Impact Factor
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Lymphocyte subsets in steady...
B-cells recruitment in heart...
Antibodies production and heart...
Cellular B-cell responses in...
Protective effects of B-cells in...
Clinical trials of B cells in...
T-cells in heart diseases
T-cells in myocardial...
T-cells in myocardial infarction
T-cells in other cardiac disease...
Therapeutic strategies of...
NK cells in heart diseases
Conclusion
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(14):4030-4046. doi:10.7150/thno.33112 This issue Cite
Review
Lymphocytic subsets play distinct roles in heart diseases
Jing Wang1, Yi Duan1, Joost PG Sluijter2,3, Junjie Xiao1 ![]()
1. Cardiac Regeneration and Ageing Lab, Institute of Cardiovascular Sciences, School of Life Science, Shanghai University, Shanghai 200444, China
2. Department of Cardiology, Laboratory of Experimental Cardiology, University Medical Center Utrecht, Utrecht 3508GA, The Netherlands
3. UMC Utrecht Regenerative Medicine Center, University Medical Center, Utrecht University, Utrecht 3508GA, The Netherlands
Received 2019-1-14; Accepted 2019-5-6; Published 2019-5-31
Abstract

Heart diseases are one of the leading causes of death for humans in the world. Increasing evidence has shown that myocardial injury induced innate and adaptive immune responses upon early cellular damage but also during chronic phases post-injury. The immune cells can not only aggravate the injury but also play an essential role in the induction of wound healing responses, which means they play a complex role throughout the acute inflammatory response and reparative response after cardiac injury. This review will summarize the current experimental and clinical evidence of lymphocytes, one of the major types of immune cells, participate in heart diseases and try to explain the possible role of these immune cells following cardiac injury.
Keywords: lymphocytes, immune response, heart diseases
Introduction
Heart diseases, known as a type of disease including coronary heart disease, heart attack, and heart failure, are one of the leading causes of death in the world [1]. The sustained increase in blood pressure, myocardial ischemia and hypoxia, and an excessive activation of the neuroendocrine system can lead to a variety of pathological changes such as the decrease in the number of myocardial cells, energy metabolism disorder, contraction and/or diastolic decrease, which will lead to decompensated myocardial hypertrophy and ventricular remodeling, and eventually to heart failure [2-4].
The immune system helps protect against pathogens such as viruses, bacteria, and parasitic worms but also plays a key role in heart diseases. After myocardial damage, the immune system becomes activated, and subsequently followed by an infiltration into the cardiac tissue [5]. The recruitment and activation of immune cells of the adaptive and innate immune systems causes a sterile inflammation in damaged cardiac tissue. These immune cells participate in clearance of necrotic debris, but also in initiating the reparative response and regenerative signaling in the myocardium [6-13]. However, immune cells not always function like the 'reparative' counterparts. The over-activation or prolonged inflammation of immune cells in the cardiac tissue may lead to aberrant expansion of cardiomyocytes apoptosis and fibrosis. This out-of-control activation of immune cells will induce additional dysfunction of cardiomyocytes [11, 13]. In this manner, the immune cell participation can be both 'reparative' and 'destructive' after myocardial damage. Thus, a better understanding of the role of immune cells in heart diseases could provide experimental evidence and theoretical support for future immunotherapy in this area. However, till now, we still lack the understanding of the role of immune cells upon cardiac injury and during wound healing.
The immune system is a complex network of cells and tissue that work together to protect the body. Leukocytes are cell types that play a key role in the immune system. Based on cell lineage, leukocytes can be classified into two categories: myeloid cells and lymphoid cells. Myeloid cells include both 1) granulocytes, classified into mast cells, eosinophils, basophils, and neutrophils, and 2) mononuclear phagocytes, which contain monocytes, macrophages, and dendritic cells, with each of them being comprised of several subsets. B-, T- and NK cells belong to lymphoid cells [14]. In this review, we will identify the role of lymphocytes in response to cardiac injury. We hope this review can call for further research into the function of these cell populations in myocardial injury and repair.
Lymphocyte subsets in steady state
As a solid and non-immunogenic organ, there are very limited leukocytes in heart at baseline. Due to limited reports of leukocytes in the human heart, most evidence is based on mouse studies. In comparison to lymphoid organs, like the thymus, spleen, tonsils, and bone marrow, the adult mouse heart contains no more than 100 thousand leukocytes in steady state. Among all the heart leukocytes, 21% are lymphocytes, and 74% are myeloid cells of which the macrophage is being the most prevalent subset in the myeloid cell population [5, 15].
B-cells recruitment in heart diseases
In a healthy adult mouse myocardium, there are less than 21000 lymphocytes (B-, T- and NK cells), of which 45% are B-cells [15]. After myocardial injury, the number of B-cells increases 5-10 times and peaks around day 7 post-MI in a permanent myocardial infarction model and around day 3 post-MI after ischemia-reperfusion [5]. The B-cells have also been detected in myocardial infarction patients' heart biopsy at day 1 and day 6, following coronary artery occlusion [16]. The mechanisms that drive B-cells recruitment and activation in heart diseases are still under investigation, but the functions of these myocardial B-cells in the progress of heart diseases are appear to be gradually revealed.
Antibodies production and heart diseases
Experimental studies
A direct link was shown between over-activated B-cells and myocardial disruption. PD-1-/- mice (programmed cell death protein-1, PD-1, B-cell differentiation factor) expressed high level of circulating IgG antibodies, produced by B-cells, that bind specifically to cardiomyocytes and thereby developed a spontaneous dilated cardiomyopathy (DCM) [17]. Meanwhile, the effect of these IgG antibodies absolutely disappeared in RAG2 deficient PD-1-/- mice (RAG2-/-, Recombination Activation Gene. B and T-cells deficient mice), due to the lack of antibody producing B-cells in these mice. This aggressive IgG class, which was detected by an independent group, was shown to attack troponin I [18]. B-cells were also detected to be activated in a murine model of nonischemic cardiomyopathy (CMP, an angiotensin-II (Ang-II)-induced heart failure model). A significantly increase in IgG3 subclass antibody depositions were detected in myocardial tissue. The absence of B-cells in this model of heart failure resulted in less hypertrophy, less collagen deposition, and preservation of left ventricular function, which further demonstrated that B-cells and their secreted antibodies play a contributory role in an Ang-II induced heart failure model [19].
In murine models of post-ischemic heart failure, the expression of B-cell-produced IgM and IgG antibodies were increased 3-fold in the post-ischemic state compared to controls [20]. Using N2 or 21G6 F(ab)2 peptides specific IgM binding to ischemic antigens in the heart could be prevented, which resulted in a significant reduction in cardiac I/R injury [21]. Mice bearing an altered natural IgM repertoire (Cr2-/-), which have an impaired humoral response to T-dependent and T-independent antigens, upon coronary artery ischemia showed a significantly protected phenotype: the infarct size was reduced, cardiomyocyte apoptosis was limited and associated with a decreased neutrophil infiltration [22]. The IgM antibodies could also function in the progression toward heart failure upon myocardial infarction. In the murine model of acute myocardial infarction, blocking of self-reactive IgM by synthetic peptide mimotopes or monoclonal antibodies significantly reduced cardiac injury [21]. More recently, agammaglobulinemic AID-/-μS-/- mice that can produce functional B-cells, but cannot synthesize secretory IgM or class-switched immunoglobulins were used in order to selectively study the role of (auto-) antibodies during post-ischemic heart context [23]. A significant reduction in infarct size and left ventricle dilation were detected in agammaglobulinemic mice when compared to immunocompetent animals at day 56 post-MI. The cardiac function was also improved in agammaglobulinemic mice, suggesting that antibodies were directly involved in ischemic heart failure [23] (Table 1, Figure 1). All these evidences supported a model in which immunoglobulins involved in the acute inflammatory response in the myocardium following ischemia and reperfusion. However, further studies were still required before providing definitive conclusion regarding the role of immunoglobulins in heart diseases.
Study characteristics of B-cells in experimental heart diseases
| Diseases | Cell type/ Species | Follow-up | Results | Conclusions | References |
|---|---|---|---|---|---|
| Dilated Cardiomyopathy (DCM) | B-cells/PD-1-/- mice | 30-weeks | High level of circulating IgG antibodies was detected in PD-1-/- mice. These antibodies bind specifically to cardiomyocytes and attack troponin I, and thereby developed a spontaneous DCM. | PD-1 was an important factor contributing to the prevention of autoimmune diseases. The IgG antibodies produced by B-cells exacerbated disease progression. | [17] |
| Nonischemic Cardiomyopathy | B-cells/Ang-II-induced heart failure mice | 35-days | An increased IgG3 depositions were detected in myocardial tissue. Absence of B-cells resulted in less hypertrophy, less collagen deposition, and preservation of left ventricular function. | B-cells and their secreted antibodies played a contributory role in an Ang-II induced heart failure model. | [19] |
| Post-Ischemic Heart Failure | B-cells/mice suffered from reperfusion injury | 5-days | Using N2 or 21G6 F(ab)2 peptides specific IgM binding to ischemic antigens in the heart resulted in a significant reduction in cardiac ischemic-reperfusion injury. | IgM antibodies functioned in the progression towards heart failure. | [21] |
| Post-Ischemic Heart Context | B-cells/AID-/-μS-/- mice | 56-days | AID-/-μS-/- mice showed a significant reduction in infarct size and left ventricle dilation at day 56 post-myocardial infarction. | Autoantibodies that were produced by B-cells were directly involved in ischemic heart failure. | [23] |
| Acute Cardiomyopathy (CMP)/Heart Failure (HF)/Cardiac Pathogenesis | B-cells/mice | 2-4 weeks | The interactions of B-cells with the innate immune system, which was mediated by the TLRs, MyD88, IRAK-4, and IRF-3, were demonstrated to control the acute CMP and HF phenotypes. | Maladaptive signaling mechanisms through the innate immune system could switch B-cells from tolerance to responsiveness, which might promote cardiac fibrosis during CMP and HF. | [35-38] |
| Myocardial Infarction (MI) | B-cells/ Ccl7-/-, Tnfrsf13c-/-, RAG1-/- and Myd88-/- mice | 1-3 weeks | Specific deletion of CCL-7 production by B cells limited monocyte/macrophage infiltration in the ischemic heart, collagen deposition and reduced deleterious left ventricular remodeling. | The decreased levels of the chemokine CCL7, due to B-cell depletion, was associated with lower proinflammatory Ly-6Chigh monocyte infiltration in the ischemic heart, which further limited myocardial injury and improved heart function. | [16] |
| Myocardial Infarction (MI) | Bone marrow cells (BMCs)/rats and mice | 28-days | BMCs were therapeutically injected into early post-ischemic myocardium that improved cardiac function by cardiomyocyte salvage. The therapeutic effect resulted from a single injection of whole BMC lysate or B-cell lysate. | B cells played an important paracrine role in effective BMCs therapy for myocardial infarction. | [40, 41] |
B-cells response to heart injury in experimental and clinical studies. B-cells infiltrate into the myocardium after myocardial damage. Proteins and cytokines released from necrotic cardiomyocytes recognized by circular and cardiac infiltrated B-cells, causing B-cells activation. Then the activated B-cells induce direct cardiac injury via complement-mediated cytotoxicity and apoptotic signaling pathway (A). While, some activated circular B-cells transform into plasma and memory B-cells that produce autoantibodies. The recognition of autoantibodies and cardiomyocytes further cause deterioration of heart function. These include apoptosis of cardiomyocytes, increased collagen deposition, the release of pro-inflammatory cytokines, and rational hypertrophy (B). Meanwhile, activated B-cells, with the participation of Th1 cells, regulate the recruitment of Ly-6Chigh monocytes from the blood into injured myocardium through a CCL-7 dependent fashion. The supplement of cardiac monocytes aggravates the damage to heart function (C).
