Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(12):3622-3638. doi:10.7150/thno.32935 This issue Cite
Research Paper
SCARNA10, a nuclear-retained long non-coding RNA, promotes liver fibrosis and serves as a potential biomarker
Kun Zhang1*, Yawei Han1*, Zhimei Hu1*, Zhen Zhang1*, Shuai Shao2, Qingbin Yao1, Lina Zheng1, Jingzhao Wang1, Xiaohui Han1, Yu Zhang2, Ting Chen1, Zhi Yao3 ![]() , Tao Han2
, Tao Han2 ![]() , Wei Hong1
, Wei Hong1 ![]()
1. Department of Histology and Embryology, Tianjin Key Laboratory of Cellular and Molecular Immunology, Key Laboratory of Immune Microenvironment and Disease of Ministry of Education, School of Basic Medical Sciences, Tianjin Medical University, Tianjin, China
2. The Third Central Clinical College of Tianjin Medical University, Department of Hepatology and Gastroenterology, Tianjin Third Central Hospital affiliated to Nankai University, Tianjin Key Laboratory of Artificial Cells, Artificial Cell Engineering Technology Research Center of Public Health Ministry, Tianjin, China
3. Department of Immunology, Tianjin Key Laboratory of Cellular and Molecular Immunology, Key Laboratory of Immune Microenvironment and Disease of Ministry of Education, School of Basic Medical Sciences, Tianjin Medical University, Tianjin, China
*These authors contributed equally to this work.
Received 2019-1-8; Accepted 2019-4-29; Published 2019-5-26
Abstract

Long non-coding RNAs (lncRNAs) are involved in numerous biological functions and pathological processes. However, the clinical significance of lncRNAs and their functions in liver fibrosis remain largely unclear.
Methods: The transcript of lncRNA SCARNA10 in serum and liver samples from patients with advanced hepatic fibrosis, liver tissues from two fibrosis mouse models, and cultured hepatic stellate cells (HSCs) was determined by real-time RT-PCR. The effects of lentivirus-mediated knockdown or over-expression of SCARNA10 in liver fibrosis were examined in vitro and in vivo. Moreover, the effects and mechanisms of down-regulation or over-expression of SCARNA10 on the expression of the genes involved in TGFβ pathway were determined.
Results: It was found lncRNA ENSMUST00000158992, named as Scarna10, was remarkably up-regulated in mouse fibrotic livers according to the microarray data. We observed that the transcript of SCARNA10 was increased in the serum and liver from patients with advanced hepatic fibrosis. Furthermore, we found that SCARNA10 promoted liver fibrosis both in vitro and in vivo through inducing hepatocytes (HCs) apoptosis and HSCs activation. Mechanistically, RNA immunoprecipitation (RIP) assays demonstrated that SCARNA10 physically associated with polycomb repressive complex 2 (PRC2). Additionally, our results demonstrated that SCARNA10 functioned as a novel positive regulator of TGFβ signaling in hepatic fibrogenesis by inhibiting the binding of PRC2 to the promoters of the genes associated with ECM and TGFβ pathway, thus promoting the transcription of these genes.
Conclusions: Our study identified a crucial role of SCARNA10 in liver fibrosis, providing a proof of this molecule as a potential diagnostic marker and a possible therapeutic target against liver fibrosis.
Keywords: lncRNA, Liver Fibrosis, SCARNA10, TGFβ, PRC2
Introduction
Hepatic fibrosis is characterized by the accumulation of excessive amounts of extracellular matrix (ECM) components in the liver. This process is caused by the persistent liver damage and wound healing reaction induced by various chronic liver diseases including hepatitis B and C, biliary obstruction, alcohol abuse, non-alcoholic steatohepatitis (NASH) and several other etiologies [1, 2]. If unresolved, the fibrotic process results in organ failure, and eventually death after the development of cirrhosis. Currently, the only effective treatment for end stage liver disease is liver transplantation. Liver biopsy is considered as a gold-standard method for the assessment of liver fibrosis [1, 3, 4]. Histologic examination is useful in identifying the underlying cause of liver disease and assessing the necro-inflammatory grade and the stage of fibrosis. However, liver biopsy is an invasive procedure, with pain and many complications. Additionally, sampling error can occur, especially when small biopsies are analyzed. Histologic examination is prone to intra- and inter-observer variation and does not predict disease progression [1-4]. Therefore, developing simple and reliable noninvasive markers of liver fibrosis is an important goal in clinical hepatology and will facilitate the design of clinical trials.
Substantial progress has been made over the last ten to twenty years in understanding the pathophysiological mechanisms that underlie liver fibrosis. Activated HSCs have been commonly recognized as the principal cellular players that can promote synthesis and deposition of ECM proteins in response to accumulated levels of inflammatory signals derived from damaged parenchymal cells [5, 6]. Activated HSCs respond to and secrete a variety of pro-fibrogenic cytokines including TGFβ, CTGF, and PDGF, all of which are the potent cytokines resulting in liver fibrosis. Additionally, Kruppel-like factor 6 (KLF6) that acts as an immediate-early gene in hepatic fibrosis, has emerged as an alternative candidate for therapeutic interventions because it often activates TGFβ pathway and up-regulates other pro-fibrotic genes such as collagen I [7, 8]. Despite the fact that HSCs play a pivotal role in liver fibrosis, HCs are the dominant cells residing in the liver and the apoptosis and impaired proliferation of this cell type also have been commonly recognized as critical initiators of fibrosis by activating HSCs in persistent liver injury [1, 3]. Therefore, the inactivation of HSCs and inhibition of HCs apoptosis have been currently accepted for the resolution of liver fibrosis.
The number of human protein-coding genes is less than 2% of the whole genome sequence, whereas the vast majority of transcripts consist of the non-coding RNAs (ncRNAs), including microRNA (miRNA), small nucleolar RNA (snoRNA) and long non-coding RNA (lncRNA) [9, 10]. Recent reports have demonstrated that ncRNAs participate in modulating biological processes through regulating gene expression at the transcriptional and posttranscriptional levels [11-17]. With multiple and diverse targets, ncRNAs are involved in numerous biological functions and pathological processes, including development, proliferation, apoptosis, differentiation and carcinogenesis [18-21]. Moreover, emerging evidence is accumulating that disease cells release substantial amounts of RNAs, including miRNA, snoRNA and lncRNA, into the bloodstream that strongly resist RNases in the blood and are present at sufficient levels for quantitative analyses [22-26]. Circulating biomarkers are useful screening tools because of the ease and relatively non-invasive nature of their collection [24, 26, 27]. Therefore, it is interesting to identify circulating lncRNAs which are directly related to the hepatic fibrogenesis and can be used as diagnostic markers of liver fibrosis.
In the present study, we report an lncRNA ENSMUST00000158992, named as Scarna10, is up-regulated in the serum and liver tissue samples from patients with advanced hepatic fibrosis as well as in liver tissues of mice developing liver fibrosis. We show that SCARNA10 promotes liver fibrosis both in vitro and in vivo through inducing HCs apoptosis and HSCs activation. Mechanistically, we demonstrate that SCARNA10 functions as a novel positive regulator of TGFβ signaling in liver fibrogenesis by inhibiting the binding of PRC2 to the promoters of the genes involved in TGFβ pathway, thus promoting the transcription of these genes. All these data suggest that SCARNA10 may not only be a therapeutic target but also may have potential as a biomarker to monitor liver fibrosis in humans.
