Impact Factor
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Materials and Methods
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(14):3980-3991. doi:10.7150/thno.32352 This issue Cite
Research Paper
Blocking interleukin-6 trans-signaling protects against renal fibrosis by suppressing STAT3 activation
Wei Chen1*, Hui Yuan1*, Wenmin Cao1*, Tianwei Wang1, Wei Chen1, Hang Yu1, Yao Fu2, Bo Jiang1, Hong Zhou3, Hongqian Guo1 ![]() , Xiaozhi Zhao1
, Xiaozhi Zhao1 ![]()
1. Department of Urology, Drum Tower Hospital, Medical School of Nanjing University, Institute of Urology, Nanjing University, Nanjing, Jiangsu, China
2. Department of Pathology, Drum Tower Hospital, Medical School of Nanjing University, Nanjing, Jiangsu, China
3. Department of Immunology, Nanjing Medical University, Nanjing, Jiangsu, China.
*These authors contributed equally to this study.
Received 2018-12-17; Accepted 2019-4-9; Published 2019-5-31
Abstract

Rationale: Renal fibrosis is the terminal manifestation of chronic and irreversible renal disease. Effective therapies other than dialysis are extremely limited. In this study, we investigated the potential effects of targeting elevated interleukin-6 (IL-6) levels in the treatment of renal fibrosis.
Methods: Fc-gp130 was used to specifically block IL-6 trans-signaling. Unilateral ureteral occlusion (UUO) and ischemia reperfusion (IR) mouse models were constructed to investigate the therapeutic effect of Fc-gp130 on renal fibrosis. The role of IL-6 trans-signaling and phosphorylation of signal transducer and activator of transcription (STAT) 3 in regulating fibroblast accumulation and extracellular matrix protein deposition were evaluated in cell experiments and mouse models.
Results: The kidneys of mice with UUO were found to have elevated soluble IL-6 receptor (sIL-6R) levels in the progression of fibrosis. Fc-gp130 attenuated renal fibrosis in mice, as evidenced by reductions in tubular atrophy and the production of extracellular matrix protein. Blockade of IL-6 trans-signaling with Fc-gp130 also reduced inflammation levels, immune cell infiltration, and profibrotic cytokines expression in renal tissue, with decreased STAT3 phosphorylation and reduced fibroblast accumulation in the renal tissue. In vitro, Fc-gp130 also reduced the phosphorylation of STAT3 induced by transforming growth factor (TGF)-β1 in fibroblasts. Furthermore, the therapeutic effect of Fc-gp130 was confirmed in a model of acute kidney injury-chronic kidney disease.
Conclusion: Overall, IL-6 trans-signaling may contribute to crucial events in the development of renal fibrosis, and the targeting of IL-6 trans-signaling by Fc-gp130 may provide a novel therapeutic strategy for the treatment of renal fibrosis.
Keywords: chronic kidney disease, renal fibrosis, IL-6 trans-signaling, STAT3, Fc-gp130
Introduction
Chronic kidney disease (CKD), including renal interstitial fibrosis, is a worldwide public health problem causing high morbidity and mortality [1]. Renal fibrosis, characterized by fibroblasts activation and extracellular matrix (ECM) deposition, is the terminal manifestation and common irreversible pathway by which CKD progresses to end stage [2]. Renal fibrosis results in trauma of renal parenchyma and loss of renal function [3]. The incidence of end stage renal disease is rapidly increasing in the prevalence of diabetes and hypertension [4]. Clinically, there are few effective therapies for renal fibrosis other than dialysis. Therefore, the development of new strategies for antifibrotic therapy is required.
Interleukin-6 (IL-6) regulates the innate and adaptive immune responses, and it has become a major target for clinical intervention in many proinflammatory conditions [5]. IL-6 signaling occurs through two main pathways. In classical pathway, IL-6 binds to the membrane-bound IL-6 receptor (mIL-6R), then recruits signal transducer glycoprotein 130 (gp130) [6]; this leads to its homodimerization, which further activates the Janus kinase (JAK)/Tyk tyrosine kinases and leads to the phosphorylation of signal transducer and activator of transcription (STAT) 3. STAT3 is a transcription factor that regulates many downstream target genes [7]. In trans-signaling pathway, soluble IL-6R (sIL-6R) detected in body fluids binds to IL-6 with comparable affinity to that of membrane-bound IL-6R. The IL-6/sIL-6R complex also activates membrane gp130, causing subsequent phosphorylation of JAK/STAT3. IL-6 is therefore involved in intracellular signal transduction. It is generally believed that the anti-inflammatory activity of IL-6 is mediated by classical signaling, whereas the proinflammatory property is mediated by the trans-signaling pathway [8].
The role of IL-6 in renal fibrosis remains controversial. Elevated plasma levels of IL-6 are commonly observed in CKD patients [9]. However, IL-6 knockout (KO) mice showed comparable expression of ECM proteins in the obstructed kidney compared to that in wild-type (WT) mice [10]. IL-6 classic signaling can protect renal tissue from overshooting immune responses [11]. IL-6 trans-signaling has been shown to play a central role in crescentic nephritis [12]. As a specific antagonist of IL-6 trans-signaling in vivo, soluble gp130 is elevated in many pathological conditions [13, 14]. Gp130 was suggested to be a novel specific antagonist of IL-6 trans-signaling [15]. Fc-linked gp130 was produced, and the homodimer was found more effective. Furthermore, recombinant gp130 is currently in clinical trials [16]. Therefore, soluble gp130, a specific IL-6 trans-signaling inhibitor that does not affect classic signaling, could be an ideal therapeutic drug for renal inflammation [17].
In our previous work, we constructed and purified recombinant Fc-gp130 [18]. The Fc-gp130 homodimer was more effective than monomer gp130 at blocking IL-6 trans-signaling, as each functional IL-6/sIL-6R complex associates with two molecules of gp130 to transduce the intracellular downstream signal. In this study, we aimed to determine whether IL-6 trans-signaling is important in the development of renal fibrosis. Our results show that treatment with Fc-gp130, by interfering with IL-6 trans-signaling, attenuates renal fibrosis, therefore indicating that IL-6 trans-signaling blockade could be a promising and novel therapeutic approach for the treatment of renal fibrosis.
Results
Fc-gp130 attenuated renal fibrosis caused by unilateral ureteral occlusion (UUO)
In UUO-ligated mice, the levels of sIL-6R in the kidney (Figure 1A), IL-6 and sIL-6R in plasma (Supplementary Figure S1A-B) were significantly increased on day 14 after UUO as compared with levels in those receiving sham treatment. In addition, significant increases in IL-6, sIL-6R, mIL-6R, and gp130 in UUO kidneys were also observed by day 14 (Figure 1B, Supplementary Figure S1C). It is worth noting that sIL-6R levels in the kidney were elevated from day 3 to day 14, suggesting a role for IL-6 trans-signaling in the progression of renal fibrosis (Figure 1B). To confirm this, we pre-treated mice with Fc-gp130 to investigate whether specific blockade of IL-6 trans-signaling would protect against renal fibrosis. Interestingly, Fc-gp130 treatment in UUO mice significantly reduced parenchymal loss and tubular atrophy (Figure 1C), whereas genetic deletion of IL-6 had no substantial protective effect on UUO-induced fibrosis. Masson's staining also revealed that UUO mice showed fibrotic renal tissue with extensive collagen deposition, whereas Fc-gp130 treatment significantly reduced collagen deposition in UUO mice (Figure 1D). Next, we analyzed the effect of Fc-gp130 on the production of major components of the ECM, in UUO renal tissue. Immunohistochemical (IHC) staining showed increased accumulation of fibronectin, collagen I, collagen Ⅲ and vimentin following UUO, whereas Fc-gp130-treated mice exhibited reduced ECM content (Figure 1E-F, Supplementary Figure S1D-E). However, there were no significant differences in renal interstitial fibrosis or deposition of ECM between WT and IL-6 KO mice following UUO injury. Collagen I and fibronectin proteins were also significantly increased in UUO kidneys, and Fc-gp130 treatment significantly reduced collagen I and fibronectin protein levels in the kidneys of UUO mice (Figure 3D). Overall, these data suggest that IL-6 trans-signaling promotes renal fibrosis.
