Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(13):3879-3902. doi:10.7150/thno.31716 This issue Cite
Research Paper
Forkhead box K2 promotes human colorectal cancer metastasis by upregulating ZEB1 and EGFR
Feng Du1,#, Chenyang Qiao1,#, Xiaowei Li1,2,#, Zhangqian Chen1,3,#, Hao liu1, Shengda Wu4, Sijun Hu1, Zhaoyan Qiu5, Meirui Qian1, Dean Tian6, Kaichun Wu1, Daiming Fan1, ![]() , Yongzhan Nie1,
, Yongzhan Nie1, ![]() , Limin Xia1,6,
, Limin Xia1,6, ![]()
1. State key Laboratory of Cancer Biology, National Clinical Research Center for Digestive Diseases and Xijing Hospital of Digestive Diseases, Fourth Military Medical University, Xi'an 710032, Shaanxi, China.
2. Department of Gastroenterology, Navy General Hospital, Beijing, 100048, China.
3. Department of Infectious Diseases, Xijing Hospital, Fourth Military Medical University, Xi'an 710032, Shaanxi, China.
4. The Department of Orthopedics, Tangdu Hospital, Fourth Military Medical University, 1 Xinsi Road, Xi'an, Shaanxi, 710038, China.
5. Department of General Surgery, Chinese People's Liberation Army General Hospital, Fuxing Road No. 28, Haidian District, Beijing, 100853, China.
6. Department of Gastroenterology, Tongji Hospital of Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430030, Hubei, China
#These authors contributed equally to this work.
Received 2018-11-22; Accepted 2019-3-23; Published 2019-5-31
Abstract

Background: Metastasis is the major reason for high recurrence rates and poor survival among patients with colorectal cancer (CRC). However, the underlying molecular mechanism of CRC metastasis is unclear. This study aimed to investigate the role of forkhead box K2 (FOXK2), one of the most markedly increased FOX genes in CRC, and the mechanism by which it is deregulated in CRC metastasis.
Methods: FOXK2 levels were analyzed in two independent human CRC cohorts (cohort I, n = 363; cohort II, n = 390). In vitro Transwell assays and in vivo lung and liver metastasis models were used to examine CRC cell migration, invasion and metastasis. Chromatin immunoprecipitation and luciferase reporter assays were used to measure the binding of transcription factors to the promoters of FOXK2, zinc finger E-box binding homeobox 1 (ZEB1) and epidermal growth factor receptor (EGFR). Cetuximab was utilized to treat FOXK2-mediated metastatic CRC.
Results: FOXK2 was significantly upregulated in human CRC tissues, was correlated with more aggressive features and indicated a poor prognosis. FOXK2 overexpression promoted CRC migration, invasion and metastasis, while FOXK2 downregulation had the opposite effects. ZEB1 and EGFR were determined to be direct transcriptional targets of FOXK2 and were essential for FOXK2-mediated CRC metastasis. Moreover, activation of EGFR signaling by EGF enhanced FOXK2 expression via the extracellular regulated protein kinase (ERK) and nuclear factor (NF)-κB pathways. The EGFR monoclonal antibody cetuximab significantly inhibited FOXK2-promoted CRC metastasis. In clinical CRC tissues, FOXK2 expression was positively correlated with the expression of p65, ZEB1 and EGFR. CRC patients who coexpressed p65/FOXK2, FOXK2/ZEB1 and FOXK2/EGFR had poorer prognosis.
Conclusions: FOXK2 serves as a prognostic biomarker in CRC. Cetuximab can block the EGF-NF-κB-FOXK2-EGFR feedback loop and suppress CRC metastasis.
Keywords: Forkhead box K2, colorectal cancer, metastasis, epidermal growth factor receptor, cetuximab
Introduction
Colorectal cancer (CRC) represents a major contributor to worldwide cancer-related deaths [1]. Metastasis is the main reason for the high recurrence rates and poor survival of CRC patients after curative resection [2]. Despite increased public awareness of the dangers of CRC and improved early detection, which have improved the survival rate of CRC at the early stage, knowledge of the underlying molecular mechanisms and therapeutics for advanced metastatic CRC are lacking [2, 3]. Therefore, elucidating the molecular mechanism of CRC metastasis is important.
The forkhead box (FOX) family of transcription factors (TFs) has expanded to more than 40 members in mammals and has acquired specialized functions in many key biological processes [4]. Deregulation of FOX genes (FOXs) has a profound effect on human disease, including cancer [4, 5]. FOX family members, such as FOXA, FOXC, FOXM, FOXO and FOXP genes, have been strongly implicated in the initiation, maintenance, progression and drug resistance in multiple types of cancers 5. However, in human CRC, the expression profile of the FOXs remains unknown. Therefore, we screened for the FOX gene family expression signature in two independent sets of CRC tissues and found that FOXK2 was the most markedly increased FOX gene in CRC tissue compared with normal tissue (Figure S1-S2).
FOXK2 is a member of the forkhead TF family that binds to DNA containing the RYMAAYA (R = A or G; Y = C or T; M = A or C) core motif [4, 5]. FOXK2 in human cancers has been found to act as either an oncogene or a tumor suppressor [6-10]. A previous study reported that FOXK2 activates Wnt/β-catenin signaling by enhancing the nuclear localization of Dishevelled proteins to promote carcinogenesis and disease progression [11]. Qian et al. reported that SOX9-mediated transcriptional activation of FOXK2 is critical for CRC cell proliferation and correlates with poor survival [12]. On the other hand, FOXK2 interacts with the transcription corepressor complex nuclear receptor corepressor 1 (NCoR) to repress hypoxia-inducible factor 1 (HIF1)-β, thereby suppressing hypoxia-induced progression in breast cancer [13]. These conflicting findings indicate that the ambiguous role of FOXK2 in regulating oncogenes and tumor suppressors in different tumor types requires further investigation.
In this study, we found that FOXK2 was significantly upregulated in CRC and indicated a poor prognosis. FOXK2 promoted CRC metastasis by transactivating epidermal growth factor receptor (EGFR) and zinc finger E-box binding homeobox 1 (ZEB1). FOXK2 was induced by epidermal growth factor (EGF)/extracellular regulated protein kinase (ERK)/nuclear factor (NF)-κB signaling. The EGFR monoclonal antibody cetuximab inhibited FOXK2-promoting CRC metastasis, providing a promising biomarker and therapeutic target for the treatment of CRC metastasis.
Materials and methods
Cell culture
All CRC cells used in this study were purchased from American Tissue Type Culture Collection (Rockville, MD). Each cell line was tested, authenticated and confirmed to be mycoplasma contamination-free by the manufacturer. Cells were cultured in Dulbecco's modified Eagle's medium (DMEM, Gibco, ThermoFisher Scientific, Cambridge, MA, USA) supplemented with 10% bovine growth serum (FBS, Gibco), nonessential amino acids, glutamine, 100 μg/mL streptomycin and 100 U/mL penicillin (Gibco) under 5% CO2 at 37 °C.
Human tissue collection
Samples of CRC tissues and adjacent normal tissues were collected from patients who had undergone CRC surgery at Xijing Hospital of Digestive Diseases and were frozen in liquid nitrogen. All samples were clinically and pathologically shown to be correctly labeled. This study was approved by Xijing Hospital's Protection of Human Subjects Committee. Informed consent was obtained from each patient.
In vivo metastatic model and bioluminescence imaging
Six-week-old BALB/C nude mice were cared for and maintained based on our institution's protocols for ethical animal care. The Committee on the Use of Live Animals in Teaching and Research (CULATR) of the Fourth Military Medical University approved all animal experiments. In the tail vein injection-based in vivo metastasis assays, 10 mice in each group received tail vein injections of 1×106 cells in 100 μL of phosphate-buffered saline (PBS). In the intrasplenic injection-based in vivo metastasis assays, the mice were first anesthetized by intraperitoneal injection (0.01 mL/mg) of a mixture of Zoletil (30 mg/kg) and Rompun (10 mg/kg). Spleens were exteriorized via a small left abdominal flank incision. A single intrasplenic injection of 2×106 luciferase-labeled cells in 50 μL of Hank's balanced salt solution (HBSS) (Gibco) was administered with a 30-gauge needle. Gentle pressure was applied to the injection site with a cotton swab for one minute to staunch bleeding and to prevent leakage of tumor cells. Spleens were carefully reinserted into the abdominal cavity, and the wound was sutured using 6-0 black silk (10 mice per group). Every week, the mice received intraperitoneal injections of 150 mg/kg of D-luciferin, and images were acquired 10 minutes after injection with an IVIS 100 Imaging System (Xenogen, Hopkinton, MA, USA). Each image was acquired within 2 minutes. The survival durations of the mice were monitored, and at 9 weeks after the initial injections, all mice were sacrificed for further histological examination for lung and liver metastases.
Patients and follow-up
Written informed consent was obtained from each patient, and ethical approval was obtained from the Ethics Committee of the Fourth Military Medical University. Cohort I included freshly sampled CRC tissues with healthy adjacent tissues collected between January 2005 and December 2007 from 363 adult patients who underwent surgery at Xijing Hospital of the Fourth Military Medical University (Xi'an, China). Cohort II included CRC tissue samples that were surgically resected from 390 adult CRC patients between January 2005 and December 2007 at the Tongji Hospital of Tongji Medical College (Wuhan, China). All patients were staged pathologically based on the American Joint Committee on Cancer (AJCC)/International Union against Cancer criteria. All patients were preoperative radiotherapy- and chemotherapy-naïve; however, those with stage II-IV disease received postoperative adjuvant chemotherapy. No patients were treated with postoperative radiotherapy. Primary tumor samples along with dissected regional lymph nodes were subjected to histomorphological analysis via hematoxylin-eosin (H&E) staining performed by the Department of Pathology of Xijing and Tongji Hospital.
The information collected during the follow-up period included the incidence of disease recurrence and the presence of distant metastasis as confirmed by imaging and procedural data (position emission tomography, ultrasonography, magnetic resonance imaging, computed tomography and endoscopy) or pathological data (biopsies and cytologic analysis). Overall survival time was defined as the period between surgical resection and death. The duration of disease-free survival was defined as the period between surgical resection and the emergence of either distant CRC metastasis or CRC recurrence, the occurrence of another noncolorectal cancer (with the exception of carcinoma in situ of the cervix and skin basal cell carcinoma) or death from any cause without documentation of a cancer-related event. Patients were followed up for a minimum of 8 years, with follow-up data collected via questionnaire letters and telephone inquiry; patient databases were updated every 3 months. Patient deaths were determined by a corroborative history from the family and verified by reviewing public records.
Immunohistochemical (IHC) staining
IHC staining was performed as described in our previous study [14]. Briefly, 4-μm thick slides were first incubated on a 60 °C heating panel for one hour before the paraffin was removed with xylene, and the sections were rehydrated using the gradient ethanol immersion technique. The sections were exposed to 3% (vol/vol) hydrogen peroxide for 12 minutes in methanol to quench endogenous peroxidase activity. The sections were then washed with PBS thrice for three minutes each time. Subsequently, the slides were placed into a microwave for half an hour while submerged in a 0.01 mol/L citrate buffer solution (pH 6.0). After the final PBS (pH 7.4, 0.01 mol/L) wash, the slides were incubated overnight with their primary antibodies diluted in PBS containing 1% (wt/vol) bovine serum albumin in a damp chamber at 4 °C. To produce negative controls, the same procedure was repeated on a separate set of slides, but preimmune mouse serum was used in place of the primary antibody. The following day, the slides were first washed thrice for 5 minutes each with PBS and exposed to a peroxidase-conjugated second antibody for half an hour (Dako, Carpinteria, CA, USA) at room temperature, followed by another three cycles of 5-minute PBS washes. Diaminobenzidine exposure for 2 minutes was used to visualize the reaction product, and images were captured with a DP70 digital camera-equipped light microscope (Olympus, Japan). Two independent observers blinded to the expected results performed the subsequent analysis. The percentage of positive cells was scored on a scale of 0 to 4: 0 (negative), 1 (1%-25%), 2 (26%-50%), 3 (51%-75%), or 4 (76%-100%). The intensity of the immunostaining was scored on a scale of 0 to 3: 0 (negative), 1 (weak), 2 (medium) or 3 (strong). The product of the above two scores was used as the total immuno-activity score, which included 0, 1, 2, 3, 4, 6, 8, 9, 12. The cut-off point for a 'high' score was a final score equal to or greater than 4 (4, 6, 8, 9, 12), while scores smaller than 4 (0, 1, 2, 3) were considered 'low'.
Plasmid construction
All plasmid generation was carried out based on previously published standard procedures [15]. Briefly, for example, human genomic DNA was used to produce the ZEB1 promoter construct (-1947/+66) ZEB1, which corresponds to the -1947 to +66 sequence (relative to the transcriptional start site) of the 5'-flanking region of the human ZEB1 gene. Forward and reverse primers containing the HindIII and KpnI sites at the 3' and 5' ends, respectively, were used to build the construct. The final PCR product was cloned into the KpnI and HindIII sites of the pGL3-Basic vector (Promega). Similarly, constructs containing a deletion of the 5'-flanking region of the ZEB1 promoter, [(-1655/+66) ZEB1, (-1262/+66) ZEB1, and (-687/+66) ZEB1], were built based on the (-1947/+66) ZEB1 construct as the template. The QuikChange II Site-Directed Mutagenesis Kit (Stratagene, CA, USA) was used to mutate the FOXK2 binding sites of the ZEB1 promoter. The sequence integrity of all constructs was verified by gene sequencing. All promoter constructs used in this experiment were generated similarly. All primers are listed in Table S5.