Materials and methods
Study population
In total, 53 human fibrotic liver tissues and 12 human healthy liver tissues from patients with hepatic haemangioma were obtained from surgical resections without preoperative treatment at Tianjin Third Central Hospital (Tianjin, China). For histological scoring of liver fibrosis, we stained paraffin embedded 5 μm liver sections with Sirius red, hematoxylin and eosin (H&E) and Masson's trichrome. Hepatic fibrosis was scored (stages F0-F4) according to the METAVIR fibrosis staging system [28] by three hepatopathologists blinded to the study protocol and stratified as normal liver (F0), portal fibrosis (F1), few septal fibrosis (F2), advanced septal (bridging) fibrosis (F3) or cirrhosis (F4). In addition, we collected 38 serum samples from patients diagnosed for liver fibrosis and 48 serum samples from patients diagnosed for cirrhosis at Tianjin Third Central Hospital (Tianjin, China), and 35 matched blood donor volunteers recruited from the same hospital with no previous history of any disease. All subjects were of the same ethnicity. Clinical and pathological characteristics including age, gender, ALT, AST and etiologies were recorded and summarized in Table S1 and Table S2. The study has been approved by the local Ethical Committee of Tianjin Third Central Hospital (Tianjin, China). Written informed consent was obtained from each patient according to the policies of the committee. The study methodologies were conformed to the standards set by the Declaration of Helsinki.
Cell culture and antibodies
The non-tumorigenic mouse hepatocyte cell line AML12 was maintained in Dulbecco's modified Eagle's medium (DMEM, Invitrogen, Camarillo, CA) supplemented with 10% fetal bovine serum (FBS, Gibco, Gaithersburg, MD, USA), 1 × insulin-transferrin-sodium selenite media supplement (ITS; Sigma-Aldrich), dexamethasone (40 ng/ml), penicillin (100 U/ml) and streptomycin (100 μg/ml). The human hepatic stellate cell line LX-2 (obtained from Merck Millipore (Beijing, China)) and HEK293T, were cultured in DMEM supplemented with 10% FBS, penicillin (100 U/ml) and streptomycin (100 μg/ml). All cells were cultured at 37°C in an atmosphere containing 5% CO2. The antibodies for α-SMA (rabbit polyclonal, Abcam, ab5694), Collagen1 (rabbit polyclonal, Abcam, ab34710), TGFβ (rabbit polyclonal, Abcam, ab66043), MMP2 (rabbit monoclonal, Abcam, ab92536), PCNA (rabbit monoclonal, Cell Signaling Technology, 13110), Smad2/3 (rabbit polyclonal, Cell Signaling Technology, 5678s and rabbit monoclonal, Cell Signaling Technology, 8685), pSmad2/3 (rabbit monoclonal, Cell Signaling Technology, 8828), KLF6 (rabbit polyclonal, Santa Cruz, sc-7158), SUZ12 (rabbit polyclonal, Abcam, ab12073), EZH2 (rabbit polyclonal, Abcam, ab3748), H3K27 (mouse monoclonal, Abcam, ab6002), rabbit IgG (Millipore, PP64B), goat anti rabbit IgG (Invitrogen, Alexa Fluor 488), goat anti rabbit IgG (Invitrogen, Alexa Fluor 594) were purchased.
Lentivirus production and construction
Oligos encoding shRNA specific for Scarna10 and the negative control shRNA were ligated into pSUPER.retro.puro, and the fragment containing the H1 promoter and hairpin sequences was subcloned into the lentiviral shuttle pCCL.PPT.hPGK.GFP.Wpre (lenti-shScarna10 and lenti-NC). The full-length Scarna10 cDNA was sequentially amplified by PCR and ligated into the lentiviral shuttle pCCL.PPT.hPGK.IRES.eGFP/pre to generate the over-expression plasmid (LV-Scarna10 and the empty plasmid as the LV-Control). These plasmids were used to produce lentivirus in HEK-293T cells with the packaging plasmids pMD2.BSBG, pMDLg/pRRE and pRSV-REV. Infectious lentiviruses were harvested at 36 h and 60 h after transfection and filtered through 0.45 μm PVDF filters. Recombinant lentiviruses used in vivo were concentrated 100-fold by ultracentrifugation (2 h at 120,000 g). The virus-containing pellet was dissolved in PBS and injected in mice within 48 h. The primer sets used are shown in Table S3.
Animals' in vivo study
Animal protocols were approved by Tianjin Medical University Animal Care and Use Committee. The methods were carried out in accordance with the approved guidelines. All Balb/c mice aged at 8 weeks were obtained from Institute of Laboratory Animal Sciences, CAMS & PUMC (Beijing , China). After acclimatization for one week, the hepatic fibrosis mice model was produced by three injections of carbon tetrachloride (CCl4, Sigma-Aldrich, St. Louis, MO, USA) per week for 2 to 10 weeks or by bile duct ligation (BDL, 3-21 days). For CCl4-induced mouse liver fibrosis model, eighty Balb/c mice were randomly divided into two groups. Subsequently, the forty Balb/c mice were randomly divided into four groups: Mice were treated with olive oil in combination with injection of lenti-NC (NC; n = 10), CCl4 in combination with injection of lenti-NC (NC + CCl4, n = 10), oil in combination with injection of lenti-shScarna10 (shScarna10, n = 10) and CCl4 in combination with injection of lenti-shScarna10 (shScarna10 + CCl4, n = 10). Another forty Balb/c mice were also randomly divided into four groups: Mice were treated with oil in combination with injection of lenti-Control (LV-Control, n = 10), CCl4 in combination with injection of lenti-Control (LV-Control + CCl4, n = 10), oil in combination with injection of lenti-Scarna10 (LV-Scarna10, n = 10) and CCl4 in combination with injection of lenti-Scarna10 (LV-Scarna10 + CCl4, n = 10). The lentivirus was injected via the tail vein 2 weeks after the first injection of CCl4 (1 × 109 pfu/mouse) [29]. Mice in the NC + CCl4 group, the shScarna10 + CCl4 group, the LV-Control + CCl4 group and the LV-Scarna10 + CCl4 group, respectively, were administered 5% CCl4 (v/v) dissolved in olive oil (0.3 ml/kg body weight) thrice per week for additional 6 weeks via intraperitoneal (ip) injection after the lentivirus was injected. NC, shScarna10, LV-Control and LV-Scarna10 group animals were injected with an equivalent volume of olive oil. After treatment with CCl4 for 8 weeks, all of mice were sacrificed under anesthesia with 3% sodium pentobarbital (45 mg/kg, ip). For BDL-induced mouse liver fibrosis model, Sixty Balb/c mice were randomly divided into four groups: Mice were treated with sham operation in combination with injection of lenti-NC (NC, n = 15), BDL operation in combination with injection of lenti-NC (NC + BDL, n = 15), sham operation in combination with injection of lenti-shScarna10 (shScarna10, n = 15) and BDL operation in combination with injection of lenti-shScarna10 (shScarna10 + BDL, n = 15). The lentivirus was injected via the tail vein 1 day before the surgical operation (1 × 109 pfu/mouse) [29]. 21 days later, all of mice were sacrificed under anesthesia with 3% sodium pentobarbital (45 mg/kg, ip). Liver specimens and serums were obtained for analyses.