Renal fibrosis was reduced in mice with unilateral ureteral obstruction (UUO) treated with Fc-gp130. (A) ELISA of sIL-6R in renal lysates from mice on day 14 after UUO and sham control (CON) surgery. (B) Western blotting analysis of IL-6, sIL-6R and tubulin in renal lysates on days 3, 7, and 14 after UUO. (C) Samples from WT mice pretreated with or without Fc-gp130 and IL-6 KO C57BL/6 mice were collected on day 14 after UUO or CON surgery. Renal sections were stained with H&E for visualizing parenchymal loss and tubular atrophy, and renal lesions were scored. (D) Masson trichrome staining for tubulointerstitial fibrosis and collagen deposition (blue), and quantitative analysis of fibrotic area. (E-F) Immunohistochemistry and quantitative analysis of collagen I (E) and fibronectin (F) content in renal tissue. Scale bar, 50 μm. Data are means ± SD, n = 6 per group; ns, not significant, *p < 0.05, **p < 0.01, ***p < 0.001. Similar results from three independent experiments are shown.
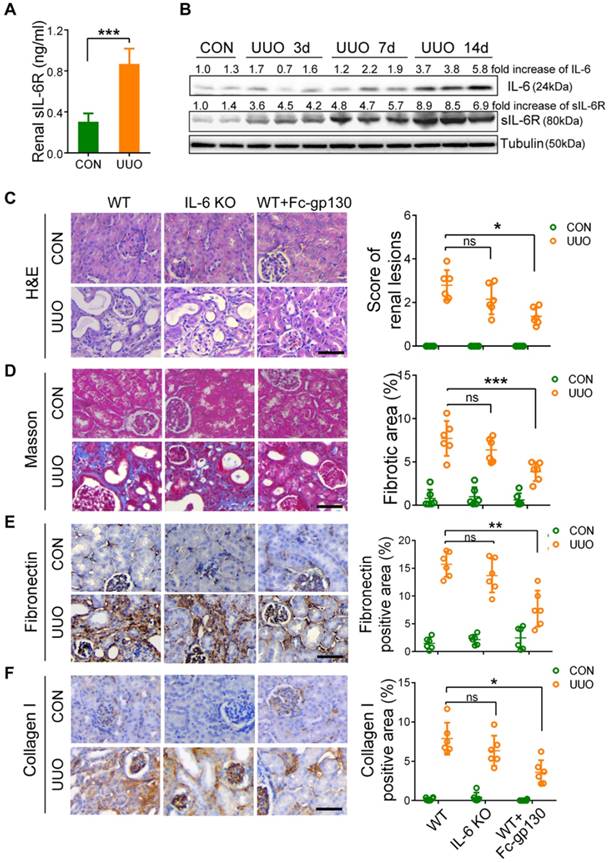
Fc-gp130 suppressed STAT3 activation in renal tissues of UUO mice. (A) Immunohistochemistry and quantitative analysis of α-SMA content in renal tissue. (B) Immunohistochemistry of p-STAT3 (brown) in fibroblasts (α-SMA, red) from renal sections. Red arrows denote p-STAT3 and α-SMA co-positive interstitial cells, black arrows denote p-STAT3 positive epithelial cells (B-1), and quantitative analysis of positive cells (B-2). Scale bar, 50 μm. Data are means ± SD, n = 6 pre group; *p < 0.05, ** p < 0.01. (C-D) Renal tissue samples were collected on day 14 after UUO and Fc-gp130 treatment. Western blotting analysis of phosphorylated (p)-STAT3, total STAT3, p-Smad3, Smad3, p-Smad2, Smad2, p-AKT, AKT, TGF-β, IL-6, sIL-6R, α-SMA (C), Fibronectin, Collagen I, p-Smad1/5/8, Smad1 and tubulin in renal lysates (D). Each experiment was performed at least in triplicate.
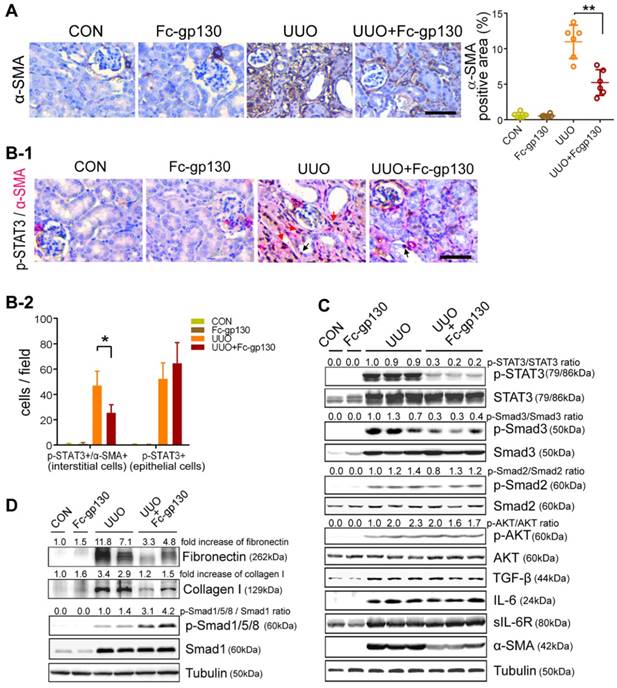
Fc-gp130 reduced inflammation in UUO mice
Inflammation is an important driver in the progression of renal fibrosis [19, 20]. Upon injury, renal tubular cells produce chemokines to recruit immune cells and inflammatory cytokines to activate myofibroblasts to produce ECM [21]. Therefore, we next evaluated the impacts of Fc-gp130 on the inflammatory responses in UUO mice. NF-κB p65 activation and nuclear translocation in renal tubular epithelial cells were significantly reduced in Fc-gp130-treated UUO mice compared with that in UUO mice alone (Figure 2A, Supplementary Figure S2A). IHC analysis also showed that Fc-gp130 inhibited the recruitment of F4/80-positive macrophages (Figure 2B), CD4+ (Figure 2C) and CD8+ T cells (Figure 2D) in the kidneys of UUO mice. In addition, the mRNA levels of proinflammatory mediator, such as tumor necrosis factor-α (Tnfa) (Figure 2E), monocyte chemoattraction protein 1 (Mcp1) (Figure 2F), and CXC chemokine ligand 16 (Cxcl16) (Figure 2G), were also significantly reduced upon Fc-gp130 treatment. The mRNA levels of Il6 (Figure 2H) and Tgfb (Figure 2I) were not significantly reduced by Fc-gp130. Therefore, blockade of IL-6 trans-signaling with Fc-gp130 attenuated renal inflammation in the UUO mouse model.
