Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and methods
Results
Discussion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(13):3918-3939. doi:10.7150/thno.31461 This issue Cite
Research Paper
Temporal inhibition of mouse mammary gland cancer metastasis by CORM-A1 and DETA/NO combination therapy
Kseniia Porshneva1, Diana Papiernik1, Mateusz Psurski1, Agnieszka Łupicka-Słowik2, Rafał Matkowski3,4, Marcin Ekiert3,4, Marcin Nowak5, Joanna Jarosz1, Joanna Banach1, Magdalena Milczarek1, Tomasz M. Goszczyński1, Marcin Sieńczyk2, Joanna Wietrzyk1 ![]()
1. Department of Experimental Oncology, Hirszfeld Institute of Immunology and Experimental Therapy, Polish Academy of Sciences, Wroclaw, Poland
2. Faculty of Chemistry, Division of Medicinal Chemistry and Microbiology, Wroclaw University of Science and Technology, Wroclaw, Poland
3. Division of Surgical Oncology and Clinical Oncology; Department of Oncology, Wroclaw Medical University, Wroclaw, Poland
4. Lower Silesian Oncology Center, Wroclaw, Poland
5. Faculty of Veterinary Medicine, Wroclaw University of Environmental and Life Sciences, Wroclaw, Poland
Received 2018-11-13; Accepted 2019-3-17; Published 2019-5-31
Abstract

Carbon monoxide and nitric oxide are two of the most important vasoprotective mediators. Their downregulation observed during vascular dysfunction, which is associated with cancer progression, leads to uncontrolled platelet activation. Therefore, the aim of our studies was to improve vasoprotection and to decrease platelet activation during progression of mouse mammary gland cancer by concurrent use of CO and NO donors (CORM-A1 and DETA/NO, respectively).
Methods: Mice injected intravenously with 4T1-luc2-tdTomato or orthotopically with 4T1 mouse mammary gland cancer cells were treated with CORM-A1 and DETA/NO. Ex vivo aggregation and activation of platelets were assessed in the blood of healthy donors and breast cancer patients. Moreover, we analyzed the compounds' direct effect on 4T1 mouse and MDA-MB-231 human breast cancer cells proliferation, adhesion and migration in vitro.
Results: We have observed antimetastatic effect of combination therapy, which was only transient in orthotopic model. During early stages of tumor progression concurrent use of CORM-A1 and DETA/NO demonstrated vasoprotective ability (decreased endothelin-1, sICAM and sE-selectin plasma level) and downregulated platelets activation (decreased bound of fibrinogen and vWf to platelets) as well as inhibited EMT process. Combined treatment with CO and NO donors diminished adhesion and migration of breast cancer cells in vitro and inhibited aggregation as well as TGF-β release from breast cancer patients' platelets ex vivo. However, antimetastatic effect was not observed at a later stage of tumor progression which was accompanied by increased platelets activation and endothelial dysfunction related to a decrease of VASP level.
Conclusion: The therapy was shown to have antimetastatic action and resulted in normalization of endothelial metabolism, diminution of platelet activation and inhibition of EMT process. The effect was more prominent during early stages of tumor dissemination. Such treatment could be applied to inhibit metastasis during the first stages of this process.
Introduction
The migration capacity allows tumor cells to disseminate from primary tumor to distant organs by entering blood and lymphatic vessels. The survival of circulating tumor cells (CTCs) in the blood stream depends on the immune system defense mechanisms where CTCs are targeted mainly by leukocytes. Moreover, they must resist hemodynamic forces. On the other hand, platelets and monocytes promote tumor cell survival. As CTCs need to attach to blood vessel walls to extravasate, interactions between CTCs and endothelium are of great importance. Activated platelets are known to play a crucial role in such interactions, contributing to CTCs' adhesion to the endothelium [1]. Due to high reactivity of the endothelium and its strong secretory ability, this organ is able to control blood coagulation, platelet activation, vessel repair and cell transmigration through blood vessels [2]. Thus, activation loops are established between platelets, tumor cells and the endothelium during metastasis [2,3]. This activity of the endothelium is driven by the production of vasoprotective mediators like prostacyclin (PGI2), nitric oxide (NO) and carbon monoxide (CO) which typically sustain hemostasis, whereas their downregulation contributes to uncontrolled platelet activation. PGI2, the most potent antiplatelet agent, is usually produced by the endothelium along with NO in a coupled manner. PGI2 and NO synergistically inhibit platelet aggregation by cAMP and cGMP-dependent mechanisms respectively. It has been recently demonstrated that CO also displays an important anti-platelet activity that complements the action of PGI2 and NO [4,5].
However, NO has a dual role in the tumor progression and metastasis. It is produced from L-arginine by three NO synthase isoforms: inducible (iNOS), neuronal (nNOS) and endothelial (eNOS) [6]. Clinical studies showed that patients with iNOS-positive tumors had significantly lower survival compared to patients with iNOS-negative tumors [7]. Furthermore, tumor associated macrophages (TAMs) are known to synthetize NO through iNOS from L-arginine. NO of such origin contributes to TAMs tumorigenic action, thus NO generated by iNOS is one of the crucial factors facilitating tumor progression [8]. On the other hand, NO produced by the endothelium is one of the antiplatelet agents which inhibit platelet aggregation [9].
Endogenous CO formation is catalyzed by heme oxygenase (HO) that decomposes heme into CO, ferrous iron, and biliverdin [10,11]. CO is a signaling molecule that exerts anti-inflammatory, anti-apoptotic and anti-platelet effects [12]. It is important that anti-aggregatory effect of CO is relatively low compared to the effect of NO. On the other hand, CO and Fe are two inhibitors of NO generation by iNOS which is upregulated during the metastatic process [13]. Moreover, it was demonstrated that endogenously produced NO or CO can stimulate colon cancer cells proliferation, whereas iNOS or HO inhibition, as well as exogenous delivery of NO or CO, suppress the growth of these cells [14].
Exogenous NO can be delivered through the specific donors: diazeniumdiolates (also known as “NONOates”). NONOates undergo decomposition spontaneously in a solution under physiological conditions yielding two molar equivalents of NO [15]. The half-life of DETA/NO, nitric oxide slow releasing molecule is 21 hours. DETA/NO attenuated the platelet - endothelial cell adhesion response to lipopolysaccharide (LPS), whereas inhibition of soluble guanylate cyclase (sGC) abolished these effects of DETA/NO [16]. Another experiment revealed that DETA/NO downregulates endothelial expression of P-selectin, vascular cell adhesion molecule (VCAM-1) and intercellular adhesion molecule (ICAM-1) in mice with the dextran sulfate sodium-induced colitis [17]. Moreover, DETA/NO induces cell cycle arrest and apoptotic cell death in the model of human breast cancer MDA-MB-231 [18]. Another interesting NO-donor is PAPA/NO. Its half-life is 15 minutes which makes it more suitable for in vitro investigation. PAPA/NO revealed strong anti-platelet activity in the experiment of human platelet aggregation on platelet reach plasma [4]. Similarly, carbon monoxide releasing molecules (CO-RMs) liberate CO in biological systems under physiological conditions. Only few of them are totally water-soluble, for instance CORM-A1 [19-21]. The anti-platelet action of CO-RMs does not involve sGC activation [4], whereas other authors showed that gaseous CO inhibited platelets aggregation through this mechanism [22]. It was also observed that CORM-3 downregulated VCAM and E-selectin expression independently of HO-1 upregulation but through inhibition of sustained NF-κB activation [23].
Thus, these donors represent promising substances in the antimetastatic approach due to their antiplatelet, anti-aggregative and cytotoxic activity. It has been shown, that exogenous NO and CO act via distinct mechanisms whose complementary action may improve vasoprotection and inhibition of platelets activation [5,24]. Recent studies of Stojak et al. have shown that concomitant use of CO and NO donors significantly decreased mitochondrial respiration and glycolysis in cancer and endothelial cells in vitro. CO with NO also additively inhibited endothelial-cancer cells interaction [25]. To confirm the hypothesis that synergistic interaction between NO and CO donors is possible in vivo, we have chosen a model of mouse mammary gland carcinoma implanted intravenously (4T1-luc2-tdTomato) or orthotopically (4T1). We have also evaluated the effect of a combination of NO and CO donors on the aggregation of platelets obtained from breast cancer patients. In addition, to assess the possible direct effect of NO and CO donors on cancer cells, we have evaluated their proliferative, adhesive and migration potential in vitro after treatment with NO and CO donors.
Materials and methods
Compounds
Nitric oxide donors DETA/NO and PAPA/NO (both from Cayman Chemical, Michigan, USA) and water-soluble carbon monoxide-releasing molecule CORM-A1 (Sigma-Aldrich, Saint Louis, USA) were used as exogenous source of NO and CO. DETA/NO, as a slow releasing molecule, was applied in in vivo experiments, while PAPA/NO was investigated in vitro due to its short half-life. CORM-A1 was used both in in vivo and in vitro assays.
Mice
In this study, we used 7/8-week-old BALB/c female mice purchased from the Center of Experimental Medicine, Medical University of Bialystok, Poland. Experimental procedures were conducted according to the Interdisciplinary Principles and Guidelines for the Use of Animals in Research, Marketing and Education issued by the New York Academy of Sciences' Ad Hoc Committee on Animal Research, and Directive 2010/63/UE of the European Parliament and of the Council of 22 September 2010 on the protection of animals used for scientific purposes. All experiments were approved by the Local Committee for Experiments with the Use of Laboratory Animals, Wroclaw, Poland (permission numbers: 46/2013, 78/2015, 63/2016, 23/2017, and 109/2018).
Cells
4T1-luc2-tdTomato cell line expressing the firefly luciferase gene and tdTomato fluorescent protein (Caliper Life Sciences Inc., USA) was cultured in RPMI 1640 medium (HIIET, Wroclaw, Poland) supplemented with 10% fetal bovine serum (FBS; Sigma-Aldrich, Chemie GmbH, Steinheim, Germany). Mouse 4T1 mammary adenocarcinoma and human MDA-MB-231 breast cancer cell lines were obtained from the American Type Culture Collection (ATCC, USA). Cells were cultured in RPMI 1640 + HEPES (HIIET, Wroclaw, Poland) medium with 10% FBS (HyClone, Thermo Fisher Scientific Inc., UK or Sigma-Aldrich, Chemie GmbH, Steinheim, Germany, respectively). All culture media were supplemented with 2 mM glutamine (Sigma-Aldrich, Germany), 100 U/ml penicillin and 100 µg/ml streptomycin (both from Polfa Tarchomin S.A. Warsaw, Poland). Cell cultures were maintained at 37°C in a humidified atmosphere with 5% CO2.
For the model of experimental metastasis (intravenous; i.v.) cells were suspended in Hank's Balanced Salt Solution (HBSS; HIIET, Wroclaw, Poland) and 7.5×104 4T1-luc2-tdTomato cells were injected into the tail vein. For the model of spontaneous metastasis (orthotopic), 1×104 4T1 cells were inoculated into the mammary fat pad [26]. In all experiments the day of cell inoculation is determined as day “0”.
The course of experiments
Administration of CO and NO donors. CORM-A1 and DETA/NO were administered intraperitoneally (i.p.) with CORM-A1 administered every 12 h and DETA/NO every 24 h. We used two different dosages of the compounds with different molar ratio of CORM-A1 to DETA/NO:
1. 0.5 mg/kg/12 h CORM-A1 and 1.570 mg/kg/24 h DETA/NO (molar ratio 1:1)
2. 1.5 mg/kg/12 h CORM-A1 and 2.358 mg/kg/24 h DETA/NO (molar ratio 1:0.5)
The doses of CORM-A1 based on previously described results showing the effective platelet aggregation inhibition after the use of 3 or 1 mg/kg/24 h of CORM-A1 [27]. Under physiological conditions, the immediate production of endogenous NO by the vascular endothelium may be aggravated by various factors; on the other hand, small amounts of CO are released constantly during the heme oxygenases (HO-1 and HO-2) mediated heme degradation. Therefore, as suggested by Chlopicki et al., in order to effectively imitate the physiological properties of CO, its slow but continuous release is required [4]. Hence, considering the half-life of CORM-A1 (21 min) [20], the 12 h administration schedule was selected. DETA/NO with long half-life (21 h) [28] was administered every 24 h as the molar equivalent or half lower dose compared to CORM-A1.
