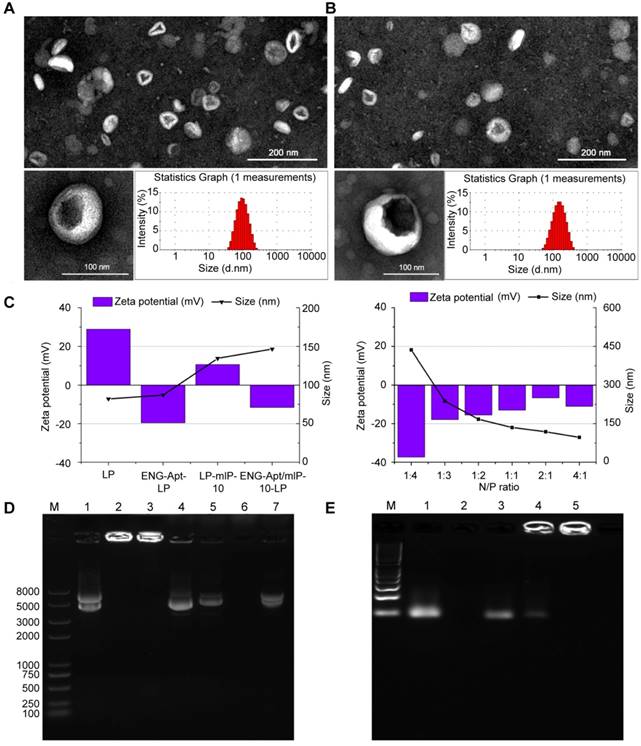
Similarly, EMSA result demonstrated that the LPs eliminated the band for the GV 143 mIP-10 plasmid in the gel with a single band appearing at the molecular weight of 5000 (Figure 1D, line 1). A bright band for ENG-Apt/mIP-10-LP nanocapsules accumulated around the inner walls of the gel wells without any further migration. The appearance of the same band after disruption of the LP membrane with Triton X-100 (Figure 1D, line 5) indicated that the GV 143 mIP-10 plasmid was encapsulated in the LPs.
ENG-Apt/mIP-10-LP presented the same band as GV143 mIP-10 (Figure S2, line 1) when the N/P ratio was changed from 1:4 to 1:2 (Figure S2, line 2-4). When the N/P ratios were 1:1, 2:1 and 4:1, ENG-Apt/mIP-10 retardation bands appeared around the inner walls of sampling wells (Figure S2, line 5-7). These results further proved that when the N/P ratio was 1:1, the liposome could successfully load and encapsulate the plasmid GV143 mIP-10 with an encapsulation rate of 77.8%.
To confirm the successful binding of ENG-Apt to the surface of the liposome, 3% agarose gel electrophoresis was utilized (Figure 1E). ENG-Apt showed clear bands on gel electrophoresis (Figure 1E, line 2), the liposome by itself was not able to display bands on gel (Figure 1E, line 3). The direct mixture of liposome and ENG-Apt showed the same electrophoresis bands as ENG-Apt (Figure 1E, line 4). Before and after ENG-Apt-LP dialysis (Figure 1E, line 5 and 6), there were obvious bright bands in around the walls area of the gel wells. Compared to ENG-Apt-LP before dialysis, the weak gel bands of ENG-Apt-LP disappeared after dialysis (Figure 1E, line 6), which indicates that the free unbound ENG-Apt was successfully removed.
Regarding with the stability test, ENG-Apt/mIP-10-LP stored in DMEM medium at 37 °C for 10 min, 0.5 h, 1.5 h, 4 h, 6 h or 8 h was examined, and observed with almost zero plasmid release. With similar settings, ENG-Apt/mIP-10-LP incubated with human serum and high glucose DMEM containing 20% FBS was observed to hold a low release rate. Additionally, after purification by ultrafiltration and precipitation, the release rate turned lower than 30% after observation for 8 h, indicating good stability (Figure S3).
ENG-Apt/mIP-10-LP nanocapsules showed good biocompatibility based on MTT assay results, demonstrating relatively low toxicity upon incubation with 293T-mE, 293T, B16, and mTEC cells with an 'almost 100 %' cellular viability (Figure S4).
Specific binding of ENG-Apt/mIP-10-LP nanocapsules to mTECs
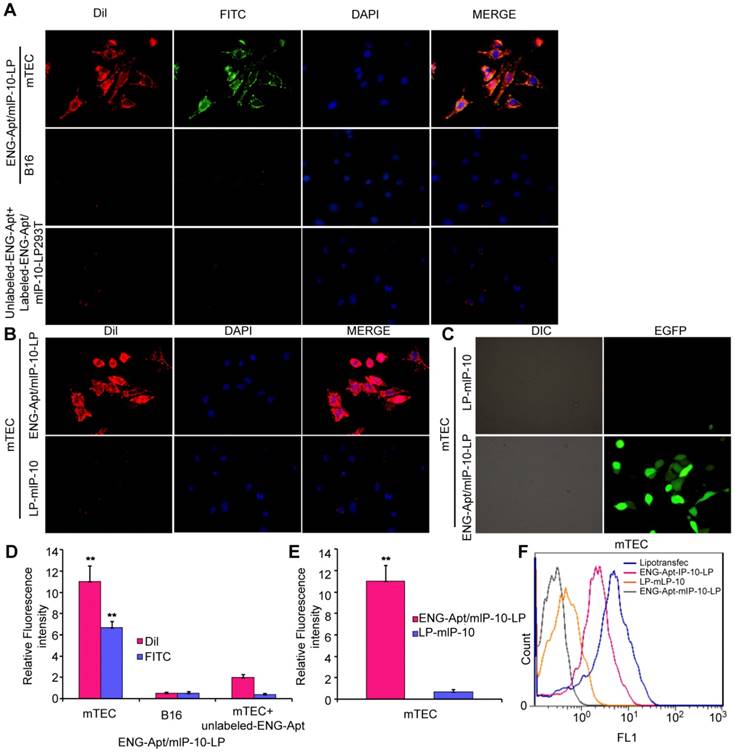
After incubation with FITC/DiI-labeled ENG-Apt/mIP-10-LP nanocapsules, all mTECs showed strong fluorescence intensity, while B16 cells showed a comparably weaker signal (Figure 2A, upper and middle panels; Figure 2D). Similarly, when free ENG-Apt was pre-added in the culture medium, and then FITC/DiI-labeled ENG-Apt/mIP-10-LP nanocapsules were incubated with mTECs, a weak fluorescence was observed in mTECs. This could be due to the binding of the pre-added free ENG-Apt with ENG receptors thereby inhibiting the binding of ENG-Apt/mIP-10-LP nanocapsules and mTECs (Figure 2A, lower panel; Figure 2D). Evaluating the targeting effects of ENG-Apt to mTECs, when ENG-Apt/mIP-10-LP and mIP-10-LPs were incubated with mTECs separately, ENG-Apt/mIP-10-LP nanocapsules were found to be internalized within the mTECs more than mIP-10-LPs (Figure 2B, E). Quantitative analysis of the fluorescence intensities of mTECs showed that in comparison to mIP-10-LPs, approximately 16 times more ENG-Apt/mIP-10-LP nanocapsules bound to mTECs, which indicates the excellent targeting effect of ENG-Apt to mTECs (Figure 2E). Similarly, the binding of ENG-Apt/mIP-10-LP nanocapsules with 293T-mEs was significantly stronger than that with 293T cells (Figure S5). These results suggest that ENG-Apt/mIP-10-LP nanocapsules could specifically target the mTECs or 293T-mEs. Upon exposure of mTECs to nanocapsules encapsulating an EGFP vector, IP-10-EGFP expression was significantly greater in mTECs exposed to ENG-Apt/mIP-10-LP nanocapsules compared to those exposed to mIP-10-LPs (Figure 2C). Flow cytometric analysis also indicated that the transfection efficiency of vector delivered by ENG-Apt/mIP-10-LP nanocapsules was close to that of Lipofectamine 2000 (Figure S6), and superior to that with the non-targeting mIP-10-LPs (Figure 2F).