Renal inflammatory responses were reduced in UUO mice treated with Fc-gp130. (A-D) Samples from mice pretreated with or without Fc-gp130 were collected on day 7 after UUO or sham control (CON) surgery. Immunohistochemistry of NF-κB p65 and quantitative analysis of p65 nuclear localization (A). Immunohistochemistry of F4/80 (B), CD4 (C), and CD8 (D) and positive cells in renal tissue. (E-I) RT-qPCR analysis of mRNA levels of Tnfa (E), Mcp1 (F), Cxcl16 (G), Il6 (H), and Tgfb (I). Data are means ± SD, n = 6 pre group; ns, not significant, *p < 0.05, ** p < 0.01, *** p < 0.001. Similar results from three independent experiments are shown.
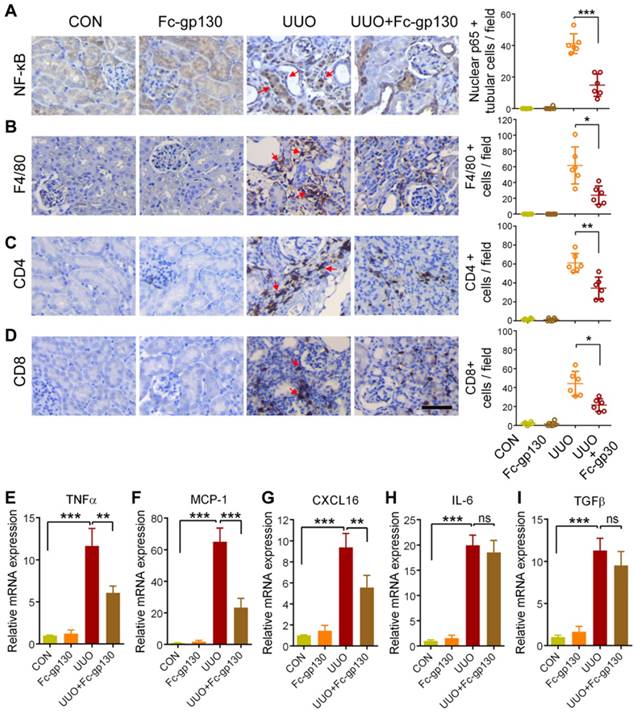
Fc-gp130 influenced STAT3 activation in fibroblasts
Immunostaining of α-smooth muscle actin (α-SMA) was used to investigate fibroblast accumulation. In UUO mice, Fc-gp130 treatment diminished fibroblast accumulation, as evidenced by reduced numbers of α-SMA-positive cells (Figure 3A). More specifically, p-STAT3-positive interstitial fibroblasts were abundant in UUO mice but were reduced in number after Fc-gp130 treatment, while p-STAT3-positive tubular epithelial cells were observed in both groups (Figure 3B-1 and 3B-2). The profibrogenic effect of IL-6 trans-signaling depends on the activation of STAT3 [22]. In UUO mice, phosphorylation of STAT3 (p-STAT3) and total STAT3 increased significantly over time, peaking on day 14 after UUO injury (Supplementary Figure S2B). Western blotting analysis on day 14 renal lysates also revealed that the ratios of p-STAT3/STAT3 and p-Smad3/Smad3, the levels of α-SMA, fibronectin and collagen I were attenuated, and the ratios of p-Smad1/5/8/Smad1 were increased in Fc-gp130-treated UUO mice compared to those in UUO mice alone; however, the ratios of p-Smad2/Smad2 and p-AKT/AKT, the levels of TGF-β, IL-6 and sIL-6R in Fc-gp130-treated and non-treated UUO mice were not significantly different (Figure 3C-D).
In NRK-49F cells, Fc-gp130 attenuated phosphorylation of STAT3 induced by IL-6 plus IL-6R treatment, whereas the p-STAT3 induced by low or high dose of IL-6 alone was not affected (Supplementary Figure S2C). We confirmed that Fc-gp130 specifically inhibits IL-6 trans-signaling in renal fibroblasts. IL-6 alone did not alter cell proliferation, but stimulation with IL-6 and IL-6R resulted in an increase in proliferation, while Fc-gp130 treatment reduced it (Figure 4A). Cells treated with IL-6 in the absence of sIL6R had a background of phosphorylated STAT3 due to signaling via the membrane bound IL-6R (classical signaling), and IL-6 plus sIL-6R induced phosphorylation of STAT3 (classical and trans-signaling) was significantly suppressed by Fc-gp130 (Figure 4B, Supplementary Figure S2D-E). This suggests that Fc-gp130 reduced fibroblast proliferation and p-STAT3 activated by IL-6 trans-signaling. We confirmed the result by examining the effect of stattic, an inhibitor of STAT3 activation. The cell proliferation activated by IL-6 trans-signaling was reduced by stattic (Figure 4C). Stattic inhibited both IL-6 induced and IL-6 plus IL-6R induced phosphorylation of STAT3 (Figure 4D). IL-6 treatment induced a slight increase in p-STAT3 in NRK-49F. IL-6 plus sIL-6R induced a prominent increase in p-STAT3 levels, comparable to that observed in fibroblasts stimulated by transforming growth factor (TGF)-β1 (Figure 4E). As expected, TGF-β1-induced increases in p-STAT3 and fibronectin levels were significantly attenuated by Fc-gp130 treatment at 6 and 12 h, and p-Smad3 was reduced by Fc-gp130 at 12 h (Figure 4F). Moreover, the levels of p-Smad1/5/8 was increased by Fc-gp130 at 12 h (Figure 4G). Therefore, Fc-gp130 attenuated the activation of renal fibroblasts in response to TGF-β1 in vitro. These data support the contribution of IL-6 trans-signaling to fibroblast and STAT3 activation in renal fibrosis.
Fc-gp130 inhibited proliferation of fibroblasts by suppressing STAT3 activation. (A-B) NRK-49F cells were incubated with vehicle (CON), IL-6 (50 ng/ml), IL-6R (200 ng/ml) and Fc-gp130 (500 ng/ml) for the indicated durations. Changes of cell proliferation (A), and western blotting analysis of p-STAT3, STAT3 and tubulin (B). (C-D) NRK-49F cells were incubated with vehicle (CON), IL-6 (50 ng/ml), IL-6R (200 ng/ml) and Stattic (2 μM) for the indicated plans and durations. Changes of cell proliferation (C), and western blotting analysis of p-STAT3, STAT3 and tubulin at 12h (D). Data are means ± SD, 6 duplicate samples in each group for (A) and (C); *p < 0.05, ** p < 0.01. (E) NRK-49F cells were incubated with vehicle (CON), TGF-β1 (5 ng/ml), IL-6 (50 ng/ml) plus IL-6R (200 ng/ml), or IL-6 only for the indicated durations. Western blotting analysis of p-STAT3, STAT3 and tubulin. (F-G) NRK-49F cells were incubated with vehicle (CON), TGF-β1 (5 ng/ml) only, or TGF-β1 (5 ng/ml) plus Fc-gp130 (500 ng/ml) for 3, 6, 12 h. Western blotting analysis of fibronectin, p-STAT3, total STAT3, p-Smad3, total Smad3 (F), p-Smad1/5/8, total Smad1 and tubulin (G). Each experiment was performed at least in triplicate.