Experimental and spontaneous models of metastasis. To properly mimic the metastasis process, we used several different models of experimental as well as spontaneous metastases. The orthotopic (spontaneous) model gives a wide view of the metastasis cascade, while the experimental one allows to assess the ability of cancer cells to survive in the circulation, arrest, extravasate and growth in a specific organ (in the case of the tail vein model - in the lungs) [29]. Using divergent metastasis models we could verify which steps of metastatic cascade are affected the most by the treatment and also how important are tumor-derived factors in modulation of the response to treatment used. Orthotopic 4T1 cancer progression is accompanied by large inflammatory response of the body [30]. LPS-induced 4T1 experimental metastases are effectively inhibited by NO synthase inhibitor N-methyl‐L‐arginine (NMA) what was not observed without LPS injection [31]. Therefore, to model the systemic inflammation in selected intravenous models, mice were injected i.p. with 1 mg/kg of LPS (Salmonella typhosa; Sigma-Aldrich, Saint Louis, USA) five hours before cell inoculation.
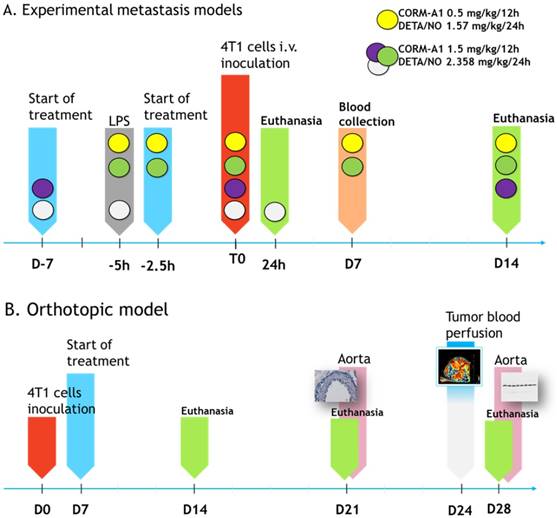
The steps of intravenous experiments are presented in Scheme 1A and Figure 1A. In the model of intravenous cells inoculation mice were observed for 24 h (model of extravasation) or 14 days (model of lung settlement); in the orthotopic model (spontaneous metastasis) for 14, 21 and 28 days (Scheme 1B). Lungs, hearts and blood were harvested. In the orthotopic model tumors and aortas were collected additionally.
The course of in vivo experiments. (A) Experimental models of metastasis. Colored circles indicate schedules of every single experiment. Single doses of compounds are indicated. T0 means the time of tumor cells inoculation. (B) Orthotopic model of spontaneous metastasis. Single doses of compounds in orthotopic model are as marked by yellow circle in Scheme A legend.
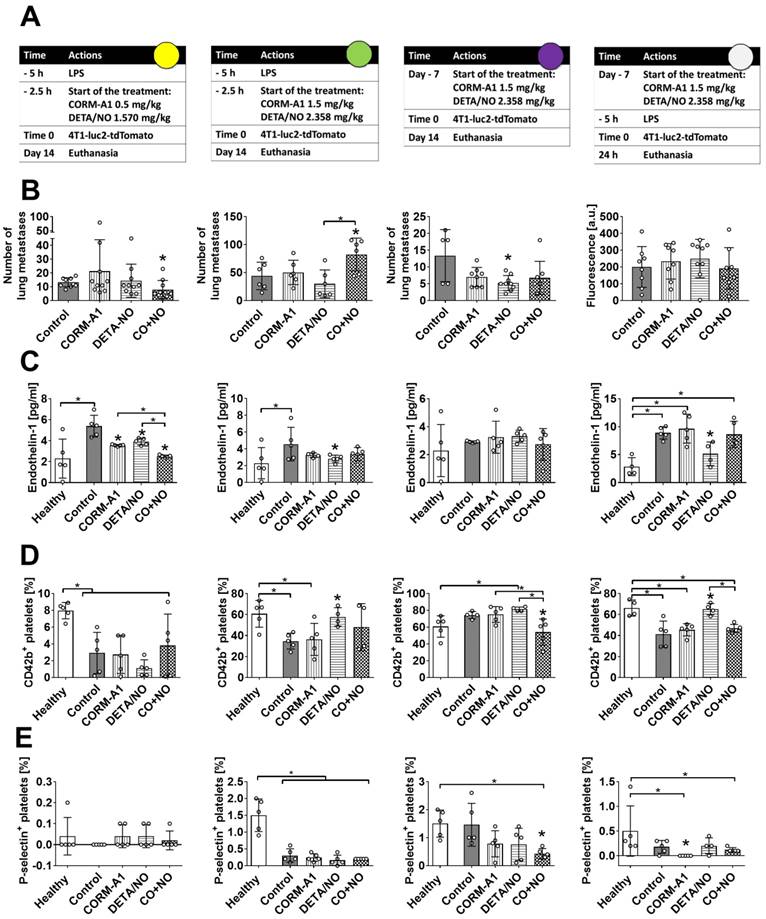
The effect of CORM-A1 and DETA/NO in the experimental models of metastasis of 4T1-luc2-tdTomato cells. (A) Experiment schedules. Single doses of donors are indicated (CORM-A1 every 12h, DETA/NO every 24h). Colored circles correspond to indications in Scheme 1A. (B) Number of lung metastatic foci or fluorescence intensity of the lung (last column). (C) Endothelin 1 (ET-1) plasma level. (D) CD42b and (E) P-selectin positive platelets assessed by flow cytometry. Data presented as mean with standard deviation with points for individual measurements. Number of mice: 5-10 per group, some analyses were performed on randomly selected mice within the group. Healthy mice used as a control: 4-5. Statistical analysis: (B) Sidak's, (C) Dunnett's, (D) and (E) Dunn's multiple comparison tests. *p< 0.05 as compared to control or as indicated.
Tumor growth measurement
Maximum length and width of tumors were measured three times a week. Tumor volume was calculated according to the following formula:
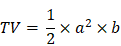
where TV - tumor volume; a - shorter diameter; b - longer diameter.
Anti-metastatic effect evaluation
Lungs were excised and fixed in 4% solution of phosphate buffered formalin and metastatic foci were counted visually. In the 24-h model of experimental metastasis, ex vivo visualizations of the metastatic foci localized in lungs were performed using an In vivo MS FX PRO system (Carestream Health INC., USA). Images were analyzed with Carestream MI SE software (Carestream Health INC., USA) as previously described [32]. The intensity of the fluorescent signal is presented as the mean intensity of the region of interest and is expressed in arbitrary units [a.u.].
Tumor blood perfusion analysis
Tumor perfusion analysis was performed using the Vevo 2100 ultrasound imaging system (VisualSonics, Ontario, Canada) on day 24 of the experiment. Marker for real-time observation of tumor blood perfusion was prepared by dissolving a vial containing MicroMarkerTM Contrast Agent (VisualSonics, Ontario, Canada) in 1 ml of sterile 0.9% saline (HIIET, Wroclaw, Poland). Mice were subjected to general anesthesia by intraperitoneal injection of medetomidine (0.5 mg/kg) and a continuous administration of 2-3% isoflurane (Baxter, Deerfield, Germany) in synthetic air (200 ml/min). Immobilized animals in anesthetic state were placed on the treatment table so that their position would allow the imaging of the central cross-section of the tumor tissue. The examination was carried out with the aid of ultrasound head with the 13-24 MHz frequency of emitted ultrasonic waves (MS250, VisualSonics, Ontario, Canada). 50 μl of the contrast medium was administered i.v. and at this time point recording of the image started and continued until the tissue was filled with contrast. Once imaging was complete, mice were awakened by intraperitoneal injection of atipam (1 mg/kg). Data analysis was performed using the Vevo LAB 1.7.1 Software (VisualSonics, Ontario, Canada).
Analysis of blood morphological parameters and platelet activation
Blood samples were analyzed using the Mythic 18 hematology analyzer (PZ Cormay S.A., Lomianki, Poland). Next, blood samples were centrifuged at 4°C, 15 min, 2000×g to obtain the plasma, which was subsequently stored at -80°C and used for further investigations.
10 μl of whole blood was diluted in 1 ml of Tyrode buffer (HIIET, Wroclaw, Poland) mixed with 50 μl of TBS (HIIET, Wroclaw, Poland). Diluted blood was centrifuged at 900×g, 5 min, (RT). Pellets were suspended in 1 ml of Tyrode buffer and CaCl2 was added to the final concentration of 1 mM. Washed samples at the volume of 100 μl were incubated with 1 μl of antibodies: DyLight649-labeled anti-mouse CD41/61, PE-labeled anti-mouse GPIIbIIIa (CD41/61) JON/A, PE-labeled anti-mouse GPIbα (CD42b), PE-labeled anti-mouse P-selectin (CD62P), FITC-labeled anti-mouse vWf, FITC-labeled anti-mouse fibrinogen (EMFRET Analytics GmbH Co. KG, Eibelstadt, Germany). After incubation, samples were centrifuged and diluted in PBS. LSR Fortessa cytometer and FACSDiva software (BD Bosciences, San Jose, USA) were used for data analysis.
Protein expression
The analysis of protein expression was carried out on the tumor, lung, aorta and heart lysates using western blot method or by ELISA test on tissue lysates and plasma. Lysate preparation was performed as previously described in Fast Prep®-24 MP Bio homogenizer (MP Biomedicals LLC., Santa Ana, USA) using RIPA buffer with phosphatase and protease inhibitor cocktail [26,33]. Protein concentration was determined by Bradford dye-binding method.
Western-blot analysis was performed as previously described [26,33]. Membranes were blocked and incubated with antibodies solutions (1:1,000) overnight at 4°C. The list of antibodies used: (1) rabbit polyclonal antibodies anti-E-cadherin and anti-N-cadherin (Proteintech Group Inc., Chicago, USA); (2) rabbit polyclonal antibody anti-α-SMA (Abcam, Cambrige, UK); (3) rabbit monoclonal antibodies anti-VASP, anti-PKG-1α and rabbit polyclonal antibodies anti-Phospho-VASP and anti-HO-1 (Cell Signaling Technology, Warsaw, Poland). The next day membranes incubated with anti-E-cadherin, anti-N-cadherin and anti-α-SMA antibodies (group A) were washed three times for 10 min with PBST while membranes incubated with anti-VASP, anti-PKG-1α, anti-Phospho-VASP and anti-HO-1 (group B) were washed three times for 5 min in TBST, followed by incubation with a solution of an anti-rabbit IgG-HRP secondary antibody [1:10,000; Santa Cruz Biotechnology, Dallas, USA (group A) or 1:1,000 Sigma-Aldrich, Warsaw, Poland (group B)] at RT for 1 h. Signal detection was performed using ECL method. The chemiluminescence reading was performed using the Image Station 4000MM PRO or GelLogic 1500 gel system (both from Carestream, Rochester, USA). The membranes from the group A were washed three times for 10 min with PBST after reading, and then incubated for 1 h (RT) with an anti-β-actin-HRP monoclonal antibody (1:1,000; Santa Cruz Biot., Dallas, USA; group A). Then, the membranes were processed as described above. The membranes from the group B were washed three times for 5 min with TBST followed by 1 h (RT) incubation with mouse monoclonal anti-β-actin antibody (1:5,000; Cell Signaling Technology, Warsaw, Poland). Membranes were washed three times for 5 min with TBST and incubated for 1 h (37°C) with anti-mouse-HRP monoclonal antibodies (1:2,500, Fitzgerald, Acton, USA). After washing signal was developed as previously described. Densitometric analysis was performed using the Carestream MI Software 5.0.6.20 (Carestream, Rochester, USA) and the chemiluminescence intensity results of examined proteins were presented in relation to those obtained for β-actin or for the heart lysates samples in relation to the total protein content determined after Coomassie staining of the gels.
ELISA tests were performed according to the producer's protocols. The result of the analysis was read using the Synergy H4 universal plate reader (Bio-Tek Instruments Inc., Winooski, USA), measuring the optical density of the solutions at 450 nm. Standard curve was prepared and used to determine the concentration of the tested proteins. ELISA kits for the detection of TXB2, vWf, P-selectin, E-selectin, TGF-β, ICAM, VCAM, 6-keto-PGF1α, iNOS, COX-2 were purchased from Elabscience, Houston, USA. Kits for detection of VEGF and myeloperoxidase (MPO) were acquired from Thermo Fisher Scientific, Waltham, USA or R&D SYSTEMS, Minneapolis, USA. ET-1 detection was performed using ELISA from Elabscience, Houston, USA or R&D SYSTEMS, Minneapolis, USA.
Spectrophotometric determination of carboxyhemoglobin content
Previously described method of carboxyhemoglobin blood level analysis by Beutler and West was applied [34]. Briefly, freshly obtained whole blood samples were lysed with 10 mM potassium phosphate buffer (PB; pH 6.85). After 10 minutes incubation at RT, samples were mixed with sodium hydrosulfite solution (1.25 mg/ml in 100 mM PB; pH 6.85) and followed by 15 minutes incubation (RT). The spectral analysis was performed on Biotek Synergy H4 Hybrid Reader. Each sample was analyzed in duplicate. The calibration curve was prepared using blood samples from healthy mice followed by samples carbon monoxide saturation. The absorbance ratio at 420 and 432 nm was used to calculate the carboxyhemoglobin fraction in the samples.