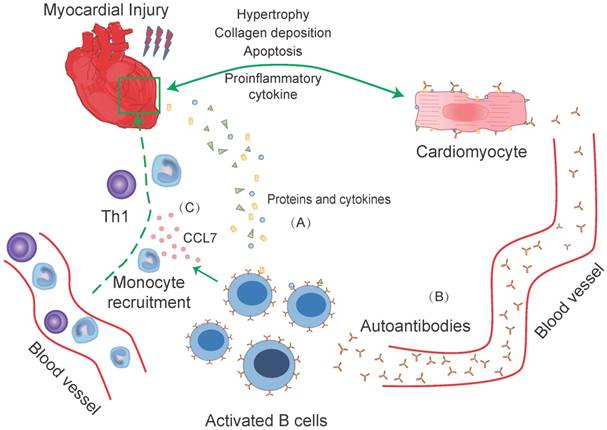
Clinical evidences
In human, during heart failure-induced immune responses, B-cells can interact with T-cells, more specifically the T helper (Th1) cells, and thereby impair heart contractility and promote adverse remodeling via producing circulating cytokines and antibodies, which had a great impact on outcomes and prognosis in congestive heart failure patients [24, 25]. In some forms of heart diseases, such as DCM, B-cells are stimulated and transform into plasma and memory B-cells. The plasma cells generated autoantibodies, which could specific recognize cardiac antigens through their F(ab) region or bind to the Fc gamma receptor (Fcγ) on cardiomyocytes through their Fc fragments [26-28]. The binding of the F(ab) region could cause an agonistic or antagonistic effect towards the specific cardiac antigens that further induce apoptosis in isolated myocytes, while binding of the Fc fragment to the Fcγ receptor could cause cells apoptosis [28]. Otherwise, memory B-cells could form a secondary response when they recognize the same antigen again. This response is much greater, much stronger, and is causing more damage to the individual cardiomyocytes [28]. Also, other antibodies, produced by the B-cells could bind several different proteins or receptors, further accelerating the process of heart diseases: Anti-B1-adrenergic receptor antibodies are directed against the cardiomyocyte inotropic responsiveness to beta-agonists and could trigger cardiomyocyte apoptosis [29, 30]. The anti-M2-receptor antibodies functioned in a similar fashion as the anti-B1-adrenergic receptor [31]. Anti-myosin antibodies could impair cardiomyocyte contractility. The anti-Kv channel antibodies might induce cardiomyocyte cell death, and antibodies against the Na+/K+- ATPase contributed to electrical instability in the heart, making it prone to arrhythmias [32-34] (Table 2, Figure 1). The association between B-cell antibody production and myocardial cell death might demonstrate a major pathogenic role in the progression of the disease. Therefore, accurate blocking the auto-antibodies production by B-cells could be an effective way to prevention and treatment heart diseases. Further experimental and clinical investigations in this area are needed.
Study characteristics of lymphocytes in clinical heart diseases
| Diseases | Cell type | Follow-up/No. patients | Results | Conclusion | Reference |
|---|---|---|---|---|---|
| Dilated Cardiomyopathy (DCM) | Plasma cells | 11 patients | The plasma cells generated autoantibodies, which could specific recognize cardiac antigens through their F(ab) region or bind to the Fcγ on cardiomyocytes through their Fc fragments. | Autoantibodies bind to their antigen via the DCM-IgG-F(ab')2 part and then induce cardiac dysfunction by activating Fcγ receptors II a on cardiomyocytes. | [26-28] |
| Ischemic Cardiomyopathy (ICM)/ Dilated Cardiomyopathy (DCM)/Myocarditis/ Cardiomyopathy | Plasma cells | Patient number from 26-98 | Anti-B1-adrenergic receptor antibodies triggered cardiomyocyte apoptosis. Anti-M2-receptor antibodies functioned in a similar fashion as the anti-B1-adrenergic receptor. Anti-myosin antibodies impaired cardiomyocyte contractility. The anti-Kv channel antibodies induced cardiomyocyte cell death, and antibodies against the Na+/K+- ATPase contributed to electrical instability in the heart, making it prone to arrhythmias. | The antibodies that were produced by the B-cells could bind several different proteins or receptors on cardiomyocytes, further accelerating the process of heart diseases. | [29-34] |
| Sudden Coronary Death/ Acute Myocardial Infarction (AMI)/ Myocarditis and patients with non-Ischemic Heart Failure (HF) | CD3+ T-cells | 72 patients | Activated T-cells infiltrated in both remote and peri-infarction regions in patients suffering from acute myocardial infarction. Activated T-cells within the epicardial coronary artery wall of both the infarct- and non-infarct-related arteries were also found in dying patients suffering from AMI but not in active lymphocytic myocarditis. | Myocardial injury in patients might activate T-cells which subsequently home to the injured myocardium and likely modulate local inflammatory activity in patients dying suddenly, shortly, or even late after coronary thrombosis. | [68] |
| Microvascular Obstruction (MVO)/Myocardial Ischemia-Reperfusion Injury (I/RI) | CD4+ and CD8+ effector T cells | 3-years/1377 patients | Transient T cell depletion was detected from the bloodstream in ST elevation myocardial infarction (STEMI) patients and reaching nadir at 90 minutes, with recovery almost complete by 24 hours following reperfusion after primary percutaneous coronary intervention (PPCI). | Effector T cells contributed to MVO through direct trapping within the myocardial microvasculature, where they released inflammatory mediators, contributing to further leukocyte infiltration and myocardial damage. | [41] |
| Coronary Artery Disease (CAD) | CD4+ CD28null T-cells | 24-months/120 patients | CD4+CD28null T-cells were present in the blood in the context of acute coronary events during several months and secreted IFN-γ and TNF-α, as well as cytotoxic mediators in the blood. | Targeting the CD4+CD28null T-cell subset in CAD could provide novel therapeutic strategies to prevent acute life-threatening coronary events. | [69-71] |
| Cytomegalovirus (CMV) patients with Myocardial Ischemia-Reperfusion | CD8 memory T-cells | 3-months/88 patients | A long-lasting fall in the peripheral frequency of terminally differentiated CD8 effector memory T-cells in CMV patients was found. PD-1 was involved in the persistent loss of CD8 memory T-cells in CMV seropositive patients. | Acute myocardial infarction and reperfusion accelerated immunosenescence in CMV-seropositive patients. | [50] |
| Acute Coronary Syndromes (ACS)/ ST-Segment Elevation Myocardial Infarction (STEMI)/ | Tregs | 48-hours to 6-days/28 to 30 patients | A decrease in circulating Treg numbers were detected in patients with ACS and STEMI. | Tregs were associated with ACS and STEMI and might play a key role in the progression of these diseases. | [72, 73] |
| Acute Myocardial Infarction (AMI) | Tregs | At least 15-years/48 AMI and 66 stroke patients | Lower fraction of Tregs and higher release of proinflammatory cytokines from activated mononuclear leukocytes were detected in myocardial infarction patients. | Low levels of Tregs were associated with an increased risk for the development of myocardial infarction but not stroke. | [74] |
| Non-ST Segment Elevation Acute Coronary Syndrome (NSTACS)/Chronic Stable Angina (CSA)/Chest Pain Syndrome (CPS) | Tregs | 1-2 days/ 84 NSTACS patients, 38 CSA patients, and 60 CPS controls | The spontaneous apoptosis of Tregs was increased in the NSTACS patients compared with the CSA and CPS groups. The level of oxidized LDL, which could induce Tregs apoptosis, were significantly higher in the NSTACS patients than in the CSA and CPS groups. | Tregs defects observed in the NSTACS patients was partially due to the impaired thymic output and their enhanced susceptibility to apoptosis in the periphery. | [75] |
| Chronic Heart Failure (CHF) | Tregs | 4-months/99 patients | Tregs suppressed CD4+ CD25- T-cells proliferation and pro-inflammatory cytokines production in CHF patients. | Tregs were involved in the progression of CHF and showed a protective effect. The number of Tregs was detected to be decreased in CHF and the defects in CHF patients were likely caused by decreased thymic output of nascent Tregs and increased susceptibility to apoptosis in the periphery. | [98-100] |
| Cardiac Allograft Vasculopathy (CAV) | CD16-dependent NK cells | 12-hours/103 patients | Enhanced the levels of CD16 expression and antibody-dependent NK cell cytotoxic function of heart transplant recipients were associated with the FCGR3A-VV genotype, which was identified as a baseline-independent predictor of CAV risk. | NK cells, with high levels of CD16 expression, contributed to acute rejection and induced ACV. | [115] |
| Coronary Heart Disease (CHD) | NK cells | 95 patients | Lower NK cytotoxic activity and a decreased absolute number and percentage of CD3-CD56dim cytotoxic NK subset were existed in patients with CHD. | CHD were associated with a redistribution of circulating lymphocytes, comprising a significant reduction of CD3-CD56dim NK cells and a concomitant loss of NK cell function. | [116, 117] |
| Coronary Artery Disease (CAD) | NK cells | 12-months/65 patients | The proportions of NK cells were reduced significantly in CAD patients. During a 12-month follow-up, the number of NK cells increased in part of the CAD patients, while the failed NK cells reconstituted patients exhibited a persistent low-grade inflammation. | The failure of reconstitute NK cell levels was associated with a persistent low-grade inflammation, suggesting a protective role of NK cells in CAD. | [118] |
Cellular B-cell responses in heart diseases
The B-cells participated in the process of heart diseases not only through the antibodies that they produced. The interactions of B-cells with the innate immune system, which was mediated by the toll-like receptors (TLRs), myeloid differentiation factor 88 (MyD88), interleukin-1 receptor-associated kinase 4 (IRAK-4), and interferon regulatory factor-3 (IRF-3), were demonstrated to control the acute CMP phenotype [35-39]. Inflammation and myocardial dysfunction can be induced by viral myocarditis, septic CMP, atherosclerosis, as well as maladaptive ventricular remodeling upon myocardial infarction in mice [39]. A TLR that have been studied the most was TLR-4, which was up-regulated in a murine model of heart failure. This up-regulated TLR-4 determines the formation of mature antibodies and B-cells valid interactions with the innate immune system [36]. Maladaptive signaling mechanisms through the innate immune system could switch B-cells from tolerance to responsiveness, which might promote cardiac fibrosis during heart failure [35, 37].
A critical role for mature B-cells in left ventricular remodeling and function was investigated using several B-cell ablation strategies in a myocardial infarction murine model [16]. Genetic deletion of mature B-cells through Baff receptor deficiency or antibody-mediated depletion by anti-Baff or anti-CD20 specific antibody, impeded CCL7 production. The decreased levels of the chemokine CCL7, due to B-cell depletion, was associated with lower proinflammatory Ly-6Chigh monocyte infiltration in the ischemic heart. Fourteen days after myocardial infarction, the deletion of B-cells reduced infarct size, lessened left ventricular dilation, and improved recovery of left ventricular function (Table 1, Figure 1).
Although the recruitment of B-cells to myocardium after myocardial injury is clear, the mechanism of recruitment is still unclear. In addition, the possible interaction between B-cells and congenital inflammation and the relationship between B-cells and adaptive immunity of damaged myocardium are worthy of further study.
Protective effects of B-cells in heart diseases
In some experimental studies, B-cells have also been reported as tissue repair factors that play a key role in treatment through paracrine action. A pilot study in rat showed that intramyocardial injection of B-cells into early post-ischemic myocardium preserved cardiac function by cardiomyocyte salvage, whereas other bone marrow (BM) mononuclear cell (MNC) subtypes were either ineffective or suppressed cardioprotection conferred by an enriched B-cell population [40]. Further research in a mouse model where BM-cells were therapeutically injected into the myocardium via a single dose improved post-MI cardiac function. The therapeutic effect resulted from a single injection of whole BMC lysate or B-cell lysate, which means B-cells play a key role in the therapy through the paracrine role. This BMCs therapeutic efficacy depends on the age of the donor mice, probably due to a reduction of B-cell numbers in older donors [41]. However, these studies do not describe the exact mechanism by which B-cells have beneficial effects in heart diseases.