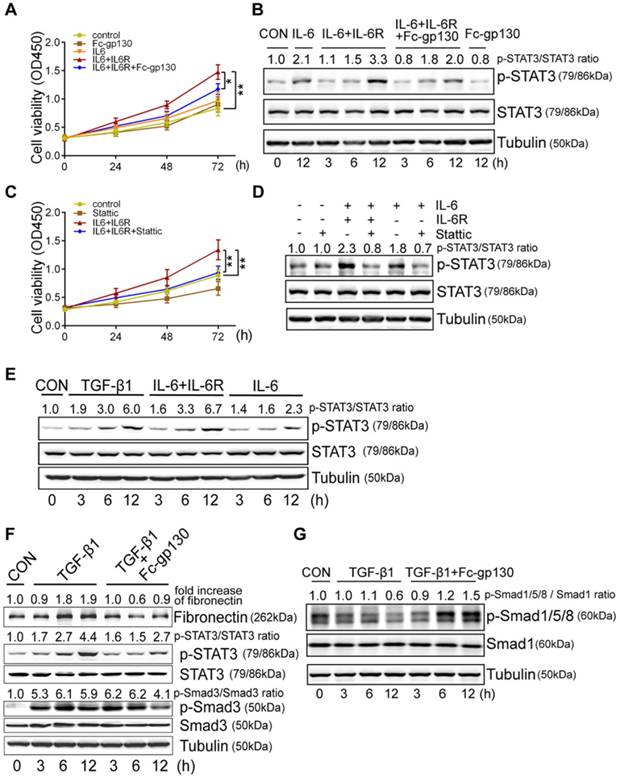
Fc-gp130 protected against fibrosis after ischemia reperfusion (IR) injury
To further confirm the protective effect of Fc-gp130, we constructed a different model of renal fibrosis, the acute kidney injury (AKI)-CKD mouse model, which is more consistent with the clinicopathological process. In AKI-CKD model, the levels of sIL-6R and IL-6 in plasma were significantly increased on day 14 after IR injury (the left renal artery was clamped for 45 min) as compared with levels in those receiving sham control (CON) treatment (Supplementary Figure S3A-B). In addition, significant increases in IL-6, sIL-6R, mIL-6R, and gp130 in IR kidneys were also observed by day 14 (Figure 5A, Supplementary Figure S3C). After IR injury, the concentration of blood urea nitrogen (BUN) and creatinine increased rapidly in one day, then return to normal gradually. The concentrations of BUN and creatinine were significantly decreased in Fc-gp130 treated IR group on day 14 and 28 (Supplementary Figure S3D-E). This indicated that Fc-gp130 can accelerate the recovery of IR injured kidneys.
Renal fibrosis was reduced in AKI-CKD mice treated with Fc-gp130. (A) Renal tissue samples were collected on day 14 after renal ischemia reperfusion (IR) or sham control (CON) surgery. Western blotting analysis of IL-6, sIL-6R and tubulin in renal lysates. (B) Mice were treated with vehicle (saline) or Fc-gp130, and samples were collected on day 42 after renal IR or sham control surgery. Western blotting analysis of fibronectin, α-SMA, p-STAT3, STAT3 and tubulin in renal lysates. (C-F) Renal sections were stained with H&E for visualizing parenchymal loss and tubular atrophy, and renal lesions were scored (C). Masson trichrome staining for tubulointerstitial fibrosis and collagen deposition (blue), and quantitative analysis of fibrotic area (D). Immunohistochemistry and quantitative analysis of α-SMA (E) and fibronectin (F) content. Scale bar, 50 μm. Data are means ± SD, n = 6 per group; ns, not significant, *p < 0.05. Similar results from three independent experiments are shown.
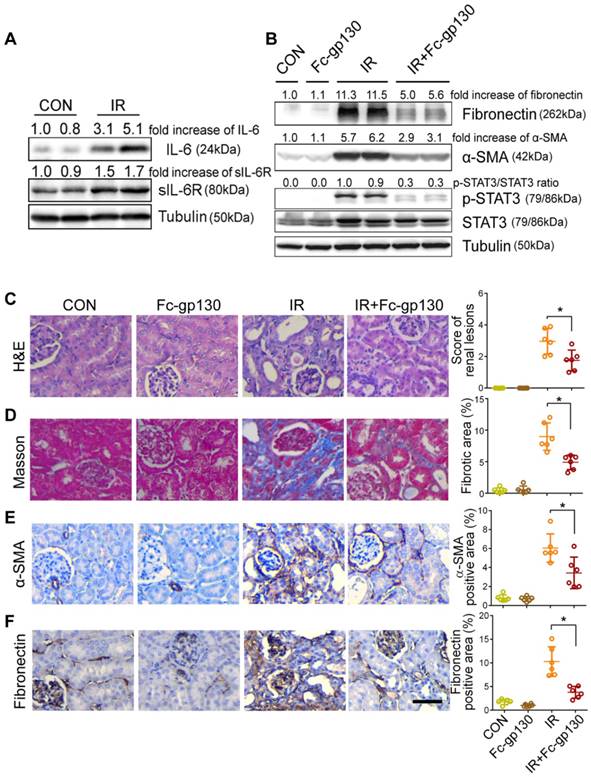
Mice treated with or without Fc-gp130 were exposed to severe renal IR injury and sacrificed at day 42 after injury. Protein levels of fibronectin, α-SMA, p-STAT3, and STAT3 were also significantly increased in IR kidneys, and Fc-gp130 treatment reduced fibronectin, α-SMA, and p-STAT3 levels (Figure 5B). IR mouse sections showed extensive renal damage as compared with that in sham control sections. Fc-gp130 treatment reduced lesions in IR mice (Figure 5C). On day 42 after IR injury, both glomeruli and interstitial areas between tubules showed significant fibrosis (Figure 5D). IR kidneys showed significantly increased α-SMA, fibronectin, collagen Ⅲ and vimentin staining (Figure 5E-F, Supplementary Figure S3F-G). Fc-gp130-treated IR mice showed significant reductions in ECM deposition and α-SMA content relative to levels in IR mice alone. These results confirmed that Fc-gp130 attenuates chronic renal fibrosis induced by IR in mice.
Discussion
IL-6 trans-signaling, mediated by sIL-6R, has been implicated in various chronic inflammatory conditions: rheumatoid arthritis [23], asthma [24] and neurodegenerative disorders [25], for example. However, its role in renal fibrosis has not yet been elucidated. In this study, we showed that targeting of the IL-6/sIL-6R complex using Fc-gp130 attenuated ECM protein production and inflammation in renal tissues of UUO and AKI-CKD mouse models, suggesting that IL-6 trans-signaling plays an essential role in the development and progression of renal interstitial fibrosis. Therefore, blocking of IL-6 trans-signaling with Fc-gp130 provides a promising new therapeutic strategy for the treatment of renal fibrosis.