Spectrophotometric determination of lipid peroxidation (MDA)
We used widely recognized thiobarbituric acid (TBA)-based method, adapted for 384-well plate format [35]. 50 µl of heart or lung homogenate was mixed with 50 µL of 10% (w/v) trichloroacetic acid aqueous solution. After 15 min incubation (RT) the samples were centrifuged (15 min, 10,000×g, 4°C). The collected supernatant was mixed with 0.67% (w/v) TBA solution and heated for 45 minutes (100°C). After cooling to room temperature, the samples were pipetted on a 384-well plate and the absorbance was read at 532 nm. Malonodialdehyde (MDA)-TBA adduct concentration was calculated based on its absorbance coefficient (155 mM-1 cm-1) and was normalized to a total protein concentration in a sample.
Spectroscopic determination of nitrite ions in plasma
Griess diazotization reaction-based test, adapted for 384-well plates format was applied [36]. Briefly, plasma samples (20 µl) were transferred to 384-well plate and 80 µl of Griess reagent was added (Sigma-Aldrich, Germany). After 15 minutes incubation in the dark (RT) nitrite concentration was calculated using the sodium nitrite standard curve (raw absorbance values measured at 530 nm were corrected by 640 nm absorbance subtraction).
Plasma analysis of liver, kidney and heart specific enzymes
Alanine aminotransferase (ALT), aspartate aminotransferase (AST), urea (URE), creatinine (CRE2), creatinine kinase (CK) and creatinine kinase MB isoform (CK-MB) were measured in plasma samples in Cobas C 111 analyzer (Roche Diagnostics, Switzerland) using reagents and procedures provided by the manufacturer.
Immunohistochemical analyses of aorta endothelium
Aortas were harvested from mice on day 21 of the experiment (Scheme 1B) and analyzed using standard methods. Briefly, aorta slides were stained with anti-eNOS and anti-VCAM rabbit antibodies (Abcam, Cambrige, UK), counterstained with hematoxylin and analyzed under Olympus BX53 microscope (Olympus, Tokyo, Japan) using immunoreactive score (IRS) taking into account the percentage of stained cells and the intensity of reaction product [26,37].
In vitro experiments
To assess the direct effect of the combination of CO and NO donors on tumor cells, in vitro studies were carried out using mouse 4T1 as well as human MDA-MB-231 mammary gland cancer cells (triple negative breast cancer cell lines).
Proliferation inhibition. Investigation of inhibition of tumor cell proliferation was carried out using SRB method as previously described [38]. Cell proliferation was evaluated after 72 h of incubation and determination of the IC50 parameter (i.e. the concentration of the compound that inhibits cell proliferation at 50%) was performed.
Fibronectin cell adhesion assay. 96-well Nunc-Immuno™ MicroWell™ solid plate was coated with 10 µg/ml of fibronectin from bovine plasma (Sigma-Aldrich, Saint Louis, USA) dissolved in PBS (HIIET, Wroclaw, Poland), then incubated at 4°C for 24 h. Next, the plate was blocked with 1% BSA (Sigma-Aldrich, Saint Louis, USA) in TSM buffer (HIIET, Wroclaw, Poland) for 30 min at 37°C. At the same time, cells were dissociated with non-enzymatic cell dissociation solution (Sigma-Aldrich, Saint Louis, USA) and suspended in TSM buffer. Then, the cells were pre-incubated with 10, 30, 50 and 100 µM solutions of CORM-A1 and PAPA/NO for 20 min at 37°C. Afterwards, the cells were seeded in the pre-coated plate at the density of 50×103 cells per well, followed by the incubation for 1 h. Adhered cells were fixed with the 200 µl of 96% ethanol and incubated at room temperature for 10 min. The cells were stained with 0.2% (w/v) crystal violet (2% crystal violet solution, GEMI P.P.F., Karczew, Poland) and dissolved in methanol (Avantor, Gliwice, Poland). The plate was incubated for 30 min at 4°C. Absorbance was measured at 570 nm using a Synergy H4 universal reader and GEN 5 software (both from Bio-Tek Instruments Inc., Winooski, USA). Experiments were performed in triplicate and repeated three times. The mean inhibition of cancer cells adhesion to fibronectin was calculated based on the following formula: A=100×(As/Ac), where A - cell adhesion (%), As - value of the absorbance measured for cells treated with test substances, Ac - value of the absorbance measured for untreated cells.
Cell Migration Assay. Cell migration was assayed in 24-well plates, on 6.5 mm Transwell® with 8.0 µm Pore Polycarbonate Membrane Inserts (Corning Inc., New York, USA) coated with 10 µg/ml of fibronectin from bovine plasma (Sigma-Aldrich, Saint Louis, USA). 7×103 MDA-MB-231 or 2×104 4T1 cells/insert suspended in DMEM (Thermo Fisher Scientific, Waltham, USA) without FBS were added to the apical chamber at the volume of 150 µl with the solution of tested compounds at the concentration of 5, 10, 50, 100 µM. DMEM with 10% FBS (Sigma-Aldrich, Saint Louis, USA) was added to the bottom chamber at the volume of 0.5 ml as a chemoattractant. After 10 h of incubation, cells on the upper surfaces of the inserts were removed by wiping with a cotton swab. Migrated cells on the undersides of the insert membranes were fixed and stained using Diff-Quick Stain kit (Medion Diagnostics, Miami, USA). Staining was performed according to the manufacturer's protocols. The number of cells on the lower face of the filter was counted in the whole space of insert membrane using Olympus CKX 41 microscope (Olympus, Tokyo, Japan). The mean inhibition of cell migration was evaluated on the basis of the % of migrated cells using the following formula: M=100×(Ms/Mc), where M - migrated cells (%); Ms - the number of migrated cells in treated samples; Mc - the number of migrated cells in the control group.
Ex vivo experiments on platelets obtained from breast cancer patients
48 breast cancer patients diagnosed from July to October 2017 at Lower Silesian Oncology Center (Wroclaw, Poland) and 14 healthy donors were included into the study. Blood was collected on 3.2% buffered sodium citrate (BD Biosciences, San Jose, USA). The patients were divided into three groups according to the breast cancer stage: 27 patients without metastasis, 13 patients with lymph node metastasis and 8 patients with distant metastasis. Studies were carried out in line with the ethical standards of the national and institutional research committee and with the latest version of the 1964 Helsinki Declaration. The study protocol was approved by the local Institutional Review Board, Wroclaw, Poland (permission numbers 71/2017 and 286/2017). Informed consent was obtained from every study participant.
Platelets aggregation analysis. Platelet-rich plasma (PRP) was obtained by centrifugation of blood (150×g/15 min) and supernatant was harvested for further investigation. To obtain platelet poor plasma (PPP), the remaining blood was centrifuged at 2,000×g for 15 min. The number of platelets in PRP was counted (Mythic 18; PZ Cormay S.A., Lomianki, Poland). The final concentration of platelets used was 180×103 platelets/µl. PRP samples were incubated with CORM-A1 and DETA/NO or PAPA/NO (100 µM) for 15 min at 37°C in a humidified atmosphere with 5% CO2. Aggregation of platelets in PRP was induced with collagen (1 µg/ml) at 37°C and estimated using optical method measuring light transmission (Chrono-Log Model 700 Agregometer; Crono-Log, Havertown, USA). PPP was used as optical control of 100% of light transmission, whereas PRP alone was considered 0% of light transmission.
Assessment of platelets activation. Seven samples from the group of patients with lymph node metastasis and with distant metastasis were obtained for analysis. Six samples of blood from healthy donors were used as a control. PRP was obtained as described above and incubated with 100 µM of CORM-A1 and PAPA/NO and their combination at 37°C in a humidified atmosphere with 5% CO2 for 15 min. Then, samples were centrifuged (2,000×g for 15 min) to obtain PPP which was used for further analysis. Platelet activation was estimated by quantitative evaluation of TGF-β and TXB2 concentration in the obtained PPP by ELISA (Elabscience, Houston, USA).
Statistical analysis
Statistical analysis was carried out using the GraphPad Prism 7.1 software. The Shapiro-Wilk data normality test was used for analysis of data distribution. Specific tests used for data analysis have been presented in the figure legends. Differences between groups for which p <0.05 were considered statistically significant.
Results
Low doses of CORM-A1 combined with DETA/NO inhibit experimental lung metastasis in mice with induced endothelial dysfunction
Using intravenous model of experimental metastasis and different scheme and dosages of treatment (Figure 1A, Scheme 1A) we have demonstrated the antimetastatic effect of DETA/NO combined with CORM-A1 in mice with induced endothelial dysfunction (pretreatment with LPS) and treated with lower doses of both compounds (Figure 1B, first column). Endothelin-1 (ET-1) level was significantly reduced in the combined treatment group (Figure 1C), whereas platelet activation parameters were not affected (Figure 1D and E, first column).
Higher doses of compounds (second column of Figure 1) have led to the stimulation of lung settlement by cancer cells in the combined treatment group (Figure 1B). ET-1 level was diminished only by DETA/NO treatment, i.e. in the group of mice with a tendency to decrease lung metastasis (Figure 1C). Moreover, CD42b on the surface of platelets in mice treated with a higher dose of DETA/NO was increased (Figure 1D, second column). The same dosages of compounds did not affect the cells extravasation measured 24 h after the cell inoculation (Figure 1, last column). Nevertheless, diminution of ET-1 and increase of CD42b on platelets were observed in mice treated with DETA/NO (Figure 1C, last column). In this short-term experiment 3 mg/kg/day of CORM-A1 reduced the percentage of P-selectin positive platelets (Figure 1 E, last column). Only DETA/NO significantly reduced lung foci formation in mice not pretreated with LPS after 14 days of cells inoculation (Figure 1 B, third column). The plasma level of ET-1 was not affected in these mice as a result of the treatments used (Figure 1C). However, significant decrease of CD42b and P-selectin positive platelets was observed in the combined treatment group (Figure 1D and E third column).
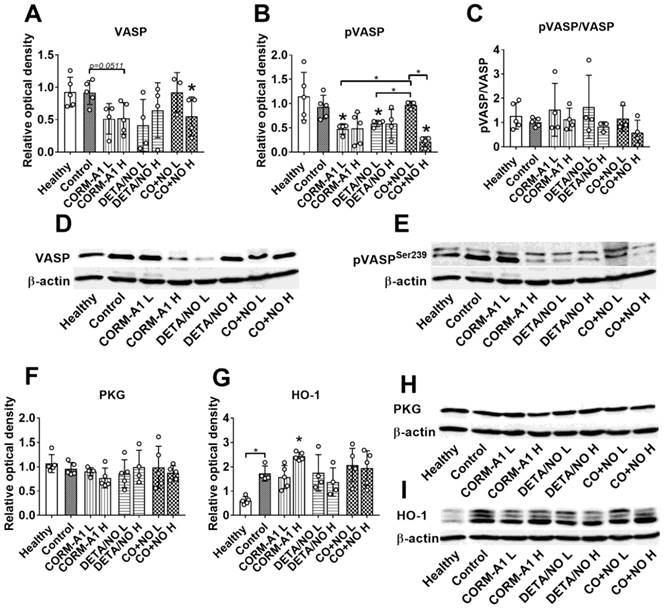
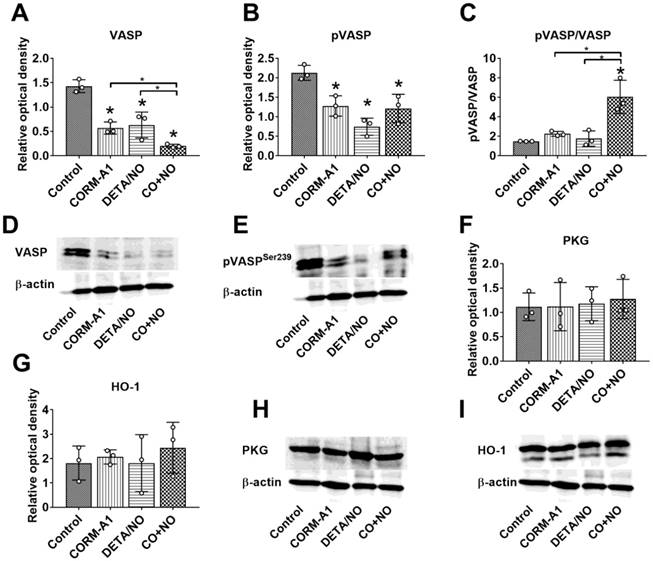
High doses of CORM-A1 combined with DETA/NO decrease total and phosphorylated VASP protein level
Western blot analysis of lung tissue harvested from mice with induced endothelial dysfunction (pretreated with LPS) and treated with lower (L) and higher (H) doses of CORM-A1 and DETA/NO revealed that in combination therapy group treated with higher doses, significant decrease of vasodilator-stimulated phosphoprotein (VASP) (Figure 2A) and VASP phosphorylated at Ser-239 (pVASPSer239) (Figure 2B) level was noticed. Moreover, the level of pVASPSer239 in CO+NO group treated with higher doses of donors was significantly diminished as compared to group of mice treated with lower doses of tested compounds (Figure 2B). The ratio of pVASPSer239/VASP was the lowest among all groups in mice treated with higher doses of CO+NO, but the results were not statistically significant (Figure 2C). cGMP-dependent protein kinase (PKG) level was not affected by the treatments used (Figure 2F). HO-1 measured in lung tissue was increased significantly after treatment with higher dose of CORM-A1 (Figure 2G). All these proteins level in mice hearts did not change regardless of the type of treatment (Figure S1).