Construction of lentivirus and stable cell lines
Lentivirus production was performed according to a previously described protocol 10. The pLKO.1-puro Non-Target shRNA Control Plasmid DNA (SHC016V, Sigma-Aldrich, CA, USA) contains a shRNA insert that does not target any known gene from any species, rendering it useful as a negative control. The shRNA sequences are listed in Table S6. Lentivirus generation and infection of cells were carried out per the recommended Addgene pLKO.1 lentiviral vector protocol. Briefly, a combination of OPTI-MEM medium (Invitrogen, MA, USA) and transfection reagent (Lipofectamine®3000, Thermo Fisher Scientific, Cambridge, MA, USA) was used to generate pMD2.G lentiviral plasmid, and the psPAX2 packaging plasmids (Addgene plasmids #12259 and #12260) were transfected into HEK-293T cells. Lentiviral harvesting was performed on the fourth and fifth days. The viruses were filtered with a 0.45-μm filter and stored at -80 °C. Target cells were infected with the produced lentiviruses in cell culture medium with 5 μg/mL polybrene (Sigma H9268). Seventy-two hours after infection, 2.5 μg/mL puromycin (OriGene, MD, USA) was used to select cells for the next 14 d, and the selected cells were used for all subsequent experiments.
In vitro migration and invasion assays
The invasive and migratory capabilities of each cell line were assessed with an 8-µm pore, 24-well Transwell plate (Corning Inc., NY, USA). For invasion assays, chamber inserts were first coated with 60 μL of Matrigel (Corning, 200 mg/mL) and left to dry overnight under sterile conditions. The next day, the uppermost chamber was plated at a cell density of 1×105. For cell migration assays, the upper chamber, which was lined with a noncoated membrane, was plated with cells at a density of 5×104. Each assay was repeated thrice, and three different inserts were used to obtain a mean cell number in three fields per membrane. The degree of invasion and migration was described as a ratio of the number of treated cells to control cells.
Transient transfection
A total of 1×105 serum-starved cells were plated in each well of a 24-well plate and allowed to attach for 12-24 hours. Then, a mixture of Lipofectamine 3000 (Invitrogen, ThermoFisher Scientific, Cambridge, MA, USA) containing 0.02 μg of the pRL-TK plasmids, 0.18 μg of the promoter reporter plasmids and 0.6 μg of the expression vector plasmids was used to cotransfect cells for 5 hours based on the manufacturer's instructions. The cells were then washed and incubated with 1% FBS-supplemented fresh medium for 48 hours.
For siRNA transfections, all the siRNAs in this study were purchased from Sigma-Aldrich (CA, USA) and were used according to the manufacturer's protocol. Cells were transfected with pooled siRNAs (ERK1 siRNA [siERK1, NM_001040056] and ERK2 siRNA [siERK2, NM_002745]) that target the two ERK genes to downregulate ERK1/2 expression. Also, the mixed siRNAs against p38α (sip38α, NM_001315) and p38β (sip38β, NM_002751) were used for transfection to knockdown p38 MAPK. siRNAs against JNK1 (siJNK1, NM_002750), JNK2 (siJNK2, NM_002752) and JNK3 (siJNK3, NM_002753) were pooled to silence the expression of JNKs. The product numbers of siRNAs for the down-regulation of the different AKT isoforms is siAKT1 NM_005163, siAKT2, NM_001626 and siAKT3 NM_005465, respectively.
Real-time qPCR
The RNeasy Plus Mini Kit (50) kit (Qiagen, Hilden, Germany) was used to extract total RNA, which was then reverse transcribed with the Advantage RT-for-PCR Kit (Qiagen) in accordance with the manufacturer's protocols. The target sequence was amplified with real-time PCR with the SYBR Green PCR Kit (Qiagen). The cycling parameters used were 95 °C for 15 s, 55-60 °C for 15 s, and 72 °C for 15 s for 45 cycles. Melting curve analyses were performed, and Ct values were determined during the exponential amplification phase of real-time PCR. SDS 1.9.1 software (Applied Biosystems, Massachusetts, USA) was used to evaluate amplification plots. The 2-ΔΔCt method was used to determine relative fold changes in target gene expression in cell lines, which was normalized to expression levels in corresponding control cells (defined as 1.0). The equation used was 2-ΔΔCt (ΔCt = ΔCt target - ΔCt GAPDH; ΔΔCt = ΔCt expressing vector - ΔCt control vector). When calculating relative expression levels in surgically extracted CRC samples, relative fold changes in target gene expression were normalized to expression values in normal colon epithelial tissues (defined as 1.0) using the following equation: 2 -ΔΔCt (ΔΔCt = ΔCt tumor - ΔCt nontumor). All experiments were performed in duplicate. Table S5 lists all sequences of all primers used.
Luciferase reporter assay
The Dual Luciferase Assay (Promega, USA) was used to quantify luciferase activity following the manufacturer's instructions. Transfected cells were subjected to cell lysis in a culture dish with lysis buffer. Subsequent lysates were transferred to an Eppendorf microcentrifuge before being centrifuged for 1 minute at maximum speed. The efficiency of transfection was normalized to Renilla activity, and relative luciferase activity was quantified with a Modulus TM TD20/20 Luminometer (Turner Biosystems, USA).
Chromatin immunoprecipitation (ChIP) assays
ChIP assays were performed as previously descripted [14]. Cells were immersed in 1% formaldehyde for 10 minutes at 37 °C to stimulate cross-linking. Then, glycine was used to quench the formaldehyde after cross-linking to stop formaldehyde fixation. After washing with PBS, the cells were resuspended in lysis buffer (1 mM PMSF, 1% SDS, 10 mM EDTA and 50 mM Tris (pH 8.1) - total volume 300 μl). Sonication was then performed to produce fragmented DNA. A slurry of protein G-Sepharose and herring sperm DNA (Sigma-Aldrich) was used to clear the supernatant. The recovered supernatant was then subjected to a 2-hour incubation period with specific antibodies or an isotype control IgG in the presence of protein G-Sepharose beads and herring sperm DNA, followed by antibody denaturation with 1% SDS in lysis buffer. Precipitated DNA was extracted from the beads by immersing them in a 1.1 M NaHCO3 solution and 1% SDS solution at 65 °C for 6 hours. Immunoprecipitated DNA was retrieved from the beads by immersion in 1% SDS and a 1.1 M NaHCO3 solution at 65 °C for 6 hours. DNA purification was performed with a PCR Purification Kit (Qiagen). Table S5 lists the primers utilized in this experiment.
For ChIP assays using cells from tissues, cells were first separated from six pairs of fresh frozen CRC tissues and normal colorectal epithelial tissues collected after surgical resection. In detail, surgically extracted tumor tissues were first washed thrice in cold PBS and added to medium supplemented with antibiotic and antifungal agents. Enzymatic digestion was performed by adding DNase I (20 mg/mL; Sigma-Aldrich) and collagenase (1.5 mg/mL; Sigma-Aldrich). The resultant mixture was pipetted every 15 minutes to evenly distribute cells for one hour at 37 °C. At the end of the hour, the dissociated cells were filtered through 100-mm-pore filters and rinsed with fresh media. Red blood cell lysis was performed by exposing the membranes briefly to ammonium chloride, followed by another rinse. The dissociated cells were collected for further investigation. The endogenous levels of FOXK2 in the six CRC tissues and paired adjacent nontumor tissues were analyzed by Western blotting (Figure S7).
Western blotting analysis
Whole cell lysates were prepared using RIPA buffer supplemented with protease inhibitor cocktail (Sigma-Aldrich) and phosphatase inhibitor (Roche). The nuclear extract was isolated using NE-PER nuclear and cytoplasmic extraction reagents (Thermo Fisher Scientific). Protein concentration was determined using the BCA Protein Assay Kit (Thermo Fisher Scientific). Extracted proteins underwent SDS-PAGE fractionation and were then transferred onto nitrocellulose membranes. A mixture of Tween 20 (TBST) (0.05% Tween 20, 150 mM NaCl and 120 mM Tris-HCl (pH 7.4)) and 5% milk in Tris-buffered saline at room temperature was incubated for 1 hour to attenuate nonspecific binding reactions. Then, all blots underwent overnight incubation with their respective antibodies at 4 °C, with β-actin used on the same membrane as a control. Antibodies against FOXK2 (ab50946), ZEB1 (ab203829), EGFR (ab52894), Phospho-EGFR (Y1068) (ab40815), EGF (ab184265), FOXK1 (ab18196) and NF-κB p65 (ab32536) were procured from Abcam. E-cadherin (#3195), Vimentin (#5741), Slug (#9585), AKT (#4691), Phospho-Akt (Ser473) (#4060), p44/42 MAPK (Erk1/2) (#4695), Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) (#4370), JNK (#9252), Phospho-SAPK/JNK (Thr183/Tyr185) (#4668), p38 MAPK (#8690), Phospho-p38 MAPK (Thr180/Tyr182) (#4511) and Phospho-NF-κB p65 (Ser536) (#3033) were purchased from Cell Signaling Technology (CST, Beverly, MA, USA). The β-actin antibody (sc-47778) was purchased from Santa Cruz Biotechnology. The next day, the membranes were washed thrice with PBS before a final incubation with horseradish peroxidase (HRP)-conjugated secondary antibody. The Dura SuperSignal Substrate (Pierce, ThermoFisher Scientific, Cambridge, MA, USA) was used to visualize proteins.
Immunofluorescence
Following 10-minute fixation with 4% paraformaldehyde in PBS, PBS was used to wash the cells twice. The cells were incubated overnight at 4 °C with primary antibodies targeting vimentin (CST, #3932) and E-cadherin (CST, #3195). The next day, the cells were washed thoroughly before being exposed to fluorescein isothiocyanate (FITC)-conjugated goat antimouse or antirabbit IgG (CST, #2985) as appropriate. Lastly, the cells underwent a final round of washing and were mounted with Mounting Medium containing 4',6-diamidino-2-phenylindole (DAPI) (Vector Laboratories). A Leica DMRA fluorescence microscope (Rueil-Malmaison) was used to capture images.
Agents
The ERK inhibitor SCH772984 (#S7101), JNK inhibitor SP600125 (#S1460), p38 inhibitor SB202190 (#S1077) and PI3K inhibitor LY294002 (#S1105) were purchased from Selleck Chemicals (Houston, TX, USA). Recombinant human EGF protein was purchased from Bio-Techne (#236-EG-200, R&D Systems, MN, USA). All the agents were used according to the manufacturer's instructions.
Databases
Oncomine (http://www.oncomine.org) and The Cancer Genome Atlas (TCGA) (https://cancergenome.nih.gov) datasets were used to determine the mRNA expression of FOXK2 in human cancer specimens compared with that in normal tissues.
Quantification and statistical analysis
All statistical analyses were carried out with SPSS software (version 19.0). Fisher's exact test was used for categorical data. Student's t-test was used for intergroup comparisons of quantitative data. Kaplan-Meier survival analysis and the log-rank test were used to determine cumulative survival rates and recurrence rates. To discern factors that independently influenced recurrence and survival, we subjected predetermined variables found to be significant on univariate analysis to further analysis with the Cox proportional hazards model. Statistical significance was ascertained when p < 0.05.
Results
Expression of FOX family genes in human CRC tissues
We first screened the expression profile of FOXs in CRC tissues compared to that of normal colorectal epithelial tissues by RT-qPCR. Among these FOXs, we found that the expression levels of FOXA2, FOXC1, FOXD1, FOXD4, FOXK1, FOXK2, FOXM1, FOXN2, FOXP3, FOXP4, FOXQ1, and FOXS1 were significantly increased in CRC tissues; in contrast, the expression levels of FOXA1, FOXD2, FOXD3, FOXE1, FOXE3, FOXF1, FOXF2, FOXH1, FOXI2, FOXJ3, FOXN3, FOXO1, FOXO4, FOXP1, and FOXP2 were notably reduced in CRC tissues (Figure S1-S2). In addition, the expression levels of FOXA3, FOXC2, FOXG1, FOXI1, FOXJ1, FOXJ2, FOXL1, FOXL2, FOXN1, FOXN4, and FOXO3 showed no obvious changes in CRC tissues compared with those in normal tissues (Figure S1-S2). Several FOXs were undetectable in both CRC and normal colorectal epithelial tissues, including FOXB1, FOXB2, FOXI3, FOXR1, and FOXR2 (data not shown). Among these increased FOXs, FOXK2 exhibited the largest fold change, indicating a potential pivotal role of FOXK2 in CRC. Moreover, we investigated FOXK2 expression in different public CRC datasets. Both the TCGA and Oncomine datasets revealed that the mRNA expression of FOXK2 was significantly increased in CRC and multiple cancer specimens compared to the levels in normal tissues, including colon adenocarcinoma (COAD) (Figure S3), suggesting that FOXK2 may act as an important transcriptional factor driving CRC progression.
FOXK2 is significantly upregulated in human CRC tissues and indicates a poor prognosis
FOXK2 expression was analyzed in a cohort of 120 pairs of CRC and normal specimens using RT-qPCR. CRC tissues displayed marked upregulation of FOXK2 mRNA compared with the levels in adjacent nontumorous tissue and normal colorectal epithelial tissue (Figure 1A). Primary CRC tissues from patients with recurrence or metastasis showed higher FOXK2 mRNA expression than primary CRC tissues from patients who did not exhibit recurrence or metastasis (Figure 1A). Metastatic CRC tissues had higher FOXK2 mRNA levels than primary CRC tissues and normal colorectal epithelial tissues (Figure 1A). Similarly, we observed significantly elevated FOXK2 protein expression in CRC tissues compared with that in paired adjacent nontumorous tissues using Western blotting and immunohistochemical staining (Figure 1B).