Elevated levels of the cytokine IL-6 are linked to renal fibrosis. However, previously, WT and IL-6 KO mice under UUO showed no significant phenotypic differences [10], probably because the effects of ablating both the anti-inflammatory classical signaling and proinflammatory trans-signaling pathways may compensate for each other. Similarly, our results indicated that WT and IL-6 KO mice showed no significant changes in renal tissue fibrosis. In the IL-6 KO mice, the absent of pro-inflammatory trans-signaling probably alleviated fibrosis; but the anti-inflammatory of classic IL-6 signaling is also absent in injured renal tissue, and the increase of other inflammatory processes may aggravate renal fibrosis. Specifically neutralizing the pro-inflammatory IL-6 trans-signaling would not compromise other beneficial activities of IL-6 [26, 27]. In this study, we showed that IL-6 trans-signaling plays a key role in the renal fibrotic process. This is consistent with recent studies in pulmonary and cardiac fibrosis [28, 29]. Soluble gp130 is the natural inhibitor of IL-6 trans-signaling [30]. Using Fc-gp130, an antagonist of IL-6 signaling [31, 32], we specifically blocked IL-6 trans-signaling without interfering with the classical signaling [33, 34]. We speculated that this is the cause for better treatment effect of Fc-gp130. In the construction of this molecule, an IgG Fc fragment was added before the N terminus of gp130, producing a homodimer that is more effective than soluble gp130 [18]. Fc-fused proteins that have been found clinically efficacious include abatacept [35], aflibercept [36], and romiplostim [37]. Fc-linked gp130 is currently in clinical trials [16]. Overall, Fc-gp130 attenuated renal fibrosis in mouse models.
Various cells and molecules are involved in the process of damage repair and reconstruction [38]. IL-6 signaling is not the sole mechanism for renal fibrosis. Complex cross-talk in vivo might contribute to the pro- and anti-inflammatory/regenerative properties of IL-6 [39]. Cells stained for p-STAT3 were observed in tubular epithelial cells and fibroblasts in obstructed kidneys. Quantitative analysis showed that Fc-gp130 decreased the number of p-STAT3 activated fibroblast, but not the number of p-STAT3-positive tubular epithelial cells. Some studies have implicated STAT3 activation in tubular epithelial cells with the progression of kidney interstitial fibrosis [40]. Previous studies have demonstrated that STAT3 signaling in kidney tubular epithelial cells inhibits cells apoptosis and plays an important protective role in limiting kidney dysfunction after acute injury [41-43]. A previous study demonstrated that a receptor-independent gp130/STAT3 agonist prevented renal fibrosis [44]. That might be because hyperactive STAT3 plays an important role in protecting the kidneys. Classic signaling can be activated by IL-6 alone in cells with membrane-bound IL-6R and gp130. In contrast, virtually all cells express membrane-bound gp130, and a receptor-independent gp130/STAT3 agonist can activate STAT3 in almost all cells. STAT3 is mechanistically involved in the communication between epithelial cells and fibroblasts [45]. Accordingly, activated STAT3 in renal fibroblast is the main driver of renal fibrosis, and tubular epithelial cells probably make a protective contribution.
Activated STAT3 has been found to be an important transcription factor in the pathogenesis of pulmonary and kidney fibrosis [46, 47]. After ligand-induced dimerization of IL-6R, STAT3 binds to associated JAKs and can be phosphorylated. In addition, a recent study reported that STAT3 could be activated by TGF-β and plays an essential role in UUO-induced renal interstitial fibrosis [22]. Therefore, cross-talk between IL-6-STAT3 and TGF-β signaling may exist. The fact that Fc-gp130 treatment affected the phosphorylation of STAT3 further supports this hypothesis. It has also been reported that IL-6-STAT3 signaling enhances the expression of TGF-β1 to promote fibrosis [48]. However, in our study, we did not observe any reduction in Tgfb mRNA levels in Fc-gp130-treated animals. Furthermore, Fc-gp130 treatment inhibited p-STAT3 levels and ECM production in NRK-49F fibroblasts with sufficient TGF-β1 supplementation in the culture. Fc-gp130 could not only inhibit TGF-β downstream effector p-SMAD3, but upregulate the anti-fibrotic molecules p-SMAD1/5/8, suggesting that Fc-gp130 may interfered with TGF-β signaling through regulation of downstream molecules, independent of an effect on its expression levels. IL-6 trans-signaling plays a major role inflammation [49]. Fc-gp130 also affected the activation of NF-κB, another key transcription factor in the development of fibrosis and inflammation. However, the detailed mechanism underlying this cross-talk is unclear, and additional studies are needed to provide further insight.
After injury, inflammatory response and IL-6 signaling are rapidly activated in renal tissue. Due to the fact that the structure and function are affected in damaged and fibrotic kidneys, administered drugs are often not efficiently distributed to the renal target cells [50]. Fc-gp130 treatment in advance could confirm the effective concentration in the body, and timely reduce the inflammation. In present study the Fc-gp130 treatment started one week prior. In fact, the disease process of human renal fibrosis can go on a long time. Fc-gp130 treatment thus may have a therapeutic effect after renal injury or fibrosis occurs. However, the opportunity and methods of Fc-gp130 for human kidney disease need further study. There may be side effects when using systemic Fc-gp130 administration. Stability, immunogenicity, and the lack of absorption are limitations of therapeutic proteins [51]. Therefore, high concentration of Fc-gp130 is required, which might inhibit both classic and trans-signaling of IL-6 and evoke adverse outcomes [52]. Kidney or target cell specific delivery systems might overcome these limitations in fibrosis therapy [53]. Subsequently, specific delivery methods could be explored to improve the targeting of Fc-gp130 in the kidney.
In conclusion, the present study found that specifically neutralizing IL-6 trans-signaling attenuated renal fibrosis in UUO and AKI-CKD mouse models. IL-6 trans-signaling may thus represent a critical therapeutic target for managing fibrosis, and Fc-gp130 may be a candidate therapeutic agent for renal fibrosis.
Materials and Methods
Animals and treatment
Animal experiments were approved by the Ethics Committee of Drum Tower Hospital, Medical School of Nanjing University. WT C57BL/6 mice were purchased from the Animal Core Facility of Nanjing Medical University. IL-6 KO mice on a C57BL/6 background were purchased from The Jackson Laboratory (Bar Harbor, ME USA). Since males have better consistency and greater susceptibility to IR than females [54], we performed all animal experiments using male mice only. Under specific pathogen-free conditions, male mice (8 weeks old) weighing 20-25 g were housed in standard rodent cages. Mice were divided into four groups for treatment: sham [phosphate-buffered saline (PBS) treatment and sham surgery]; Fc-gp130 (Fc-gp130 treatment and sham surgery); UUO/IR (PBS treatment and induction of renal fibrosis); UUO/IR+Fc-gp130 (Fc-gp130 treatment and induction of renal fibrosis). Recombinant mouse Fc-gp130 (Vazyme, Nanjing, China) was used to block IL-6 trans-signaling. Fc-gp130 (20 μg/kg/day) [18] or vehicle (PBS) was intraperitoneally injected into mice every other day from one week before the surgery.
UUO model
WT or IL-6 KO mice were anesthetized intraperitoneally with 2% pentobarbital sodium (4 ml/kg) and then placed on the test bench in the prone position. The left ureter was exposed and ligated with sutures at two points near the renal hilum, followed by excision [55]. After the incision was closed, mice were given penicillin by intramuscular injection and were allowed to recover in standard rodent cages. Sham-operated control mice underwent abdominal laparotomy without excision of the right ureter. Mice were sacrificed at 3, 7, and 14 days, and renal tissue samples were collected for analysis.