Clinical trials of B cells in heart diseases
The experimental study of mature B-cells in myocardial infarction demonstrated a critical role of these B-cells in left ventricular remodeling and function. The CCL7 production by these B-cells orchestrated monocyte mobilization from the bone marrow to the blood and then recruited monocyte into the ischemic heart. The same as animal studies, plasma levels of CCL7 and BAFF were predictive of major adverse cardiovascular events in myocardial infarction patients (a French cohort of myocardial infarction patients, FAST-MI) [16]. Thus, these clinical results open a promising new therapeutic area of ischemic heart failure through antibody-mediated depletion of CD20 or BAFF in myocardial infarction patients [16, 42]. Recently, an anti-CD20 antibody Rituximab, which was used clinically to treat inflammatory autoimmune disease and B-cell cancers, was tested in patients with acute ST-elevation myocardial infarction. The study is currently recruiting participants to test the clinical efficacy and safety of this anti-CD20 antibody in myocardial infarction patients [43, 44]. However, the clinical exploration of B-cells in the therapeutic of myocardial infarction and other heart diseases is still in its infancy, which need more clinical investigation.
T-cells in heart diseases
The central organ for the generation of T-cells is the thymus. Naïve CD4+ and CD8+ T-cells are released from thymus and decline with age [45]. Even though the number of T-cells is lower than B-cells in steady and impaired state of the adult heart, there are more studies reporting on T-cells in heart diseases than B-cells. Like B-cells, T-cells number also increases 5-10 times and peaks around day 7 post-MI in the permanent myocardial infarction heart and around day 3 post-MI after ischemia-reperfusion, similar for CD4+ and CD8+ T-cells [5]. The same as experimental studies, pre-clinical studies have also discovered that T-cells accumulated in the infarct myocardium within minutes following cardiac I/R injury [46].
T-cells in myocardial ischemia-reperfusion injury
Conventional T-cells
In animal studies, CD4+ T-cells were reported to contribute to myocardial ischemia-reperfusion injury. The CD4+ T-cells deficient mice showed a prominently decreased infarct size compared with control mice. The infarct size in T-cells deficient RAG1 KO mice was increased to the level of control mice by adoptive transferring of CD4+ T-cells derived from WT control mice [47]. However, myocardial infarct size was not increased in CD4+ T-cells rescued RAG1 and IFN-γ double KO mice, indicating that CD4+ T-cells promote ischemia-reperfusion injury through IFN-γ expression [47]. The same as CD4+ αβ T-cells, γδ T-cells also infiltrated into the reperfused myocardium, albeit in low absolute number. Interleukin-17A (IL-17A) was elevated after mouse left coronary artery ligation and reperfusion, which was mainly produced by these infiltrated γδ T-cells. Then, the produced IL-17A in reperfused myocardium mediated cardiomyocytes apoptosis through regulating the Bax/Bcl-2 ratio [48] (Table 3, Figure 2).
Study characteristics of T-and NK-cells in experimental heart diseases
| Diseases | Cell type/ Species | Follow-up | Results | Conclusion | References |
|---|---|---|---|---|---|
| Myocardial Ischemia-Reperfusion Injury (I/RI) | CD4+ T-cells/ RAG1 KO mice | 24-hours | The CD4+ T-cell deficient mice showed a prominently decreased infarct size after coronary artery and reperfusion. Adoptive transferring of CD4+ T-cells in RAG1 KO but not RAG1 and IFN-γ double KO mice increased the infarct size to the level of WT mice. | CD4+ T-cells were contributed to myocardial ischemia-reperfusion injury, which was partially dependent on the IFN-γ expression. | [47] |
| Myocardial Ischemia-Reperfusion Injury (I/RI) | γδT-cells/mice | 1-3 days | IL-17A, which was mainly produced by γδT-cells in reperfused myocardium, mediated cardiomyocytes apoptosis through regulating the Bax/Bcl-2 ratio. | γδT-cells played a pathogenic role in myocardial ischemia-reperfusion injury by inducing cardiomyocytes apoptosis and neutrophils infiltration. | [48] |
| Myocardial Ischemia-Reperfusion Injury (I/RI) | CD4+ Tregs/mice | 3h-1 day | CD4+ Tregs rapidly accumulated in mouse heart following ischemia-reperfusion. The adoptive transfer of in vitro-activated Tregs attenuated cardiomyocyte apoptosis, activated a pro-survival pathway involving Akt, ERK and inhibited neutrophil infiltration, which was compromised by CD39 deficiency. | Tregs played a protective role in myocardial ischaemia-reperfusion injury and the protective effect of Tregs was at least partially related to CD39 expression. | [52] |
| Myocardial Ischemia-Reperfusion Injury (I/RI) | Tregs/ ApoeR61h/h/ SRB1-/- mice | 4-weeks | Long-term FTY720 feeding mice significantly improved left ventricular developed pressure and reduced infarct size compared with controls after ex vivo cardiac ischemia-reperfusion injury. | The benefits of FTY720 treatment in I/RI patients through increasing the percentage of Tregs and nature Tregs activity in the circulation, spleen, and lymph nodes. | [53] |
| Myocardial Infarction (MI) | T cells/CD4+ T-cell-deficient mice | 56-days | CD4 KO mice displayed higher total numbers of leukocytes and proinflammatory monocytes. Collagen matrix formation in the infarct zone was severely disturbed in CD4 KO mice. | CD4+ T-cells become activated after MI, presumably driven by recognition of cardiac autoantigens, and facilitated wound healing of the myocardium. | [54] |
| Myocardial Infarction (MI) | T-cells/CD73-/-, CD4-CD73-/- mice | 4-weeks | The CD73, which were expressed on T-cells, degraded the damaged cells and generated the proinflammatory danger signal ATP to the anti-inflammatory mediator adenosine. An accelerated secretion of IL-2, INF-γ, and IL-17 were detected in CD73 deficient mice after MI. | CD73 on T-cells orchestrated cardiac wound healing after myocardial infarction via purinergic metabolic reprogramming. | [55] |
| Myocardial Infarction (MI) | γδT-cells/ IL-17A-KO and TCRγδ-KO mice | 7-28 days | A deficiency in IL-23, IL-17A, or γδT cells improved mice survival after 7 days post-MI. These deficiencies also limited infarct expansion and fibrosis in noninfarcted myocardium, alleviated left ventricular dilatation and systolic dysfunction on day 28 post-MI. | The IL-23/IL-17A immune axis and γδT cells were potentially promising therapeutic targets after MI to prevent progression to end-stage dilated cardiomyopathy. | [56] |
| Myocardial Infarction (MI) | iNK cells/mice | 7-28 days | The infiltration of iNKT cells were increased during early phase in noninfarcted left ventricular from MI. αGC injection further enhanced iNKT cells infiltration, decreased myocyte hypertrophy, interstitial fibrosis, and apoptosis, increased survival rate and attenuated left ventricular cavity dilatation and dysfunction after MI. | iNKT cells played a protective role against post-MI left ventricular remodeling and failure through the enhanced expression of cardioprotective cytokines such as IL-10. | [57] |
| Myocardial Infarction (MI) | CD8+AT2R+ T-cells or CD4+AT2R+ T-cells /rats | 7-days or 2-weeks | CD8+AT2R+ T-cell and CD4+AT2R+ T-cell populations were increased during ischemic heart injury and contributed to maintaining cardiomyocytes viability, providing IL-10 and reducing infarct size. | AT2R activation enhances cardioprotective CD8+AT2R+/ CD4+AT2R+ T-cells and IL-10 production in the infarcted myocardium, revealing an AT2R-mediated cellular mechanism in reducing inflammatory injury in the heart. | [58, 59] |
| Myocardial Infarction (MI) | Tregs/Foxp3(DTR) mice | 7-days | Therapeutic Tregs activation led to improve healing and survival and showed increased collagen de novo expression within the scar, correlating with decreased rate of left ventricular ruptures. | Tregs beneficially influenced wound healing after MI by modulating monocyte/macrophage differentiation through secreting factors, including IL-10, IL-13, and TGF-β. Therapeutic activation of Tregs constituted a novel approach to improve healing post-MI. | [60, 62] |
| Myocardial Infarction (MI) | Tregs/FoxP3(EGFP) reporter mice | 1-7 days | Tregs depletion in infarcted mice accelerated ventricular dilation and accentuated apical remodeling. In vitro, Tregs modulated the cardiac fibroblast phenotype, reducing expression of α-smooth muscle actin, decreasing expression of matrix metalloproteinase-3, and attenuating contraction of fibroblast-populated collagen pads. | Tregs directly modulated cardiac fibroblasts phenotype and function and played a therapy role in the attenuation of adverse postinfarction remodeling. | [64] |
| Myocardial Infarction (MI) | Tregs/rats | 4-weeks | Interstitial fibrosis, myocardial matrix metalloproteinase-2 activity and cardiac apoptosis were attenuated in the rats that received Tregs transfer. Infiltration of neutrophils, macrophages and lymphocytes as well as TNF-α and IL-1β were also significantly decreased, and the CD8+ cardiac-specific cytotoxic T-cell response were also inhibited by Tregs transfer. | Tregs served to protect against adverse ventricular remodeling and contributed to improve cardiac function after myocardial infarction via inhibition of inflammation and direct protection of cardiomyocytes. | [66] |
| Myocardial Infarction (MI) | Tregs/CCR5-/- mice | 1-7 days | Deficiency of CCR5 was associated with low recruitment of CD4+ FoxP3+ Tregs. Reduced IL-10 expression, TIMP levels, and enhanced MMP-2 and MMP-9 activity were detected in CCR5-/- mice and thereby resulting in worse cardiac dilation. | CCR5-mediated Treg recruitment could restrain postinfarction inflammation, prevent excessive matrix degradation and attenuate adverse remodeling. | [67] |
| Myocardial Infarction (MI) | Bone marrow-derived NK cells/mice | 35-days | The transferring c-kit+ bone marrow derived NK cells into the myocardial infarcted heart reduced cardiomyocytes apoptosis and collagen deposition, along with an increased in neovascularization. | c-kit+ bone marrow-derived NK cells contributed to improved remodeling and cardiac function after MI. | [113, 114] |
| Cardiac Hypertrophy and Heart Failure (HF) | CD4+ T-cells/ RAG1/2-/- mice | 6-weeks for TAC/2-weeks for Ang-II | RAG1/2-/- mice did not develop cardiac dilation and showed improved contractile function and blunted adverse remodeling in TAC or Ang-II induced heart disease mouse models. The adoptive transfer of T, but not B-cells, worsened the hypertension in these models. | CD4+ T cells promoted the progression of TAC or Ang-II induced cardiac hypertrophy and HF. | [79, 80] |
| Cardiac Hypertrophy | CD4+CD25+ Tregs/Ang-II infused hypertensive mice | 2-weeks | Adoptive transferring Tregs into Ang-II infused hypertensive or aortic constriction mice improved cardiac hypertrophy and reduced cardiac fibrosis. | Tregs played an immunosuppressive effect to meliorate cardiac damage in Ang-II induced cardiac hypertrophy. | [85-87] |
| Cardiac Hypertension | CD8+ T-cells/ CD8 KO mice | 7-days | The infiltration and activation of macrophages were hallmarks of acute cardiac inflammatory during high blood pressure. CD8+ T-cells depletion showed significantly reduced macrophages infiltration and cardiac inflammatory response to the elevation of blood pressure after Ang-II infusion. | CD8+ T cells were required for macrophage infiltration in myocardium and were required to initiate and augment acute cardiac inflammatory response to high blood pressure. | [81] |