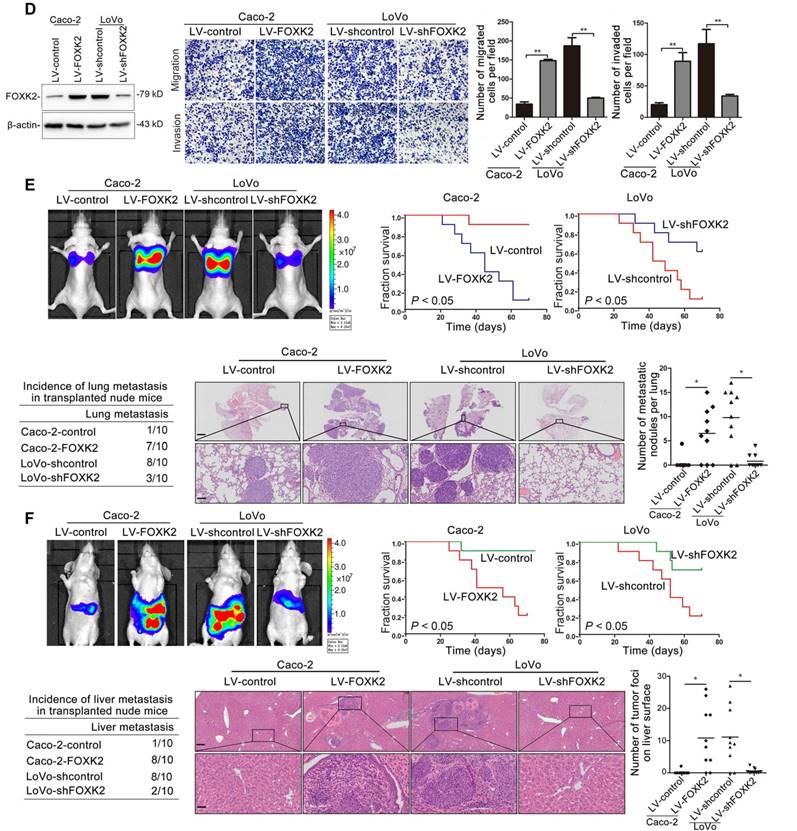
Elevated FOXK2 expression promotes CRC migration, invasion and metastasis and indicates a poor prognosis in human CRC. (A) RT-qPCR analysis of FOXK2 mRNA expression in 120 pairs of CRC tissues and adjacent nontumor tissues and in 20 normal colorectal epithelial tissues. Relative FOXK2 mRNA expression in patients with (n = 59) or without (n = 61) CRC recurrence. Relative FOXK2 mRNA expression in patients with (n = 55) or without (n = 63) CRC metastasis. RT-qPCR analysis of FOXK2 mRNA levels in normal colorectal epithelial tissues, primary colon cancer tissues and paired metastatic CRC tissues (n = 30). ** P < 0.01; *** P < 0.001 compared with the control. The data are presented as the mean±s.d. (B) Analysis of FOXK2 protein levels in CRC and paired adjacent nontumor tissues (n = 10) by Western blotting and IHC staining. Scale bars: top, 100 μm; bottom, 20 μm. (C) A Kaplan-Meier analysis of the correlation of FOXK2 expression with recurrence and overall survival in cohort I and cohort II. + High; - Low. (D) Western blotting analysis of FOXK2 protein levels in the indicated CRC cells. Transwell assay analysis of cell migration and invasion. n = 3 independent experiments performed in triplicate. ** P < 0.01 compared with the control. The data are presented as the mean±s.d. (E) In vivo lung metastatic assays. Cells were injected into the tail veins of mice (n = 10 mice per group). Bioluminescence imaging 9 weeks after implantation, the incidence of lung metastasis, overall survival times, the number of metastatic lung nodules, and H&E staining of lung tissues from the different groups are shown. Scale bars: top, 500 μm; bottom, 40 μm. * P < 0.05 compared with the control. The data are presented as the mean±s.d. (F) In vivo liver metastasis assays. Cells were injected into the spleens of mice (n = 10 mice per group). Bioluminescence imaging 9 weeks after implantation, the incidence of liver metastasis, overall survival times, the number of metastatic liver nodules, and H&E staining of liver tissues from the different groups are shown. Scale bars: top, 200 μm; bottom, 40 μm. * P < 0.05 compared with the control. The data are presented as the mean±s.d.
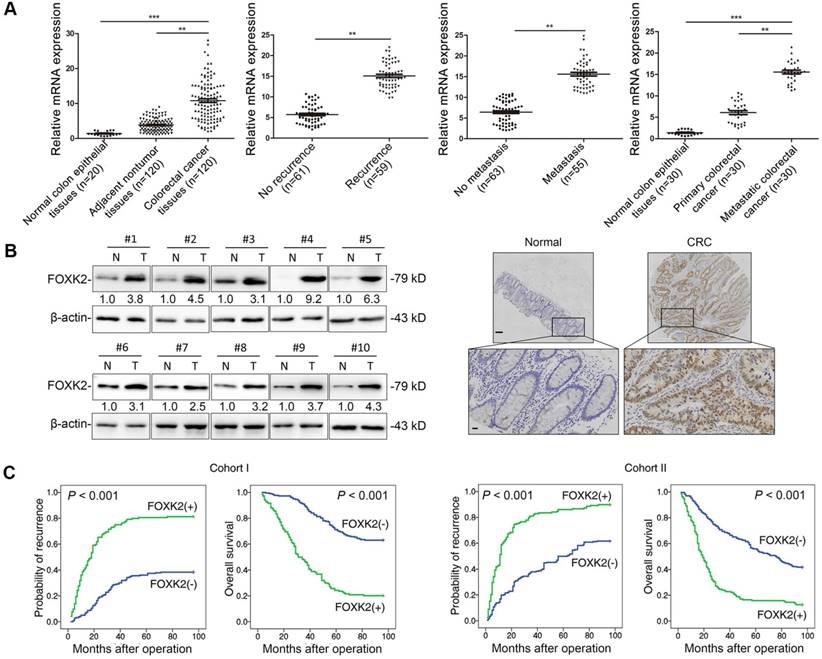
Moreover, we then analyzed correlation between FOXK2 expression and clinicopathological characteristics of CRC in two independent cohorts of human CRC tissues (cohort I, n = 363; cohort II, n = 390) (Table 1). Significant correlations were found between FOXK2 overexpression and poor tumor differentiation, greater tumor invasion, lymph node metastasis, distant metastasis and AJCC stage (Table 1). Overall survival times were shorter and recurrence rates were increased in patients with high FOXK2 expression compared to those in patients with low FOXK2 expression in both cohort I and cohort II (Figure 1C).
Correlation between FOXK2 expression and clinicopathological characteristics of CRC in two independent cohorts of human CRC tissues
| Clinicopathological variables | Cohort I (n=363) | P value | Cohort II (n=390) | P value | ||||
|---|---|---|---|---|---|---|---|---|
| Tumor FOXK2 expression | Tumor FOXK2 expression | |||||||
| Low (n=219) | High (n=144) | Low (n=232) | High (n=158) | |||||
| Age | 66.57(11.12) | 65.88(11.70) | 0.473 | 67.50(12.02) | 67.47(10.87) | 0.356 | ||
| Sex | female | 91 | 70 | 0.196 | 101 | 76 | 0.408 | |
| male | 128 | 74 | 131 | 82 | ||||
| Tumor location | right colon | 98 | 75 | 0.006 | 87 | 78 | 0.025 | |
| left colon | 71 | 57 | 107 | 61 | ||||
| rectum | 50 | 12 | 38 | 19 | ||||
| Tumor size | <5 cm | 93 | 61 | 1 | 93 | 56 | 0.396 | |
| ≥5 cm | 126 | 83 | 139 | 102 | ||||
| Tumor differentiation | well or moderate | 184 | 66 | <0.001 | 159 | 65 | <0.001 | |
| poor | 35 | 78 | 73 | 93 | ||||
| Tumor invasion | T1 | 6 | 1 | 0.014 | 14 | 2 | 0.021 | |
| T2 | 25 | 5 | 13 | 11 | ||||
| T3 | 141 | 101 | 166 | 107 | ||||
| T4 | 47 | 37 | 39 | 38 | ||||
| Lymph node metastasis | absent | 162 | 37 | <0.001 | 185 | 41 | <0.001 | |
| present | 57 | 107 | 47 | 117 | ||||
| Distant metastasis | absent | 208 | 90 | <0.001 | 222 | 96 | <0.001 | |
| present | 11 | 54 | 10 | 62 | ||||
| AJCC stage | Stage I | 31 | 4 | <0.001 | 18 | 2 | <0.001 | |
| Stage II | 131 | 28 | 167 | 32 | ||||
| Stage III | 46 | 58 | 38 | 63 | ||||
| Stage IV | 11 | 54 | 9 | 61 | ||||
Multivariate analysis identified that the FOXK2 overexpression, lymph node metastasis, distant metastasis and advanced AJCC stage were independent and significant risk factors for reduced survival and recurrence (Tables 2-3). These clinical data suggest that FOXK2 is a promising prognostic biomarker in CRC.
Univerate and multivariate analysis of factors associated with survival and recurrence in Cohort I human CRCs
| Variables | Recurrence | Survival | ||||
|---|---|---|---|---|---|---|
| HR (95% CI) | P value | HR (95% CI) | P value | |||
| Univariate analysis | ||||||
| Age | 0.999 (0.986-1.011) | 0.819 | 0.998 (0.985-1.011) | 0.772 | ||
| Sex (female versus male) | 1.272 (0.964-1.677) | 0.089 | 1.212 (0.916-1.604) | 0.179 | ||
| Tumor size (≤5 versus >5 cm) | 0.833 (0.628-1.105) | 0.205 | 0.827 (0.621-1.101) | 0.193 | ||
| Tumor differentiation(well/moderate versus poor) | 0.184 (0.138-0.245) | <0.001 | 0.186 (0.139-0.249) | <0.001 | ||
| Tumor invasion(T1-T3 versus T4) | 0.350 (0.261-0.469) | <0.001 | 0.357 (0.265-0.480) | <0.001 | ||
| Lymph node metastasis (absent versus present) | 0.138 (0.100-0.190) | <0.001 | 0.136 (0.098-0.189) | <0.001 | ||
| Distant metastasis (absent versus present) | 0.114 (0.081-0.159) | <0.001 | 0.113 (0.081-0.157) | <0.001 | ||
| AJCC stage(I-II versus III-Ⅳ) | 0.134 (0.097-0.187) | <0.001 | 0.133 (0.095-0.186) | <0.001 | ||
| FOXK2 expression (low versus high) | 0.256 (0.192-0.340) | <0.001 | 0.250 (0.187-0.333) | <0.001 | ||
| Multivariate analysis | ||||||
| Tumor differentiation(well/moderate versus poor) | 0.760 (0.515-1.122) | 0.168 | 0.809 (0.544-1.202) | 0.294 | ||
| Tumor invasion(T1-T3 versus T4) | 0.638 (0.214-1.910) | 0.421 | 0.669 (0.218-2.058) | 0.484 | ||
| Lymph node metastasis (absent versus present) | 0.354 (0.123-1.013) | 0.053 | 0.336 (0.114-0.984) | 0.047 | ||
| Distant metastasis (absent versus present) | 0.527 (0.343-0.812) | 0.004 | 0.485 (0.314-0.749) | 0.001 | ||
| AJCC stage(I-II versus III-Ⅳ) | 0.475 (0.337-0.669) | <0.001 | 0.489 (0.344-0.694) | <0.001 | ||
| FOXK2 expression (low versus high) | 0.421 (0.304-0.582) | <0.001* | 0.415 (0.300-0.576) | <0.001* | ||
Univerate and multivariate analysis of factors associated with survival and recurrence in Cohort II human CRCs
| Variables | Recurrence | Survival | ||||
|---|---|---|---|---|---|---|
| HR (95% CI) | P value | HR (95% CI) | P value | |||
| Univariate analysis | ||||||
| Age | 0.998 (0.988-1.009) | 0.774 | 1.000 (0.989-1.011) | 0.951 | ||
| Sex (female versus male) | 1.070 (0.847-1.351) | 0.569 | 1.116 (0.880-1.415 | 0.367 | ||
| Tumor size (≤5 versus >5 cm) | 0.901 (0.709-1.146) | 0.396 | 0.877( 0.686-1.122) | 0.297 | ||
| Tumor differentiation(well/moderate versus poor) | 0.469 (0.370-0.593) | <0.001 | 0.449 (0.353-0.571) | <0.001 | ||
| Tumor invasion(T1-T3 versus T4) | 0.605 (0.461-0.796) | <0.001 | 0.607 (0.459-0.803) | <0.001 | ||
| Lymph node metastasis (absent versus present) | 0.193 (0.150-0.248) | <0.001 | 0.172 (0.132-0.222) | <0.001 | ||
| Distant metastasis (absent versus present) | 0.130 (0.095-0.178) | <0.001 | 0.111 (0.081-0.154) | <0.001 | ||
| AJCC stage(I-II versus III-Ⅳ) | 0.179 (0.138-0.230) | <0.001 | 0.159 (0.122-0.207) | <0.001 | ||
| FOXK2 expression (low versus high) | 0.349 (0.275-0.443) | <0.001 | 0.343 (0.270-0.438) | <0.001 | ||
| Multivariate analysis | ||||||
| Tumor differentiation(well/moderate versus poor) | 0.798 (0.614-1.039) | 0.094 | 0.802 (0.612-1.050) | 0.109 | ||
| Tumor invasion(T1-T3 versus T4) | 0.685 (0.516-0.910) | 0.009 | 0.765 (0.575-1.018) | 0.066 | ||
| Lymph node metastasis (absent versus present) | 1.266 (0.587-2.730) | 0.547 | 1.075 (0.499-2.320) | 0.853 | ||
| Distant metastasis (absent versus present) | 0.378 (0.266-0.538) | <0.001 | 0.331 (0.231-0.474) | <0.001 | ||
| AJCC stage(I-II versus III-Ⅳ) | 0.210 (0.093-0.473) | <0.001 | 0.216 (0.095-0.490) | <0.001 | ||
| FOXK2 expression (low versus high) | 0.740 (0.559-0.980) | 0.036* | 0.688 (0.515-0.919) | 0.011* | ||
FOXK2 promotes CRC invasion and metastasis
To clarify the potential role of FOXK2 in CRC invasion and metastasis, we first detected the expression levels of FOXK2 in panel of CRC cell lines and found that Caco-2 and DiFi cells had relatively low levels of FOXK2, while LoVo and DLD-1 cells had high levels of FOXK2 (Figure S4). For gain- and loss-of-function analyses, we overexpressed FOXK2 in Caco-2 cells and downregulated the expression of FOXK2 in LoVo cells (Figure 1D). Transwell assay showed that upregulating FOXK2 expression increased the invasion and migration capabilities of Caco-2 cells, whereas downregulating FOXK2 expression decreased the invasion and migration capabilities of LoVo cells (Figure 1D). To preclude the possibility of off-target effects, we also used a second lentivirus shRNA to knock down FOXK2 expression and then rescued FOXK2 expression with a Flag-tagged shRNA-resistant wild-type FOXK2. Immunoblot analyses confirmed the depletion of endogenous FOXK2 and the expression of Flag-tagged exogenous FOXK2. FOXK2 knockdown with two shRNAs significantly reduced the migration and invasion capacities of the highly metastatic LoVo cells, which could be rescued by the expression of shRNA-resistant FOXK2, precluding the possibility of off-target effects of FOXK2 shRNAs (Figure S5A).