Isolation and culture of primary HSCs and HCs
Primary mouse HSCs and HCs were isolated by pronase/collagenase perfusion digestion followed by subsequent density gradient centrifugation, as previously described [30]. In brief, 40-week-old male Balb/c mice used in the study received human cares. Liver was initially in situ digested with 0.05% pronase E (Roche, Mannheim, Germany), 0.03% collagenase type IV (Sigma-Aldrich, St. Louis, MO, USA) and then further digested with collagenase type IV, pronase E and DNase I (Roche, Mannheim, Germany) solution at 37°C bath shaking for 20 minutes. Subsequently, HSCs were isolated from nonparenchymal cells using 8.2%, 12% and 18% Nycodenz solution (Sigma-Aldrich, St. Louis, MO, USA) at 1450 g and 4°C without brake for 22 minutes. In addition, the purity of the isolated population was tested by the characteristics of star-like shape, perinuclear lipid droplets and α-SMA staining. HCs were isolated from the 10-week-old Balb/c mice by in situ perfusion with 30 ml SC1 solution and 30 ml 0.05% Collagenase IV solution sequentially. HCs were then pelleted by centrifugation 50 g for 4 minutes three times. Cell viability was determined by the trypan blue exclusion method.
Nuclear-cytoplasmic fractionation
Cytoplasmic and nuclear RNA isolation were performed with PARIS™ Kit (Invitrogen, Grand Island, NY, USA) following the manufacturer's instruction. Briefly, the cells (LX-2, AML-12, primary HSCs, primary HCs) were digested to individual cell with trypsin that was subsequently inactivated with complete medium followed by centrifugation at 1100 rpm for 3min. The collected cells were washed twice with ice-cold PBS and lysed with 500 μl ice-cold cell fractionation buffer. Cells were gently re-suspended by vortex or pipetting and incubated on ice for 10 min. Centrifuge the samples 3 min at 500 g to separate the nuclear and cytoplasmic cell fractions. The supernatant was transferred to a fresh RNase-free tube. In addition, the remaining lysate was washed with ice-cold cell fractionation buffer and centrifuged at 500 g for 1 min. Then, add 450 μl of ice-cold cell disruption buffer to lyse the nuclei until the lysate is homogenous. Mix the lysate and the supernatant above with 2 × lysis/binding solution and add equal volume of 100% ethanol to the mixture followed by drawing the mixture through a filter cartridge. Wash the sample sequentially with 700 μl wash solution, 1.5 ml wash solution 2/3. The cytoplasmic and nuclear RNA was eluted with 40-60 μl of 95 °C elution solution. All fractionation steps were performed at 4 °C or on ice.
5' and 3' rapid amplification of cDNA ends (RACE)
We used the 5'-RACE and 3'-RACE analyses to determine the transcriptional initiation and termination sites of SCARNA10 using a SMARTer™ RACE cDNA Amplification Kit (Clontech, Palo Alto, CA) according to the manufacturer's instructions. In brief, RNA was isolated from mouse liver and LX-2 cells, and 3'- and 5'-RACE-Ready cDNA were synthesized using SMARTScribe Reverse Transcriptase. The obtained band was gel purified and cloned with the lineareized pRACE vector. The obtained band was then sequenced. The gene -specific primers used for the PCR of the RACE analysis are provided in Table S3.
Chromatin immunoprecipitation (ChIP)
ChIP assays were performed essentially as described previously [31, 32]. Briefly, LX-2 and AML12 cells were infected with shRNA-control or LV-shSCARNA10 for 72 h, or treated with or without TGFβ for 24 h, and seeded in cell cultures. Chromatin was cross-linked with 15 ml pre-heated 10% FBS media / 1% formaldehyde for 10 min at RT. Fixation was terminated by addition of glycine to final concentration of 0.125 M and incubated for 5 min at RT. The cells were washed twice with 1 × PBS and harvested in 670 μl SDS Buffer containing protease inhibitors (PMSF), and centrifuged at 1200 rpm followed by re-suspending with ice-cold IP Buffer (1 volume SDS Buffer and 0.5 volume Triton Dilution Buffer). Samples were sheared by sonication with a 5 sec / 15 sec cycle at power setting 30% for 40 times and centrifuged at 20,000 g for 30 min. The supernatant was transferred to new tubes and quantified the protein content from each sample by the BCATM Protein Assay Kit (Bio-Rad Laboratories, Hercules, CA, USA) using BSA as standard. Next, dilute samples with IP Buffer to a desired concentration and remove 10 μl (1%) of lysate used as total control. Each single IP was performed in 1 ml volume. Lysates were incubated with primary antibodies (5 μg SUZ12, 5 μg EZH2, 5 μg H3K27 or 5 μg IgG) overnight rotating at 4 °C. 50 μl beads was added and incubated on a rotating wheel 4 h at 4 °C. Next, immunoprecipitated complexes were sequentially washed two times with wash buffer (1 ml 150 mM wash buffer and 500 mM wash buffer) followed by adding 120 μl of 1% SDS, 0.1 M NaHCO3 and incubating overnight at 65°C. Finally, DNA was purified with PCR purification kit (Qiagen) and used as templates for PCR reactions. Primers used for PCR in ChIP experiments are described in Table S3.
RNA immunoprecipitation (RIP)
RIP was performed using the Magna RIP RNA-Binding Protein Immunoprecipitation Kit (Millipore, Bedford, MA) according to the manufacturer's instructions. Briefly, the single cell suspensions isolated from mouse liver and 90% confluent LX-2 cells were sequentially washed twice with ice-cold PBS, harvested into 15 ml conical tubes with 10 ml ice-cold PBS and collected by centrifugation at 1500 rpm for 5 min at 4 °C. Subsequently, re-suspended the cell pellet in an equal pellet volume of complete RIP lysis buffer and incubated on ice for 5 min, and centrifuged at 14,000 rpm for 10 min at 4 °C, transferred 10 μl supernatant to new tubes as input and another 100 μl supernatant to the beads-antibody complex followed by adding 900 μl RIP buffer for each test. The antibodies used for RIP were 5 μg SUZ12, 5 μg EZH2 or 5 μg IgG, and all the tubes were incubated with rotating overnight at 4 °C. Next, centrifuge the immunoprecipitation tubes briefly and discard the supernatant using a magnetic separator. The beads were then washed and purified the RNA. The purified RNAs were detected by qRT-PCR. The gene-specific primers used for detecting SCARNA10 are presented in Table S3.
Histology and immunohistochemistry; Liver enzyme measurement; Hydroxyproline assay; Cell proliferation assay; EdU incorporation assay; Apoptosis assay; Confocal microscopy; Quantitative real-time polymerase chain reaction; Western blot analysis, RNA interference.
These methods are detailed in Supplementary Materials and Methods.
Statistical analysis
Data were expressed as mean ± SD. All the statistical analyses were performed with the SPSS 13.0 (IBM, Armonk, NY, USA). Statistical analyses were performed using either Student's t-test (two-group comparison) or one-way analysis of variance (more than two groups) followed by post hoc comparison, and differences with p < 0.05 were considered significantly.