| Hypertension | Th17 cells/ IL-17-/- mice | 28-days | Vessels from IL-17-/- mice displayed preserved vascular function, decreased superoxide production, and reduced T-cell infiltration in response to Ang-II. | IL-17 that was produced by Th17 cells was critical for the maintenance of Ang-II induced hypertension and vascular dysfunction. | [82-84] |
| Myocarditis | CD4+ T-cells/mice | 1-3 weeks | Depletion of CD4+ T cells showed protective effect against the induction of myocarditis, while depletion of CD8+ T cells reduced the severity of inflammation but did not prevent induction of myocarditis. | CD4+ T-cells were required for the initiation of myocarditis and systolic dysfunction during disease progression. | [88-90] |
| Myocarditis | Tregs/murine models | 1-3 weeks | In virus-induced myocarditis, Tregs limited the immunopathology and prevented tissue damage. Adoptive transfer Tregs suppressed the immune response to cardiac tissue, protected against CVB3-induced cardiac fibrosis via secreting IL-10 and maintaining the anti-viral immune responses and function through the TGF-β-coxsackie-adenovirus receptor pathway. | The infiltration and activation of Tregs could decrease the myocardial inflammation and prevent progression of myocarditis. | [78, 92, 93] |
| Myocarditis | NK cells/CD-1 mice | 3-days | NK cells deficient mice increased CVB3m titers in heart tissues, exacerbated myocarditis, and exhibited increased myocyte degeneration in mice that were inoculated with CVB3m. | NK cells provided some protection against CVB3m-induced myocarditis by limiting virus replication in heart tissues. | [111] |
| Experimental Autoimmune Myocarditis (EAM) | NK cells/ RAG1-/- mice, IFN-γ-/- mice, and Ccr3-/- mice | 21-days | The depletion of NK cells during EAM significantly increased myocarditis severity, elevated fibrosis and increased the percentage of cardiac-infiltrating eosinophils, which directly responsible for the increased disease severity. | NK cells protected against the development of cardiac fibrosis by preventing the accumulation of certain inflammatory populations in the myocardium, limiting collagen formation in cardiac fibroblasts and through halting eosinophil infiltration. | [112] |
| Heart Failure (HF) | CD73 expressing T-cells/ CD73-/- mice | 16-weeks | CD73 expressing T-cells were enhanced in TAC-induced heart failure. The expression of CD73 on T-cells could form adenosine, which further inhibited cardiac inflammation and fibrosis and preserved contractile function. | CD73 on T-cells played an important anti-inflammatory role in TAC-induced heart failure. | [97] |
| Nonischemic Heart Failure (HF) | IFN-γ+/+ Th1 cells/TCR-α-/- mice and IFN-γ-/-mice | 4-weeks | IFN-γ+/+ Th1 cells infiltrated into myofibroblasts after nonischemic HF and selectively drove cardiac fibrosis both in vitro and in vivo. Adoptive transfer of Th1 cells, opposite to activated IFN-γ-/- Th cells, partially reconstituted cardiac fibrosis and HF in TCR-α-/- recipient mice. | Th1 cells were integrators of perivascular cardiac fibrosis and cardiac dysfunction in nonischemic HF. | [94, 95] |
| Ischemic Heart Failure (HF) | CD4+ T-cells/mice | 8-weeks | CD4+ T-cells were globally expanded and activated in ischemic HF. Antibody-mediated CD4+ T-cells depletion in HF mice prevented progressive left ventricular dilatation and hypertrophy, whereas adoptive transfer of splenic CD4+ T-cells from donor HF mice induced long-term left ventricular dysfunction, fibrosis, and hypertrophy in naïve recipient mice. | Cardiac and splenic T-cells in HF were primed to induce cardiac injury and remodeling, and retained this memory upon adoptive transfer. | [96] |
Role of T-cells in experimental models of heart injury. T-cells infiltrate into the myocardium after myocardial injury. The infiltrated γδT cells in injured myocardium promote the production of IL-17A, which mediate the apoptosis of cardiomyocytes by regulating the ratio of Bax/Bcl-2. Conventional T (Tconv) cells in damaged myocardium promote myocardium injury, induce proinflammatory monocyte recruitment and a proinflammatory milieu. IFN-γ that produced by T cells, aggravates cardiac injury and apoptosis of cardiomyocytes. Tregs promote anti-inflammatory milieu through inhibit the accumulation of inflammatory cells (such as neutrophils, monocytes and Tconv cells) and the production of inflammatory cytokines (TNF-α, IL-1β, IFN-γ, etc.). In addition, Tregs inhibit the M1-macrophages differentiation from Ly-6Chigh monocytes and derived cytokines (IL-10, IL-13, TGFβ) induce an M2-like differentiation state of monocyte-derived cells, which are characterized to promote angiogenesis and collagen formation. Moreover, the IL-10, an anti-inflammatory cytokine that is secreted by Tregs and iNK cells, modulate cardiac fibroblasts and protect left ventricular remodeling.
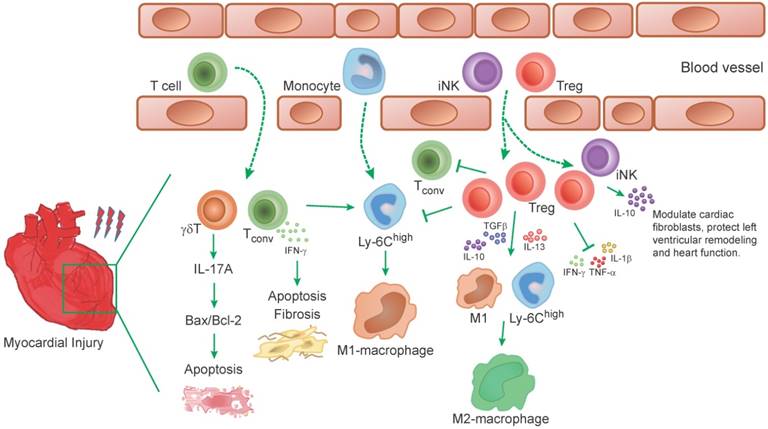
A considerable number of clinical studies have analyzed T-cells in peripheral blood in patients with acute coronary syndromes. The data partly supports a pro-atherogenic role of T-cells subset on top of the traditional cardiovascular risk factors [49]. A recent clinical study reported the role of effector T-cells in the development of microvascular obstruction (MVO) and myocardial ischemia-reperfusion injury. That study suggested that effector T-cells may contribute to MVO through direct trapping within the myocardial microvasculature, where they release inflammatory mediators, contributing to further leukocyte infiltration and progressive myocardial damage [46] (Figure 3). Meanwhile, the same group reported data on the CD8 memory T-cells compartment in patients with cytomegalovirus-seropositive (CMV) myocardial ischemia and reperfusion. A long-lasting fall in the peripheral frequency of terminally differentiated CD8 effector memory T-cells in CMV patients was found. Further studies indicated that PD-1 might be involved in the persistent loss of CD8 memory T-cells in CMV positive patients [50].
Characters of T and NK cells in clinical studies of heart injury. T and NK cells are increased in myocardium after myocardial injury. Myocardial injury in patients activate T-cells which subsequently home to the injured myocardium and modulate local inflammatory activity. Effector T-cells are detected to be increased within the myocardial microvasculature and release inflammatory mediators that contribute to further leukocyte infiltration. In cardiovascular diseases such as acute coronary syndromes and atherosclerosis, CD4+CD28null T-cells present in the blood in the context of acute coronary events during several months and secreted IFN-γ and TNF-α, as well as cytotoxic mediators in the blood. Which may cause vascular damage either directly by killing endothelial cells or indirectly through macrophage activation. During the myocardial injury, Tregs are established to migrate from peripheral to the inflamed tissue to suppress diverse types of immune reactions and the inflammatory responses, such as in suppressing CD4+ CD25- T-cells proliferation and pro-inflammatory cytokines production. However, these protective Tregs show higher frequency of apoptosis than health controls (detected in non-ST-segment elevation acute coronary syndrome (NSTEACS) patients only). The same as Tregs, a decreased NK cell number is detected in coronary artery disease patients, which further induce a persistent low-grade inflammation in these patients.
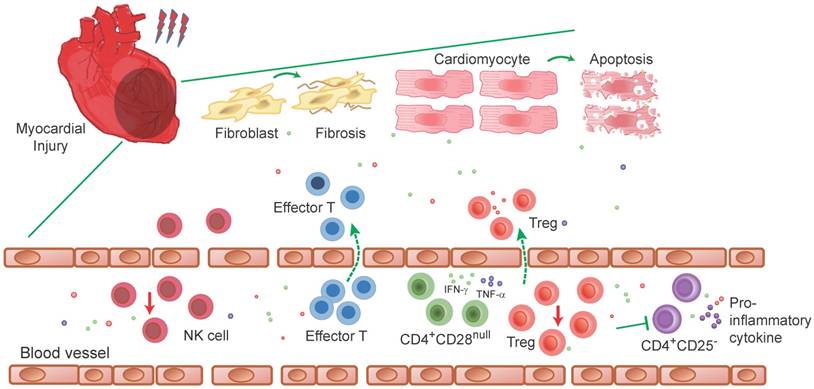
Regulatory T cells (Tregs)
The T-cells subsets with immunosuppressive functions have also been described in experimental studies. The most important one was nature regulatory T cells (Tregs), which can directly or through inhibition of antigen-presenting cells to suppress activated effector T-cells and limit autoimmunity and maintain self-tolerance. Tregs were reported to protect multiple organs from ischemia-reperfusion injury, such as intestine, liver, kidney etc., through regulating immune and inflammatory responses [51]. A recent study reported that CD4+ Tregs rapidly accumulated in mouse heart following ischemia-reperfusion. The adoptive transfer of in vitro activated Tregs significantly decrease myocardial injury but not the non-activated Tregs. This protective effect of activated Tregs was related to ectonucleoidase (CD39) expression, which means the protective effect of Tregs was at least partially related to CD39 expression [52]. The FTY720, an analog of sphingosine-1-phosphate, further confirmed the protective effect of Tregs on mouse myocardial ischemia-reperfusion injury. Long-term FTY720 treatment enhanced left ventricular function and increased longevity in ischemia-reperfusion injury mice through increasing the percentage of Tregs in the circulation, spleen, and lymph nodes [53] (Table 3, Figure 2). All these findings indicated that Tregs specific expanding could be promising in attenuating myocardial ischemia-reperfusion injury. However, the direct healing mechanism of Tregs in myocardial ischemia-reperfusion injury has not yet been fully described, which demanded further investigation.
T-cells in myocardial infarction
CD4+ T-cells
Experimental animal models provided convincing evidence that conventional T-cells involved in post-myocardial infarction and heart remodeling. CD4+ T-cells were first been demonstrated the deleterious role in modulating infarct size after myocardial infarction by using anti-CD4 specific depleting antibody in a WT mouse model [47].
In contrast to its deleterious role in myocardial infarction, T-cells were also needed for proper healing after myocardial infarction. In some studies, CD4+ T-cells were reported to become activated in mediastinal lymph nodes within few days after permanent artery coronary ligation induced myocardial infarction mouse model and were required for collagen deposition, a protective mechanism against left cavity dilation and rupture. CD4+ T cell-deficient mice subjected to myocardial infarction showed increased left ventricular dilatation, increased leukocytes, and a higher prevalence of proinflammatory monocytes [13, 54]. The CD73, which were expressed on T-cells, could degrade the damaged cells and generate the proinflammatory danger signal ATP to the anti-inflammatory mediator adenosine. After myocardial infarction, an accelerated secretion of proinflammatory and profibrotic cytokine, such as IL-2, INF-γ, and IL-17, would be detected in CD73 deficient mice. The CD73 on T-cells orchestrated cardiac wound healing after myocardial infarction via purinergic metabolic reprogramming [55] (Table 3).
γδ T-cells and NKT-cells
Along with conventional CD4+ T-cells, γδ T-cells and NKT-cells were also detected in the mouse myocardium after myocardial infarction [5]. γδ T-cells were recruited to the myocardium within days after myocardial infarction in murine models and produced IL-17A in the myocardium. The infarct size of IL-17A deficient mice was like that in WT mice 24 hours after surgery, however, a deficiency in IL-17A, or γδ T-cells improved survival after 7 days. These deficient mice limited infarct expansion and fibrosis in non-infarcted myocardium and alleviated left ventricular dilatation and systolic dysfunction on day 28 post-myocardial infarction [56]. Thus, IL-17A was involved in late-stage ventricular remodeling after myocardial infarction.