In vivo metastasis assays showed that upregulating FOXK2 increased the number of metastatic lung nodules and the incidence of lung metastasis, resulting in an overall decrease in Caco-2-FOXK2 group survival times. Conversely, downregulation of FOXK2 decreased the number of metastatic lung nodules and the incidence of lung metastasis, resulting in an overall increase in LoVo-shFOXK2 group survival times (Figure 1E). Similar results were observed in the intrasplenic injection metastasis assays, where FOXK2 upregulation increased the number of metastatic liver nodules and the incidence of liver metastasis, resulting in an overall decrease in Caco-2-FOXK2 group survival times. However, downregulation of FOXK2 decreased the number of metastatic liver nodules and the incidence of liver metastasis, resulting in an overall increase in LoVo-shFOXK2 group survival times (Figure 1F). These findings suggest that FOXK2 promotes CRC migration, invasion and metastasis in vitro and in vivo.
FOXK2 induces CRC cell epithelial-mesenchymal transition (EMT) by transactivating ZEB1
Of note, immunofluorescence (IF) showed that FOXK2 upregulation decreased epithelial marker E-cadherin expression and increased mesenchymal marker vimentin expression in Caco-2 cells, while downregulation of FOXK2 increased E-cadherin expression and decreased vimentin expression in LoVo cells (Figure 2A). Because of the pivotal role of the EMT in metastasis [16, 17], we then compared the mRNA expression profiles of Caco-2-FOXK2 and Caco-2-control cells using an EMT RT2 profiler polymerase chain reaction array. FOXK2 overexpression resulted in upregulated expression of several metastasis-related genes, including EGFR, ZEB1, VIM, FN1, MMP2, MMP9, ERBB3, MSN, VCAN, ITGAV, KRT7, and AKT1 (Table S1). EGFR and ZEB1, which represented the top two genes with increased expression in response to FOXK2 overexpression, were of our particular interest for their important roles in cancer invasion and metastasis.
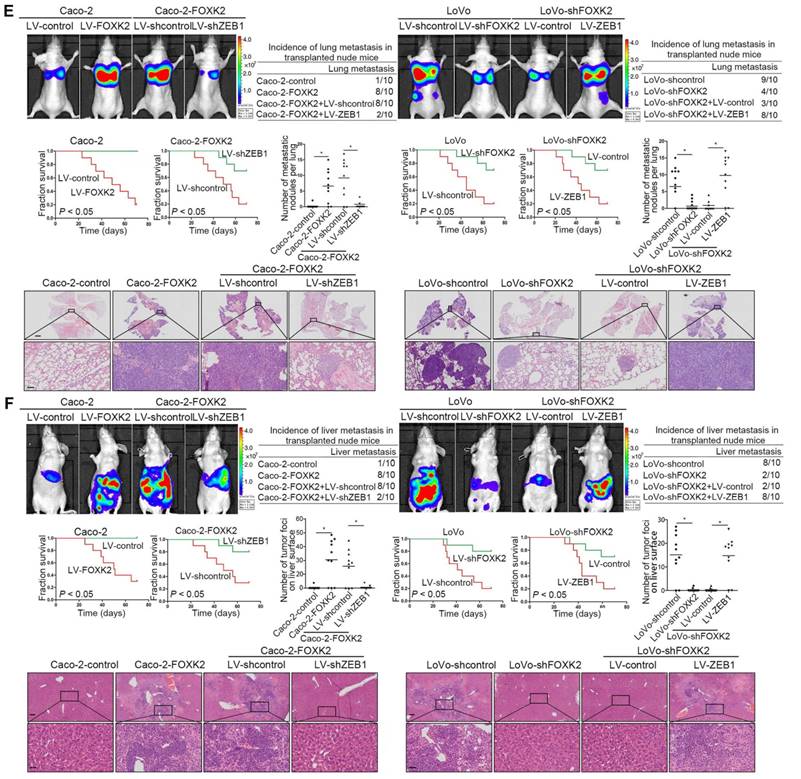
FOXK2 overexpression induces the EMT by transactivating ZEB1. (A) Immunofluorescence staining of E-cadherin and vimentin. n = 3 independent experiments performed in triplicate. Scale bars: 20 μm. (B) RT-qPCR and Western blotting analysis of FOXK2 and ZEB1 expression in CRC cells. n = 3 independent experiments performed in triplicate. * P < 0.05 compared with the control. The data are presented as the mean±s.d. (C) Luciferase reporter assays of the indicated cells cotransfected with pCMV-FOXK2 and the ZEB1 promoter luciferase construct. ChIP-qPCR assay demonstrated that FOXK2 directly binds to the ZEB1 promoter in human CRC tissues and cells. * P < 0.05; ** P < 0.01 compared with the control. The data are presented as the mean±s.d. (D) Western blotting and RT-qPCR evaluation of the expression of FOXK2, ZEB1, vimentin and E-cadherin in the indicated cells. n = 3 independent experiments performed in triplicate. (E) In vivo lung metastatic assays. Cells were injected into the tail veins of mice (n = 10 mice per group). Bioluminescence imaging 9 weeks after implantation, the incidence of lung metastasis, overall survival times, the number of metastatic lung nodules, and H&E staining of lung tissues from the different groups are shown. Scale bars: top, 500 μm; bottom, 40 μm. * P < 0.05 compared with the control. The data are presented as the mean±s.d. (F) In vivo liver metastasis assays. Cells were injected into the spleens of mice (n = 10 mice per group). Bioluminescence imaging 9 weeks after implantation, the incidence of liver metastasis, overall survival times, the number of metastatic liver nodules, and H&E staining of liver tissues from the different groups are shown. Scale bars: top, 200 μm; bottom, 40 μm. * P < 0.05 compared with the control. The data are presented as the mean±s.d.
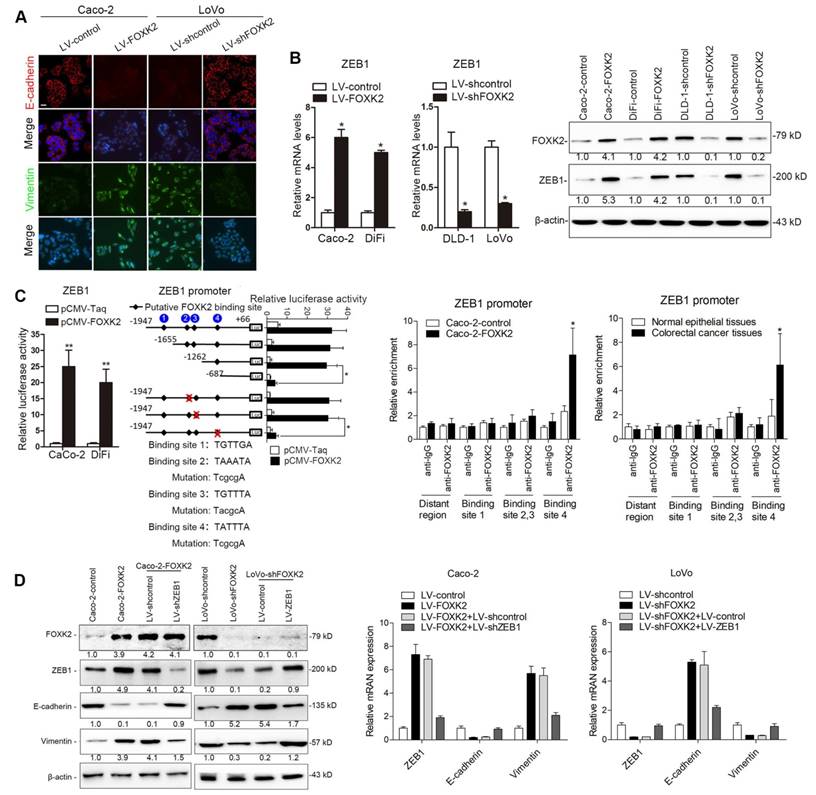
ZEB1 is a key EMT driver that directly binds to the CDH1 promoter and inhibits E-cadherin transcription [16, 18]. We questioned whether ZEB1 is direct target gene of FOXK2 and is involved in the FOXK2-induced EMT. We first established four stable cell lines, Caco-2-FOXK2, DiFi-FOXK2, LoVo-shFOXK2 and DLD-1-shFOXK2, and found that FOXK2 overexpression increased ZEB1 expression, while FOXK2 knockdown had the opposite effect (Figure 2B). Luciferase reporter assays showed that FOXK2 enhanced ZEB1 promoter activity (Figure 2C). Upon sequence analysis, the ZEB1 promoter region was found to have 4 putative FOXK2 binding sites. Serial deletion and site-directed mutagenesis indicated that the fourth FOXK2 binding site on the ZEB1 promoter region was vital for the transactivation of ZEB1 expression by FOXK2 (Figure 2C). ChIP-qPCR assay further confirmed that FOXK2 bound directly to the fourth binding site on the ZEB1 promoter in CRC cells and tissues (Figure 2C and Figure S7A). Moreover, ZEB1 downregulation in Caco-2-FOXK2 cells significantly increased E-cadherin expression but decreased vimentin expression. In contrast, ZEB1 upregulation in LoVo-shFOXK2 cells markedly decreased E-cadherin expression but increased vimentin expression (Figure 2D). These studies suggested that FOXK2 promotes the EMT by transactivating ZEB1 in human CRC cells.
ZEB1 is essential for FOXK2-mediated CRC migration, invasion and metastasis
Considering the important role of ZEB1 in invasion and metastasis, we questioned whether ZEB1 is involved in FOXK2-mediated CRC metastasis. ZEB1 upregulation rescued the decreased migration and invasion abilities induced by FOXK2 knockdown, while ZEB1 downregulation significantly reduced FOXK2-enhanced cell migration and invasion (Figure S6A). In addition, we used a second lentiviral shRNA to knock down endogenous ZEB1 expression and rule out the possibility of an off-target effect (Figure S5B).
In vivo metastasis assays demonstrated that downregulation of ZEB1 decreased the number of metastatic lung nodules and the incidence of lung metastasis, resulting in an overall increase in Caco-2-FOXK2 group survival times (Figure 2F). By contrast, upregulation of ZEB1 increased the number of metastatic lung nodules and the incidence of lung metastasis, resulting in an overall decrease in LoVo-shFOXK2 group survival times (Figure 2E). The in vivo liver metastasis assay showed similar effects (Figure 2F). These findings indicate that ZEB1 is essential for FOXK2-mediated CRC migration, invasion and metastasis.
FOXK2 promotes CRC metastasis by upregulating EGFR expression
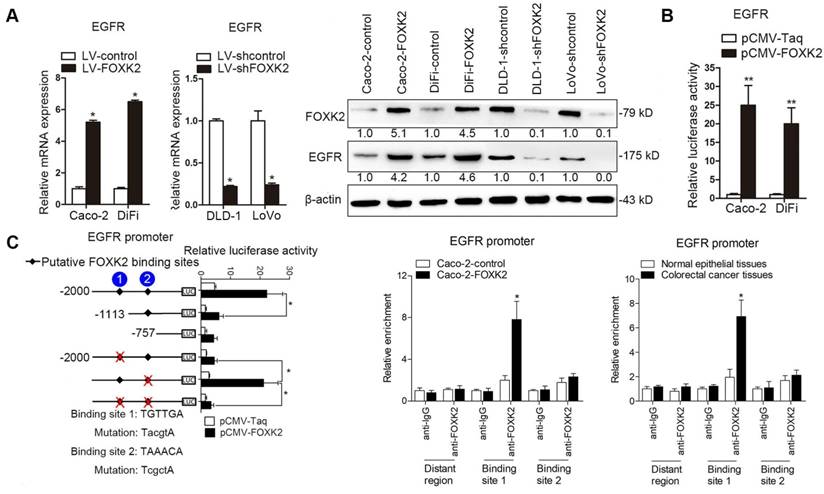
Because EGFR expression increased the most after FOXK2 overexpression (Table S1), we examined whether EGFR is a target gene of FOXK2. Western blotting showed that FOXK2 upregulation increased EGFR mRNA and protein levels, while FOXK2 downregulation decreased EGFR expression (Figure 3A). In line with this finding, EGFR expression in a panel of CRC cell lines correlated with FOXK2 levels (Figure S4). Luciferase reporter assay further confirmed that FOXK2 increased EGFR promoter activity (Figure 3B), indicating that FOXK2 may transactivate EGFR expression. Sequence analysis showed that two putative FOXK2 binding sites exist in the EGFR promoter region. Serial deletion and site-directed mutagenesis revealed that the first FOXK2 binding site on the EGFR promoter region is vital for transactivation of EGFR by FOXK2 (Figure 3C). ChIP assays revealed the direct binding of FOXK2 to the EGFR promoter in CRC cells and tissues (Figure 3C and Figure S7B). These findings suggested that FOXK2 directly binds to the EGFR promoter and transactivates EGFR expression.