Renal IR model
WT mice were anesthetized intraperitoneally with 2% pentobarbital sodium (4 ml/kg) and placed on the test bench. After abdominal laparotomy, the left renal pedicle was exposed and clamped for 45 min (ischemia); then, the clamp was removed (reperfusion). After closing the incision, mice were given penicillin by intramuscular injection and were allowed to recover in standard rodent cages. Sham-operated control mice underwent abdominal laparotomy without the clamp. Mice were sacrificed on day 42, and renal tissue samples were collected for analysis. The concentrations of blood serum urea nitrogen (BUN) and creatinine were assessed in duplicated with a commercially available assay kit (BioAssay System, Hayward, CA, USA) according to the instructions.
Enzyme-linked immunosorbent assay (ELISA)
Soluble IL-6R was quantified in plasma and tissue lysates from sham and UUO mouse kidneys using a mouse IL-6R ELISA kit (Raybiotech, Norcross, GA, USA). Plasma IL-6 was quantified using a mouse IL-6 ELISA kit (Raybiotech, Norcross, GA, USA). Renal lysate and plasma samples were diluted in reagent diluent and subjected to ELISA analysis according to the manufacturer's protocol.
Antibodies
Antibodies against Fibronectin (ab2413), Collagen I (ab34710), α-SMA (ab5694), F4/80 (ab6640), CD4 (ab183685), CD8 (ab209775), p-Smad3 (ab52903) and pan Cadherin (ab51034) were from Abcam (Cambridge, UK). Antibodies against p-STAT3 (9145), STAT3 (9139), NF-κB p65 (8242), p-p65 (3033), p-AKT (4060), p-Smad2 (3108), Smad2 (5339), Smad3 (9523), p-Smad1/5/8 (13820), Smad1 (6944), Vimentin (5741), Tubulin (2148), rabbit IgGs (7074) and mouse IgGs (7076) were from Cell Signaling Technology (Danvers, MA, USA). Antibodies against IL-6 (21865), IL-6R (23457), AKT (10176), TGF-β (21898), gp130(21175) and Collagen Ⅲ (22734) were from Proteintech (Rosemont, IL, USA).
Histological analysis
Renal tissues were fixed in 4% formaldehyde, dehydrated, embedded, and sectioned at 4 μm thickness. Renal sections were stained with hematoxylin and eosin (H&E) or Masson's trichrome (Solarbio Life Sciences, Beijing, China) to evaluate the degree of fibrosis. Sections were observed and scored in a blinded manner. HE slides were scored on a scale from 0 to 4: 0, normal; 1, mild (<25% of the cortex); 2, moderate (25-50%); 3, severe (50-75%); 4, extensive damage (>75%) [56, 57]. The scoring and the positive area of collagen fibers (Masson's trichrome, blue) was calculated in 10 bright fields (×200) in the eyepiece of the microscope. Images were acquired using a Leica DM 1000 microscope image system with a digital camera (Wetzlar, Germany).
Immunohistochemistry
After dewaxing and rehydration, slides were incubated with 3% H2O2 for quenching endogenous peroxidase activity and then heated in boiling antigen unmasking solution for antigen retrieval. Slides were blocked with 5% goat serum (Vector Laboratories, UK) for 1 h, then incubated with primary antibody in a humidified chamber at 4℃ overnight. Positive signal was visualized by DAB solution (ZSGB-BIO, Beijing, China). The images were visualized and acquired using the Leica DM 1000 microscope image system. Quantitative evaluation involved the use of Image-Pro Plus 6.0 (Media Cybernetics, Rockville, MD, USA) [58]. Positive signals and the number of positive cells in the kidney were quantified in 10 bright fields (×200) in the eyepiece of the microscope.
Cell culture and treatment
Renal fibroblast cell line NRK-49F was from American Type Culture Collection (ATCC, Manassas, USA), and cultured with Dulbecco's modified Eagle's medium (DMEM, Wisent, Nanjing, China) containing 10% (v/v) fetal bovine serum (FBS, Gibco, Gaithersburg, MD, USA) at 37°C with 5% CO2. Fibroblasts were pretreated with serum-free medium for 12 h and then exposed to 5 ng/ml TGF-β1, 50 ng/ml IL-6, 200 ng/ml IL-6R (R&D Systems, MN, USA), or 500 ng/ml Fc-gp130 for 3, 6, or 12 h.
Cell proliferation assay
Cell viability was measured at 0, 24, 48, 72 h using a CCK-8 assay kit (Vazyme Biotech, Nanjing, China). Cells were washed and then incubated with a working solution at 37°C for 30 min. Wst-8 in working solution can be reduced to highly soluble formazan (orange product) by various dehydrogenases in the mitochondria. The absorption value of each well, which is proportional to cell viability, was detected at 450 nm using a spectrophotometer system (TECAN, Männedorf, Switzerland).
Western blotting analysis
Renal tissues or cell samples were lysed on ice with lysis buffer containing cocktail proteinase inhibitors and protein phosphatase inhibitors (Thermo Fisher, Rockford, IL, USA). The extracted protein concentrations were quantified via BCA assay, mixed with loading buffer and boiled. Proteins were resolved on 10% SDS-PAGE in running buffer, transferred to polyvinylidene difluoride (PVDF) membranes (Bio-Rad, Hercules, USA), blocked with blocking buffer, and blotted with the primary antibodies at 4℃ overnight. Blots were washed and followed by incubation with secondary antibodies. After washing three times with PBST, signals were detected by chemiluminescence (Tanon, Shanghai, China), and visualized by ChemiScope 3300 Mini Imaging System (CLiNX, Shanghai, China). The western blot data was quantified by IMAGE J.
Real-time quantitative polymerase chain reaction (RT-qPCR)
Total RNA samples were extracted with TRIzol reagent (Invitrogen, Carlsbad, USA). Reverse transcription were performed using the PrimeScript RT Reagent Kit (Takara, Otsu, Japan). PCR reaction systems were performed using AceQ qPCR SYBR Green Master Mix (Vazyme). Reactions were run on a StepOne Plus (Applied Biosystems, Foster City, USA). The mRNA levels were normalized to that of actin mRNA and calculated using the 2-△△CT method. The specific primers used for analysis were as follows: MCP-1 forward, 5'-TTAAAAACCTGGATCGGAACCAA-3'; MCP-1 reverse, 5'-GCATTAGCTTCAGATTTACGGGT-3'; TNFα forward, 5'-ACCCTCACACTCAGATCATCTTC-3'; TNFα reverse, 5'-TGGTGGTTTGCTACGACGT-3'; CXCL16 forward, 5'-AGTGTCGCTGGAAGTTGTTCT-3'; CXCL16 reverse, 5'-GTTCCACACTCTTTGCGCTC-3'; IL-6 forward, ACAAAGCCAGAGTCCTTCAGAGA; IL-6 reverse, CTGTTAGGAGAGCATTGGAAATTG; TGF-β forward, CGGTGCTCGCTTTGTA; TGF-β reverse, GCCACTCAGGCGTATC; Actin forward, 5'-TAAAGACCTCTATGCCAACACAGT-3'; Actin reverse, 5'-CACGATGGAGGGGCCGGACTCATC-3'.