ENG-Apt/mIP-10-LP nanocapsules target mTECs to increase mTEC expression of mIP-10. (A, B) ENG-Apt/mIP-10-LP nanocapsules specifically bind to mTECs through ENG receptors as is evident by the fluorescence of DiI-labeled liposomes (red), DAPI-stained nuclei (blue), and FITC-labeled ENG-Apt (green), at a magnification of 400×. (C) the fluorescence intensity in mTECs after binding of ENG-Apt/mIP-10-LP nanocapsules is 16 times greater than that with mIP-10-LPs. (D) the fluorescence intensity analysis in mTECs and B16 after binding of ENG-Apt/mIP-10-LP nanocapsules. (E) Fluorescence intensity analysis showing ENG-Apt/mIP-10-LP nanocapsules has high binding rate with mTECs. (F) Flow cytometric analysis showing ENG-Apt/mIP-10-LP nanocapsules has high binding rate with mTECs rather than the controls. Mean ± SE of three independent experiments is shown. ** p < 0.01.
In vivo distribution of ENG-Apt/mIP-10-LP nanocapsules in melanoma-bearing mice
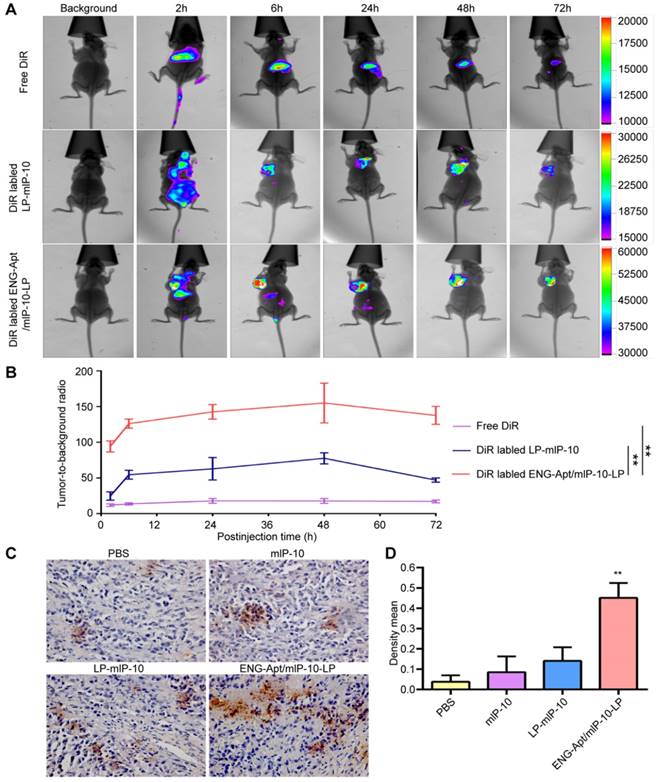
The distribution of DiR-labeled nanocapsules in various organs and tissues of melanoma-bearing mice in vivo, was detected using a fluorescence imaging system. ENG-Apt/mIP-10-LP nanocapsules were rapidly deposited at tumor sites with obvious fluorescence appearing within 6 h and reaching peak intensity at 48 h. By contrast, injected DiR dye molecules were rapidly migrated in vivo, as indicated by the decreased fluorescence after 2 h. The fluorescence intensity of ENG-Apt/mIP-10-LP nanocapsules at tumor sites was higher than that of the control mIP-10-LPs at each time point. At 48 h after injection, the fluorescence intensity of ENG-Apt/mIP-10-LP nanocapsules at tumor sites was 2.5 times greater than that of mIP-10-LPs, indicating that a large number of ENG-Apt/mIP-10-LP nanocapsules continued to be present in tumor tissues whereas mIP-10-LPs had been removed (Figure 3A, B). Similarly, anti-mIP-10 antibody staining showed that IP-10 expression in tumor tissues after treatment with ENG-Apt/mIP-10-LP nanocapsules was significantly greater than that after treatment with the control mIP-10-LPs (Figure 3C, D).
ENG-Apt/mIP-10-LP nanocapsules targeted to tumor tissues result in high expression of IP-10 protein. B16 cells (5×105 cells/mouse) were implanted via subcutaneous injection in the left armpit of 4-week-old female inbred BALB/c athymic nude mice. Once the tumors reached a volume of 500 mm3, tumor-bearing mice received one of three treatments via intravenous tail injections: 0.6 mg DilC18(7) (DiR)/kg body weight (DiR control), DiR-labeled mIP-10-LPs, and ENG-Apt/mIP-10-LP nanocapsules. The distribution of the fluorescent particles in each group was detected using an in vivo imaging system (In Vivo FX PRO, Bruker, Billerica, MA, USA) (excitation at 720 nm, emission at 790 nm) at 2, 6, 24, 48 and 72 hours after injection. (A) In vivo distribution of DiR-labeled nanocapsules in tumor-bearing mice. (B) Relative fluorescent intensity (RFI) of nanocapsules in tumor tissues, RFI in tumor tissues 48 h after delivery of DiR-labeled ENG-Apt/mIP-10-LPs is 2.5 times more than that achieved with mIP-10-LPs. (C) The expression of mIP-10 protein in tumor tissues of various groups detection by immunohistochemical staining (magnification, 400×). (D) Statistical quantification of mIP-10 protein expression in tumor tissues of various groups. The mean ± SE of three independent experiments is shown. ** p < 0.01 for the comparison of the ENG-Apt/mIP-10-LP group to control groups.