Results
SCARNA10 is up-regulated in human and mouse liver fibrosis
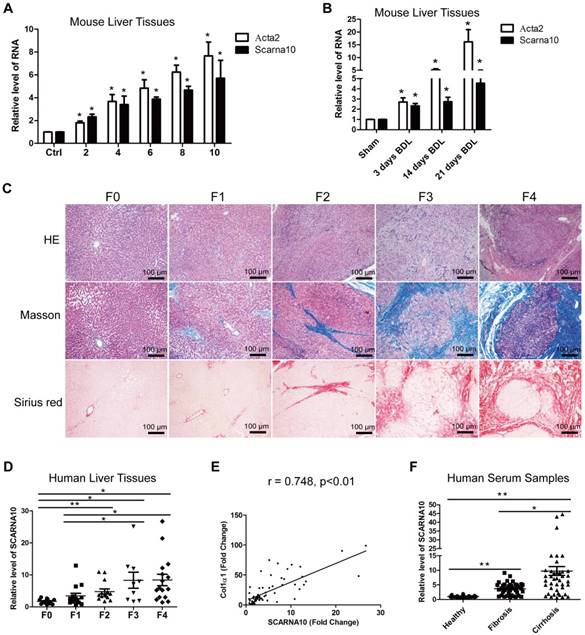
We previously identified systemic variations in the expression of lncRNAs between mouse fibrotic and normal livers using a microarray analysis [33]. From that study, we noted that lncRNA ENSMUST00000158992, named as Scarna10, was remarkably up-regulated in liver fibrosis according to the microarray data. (The microarray data discussed in that article have been deposited in NCBI Gene Expression Omnibus and are accessible through GEO Series accession number GSE80601; http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE80601.). The results in the array analysis were confirmed by qRT-PCR. We performed 5'- and 3'- rapid amplification of cDNA ends (RACE) assay in mouse liver tissues and found Scarna10 is a 325-nucleotide transcript without poly (A) tail, consistent with our microarray analysis (Figure S1A). Moreover, sequence homology searches revealed a clear orthologous SCARNA10 transcript in the human genome. RACE assay in immortalized human HSC LX-2 cells showed that the sequence of SCARNA10 is consistent with the NCBI database (Figure S1B). Cell fractionation followed by qRT-PCR showed that SCARNA10 is mainly located in the nucleus of HCs and HSCs (Figure S1C). Scarna10 expression was then measured in total RNA extracts from fibrotic livers of Balb/c mice treated with CCl4 for various weeks, and the results showed that the transcripts of Scarna10 and α-SMA (Acta2) are gradually increased with persistent injury (Figure 1A). The up-regulation of Scarna10 during fibrosis progression was also observed in another liver fibrosis mouse model of BDL (Figure 1B). To correlate these findings with data from patients, 12 healthy liver tissues and 53 fibrotic liver tissues were obtained from surgical resections without preoperative treatment and divided into five stages according to the HE, Masson and Sirius red staining by three pathologists independently (Figure 1C). The expression of SCARNA10 in patients with fibrosis/cirrhosis was much higher and positively correlated with the level of Col1α1 but not with α-SMA, ALT and AST (Figure 1D, E and Figure S1D-H). In addition, we also isolated total RNAs from the serum of 38 patients with liver fibrosis and 45 patients with liver cirrhosis and compared the level of SCARNA10 in these patients with that of 35 healthy volunteers. Our results indicated that the level of SCARNA10 was increased in the serum from fibrotic and cirrhotic patients, and the patients with advanced liver cirrhosis displayed significantly higher SCARNA10 level than those with liver fibrosis (Figure 1F). All these data suggested that SCARNA10 might have potential as a biomarker to monitor liver fibrosis.
SCARNA10 is up-regulated in human and mouse fibrotic liver. (A) qRT-PCR analysis of the expression of Scarna10 and Acta2 (α-SMA) in livers from mice treated with CCl4 for 2, 4, 6, 8 or 10 weeks. (B) qRT-PCR analysis of the expression of Scarna10 and Acta2 in livers from mice that underwent BDL for 3, 14 or 21 days. (C) Human liver tissues were divided into five stages according to the HE, Masson and Sirius red staining. (D) qRT-PCR analysis of the transcript of SCARNA10 in liver samples of healthy, fibrotic and cirrhotic patients. (E) The correlation between SCARNA10 level and Col1α1 expression in patient liver tissues was assessed using Pearson' correlation analysis, n=65. (F) qRT-PCR analysis of the SCARNA10 transcript in serum samples of healthy, fibrotic and cirrhotic patients. The data are expressed as the mean ± SD for at least triplicate experiments, *p< 0.05.
Knockdown of Scarna10 suppresses, while over-expression of Scarna10 aggravates CCl4-induced mouse liver fibrosis in vivo
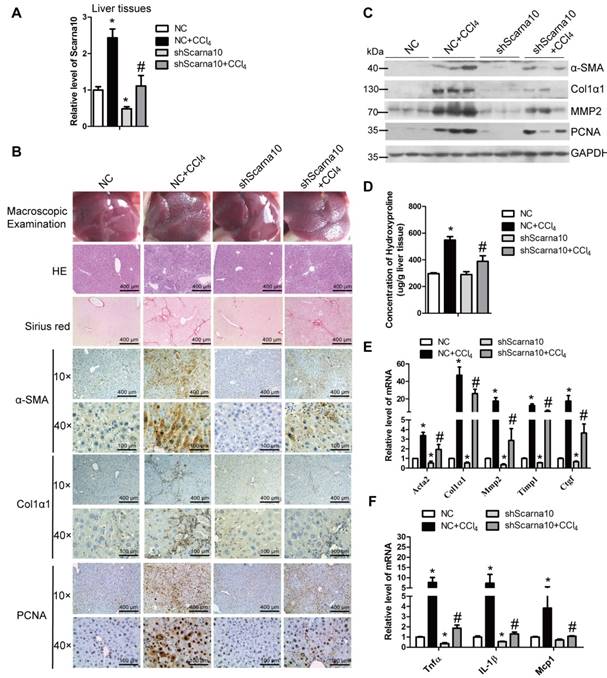
To investigate the role of Scarna10 in the progression of liver fibrosis in vivo, lenti-shScarna10 or lenti-NC was intravenously injected into CCl4-treated or oil-treated mice via the tail vein 2 weeks after the first injection of CCl4. After 8 weeks of CCl4 treatment, Scarna10 silencing was confirmed by qRT-PCR in whole liver extracts (Figure 2A), and the CCl4 group mice infected with lenti-NC developed severe liver fibrosis. However, administration of lenti-shScarna10 greatly reduced CCl4-induced liver fibrosis, as demonstrated by macroscopic examination, H&E staining, Sirius red staining, IHC for α-SMA, Collagen1 and PCNA, and western blot for α-SMA, Collagen1, MMP2 and PCNA (Figure 2B, C). Quantification of liver hydroxyproline content and serum level of ALT and AST in lenti-shScarna10-infected mice were also significantly decreased in comparison with the CCl4 group mice infected with lenti-NC (Figure 2D and Table S4). In addition, knockdown of Scarna10 resulted in significant reduced expression of pro-fibrogenic and pro-inflammation genes, simultaneously the CCl4-induced up-regulation of these genes were also greatly inhibited (Figure 2E, F). On the other hand, to test whether over-expression of Scarna10 would aggravate fibrosis, lenti-Scarna10 that can effectively express Scarna10 or lenti-Control was intravenously injected into CCl4-treated or oil-treated mice via the tail vein 2 weeks after the first injection of CCl4. After 8 weeks of CCl4 treatment Scarna10 was over-expressed (Figure S2A), and the CCl4 group mice infected with lenti-Control developed severe liver fibrosis. However, administration of lenti-Scarna10 greatly enhanced the severity of liver fibrosis, as demonstrated by macroscopic examination, H&E staining, Sirius red staining, IHC for α-SMA, Collagen1 and PCNA, western blot for α-SMA, MMP2, Collagen1 and PCNA, quantification of liver hydroxyproline content and serum levels of ALT and AST, and the expression of the pro-fibrogenic and pro-inflammation genes (Figure S2B-F and Table S5). Taken together, these results indicated that knockdown of Scarna10 suppresses, while over-expression of Scarna10 aggravates the progression of liver fibrosis in vivo.