Statistical analysis
All data are expressed as mean ± SD. Statistical analysis involving two groups was performed by means of Student's t test. Multiple group comparisons were performed by one-way ANOVA statistical test. All data were processed with IBM SPSS Statistics 21 and GraphPad Prism 6. A p-value < 0.05 was considered statistically significant.
Abbreviations
UUO: unilateral ureteral occlusion; IL-6: interleukin-6; sIL-6R: soluble IL-6 receptor; gp130: glycoprotein 130; STAT3: signal transducer and activator of transcription 3; ECM: extracellular matrix; α-SMA: α-smooth muscle actin; TGF-β1: transforming growth factor-β1; TNFα: tumor necrosis factor-α; MCP-1: monocyte chemoattraction protein 1; CXCL16: CXC chemokine ligand 16; AKI: acute kidney injury; CKD: chronic kidney disease; IR: ischemia reperfusion.
Supplementary Material
Supplementary figures.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (81772710, 81502203), the Project of Invigorating Health Care through Science, Technology and Education Jiangsu Provincial Key Medical Discipline (ZDXKB2016014) and Key Medical Talents (QNRC2016015), the General Financial Grant from the China Postdoctoral Foundation (2018M640477, 2017M621729), Nanjing Health Distinguished Youth Fund (JQX15006), Nanjing Science and Technology Project (201715025, QRX17139), the Postdoctoral Research Foundation of Jiangsu Province (1701021B, 2018Z079).
Contributions
WC and XZ designed the study. HY, TW, WC and HY performed animal experiments. WC and YF performed histological analysis and IHC. WC and BJ performed WB and RT-qPCR. WC performed cell experiments. WC and XZ analyzed data. WC, HZ and XZ wrot the manuscript. XZ and HG supervised research.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Mehta RL, Cerda J, Burdmann EA, Tonelli M, Garcia-Garcia G, Jha V. et al. International Society of Nephrology's 0by25 initiative for acute kidney injury (zero preventable deaths by 2025): a human rights case for nephrology. Lancet. 2015;385:2616-43
2. Nath KA. The tubulointerstitium in progressive renal disease. Kidney Int. 1998;54:992-4
3. Liu Y. Renal fibrosis: new insights into the pathogenesis and therapeutics. Kidney Int. 2006;69:213-7
4. Sharma SK, Zou H, Togtokh A, Ene-Iordache B, Carminati S, Remuzzi A. et al. Burden of CKD, proteinuria, and cardiovascular risk among Chinese, Mongolian, and Nepalese participants in the International Society of Nephrology screening programs. Am J Kidney Dis. 2010;56:915-27
5. Jones SA, Scheller J, Rose-John S. Therapeutic strategies for the clinical blockade of IL-6/gp130 signaling. J Clin Invest. 2011;121:3375-83
6. Murakami M, Hibi M, Nakagawa N, Nakagawa T, Yasukawa K, Yamanishi K. et al. IL-6-induced homodimerization of gp130 and associated activation of a tyrosine kinase. Science. 1993;260:1808-10
7. Heinrich PC, Behrmann I, Muller-Newen G, Schaper F, Graeve L. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem J. 1998;334( Pt 2):297-314
8. Scheller J, Chalaris A, Schmidt-Arras D, Rose-John S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim Biophys Acta. 2011;1813:878-88
9. Pecoits-Filho R, Lindholm B, Axelsson J, Stenvinkel P. Update on interleukin-6 and its role in chronic renal failure. Nephrol Dial Transplant. 2003;18:1042-5
10. Yang J, Chen J, Yan J, Zhang L, Chen G, He L. et al. Effect of interleukin 6 deficiency on renal interstitial fibrosis. PLoS One. 2012;7:e52415
11. Luig M, Kluger MA, Goerke B, Meyer M, Nosko A, Yan I. et al. Inflammation-induced IL-6 functions as a natural brake on macrophages and limits GN. J Am Soc Nephrol. 2015;26:1597-607
12. Braun GS, Nagayama Y, Maruta Y, Heymann F, van Roeyen CR, Klinkhammer BM. et al. IL-6 trans-signaling drives murine crescentic GN. J Am Soc Nephrol. 2016;27:132-42
13. Nowell MA, Williams AS, Carty SA, Scheller J, Hayes AJ, Jones GW. et al. Therapeutic targeting of IL-6 trans signaling counteracts STAT3 control of experimental inflammatory arthritis. J Immunol. 2009;182:613-22
14. Rose-John S, Scheller J, Elson G, Jones SA. Interleukin-6 biology is coordinated by membrane-bound and soluble receptors: role in inflammation and cancer. J Leukoc Biol. 2006;80:227-36
15. Fischer M, Goldschmitt J, Peschel C, Brakenhoff JP, Kallen KJ, Wollmer A. et al. I. A bioactive designer cytokine for human hematopoietic progenitor cell expansion. Nat Biotechnol. 1997;15:142-5
16. Garbers C, Heink S, Korn T, Rose-John S. Interleukin-6: designing specific therapeutics for a complex cytokine. Nat Rev Drug Discov. 2018;17:395-412
17. Dai Y, Zhang W, Wen J, Zhang Y, Kellems RE, Xia Y. A2B adenosine receptor-mediated induction of IL-6 promotes CKD. J Am Soc Nephrol. 2011;22:890-901
18. Zhao X, Chen W, Ge L, Jiang W, Tang B, Zhang Q. et al. Transient expression of Fc-fused human glycoprotein 130 in Expi293F suspension cells. Protein Expr Purif. 2016;124:41-7
19. Chung AC, Lan HY. Chemokines in renal injury. J Am Soc Nephrol. 2011;22:802-9
20. Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140:821-32
21. Bonventre JV, Yang L. Cellular pathophysiology of ischemic acute kidney injury. J Clin Invest. 2011;121:4210-21
22. O'Reilly S, Ciechomska M, Cant R, van Laar JM. Interleukin-6 (IL-6) trans signaling drives a STAT3-dependent pathway that leads to hyperactive transforming growth factor-beta (TGF-beta) signaling promoting SMAD3 activation and fibrosis via Gremlin protein. J Biol Chem. 2014;289:9952-60
23. Cronstein BN. Interleukin-6-a key mediator of systemic and local symptoms in rheumatoid arthritis. Bull NYU Hosp Jt Dis. 2007;65(Suppl 1):S11-5
24. Finotto S, Eigenbrod T, Karwot R, Boross I, Doganci A, Ito H. et al. Local blockade of IL-6R signaling induces lung CD4+ T cell apoptosis in a murine model of asthma via regulatory T cells. Int Immunol. 2007;19:685-93
25. Campbell IL, Erta M, Lim SL, Frausto R, May U, Rose-John S. et al. Trans-signaling is a dominant mechanism for the pathogenic actions of interleukin-6 in the brain. J Neurosci. 2014;34:2503-13
26. Waetzig GH, Rose-John S. Hitting a complex target: an update on interleukin-6 trans-signalling. Expert Opin Ther Targets. 2012;16:225-36