Effects of ENG-Apt/mIP-10-LP nanocapsules combined with TRP2CD8+ T cells in vivo
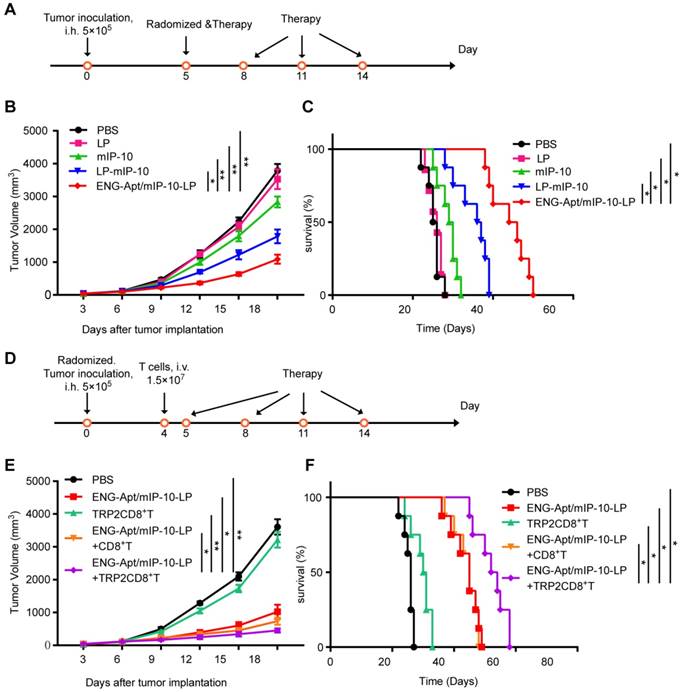
HE staining showed that the ENG-Apt/mIP-10-LP had no inflammatory cell infiltration and toxicity damage to the heart, liver, spleen, lung and kidney of mice (200×) (Figure S7). Comparison of the survival curves for melanoma-bearing mice treated with ENG-Apt/mIP-10-LP nanocapsules or mIP-10-LPs indicated that ENG-Apt/mIP-10-LP nanocapsule delivery resulted in significantly greater inhibition of tumor growth and considerably longer survival than non-targeting mIP-10-LPs delivery (Figure 4A, B, C). Additionally, TRP2CD8+ T cells combined with ENG-Apt/mIP-10-LP nanocapsules treatment demonstrated remarkably enhanced anti-tumor efficacy, conversely, treatment with TRP2CD8+ T cells displayed weak anti-tumor effects. These results translated into significantly decreased tumor growth and prolonged survival time of melanoma-bearing mice receiving the combination treatment (Figure 4D, E, F).
Tumor volume and survival time in melanoma-bearing mice in different treatment groups. (A, B, C) Anti-tumor effect of the treatment with ENG-Apt/mIP-10-LP nanocapsules in melanoma-bearing mice. (A) Experiment design for the treatment of melanoma-bearing mice with ENG-Apt/mIP-10-LP nanocapsules. B16 cells (5×105 cells/mouse) were inoculated into the right groin area of the C57BL/6 mice via subcutaneous injection. On the fifth day after the tumor cell inoculation, these tumor-bearing mice were then randomized into four groups (n=5), and respectively treated with PBS, mIP-10 plasmid, mIP-10-LPs, or ENG-Apt/mIP-10-LP nanocapsules at 5, 8, 11, and 14 days after tumor cells inoculation via tail vein intravenous injections. Another 32 mice subjected to the same treatments (n=8) were monitored to generate survival curves. (B) Showing tumor growth curves in mm3 among groups over the days after the treatment (n=5). (C) showing the survival rates of mice among groups over the days after the treatment (n=8). * p < 0.05, ** p <0.01 for the comparison of survival percentage in the ENG-Apt/mIP-10-LP group and control groups. (D, E, F) Anti-tumor effect of combination treatment with ENG-Apt/mIP-10-LP nanocapsules and adoptive TRP2CD8+ T cells in melanoma-bearing mice. (D) Experiment design for the combination treatment of melanoma-bearing mice with ENG-Apt/mIP-10-LP and TRP2-specific CD8+ T cells. C57BL/6 mice were randomized into five groups (n=5). Four days after subcutaneous inoculation of B16 cells, CD8+ T cells or TRP2-specific CD8+ T cells were resuspended in PBS at 1.5×107 cells/mL (200 µL/mouse) via the tailvein injection in vivo. The next day, PBS and ENG-Apt/mIP-10-LP nanocapsules (mIP-10 plasmid, 50 µg/mouse) were also injected intravenously in the corresponding groups. Mice of different groups were treated at 5, 8, 11, and 14 days after tumor cells inoculation via tail vein intravenous injections. Another 40 mice subjected to the same treatments (n=8) were monitored to generate survival curves. (E) showing tumor growth curves in mm3 among groups over the days after the treatment (n=5). (F) showing the survival rates of mice among groups over the days after the treatment. * p < 0.05, ** p < 0.01 for the comparison of survival percentage between the ENG-Apt/mIP-10-LP + TRP2CD8+ T group and control groups. Mean ± SE of three independent experiments is shown.
ISTS and Flow cytometric analyses were to determine the presence of TRP2CD8+ T cells among TILs in melanoma-bearing mice after the treatments. The amount of TRP2CD8+ T cells at tumor sites in mice treated with both ENG-Apt/mIP-10-LP nanocapsules and TRP2CD8+ T cells was significantly greater compared to that of the other treatments. (Figure 5A, B, D). Furthermore, we observed that, in addition to a large number of TRP2CD8+ T cells infiltrating the tumor tissue after the combination treatment, the number of CD8+ T cells at these sites also increased (Figure S5A-B, Figure S8A-B).
Combination of ENG-Apt/mIP-10-LP nanocapsules and TRP2CD8+ T cells increase the number and activity of tumor cell-specific CD8+ T cells in tumor tissues. (A) Frequencies of TRP2CD8+ T cells in tumor tissues of various groups detected by flow cytometry. (B) TRP2CD8+ T cells infiltration in tumor tissues of various groups detected by ISTS. Fluorescence images of tissue sections stained with CD8+ T in green, TRP2 tetramer in red and cell nuclei were labeled with DAPI (blue). (C) TRP2-specific IFN-γ-secreting cells among TILs detected by immunospot staining. (D) Requencies of TRP2CD8+ T cells among TILs. (E) Frequencies of CD8+ T cells among TILs. (F) The numbers of TRP2-specific IFN-γ-positive cells among TILs. The mean ± SE of three independent experiments is shown. ** p < 0.01 for the comparison of the ENG-Apt/mIP-10-LP + TRP2CD8+ T cell group to control groups. Mean ± SE of three independent experiments is shown.
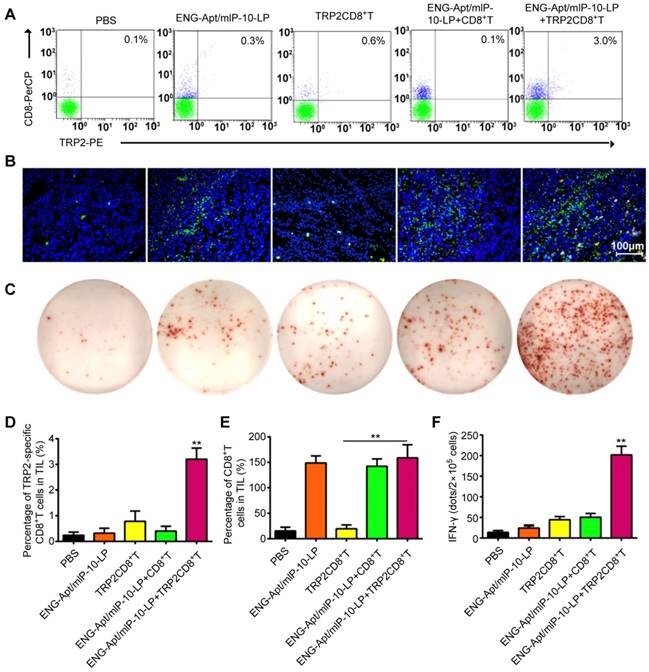
To assess the activity of TRP2CD8+ T cells in tumor tissues, the number of TRP2-specific IFN-γ-secreting T cells among TILs of each group was measured by ELISPOT assay. The dot counts of group co-treated with ENG-Apt/mIP-10-LP nanocapsules and TRP2CD8+ T cells, were significantly higher than that treated with only TRP2CD8+ T cells (Figure 5C, F, Figure S8C-D). Consistently, the results of intracellular immunochemical staining corroborated that the combination treatment resulted in a significant increase of TRP2-specific, IFN-γ-secreting CD8+ Tcells.