Knockdown of Scarna10 suppresses CCl4-induced liver fibrosis in vivo. Mice were treated with oil in combination with injection of lenti-NC (Negative Control, n = 10), or CCl4 in combination with injection of lenti-NC (NC + CCl4, n = 10), or oil in combination with injection of lenti-shScarna10 (shScarna10, n = 10), or CCl4 in combination with injection of lenti-shScarna10 (shScarna10 + CCl4, n = 10). (A) The expression of Scarna10 in liver tissues of each group was examined by qRT-PCR. (B) Liver fibrosis was evaluated by macroscopic examination, H&E staining, Sirius red staining and IHC for α-SMA, Collagen1 and PCNA; scale bar = 400 μm for 10× and 100 μm for 40×. (C) The protein level of α-SMA, Collagen1, MMP2 and PCNA was determined by western blot. GAPDH was used as an internal control. (D) Quantification of hepatic hydroxyproline content. The data are expressed as hydroxyproline (μg)/liver wet weight (g). (E, F) The mRNA level of hepatic pro-fibrogenic genes (Acta2, Col1α1, MMP2, TIMP1 and CTGF) (E) and pro-inflammation genes (TNFα, IL-1β and MCP1) (F) was determined by qRT-PCR. The data are expressed as the mean ± SD for at least triplicate experiments, */#p<0.05. *p<0.05 for vs NC. #p<0.05 for vs NC+ CCl4.
Knockdown of Scarna10 suppresses BDL-induced mouse liver fibrosis in vivo
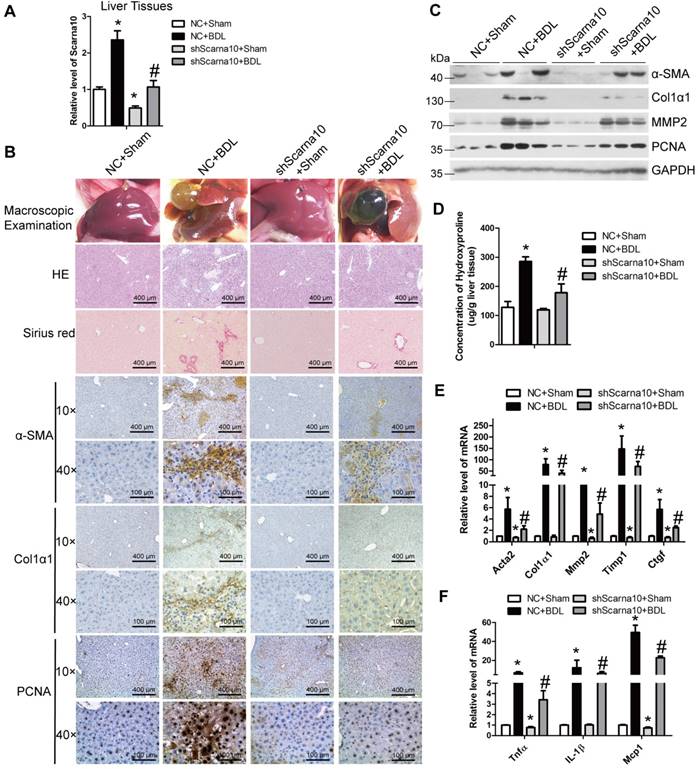
To exclude the possibility that Scarna10 alters the metabolism or toxicity of CCl4 rather than by altering cell responses, we further confirmed the results in a BDL-induced mouse liver fibrosis model. lenti-shScarna10 or lenti-NC was intravenously injected into BDL mice via the tail vein 1 day before the surgical operation. Scarna10 silencing was confirmed by qRT-PCR (Figure 3A), and the BDL group mice infected with lenti-NC developed severe liver fibrosis. However, administration of lenti-shScarna10 greatly reduced BDL-induced liver fibrosis, as demonstrated by H&E staining, Sirius red staining, IHC for α-SMA, Collagen1 and PCNA, and western blot for α-SMA, MMP2, Collagen1 and PCNA (Figure 3B, C). Quantification of liver hydroxyproline content and serum level of ALT and AST in lenti-shScarna10-infected mice were also significantly decreased in comparison with the BDL mice infected with lenti-NC (Figure 3D and Table S6). In addition, lentivirus-mediated knockdown of Scarna10 resulted in significantly reduced expression of pro-fibrogenic and pro-inflammation genes. The BDL-induced up-regulation of these genes was also greatly repressed (Figure 3E, F). Taken together, these results further confirmed that knockdown of Scarna10 represses the progression of liver fibrosis in vivo.
Knockdown of Scarna10 suppresses BDL-induced liver fibrosis in vivo. Mice were treated with sham operation in combination with injection of lenti-NC (NC + Sham, n = 15), BDL operation in combination with injection of lenti-NC (NC + BDL, n = 15), sham operation in combination with injection of lenti-shScarna10 (shScarna10 + Sham, n = 15) and BDL operation in combination with injection of lenti-shScarna10 (shScarna10 + BDL, n = 15). (A) The expression of Scarna10 in liver tissues of each group was examined by qRT-PCR. (B) Liver fibrosis was evaluated by macroscopic examination, H&E staining, Sirius red staining and IHC for α-SMA, Collagen1 and PCNA; scale bar = 400 μm for 10× and 100 μm for 40×. (C) The protein level of α-SMA, MMP2, Collagen1 and PCNA was determined by western blot. GAPDH was used as an internal control. (D) Quantification of hepatic hydroxyproline content. The data are expressed as hydroxyproline (μg)/liver wet weight (g). (E, F) The mRNA level of hepatic pro-fibrogenic genes (Acta2, Col1α1, MMP2, TIMP1 and CTGF) (E) and pro-inflammation genes (TNFα, IL-1β and MCP1) (F) was determined by qRT-PCR. The data are expressed as the mean ± SD for at least triplicate experiments, */#p<0.05. *p<0.05 for vs NC. #p<0.05 for vs NC+ CCl4.