FOXK2 promotes CRC metastasis by upregulating EGFR expression. (A) RT-qPCR and Western blotting analysis of FOXK2 and EGFR expression in CRC cells. n = 3 independent experiments performed in triplicate. * P < 0.05 compared with the control. The data are presented as the mean±s.d. (B) Luciferase reporter assays of the indicated cells cotransfected with pCMV-FOXK2 and the EGFR promoter luciferase construct. n = 3 independent experiments performed in triplicate. * P < 0.05; ** P < 0.01 compared with the control. The data are presented as the mean±s.d. (C) Relative luciferase activity was determined after serially truncated and mutated EGFR promoter constructs were cotransfected with pCMV-FOXK2. ChIP-qPCR assays demonstrated that FOXK2 directly binds to the EGFR promoter in human CRC tissues and CRC cells. * P < 0.05 compared with the control. The data are presented as the mean±s.d. (D) In vivo lung metastatic assays. Cells were injected into the tail veins of mice (n = 10 mice per group). Bioluminescence imaging 9 weeks after implantation, the incidence of lung metastasis, overall survival times, the number of metastatic lung nodules, and H&E staining of lung tissues from the different groups are shown. Scale bars: top, 500 μm; bottom, 40 μm. * P < 0.05 compared with the control. The data are presented as the mean±s.d. (E) In vivo liver metastasis assays. Cells were injected into the spleens of mice (n = 10 mice per group). Bioluminescence imaging 9 weeks after implantation, the incidence of liver metastasis, overall survival times, the number of metastatic liver nodules, and H&E staining of liver tissues from the different groups are shown. Scale bars: top, 200 μm; bottom, 40 μm. * P < 0.05 compared with the control. The data are presented as the mean±s.d.
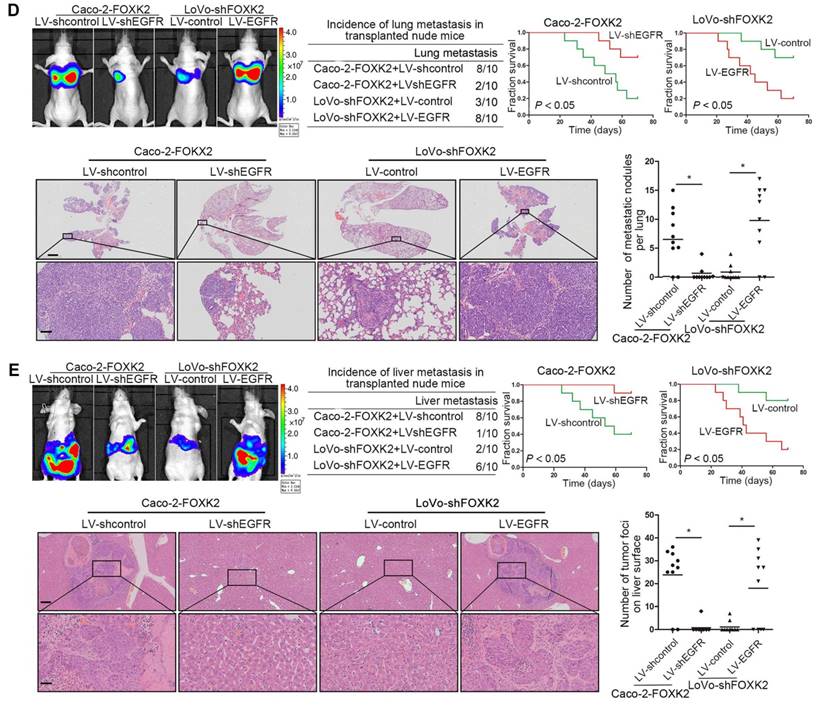
We further questioned whether EGFR is critical for FOXK2-mediated CRC metastasis. Transwell assays revealed that EGFR upregulation rescued the decreased migration and invasion abilities induced by FOXK2 knockdown, while EGFR downregulation significantly suppressed FOXK2-enhanced cell migration and invasion (Figure S6B-C). In addition, we used a second lentiviral shRNA to knock down endogenous EGFR expression and ruled out the possibility of an off-target effect of EGFR shRNA (Figure S5C). In vivo metastasis assays demonstrated that downregulation of EGFR decreased the number of metastatic lung nodules and the incidence of lung metastasis, resulting in improved survival in the Caco-2-FOXK2 group (Figure 3D). By contrast, upregulation of EGFR increased the number of metastatic lung nodules and the incidence of lung metastasis, leading to poorer survival in the LoVo-shFOXK2 group (Figure 3D). Similarly, in a liver metastasis assay, EGFR downregulation reduced liver metastasis in Caco-2-FOXK2 cells, while EGFR overexpression promoted liver metastasis in LoVo-shFOXK2 cells (Figure 3E). Our experiments indicate that EGFR is directly transactivated by FOXK2 and is essential for FOXK2-promoted CRC migration, invasion and metastasis.
Epidermal growth factor (EGF) induces FOXK2 expression via the extracellular-signal-regulated kinase (ERK)/nuclear factor κB (NF-κB) pathway
To determine whether EGFR activation induces FOXK2 expression in CRC cells, gradient concentrations of EGF were used to treat CRC cells. Interestingly, EGF treatment significantly increased FOXK2 expression at both the protein and mRNA levels in a dose-dependent manner (Figure 4A). Of note, FOXK2 promoter activity was significantly increased in response to EGF treatment, indicating that EGF induces FOXK2 expression by transactivating its promoter (Figure 4A).
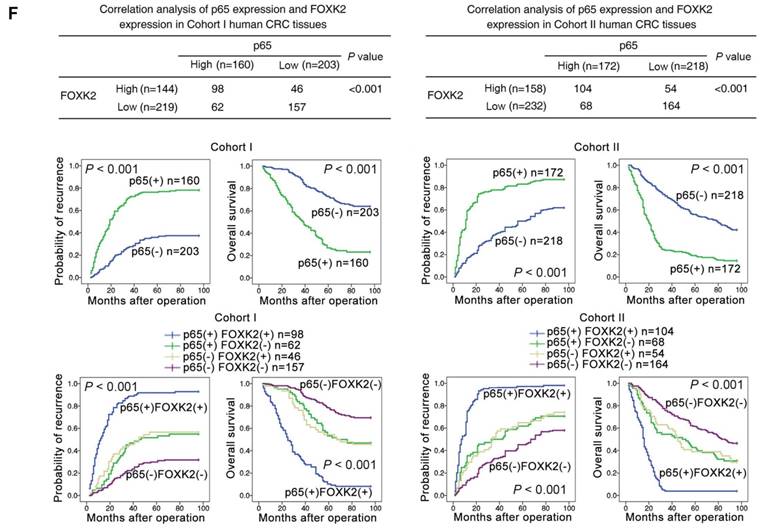
EGF induces FOXK2 expression through the ERK/NF-κB pathway. (A) CRC cells were exposed to different EGF concentrations for 24 hours, followed by Western blotting and RT-qPCR to detect FOXK2 expression. Cells were incubated with or without EGF for 24 hours after transfection with the FOXK2 promoter luciferase reporter to determine luciferase activity. n = 3 independent experiments performed in triplicate. * P < 0.05; ** P < 0.01 compared with the control. The data are presented as the mean±s.d. (B) Relative luciferase activity was determined after serially truncated and mutated FOXK2 promoter in Caco-2 cells incubated with EGF. * P < 0.05 compared with the control. The data are presented as the mean±s.d. (C) Caco-2 cells were transfected with a small-interfering RNA (siRNA) for p65 or control siRNA, vehicle or the NF-κB inhibitor BAY 11-7082, followed by EGF treatment for 24 hours. FOXK2 promoter activity and expression were measured via Western blotting, RT-qPCR and luciferase activity assays. n = 3 independent experiments performed in triplicate. * P < 0.05; ** P < 0.01 compared with the control. The data are presented as the mean±s.d. (D) Caco-2 cells were treated with EGF and potent inhibitors of PI3K, JNK, ERK and p38. Western blotting was used to quantify the protein levels of FOXK2 as well as the total and phosphorylated levels of p38, JNK, ERK and AKT, as well as the levels of nuclear and total p65. β-actin and LaminB1 served as the loading control. n = 3 independent experiments performed in triplicate. (E) ChIP assays demonstrated that EGF caused NF-κB to directly bind to the FOXK2 promoter through the ERK pathway. n = 3 independent experiments performed in triplicate. * P < 0.05 compared with the control. The data are presented as the mean±s.d. (F) The correlation between p65 and FOXK2 expression in two independent cohorts of CRC patients (cohort I, n = 363; cohort II, n = 390). Kaplan-Meier analysis of the correlations of p65 expression, FOXK2 expression, and p65/FOXK2 coexpression with overall survival and recurrence in cohort I and cohort II. + High; - Low.
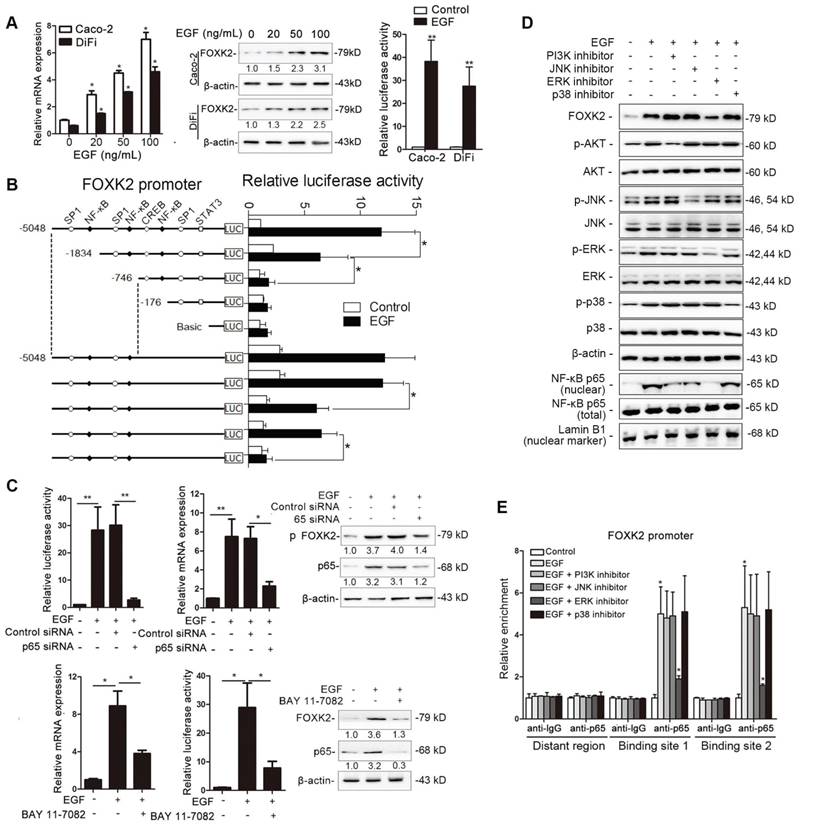
To identify the exact location of the FOXK2 promoter sequence on the cis-regulatory elements that respond to EGF, a series of truncated mutants of the FOXK2 promoter was generated (Figure 4B). Marked reductions in FOXK2 promoter activity were observed in mutants with a deletion from nt-1834 to nt-176, suggesting that this sequence is important for EGF-enhanced FOXK2 activation. This region contains two specificity protein 1 (SP1) binding sites and two NF-κB binding sites. Interestingly, site-directed mutagenesis of the NF-κB binding sites attenuated EGF-facilitated FOXK2 activity, while no effect was observed upon mutating the SP1 binding sites (Figure 4B). Furthermore, p65 knockdown significantly impaired transactivation of the FOXK2 promoter by EGF as evidenced by luciferase reporter assays, RT-qPCR and Western blotting (Figure 4C). Similarly, an NF-κB inhibitor (BAY 11-7082) significantly blocked EGF-mediated FOXK2 promoter transactivation and FOXK2 overexpression (Figure 4C). Our findings indicate that EGF increases FOXK2 transcription through NF-κB activation.
To clarify which pathway is involved in EGF-induced FOXK2 expression, we treated cells with inhibitors of phosphoinositide 3-kinase (PI3K), p38 kinases, c-Jun-N-terminal kinase (JNK) and ERK because EGF can induce activation of these pathways [19]. Pretreatment of cells with the ERK inhibitor reduced EGF-induced FOXK2 expression. However, pretreating cells with p38, JNK or PI3K inhibitors did not affect EGF-induced FOXK2 expression (Figure 4D). Furthermore, ChIP assays revealed that the ERK inhibitor markedly attenuated NF-κB binding to the FOXK2 promoter, while PI3K, JNK, and p38 inhibitors had no effect on the binding of NF-κB to the FOXK2 promoter (Figure 4E). To limit off-target effects of the PI3K, ERK, JNK or p38 inhibitors, siRNA against AKT (siAKT), ERK (siERK), JNK (siJNK) or p38 (sip38) was cotransfected with FOXK2 promoter products into Caco-2 cells upon EGF treatment. The protein levels of FOXK2, PI3K, JNK and p38 were detected after 48 hours of transfection using Western blotting (Figure S8A). In line with the experiment with inhibitors, ChIP-qPCR assays indicated that siERK, but not siAKT, siJNK or sip38, significantly abrogated EGF-induced FOXK2 expression (Figure S8A-B), indicating that ERK activation is responsible for EGF-induced FOXK2 expression in CRC cells.
The possible correlation between p65 expression and FOXK2 expression was examined in two independent cohorts of human CRC patients (cohort I, n = 363; cohort II, n = 390). Elevated expression of nuclear p65 in CRC indicated a poor prognosis and was associated with greater aggressiveness (Table S2). FOXK2 expression was positively correlated with p65 expression (Figure 4F), and Kaplan-Meier analysis showed that patients who coexpressed FOXK2 and p65 suffered from the shortest overall survival times and the highest tumor recurrence rates (Figure 4F).