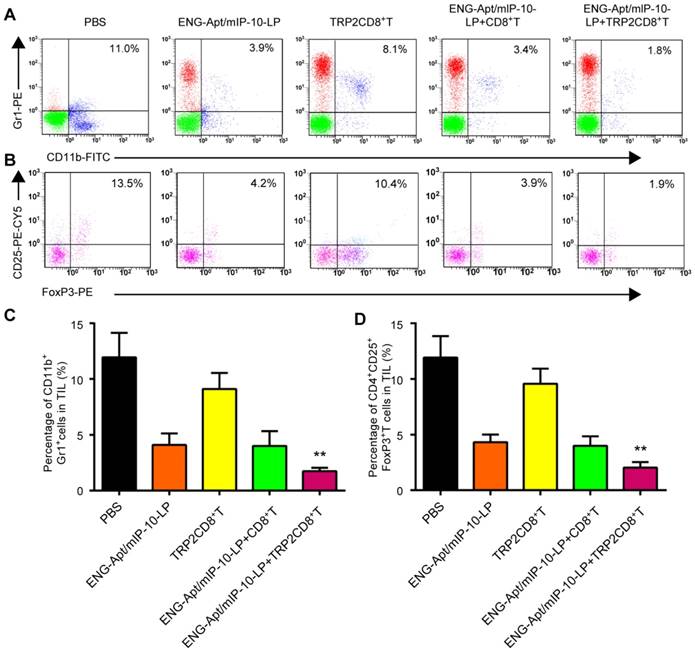
Flow cytometry was used to analyze the numbers of MDSCs and Tregs that negatively regulate the immune responses in tumor-bearing mice. After combined treatment with ENG-Apt/mIP-10-LP nanocapsules and TRP2CD8+ T cells, the number of CD11b+Gr1hi MDSCs in tumor tissues remarkably decreased rather than in the control group, associated with a rise of Gr1 expression level. The percentage of CD4+CD25+FoxP3+ Tregs among TILs was also significantly decreased (Figure 6). Similarly, treatment with ENG-Apt/mIP-10-LP alone led to considerably reduced number of MDSCs and Tregsin the tumor-bearing mice, compared to the treatment with non-targeting mIP-10-LPs (Figure S9).
Combination treatment with ENG-Apt/mIP-10-LP and TRP2CD8+ T cells reduced the amounts of MDSCs and Tregs in the tumor microenvironment. The flow cytometric analyses for MDSC and Treg showed that the percentage of CD11b+Gr1+ MDSC in single cells of tumor tissue of mice treated with ENG-Apt/mIp-10-LP combined with TRP2CD8+ T cells was significantly reduced and significantly lower than that of other control groups. The percentage of CD4+CD25+FoxP3+ Tregs in TIL was also significantly reduced. (A) The percentages of MDSCs in tumor tissues detected by flow cytometry. (B) The percentages of Tregs in tumor tissues; results with CD4+ gating are shown. (C) The quantified numbers of MDSCs in tumor tissues. (D) The quantified numbers of Tregs in tumor tissues. The mean ± SE of three independent experiments is shown. ** p < 0.01 for the comparison of ENG-Apt/mIP-10-LP + TRP2CD8+ T cell group to control groups.
Anti-tumor mechanism of the combination treatment
To explore the mechanisms responsible for the inhibition of melanoma growth in mice after ENG-Apt/mIP-10-LP nanocapsules and TRP2CD8+ T cells combination treatment, the number of PCNA-positive cells was detected by immunohistochemical (IHC) staining. The results showed that the number of PCNA-positive cells in tumor tissues was 4 times lower in the combination treatment group than that in the PBS group, whereas no significant difference in the number of PCNA-positive cells was observed between the ENG-Apt/mIP-10-LP and the ENG-Apt/mIP-10-LP + CD8+ T cell groups (Figure 7A, D). Notably, the numbers of PCNA-positive cells after treatment with ENG-Apt/mIP-10-LP were significantly lower than those after mIP-10-LP treatment (Figure S10A, C).
Combination treatment with ENG-Apt/mIP-10-LP and TRP2CD8+ T cells inhibits tumor cell proliferation, promotes tumor cell apoptosis, and suppresses tumor angiogenesis. (A) Detection for Proliferating cell nuclear antigen expression (PCNA) by IHC staining, at a magnification of 400×. (B) Results of tumor apoptosis detection by TUNEL assay; nuclei stained blue, positive cells stained green, at 200×. (B) Tumor microvascular densities detected by CD105 monoclonal antibodies, at 100×. (D) Statistical quantification of PCNA numbers in various groups. (E) Statistical quantification of tumor apoptosis numbers in various groups. (F) Statistical quantification of tumor microvasculature number in various groups. The mean ± SE of three independent experiments is shown. * p < 0.05 and ** p < 0.01 for comparison of ENG-Apt/mIP-10-LP + TRP2CD8+ T cells group to other controls.
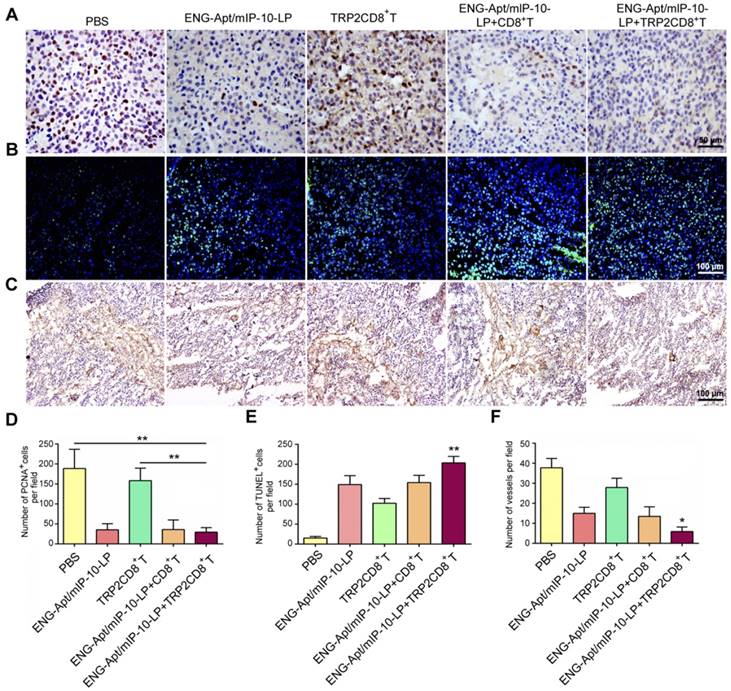
Upon treatment of melanoma-bearing mice with ENG-Apt/mIP-10-LP nanocapsules and TRP2CD8+ T cells, the increase of cellular apoptosis in tumor tissues was notably higher than that of control groups (Figure 7B, E; Figure S10B-C). IHC staining showed significantly fewer dense micro-vessels expressing ENG in tumor tissues of mice treated with ENG-Apt/mIP-10-LP nanocapsules and TRP2CD8+ T cells (Figure 7C, F). IHC staining of tumor tissues for CD31 expression illustrated that the tumor vascular density after ENG-Apt/mIP-10-LP nanocapsules treatment was remarkably lower, which was positively correlated with mIP-10 expression. Tumor vascular density was also lower than that in mIP-10-LP group and 5-6 times lower than the densities in the PBS and mIP-10 groups (Figure S11).