Scarna10 promotes the apoptosis of HCs
HCs are the dominant cells residing in the liver and HCs apoptosis has been commonly recognized as critical initiators of fibrosis in persistent liver injury [1, 3, 4]. The in vivo data showed that knockdown of Scarna10 attenuates, while over-expression of Scarna10 aggravates HCs damages (Table S4-6). Therefore, we isolated primary HCs from livers of mice treated with CCl4 or control for 6 weeks. Scarna10 showed a clear up-regulation in HCs from fibrotic livers compared with control, correlating with an up-regulation of Col1α1, Tnfα and Mcp1 (Figure 4A). In addition, stimulation of primary HCs and AML12 cells with recombinant TGFβ, recognized as a potent apoptosis inducer, resulted in a significant increased level of Scarna10 (Figure 4B, C).
Scarna10 promotes the apoptosis of HCs. (A) Primary HCs were isolated from livers of Balb/c mice treated for 6 weeks with CCl4 or oil, and the RNA level of Scarna10, Col1α1, TNFα and MCP1 was determined by qRT-PCR. (B, C) Primary HCs (B) and AML12 cells (C) were stimulated with TGF-β for 24 h and the expression of Scarna10, Col1α1, TNFα and CTGF was determined by qRT-PCR. (D, E) Primary HCs were infected with lentivirus-mediated shScarna10 for 72 h and further treated with 10 ng/ml TGF-β for additional 24 h. The expression of Scarna10, pro-fibrogenic genes (Acta2, Col1α1, MMP2, TIMP1 and CTGF), pro-inflammation genes (TNFα, IL-1β and MCP1) and pro-apoptosis genes (Bax and Bad) was detected by qRT-PCR (D). The protein level of α-SMA and Col1α1 was detected by western blot. GAPDH was used as an internal control (E). (F, G) Primary HCs were infected with lentivirus-mediated Scarna10 for 72 h and further treated with 10 ng/ml TGF-β for additional 24 h. The RNA level of Scarna10, pro-fibrogenic, pro-inflammation and pro-apoptosis genes was detected in AML12 cells infected with lenti-Scarna10 or lenti-Control by qRT-PCR (F). The protein level of α-SMA and Col1α1 was detected by western blot. GAPDH was used as an internal control (G). (H) AML12 cells were infected with lentivirus-mediated shScarna10 for 72 h and further treated with 10 ng/ml TGF-β for additional 24 h. Cell apoptosis was determined by FACS analysis. The data are shown as the mean ± SD for at least triplicate experiments, */#p<0.05. *p<0.05 for vs shRNA-control or LV-Control. #p<0.05 for vs shRNA-control + TGF-β or LV-Control + TGF-β.
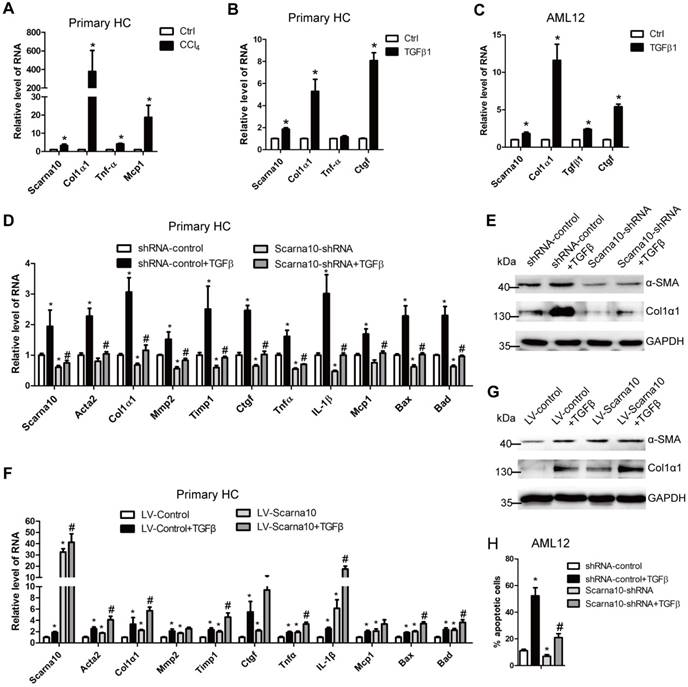
Since HCs produce and release ECM and inflammation factors with the exception of apoptosis, all of which can be induced by TGF-β in the pathogenesis of liver fibrosis [7], we investigated the role of Scarna10 in HCs, using lentivirus vector of Scarna10-shRNA to knock down its expression in primary HCs and AML12 cells and subsequently treated with TGFβ. The results showed that the expression of the pro-fibrogenic genes α-SMA, Col1α1, MMP2, TIMP1 and CTGF, the pro-inflammation genes TNFα, IL-1β and MCP1, and the pro-apoptosis genes BAX and BAD was up-regulated upon TGFβ treatment. However, knockdown of Scarna10 abrogates TGFβ-induced up-regulation of these genes in primary HCs and AML12 cells (Figure 4D, E and Figure S3A-C). On the other hand, over-expression of Scarna10 increases the expression of the pro-fibrogenic, pro-inflammation and pro-apoptosis genes, and aggravates TGFβ-induced up-regulation of these genes in primary HCs and AML12 cells (Figure 4F, G and Figure S3D-E). Flow cytometry (FACS) analysis further confirmed that TGFβ stimulation dramatically increases AML12 apoptosis, however, Scarna10 silencing significantly suppresses TGFβ-induced apoptosis (Figure 4H). Consistently, the expression of the pro-fibrogenic, pro-inflammation and pro-apoptosis genes in the HCs isolated from lenti-shScarna10-infected mice exhibited a marked decrease, while demonstrated a profound increase in those from lenti-Scarna10-infected mice, in comparison with the lenti-NC/Control-infected mice (Figure S4A-C). Taken together, these results clearly suggested that Scarna10 promotes the apoptosis of HCs.
SCARNA10 promotes HSCs activation and proliferation
Activated HSCs have been commonly recognized as the principal cellular players promoting synthesis and deposition of ECM proteins. The expression of Scarna10 showed a clear up-regulation in HSCs from fibrotic livers compared with control, correlating with an up-regulation of α-SMA and Col1α1 (Figure S5A). Furthermore, primary HSCs at day 3, day 7 and day 14 were also collected to evaluate Scarna10 expression during HSC activation. As shown in Figure S5B, activated HSCs exhibited significant up-regulation of the expression of Scarna10, α-SMA and Col1α1 at day 14, compared with day 3. In addition, stimulation of primary HSCs with recombinant TGFβ resulted in a significantly increased level of Scarna10, correlating with an enhanced expression of α-SMA and Col1α1 in these cells (Figure S5C). All these data suggested that up-regulation of Scarna10 might have important roles in HSCs.