FOXK2 is crucial for EGF-EGFR signaling-mediated CRC metastasis
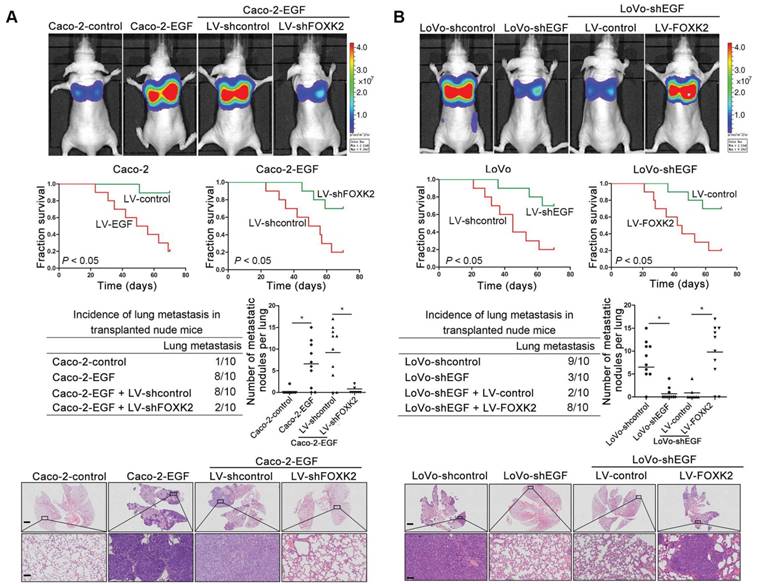
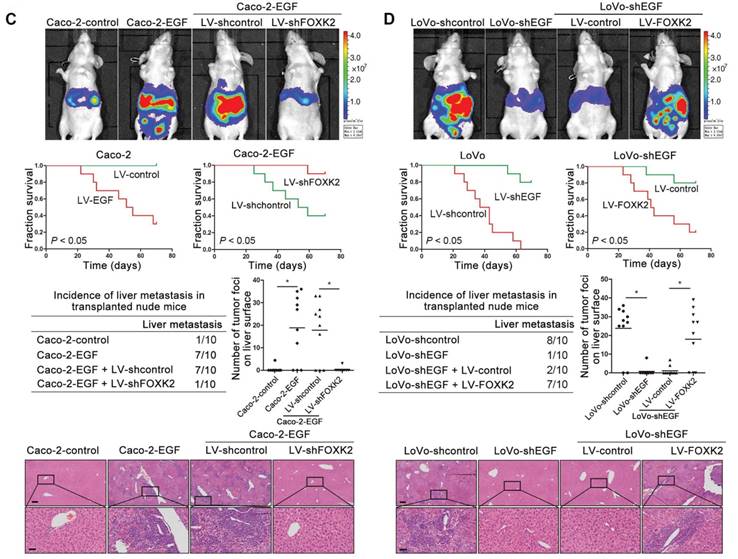
We then generated Caco-2-EGF cells and LoVo-shEGF cells to examine the role of FOXK2 in EGF-facilitated metastasis. The levels of EGF and FOXK2 were analyzed by Western blotting (Figure S9). FOXK2 downregulation attenuated the ability of Caco-2-EGF cells to invade and migrate, while FOXK2 overexpression promoted the migration and invasion abilities of LoVo-shEGF cells (Figure S9). An in vivo metastasis assay showed that FOXK2 downregulation decreased the number of metastatic lung nodules and the incidence of lung metastasis, resulting in an overall increase in survival time in the Caco-2-EGF group (Figure 5A). Conversely, FOXK2 upregulation increased the number of metastatic lung nodules and the incidence of lung metastasis, resulting in decreased overall survival in the LoVo-shEGF group (Figure 5B). Consistent with these findings, the intrasplenic injection metastasis assay confirmed that FOXK2 was indispensable for EGF-mediated CRC metastasis (Figure 5C-D).
FOXK2 is critical for EGF-EGFR signaling-mediated CRC metastasis. (A and B) In vivo lung metastatic assays. Cells were injected into the tail veins of mice (n = 10 mice per group). Bioluminescence imaging 9 weeks after implantation, the incidence of lung metastasis, overall survival times, the number of metastatic lung nodules, and H&E staining of lung tissues from the different groups are shown. Scale bars: top, 500 μm; bottom, 40 μm. * P < 0.05 compared with the control. The data are presented as the mean±s.d. (C and D) In vivo liver metastasis assays. Cells were injected into the spleens of mice (n = 10 mice per group). Bioluminescence imaging 9 weeks after implantation, the incidence of liver metastasis, overall survival times, the number of metastatic liver nodules, and H&E staining of liver tissues from the different groups are shown. Scale bars: top, 200 μm; bottom, 40 μm. * P < 0.05 compared with the control. The data are presented as the mean±s.d.
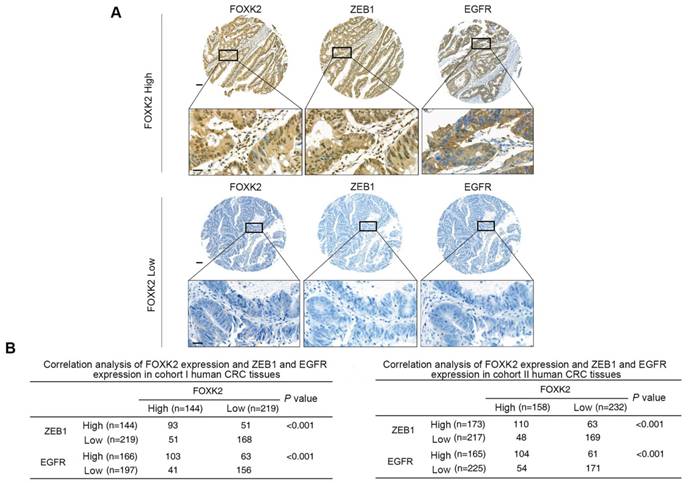
FOXK2 expression is positively correlated with ZEB1 and EGFR expression in human CRC tissues
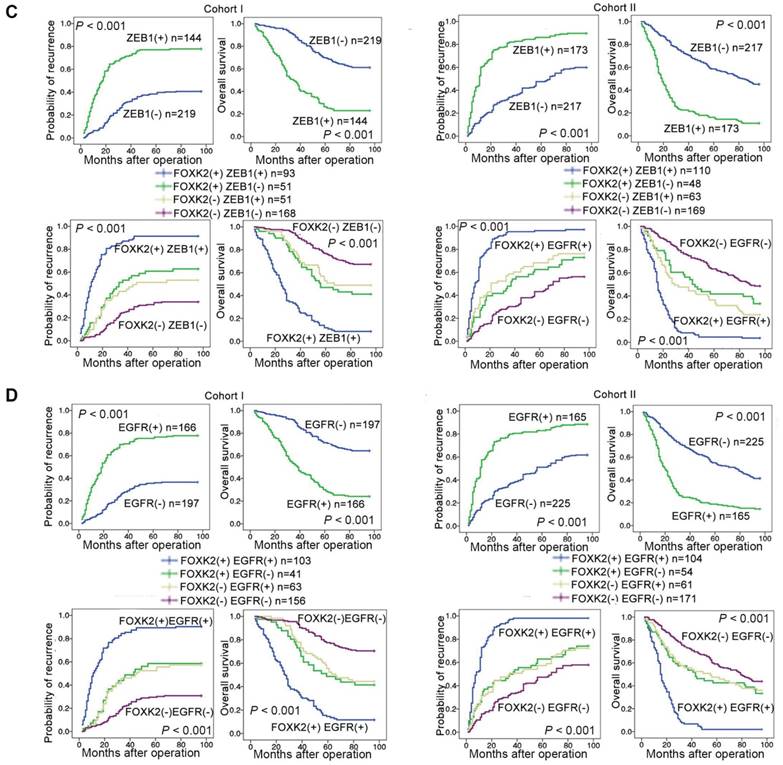
Clinical associations between FOXK2 and ZEB1 or EGFR expression were then evaluated in two independent cohorts of human CRC patients (cohort I, n = 363; cohort II, n = 390). EGFR and ZEB1 expression levels were positively correlated with FOXK2 expression (Figure 6A-B). The elevated expression levels of ZEB1 and EGFR were significantly correlated with a higher AJCC stage, distant metastasis, lymph node metastasis and poorer tumor differentiation (Tables S3-S4). Kaplan-Meier analysis revealed that CRC patients with positive expression of ZEB1 or EGFR had higher recurrence rates and a shorter overall survival time than those who were negative for ZEB1 or EGFR (Figure 6C-D). Patients were divided into four groups based on FOXK2 and ZEB1 expression or FOXK2 and EGFR expression. Those who coexpressed FOXK2 and ZEB1 or FOXK2 and EGFR had the shortest overall survival times and the highest recurrence rates (Figure 6C-D).
FOXK2 expression is positively correlated with the expression of ZEB1 and EGFR in human CRC tissues. (A) Representative IHC images of high or low FOXK2, ZEB1, and EGFR expression in CRC tissues. Scale bars: top, 100 μm; bottom, 20 μm. (B) Correlation between FOXK2 and ZEB1 or EGFR expression in CRC tissues of patients from cohort I (n = 363) and cohort II (n = 390). (C) Kaplan-Meier analysis of the correlations of FOXK2 expression, ZEB1 expression, and FOXK2/ZEB1 coexpression with overall survival and recurrence in cohort I and cohort II. + High; - Low. (D) Kaplan-Meier analysis of the correlations of FOXK2 expression, EGFR expression, and FOXK2/EGFR coexpression with overall survival and recurrence in cohort I and cohort II. + High; - Low.
The EGFR monoclonal antibody cetuximab decreases FOXK2-mediated CRC metastasis
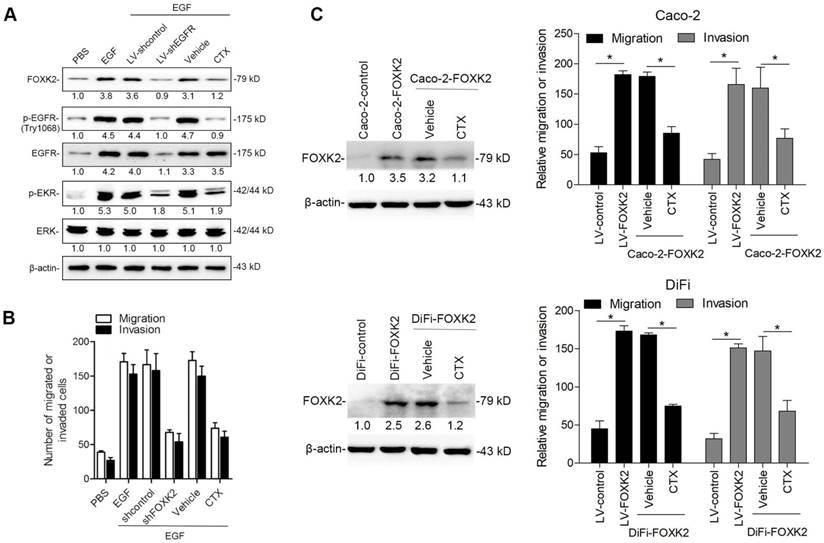
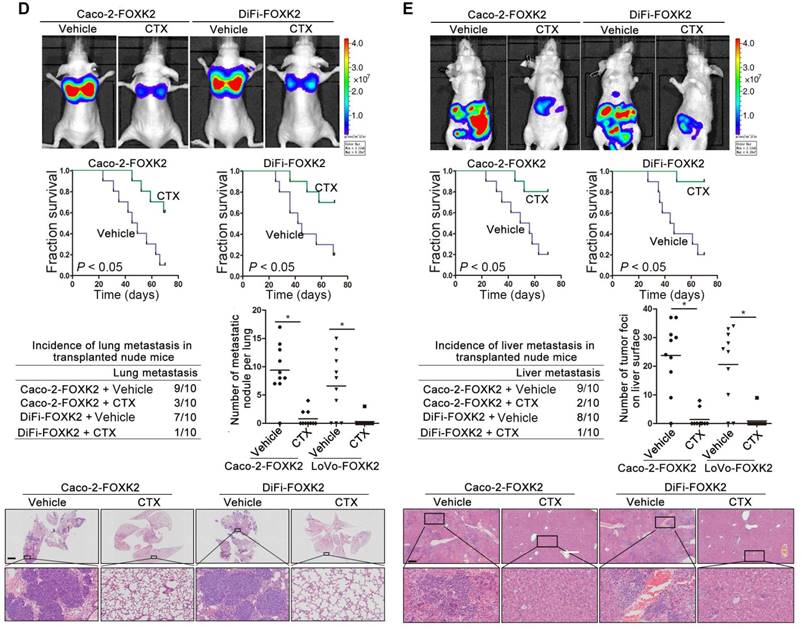
Because FOXK2 promoted CRC EMT and metastasis by transactivating ZEB1 and EGFR expression and EGF induced FOXK2 expression through the ERK/NF-κB pathway, we examined whether cetuximab, an anti-EGFR monoclonal antibody, can block the EGF-NF-κB-FOXK2-EGFR feedback loop to suppress CRC metastasis. Western blotting and Transwell analysis indicated that EGFR silencing or cetuximab significantly suppressed EGF-induced FOXK2 expression, EGFR phosphorylation, ERK signaling activation and cell migration (Figure 7A-B). To further determine whether cetuximab can suppress CRC metastasis promoted by FOXK2 upregulation, we treated FOXK2-overexpressing cells (Caco-2-FOXK2 and DiFi-FOXK2) with cetuximab. As shown in Figure 7C, cetuximab inhibited CRC cell migration and invasion promoted by FOXK2 overexpression. An in vivo lung metastasis assay showed that FOXK2 overexpression promoted the occurrence of metastatic colonization and the quantity of metastatic lung nodules and decreased the overall survival time or transplanted mice (Figure 1E), while cetuximab treatment significantly inhibited lung metastasis in the FOXK2-overexpressing group (Figure 7D). Similar results were observed in an in vivo liver metastasis model (Figure 1F and 7E), suggesting that cetuximab can be used to suppress FOXK2-promoted CRC metastasis and thus providing a therapeutic strategy for CRC patients with high FOXK2 expression.