Discussion
Blood vessels play a critical role in the evolution and progression of tumors [8, 18, 20, 34] such that tumor angiogenesis indirectly reflects tumor growth. A few studies have investigated potential strategies for attracting CTLs to the tumor environment within the context of tumor immunotherapy [2, 3]. In our previous study [36], we found that after the treatment of melanoma-specific cytotoxic CD8+CD28+ T lymphocyte combined with FA-CS-mIP-10, which specifically targeting the tumor cells, the percentage of CXCR3+CD8+ T cells was showed to increase. However, we haven't observed any melanoma-specific cytotoxic CD8+CD28+ T lymphocyte in tumor tissues by in situ tetramer staining (ISTS). In order to increase the number of melanoma-specific cytotoxic CD8+ T lymphocyte in tumor tissues, the methodology requires optimization and improvement. Here, we creatively designed and developed an ENG-Apt/mIP-10-LP nanocapsule delivery to target tumoral neovasculatures. This is the first report of a novel aptamer-based nanosystem platform in the field of T cells therapy. And this is also the first time that ENG-Apt/mIP-10-LP nanocapsules driving melanoma-specific cytotoxic CD8+ T cells in tumor tissues was observed by ISTS method.
In our study, we used ENG, a specific surface antigen of tumor neovascular endothelial cells [14, 15], as the targeted media for nanoparticle delivery. IP-10 plays an important role in the chemotaxis of activated T lymphocytes via CXCR3 [8], thereby facilitating the regression of tumor angiogenesis. To combine these advantages, surface ENG aptamer modified liposomal nanocapsules encapsulating the IP-10 plasmid (ENG-Apt/mIP-10-LP), were constructed and characterized. After specific binding to tumoral neovascular cells, the plasmid was induced intracellularly, and their ability to recruit tumor antigen-specific CTLs and form a CTL-enriched zone around tumor endothelial cells was evaluated in vitro and in vivo.
In vitro phagocytosis experiments demonstrated that ENG-Apt/mIP-10-LP nanocapsules could specifically bind to and be uptaken by mTECs, which accomplished the delivery of IP-10 plasmid and therefore induced the expression of IP-10 protein by mTECs. The higher expression of IP-10 by mTECs exposed to ENG-Apt/mIP-10-LP nanocapsules compared to those exposed to control IP-10-LPs indicated that the recognition of cell surface receptors by the ENG-Apt effectively enhanced delivery of the exogenous gene to the targeted cells. Therefore, our study indicated that modification of nanocapsules with ENG aptamer which is specifically targeting tumoral neovasculatures, has the potential to improve the clinical efficacy of cancer immunotherapy.
There was a preferential accumulation of ENG-Apt/mIP-10-LP in tumor tissues of melanoma-bearing mice as evidenced by in vivo imaging analysis. In comparing with mIP-10-LPs, the addition of ENG-Apt in ENG-Apt/mIP-10-LP nanocapsules, could significantly reduce the distribution of the nanocarriers in non-tumor tissues, thereby decreasing the toxicity of the immunotherapeutic agent in the general circulation. Similarly, IP-10 expression in tumor tissues of mice treated with ENG-Apt/mIP-10-LP nanocapsules was significantly greater than that in mice treated with mIP-10-LPs as shown by IHC staining. These findings strongly suggest that the ENG-Apt/mIP-10-LP nanocapsules can specifically recognize tumor endothelial cell surface receptors for targeted delivery and subsequent induction of IP-10 protein expression in tumor-bearing mice. Some studies have indicated that certain size of liposome (20-200 nm) could be easier to accumulate into tumors rather than healthy tissues through the enhanced permeability and retention effect (EPR) [37, 38]. In our study, the size of ENG-Apt/mIP-10-LP nanocapsules was measured 145 nm, suggesting a possible exist of EPR mechanism. The PEG moiety forms an aqueous layer on the surface of liposomes that could additionally provide stabilization of the lipid bilayer, resulting in the inhibition of particles adsorption, recognition and uptake by the mononuclear phagocytic system (MPS) in the body [39]. Thus, PEGylation of liposome as the carrier for the functional plasmid in the construction of the ENG-Apt/mIP-10-LP, provided protection from decreasing the drug depletion by the degradation or phagocytosis, and extended the plasma half-life for the liposome-based nanocapsules. Besides, the presence of ENG-Apt in the ENG-Apt/mIP-10-LP, provides targeting specificity to tumor vascular endothelium, may also decrease the possibility of being processed by mononuclear phagocyte system and causes higher accumulation of mIP-10 in the tumor site than elsewhere.
Currently, T cell-based immunotherapy has achieved a breakthrough in tumor therapy, and CTLs have been shown to be critical for achieving an efficient therapeutic effect [40, 41]. However, previous studies have shown that tumor antigen-specific CTLs have limited anti-tumor efficacy due to their insufficient infiltration of tumor tissues [42], and the cytotoxic activities of these cells are inhibited by factors in the tumor microenvironment [43, 44]. It has also been reported that chemokine IP-10 can induce migration of lymphocytes and promote their proliferation and activation, thus enhancing the cytotoxic activity of CTLs [45-47]. Our results showed that treatment with ENG-Apt/mIP-10-LP nanocapsules increased the numbers of CD8+ T cells and IFN-γ positive cells within tumor tissues, indicating the potential of these engineered nanocapsules to improve the activity of T cells.
Residues 180-188 (N-SVYDFFVWL-COOH) of the H-2Kb-restricted TRP2 compose a melanoma-specific antigen peptide [48]. TRP2-specific CD8+ T cells can be prepared in vitro by stimulating CD8+ T cells with H-2Kb:Ig/TRP2180-188 artificial antigen-presenting cells. To further determine if ENG-Apt/mIP-10-LP nanocapsules could enhance the anti-tumor effects through driving these TRP2-specific CD8+ T cell induced by H-2Kb:Ig/TRP2180-188 artificial antigen-presenting cells to tumor location, we applied adoptive immunotherapy of TRP2-specific CD8+ T cells associated with ENG-Apt/mIP-10-LP nanocapsules to treat melanoma-bearing mice. ISTS was used to detect the percentage of TRP2CD8+ T cells among TILs. Our experiment results showed that the presence of melanoma-specific TRP2CD8+ T cells in tumor tissues after the combine treatment of TRP2-specific CD8+ T cell and ENG-Apt/mIP-10-LP nanocapsules, whereas a large amount of CD8+ T cell enrichment was observed in the tumor tissues after the treatment of ENG-Apt/mIP-10-LP nanocapsules alone, as well as the combine treatment. These findings demonstrated that the ENG-Apt/mIP-10-LP nanocapsules could drive not only endogenous CD8+ T cells, but also the exogenously adoptive TRP2CD8+ T cells in the melanoma-bearing mice. In this manuscript, we originally reported the important role of ENG-Apt/mIP-10-LP nanocapsules in promoting both endogenous and exogenous tumor antigen-specific CTLs in vivo.