Next, we investigated the effect of SCARNA10 on the activation and proliferation of LX-2 cells. The cells infected with SCARNA10-shRNA virus showed a decreased mRNA level of the pro-fibrogenic genes α-SMA, Col1α1, Col1α2, Col3α1, Col4α5, MMP2, TIMP1 and the pro-proliferation genes CYCLIND1, PCNA and CTGF, compared with control. Moreover, knockdown of SCARNA10 dramatically decreased TGFβ-induced up-regulation of these genes (Figure S6A). Similarly, the protein level of α-SMA, Col1α1, MMP2 and PCNA was also markedly decreased by SCARNA10-shRNA infection (Figure S6B). Furthermore, confocal microscopy demonstrated that the expression of α-SMA, Col1α1 and MMP2 was decreased when LX-2 cells were transfected with SCARNA10 siRNA. Simultaneously, the TGFβ-induced up-regulation of α-SMA, Col1α1 and MMP2 was also greatly inhibited (Figure S6C). In addition, the results of MTT and FACS revealed that cell proliferation is significantly suppressed in SCARNA10 down-regulated LX-2 cells (Figure S6D, E). In order to further investigate the roles of SCARNA10 in regulating HSCs activation and proliferation, we over-expressed SCARNA10 in LX-2 cells. Forced expression of SCARNA10 obviously increases the expression of the pro-fibrogenic and pro-proliferation genes, and aggravates TGFβ-induced up-regulation of these genes assessed by qRT-PCR, western blot and confocal microscopy (Figure S7A-C). Moreover, the results of MTT and FACS revealed that cell proliferation is significantly increased in SCARNA10 up-regulated LX-2 cells (Figure S7D, E). These data suggested that SCARNA10 promotes LX-2 cells activation and proliferation.
To further examine whether the results observed in LX-2 cells could be confirmed in primary HSCs. We used lentivirus vector of Scarna10-shRNA to knock down its expression at early stage of primary HSCs in day 2. Recombinant TGFβ was then added to day 5 HSCs infected with Scarna10-shRNA or control virus, and total RNA was extracted for detection of the expression of the fibrosis-related and proliferation-related genes. We found that cells infected with Scarna10-shRNA expressed a lower level of α-SMA, Col1α1, Col1α2, Col4α5, Mmp2/10, Timp1, CyclinD1, Pcna and Ki67, compared with the control cells (Figure 5A). Moreover, knockdown of Scarna10 dramatically decreases TGFβ-induced up-regulation of these genes in primary HSCs (Figure 5A). These findings were further confirmed by western blot (Figure 5B). Furthermore, immunofluorescence results demonstrated that the expression of both α-SMA and Col1α1 was decreased when HSCs were transfected with Scarna10 siRNA. Simultaneously, the TGFβ-induced up-regulation of α-SMA and Col1α1 was also greatly inhibited (Figure 5C). In addition, the results of FACS assay revealed that cell proliferation is significantly suppressed in Scarna10 down-regulated HSCs, while drastically increased in Scarna10 up-regulated HSCs (Figure 5D). However, forced expression of Scarna10 obviously increases the expression of α-SMA, Col1α1, MMP2, TIMP1 and PCNA, and aggravates TGFβ-induced up-regulation of these genes assessed by qRT-PCR (Figure 5E) and western blot (Figure 5F). Consistently, the expression of these pro-fibrogenic and pro-proliferation genes in the HSCs isolated from lenti-shScarna10-infected mice showed a marked decrease, while exhibited a profound increase in those from lenti-Scarna10-infected mice, in comparison with the lenti-NC/Control -infected mice (Figure S8A-E).
Scarna10 promotes the activation and proliferation of primary HSCs. (A, B) Primary HSCs were infected with lentivirus-mediated shSCARNA10 for 72 h and further treated with 10 ng/ml TGF-β for additional 24 h. The expression of Scarna10, Acta2, Col1α1, Col1α2, Col4α5, MMP2, MMP10, TIMP1, CyclinD1, PCNA and Ki67 was detected by qRT-PCR (A). The protein level of α-SMA, Col1α1, MMP2 and PCNA was detected by western blot. GAPDH was used as an internal control (B). (C) Primary HSCs were transfected with siRNA for SCARNA10 for 48 h and further treated with 10 ng/ml TGF-β for additional 24 h. The expression of α-SMA and Col1α1 was determined by immunofluorescence. DAPI stained nuclei blue; scale bar = 50μm. (D) The numbers refer to the percentages of EdU-positive cells measured by flow cytometry. (E, F) Primary HSCs were infected with lentivirus-mediated SCARNA10 for 72 h and further treated with 10 ng/ml TGF-β for additional 24 h. The RNA level of Scarna10, Acta2, Col1α1, Col1α2, Col4α5, MMP2, MMP10, TIMP1, CyclinD1, PCNA and Ki67 was detected by qRT-PCR (E). The protein level of α-SMA, Col1α1, MMP2 and PCNA was detected by western blot. GAPDH was used as an internal control (F). The data are expressed as the mean ± SD for at least triplicate experiments, */#p<0.05. *p<0.05 for vs shRNA-control or LV-Control. #p<0.05 for vs shRNA-control + TGF-β or LV-Control + TGF-β.
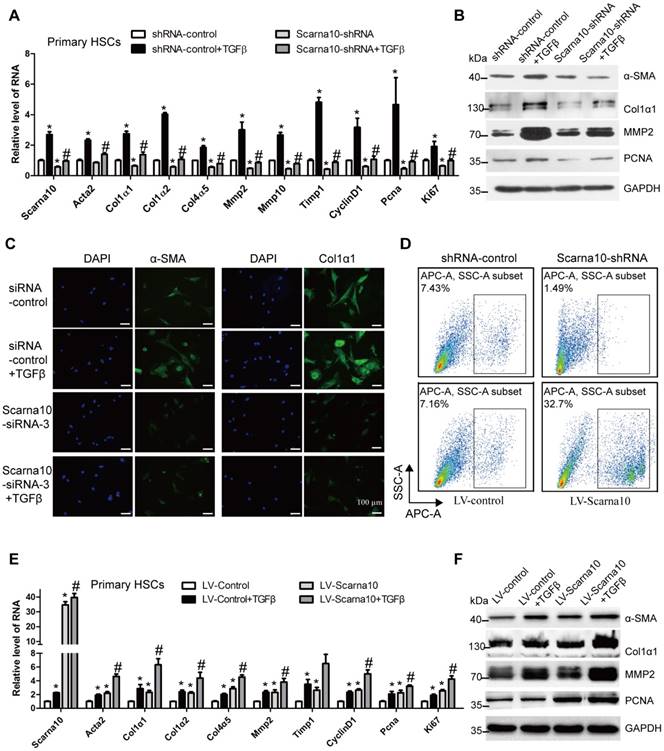
Taken together, these results demonstrated that Scarna10 promotes the activation and proliferation of HSCs.