The impact of CORM-A1 and DETA/NO on protein levels in lung tissue in the experimental models of metastasis of 4T1-luc2-tdTomato cells. (A) Total VASP, (B) pVASPSer239, (F) PKG-1α and (G) HO-1 densitometric analysis (mean optical density of bands of protein tested to β-actin) and representative blots (D), (E), (H), and (I) respectively. (C) Calculated pVASPSer239/VASP ratio. “L” means lower dose of CORM-A1 and DETA/NO (0.5 mg/kg/12h and 1.57 mg/kg/24h, respectively; treatment schedule indicated by yellow circle in Scheme 1A and Figure 1A). “H” means higher dose of CORM-A1 and DETA/NO (1.5 mg/kg/12h and 2.358 mg/kg/24h, respectively; treatment schedule indicated by green circle in Scheme 1A and Figure 1A). Data presented as mean with standard deviation with points for individual measurements. Number of mice: 4-5 per group. Healthy mice used as a control: 5. Statistical analysis: Dunnett's multiple comparison tests. *p< 0.05 as compared to control or as indicated.
VEGF and myeloperoxidase (MPO) level measured in plasma, lung and heart tissues as well as lipids peroxidation (malondialdehyde; MDA level) measured in lung and heart tissues did not change significantly between treated and control mice (Figure S2).
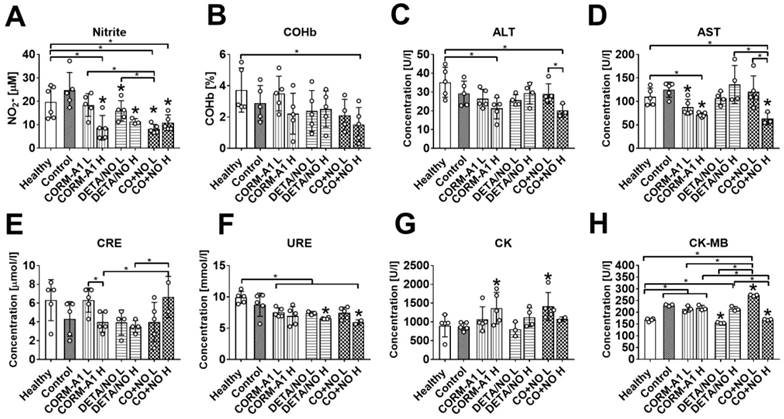
The plasma level of nitrite was diminished in mice treated with higher dose of CORM-A1, both doses of DETA/NO and both doses of combined treatment. Combined treatment with lower doses of compounds also significantly diminished nitrite plasma level as compared to both donors used alone (Figure 3A). Carboxyhemoglobin level was not affected significantly by the treatment (Figure 3B). The combined use of higher doses of compounds significantly affected plasma level of selected enzymes related to the function of liver and kidney; decrease of alanine aminotransferase (ALT), aspartate aminotransferase (AST), and urea (URE) (Figure 3C, 3D and 3F), and increase of creatinine (CRE) (Figure 3E) were noticed. Creatinine kinase (CK) was elevated after combined treatment with lower dose of compounds (Figure 3G). Cardiac MB isoform of CK (CK-MB) was also significantly increased in mice treated with lower dose compounds combination whereas high-dose treatment acts oppositely (Figure 3H).
The impact of CORM-A1 and DETA/NO on blood biochemical parameters in the experimental models of metastasis of 4T1-luc2-tdTomato cells. (A) Nitrite, (B) carboxyhemoglobin (COHb), (C) Alanine aminotransferase (ALT), (D) aspartate aminotransferase (AST), (E) creatinine (CRE2), (F) urea (URE), (G) creatinine kinase (CK) and (H) creatinine kinase MB isoform (CK-MB) plasma level. “L” means lower dose of CORM-A1 and DETA/NO (0.5 mg/kg/12h and 1.57 mg/kg/24h, respectively; treatment schedule indicated by yellow circle in Scheme 1A and Figure 1A). “H” means higher dose of CORM-A1 and DETA/NO (1.5 mg/kg/12h and 2.358 mg/kg/24h, respectively; treatment schedule indicated by green circle in Scheme 1A and Figure 1A). Data presented as mean with standard deviation with points for individual measurements. Number of mice: 4-5 per group. Statistical analysis: Sidak's multiple comparison tests. *p< 0.05 as compared to control or as indicated.
CORM-A1 combined with DETA/NO inhibits lung metastasis in 4T1 orthotopic model and improves tumor blood perfusion
Lung metastatic foci formation was diminished in mice treated with the combination of CO and NO donor, but this effect was only temporarily observed on day 21 (Figure 4A). Primary tumor growth was not affected by the treatments used (Figure S3), but blood perfusion measured as a wash-in rate (WIR) parameter increased in tumor tissue obtained from mice treated with both compounds alone and in combination. On the other hand, CO and NO donors used concurrently did not affect wash-in perfusion index (WIPI), while this parameter increased significantly when they were used alone (Figure 4B). The level of vascular endothelial growth factor (VEGF) in tumor tissue was not affected by the compounds used (Figure S4A). The plasma level of VEGF was significantly decreased by CO+NO combined treatment on day 14. On the next days of tumor progression its level was similar to that observed in control tumor bearing or healthy mice (Figure 4D).
Antimetastatic effect of CORM-A1 combined with DETA/NO in orthotopic model of mouse mammary gland cancer 4T1. (A) Number of lung metastatic foci counted on day 21 and 28. (B) Tumor blood flow analysis: Wash-in rate (WIR, maximum slope) = peak enhancement/rise time, indicated how fast peak enhancement is achieved. Wash-in perfusion index (WIPI) = Area Under the Curve (WIAUC)/rise time and is representative of blood flow. Peak enhancement is calculated as a difference between the peak and the baseline. (C) Representative images of WIR. (D) VEGF concentration in mice plasma. (E) Tumor tissue level of endothelin-1 (ET-1), COX-2 and TGF-β estimated by ELISA. Mice were inoculated with 4T1 cells into fat pad and 7 days later, when tumors were palpable, the i.p. administration of CORM-A1 (0.5 mg/kg, every 12 h) and DETA/NO (1.57 mg/kg, every 24 h) started. Data presented as mean with standard deviation with points for individual measurements. Number of mice: 8-11 per group, some analyses were performed on randomly selected mice within the group. Number of healthy mice used as a control: 5. Statistical analysis: (A) Dunn's, (B) Dunnett's, (D) and (E) Sidak's multiple comparison tests. *p< 0.05 as compared to control or as indicated.
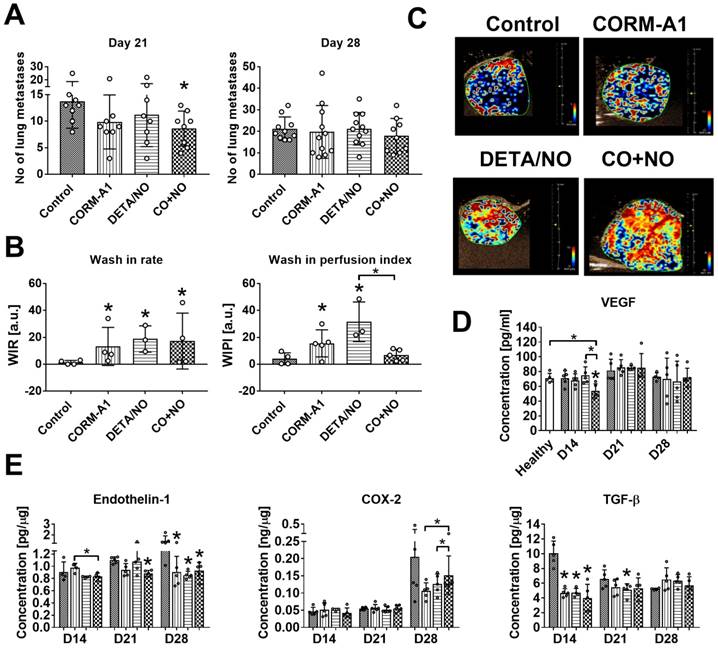
Tumor tissue was also evaluated for the level of ET-1, COX-2, TGF-β and iNOS. iNOS tumor tissue level increased on day 21 in all mice but was not changed by treatment regimens used (Figure S4B). ET-1 tumor level was diminished by combined therapy on day 21 and in all treated mice on day 28 (Figure 4E). The tendency to decrease COX-2 level was observed on day 28 in mice treated with CO and NO donors used alone or in combination, but this effect was significantly lower in the combination group (Figure 4E). TGF-β decrease was noticed on day 14 in all treated groups (Figure 4E).
Combined treatment with CORM-A1 and DETA/NO affects the plasma level of molecules involved in endothelium dysfunction and platelets activation
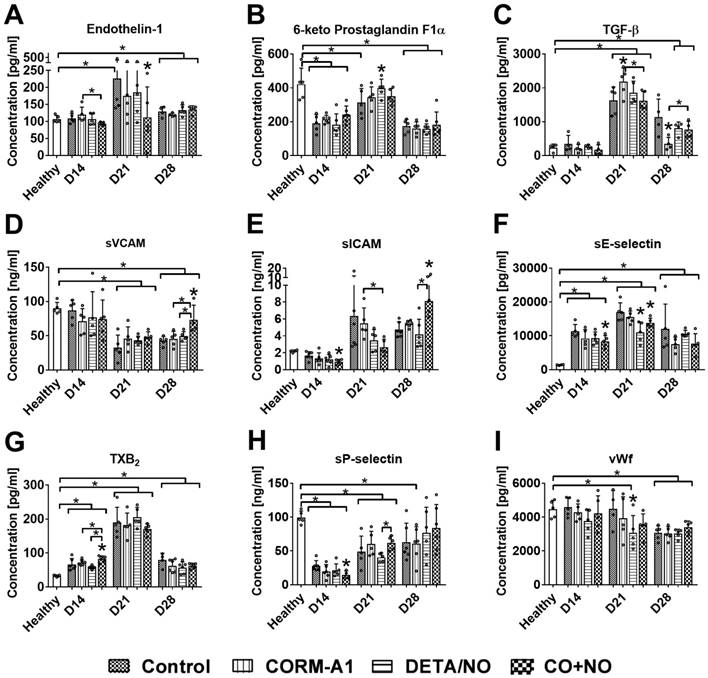
The level of ET-1 was the lowest in CO+NO group on day 14 and 21 (Figure 5A). Only DETA/NO significantly increased the plasma level of PGI2 metabolite on day 21 (Figure 5B). TGF-β level was significantly increased by CORM-A1 alone on day 21 and decreased on day 28 as compared to control mice. Combined treatment acted oppositely on these two days, first (day 21) decreasing and next (day 28) increasing TGF-β compared to CORM-A1 (Figure 5C). sVCAM and sICAM plasma level was significantly increased in mice treated with a combination of CO and NO donors on day 28. However, on days 14 and 21 sICAM level was the lowest in combined treatment groups (Figure 5D, 5E). Combined treatment decreased the level of sE-selectin particularly on day 14 and 21 (Figure 5F). TXB2 was increased and sP-selectin was decreased by compounds used in combination therapy on day 14 (Figure 5G, 5H). VWf plasma level was diminished only by DETA/NO on day 21 (Figure 5I).
Plasma level of selected molecules involved in endothelium dysfunction and platelets activation. Measured by ELISA kits: (A) Endothelin-1, (B) prostacyclin metabolite (C) TGF-β, (D) VCAM, (E) ICAM, (F) E-selectin, (G) TXB2, (H) P-selectin, (I) vWf. Mice were inoculated with 4T1 cells into fat pad and 7 days later, when tumors were palpable, the i.p. administration of CORM-A1 (0.5 mg/kg, every 12 h) and DETA/NO (1.570 mg/kg, every 24h) started. Data presented as mean with standard deviation with points for individual measurements. Number of mice: 8-11 per group, some analyses were performed on randomly selected mice within the group. Number of healthy mice used as a control: 4-5. Statistical analysis: Dunnett's multiple comparison test. *p< 0.05 as compared to control or as indicated.