It is generally believed that, in addition to suppressing the tumor-killing activity of T cells, local tumor microenvironment interferes with the ability of T cells to recognize tumor antigens and enables a state of systemic immunosuppression by directly inhibiting the function of T cells throughout the body [49]. Tregs and MDSCs both play an immunosuppressive role in vivo. The abilities of these two cell types in leading immunosuppression driven by chronic inflammation in the tumor microenvironment, have been investigated in several studies [50,51]. It has also been reported that MDSCs and Tregs are overexpressed in vivo during tumor formation [2, 52-54] that decreases the anti-tumor efficacy of CTLs [55-57], suggesting a negative correlation between the number of MDSCs and Tregs with that of CTLs during the tumor development. Our flow cytometric analysis data is promising and showed that treatment of melanoma-bearing mice with ENG-Apt/mIP-10-LP nanocapsules significantly decreased the numbers of MDSCs in bone marrow and spleen as well as in tumor tissues. The result demonstrated that percentage of tumor tissue CD11b+Gr1+ MDSCs in PBS control group was 11%, which was remarkably close to other study based on similar B16 tumor mice model [58], suggesting the reliability of flow cytometric analysis for MDSCs. MDSCs are a group of cells consisting of various cell types. In our study, CD11b+Gr1+ cells in PBS group and treatment groups might not be the same cell populations based on their Gr1 expression levels. The phenotype and functions of MDSC subgroups are not entirely conclusive, and CD11b+Gr1+ cells have been commonly accepted as MDSCs [58, 59]. Therefore, we herein determined the portion of MDSCs by roughly gating CD11b+Gr1+ cells. Interestingly, we recognized a unique population of Gr1+CD11b- cells that was present in all treatment groups but not in the PBS group and associated with the reduced number of MDSCs after each treatment. Similar cell population was also observed in some other studies [60, 61]. This cell population may possess a possible role associated with anti-tumoral functions, however, further validation is still necessary. In addition, it also reduced the numbers of Tregs in the spleen. These results are consistent with previous studies showing that depletion of Tregs and MDSCs improved the outcome of mesothelioma [62, 63].
Notably, there was a more substantial reduction in the numbers of MDSCs and Tregs after combined treatment with ENG-Apt/mIP-10-LP nanocapsules and TRP2CD8+ T cells. Because the adoptive CTLs are TRP2CD8+ T cells which were TRP2-specific and -sensitive. Thus, treatment of TRP2CD8+ T cells alone without the targeted priming of ENG-Apt/mIP-10-LP, might not induce a sufficient number of CTL recruitment in the tumor environment as the treatment combined with ENG-Apt/mIP-10-LP. This may answer the question why TRP2CD8+ T cells treatment group did not display the same functionality. Our study revealed that treatment with ENG-Apt/mIP-10-LP nanocapsules combined with TRP2CD8+ T cells is promising as a more efficient immunotherapeutic approach.
Our in vivo experiments demonstrated that treatment with ENG-Apt/mIP-10-LP nanocapsules inhibited tumor growth and increased the survival time of tumor-bearing mice, and that the combination treatment with both ENG-Apt/mIP-10-LP nanocapsules and TRP2CD8+ T cells further increased the survival time of mice. As a marker of cell proliferation, PCNA expression is associated with tumor cell differentiation and malignant behavior [64, 65], such as invasion and metastasis, and that is closely related with cancer prognosis [66]. Therefore, detection of PCNA provides insight into the degree of tumor malignancy and can guide clinical treatment and prognosis. Our experiments evaluating PCNA expression showed that treatment with ENG-Apt/mIP-10-LP nanocapsules reduced the vascular density within tumors and inhibited tumor cell proliferation. Also, TUNEL analysis showed that nanocapsule delivery promoted tumor cell apoptosis in tumor-bearing mice. Combination treatment with ENG-Apt/mIP-10-LP nanocapsules and TRP2CD8+ T cells resulted in significantly decreased microvascular density in tumors and further promoted apoptosis of tumor cells, indicating that the anti-tumor effect of the ENG-Apt/mIP-10-LP nanocapsules was, to some extent, related to reversal of the imbalance between proliferation and apoptosis among tumor cells.
Conclusion
In this study, we designed and constructed ENG-Apt-modified, IP-10 plasmid-containing liposomal (ENG-Apt/mIP-10-LP) nanocapsules, with the purpose of enhancing CTL recruitment to the tumor vasculature. These prepared ENG-Apt/mIP-10-LP nanocapsules possess a viable nanometric size, good stability, and displayed high specificity binding to mTECs and the ability to upregulate the IP-10 protein expression in vitro and in vivo. The combination treatment of ENG-Apt/mIP-10-LP nanocapsules and adoptive TRP2CD8+ T cells, increased IP-10 protein expression in tumor areas of neo-angiogenesis in the melanoma-bearing mice, and also promoted the recruitment of endogenous and exogenous tumor antigen-specific CTLs thus inhibiting tumor growth in vivo. Altogether, the ENG-Apt/mIP-10-LP nanocapsule developed in our current study, has the potential application prospect in efficient anti-melanoma treatment, and therefore provides a new promising strategy for cancer immunotherapy.
Supplementary Material
Supplementary figures.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Koning D, Costa AI, Hasrat R, Grady BP, Spijkers S, Nanlohy N. et al. In vitro expansion of antigen-specific CD8+ T cells distorts the T-cell repertoire. J Immunol Methods. 2014;405:199-203
2. Fernandez I, Ooi TP, Roy K. Generation of functional, antigen-specific CD8+ human T cells from cord blood stem cells using exogenous notch and tetramer-TCR signaling. Nat Rep Stem Cells. 2014;32:93-104
3. Newell EW, Lin W. High-dimensional analysis of human CD8+ T cell phenotype, function, and antigen specificity. Curr Top Microbiol Immunol. 2014;377:61-84
4. Gerdemann U, Vera JF, Rooney CM, Leen AM. Generation of multivirus-specific T cells to prevent/treat viral infections after allogeneic hematopoietic stem cell transplant. J Vis Exp. 2011;13:137-6
5. Chiu YL, Schneck JP, Oelke M. HLA-Ig based artificial antigen presenting cells for efficient ex vivo expansion of human CTL. J Vis Exp. 2011;12:3055-63