The EGFR inhibitor cetuximab suppresses FOXK2-induced promotion of CRC metastasis. (A) Caco-2 cells were infected with the lentivirus LV-shEGFR or were treated with the EGFR inhibitor cetuximab (CTX, 10 nM) upon EGF treatment (100 ng/mL). Twenty-four hours post-EGF treatment, FOXK2 expression was measured by Western blotting. n = 3 independent experiments performed in triplicate. (B) Migration and invasion of the indicated cells were measured by Transwell assays. n = 3 independent experiments performed in triplicate. The data are presented as the mean±s.d. (C) Caco-2 and DiFi cells were treated with vehicle or CTX (10 nM) after lentivirus transfection (LV-control or LV-FOXK2). The levels of FOXK2 proteins were detected by Western blotting. The migration and invasion of the indicated cells were detected by Transwell assays. n = 3 independent experiments performed in triplicate. * P < 0.05 compared with the control. The data are presented as the mean±s.d. (D) In vivo lung metastatic assays. Cells were injected into the tail veins of mice (n = 10 mice per group). Bioluminescence imaging 9 weeks after implantation, the incidence of lung metastasis, overall survival times, the number of metastatic lung nodules, and H&E staining of lung tissues from the different groups are shown. Scale bars: top, 500 μm; bottom, 40 μm. * P < 0.05 compared with the control. The data are presented as the mean±s.d. (E) In vivo liver metastasis assays. Cells were injected into the spleens of mice (n = 10 mice per group). Bioluminescence imaging 9 weeks after implantation, the incidence of liver metastasis, overall survival times, the number of metastatic liver nodules, and H&E staining of liver tissues from the different groups are shown. Scale bars: top, 200 μm; bottom, 40 μm. * P < 0.05 compared with the control. The data are presented as the mean±s.d.
Discussion
FOXK2 plays critical roles in carcinogenesis, and progression appears to be a double-edged sword in tumorigenesis. FOXK2 suppresses tumor initiation and metastasis in breast cancer and clear-cell renal cell carcinoma [13, 20]. By contrast, FOXK2 promotes malignant hepatocellular carcinoma cell growth and confers a worse clinical prognosis [21]. Notably, in CRC, Qian et al. also reported that FOXK2 was transcriptionally activated in CRC tissues compared to non-cancer tissues, and that high FOXK2 expression was significantly correlated with poor survival [12]. In line with these studies, we found that FOXK2 levels were significantly elevated in human CRC and indicated a poor prognosis. FOXK2 promoted CRC cell EMT, invasion and metastasis by ZEB1 and EGFR transactivation. NF-κB directly transactivated FOXK2 upon activation by the EGFR/ERK signaling pathway. Moreover, in clinical human samples, FOXK2 expression correlated positively with p65, ZEB1 and EGFR expression, and CRC patients who co-expressed p65(+)/FOXK2(+), FOXK2(+)/ZEB1(+) or FOXK2(+)/EGFR(+) had worse clinical prognosis. Although some similar information regarding the correlations of FOXK2 levels in CRC patients has already been shown in Qian et al.'s paper, several innovative points affirm the novelty of our study. First, we not only found elevated FOXK2 levels in CRC tissues compared with those in non-cancer tissues but also found that FOXK2 expression was significantly upregulated in CRC tissues from patients with recurrence and metastasis. FOXK2 overexpression was significantly correlated with the loss of tumor encapsulation, microvascular invasion, and a higher tumor-nodule-metastasis (TNM) stage, indicated a poor prognosis in CRC patients, and was an independent and significant risk factor for recurrence and reduced survival after curative resection. Second, although Qian's study reveals that FOXK2 promoted cell proliferation, we found that FOXK2 promotes CRC cell EMT, migration, invasion and metastasis. Third, Qian's study reported that oncogene SOX9 was responsible for upregulation of FOXK2 by directly binding to its promoter. However, in our study, EGF was found to activate FOXK2 expression through the ERK/NF-κB pathway, and FOXK2 was found to be essential for EGF-induced CRC metastasis. Fourth, Qian et al. did not explore the transcriptional target genes of FOXK2 responsible for FOXK2-mediated CRC proliferation.
In our study, we demonstrated that ZEB1 and EGFR were directly transactivated by FOXK2 and were essential for FOXK2-promoted CRC cell EMT, migration, invasion and metastasis, illuminating the mechanism by which FOXK2 promotes CRC progression. Finally, we also found that the anti-EGFR monoclonal antibody cetuximab can block the EGF-FOXK2-EGFR feedback loop in CRC and suppress FOXK2-mediated CRC metastasis, providing a new therapeutic strategy for metastatic CRC. Taken together, our results provide a line of novel evidence that FOXK2 functions as a metastasis-promoting gene by transactivating ZEB1 and EGFR and is critical for EGF-EGFR signaling-mediated CRC metastasis.
Of note, FOXK2 was reported as a tumor suppressor through the inhibition of EGFR in renal carcinoma [20], suggesting that FOXK2 can regulate the same gene to exert opposite effects in a context and model-dependent way. In detail, this may be attributed to that the FOXK2 polypeptide work coordinately, in a context-dependent fashion, to achieve DNA binding and transactivation. FOXK2 contains two distinct structural domains: the C-terminal forkhead (FH) domain, also known as winged helix, and the N-terminal FH-associated (FHA) domain, a phosphopeptide recognition motif found in many regulatory proteins [4]. FOXK2 proteins can both activate and repress gene expression through the recruitment of co-factors or repressors with different functional domains. In addition, FOXK2 proteins interact extensively with other factors such as DVLs and AP-1 to modulate gene expression. For example, FOXK2 interacts with transcription corepressor complexes NCoR/SMRT, SIN3A, NuRD, and REST/CoREST with FHA domain to repress a cohort of genes including HIF1b and EZH2 to suppress the hypoxic response and breast cancer carcinogenesis [13]. While its FHA-adjacent region can interact with DVLs, positively regulate Wnt/β-catenin signaling by translocating DVL into the nucleus and promote CRC tumorigenesis 11. Besides, FOXK2 participates in combinatorial transcriptional control with the AP-1 to promote efficient recruitment of AP-1 to chromatin [22]. Moreover, FOXK2 proteins are largely regulated through alterations in post-translational modifications. Post-translational control of FOXK2 protein activity is exerted through an intricate balance of phosphorylation, acetylation and ubiquitylation that influences protein interactions and sub-cellular localization, which may impart additional regulatory diversity in a context-dependent fashion [23-25]. More importantly, our study demonstrated the direct interaction between FOXK2 and EGFR promoter region. Sequence analysis showed that two putative FOXK2 binding sites exist in the EGFR promoter region. Serial deletion and site-directed mutagenesis revealed that the first FOXK2 binding site on the EGFR promoter region is vital for transactivation of EGFR by FOXK2. ChIP assays revealed the direct binding of FOXK2 to the EGFR promoter in CRC cells and tissues. These findings suggested that FOXK2 directly binds to the EGFR promoter and transactivates EGFR expression in CRC cells. However, in Zhang et al.'s study, they only observed the expression correlation between FOXK2 and EGFR, which suggest that EGFR is a potential downstream gene of FOXK2 and it is very likely that FOXK2 indirectly regulates EGFR expression in ccRCC cells, because that deregulation of FOXK2 protein activity or expression can result in changes in both direct and indirect target genes. Therefore, according to our results, we confirm that EGFR is a direct functional target gene of FOXK2 in CRC cells, while more directly regulatory relationships between FOXK2 and EGFR in ccRCC cells according to Zhang et al.'s study require further investigation.
Various human cancers are heavily dependent on EGFR signal activation, which plays a critical role in tumorigenesis and progression by promoting various cellular processes [4, 26]. Disruption of EGFR signaling, either by inhibiting intracellular tyrosine kinase activity or by blocking EGFR binding sites on the extracellular domain of the receptor, can halt the development of tumors reliant on EGFR, thus improving clinical outcomes [5, 27]. Recent studies have revealed that NF-κB activation in CRC likely involves EGFR signaling. EGFR stimulation induces protein kinase C (PKC)-dependent phosphorylation of migration and invasion inhibitory protein (MIIP) at Ser303, which facilitates RelA-MIIP nuclear interaction, thus augmenting RelA transcriptional activity and promoting tumor metastasis [28]. However, the concrete mechanisms by which NF-κB regulates CRC metastasis and progression after EGFR activation must be further clarified. To the best of our knowledge, the current study represents the first series of experiments proving that EGFR serves as a direct transcriptional target of FOXK2. FOXK2 transactivates the expression of EGFR by binding directly to its promoter, triggering EGFR overexpression in CRC. EGFR activation subsequently transactivates FOXK2 expression through the NF-κB signaling pathway. Downregulation of EGFR significantly decreased FOXK2-enhanced CRC migration, invasion, and lung and liver metastasis, whereas upregulation of EGFR restored the FOXK2-knockdown-induced attenuation of CRC migration, invasion, and lung and liver metastasis. Clinically, FOXK2 expression was positively correlated with EGFR expression as well as p65, the core transducer of the NF-κB signaling pathway. CRC patients who coexpressed FOXK2(+)/EGFR(+) or p65(+)/FOXK2(+) were also found to have poorer prognosis. Most importantly, we demonstrated that cetuximab can dramatically inhibit activation of EGFR and decrease FOXK2-enhanced CRC migration, invasion and metastasis. Collectively, our series of experiments reveal a positive feedback loop between EGFR-NF-κB/FOXK2 and suggest that cetuximab can be used for the treatment of FOXK2-mediated CRC metastasis.
Moreover, cancerous cells, particularly those of CRC, are thought to acquire their pro-metastatic phenotype primarily during the EMT [29]. Extensive evidence indicates that the EMT is involved in CRC invasion and metastasis via multiple signaling pathways [30-32]. Despite these findings, the role of the EMT and the molecular mechanisms facilitating malignant CRC has not been fully elucidated. Our investigations reveal that ZEB1 is a direct transcriptional target of FOXK2. FOXK2 transactivates the expression of ZEB1 by directly binding to its promoter. ZEB1 downregulation markedly attenuated FOXK2-augmented CRC cell EMT, migration, invasion, and metastasis, whereas overexpression of ZEB1 restored FOXK2-knockdown-induced attenuation of CRC metastasis. In two independent CRC cohorts, FOXK2 expression was positively correlated with ZEB1 expression. CRC patients who coexpressed FOXK2(+)/ZEB1(+) had poorer prognosis. Our findings indicate that CRC cell EMT and metastasis are promoted by FOXK2 transactivation of ZEB1. Of interest, we found an internal relationship that exists between ZEB1 and EGFR in addition to being target genes of FOXK2, given that promotion of metastasis by LV-FOXK2 is dependent on both ZEB1 and EGFR in Caco-2. But metastasis can be restored by either LV-ZEB1 or LV-EGFR alone in LoVo-shFOXK2. EGFR or ZEB1 gain- and loss-of-function models were established and the protein levels of EGFR and ZEB1 after lentivirus transfection in the indicated cells was examined by Western blotting (Figure S10A). EGFR overexpression promoted the expression of ZEB1, while ZEB1 overexpression did not alter EGFR levels. In contrast, downregulation of EGFR inhibited the expression of ZEB1, whereas ZEB1 knockdown had little effect on EGFR expression (Figure S10A). These observations indicated that EGFR is the upstream regulator of ZEB1 expression. Moreover, we also performed Transwell assays to determine whether ZEB1 is a functional downstream effector of EGFR. Downregulation of ZEB1 suppressed the enhanced cell migration and invasion induced by EGFR overexpression. In contrast, ectopic expression of ZEB1 restored the impaired migratory and invasive abilities of EGFR-knockdown CRC cells (Figure S10B). These findings revealed that ZEB1 is a functional downstream effector of EGFR, which may explain the more complicated regulation network between ZEB1 and EGFR in addition to being target genes of FOXK2.
Interestingly, FOXK1, the homolog of FOXK2, has also been reported to play a critical role in the EMT and cell migration in gastric cancer [33, 34], indicating that FOXK1 and FOXK2 may very likely exhibit redundancy in CRC metastasis. However, based on our findings, although both FOXK1 and FOXK2 were increased in CRC, the degree of elevation of FOXK2 expression (24.73±0.438) was markedly higher than that of FOXK1 expression (1.43±0.037), implying that FOXK2 is the main contributor of the endogenic FOXK subfamily to CRC metastasis. Furthermore, to investigate the mechanism by which FOXK1 promotes CRC cell EMT and migration, we transfected Caco-2 cells with FOXK1 or control lentivirus (LV-FOXK1 or LV-control) and then performed EMT RT2 profiler polymerase chain reaction array assays in FOXK1-overexperssing cells (Caco-2-FOXK1) and control cells (Caco-2-control). FOXK1 overexpression resulted in upregulated expression of a group of EMT- and metastasis-related genes, including FN1, TWIST1, SNAI2, SNAI1, VIM, and MMP9 (Table S7). Among them, VIM, FN1, MMP9, VCAN, IGFBP4, ITGA5, TWIST1 and SNAI1 can be upregulated by both FOXK1 and FOXK2 overexpression (Table S7 and Figure S11A). These observations may explain the redundancy between FOXK1 and FOXK2 in CRC cell EMT and cancer cell migration, which is consistent with the Western blotting and Transwell assays revealing that both FOXK1 and FOXK2 elevate vimentin expression and promote the migration and invasion of CRC cells (Figure S11B-C). Interestingly, EGFR and ZEB1, which are significantly upregulated by FOXK2 overexpression, show no obvious changes with overexpression of FOXK1. In contrast, FOXK1 overexpression activates TWIST1 and SNAI2 but has no effect on EGFR and ZEB1 expression (Figure S11B-C). These findings suggest that FOXK1 and FOXK2 can both promote the EMT of CRC cells through different groups of target genes, which may contribute to FOXK1 and FOXK2 binding to different transcription co-regulators, although such speculation requires further investigation.