We also performed the immunohistochemical analysis of endothelium in the aorta. eNOS level was increased by DETA/NO used alone and combined with CORM-A1. CORM-A1 alone did not influence eNOS level (Figure S5A, S5C). All treatments used (compounds alone or combined) decreased the level of VCAM positive endothelial cells, but only compounds used alone did so in a statistically significant manner (Figure S5B, S5D).
Prolonged CORM-A1 and DETA/NO combination therapy decrease total and phosphorylated VASP protein level in aorta but increase pVASP Ser239/VASP ratio
VASP and pVASPSer239 level in aortas harvested from mice on day 28 of the experiment was significantly diminished in combined treatment group, however the ratio of pVASP Ser239/VASP was significantly increased as compared to control tumor-bearing mice (Figure 6A-C). PKG and HO-1 level did not change in treated mice as compared to control tumor-bearing mice (Figure 6F, 6G).
CORM-A1 and DETA/NO combination therapy effect on total and phosphorylated VASP protein level in aorta from mice bearing 4T1 orthotopic tumors. (A) Total VASP, (B) pVASPSer239, (F) PKG-1α and (G) HO-1 densitometric analysis (mean optical density of bands of protein tested to β-actin) and representative blots (D), (E), (H), and (I) respectively. (C) Calculated pVASPSer239/VASP ratio. Mice were inoculated with 4T1 cells into fat pad and 7 days later, when tumors were palpable, the i.p. administration of CORM-A1 (0.5 mg/kg, every 12 h) and DETA/NO (1.570 mg/kg, every 24h) started. Tissues collected on day 28 of experiment. Data are presented as mean ±SD with individual measurements depicted. Number of samples analyzed: 3 per group. Statistical analysis: Dunnett's multiple comparison tests. *p< 0.05 as compared to control or as indicated.
Combination therapy with CORM-A1 and DETA/NO decreased epithelial to mesenchymal transition of 4T1 tumor cells
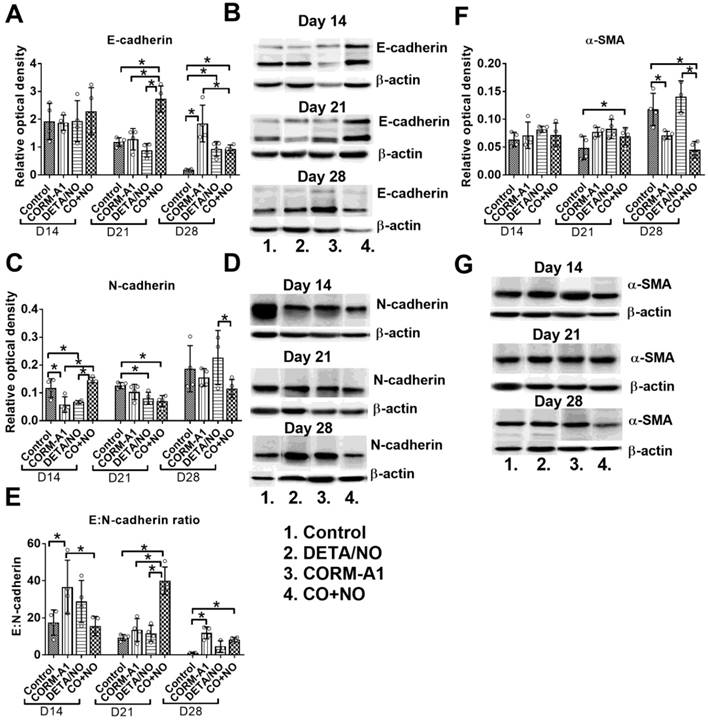
Significant increase of E-cadherin expression in tumor tissue obtained from mice treated with the combination of CO with NO donor was observed, particularly on day 21; it was accompanied by decreased expression of N-cadherin in tumor tissue (Figure 7A and 7C). This modulation of E- and N-cadherin levels led to a significant increase in E:N-cadherin ratio on day 21 and 28 in the combination treatment group (Figure 7E). α-SMA tumor tissue level was increased on day 21, but significantly decreased on day 28 in mice treated concurrently with CORM-A1 and DETA/NO (Figure 7F).
Epithelial to mesenchymal transition markers in 4T1 tumor tissue. (A) E-cadherin, (C) N-cadherin, (F) α-SMA densitometric analysis (mean optical density of bands of protein tested to β-actin) and representative blots (B), (D), (G), respectively. (E) Calculated E:N-cadherin ratio. Mice were inoculated with 4T1 cells into fat pad and 7 days later, when tumors were palpable, the i.p. administration of CORM-A1 (0.5 mg/kg, every 12 h) and DETA/NO (1.570 mg/kg, every 24h) started. Data presented as mean with standard deviation with points for individual measurements. Analyses were performed on tumors from randomly selected four mice within the group. Statistical analysis: Dunnett's multiple comparison test. *p<0.05 as indicated.
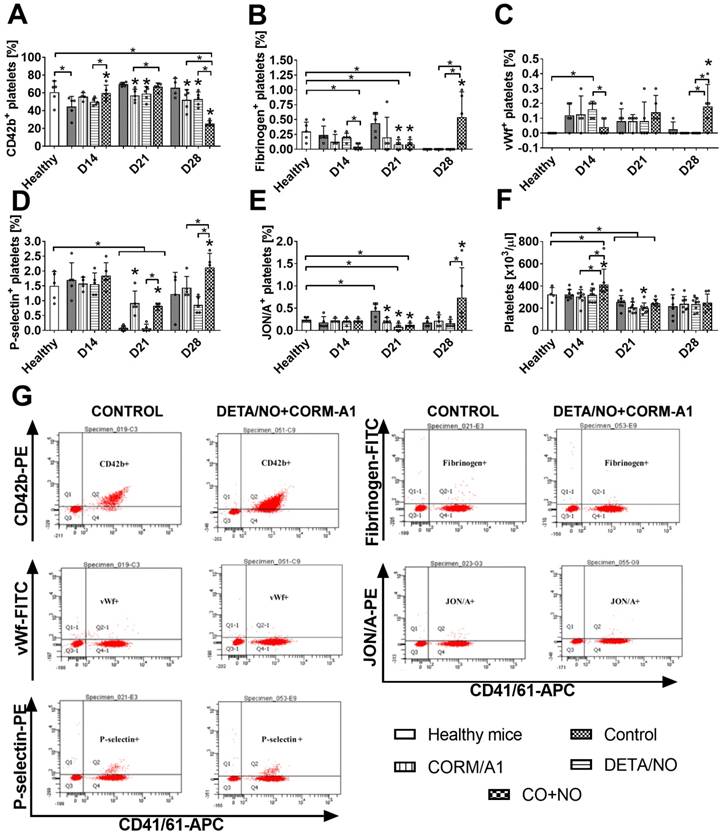
Decrease of platelets activation at the start of tumor progression and increase at the later stages by CORM-A1 combined with DETA/NO treatment
Markers of platelets activation measured on resting platelets on day 14 (Figure 8) indicated decreased activation in the combined treatment group as compared to control tumor bearing mice: increase of CD42b (Figure 8A) and decrease of fibrinogen (Figure 8B) and vWf bond (Figure 8C). On day 21 a similar tendency was observed for P-selectin (Figure 8D), however CORM-A1 used alone and combined with DETA/NO increased it. Moreover, vWf bond on platelets on day 21 has a tendency to increase in the combined treatment group, which was significant on day 28. Also, P-selectin, fibrinogen and active CD41/61 (JON/A antibody, Figure 8E) increased significantly on platelets but CD42b was significantly decreased on day 28 in mice treated with a combination of CORM-A1 and DETA/NO.
Platelets activation markers in mice bearing 4T1 tumors transplanted orthotopically and treated with CORM-A1 combined with DETA/NO. Flow cytometry analysis was performed on resting platelets collected on day 14, 21 and 28. Platelets were gated using DyLight649-labeled anti-mouse CD41/61 antibody. The following antibodies were used to assess platelets activation status: (A) PE-labeled anti-mouse GPIbα (CD42b), (B) FITC-labeled anti-mouse fibrinogen, (C) FITC-labeled anti-mouse vWf, (D) PE-labeled anti-mouse P-selectin (CD62P), (E) PE-labeled anti-mouse GPIIIbIIa (CD41/61) JON/A. (F) Number of platelets. (G) Representative dot-plots of analyses performed in control mice and mice treated with a combination of CORM-A1 and DETA/NO. Data presented as mean with standard deviation with points for individual measurements. Analyses were performed on blood from randomly selected 4-5 mice within the group. Statistical analysis: Dunnett's multiple comparison test. *p< 0.05 as compared to control or as indicated.
Decreased platelets aggregation and TGF-β release after ex vivo treatment with CORM-A1 combined with DETA/NO or PAPA/NO
CORM-A1 used alone did not significantly affect aggregation of platelets collected from breast cancer patients or from healthy donors. DETA/NO used alone or combined with CORM-A1 significantly decreased platelets aggregation. The combination of CO and NO donor tends to decrease platelet aggregation more intensively than DETA/NO used alone but only in healthy donors and in breast cancer patients with lymph node metastasis (Figure 9A). Figure 6B shows an example graph of platelet aggregation in PRP from a patient with metastatic cancer. The fastest process of platelet aggregation was observed in control group where the aggregation was 100% in six minutes. The maximal platelet aggregation after CORM-A1 incubation was 80% in five minutes. DETA/NO group reached the peak of platelet aggregation (70%) within six minutes. The combination of NO + CO had the strongest inhibitory effect on platelet aggregation for which only 10% aggregation was observed after 11 minutes. Figure 9C shows data for platelets aggregation only after incubation with PAPA/NO alone or combined with CORM-A1. All aggregation values shown in Figure 9C were significantly lower as compared to control platelets and platelets incubated with CORM/A1 (Figure 9A). The effect of PAPA/NO was increased by CORM-A1, and this tendency was highest for platelets from breast cancer patients without metastasis. However, the highest anti-aggregative effect of CORM-A1+PAPA/NO was observed for platelets obtained from breast cancer patients with lymph node metastasis (Figure 9C).
Effect of CORM-A1 combined with DETA/NO or PAPA/NO on platelets obtained from breast cancer patients and healthy donors ex vivo. (A) Platelet aggregation after the use of CORM-A1 and DETA/NO, (B) representative curves of platelet aggregation, (C) platelet aggregation after the use of CORM/A1 and PAPA/NO. CORM-A1 and DETA/NO or PAPA/NO were added to the PRP samples at the concentration of 100 µM for 15 min, next aggregation was induced with collagen (1 µg/ml). Number of patients: 27 without metastasis, 13 with lymph node metastasis (LN metastasis), 8 with distant metastasis (disseminated disease) and 14 healthy donors. (D) TGF-β and (E) TXB2 release from platelets treated ex vivo with CORM-A1 and PAPA/NO. Seven samples from patients with distant metastasis and metastasis in lymph nodes and six from healthy donors were analyzed. PRP was incubated with 100 µM of CORM-A1 and PAPA/NO and their combination for 15 min. Then, samples were centrifuged to obtain PPP in which the quantitative evaluation of TGF-β and TXB2 by ELISA test was performed. Data presented as mean with standard deviation with points for individual measurements. Statistical analysis: Dunn's multiple comparison test. *p< 0.05 as compared to control or as indicated.
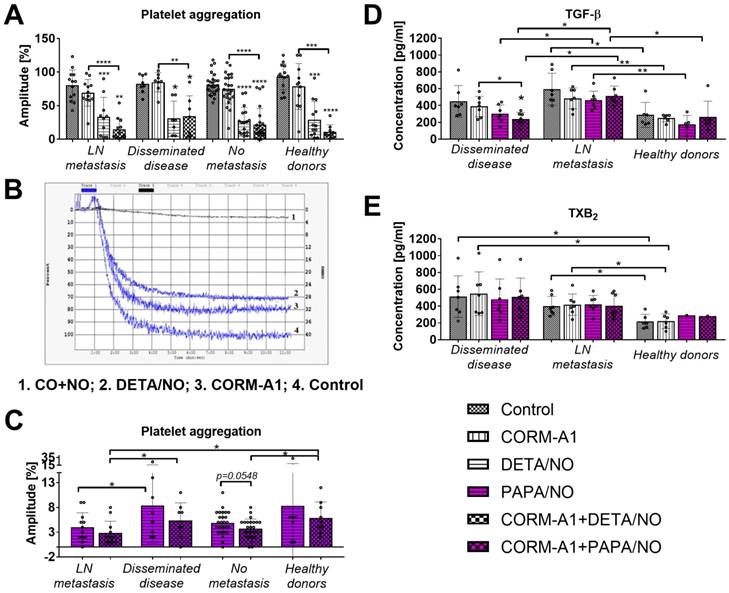
Platelets derived from patients with distant metastasis released lower levels of TGF-β after ex vivo incubation with CO+NO (Figure 9D). The treatment used did not affect the release of TXB2 from platelets (Figure 9E).