In experimental studies, the infiltration of iNK cells was detected to be increased during early phase in non-infarcted left ventricular from myocardial infarction. iNK cells could be activated by either the MHC class I-like molecule CD11d presented antigens, cytokines or the glycospingolipid α-galactosylceramide (αGC). αGC injection after myocardial infarction could further enhanced the iNK cell infiltration. Further study confirmed that iNK cells played a protective role against left ventricular remodeling and failure post-myocardial infarction via enhanced expression of anti-inflammatory cytokines such as IL-10 [57] (Table 3, Figure 2).
CD8+ T-cells
The major role of CD8+ T-cells in immune system was directly killing virally infected or damaged cells. Little was known about the pathogenic role of CD8+ T-cells in myocardial infarction. In a rat myocardial infarction model that was induced by permanent ligation of descending left coronary artery, CD8+ T-cells were detected in the peri-infarct myocardium 7 days after myocardial infarction in rats [58]. Angiotensin AT2R, which was verified having a cardioprotective role by emerging evidence, was detected in a fraction of CD8+ T-cells infiltrating in the peri-infarct myocardium. This subset CD8+ AT2R+ T-cells differed from CD8+ AT2R- T-cells in their capacity to upregulate IL-10 and downregulate IL-2 and INF-γ expression in response to Ang-II stimulation in vitro. Furthermore, the function of these CD8+ AT2R+ T-cells was demonstrated by the transfer of these cells that were harvested from a donor post-MI. These cells contribute to maintaining cardiomyocyte viability and reducing infarct size 2 weeks after experimental myocardial infarction [58]. The same beneficial action was also reported in CD4+ AT2R+ T-cells. These CD4+ AT2R+ T-cells improved heart function post-myocardial infarction corresponding with reduced infarction size in a rat myocardial infarction model [59].
Tregs
In a murine myocardial infarction model, both the conventional T-cells and the Foxp3+ CD4+ Tregs were activated and proliferated in heart draining lymph nodes. The absolute number of conventional T-cells and Tregs in myocardium and lymph nodes increased after myocardial infarction and peaked on day 7 [5, 60]. After myocardial infarction, the ischemic heart experiences many compensatory changes in ventricular shape, size and even the function to maintain the body's sufficient cardiac output, which was called ventricular remodeling. Meanwhile, the persistent inflammatory reaction in infarcted myocardium during these processes increased cardiomyocytes apoptosis and promoted interstitial fibrosis [61]. Tregs infiltrate into the myocardium after myocardial infarction and might attenuate myocardial infarction-induced ventricular remodeling [51, 54, 62-65]. By secreting factors, including IL-10, IL-13, and transforming growth factor beta (TGF-β), Tregs promoted the differentiation of recruited Ly-6Chigh monocytes and M1 macrophages toward anti-inflammatory M2 macrophages, which was associated with increased collagen de novo formation in the scar correlating with decreased rates of left ventricular ruptures. Absence of Foxp3+ CD4+ Tregs induced a preferential M1 macrophages differentiation [60] (Figure 2).
Moreover, expansion of Tregs in rats through the adoptive transfer or a CD28 super-agonistic antibody (JJ316) significantly improved cardiac function in rats after myocardial infarction. These overexpressed Tregs reduced the infiltration of neutrophils, macrophages, lymphocytes, etc, and inhibited the CD8+ cardiac specific cytotoxic T-cells response. The production of pro-inflammatory cytokines such as TNF-α and IL-1β was also reduced by Tregs transfer [66]. Furthermore, Tregs could directly modulate cardiac fibroblasts phenotype and function, whose persistent activation promoted fibrosis and adverse ventricular remodeling [64, 66]. Thus, Tregs protected against adverse ventricular remodeling and improved cardiac function after myocardial infarction not only via inhibition of inflammation but also direct modulating the phenotype and function of cardiac fibroblasts.
After myocardial infarction, Tregs were shown to traffic from peripheral blood circulation to the infarcted mouse myocardium [64, 67]. The trafficking of Tregs is partially mediated by their surface expressed specific chemokine receptors (CCRs). In the infarcted mouse myocardium, CCR5 and its ligands macrophage inflammatory protein (MIP)-1α and MIP-1β were markedly induced. Deficiency of CCR5 was associated with low recruitment of CD4+ FoxP3+ Tregs and thereby worse cardiac dilation [67]. However, the number of infiltrated Tregs was very limited in infarcted myocardium. A low number of Tregs infiltration was traced in infarcted myocardium after 1-3 days of reperfused myocardial tissue by using FoxP3-EGFP reporter mice. The depletion of Tregs through anti-CD25 antibody had no significant effects on cardiac dysfunction and scar size after reperfusion but accelerated ventricular dilation and accentuated apical remodeling, which further suggested the reparative effect of Tregs after myocardial infarction [64] (Table 3, Figure 2).
Clinical findings of T-cells
Because there were no clinically applicable non-invasive imaging techniques to detect T-cell migration and function in the human myocardium, less evidence about T-cells profiling in the context human myocardial infarction is available. Histopathological studies in necropsy specimen from patients suffering from acute myocardial infarction reported that activated T-cells infiltrate in both remote and peri-infarction regions. Activated T-cells within the epicardial coronary artery wall of both the infarct- and non-infarct-related arteries were also found in dying patients suffering from acute myocardial infarction but not in active lymphocytic myocarditis [68]. These clinical studies give a valuable hint that myocardial injury in patients may activate T-cells which subsequently home to the injured myocardium and likely modulate local inflammatory activity in patients dying suddenly, shortly, or even late after coronary thrombosis [68]. A specific human CD4+CD28null T-cells, which do not exist in mice, was investigated in cardiovascular diseases such as acute coronary syndromes and atherosclerosis. These specific human CD4+CD28null T-cells that was belong to Th1 subpopulation, was present in the blood in the context of acute coronary events during several months and secreted IFN-γ and TNF-α, as well as cytotoxic mediators in the blood. Further investigations of there cells speculated that they can cause vascular damage either directly by killing endothelial cells or indirectly through macrophage activation [69-71].
Tregs were also reported to play a key role in patients with myocardial infarction, since naturally occurring CD4+ CD25+ Treg numbers were significantly reduced in patients with acute coronary syndromes (ACS), as compared with patients with stable angina and normal coronary artery specimen. Moreover, Tregs in ACS patients were significantly compromised as their ability to suppress responder CD4+ CD25- T-cell proliferation was attenuated [72]. A similar decrease in circulating CD27+ and CD27- Treg numbers was also demonstrated in patients with ST-segment elevation myocardial infarction compared with healthy controls [73]. A protective study addressed a cox proportional hazard regression model and reported that low levels of baseline CD4+ FoxP3+ T-cells were associated with an increased risk for the development of acute coronary events, suggesting that Tregs might play a key role to protect the myocardium throughout the myocardial infarction [74]. According to a recent study, the reduced number of circulating Tregs in myocardial infarction patients might be partially due to the impaired output from the thymus, thereby increasing apoptosis and trafficking of peripheral Tregs to inflammatory sites [75]. The migratory behavior of peripheral Tregs to the inflamed tissue to suppress diverse types of immune reactions the inflammatory response has already been well established [76]. Beside that, higher frequency of Tregs apoptosis was also detected in patients with non-ST-segment elevation acute coronary syndrome (NSTEACS) than in patients with chronic stable angina (CSA) and patients with chest pain syndrome (CPS). Besides, lower frequency of recent thymic emigrant CD4+ CD25+ CD127low CD45RO- CD45RA+ CD31+ Tregs in peripheral blood was detected in patients with NSTEACS than in patients with CAS and CPS, suggesting that the production of Tregs by thymus was attenuated in NSTEACS patients [75] (Table 2, Figure 3). Unlike basic studies, which have a lot of experiential evidences, clinical studies of T-cells in myocardial infarction are still very deficient. Further clinical studies are required to elucidate the role of T-cells in post myocardial infarction.
T-cells in other cardiac disease phenotypes
Cardiac hypertrophy
Cardiac hypertrophy and remodeling are pathological features of many cardiac diseases. Hypertension, cardiomyopathy, and myocardial infarction could induce a hypertrophic response in cardiomyocytes [77]. Deleting T-cells via anti-CD3 antibody treatment in cardiac-TNF-α-overexpressing mice significantly reduced inflammatory cell recruitment and blocked hypertrophy. The same result was also detected in chronic pressure overload-induced hypertrophy [78]. B and T-cells deficient RAG2-/- mice showed reduced myocardial fibrosis and hypertrophy. Meanwhile, RAG1-/- mice (another B and T-cells deficient mice) blunted hypertension and vascular dysfunction during Ang-II or desoxycorticosterone acetate (DOCA)-slat infusion. The adoptive transfer of T, but not B-cells, worsened the hypertension in these mouse models, which meant that T-cells were involved in cardiac hypertrophy and remodeling [79, 80]. During high blood pressure, the infiltration and activation of macrophages were hallmarks of acute cardiac inflammatory. CD8+ T-cells depletion showed significantly reduced macrophages infiltration and cardiac inflammatory response to the elevation of blood pressure after Ang-II infusion. Thus, CD8+ T-cells were required to initiate and augment acute cardiac inflammatory response to high blood pressure [81]. Th17 cells were known to involve in Ang-II induced hypertension. IL-17 that was produced by Th17 cells was increased in Ang-II infusion mice and hypertension was not sustained in IL17-/- mice. Therefore, Th17-cells were critical for the maintenance of Ang-II induced hypertension and vascular dysfunction [82]. The deficiency of IL-17 has also been reported to mitigate hypertensive cardiac hypertrophy, dilated cardiomyopathy and myocardial infarction [48, 83, 84] (Table 3).
The function of Tregs in ischemia-reperfusion injury and myocardial infarction has already been mentioned in a preceding part of this review. Besides that, Tregs also plays a role in cardiac hypertrophy. Adoptive transferred Tregs into Ang-II infused hypertensive or aortic constriction mice improved cardiac hypertrophy and reduced cardiac fibrosis [85-87]. However, the mechanisms of Tregs in regulating hypertension and hypertrophic remodeling are still under investigation. Till now it is clear that Tregs can function through modulating or suppressing of other T-cells or immune cells responses in cardiac hypertrophy.
Myocarditis
The first evidence of T-cells involved in myocarditis was reported in athymic mice that lack T-cells failed to develop myocarditis [88]. Further studies in the animal models of cardiac myosin immunization-induced experimental autoimmune myocarditis (EAM) and virus-induced myocarditis demonstrated that CD4+ T-cells were required for the initiation of myocarditis and systolic dysfunction during disease progression [89, 90]. Beside conventional T-cells, Tregs were found to have a critical role in the pathogenesis of myocarditis. In coxsackievirus B3 (CVB3), herpes simplex virus or C virus-induced myocarditis, Tregs limited the immunopathology and preventing tissue damage [91, 92]. Adoptive transfer of Tregs could protect mice from CVB3-induced myocarditis. The transferred Treg suppressed the immune response to cardiac tissue, maintaining the anti-viral immune responses and function through the TGF-β-coxsackie-adenovirus receptor pathway. Furthermore, the adoptive transfer of Tregs also protected against CVB3-induced cardiac fibrosis via secreting IL-10 [78, 93] (Table 3). Thus, the infiltration and activation of Tregs can decrease the myocardial inflammation and prevent progression of myocarditis.