6. Garrido F, Perea F, Bernal M, Sánchez-Palencia A, Aptsiauri N, Ruiz-Cabello F. et al. The escape of cancer from T cell-mediated immune surveillance: HLA class I loss and tumor tissue architecture. NPJ Vaccines. 2017;5(1):7
7. Ribatti D. The concept of immune surveillance against tumors: the first theores. Oncotarget. 2017;8(4):7175
8. Zhuo H, Peng Y, Yao Q, Zhou N, Zhou S, He J. et al. Tumor imaging and interferon-gamma-inducible protein-10 gene transfer using a highly efficient transferrin-conjugated liposome system in mice. Clin Cancer Res. 2013;19:4206-17
9. Yang J, Richmond A. The angiostatic activity of interferon-inducible protein-10/CXCL10 in human melanoma depends on binding to CXCR3 but not to glycosaminoglycan. Mol Ther. 2004;9:846-55
10. Nagpal ML, Davis J, Lin T. Overexpression of CXCL10 in human prostate LNCaP cells activates its receptor (CXCR3) expression and inhibits cell proliferation. Biochim Biophys Acta Bioenerg. 2006;1762:811-8
11. Wang X, Zhang FC, Zhao HY, Lu XL, Sun Y, Xiong ZY. et al. Human IP10-scFv and DC-induced CTL synergistically inhibit the growth of glioma in a xenograft model. Tumour Biol. 2014;35:7781-91
12. Lu XL, Jiang XB, Liu RE, Zhang SM. The enhanced anti-angiogenic and antitumor effects of combining flk1-based DNA vaccine and IP-10. NPJ Vaccines. 2008;26(42):5352-7
13. Nagpal ML, Chen Y, Lin T. Effects of overexpression of CXCL10 (cytokine-responsive gene-2) on MA-10 mouse Leydig tumor cell steroidogenesis and proliferation. J Clin Endocrinol Diabetes. 2004;183(3):585-94
14. Xu Y, Wang D, Zhao LM, Zhao XL, Shen JJ, Xie Y. et al. Endoglin is necessary for angiogenesis in human ovarian carcinoma-derived primary endothelial cells. Cancer Biol Ther. 2013;14:937-48
15. Miyata Y, Sagara Y, Watanabe S, Asai A, Matsuo T. et al. CD105 is a more appropriate marker for evaluating angiogenesis in urothelial cancer of the upper urinary tract than CD31 or CD34. Virchows Arch. 2013;463:673-9
16. Dallas NA, Samuel S, Xia L, Fan F, Gray MJ, Lim SJ. et al. Endoglin (CD105): a marker of tumor vasculature and potential target for therapy. Am J Clin Cancer Res. 2008;14(7):1931-7
17. Li C, Guo B, Wilson PB, Stewart A, Byrne G, Bundred N. et al. Plasma levels of soluble CD105 correlate with metastasis in patients with breast cancer. Int J Cancer Suppl. 2000;89(2):122-126
18. Chen H, Liu D, Yang Z, Sun L, Deng Q, Yang S. et al. Adrenergic signaling promotes angiogenesis through endothelial cell-tumor cell crosstalk. Endocr Relat Cancer. 2014;21:783-95
19. Yang Y, Zhang Y, Hong H, Liu G, Bryan RL, Cai WB. In vivo near-infrared fluorescence imaging of CD105 expression during tumor angiogenesis. Eur J Nucl Med Mol Imaging. 2011;38:2066-76
20. Ria R, Reale A, Castrovilli A. et al. Angiogenesis and progression in human melanoma. Dermatol Res Pract. 2010;2010:185687
21. Flaten GE, Chang TT, Phillips WT. et al. Liposomal formulations of poorly soluble camptothecin: drug retention and biodistribution. J Liposome Res. 2013;23:70-81
22. Lim SK, Shin DH, Choi MH, Kim JS. Enhanced antitumor efficacy of gemcitabine-loaded temperature-sensitive liposome by hyperthermia in tumor-bearing mice. Drug Dev Ind Pharm. 2014;40:470-6
23. Choi KM, Kwon IC, Ahn HJ. Self-assembled amphiphilic DNA-cholesterol/DNA-peptide hybrid duplexes with liposome-like structure for doxorubicin delivery. Bioact Mater. 2013;34:4183-90
24. Alexander CM, Dabrowiak JC, Maye MM. Investigation of the drugBinding properties and cytotoxicity of DNA-Capped nanoparticles designed as delivery vehicles for the anticancer agents doxorubicin and actinomycin D. Bioconjug Chem. 2012;23(10):2061-2070
25. Morita Y, Leslie M, Kameyama H, Volk DE, Tanaka T. Aptamer therapeutics in cancer: current and future. J Cancer Res Oncobiol. 2018;10(3):80
26. Ziffels B, Grötsch A, Al-Bayati L, Neri D. Targeted delivery of calreticulin to ED-A fibronectin leads to tumor-growth retardation. J Biotechnol. 2019;290:53-58
27. Lv TT, Li ZY, Xu L, Zhang YY, Chen HJ, Gao Y. Chloroquine in combination with aptamer modified nanocomplexes for tumor vessel normalization and efficient erlotinib/survivin-shRNA co-delivery to overcome drug resistance in EGFR-mutated NSCLC. Acta Biomater. 2018;76:257-74
28. Zhang Y, Hong H, Cai W. Tumor-targeted drug delivery with Aptamers. Curr Med Chem. 2011;18(27):4185-4194
29. Reinemann C, Strehlitz B. Aptamer-modified nanoparticles and their use in cancer diagnostics and treatment. Swiss Med Wkly. 2014;144:w13908
30. Ongaro T, Matasci M, Cazzamalli S, Gouyou B, De Luca R, Neri D. et al. A novel anti-cancer L19-interleukin-12 fusion protein with an optimized peptide linker efficiently localizes in vivo at the site of tumors. localizes in vivo at the site of tumors. J Biotechnol. 2018;291:17-25
31. Mortara L, Balza E, Bruno A, Poggi A, Orecchia P, Carnemolla B. Anti-cancer therapies employing IL-2 cytokine tumor targeting: contribution of Innate, adaptive and immunosuppressive cells in the anti-tumor efficacy. Front Immunol. 2018;9:2905
32. Balza E, Carnemolla B, Orecchia P, Rubartelli A, Poggi A, Mortara L. Tumor vasculature targeted TNF-α therapy: reversion of microenvironment anergy and enhancement of the anti-tumor efficiency. Curr Med Chem. 2018
33. Yin Q, Tang L, Cai K, Yang X, Yin L, Zhang Y. et al. Albumin as a "Trojan Horse" for polymeric nanoconjugate transendothelial transport across tumor vasculatures for improved cancer targeting. Biomater Sci. 2018;6(9):10.1039.C8BM00149A
34. Pezzolo A, Marimpietri D, Raffaghello L, Cocco C, Pistorio A, Gambini C. et al. Failure of anti-tumor-derived endothelial cell immunotherapy depends on augmentation of tumor hypoxia. Oncotarget. 2014;5:10368-81
35. He J, Duan S, Yu X, Qian Z, Zhou S, Zhang Z. et al. Folate-modified chitosan nanoparticles containing the IP-10 gene enhance melanoma-specific cytotoxic cd8+cd28+ t lymphocyte responses. Theranostics. 2016;6:752-761
36. Ramot R, Kishore IK, Wilson CJ. Lactose repressor experimental folding landscape: fundamental functional unit and tetramer folding mechanisms. Biochemistry. 2012;51:7569-79
37. Maeda H. The enhanced permeability and retention (EPR) effect in tumor vasculature: the key role of tumor-selective macromolecular drug targeting. Adv Enzyme Regul. 2001;41(1):189-207
38. Lane RS, Haller FM, Chavaroche AAE, Almond A, DeAngelis PL. Heparosan-coated liposomes for drug delivery. Glycobiology. 2017