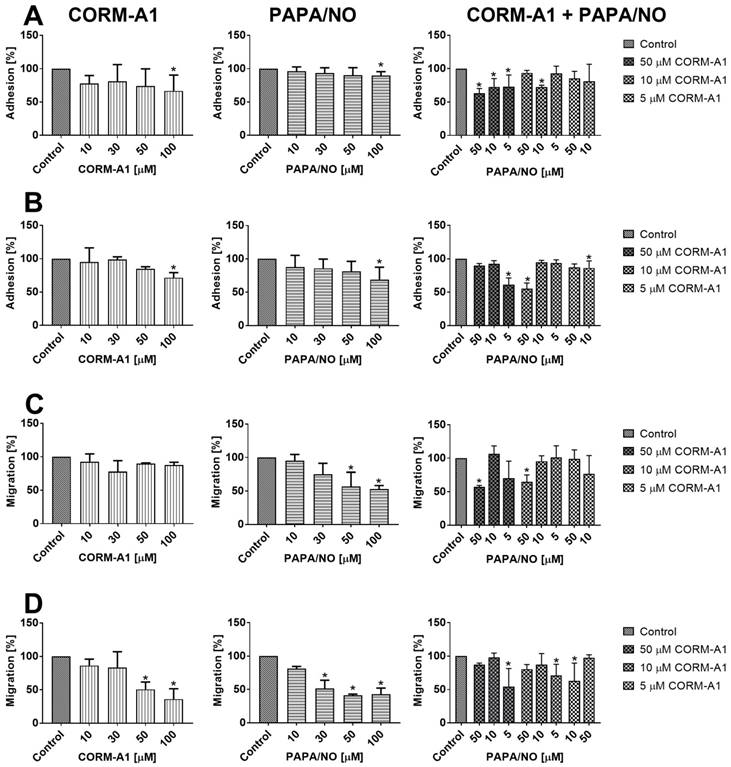
Direct effect of CORM-A1 and PAPA/NO on breast cancer cells in vitro
No inhibitory effect of CORM-A1 and PAPA/NO on the proliferation of 4T1 and 4T1-luc2-tdTomato or MDA-MB-231 cells was observed up to the concentration of 100 µM. We evaluated the adhesion of 4T1 and MDA-MB-231 cells to fibrinogen and their migration through fibrinogen after incubation with tested compounds using various concentrations (10-100 µM). CORM-A1 or PAPA/NO used alone inhibited adhesion of both cell lines only at the concentrations of 100 µM (Figure 10A and 10B). For the combined treatment we used concentrations of compounds which did not affect cell adhesion when used alone and we observed inhibition of 4T1 cells adhesion by 50 µM of CORM-A1 combined with all PAPA/NO concentrations (Figure 10A). In MDA-MB-231 cells this effect was observed only when 50 µM of CORM-A1 was combined with 5 µM PAPA/NO and 10 µM of CORM-A1 was combined with 50 µM PAPA/NO (Figure 10B). The effect of combined incubation on 4T1 cells migration was only the result of the compounds used alone (Figure 10C). In the case of MDA-MB-231 cells migration we also observed inhibition at low doses, which did not affect the migration alone: 5 µM PAPA/NO with 10 µM of CORM-A1 as well as 10 µM PAPA/NO with 5 µM of CORM-A1 (Figure 10D).
In vitro adhesion and migration of 4T1 and MDA-MB-231 cells incubated with CORM-A1 and PAPA/NO. Adhesion to fibrinogen of (A) 4T1 and (B) MDA-MB-231 cells. Migration through fibrinogen coated wells of (C) 4T1 and (D) MDA-MB-231 cells. Data presented as mean with standard deviation. First column: CORM-A1; Second column: PAPA/NO; Third column: CORM-A1 combined with PAPA/NO. Percent of migration in relation to non-treated control cells is shown. Experiments were repeated three times. Statistical analysis: Dunn's multiple comparison test. *p< 0.05 as compared to control.
Discussion
Analysis of antimetastatic effect of the combined application of CO and NO donors observed when these compounds were used at lower dosages at a 1:1 ratio shows that at least three factors that could determine the final result can be taken into account. The first one is the influence on endothelial dysfunction. It is directly correlated with the second one, namely the effect on platelet activation developing during tumor progression. The last factor is the direct effect on tumor cells. During our studies we evaluated all of these possible mechanisms of the effects of concurrent use of CORM-A1 with DETA/NO in the antimetastatic treatment of breast cancer.
The results of our studies conducted in intravenous models of experimental lung metastasis of 4T1-luc2-tdTomato suggest a major role of proper dosage regime, namely that higher doses of CORM-A1 (3 mg/kg/24h) combined with DETA/NO (2.358 mg/kg/24h) significantly stimulated lung metastatic foci formation, whereas lower doses acted oppositely. Kramkowski et al. have shown that antiplatelet properties of CORM-A1 are concentration-dependent and doses of CORMA-1 similar to those used in our studies significantly reduced platelet aggregation [27]. There is also a lot of information about various effect-concentration relationships of NO action and higher concentrations of NO having a rather stronger inhibitory effect on platelet aggregation [39]. However, it has been already reported that NO in combination with reactive oxygen species (ROS) can exert either a protective or disrupting effect on the endothelium depending on the concentration: low concentrations protect the endothelial layer, while higher amounts of NO might induce endothelial injury as well as a loss of its barrier function [40,41]. Therefore, the pro-metastatic action of higher concentrations of investigated compounds can be the result of the disrupting effect on the endothelium which prevails over its anti-aggregatory effect on platelets.
Additionally, when we combined the therapy using DETA/NO with clopidogrel in our previous studies, also only lower doses of DETA/NO combined with clopidogrel had an antimetastatic effect, but at higher doses adverse effects similar to those observed for a combination with CORM-A1 were not observed [26]. It may be explained by vasoprotective activity of clopidogrel [42] that protects endothelium against NO-induced injury. In order to verify this hypothesis, we have compared both experimental models (with high and low dosage of CO and NO donors) and analyzed the expression of proteins associated with the signaling pathways involved in the protection of the vascular endothelium dysfunction, namely cGMP downstream signaling molecules [43]. Both CO and NO are known as molecules that induce phosphorylation of vasodilator-stimulated phosphoprotein (VASP) at Ser-239 by cGMP-dependent protein kinase (PKG) [44]. VASP phosphorylation, among others, leads to reduced vascular inflammation [43]. In our studies, the lowest levels of total VASP, pVASPSer239 and the lowest ratio of pVASPSer239/VASP in the lung were observed in the combined treatment group, where the stimulation of metastasis was noted, which contributes to the confirmation of our assumption concerning increased endothelium damage in this group of mice.
One of the mechanisms involved in the endothelial dysfunction is the oxidative stress, and NO can inactivate antioxidative enzymes such as glutathione peroxidase [45,46]. On the other hand, oxidative stress inactivates NO and impairs its functions e.g. endothelium-dependent vasodilation [47]. It can be assumed that excessive supply of exogenous NO might lead to the trap of a vicious circle, resulting in the development of diverse undesired reactions. The CO (HO-1-derived or delivered by various donors) is considered as a cytoprotective and antioxidative molecule [48-50]. On the other hand, HO derived CO is able to regulate blood pressure only when NOS pathway is functional [51]. Moreover, prolonged stimulation of cGMP by CO decreases cGMP level and guanylate cyclase activity by limiting the availability of cellular heme [52]. In our studies we did not observe any significant changes in the level of molecules associated with the oxidative stress including myeloperoxidase (MPO) or lipids peroxidation in combined treatment groups. However, both doses of DETA/NO, a high dose of CORM-A1 and both doses in combined treatment groups led to a decrease in nitrite plasma levels. Moreover, only in the high-dose CORM-A1 group we observed increased HO-1 expression, whereas in the high-dose combined treatment group this effect was not visible. Partial explanation of this observation may by the fact, that chronic induction of HO impaired NOS pathway [51] and CO is an inhibitor of NO generation by iNOS [13]. In addition, during the growth of 4T1 cells an impairment of NO production by pulmonary endothelium was observed [53] which may increase the tissue requirements for this molecule. Therefore the level of nitrite determined in plasma will not correspond quantitatively to the exogenously supplied NO.
Stojak et al. have shown that CO and NO donors used simultaneously impaired bioenergetics of both cancer and endothelial cells [25]. Thus, a decreased metabolic activity may affect both cancer and normal cells in the body. It can be speculated, that the decrease in nitrite plasma level, especially in DETA/NO and combined treatment groups as well as decreased VASP lung levels may result from the impaired metabolism of cells. Interestingly, we have observed decreased aspartate aminotransferase (AST) in CORM-A1 and in the higher doses combined group. Decreased metabolic activity of hepatocytes may explain this observation; previously reported inhibitory effect of CO donor (CORM-401) on glycolysis have shown irreversibility of its effect, whereas the effect of NO donor (PAPA/NO) on mitochondrial respiration was reversible [25]. Discussed above effects on metastatic process: deleterious effects of high-dose combined treatment regime and beneficial effects of low-dose combination are reversed when cardiac injury parameter is analyzed. Increased, as compared to healthy mice, CK-MB isoform level in 4T1 cells bearing mice was decreased by high-dose treatment and in opposite - increased by low-dose combination. Duvigneau and Kozlov in an interesting review have summarized the beneficial and deleterious effects of CO and NO on mitochondria and conclude the importance of the balance between those molecules, their elimination rate, ongoing inflammation, ROS generation as well as the presence of tissue hypoxia in the final results on the mitochondria-mediated CO and NO effects [54]. Further research is necessary to explain how safe it is to exogenously provide these two molecules, especially to the organism affected by the cancer-accompanied inflammatory process, so that the beneficial effects are ultimately obtained, and the harmful ones can be omitted.
As we have shown, the inflammatory response to LPS was found to be important in our intravenous model. The stimulation of metastatic process by higher doses of compounds was observed only in mice pretreated with LPS, where the endothelium dysfunction was visible as a higher ET-1 plasma concentration in LPS and tumor cells injected mice as compared to healthy ones. In mice pretreated with LPS, the ET-1 level was significantly increased 24 h after the cancer cells inoculation and it was still observed after 14 days as compared to healthy mice. This effect was not related to cancer cells itself: it was not observed in mice not pretreated with LPS. ET-1 is the molecule whose level increased during endothelial dysfunction and can be used as a marker of this process [55]. The lowest level of ET-1 in mice treated with lower doses of CO+NO donors correlated with the lowest number of lung metastases. On the other hand, it was not correlated with platelets activation status. Both compounds used concurrently were able to significantly diminish P-selectin positive platelets percentage only in mice not pretreated with LPS. 4T1 mammary gland cancer progression is accompanied by an extensive inflammatory process [30], therefore the use of LPS as the inducer of inflammation and endothelial dysfunction [31,56] was dictated by the necessity to recreate some of the conditions prevailing in an organism with this type of cancer. On the other hand, we were able to omit the impact of therapy used on the primary tumor and indicate the importance of the combined treatment's influence on the endothelium. Therefore, we can conclude that in the intravenous model of lung metastasis, inhibition of endothelial dysfunction is the mechanism responsible for antimetastatic action of CORM-A1 combined with DETA/NO.
Orthotopic tumor models are more relevant models allowing us to follow almost all steps of tumor progression and metastasis [57]. Therefore, in our research we transplanted 4T1 mammary gland cancer cells orthotopically and mice were observed 14, 21 and 28 days after tumor cells inoculation. Mice were treated with lower doses of compounds, selected on the basis of the studies using the intravenous model. However, we could observe only transient (on day 21) antimetastatic effect of combined treatment, which disappeared on day 28. Tumor growth was not changed during treatment as compared to non-treated mice. However, wash-in rate parameter, which indicates how fast maximal concentration of contrast agent in tumor tissue is achieved, increased in all treated groups. On the other hand, wash-in perfusion index - related to blood flow was increased only in mice treated with both agents alone. Combined treatment with CO and NO donors seems not to affect blood flow significantly, despite decreased VEGF plasma level on day 14 in combined treatment group. VEGF is the most important proangiogenic molecule, whose treatment-induced reduction may lead to blood vessel normalization and consequently to improved drug delivery to the tumor tissue [58]. However, although the VEGF plasma level diminished as the effect of combined treatment, we did not observe any effects on VEGF level in tumor tissue.