Heart failure
In transverse aortic constriction (TAC)-induced heart failure, CD4+ T-cells were detected. Mice that lacking CD4+ T-cells did not undergo ventricular dilatation, and showed a drastic reduction in fibrosis by attenuating collagen accumulation after TAC. Furthermore, these mice exhibited preserved contractile function in TAC-induced heart failure [79]. In a nonischemic heart failure mouse model, infiltrating Th1 effector T-cell numbers were detected to be increased in the fibrotic myocardium in nonischemic heart failure patients. These Th1 cells acted as integrators of perivascular cardiac fibrosis and cardiac dysfunction in nonischemic heart failure [94, 95]. Unlike in nonischemic heart failure, CD4+ T-cells were globally expanded and activated in chronic ischemic heart failure. Antibody-mediated CD4+ T-cells depletion in these heart failure mice prevented progressive left ventricular dilatation and hypertrophy [96]. Besides that, T-cells have also been reported as a protector in some conditions. In TAC-induced heart failure, a significantly enhanced infiltration of CD73 expressing T-cells were detected. The expression of CD73 on T-cells could form adenosine, which further inhibited cardiac inflammation and fibrosis and preserved contractile function. T-cells isolated from TAC-treated hearts showed enhanced production of proinflammatory cytokines in CD73 deficient mice. Therefore, CD73 on T-cells plays an important anti-inflammatory role during TAC-induced heart failure, which is associated with antifibrotic activity and reduced production of proinflammatory cytokines [97] (Table 3).
In clinical studies, Treg numbers were also related with heart failure, since circulating Treg numbers in peripheral blood mononuclear cells (PBMCs) were significantly reduced in chronic heart failure patients than healthy controls. Tregs from these patients showed a compromised effect in suppressing CD4+ CD25- T-cells proliferation and pro-inflammatory cytokines production. These functions were directly related to left ventricular end-diastolic dimension and levels of N-terminal prohormone brain natriuretic peptide [98-100] (Table 2). Therefore, the protective effect of Tregs might be a therapeutic target in the field of heart failure clinical therapy.
Therapeutic strategies of T-cells in human heart diseases
Experimental and pre-clinical studies have shown that T-cells were involved in the progressing of heart diseases. The accumulation of T-cells in the infarcted myocardium was detected within minutes following I/RI and that CD4+ T-cells contributed to additional tissue injury [47]. Unlike other T-cells, experimental studies indicated an important protective role of Tregs in improving myocardium regeneration by modulating the function of other leucocytes after heart injury [51]. Dependent on the potential therapeutic ability of Tregs, several therapeutic strategies for Tregs expansion are currently being developed as a new direction for heart disease patient treatment.
Without engagement of the T cell receptor, stimulation of CD28 with specific superagonistic antibodies could induce polyclonal Tregs expansion and IL-10 overproduction in animal studies [101, 102]. However, despite promising experiments results of Tregs, the phase I trial of superagonistic anti-CD28 antibody in human was stopped due to unexpected toxicity [103]. Beside anti-CD28 antibody, other strategies like anti-CD3 monoclonal antibody injection or supplementation with low-dose IL-2 have also been reported in experimental studies [104, 105]. Thus, in clinical studies, such an IL-2 based strategy in patients with stable ischemic heart disease and acute coronary syndromes (LILACS) is currently under investigation in myocardial infarction patients [106]. However, till now, no clinical trials that specifically targeting the Tregs or other T-cells have been successfully finished yet.
In addition to the Tregs expansion therapeutic strategies for clinical heart disease patient treatment, the function of Tregs in myocardial ischemia-reperfusion injury area was described in adjuvant therapeutic strategies, such as to minimize myocardial ischemia-reperfusion injury through pharmacological interventions and the complex interactions with body repair system [107]. Using rosuvastain before myocardial ischemia-reperfusion, cardiac injury was attenuated and the inflammatory cell infiltration was reduced. This rosuvastain pretreatment was associated with Tregs accumulation, suggesting that such cardioprotection against ischemia-reperfusion was partially mediated by immunosuppression of Tregs [108]. Besides that, in post-myocardial infarction and heart failure medication studies, several drug classes also affect a range of immunological pathway and modulate the post-MI immune response, including ROS, complement, leukocyte infiltration, inflammatory cytokines IL-1, and the inhibition of B and T-cells, which is crucial to take into account when designing future immunomodulatory trails [109].
There are considerable experimental evidences that T-cells play an important role in the response to heart diseases. Experimental mouse models of myocardial infarction, ischemia-reperfusion injury, ischemic cardiomyopathy, and other cardiovascular diseases revealed that T-cells infiltrate and function as 'reparative' or 'destructive' after myocardial damage. Although the molecular mechanisms underlying the effects of T-cells in these diseases are largely unknown, T-cells might be a promising therapeutic target in cardiovascular diseases.
NK cells in heart diseases
Innate lymphoid cell (ILC) family functions in both repairing damaged tissue and maintaining tissue homeostasis. NK cells contain the largest subset of ILCs [110]. Among all the leukocytes, although only 1% of NK cells are present in a healthy heart, they still participate in regulating the process of heart diseases [15].
Experimental murine models provided convincing evidences that NK cells protected against acute viral pathogens such as CVB and murine cytomegalovirus induced myocarditis [111]. NK cells have also been reported to protect against the development of cardiac fibrosis by preventing the accumulation of certain inflammatory populations in the myocardium and directly limiting collagen formation in cardiac fibroblasts. NK cells protected against fibrosis in the heart through negative regulation of pro-fibrotic cell types by the production of IFN-γ and other mediators [112]. NK cells could also limit cardiac fibrosis through halting eosinophil infiltration. In EAM, eosinophil resultant influx to the heart directly increased disease severity. NK cells altered eosinophil-related chemokine production by cardiac fibroblasts to indirectly limit the eosinophilic infiltration [112]. Besides that, transferring c-kit+ bone marrow derived NK cells into the myocardial infarcted heart responded a reduction in cardiomyocytes apoptosis and collagen formation [113, 114] (Table 3). Thus, in some EAM and post-myocardial infarction conditions, NK cells function through its protective effects against the development of cardiac fibrosis.
Cardiac allograft vasculopathy (ACV) was known as the major cause of late mortality after cardiac transplantation. In clinical cardiac transplantation, NK cells were crucial in distinguishing self and non-self origin of the cells, which were therefore accumulated in lesions found in donor tissue after cardiac transplants. Some but not all these infiltrated NK cells, with high levels of CD16 expression, contributed to acute rejection and induced ACV. However, other NK cells functioned like protectors in cardiac transfer [115]. In other hand, a decreased NK cell number was detected in coronary artery disease patients. These patients showed lower NK cytotoxic activity and a decreased absolute number and percentage of CD3-CD56dim cytotoxic NK subset [116, 117]. During a 12-month follow-up, the number of NK cells increased in part of the coronary artery disease patients. The failed NK cells reconstituted patients exhibited a persistent low-grade inflammation, suggesting a protective role of NK cells in coronary artery disease [118] (Table 2). All these clinical results suggested that except the acute rejection that was partially induced by some high levels of CD16 expression NK cell, the vast majority NK cells played protective role in heart disease.
Although NK cells were demonstrated to be involved in heart diseases, the regulatory role of NK cells in the pathogenesis of cardiac diseases still need further investigation. More experimental and clinical studies are also required to determine exactly how NK cells and their mechanisms might be exploited for clinical therapy.
Conclusion
There are considerable experimental evidences that lymphocytes play a significant role in heart diseases. More specifically, B cells involved in the progressing of heart diseases through humoral B-cell responses or cellular B-cell responses. The absence or suppression of B-cells could alleviate the progressing of heart diseases. B-cells can also interact with T-cells to impair cardiac contractility and induce cardiac remodeling. Besides that, tons of evidences have shown that T-cells, especially Tregs play an important role in regulating inflammation and heart diseases. Low Tregs level can lead to increased infarct area upon myocardial infarction. Changing the concentration of Tregs is expected to be a new treatment method for heart diseases. In addition, NK cells have also been reported to be involved in the regulation of some heart diseases.
Although we have a better understanding of immune cells, their pro-inflammatory mediators, and their role in heart injury and repair, deeper studies are still required in this area. Future work should aim at characterizing the function of lymphocytes in clinical studies and the pathophysiological role in patients with heart diseases. A deeper understanding of lymphocytes' pathophysiological role in the heart injury will open new directions in immunomodulatory treatment options for heart disease patients [119].
Acknowledgements
This review was supported by the grants from National Natural Science Foundation of China (81722008, 91639101 and 81570362 to JJ Xiao), Innovation Program of Shanghai Municipal Education Commission (2017-01-07-00-09-E00042 to JJ Xiao), the grant from Science and Technology Commission of Shanghai Municipality (17010500100, 18410722200 to JJ Xiao), the development fund for Shanghai talents (to JJ Xiao) and the Sailing Program from Science and Technology Commission of Shanghai (19YF1415400 to J Wang). JS is supported by Horizon2020 ERC-2016-COG EVICARE (725229) and Technobeat (668724).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Sarrafzadegan N, Gotay C. CVD prevention in 2014: Advances in the prevention of cardiovascular disease. Nat Rev Cardiol. 2015;12:71-3
2. Wang L, Lv Y, Li G, Xiao J. MicroRNAs in heart and circulation during physical exercise. J Sport Health Sci. 2018;7:433-41
3. Batacan RB Jr, Duncan MJ, Dalbo VJ, Buitrago GL, Fenning AS. Effect of different intensities of physical activity on cardiometabolic markers and vascular and cardiac function in adult rats fed with a high-fat high-carbohydrate diet. J Sport Health Sci. 2018;7:109-19