The regulatory mechanism of FOXK2 overexpression in human CRC remains unknown. Our findings indicate that EGF increases FOXK2 transcription through NF-κB activation. Site-directed mutagenesis luciferase reporter assays and ChIP showed that NF-κB directly binds to the promoter of FOXK2 and transactivates its expression. p65 knockdown or NF-κB inhibition significantly impaired FOXK2 expression. We further treated cells with small molecular inhibitors or siRNA against AKT, p38, JNK and ERK and found that the activation of ERK, but not PI3K, JNK or p38, contributes to NF-kB-mediated FOXK2 expression in CRC cells. Previous studies have reported that ERK-dependent activation of NF-kB can lead to expression of TNF-α and other cytokines in HSP70-mediated inflammatory conditions [35]. Park SH et al. also reported that neutrophil elastase causes MUC5AC mucin trans-activation and synthesis via ERK and NF-kB pathways in A549 cells [36]. This evidence suggests that ERK is a universal regulator of NF-kB. In additon, it is well established that AKT, ERK, p38 and JNK are known regulators of NF-kB and EGF also activated NF-kB through pathways other than ERK. However, only the ERK inhibitor inhibited the binding of NF-kB to FOXK2 promoter and thereby suppressing FOXK2 expression. This could be explained that the binding of NF-kB to its target gene's promoter is context-dependent. Without appropriate co-factors, the transcription factor NF-kB cannot accurately binds to the promoter of target genes even though it has been translocated into the nucleus. These observations reflect dynamic spatial and temporal regulatory patterns of NF-kB-mediated FOXK2 transcription, which need further investigation. Moreover, the circuitry through which FOXK2 transactivates EGFR and FOXK2's role as a downstream target of EGF are not observed in normal cells or tissues (Figure S12). This could be explained, at least partially, that ERK and NF-kB are integral to maintaining biological equilibrium through their ability to regulate gene expression in complex networks, the target genes of a specific TF are regulated in a context-dependent and cell type-specific manner [37-40]. In some cases, the same TF targets different genes by binding to different transcription co-activators during different stages of diseases [40], reflecting dynamic spatial and temporal regulatory patterns.
In conclusion, we discovered a novel function of FOXK2 in CRC metastasis. FOXK2 overexpression was significantly correlated with more aggressive features and indicated a poor prognosis in CRC patients. FOXK2 promoted CRC metastasis by transactivating ZEB1 and EGFR expression. EGF induced FOXK2 expression via the ERK/NF-κB pathway, thereby promoting CRC metastasis. In addition, the EGFR monoclonal antibody cetuximab could block FOXK2-promoted CRC metastasis. This study provides a potential prognostic biomarker and a potential therapeutic strategy for CRC metastasis.
Abbreviations
FOXK2: forkhead box K2; CRC: colorectal cancer; EMT: epithelial-mesenchymal transition; TNM: tumor-node-metastasis; ChIP: chromatin immunoprecipitation analysis; IHC: immunohistochemical; ZEB1: zinc finger E-box binding homeobox 1; EGFR: epidermal growth factor receptor; ERK: extracellular regulated protein kinases; NF-κB: nuclear factor κB.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
Research was supported by grants from the National Key Research and Development Program of China 2018YFC1312103 (L.X.), National Natural Science Foundation of China No.81522031 (L.X.), No. 81772623 (L.X.), No. 81627807 (K.W.), No.81430072 (D.F.) and No. 81421003 (K.W.), National Key Research and Development Program of China SQ2017YFSF090132 (K.W.), and National Center for Clinical Research of Digestive Diseases 2015BAI13B07 (D.F.).
We would like to thank Qingling An and Jianhua Dou from the Fourth Military Medical University for providing excellent technical assistance.
Author contributions
Feng Du and Chenyang Qiao performed the experiments. Xiaowei Li and Zhangqian Chen assisted in immunohistochemistry staining and animal experiments. Hao Liu, Shengda Wu, Sijun Hu, Zhaoyan Qiu and Meirui Qian gave assistance in collecting tissues samples. Dean Tian, Yongzhan Nie, Kaichun Wu and Daiming Fan gave assistance in conceiving experiments and analyzing data. Limin Xia and Feng Du designed studies and wrote the paper.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68:7-30
2. Fakih MG. Metastatic colorectal cancer: current state and future directions. J Clin Oncol. 2015;33:1809-1824
3. Punt CJ, Koopman M, Vermeulen L. From tumour heterogeneity to advances in precision treatment of colorectal cancer. Nat Rev Clin Oncol. 2017;14:235-246
4. Hannenhalli S, Kaestner KH. The evolution of Fox genes and their role in development and disease. Nat Rev Genet. 2009;10:233-240
5. Myatt SS, Lam EW. The emerging roles of forkhead box (Fox) proteins in cancer. Nat Rev Cancer. 2007;7:847-859
6. Liu X, Wei X, Niu W, Wang D, Wang B, Zhuang H. Downregulation of FOXK2 is associated with poor prognosis in patients with gastric cancer. Mol Med Rep. 2018;18:4356-4364
7. Wang B, Zhang X, Wang W, Zhu Z, Tang F, Wang D. et al. Forkhead box K2 inhibits the proliferation, migration, and invasion of human glioma cells and predicts a favorable prognosis. Onco Targets Ther. 2018;11:1067-1075
8. Chen S, Jiang S, Hu F, Xu Y, Wang T, Mei Q. Foxk2 inhibits non-small cell lung cancer epithelial-mesenchymal transition and proliferation through the repression of different key target genes. Oncol Rep. 2017;37:2335-2347
9. Liu Y, Ao X, Jia Z, Bai XY, Xu Z, Hu G. et al. FOXK2 transcription factor suppresses ERalpha-positive breast cancer cell growth through down-regulating the stability of ERalpha via mechanism involving BRCA1/BARD1. Sci Rep. 2015;5:8796
10. Nestal DMG, Khongkow P, Gong C, Yao S, Gomes AR, Ji Z. et al. Forkhead box K2 modulates epirubicin and paclitaxel sensitivity through FOXO3a in breast cancer. Oncogenesis. 2015;4:e167
11. Wang W, Li X, Lee M, Jun S, Aziz KE, Feng L. et al. FOXKs promote Wnt/beta-catenin signaling by translocating DVL into the nucleus. Dev Cell. 2015;32:707-718
12. Qian Y, Xia S, Feng Z. Sox9 mediated transcriptional activation of FOXK2 is critical for colorectal cancer cells proliferation. Biochem Biophys Res Commun. 2017;483:475-481
13. Shan L, Zhou X, Liu X, Wang Y, Su D, Hou Y. et al. FOXK2 Elicits Massive Transcription Repression and Suppresses the Hypoxic Response and Breast Cancer Carcinogenesis. Cancer Cell. 2016;30:708-722
14. Huang W, Chen Z, Zhang L, Tian D, Wang D, Fan D. et al. Interleukin-8 Induces Expression of FOXC1 to Promote Transactivation of CXCR1 and CCL2 in Hepatocellular Carcinoma Cell Lines and Formation of Metastases in Mice. Gastroenterology. 2015;149:1053-1067
15. Huang W, Chen Z, Shang X, Tian D, Wang D, Wu K. et al. Sox12, a direct target of FoxQ1, promotes hepatocellular carcinoma metastasis through up-regulating Twist1 and FGFBP1. Hepatology. 2015;61:1920-1933
16. Nieto MA, Huang RY, Jackson RA, Thiery JP. EMT: 2016. Cell. 2016;166:21-45
17. Lambert AW, Pattabiraman DR, Weinberg RA. Emerging Biological Principles of Metastasis. Cell. 2017;168:670-691
18. Winder T, Lenz HJ. Vascular endothelial growth factor and epidermal growth factor signaling pathways as therapeutic targets for colorectal cancer. Gastroenterology. 2010;138:2163-2176
19. Baselga J, Arteaga CL. Critical update and emerging trends in epidermal growth factor receptor targeting in cancer. J Clin Oncol. 2005;23:2445-2459
20. Zhang F, Ma X, Li H, Zhang Y, Li X, Chen L. et al. FOXK2 suppresses the malignant phenotype and induces apoptosis through inhibition of EGFR in clear-cell renal cell carcinoma. Int J Cancer. 2018;142:2543-2557
21. Lin MF, Yang YF, Peng ZP, Zhang MF, Liang JY, Chen W. et al. FOXK2, regulted by miR-1271-5p, promotes cell growth and indicates unfavorable prognosis in hepatocellular carcinoma. Int J Biochem Cell Biol. 2017;88:155-161
22. Ji Z, Donaldson IJ, Liu J, Hayes A, Zeef LA, Sharrocks AD. The forkhead transcription factor FOXK2 promotes AP-1-mediated transcriptional regulation. Mol Cell Biol. 2012;32:385-398
23. Nestal DMG, Ji Z, Fan LY, Yao S, Zona S, Sharrocks AD. et al. SUMOylation modulates FOXK2-mediated paclitaxel sensitivity in breast cancer cells. Oncogenesis. 2018;7:29
24. Okino Y, Machida Y, Frankland-Searby S, Machida YJ. BRCA1-associated protein 1 (BAP1) deubiquitinase antagonizes the ubiquitin-mediated activation of FoxK2 target genes. J Biol Chem. 2015;290:1580-1591
25. Marais A, Ji Z, Child ES, Krause E, Mann DJ, Sharrocks AD. Cell cycle-dependent regulation of the forkhead transcription factor FOXK2 by CDK cyclin complexes. J Biol Chem. 2010;285:35728-35739
26. Ohashi K, Maruvka YE, Michor F, Pao W. Epidermal growth factor receptor tyrosine kinase inhibitor-resistant disease. J Clin Oncol. 2013;31:1070-1080
27. Arena S, Bellosillo B, Siravegna G, Martinez A, Canadas I, Lazzari L. et al. Emergence of Multiple EGFR Extracellular Mutations during Cetuximab Treatment in Colorectal Cancer. Clin Cancer Res. 2015;21:2157-2166
28. Chen T, Li J, Xu M, Zhao Q, Hou Y, Yao L. et al. PKCepsilon phosphorylates MIIP and promotes colorectal cancer metastasis through inhibition of RelA deacetylation. Nat Commun. 2017;8:939
29. Spaderna S, Schmalhofer O, Hlubek F, Berx G, Eger A, Merkel S. et al. A transient, EMT-linked loss of basement membranes indicates metastasis and poor survival in colorectal cancer. Gastroenterology. 2006;131:830-840
30. Hur K, Toiyama Y, Takahashi M, Balaguer F, Nagasaka T, Koike J. et al. MicroRNA-200c modulates epithelial-to-mesenchymal transition (EMT) in human colorectal cancer metastasis. Gut. 2013;62:1315-1326
31. Rokavec M, Oner MG, Li H, Jackstadt R, Jiang L, Lodygin D. et al. IL-6R/STAT3/miR-34a feedback loop promotes EMT-mediated colorectal cancer invasion and metastasis. J Clin Invest. 2015;125:1362
32. Lamprecht S, Kaller M, Schmidt EM, Blaj C, Schiergens TS, Engel J. et al. PBX3 Is Part of an EMT Regulatory Network and Indicates Poor Outcome in Colorectal Cancer. Clin Cancer Res. 2018;24:1974-1986
33. Zhang P, Tang WM, Zhang H, Li YQ, Peng Y, Wang J. et al. MiR-646 inhibited cell proliferation and EMT-induced metastasis by targeting FOXK1 in gastric cancer. Br J Cancer. 2017;117:525-534
34. Peng Y, Zhang P, Huang X, Yan Q, Wu M, Xie R. et al. Direct regulation of FOXK1 by c-jun promotes proliferation, invasion and metastasis in gastric cancer cells. Cell Death Dis. 2016;7:e2480
35. Somensi N, Brum PO, de Miranda RV, Gasparotto J, Zanotto-Filho A, Rostirolla DC. et al. Extracellular HSP70 Activates ERK1/2, NF-kB and Pro-Inflammatory Gene Transcription Through Binding with RAGE in A549 Human Lung Cancer Cells. Cell Physiol Biochem. 2017;42:2507-2522
36. Song JS, Cho KS, Yoon HK, Moon HS, Park SH. Neutrophil elastase causes MUC5AC mucin synthesis via EGF receptor, ERK and NF-kB pathways in A549 cells. Korean J Intern Med. 2005;20:275-283
37. Briggs JA, Weinreb C, Wagner DE, Megason S, Peshkin L, Kirschner MW. et al. The dynamics of gene expression in vertebrate embryogenesis at single-cell resolution. Science. 2018;360:eaar5780
38. Kernfeld EM, Genga R, Neherin K, Magaletta ME, Xu P, Maehr R. A Single-Cell Transcriptomic Atlas of Thymus Organogenesis Resolves Cell Types and Developmental Maturation. Immunity. 2018;48:1258-1270
39. Wang J, Jenjaroenpun P, Bhinge A, Angarica VE, Del SA, Nookaew I. et al. Single-cell gene expression analysis reveals regulators of distinct cell subpopulations among developing human neurons. Genome Res. 2017;27:1783-1794
40. Jolma A, Yin Y, Nitta KR, Dave K, Popov A, Taipale M. et al. DNA-dependent formation of transcription factor pairs alters their binding specificity. Nature. 2015;527:384-388
Author contact
![]() Corresponding authors: Dr. Limin Xia, Dr. Yongzhan Nie and Dr. Daiming Fan, State Key Laboratory of Cancer Biology and Xijing Hospital of Digestive Diseases, Fourth Military Medical University, Xi'an 710032, Shaanxi Province, China; Phone: 86 29 8477 1502; Fax: 86 29 8253 9041; Email: xialiminedu.cn, yongznieedu.cn and fandaimedu.cn
Corresponding authors: Dr. Limin Xia, Dr. Yongzhan Nie and Dr. Daiming Fan, State Key Laboratory of Cancer Biology and Xijing Hospital of Digestive Diseases, Fourth Military Medical University, Xi'an 710032, Shaanxi Province, China; Phone: 86 29 8477 1502; Fax: 86 29 8253 9041; Email: xialiminedu.cn, yongznieedu.cn and fandaimedu.cn