TGF-β is another molecule that is known to affect tumor angiogenesis [59,60]. Tumor tissue level of TGF-β was significantly diminished in mice from all treated groups, whereas its plasma level was significantly increased by CORM-A1 on day 21 which was attenuated by DETA/NO in the combined treatment group. Decrease of TGF-β level in tumor tissue may be the reason of decreased epithelial to mesenchymal transition (EMT) process observed in tumor tissue of mice treated with both compounds combined, as TGF-β is one of the most important molecules involved in EMT process [61]. EMT is one of the first steps of metastatic progression of tumor cells [62] and its significant inhibition, especially on day 21 may be responsible for decreased metastatic potential of 4T1 tumor cells. Platelets are a major source of TGF-β also within the tumor tissue. Platelets affected by CORM-A1 and DETA/NO may indirectly change the progression of EMT. Our ex vivo studies analyzing the aggregation of platelets from healthy donors or breast cancer patients showed some improvement of antiaggregatory effect of DETA/NO or PAPA/NO by CORM-A1. Moreover, platelets from breast cancer patients with disseminated disease incubated with CORM-A1 combined with PAPA/NO released the lowest TGF-β level. Unfortunately, we did not observe improvement of TGF-β diminishing by combined treatment in 4T1 tumor tissue, probably due to the fact that platelets are not the only source of this molecule. Cancer cells themselves, cancer associated fibroblasts, and various inflammatory response cells are the source of TGF-β [63,64] and not all of them may be modulated by the therapy used in our studies. Additionally, the agents used together may affect EMT process by direct influence on tumor cells. This effect was suggested by the results of in vitro studies, where both compounds decreased adhesion and migration properties of 4T1 as well as MDA-MB-231 cells. The recent studies by Stojak et al. support these conclusions, showing that the CO donor used together with the NO donor inhibits breast cancer cells transmigration through endothelial cells in vitro [25]. Moreover, improved motility is a characteristic feature of cancer cells that underwent EMT process [62].
As we have shown in intravenous model, CORM-A1 combined with DETA/NO diminished ET-1 plasma level indicating the endothelium as the target of the combined treatment used. A similar effect was observed temporarily in orthotopic model: diminishing of ET-1 level was observed only on day 14 and 21, and then the effect on ET-1 or on metastasis was not observed. Also other molecules informing about endothelial activation, like sICAM and sE-selectin [9,65] were diminished in the beginning of tumor progression. However, we observed a significant elevation of sICAM and sVCAM plasma level in mice treated with CORM-A1 combined with DETA/NO on the last day of observation, which was accompanied by the lack of influence on metastases. Moreover, VCAM positive endothelial cells in the aorta were diminished significantly only in mice treated with donors used alone. NO was previously reported to limit endothelium activation through diminution of VCAM [66]; Stanford et al. reported that CO-releasing molecule can succesfully inhibit ET-1 production in human pulmonary arteria [67]. Moreover, Kaczara et al. have recently shown that CO-RMs increased the production of NO by eNOS in endothelial cells [24]. Paradoxically, gaseous CO rapidly enhanced mitochondria activity of cancer cells that resulted in metabolic exhaustion and cellular collapse causing tumor regression. Furthermore, CO increased cancer cell sensitivity to chemotherapeutics, simultaneously protecting normal cell growth and viability [68]. However, CORM-A1 used alone in our studies did not significantly influence eNOS expression in the aorta endothelium. Only NO-donor alone or combined with CORM-A1 significantly upregulated eNOS expression. These data suggest that it is exogenous NO and not CO that regulates eNOS expression. Yuhanna et al. showed that exogenous NO is able to directly modulate eNOS expression [69]. Thus, exogenous NO is able both to compensate the lack of endogenous NO and to stimulate its production by elevating eNOS expression. This effect may support protective effects of combined treatment on the endothelium in mice bearing 4T1 mammary gland cancer at earlier stages of tumor progression, because it was reported that development of endothelium dysfunction in this tumor model is accompanied by impaired NO production [70].
Also, the action of both compounds on platelet activation changed during tumor progression. On day 14 in the combined treatment group we observed diminished bound of fibrinogen and vWf on platelets, whereas on day 28 all measured parameters of platelet activation significantly increased. This increased platelets activation is directly related to the effects of the treatment on endothelium. It is recognized, that endothelium dysfunction lead to platelet activation manifested, e.g. by an increased binding of vWF and fibrinogen to platelets [71]. Although we have observed described above signs of endothelium activation on 28 day of experiment, we have also noticed significantly increased phosphorylation of VASP, measured as a ratio of pVASPSer239 to total VASP, synergistically induced by combined treatment. However, the importance of this valuable process for the protection of the vascular endothelium induced by CO and NO is reduced by the observed significant decrease in the overall level of VASP in this group. Interestingly, in the model of experimental metastases higher doses of compounds used also diminished total VASP level. It can therefore be concluded that both long-term and high doses of CORM-A1 together with DETA/NO exposure leads to the reduction in the total VASP level. Recent studies by Zhikui et al. have shown, that VASP overexpression in cancer cells leads to an increased invasive potential and motility [72]. These decreasing effects of CORM-A1 and DETA/NO combined treatments on VASP level in aorta or lung tissue (with present metastatic foci) may occurs also in tumor tissue and contribute to antimetastatic effects (including EMT inhibition [72]) observed in early step of tumor progression.
To sum up, the applied therapy has shown antimetastatic action preferably at the early stages of tumor dissemination i.e.: it inhibited both EMT and tumor cell migration in fibronectin. This is also confirmed by the results of in vivo studies which showed stronger inhibitory effect of the applied therapy on the platelet activation and endothelial dysfunction at the earlier stages of cancer progression and a weaker or opposite effect of the compounds at the last time points (Scheme 2).
Mechanism of antimetastatic action of CORM-A1 + DETA/NO combined therapy. In the diagram, the upper arrow (I) indicates early stages of progression of 4T1 mammary gland cancer, the lower arrow (II) shows a later stage (after day 21). The descriptions in the bottom panel refer to the changes observed at earlier stages of cancer progression under the influence of combined therapy using CO and NO donors.
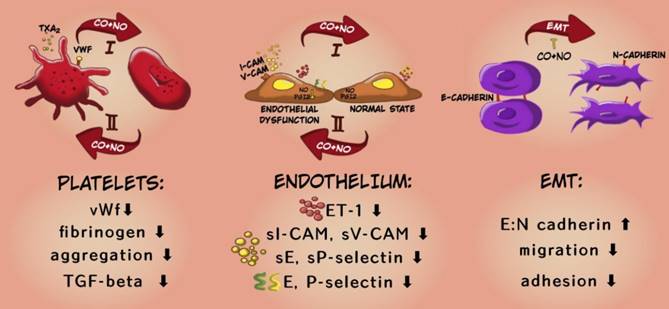
Therefore, the investigated treatment could be applied to prolong the time period over which primary tumor does not metastasize, or to inhibit metastasis formation at the early stages of this process. Although it is less effective at advanced stages of cancer development, this effectiveness may be sufficient when used in combination with standard treatment strategies. However, to define the safety of CO and NO exogenous delivery, especially when cancer progression is accompanied by inflammation, further studies are necessary.
Supplementary Material
Supplementary figures.
Acknowledgements
This study was supported by the National Centre for Research and Development under the Polish Strategical Framework Program STRATEGMED (grant coordinated by JCET-UJ No. STRATEGMED1/233226/11/NCBR/2015). AŁ-S and MS are thankful to the Wroclaw University of Science and Technology for support (Statute Funds 0401/0145/18). The funding bodies did not participate in the design of the study, collection, analysis and interpretation of data or in writing the manuscript.
Author Contribution
Concept of the study: JW. Design of the study: KP, JW and MP. Data acquisition and analysis: KP, DP, MP, AŁ-S, MM, MN, TMG, JJ, JB, MS, RM and JW. Collection of patient samples: RM, ME. Writing the main part of the manuscript: JW. All the authors reviewed and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Rejniak KA. Circulating Tumor Cells: When a Solid Tumor Meets a Fluid Microenvironment. Adv Exp Med Biol. 2016;936:93-106
2. Blazejczyk A, Papiernik D, Porshneva K, Sadowska J, Wietrzyk J. Endothelium and cancer metastasis: Perspectives for antimetastatic therapy. Pharmacol Reports. 2015;67:711-8
3. Wojtukiewicz MZ, Sierko E, Hempel D, Tucker SC, Honn K V. Platelets and cancer angiogenesis nexus. Cancer Metastasis Rev. 2017;36:249-62
4. Chlopicki S, Lomnicka M, Fedorowicz A, Grochal E, Kramkowski K, Mogielnicki A. et al. Inhibition of platelet aggregation by carbon monoxide-releasing molecules (CO-RMs): Comparison with NO donors. Naunyn Schmiedebergs Arch Pharmacol. 2012;385:641-50
5. Chlopicki S, Olszanecki R, Marcinkiewicz E, Lomnicka M, Motterlini R. Carbon monoxide released by CORM-3 inhibits human platelets by a mechanism independent of soluble guanylate cyclase. Cardiovasc Res. 2006;71:393-401
6. Tousoulis D, Kampoli A-M, Tentolouris C, Papageorgiou N, Stefanadis C. The role of nitric oxide on endothelial function. Curr Vasc Pharmacol. 2012;10:4-18
7. Zafirellis K, Zachaki A, Agrogiannis G, Gravani K. Inducible nitric oxide synthase expression and its prognostic significance in colorectal cancer. APMIS. 2010;118:115-24
8. Massi D, Marconi C, Franchi A, Bianchini F, Paglierani M, Ketabchi S. et al. Arginine metabolism in tumor-associated macrophages in cutaneous malignant melanoma: evidence from human and experimental tumors. Hum Pathol. 2007;38:1516-25
9. Lu Y, Yu T, Liang H, Wang J, Xie J, Shao J. et al. Nitric oxide inhibits hetero-adhesion of cancer cells to endothelial cells: Restraining circulating tumor cells from initiating metastatic cascade. Sci Rep. 2014;4:1-9
10. Motoya A, Masayuki T, Toshiyuki K, Masahiko K. Differential inhibition of platelet aggregation and calcium mobilization by nitroglycerin and stabilized nitric oxide. J Cardiovasc Pharmacol. 1994;24:860-6
11. Chegaev K, Riganti C, Lazzarato L, Rolando B, Guglielmo S, Campia I. et al. Nitric oxide donor doxorubicins accumulate into Doxorubicin-resistant human colon cancer cells inducing cytotoxicity. ACS Med Chem Lett. 2011;2:494-7
12. Sharma VS, Magde D. Activation of Soluble Guanylate Cyclase by Carbon Monoxide and Nitric Oxide: A Mechanistic Model. Methods. 1999;19:494-505
13. Datta PK, Koukouritaki SB, Hopp KA, Lianos EA. Heme oxygenase-1 induction attenuates inducible nitric oxide synthase expression and proteinuria in glomerulonephritis. J Am Soc Nephrol. 1999;10:2540-50
14. Oláh G, Módis K, Törö G, Hellmich MR, Szczesny B, Szabo C. Role of endogenous and exogenous nitric oxide, carbon monoxide and hydrogen sulfide in HCT116 colon cancer cell proliferation. Biochem Pharmacol. 2018;149:186-204
15. Miller MR, Megson IL. Recent developments in nitric oxide donor drugs. Br J Pharmacol. 2009;151:305-21
16. Cerwinka WH, Cooper D, Krieglstein CF, Feelisch M, Granger DN. Nitric oxide modulates endotoxin-induced platelet-endothelial cell adhesion in intestinal venules. Am J Physiol Circ Physiol. 2002;282:H1111-7