4. Hashimoto H, Olson EN, Bassel-Duby R. Therapeutic approaches for cardiac regeneration and repair. Nat Rev Cardiol. 2018;15:585-600
5. Yan X, Anzai A, Katsumata Y, Matsuhashi T, Ito K, Endo J. et al. Temporal dynamics of cardiac immune cell accumulation following acute myocardial infarction. J Mol Cell Cardiol. 2013;62:24-35
6. Anzai A, Anzai T, Nagai S, Maekawa Y, Naito K, Kaneko H. et al. Regulatory role of dendritic cells in postinfarction healing and left ventricular remodeling. Circulation. 2012;125:1234-45
7. Arslan F, de Kleijn DP, Pasterkamp G. Innate immune signaling in cardiac ischemia. Nat Rev Cardiol. 2011;8:292-300
8. Nahrendorf M, Pittet MJ, Swirski FK. Monocytes: protagonists of infarct inflammation and repair after myocardial infarction. Circulation. 2010;121:2437-45
9. Marchant DJ, Boyd JH, Lin DC, Granville DJ, Garmaroudi FS, McManus BM. Inflammation in myocardial diseases. Circ Res. 2012;110:126-44
10. Wang YP, Xie Y, Ma H, Su SA, Wang YD, Wang JA. et al. Regulatory T lymphocytes in myocardial infarction: A promising new therapeutic target. Int J Cardiol. 2016;203:923-8
11. Frangogiannis NG. Regulation of the inflammatory response in cardiac repair. Circ Res. 2012;110:159-73
12. Cipryan L. The effect of fitness level on cardiac autonomic regulation, IL-6, total antioxidant capacity, and muscle damage responses to a single bout of high-intensity interval training. J Sport Health Sci. 2018;7:363-71
13. Hofmann U, Frantz S. Role of lymphocytes in myocardial injury, healing, and remodeling after myocardial infarction. Circ Res. 2015;116:354-67
14. Myrphy K, Travers P, Walport M, Janeway CA. Janeway's Immunobiology. 8th ed. New York: Garland Science. 2012:249
15. Yu YR, O'Koren EG, Hotten DF, Kan MJ, Kopin D, Nelson ER. et al. A Protocol for the Comprehensive Flow Cytometric Analysis of Immune Cells in Normal and Inflamed Murine Non-Lymphoid Tissues. PLoS One. 2016;11:e0150606
16. Zouggari Y, Ait-Oufella H, Bonnin P, Simon T, Sage AP, Guerin C. et al. B lymphocytes trigger monocyte mobilization and impair heart function after acute myocardial infarction. Nat Med. 2013;19:1273-80
17. Nishimura H, Okazaki T, Tanaka Y, Nakatani K, Hara M, Matsumori A. et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science. 2001;291:319-22
18. Kaya Z, Katus HA, Rose NR. Cardiac troponins and autoimmunity: their role in the pathogenesis of myocarditis and of heart failure. Clin Immunol. 2010;134:80-8
19. Cordero-Reyes AM, Youker KA, Trevino AR, Celis R, Hamilton DJ, Flores-Arredondo JH. et al. Full Expression of Cardiomyopathy Is Partly Dependent on B-Cells: A Pathway That Involves Cytokine Activation, Immunoglobulin Deposition, and Activation of Apoptosis. J Am Heart Assoc. 2016:5
20. Lachtermacher S, Esporcatte BL, Montalvao F, Costa PC, Rodrigues DC, Belem L. et al. Cardiac gene expression and systemic cytokine profile are complementary in a murine model of post-ischemic heart failure. Braz J Med Biol Res. 2010;43:377-89
21. Haas MS, Alicot EM, Schuerpf F, Chiu I, Li J, Moore FD. et al. Blockade of self-reactive IgM significantly reduces injury in a murine model of acute myocardial infarction. Cardiovasc Res. 2010;87:618-27
22. Zhang M, Michael LH, Grosjean SA, Kelly RA, Carroll MC, Entman ML. The role of natural IgM in myocardial ischemia-reperfusion injury. J Mol Cell Cardiol. 2006;41:62-7
23. Keppner L, Heinrichs M, Rieckmann M, Demengeot J, Frantz S, Hofmann U. et al. Antibodies aggravate the development of ischemic heart failure. Am J Physiol Heart Circ Physiol. 2018;315:H1358-h67
24. Cappuzzello C, Di Vito L, Melchionna R, Melillo G, Silvestri L, Cesareo E. et al. Increase of plasma IL-9 and decrease of plasma IL-5, IL-7, and IFN-gamma in patients with chronic heart failure. J Transl Med. 2011;9:28
25. Deswal A, Petersen NJ, Feldman AM, White BG, Mann DL. Effects of vesnarinone on peripheral circulating levels of cytokines and cytokine receptors in patients with heart failure: a report from the Vesnarinone Trial. Chest. 2001;120:453-9
26. Torre-Amione G, Kapadia S, Benedict C, Oral H, Young JB, Mann DL. Proinflammatory cytokine levels in patients with depressed left ventricular ejection fraction: a report from the Studies of Left Ventricular Dysfunction (SOLVD). J Am Coll Cardiol. 1996;27:1201-6
27. Staudt A, Eichler P, Trimpert C, Felix SB, Greinacher A. Fc(gamma) receptors IIa on cardiomyocytes and their potential functional relevance in dilated cardiomyopathy. J Am Coll Cardiol. 2007;49:1684-92
28. Kaya Z, Leib C, Katus HA. Autoantibodies in heart failure and cardiac dysfunction. Circ Res. 2012;110:145-58
29. Staudt Y, Mobini R, Fu M, Felix SB, Kuhn JP, Staudt A. Beta1-adrenoceptor antibodies induce apoptosis in adult isolated cardiomyocytes. Eur J Pharmacol. 2003;466:1-6
30. Limas CJ, Goldenberg IF, Limas C. Autoantibodies against beta-adrenoceptors in human idiopathic dilated cardiomyopathy. Circ Res. 1989;64:97-103
31. Fu LX, Magnusson Y, Bergh CH, Liljeqvist JA, Waagstein F, Hjalmarson A. et al. Localization of a functional autoimmune epitope on the muscarinic acetylcholine receptor-2 in patients with idiopathic dilated cardiomyopathy. J Clin Invest. 1993;91:1964-8
32. Warraich RS, Griffiths E, Falconar A, Pabbathi V, Bell C, Angelini G. et al. Human cardiac myosin autoantibodies impair myocyte contractility: a cause-and-effect relationship. FASEB J. 2006;20:651-60
33. Baba A, Yoshikawa T, Ogawa S. Autoantibodies produced against sarcolemmal Na-K-ATPase: possible upstream targets of arrhythmias and sudden death in patients with dilated cardiomyopathy. J Am Coll Cardiol. 2002;40:1153-9
34. Landsberger M, Staudt A, Choudhury S, Trimpert C, Herda LR, Klingel K. et al. Potential role of antibodies against cardiac Kv channel-interacting protein 2 in dilated cardiomyopathy. Am Heart J. 2008;156:92-9 e2
35. Crampton SP, Voynova E, Bolland S. Innate pathways to B-cell activation and tolerance. Ann N Y Acad Sci. 2010;1183:58-68
36. Timmers L, Sluijter JP, van Keulen JK, Hoefer IE, Nederhoff MG, Goumans MJ. et al. Toll-like receptor 4 mediates maladaptive left ventricular remodeling and impairs cardiac function after myocardial infarction. Circ Res. 2008;102:257-64
37. Blyszczuk P, Kania G, Dieterle T, Marty RR, Valaperti A, Berthonneche C. et al. Myeloid differentiation factor-88/interleukin-1 signaling controls cardiac fibrosis and heart failure progression in inflammatory dilated cardiomyopathy. Circ Res. 2009;105:912-20
38. Tsushima K, Osawa T, Yanai H, Nakajima A, Takaoka A, Manabe I. et al. IRF3 regulates cardiac fibrosis but not hypertrophy in mice during angiotensin II-induced hypertension. FASEB J. 2011;25:1531-43
39. Mann DL. The emerging role of innate immunity in the heart and vascular system: for whom the cell tolls. Circ Res. 2011;108:1133-45
40. Goodchild TT, Robinson KA, Pang W, Tondato F, Cui J, Arrington J. et al. Bone marrow-derived B cells preserve ventricular function after acute myocardial infarction. JACC Cardiovasc Interv. 2009;2:1005-16
41. An S, Wang X, Ruck MA, Rodriguez HJ, Kostyushev DS, Varga M. et al. Age-Related Impaired Efficacy of Bone Marrow Cell Therapy for Myocardial Infarction Reflects a Decrease in B Lymphocytes. Mol Ther. 2018;26:1685-93
42. Santos-Zas I, Lemarie J, Tedgui A, Ait-Oufella H. Adaptive Immune Responses Contribute to Post-ischemic Cardiac Remodeling. Front Cardiovasc Med. 2018;5:198
43. Panahi M, Papanikolaou A, Torabi A, Zhang JG, Khan H, Vazir A. et al. Immunomodulatory interventions in myocardial infarction and heart failure: a systematic review of clinical trials and meta-analysis of IL-1 inhibition. Cardiovasc Res. 2018;114:1445-61
44. Rituximab in Patients with Acute ST-elevation Myocardial Infarction Study—Full Text View—ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT03072199
45. Boehm T, Swann JB. Thymus involution and regeneration: two sides of the same coin? Nat Rev Immunol. 2013;13:831-8
46. Boag SE, Das R, Shmeleva EV, Bagnall A, Egred M, Howard N. et al. T lymphocytes and fractalkine contribute to myocardial ischemia/reperfusion injury in patients. J Clin Invest. 2015;125:3063-76
47. Yang Z, Day YJ, Toufektsian MC, Xu Y, Ramos SI, Marshall MA. et al. Myocardial infarct-sparing effect of adenosine A2A receptor activation is due to its action on CD4+ T lymphocytes. Circulation. 2006;114:2056-64
48. Liao YH, Xia N, Zhou SF, Tang TT, Yan XX, Lv BJ. et al. Interleukin-17A contributes to myocardial ischemia/reperfusion injury by regulating cardiomyocyte apoptosis and neutrophil infiltration. J Am Coll Cardiol. 2012;59:420-9
49. Ammirati E, Moroni F, Magnoni M, Camici PG. The role of T and B cells in human atherosclerosis and atherothrombosis. Clin Exp Immunol. 2015;179:173-87
50. Hoffmann J, Shmeleva EV, Boag SE, Fiser K, Bagnall A, Murali S. et al. Myocardial ischemia and reperfusion leads to transient CD8 immune deficiency and accelerated immunosenescence in CMV-seropositive patients. Circ Res. 2015;116:87-98
51. Meng X, Yang J, Dong M, Zhang K, Tu E, Gao Q. et al. Regulatory T cells in cardiovascular diseases. Nat Rev Cardiol. 2016;13:167-79
52. Xia N, Jiao J, Tang TT, Lv BJ, Lu YZ, Wang KJ. et al. Activated regulatory T-cells attenuate myocardial ischaemia/reperfusion injury through a CD39-dependent mechanism. Clin Sci (Lond). 2015;128:679-93
53. Wang G, Kim RY, Imhof I, Honbo N, Luk FS, Li K. et al. The immunosuppressant FTY720 prolongs survival in a mouse model of diet-induced coronary atherosclerosis and myocardial infarction. J Cardiovasc Pharmacol. 2014;63:132-43
54. Hofmann U, Beyersdorf N, Weirather J, Podolskaya A, Bauersachs J, Ertl G. et al. Activation of CD4+ T lymphocytes improves wound healing and survival after experimental myocardial infarction in mice. Circulation. 2012;125:1652-63
55. Borg N, Alter C, Gorldt N, Jacoby C, Ding Z, Steckel B. et al. CD73 on T Cells Orchestrates Cardiac Wound Healing After Myocardial Infarction by Purinergic Metabolic Reprogramming. Circulation. 2017;136:297-313
56. Yan X, Shichita T, Katsumata Y, Matsuhashi T, Ito H, Ito K. et al. Deleterious effect of the IL-23/IL-17A axis and gammadeltaT cells on left ventricular remodeling after myocardial infarction. J Am Heart Assoc. 2012;1:e004408
57. Sobirin MA, Kinugawa S, Takahashi M, Fukushima A, Homma T, Ono T. et al. Activation of natural killer T cells ameliorates postinfarct cardiac remodeling and failure in mice. Circ Res. 2012;111:1037-47
58. Curato C, Slavic S, Dong J, Skorska A, Altarche-Xifro W, Miteva K. et al. Identification of noncytotoxic and IL-10-producing CD8+AT2R+ T cell population in response to ischemic heart injury. J Immunol. 2010;185:6286-93
59. Skorska A, von Haehling S, Ludwig M, Lux CA, Gaebel R, Kleiner G. et al. The CD4(+) AT2R(+) T cell subpopulation improves post-infarction remodelling and restores cardiac function. J Cell Mol Med. 2015;19:1975-85
60. Weirather J, Hofmann UD, Beyersdorf N, Ramos GC, Vogel B, Frey A. et al. Foxp3+ CD4+ T cells improve healing after myocardial infarction by modulating monocyte/macrophage differentiation. Circ Res. 2014;115:55-67
61. Sutton MG, Sharpe N. Left ventricular remodeling after myocardial infarction: pathophysiology and therapy. Circulation. 2000;101:2981-8
62. Matsumoto K, Ogawa M, Suzuki J, Hirata Y, Nagai R, Isobe M. Regulatory T lymphocytes attenuate myocardial infarction-induced ventricular remodeling in mice. Int Heart J. 2011;52:382-7