39. Hatakeyama H, Akita H, Harashima H. The polyethyleneglycol dilemma: advantage and disadvantage of PEGylation of liposomes for systemic genes and nucleic acids delivery to tumors. Biol Pharm Bull. 2013;36(6):892-9
40. Park JS, Park SY, Cho HI, Sohn HJ, Kim TG. Enhanced induction of t cell immunity using dendritic cells pulsed with HIV tat and HCMV-pp65 fusion protein in vitro. Immune Netw. 2011;11:182-9
41. Hosoi A, Matsushita H, Shimizu K, Fujii S, Ueha S, Abe J. et al. Adoptive cytotoxic tlymphocyte therapy triggers a counter-regulatory immunosuppressive mechanism via recruitment of myeloid-derived suppressor cells. Int J Cancer Suppl. 2014;134:1810-22
42. Chia WK, Teo M, Wang WW, Lee B, Ang SF, Tai WM. et al. Adoptive T-cell transfer and chemotherapy in the first-line treatment of metastatic and/or locally recurrent nasopharyngeal carcinoma. Mol Ther. 2014;22:132-9
43. Seidel UJ, Oliveira CC, Lampen MH, Hall TV. A novel category of antigens enabling CTL immunity to tumor escape variants: cinderella antigens. Cancer Immunol Immunother. 2012;61:119-25
44. Luo F, Song X, Zhang Y, Chu Y. Low-dose curcumin leads to the inhibition of tumor growth via enhancing CTL-mediated antitumor immunity. Int Immunopharmacol. 2011;11:1234-40
45. Nagpal ML, Davis J, Lin T. Overexpression of CXCL10 in human prostate LNCaP cells activates its receptor (CXCR3) expression and inhibits cell proliferation. Biochim Biophys Acta. 2006;1762:811-8
46. Wang X, Lu XL, Zhao HY, Zhang FC, Jiang XB. A novel recombinant protein of IP10-EGFRvIIIscFv and CD8+ cytotoxic T lymphocytes synergistically inhibits the growth of implanted glioma in mice. Cancer Immunol Immunother. 2013;62:1261-72
47. Zhang Y, Gao Z, Wang D, Zhang T, Sun B, Mu L. et al. Accumulation of natural killer cells in ischemic brain tissues and the chemotactic effect of IP-10. J Neuroinflammation. 2014;11:1132-38
48. Vasievich EA, Ramishetti S, Zhang Y, Huang L. Trp2 peptide vaccine adjuvanted with (R)-DOTAP inhibits tumor growth in an advanced melanoma model. Mol Pharm. 2012;9:261-8
49. Alvarez Arias DA, Kim HJ, Zhou P, Holderried TA, Wang X, Dranoff G. et al. Disruption of CD8+ Treg activity results in expansion of T follicular helper cells and enhanced antitumor immunity. Cancer Immunol Res. 2014;2:207-16
50. Filipazzi P, Valenti R, Huber V, Pilla L, Canese P, Iero M. et al. Identification of a new subset of myeloid suppressor cells in peripheral blood of melanoma patients with modulation by a granulocyte-macrophage colony-stimulation factor-based antitumor vaccine. J Clin Oncol. 2007;25:2546-2553
51. Poschke I, Mao Y, Adamson L, Salazar-Onfray F, Masucci G, Kiessling R. Myeloid-derived suppressor cells impair the quality of dendritic cell vaccines. Cancer Immunol Immunother. 2012;61:827-838
52. Weiss JM, Subleski JJ, Back T, Chen X, Watkins SK. et al. Regulatory t cells and myeloid-derived suppressor cells in the tumor microenvironment undergo fas-dependent cell death during IL-2/αCD40 therapy. J Immunol. 2014;19:1230-9
53. Weide B, Martens A, Zelba H, Stutz C, Derhovanessian E, Di Giacomo AM. et al. Myeloid-derived suppressor cells predict survival of patients with advanced melanoma: comparison with regulatory Tcells and NY-ESO-1- or melan-a-specific T cells. Clin Cancer Res. 2014;20:1601-9
54. Pereira S, Teixeira L, Aguilar E, Oliveira M, Savassi-Rocha A, Pelaez JN. et al. Modulation of adipose tissue inflammation by FoxP3+ Treg cells, IL-10, and TGF-beta in metabolically healthy class III obese individuals. Nutrition. 2014;30:784-90
55. Zou Q, Wu B, Xue J, Fan X, Feng C, Geng S, Wang M, Wang B. et al. CD8+ Treg cells suppress CD8+ T cell-responses by IL-10-dependent mechanism during H5N1 influenza virus infection. Eur J Immunol. 2014;44:103-14
56. Verneris MR. Natural killer cells and regulatory T cells: how to manipulate a graft for optimal GVL. Hematology Am Soc Hematol Educ Program. 2013:335-41
57. Hermans C, Anz D, Engel J, Kirchner T, Endres S, Mayr D. Analysis of FoxP3+ T-regulatory cells and CD8+ T-cells in ovarian carcinoma: location and tumor infiltration patterns are key prognostic markers. PloS One. 2014;9:e111757
58. Yang ZK, Qi YX, Lai N, Zhang JH, Chen ZH, Liu MY. et al. Notch1 signaling in melanoma cells promoted tumor-induced immunosuppression via upregulation of TGF-β1. J Exp Clin Cancer Res. 2018;37(1):1
59. Sendo S, Saegusa J, Okano T, Takahashi S, Akashi K, Morinobu A. CD11b+Gr1dim tolerogenic dendritic cell-like cells are expanded in interstitial lung disease in SKG mice. Arthritis Rheumatol. 2017;69(12):2314-27
60. Gamvrellis A, Leong D, Hanley JC, Xiang SD, Mottram P, Plebanski M. Vaccines that facilitate antigen entry into dendritic cells. Immunol Cell Biol. 2004;82(5):506-516
61. Dumas A, Drévo MAL, Moreau MF, Guillet C, Baslé MF, Chappard D. Isolation of osteoprogenitors from murine bone marrow by selection of CD11b negative cells. Cytotechnology. 2008;58(3):163
62. Wu L, Yun Z, Tagawa T, Rey-McIntyre K, Anraku M, de Perrot M. Tumor cell repopulation between cycles of chemotherapy is inhibited by regulatory T-cell depletion in a murine mesothelioma model. J Thorac Oncol. 2011;6:1578-1586
63. Sevko A, Michels T, Vrohlings M, Umansky L, Beckhove P, Kato M. et al. Antitumor effect of paclitaxel is mediated by inhibition of myeloid-derived suppressor cells and chronic inflammation in the spontaneous melanoma model. Open J Immunol. 2013;190(5):2464-2471
64. Jia Z, Zhao W, Fan L, Sheng W. et al. The expression of PCNA, c-erbB-2, p53, ER and PR as well as atypical hyperplasia in tissues nearby the breast cancer. J Mol Histol. 2012;43:115-20
65. Mukhtar RA, Moore AP, Nseyo O, Baehner FL, Au A, Moore DH. et al. Elevated PCNA+ tumor-associated macrophages in breast cancer are associated with early recurrence and non-Caucasian ethnicity. Breast Cancer Res Treat. 2011;130:635-44
66. Kato K, Kawashiri S, Yoshizawa K, Kitahara H, Okamune A, Sugiura S. et al. Expression form of p53 and PCNA at the invasive front in oral squamous cell carcinoma: correlation with clinicopathological features and prognosis. J Oral Pathol Med. 2011;40:693-8
Author contact
![]() Corresponding author: Xiaoling Lu, Ph.D., Professor, Nanobody Research Center, Guangxi Medical University, Nanning, Guangxi, 530021, China. Tel: (86) 771-5317 061; Fax: (86) 771-5317 061. E-mail: luxiaolingedu.cn
Corresponding author: Xiaoling Lu, Ph.D., Professor, Nanobody Research Center, Guangxi Medical University, Nanning, Guangxi, 530021, China. Tel: (86) 771-5317 061; Fax: (86) 771-5317 061. E-mail: luxiaolingedu.cn