17. Panés J, Soriano A, Salas A, Sans M, Gironella M, Piqué JM. et al. Nitric Oxide Supplementation Ameliorates Dextran Sulfate Sodium-Induced Colitis in Mice. Lab Investig. 2004;82:597-608
18. Pervin S, Singh R, Chaudhuri G. Nitric oxide-induced cytostasis and cell cycle arrest of a human breast cancer cell line (MDA-MB-231): potential role of cyclin D1. Proc Natl Acad Sci U S A. 2001;98:3583-8
19. Clark JE, Naughton P, Shurey S, Green CJ, Johnson TR, Mann BE. et al. Cardioprotective Actions by a Water-Soluble Carbon Monoxide-Releasing Molecule. Circ Res. 2003;93:e2-8
20. Motterlini R, Sawle P, Hammad J, Bains S, Alberto R, Foresti R. et al. CORM-A1: a new pharmacologically active carbon monoxide-releasing molecule. FASEB J. 2005;19:284-6
21. Motterlini R, Clark JE, Foresti R, Sarathchandra P, Mann BE, Green CJ. Carbon monoxide-releasing molecules: characterization of biochemical and vascular activities. Circ Res. 2002;90:E17-24
22. Brüne B, Schmidt KU, Ullrich V. Activation of soluble guanylate cyclase by carbon monoxide and inhibition by superoxide anion. Eur J Biochem. 1990;192:683-8
23. Song H, Bergstrasser C, Rafat N, Höger S, Schmidt M, Endres N. et al. The carbon monoxide releasing molecule (CORM-3) inhibits expression of vascular cell adhesion molecule-1 and E-selectin independently of haem oxygenase-1 expression. Br J Pharmacol. 2009;157:769-80
24. Kaczara P, Proniewski B, Lovejoy C, Kus K, Motterlini R, Abramov AY. et al. CORM-401 induces calcium signalling, NO increase and activation of pentose phosphate pathway in endothelial cells. FEBS J. 2018;285:1346-58
25. Stojak M, Kaczara P, Motterlini R, Chlopicki S. Modulation of cellular bioenergetics by CO-releasing molecules and NO-donors inhibits the interaction of cancer cells with human lung microvascular endothelial cells. Pharmacol Res. 2018;136:160-71
26. Porshneva K, Papiernik D, Psurski M, Nowak M, Matkowski R, Ekiert M. et al. Combination Therapy with DETA/NO and Clopidogrel Inhibits Metastasis in Murine Mammary Gland Cancer Models via Improved Vasoprotection. Mol Pharm. 2018;15:5277-90
27. Kramkowski K, Leszczynska A, Mogielnicki A, Chlopicki S, Fedorowicz A, Grochal E. et al. Antithrombotic Properties of Water-Soluble Carbon Monoxide-Releasing Molecules. Arterioscler Thromb Vasc Biol. 2012;32:2149-57
28. Mooradian DL, Hutsell TC, Keefer LK. Nitric oxide (NO) donor molecules: effect of NO release rate on vascular smooth muscle cell proliferation in vitro. J Cardiovasc Pharmacol. 1995;25:674-8
29. Gómez-Cuadrado L, Tracey N, Ma R, Qian B, Brunton VG. Mouse models of metastasis: progress and prospects. Dis Model Mech. 2017;10:1061-74
30. DuPre' SA, Hunter KW. Murine mammary carcinoma 4T1 induces a leukemoid reaction with splenomegaly: Association with tumor-derived growth factors. Exp Mol Pathol. 2007;82:12-24
31. Harmey JH, Bucana CD, Lu W, Byrne AM, McDonnell S, Lynch C. et al. Lipopolysaccharide-induced metastatic growth is associated with increased angiogenesis, vascular permeability and tumor cell invasion. Int J Cancer. 2002;101:415-22
32. Denslow A, Świtalska M, Jarosz J, Papiernik D, Porshneva K, Nowak M. et al. Clopidogrel in a combined therapy with anticancer drugs—effect on tumor growth, metastasis, and treatment toxicity: Studies in animal models. Rich BE, redakteur. PLoS One. 2017;12:e0188740
33. Anisiewicz A, Pawlik A, Filip-Psurska B, Turlej E, Dzimira S, Milczarek M. et al. Unfavorable effect of calcitriol and its low-calcemic analogs on metastasis of 4T1 mouse mammary gland cancer. Int J Oncol. 2018;52:103-26
34. Beutler E, West C. Simplified determination of carboxyhemoglobin. Clin Chem. 1984;30:871-4
35. Janero DR. Malondialdehyde and thiobarbituric acid-reactivity as diagnostic indices of lipid peroxidation and peroxidative tissue injury. Free Radic Biol Med. 1990;9:515-40
36. Giustarini D, Rossi R, Milzani A, Dalle-Donne I. Nitrite and Nitrate Measurement by Griess Reagent in Human Plasma: Evaluation of Interferences and Standardization. Methods Enzymol. 2008;440:361-80
37. Remmele W, Stegner HE. [Recommendation for uniform definition of an immunoreactive score (IRS) for immunohistochemical estrogen receptor detection (ER-ICA) in breast cancer tissue]. Pathologe. 1987;8:138-40
38. Wietrzyk J, Chodyński M, Fitak H, Wojdat E, Kutner A, Opolski A. Antitumor properties of diastereomeric and geometric analogs of vitamin D3. Anticancer Drugs. 2007;18:447-57
39. Sogo N, Magid KS, Shaw CA, Webb DJ, Megson IL. Inhibition of human platelet aggregation by nitric oxide donor drugs: Relative contribution of cGMP-independent mechanisms. Biochem Biophys Res Commun. 2000;279:412-9
40. Darley-Usmar V, White R. Physiological society symposium: impaired endothelial and smooth muscle cell function in oxidative stress. Disruption of vascular signalling by the reaction of nitric oxide with superoxide: implications for cardiovascular disease. ExperPhysiol. 1997;82:305-16
41. Palmer RM, Bridge L, Foxwell NA, Moncada S. The role of nitric oxide in endothelial cell damage and its inhibition by glucocorticoids. Br J Pharmacol. 1992;105:11-2
42. Yang H, Zhao P, Tian S. Clopidogrel Protects Endothelium by Hindering TNFalpha-Induced VCAM-1 Expression through CaMKKbeta/AMPK/Nrf2 Pathway. J Diabetes Res. 2015;2016:e9128050
43. Cheng AM, Rizzo-DeLeon N, Wilson CL, Lee WJ, Tateya S, Clowes AW. et al. Vasodilator-stimulated phosphoprotein protects against vascular inflammation and insulin resistance. Am J Physiol Endocrinol Metab. 2014;307:E571-9
44. Calzi SL, Purich DL, Chang KH, Afzal A, Nakagawa T, Busik J V. et al. Carbon monoxide and nitric oxide mediate cytoskeletal reorganization in microvascular cells via vasodilator-stimulated phosphoprotein phosphorylation: Evidence for blunted responsiveness in diabetes. Diabetes. 2008;57:2488-94
45. Asahi M, Fujii J, Takao T, Kuzuya T, Hori M, Shimonishi Y. et al. The oxidation of selenocysteine is involved in the inactivation of glutathione peroxidase by nitric oxide donor. J Biol Chem. 1997;272:19152-7
46. Asahi M, Fujii J, Suzuki K, Seo HG, Kuzuya T, Hori M. et al. Inactivation of Glutathione Peroxidase by Nitric Oxide. J Biol Chem. 1995;270:21035-9
47. Higashi Y, Noma K, Yoshizumi M, Kihara Y. Endothelial function and oxidative stress in cardiovascular diseases. Circ J. 2009;73:411-8
48. Motterlini R, Nikam A, Manin S, Ollivier A, Wilson JL, Djouadi S. et al. HYCO-3, a dual CO-releaser/Nrf2 activator, reduces tissue inflammation in mice challenged with lipopolysaccharide. Redox Biol. 2019;20:334-48
49. Ryter SW, Otterbein LE, Morse D, Choi AMK. Heme oxygenase/carbon monoxide signaling pathways: Regulation and functional significance. Mol Cell Biochem. 2002:234 /235: 249-63
50. Kaczara P, Motterlini R, Rosen GM, Augustynek B, Bednarczyk P, Szewczyk A. et al. Carbon monoxide released by CORM-401 uncouples mitochondrial respiration and inhibits glycolysis in endothelial cells: A role for mitoBKCa channels. Biochim Biophys Acta - Bioenerg. 2015;1847:1297-309
51. Polizio AH, Santa-Cruz DM, Balestrasse KB, Gironacci MM, Bertera FM, Höcht C. et al. Heme oxygenase-1 overexpression fails to attenuate hypertension when the nitric oxide synthase system is not fully operative. Pharmacology. 2011;87:341-9
52. Abraham NG, Quan S, Mieyal PA, Yang L, Burke-Wolin T, Mingone CJ. et al. Modulation of cGMP by human HO-1 retrovirus gene transfer in pulmonary microvessel endothelial cells. Am J Physiol Cell Mol Physiol. 2002;283:L1117-24
53. Smeda M, Kieronska A, Adamski MG, Proniewski B, Sternak M, Mohaissen T. et al. Nitric oxide deficiency and endothelial-mesenchymal transition of pulmonary endothelium in the progression of 4T1 metastatic breast cancer in mice. Breast Cancer Res. 2018;20:86
54. Duvigneau JC, Kozlov A V. Pathological Impact of the Interaction of NO and CO with Mitochondria in Critical Care Diseases. Front Med. 2017;4:223
55. Iglarz M, Clozel M. Mechanisms of ET-1-induced endothelial dysfunction. J Cardiovasc Pharmacol. 2007;50:621-8
56. Li S, Xu X, Jiang M, Bi Y, Xu J, Han M. Lipopolysaccharide induces inflammation and facilitates lung metastasis in a breast cancer model via the prostaglandin E2-EP2 pathway. Mol Med Rep. 2015;11:4454-62
57. Rashid OM, Nagahashi M, Ramachandran S, Dumur C, Schaum J, Yamada A. et al. An improved syngeneic orthotopic murine model of human breast cancer progression. Breast Cancer Res Treat. 2014;147:501-12
58. Maj E, Papiernik D, Wietrzyk J. Antiangiogenic cancer treatment: The great discovery and greater complexity (Review). Int J Oncol. 2016;49:1773-84
59. Ferrari G, Cook BD, Terushkin V, Pintucci G, Mignatti P. Transforming growth factor-beta 1 (TGF-beta1) induces angiogenesis through vascular endothelial growth factor (VEGF)-mediated apoptosis. J Cell Physiol. 2009;219:449-58
60. Viñals F, Pouysségur J. Transforming growth factor beta1 (TGF-beta1) promotes endothelial cell survival during in vitro angiogenesis via an autocrine mechanism implicating TGF-alpha signaling. Mol Cell Biol. 2001;21:7218-30
61. Santibanez JF, Obradović H, Kukolj T, Krstić J. Transforming growth factor-β, matrix metalloproteinases, and urokinase-type plasminogen activator interaction in the cancer epithelial to mesenchymal transition. Dev Dyn. 2018;247:382-95
62. Zeisberg M, Neilson EG. Biomarkers for epithelial-mesenchymal transitions. J Clin Invest. 2009;119:1429-37
63. Costanza B, Umelo I, Bellier J, Castronovo V, Turtoi A. Stromal Modulators of TGF-β in Cancer. J Clin Med. 2017;6:7
64. Yu Y, Xiao C-H, Tan L-D, Wang Q-S, Li X-Q, Feng Y-M. Cancer-associated fibroblasts induce epithelial-mesenchymal transition of breast cancer cells through paracrine TGF-β signalling. Br J Cancer. 2014;110:724-32
65. P. Mann A, Tanaka T. E-selectin: Its Role in Cancer and Potential as a Biomarker. Transl Med. 2011;01:002
66. De Caterina R, Libby P, Peng HB, Thannickal VJ, Rajavashisth TB, Gimbrone MA. et al. Nitric oxide decreases cytokine-induced endothelial activation. Nitric oxide selectively reduces endothelial expression of adhesion molecules and proinflammatory cytokines. J Clin Invest. 1995;96:60-8
67. Stanford SJ, Walters MJ, Mitchell JA. Carbon monoxide inhibits endothelin-1 release by human pulmonary artery smooth muscle cells. Eur J Pharmacol. 2004;486:349-52
68. Wegiel B, Gallo D, Csizmadia E, Harris C, Belcher J, Vercellotti GM. et al. Carbon monoxide expedites metabolic exhaustion to inhibit tumor growth. Cancer Res. 2013;73:7009-21
69. Yuhanna IS, MacRitchie AN, Lantin-Hermoso RL, Wells LB, Shaul PW. Nitric Oxide (NO) Upregulates NO Synthase Expression in Fetal Intrapulmonary Artery Endothelial Cells. Am J Respir Cell Mol Biol. 1999;21:629-36
70. Smeda M, Kieronska A, Proniewski B, Jasztal A, Selmi A, Wandzel K. et al. Dual antiplatelet therapy with clopidogrel and aspirin increases mortality in 4T1 metastatic breast cancer-bearing mice by inducing vascular mimicry in primary tumour. Oncotarget. 2018;9:17810-24
71. Buerkle MA, Lehrer S, Sohn HY, Conzen P, Pohl U, Krötz F. Selective inhibition of cyclooxygenase-2 enhances platelet adhesion in hamster arterioles in vivo. Circulation. 2004;110:2053-9
72. Yang W, Wang Y, Wang L, Tu K, Liu Z, Dou C. et al. Hypoxia-induced up-regulation of VASP promotes invasiveness and metastasis of hepatocellular carcinoma. Theranostics. 2018;8:4649-63
Author contact
![]() Corresponding author: Prof. Joanna Wietrzyk. Email: wietrzykpan.wroc.pl
Corresponding author: Prof. Joanna Wietrzyk. Email: wietrzykpan.wroc.pl