27. Rose-John S, Waetzig GH, Scheller J, Grotzinger J, Seegert D. The IL-6/sIL-6R complex as a novel target for therapeutic approaches. Expert Opin Ther Targets. 2007;11:613-24
28. Le TT, Karmouty-Quintana H, Melicoff E, Weng T, Chen NY, Pedroza M. et al. Blockade of IL-6 Trans signaling attenuates pulmonary fibrosis. J Immunol. 2014;193:3755-68
29. Chou CH, Hung CS, Liao CW, Wei LH, Chen CW, Shun CT. et al. IL-6 trans-signalling contributes to aldosterone-induced cardiac fibrosis. Cardiovasc Res. 2018;114:690-702
30. Jostock T, Mullberg J, Ozbek S, Atreya R, Blinn G, Voltz N. et al. Soluble gp130 is the natural inhibitor of soluble interleukin-6 receptor transsignaling responses. Eur J Biochem. 2001;268:160-7
31. Lee JJ, Kim HJ, Yang CS, Kyeong HH, Choi JM, Hwang DE. et al. A high-affinity protein binder that blocks the IL-6/STAT3 signaling pathway effectively suppresses non-small cell lung cancer. Mol Ther. 2014;22:1254-65
32. Boettger MK, Leuchtweis J, Kummel D, Gajda M, Brauer R, Schaible HG. Differential effects of locally and systemically administered soluble glycoprotein 130 on pain and inflammation in experimental arthritis. Arthritis Res Ther. 2010;12:R140
33. Garbers C, Thaiss W, Jones GW, Waetzig GH, Lorenzen I, Guilhot F. et al. Inhibition of classic signaling is a novel function of soluble glycoprotein 130 (sgp130), which is controlled by the ratio of interleukin 6 and soluble interleukin 6 receptor. J Biol Chem. 2011;286:42959-70
34. Garbers C, Aparicio-Siegmund S, Rose-John S. The IL-6/gp130/STAT3 signaling axis: recent advances towards specific inhibition. Curr Opin Immunol. 2015;34:75-82
35. Moreland L, Bate G, Kirkpatrick P. Abatacept. Nat Rev Drug Discov. 2006;5:185-6
36. Semeraro F, Morescalchi F, Duse S, Parmeggiani F, Gambicorti E, Costagliola C. Aflibercept in wet AMD: specific role and optimal use. Drug Des Devel Ther. 2013;7:711-22
37. Shimamoto G, Gegg C, Boone T, Queva C. Peptibodies: A flexible alternative format to antibodies. MAbs. 2012;4:586-91
38. Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214:199-210
39. Garbers C, Hermanns HM, Schaper F, Muller-Newen G, Grotzinger J, Rose-John S. et al. Plasticity and cross-talk of interleukin 6-type cytokines. Cytokine Growth Factor Rev. 2012;23:85-97
40. Kuratsune M, Masaki T, Hirai T, Kiribayashi K, Yokoyama Y, Arakawa T. et al. Signal transducer and activator of transcription 3 involvement in the development of renal interstitial fibrosis after unilateral ureteral obstruction. Nephrology (Carlton). 2007;12:565-71
41. Wang J, Ouyang C, Chen X, Fu B, Lu Y, Hong Q. STAT3 inhibits apoptosis of human renal tubular epithelial cells induced by ATP depletion/recovery. Nephron Exp Nephrol. 2008;108:e11-8
42. Chen J, Chen JK, Conway EM, Harris RC. Survivin mediates renal proximal tubule recovery from AKI. J Am Soc Nephrol. 2013;24:2023-33
43. Xu MJ, Feng D, Wang H, Guan Y, Yan X, Gao B. IL-22 ameliorates renal ischemia-reperfusion injury by targeting proximal tubule epithelium. J Am Soc Nephrol. 2014;25:967-77
44. Matsumoto K, Xavier S, Chen J, Kida Y, Lipphardt M, Ikeda R. et al. Instructive role of the microenvironment in preventing renal fibrosis. Stem Cells Transl Med. 2017;6:992-1005
45. Bienaime F, Muorah M, Yammine L, Burtin M, Nguyen C, Baron W. et al. Stat3 controls tubulointerstitial communication during CKD. J Am Soc Nephrol. 2016;27:3690-705
46. O'Donoghue RJ, Knight DA, Richards CD, Prele CM, Lau HL, Jarnicki AG. et al. Genetic partitioning of interleukin-6 signalling in mice dissociates Stat3 from Smad3-mediated lung fibrosis. EMBO Mol Med. 2012;4:939-51
47. Pechkovsky DV, Prele CM, Wong J, Hogaboam CM, McAnulty RJ, Laurent GJ. et al. STAT3-mediated signaling dysregulates lung fibroblast-myofibroblast activation and differentiation in UIP/IPF. Am J Pathol. 2012;180:1398-412
48. Ogata H, Chinen T, Yoshida T, Kinjyo I, Takaesu G, Shiraishi H. et al. Loss of SOCS3 in the liver promotes fibrosis by enhancing STAT3-mediated TGF-beta1 production. Oncogene. 2006;25:2520-30
49. Rabe B, Chalaris A, May U, Waetzig GH, Seegert D, Williams AS. et al. Transgenic blockade of interleukin 6 transsignaling abrogates inflammation. Blood. 2008;111:1021-8
50. Eirin A. Hitching a ride to renal repair. Am J Physiol Renal Physiol. 2017;312:F276-F7
51. Sharma AR, Kundu SK, Nam JS, Sharma G, Priya Doss CG, Lee SS. et al. Next generation delivery system for proteins and genes of therapeutic purpose: why and how? Biomed Res Int. 2014;2014:327950
52. Bartoli E. Adverse effects of drugs on the kidney. Eur J Intern Med. 2016;28:1-8
53. Breyer MD, Susztak K. The next generation of therapeutics for chronic kidney disease. Nat Rev Drug Discov. 2016;15:568-88
54. Wei Q, Wang MH, Dong Z. Differential gender differences in ischemic and nephrotoxic acute renal failure. Am J Nephrol. 2005;25:491-9
55. Chen G, Lin SC, Chen J, He L, Dong F, Xu J. et al. CXCL16 recruits bone marrow-derived fibroblast precursors in renal fibrosis. J Am Soc Nephrol. 2011;22:1876-86
56. Shih W, Hines WH, Neilson EG. Effects of cyclosporin A on the development of immune-mediated interstitial nephritis. Kidney Int. 1988;33:1113-8
57. Pulskens WP, Rampanelli E, Teske GJ, Butter LM, Claessen N, Luirink IK. et al. TLR4 promotes fibrosis but attenuates tubular damage in progressive renal injury. J Am Soc Nephrol. 2010;21:1299-308
58. Li X, Nie Y, Lian H, Hu S. Histopathologic features of alcoholic cardiomyopathy compared with idiopathic dilated cardiomyopathy. Medicine. 2018;97:e12259
Author contact
![]() Corresponding authors: Xiaozhi Zhao (E-mail: zhaoxzedu.cn) and Hongqian Guo (E-mail: dr.ghqedu.cn), Department of Urology, Drum Tower Hospital, Medical School of Nanjing University, Institute of Urology, Nanjing University, Nanjing, Jiangsu, China. Tel +86 25 68182222, Fax +86 25 68185987.
Corresponding authors: Xiaozhi Zhao (E-mail: zhaoxzedu.cn) and Hongqian Guo (E-mail: dr.ghqedu.cn), Department of Urology, Drum Tower Hospital, Medical School of Nanjing University, Institute of Urology, Nanjing University, Nanjing, Jiangsu, China. Tel +86 25 68182222, Fax +86 25 68185987.