63. Chan L. Harnessing regulatory T cells for therapeutic purposes. Kidney Int. 2012;81:935-6
64. Saxena A, Dobaczewski M, Rai V, Haque Z, Chen W, Li N. et al. Regulatory T cells are recruited in the infarcted mouse myocardium and may modulate fibroblast phenotype and function. Am J Physiol Heart Circ Physiol. 2014;307:H1233-42
65. Hofmann U, Frantz S. Role of T-cells in myocardial infarction. Eur Heart J. 2016;37:873-9
66. Tang TT, Yuan J, Zhu ZF, Zhang WC, Xiao H, Xia N. et al. Regulatory T cells ameliorate cardiac remodeling after myocardial infarction. Basic Res Cardiol. 2012;107:232
67. Dobaczewski M, Xia Y, Bujak M, Gonzalez-Quesada C, Frangogiannis NG. CCR5 signaling suppresses inflammation and reduces adverse remodeling of the infarcted heart, mediating recruitment of regulatory T cells. Am J Pathol. 2010;176:2177-87
68. Abbate A, Bussani R, Liuzzo G, Biondi-Zoccai GG, Barresi E, Mellone P. et al. Sudden coronary death, fatal acute myocardial infarction and widespread coronary and myocardial inflammation. Heart. 2008;94:737-42
69. Dumitriu IE, Araguas ET, Baboonian C, Kaski JC. CD4+ CD28 null T cells in coronary artery disease: when helpers become killers. Cardiovasc Res. 2009;81:11-9
70. Liuzzo G, Biasucci LM, Trotta G, Brugaletta S, Pinnelli M, Digianuario G. et al. Unusual CD4+CD28null T lymphocytes and recurrence of acute coronary events. J Am Coll Cardiol. 2007;50:1450-8
71. Dumitriu IE. The life (and death) of CD4+ CD28(null) T cells in inflammatory diseases. Immunology. 2015;146:185-93
72. Mor A, Luboshits G, Planer D, Keren G, George J. Altered status of CD4(+)CD25(+) regulatory T cells in patients with acute coronary syndromes. Eur Heart J. 2006;27:2530-7
73. Sardella G, De Luca L, Francavilla V, Accapezzato D, Mancone M, Sirinian MI. et al. Frequency of naturally-occurring regulatory T cells is reduced in patients with ST-segment elevation myocardial infarction. Thromb Res. 2007;120:631-4
74. Wigren M, Bjorkbacka H, Andersson L, Ljungcrantz I, Fredrikson GN, Persson M. et al. Low levels of circulating CD4+FoxP3+ T cells are associated with an increased risk for development of myocardial infarction but not for stroke. Arterioscler Thromb Vasc Biol. 2012;32:2000-4
75. Zhang WC, Wang J, Shu YW, Tang TT, Zhu ZF, Xia N. et al. Impaired thymic export and increased apoptosis account for regulatory T cell defects in patients with non-ST segment elevation acute coronary syndrome. J Biol Chem. 2012;287:34157-66
76. Huehn J, Hamann A. Homing to suppress: address codes for Treg migration. Trends Immunol. 2005;26:632-6
77. Nakamura M, Sadoshima J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat Rev Cardiol. 2018;15:387-407
78. Huber SA, Feldman AM, Sartini D. Coxsackievirus B3 induces T regulatory cells, which inhibit cardiomyopathy in tumor necrosis factor-alpha transgenic mice. Circ Res. 2006;99:1109-16
79. Laroumanie F, Douin-Echinard V, Pozzo J, Lairez O, Tortosa F, Vinel C. et al. CD4+ T cells promote the transition from hypertrophy to heart failure during chronic pressure overload. Circulation. 2014;129:2111-24
80. Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S. et al. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med. 2007;204:2449-60
81. Ma F, Feng J, Zhang C, Li Y, Qi G, Li H. et al. The requirement of CD8+ T cells to initiate and augment acute cardiac inflammatory response to high blood pressure. J Immunol. 2014;192:3365-73
82. Madhur MS, Lob HE, McCann LA, Iwakura Y, Blinder Y, Guzik TJ. et al. Interleukin 17 promotes angiotensin II-induced hypertension and vascular dysfunction. Hypertension. 2010;55:500-7
83. Liu W, Wang X, Feng W, Li S, Tian W, Xu T. et al. Lentivirus mediated IL-17R blockade improves diastolic cardiac function in spontaneously hypertensive rats. Exp Mol Pathol. 2011;91:362-7
84. Baldeviano GC, Barin JG, Talor MV, Srinivasan S, Bedja D, Zheng D. et al. Interleukin-17A is dispensable for myocarditis but essential for the progression to dilated cardiomyopathy. Circ Res. 2010;106:1646-55
85. Kvakan H, Kleinewietfeld M, Qadri F, Park JK, Fischer R, Schwarz I. et al. Regulatory T cells ameliorate angiotensin II-induced cardiac damage. Circulation. 2009;119:2904-12
86. Kanellakis P, Dinh TN, Agrotis A, Bobik A. CD4(+)CD25(+)Foxp3(+) regulatory T cells suppress cardiac fibrosis in the hypertensive heart. J Hypertens. 2011;29:1820-8
87. Barhoumi T, Kasal DA, Li MW, Shbat L, Laurant P, Neves MF. et al. T regulatory lymphocytes prevent angiotensin II-induced hypertension and vascular injury. Hypertension. 2011;57:469-76
88. Woodruff JF, Woodruff JJ. Involvement of T lymphocytes in the pathogenesis of coxsackie virus B3 heart disease. J Immunol. 1974;113:1726-34
89. Smith SC, Allen PM. Myosin-induced acute myocarditis is a T cell-mediated disease. J Immunol. 1991;147:2141-7
90. Lee JH, Kim TH, Park HE, Lee EG, Jung NC, Song JY. et al. Myosin-primed tolerogenic dendritic cells ameliorate experimental autoimmune myocarditis. Cardiovasc Res. 2014;101:203-10
91. Rouse BT, Sarangi PP, Suvas S. Regulatory T cells in virus infections. Immunol Rev. 2006;212:272-86
92. Wei L, Wei-Min L, Cheng G, Bao-Guo Z. Upregulation of CD4+CD25+ T lymphocyte by adenovirus-mediated gene transfer of CTLA4Ig fusion protein in experimental autoimmune myocarditis. Autoimmunity. 2006;39:289-98
93. Shi Y, Fukuoka M, Li G, Liu Y, Chen M, Konviser M. et al. Regulatory T cells protect mice against coxsackievirus-induced myocarditis through the transforming growth factor beta-coxsackie-adenovirus receptor pathway. Circulation. 2010;121:2624-34
94. Nevers T, Salvador AM, Velazquez F, Ngwenyama N, Carrillo-Salinas FJ, Aronovitz M. et al. Th1 effector T cells selectively orchestrate cardiac fibrosis in nonischemic heart failure. J Exp Med. 2017;214:3311-29
95. Nevers T, Salvador AM, Grodecki-Pena A, Knapp A, Velazquez F, Aronovitz M. et al. Left Ventricular T-Cell Recruitment Contributes to the Pathogenesis of Heart Failure. Circ Heart Fail. 2015;8:776-87
96. Bansal SS, Ismahil MA, Goel M, Patel B, Hamid T, Rokosh G. et al. Activated T Lymphocytes are Essential Drivers of Pathological Remodeling in Ischemic Heart Failure. Circ Heart Fail. 2017;10:e003688
97. Quast C, Alter C, Ding Z, Borg N, Schrader J. Adenosine Formed by CD73 on T Cells Inhibits Cardiac Inflammation and Fibrosis and Preserves Contractile Function in Transverse Aortic Constriction-Induced Heart Failure. Circ Heart Fail. 2017:10
98. Tang TT, Ding YJ, Liao YH, Yu X, Xiao H, Xie JJ. et al. Defective circulating CD4CD25+Foxp3+CD127(low) regulatory T-cells in patients with chronic heart failure. Cell Physiol Biochem. 2010;25:451-8
99. Tang TT, Zhu ZF, Wang J, Zhang WC, Tu X, Xiao H. et al. Impaired thymic export and apoptosis contribute to regulatory T-cell defects in patients with chronic heart failure. PLoS One. 2011;6:e24272
100. Okamoto N, Noma T, Ishihara Y, Miyauchi Y, Takabatake W, Oomizu S. et al. Prognostic value of circulating regulatory T cells for worsening heart failure in heart failure patients with reduced ejection fraction. Int Heart J. 2014;55:271-7
101. Tacke M, Hanke G, Hanke T, Hunig T. CD28-mediated induction of proliferation in resting T cells in vitro and in vivo without engagement of the T cell receptor: evidence for functionally distinct forms of CD28. Eur J Immunol. 1997;27:239-47
102. Hunig T. Manipulation of regulatory T-cell number and function with CD28-specific monoclonal antibodies. Adv Immunol. 2007;95:111-48
103. Suntharalingam G, Perry MR, Ward S, Brett SJ, Castello-Cortes A, Brunner MD. et al. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med. 2006;355:1018-28
104. Penaranda C, Tang Q, Bluestone JA. Anti-CD3 therapy promotes tolerance by selectively depleting pathogenic cells while preserving regulatory T cells. J Immunol. 2011;187:2015-22
105. Saadoun D, Rosenzwajg M, Joly F, Six A, Carrat F, Thibault V. et al. Regulatory T-cell responses to low-dose interleukin-2 in HCV-induced vasculitis. N Engl J Med. 2011;365:2067-77
106. Zhao TX, Kostapanos M, Griffiths C, Arbon EL, Hubsch A, Kaloyirou F. et al. Low-dose interleukin-2 in patients with stable ischaemic heart disease and acute coronary syndromes (LILACS): protocol and study rationale for a randomised, double-blind, placebo-controlled, phase I/II clinical trial. BMJ open. 2018;8:e022452
107. Bernink FJ, Timmers L, Beek AM, Diamant M, Roos ST, Van Rossum AC. et al. Progression in attenuating myocardial reperfusion injury: an overview. Int J Cardiol. 2014;170:261-9
108. Ke D, Fang J, Fan L, Chen Z, Chen L. Regulatory T cells contribute to rosuvastatin-induced cardioprotection against ischemia-reperfusion injury. Coron Artery Dis. 2013;24:334-41
109. Panahi M, Vadgama N, Kuganesan M, Ng FS, Sattler S. Immunopharmacology of Post-Myocardial Infarction and Heart Failure Medications. J Clin Med. 2018:7
110. Vosshenrich CA, Di Santo JP. Developmental programming of natural killer and innate lymphoid cells. Curr Opin Immunol. 2013;25:130-8
111. Godeny EK, Gauntt CJ. Involvement of natural killer cells in coxsackievirus B3-induced murine myocarditis. J Immunol. 1986;137:1695-702
112. Ong S, Ligons DL, Barin JG, Wu L, Talor MV, Diny N. et al. Natural killer cells limit cardiac inflammation and fibrosis by halting eosinophil infiltration. Am J Pathol. 2015;185:847-61
113. Ayach BB, Yoshimitsu M, Dawood F, Sun M, Arab S, Chen M. et al. Stem cell factor receptor induces progenitor and natural killer cell-mediated cardiac survival and repair after myocardial infarction. Proc Natl Acad Sci U S A. 2006;103:2304-9
114. Boukouaci W, Lauden L, Siewiera J, Dam N, Hocine HR, Khaznadar Z. et al. Natural killer cell crosstalk with allogeneic human cardiac-derived stem/progenitor cells controls persistence. Cardiovasc Res. 2014;104:290-302
115. Paul P, Picard C, Sampol E, Lyonnet L, Di Cristofaro J, Paul-Delvaux L. et al. Genetic and Functional Profiling of CD16-Dependent Natural Killer Activation Identifies Patients at Higher Risk of Cardiac Allograft Vasculopathy. Circulation. 2018;137:1049-59
116. Hak L, Mysliwska J, Wieckiewicz J, Szyndler K, Trzonkowski P, Siebert J. et al. NK cell compartment in patients with coronary heart disease. Immun Ageing. 2007;4:3
117. Jonasson L, Backteman K, Ernerudh J. Loss of natural killer cell activity in patients with coronary artery disease. Atherosclerosis. 2005;183:316-21
118. Backteman K, Ernerudh J, Jonasson L. Natural killer (NK) cell deficit in coronary artery disease: no aberrations in phenotype but sustained reduction of NK cells is associated with low-grade inflammation. Clin Exp Immunol. 2014;175:104-12
119. van den Akker F, Deddens JC, Doevendans PA, Sluijter JP. Cardiac stem cell therapy to modulate inflammation upon myocardial infarction. Biochim Biophys Acta. 2013;1830:2449-58
Author contact
![]() Corresponding author: Junjie Xiao. Cardiac Regeneration and Ageing Lab, Institute of Cardiovascular Sciences, School of Life Science, Shanghai University, 381 Nan Chen Road, Shanghai 200444, China. Tel.: 0086-21-66138131, Fax: 0086-21-66138131, E-mail: junjiexiaoedu.cn
Corresponding author: Junjie Xiao. Cardiac Regeneration and Ageing Lab, Institute of Cardiovascular Sciences, School of Life Science, Shanghai University, 381 Nan Chen Road, Shanghai 200444, China. Tel.: 0086-21-66138131, Fax: 0086-21-66138131, E-mail: junjiexiaoedu.cn