SCARNA10 promotes liver fibrogenesis via TGFβ signaling
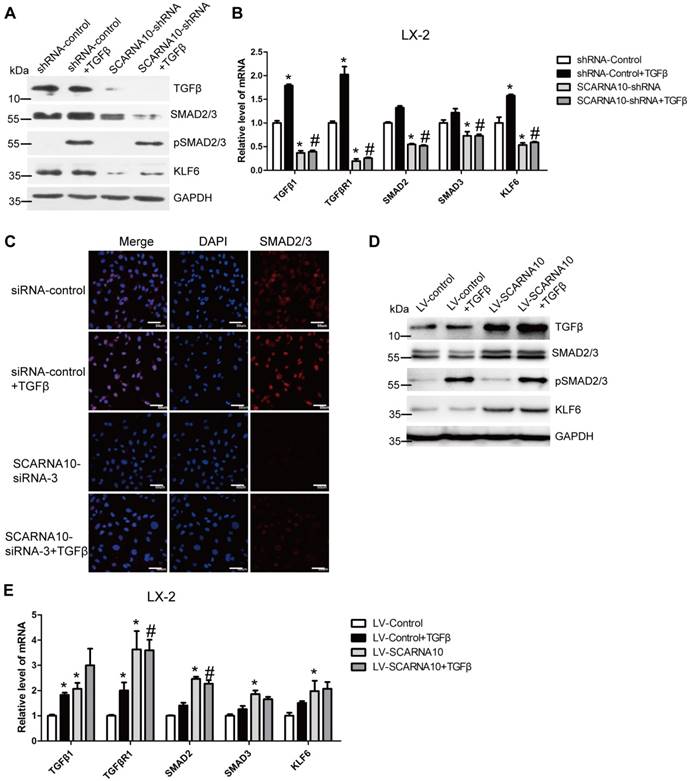
As TGFβ pathway is one of the well-investigated signaling cascades in liver fibrosis and Smad2/3 are involved in this signaling [34], we firstly investigated whether SCARNA10 regulates the TGFβ-Smad2/3 signaling. LX-2 cells were infected with lenti-SCARNA10-shRNA or control virus and followed by TGFβ treatment. The results revealed that SCARNA10 silencing only slightly inhibits TGFβ-induced SMAD2/3 phosphorylation, however, the protein level of total SMAD2/3 and TGFβ was decreased in lenti-SCARNA10-shRNA-infected LX-2 cells (Figure 6A). Additionally, KLF6, a transcription factor which up-regulates TGFβ, TGFβRI, TGFβRII and collagen I, was also down-regulated by knockdown of SCARNA10 (Figure 6A). Furthermore, qRT-PCR results showed that SCARNA10 silencing down-regulates the mRNA level of TGFβ, TGFβRI, SMAD2, SMAD3 and KLF6 (Figure 6B). Consistently, the results of confocal microscopy in LX-2 cells revealed that knockdown of SCARNA10 dramatically decreased the expression of SMAD2/3 (Figure 6C). On the other hand, over-expression of SCARNA10 increases the expression of TGFβ, TGFβRI, SMAD2, SMAD3 and KLF6 in LX-2 cells as shown by qRT-PCR and western blot (Figure 6D, E). Similar results were observed in primary HSCs (Figure S9A-D), AML12 and primary HCs (Figure S10A-H). To confirm the results that SCARNA10 increases the expression of TGFβ, SMAD2, SMAD3 and KLF6 in vitro, we validated the differential expression of these genes in livers from lenti-shScarna10, lenti-Scarna10 or lenti-NC/Control -infected mice treated with or without CCl4 or BDL. As shown in Figure S11A-C, Figure S12A-C and Figure S13A-C, lentivirus-mediated knockdown of Scarna10 resulted in reduced expression of TGFβ, Smad2, Smad3 and KLF6, and inhibited CCl4- or BDL-induced up-regulation of these genes, whereas over-expression of Scarna10 enhanced the expression of TGFβ, Smad2, Smad3 and KLF6, and aggravated CCl4-induced up-regulation of these genes. Taken together, all these data suggested that SCARNA10 promotes liver fibrogenesis through TGFβ signaling.
SCARNA10 promotes liver fibrogenesis through TGF-β signaling. LX-2 cells were infected with lentivirus-mediated shSCARNA10 for 72 h and further treated with 10 ng/ml TGF-β for additional 24 h. The protein level of pSmad2/3, total Smad2/3, TGF-β and KLF6 was detected by western blot. GAPDH was used as an internal control (A). The mRNA level of TGF-β, TGF-βR1, SMAD2, SMAD3 and KLF6 was determined by qRT-PCR (B). (C) LX-2 cells were transfected with siRNA for SCARNA10 for 48 h and further treated with 10 ng/ml TGF-β for additional 24 h. The expression and location of total Smad2/3 were determined by confocal microscopy. DAPI stained nuclei blue; scale bar = 50μm. (D, E) LX-2 cells were infected with lentivirus-mediated SCARNA10 for 72 h and further treated with 10 ng/ml TGF-β for additional 24 h. The protein level of pSmad2/3, total Smad2/3, TGF-β and KLF6 was detected by western blot. GAPDH was used as an internal control (D). The mRNA level of TGF-β, TGF-βR1, SMAD2, SMAD3 and KLF6 was determined by qRT-PCR (E). The data are expressed as the mean ± SD for at least triplicate experiments, *,#p<0.05. *p<0.05 for vs shRNA-control or LV-Control. #p<0.05 for vs shRNA-control + TGF-β or LV-Control + TGF-β.
SCARNA10 interacts PRC2 and releases PRC2-repressed genes expression
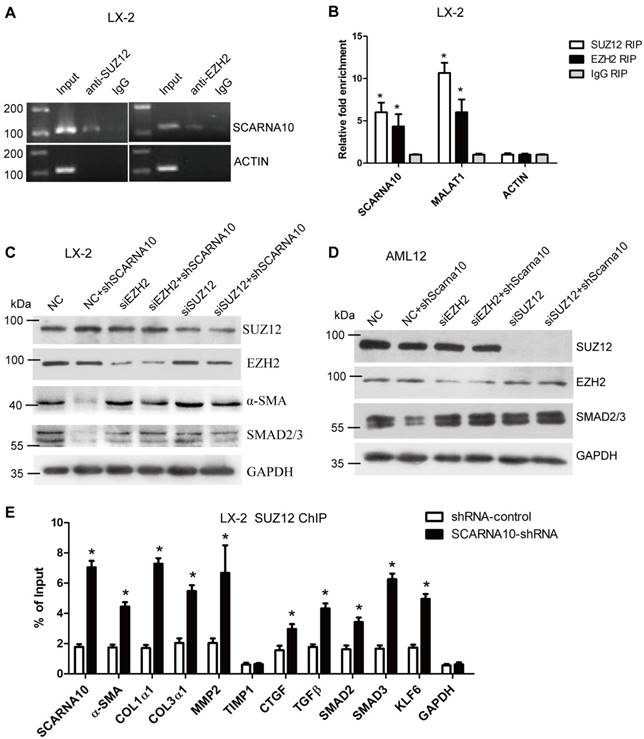
Recent studies have demonstrated that a significant number of lncRNAs involved in transcription regulation often function through binding chromatin modifying enzymes to promote epigenetic activation or silencing of gene expression [19, 35-37]. It has been reported that twenty percent of all human lncRNAs physically associate with the PRC2 [18], thus, we hypothesized that SCARNA10 might affect gene expression in such a manner. To test this, we performed RIP assay in LX-2 cells to pull down endogenous RNAs associated with SUZ12 or EZH2, an important subunit of PRC2. The results showed that a significant enrichment of both SCARNA10 and lnc-MALAT1, a positive control lncRNA, but no enrichment of β-actin with the SUZ12 and EZH2, compared with control (Figure 7A, B), suggesting that SCARNA10 physically interacts with PRC2. Consistently, these findings were also confirmed in mouse liver tissue (Figure S14A). To further examine whether SCARNA10 increases the expression of α-SMA and Smad2/3 via binding with PRC2, we knocked down SUZ12 and EZH2 in lenti-SCARNA10-shRNA -infected LX-2 and AML12 cells. Immunoblot showed that the reduced protein level of α-SMA and Smad2/3 by silencing SCARNA10 was reversed when SUZ12 or EZH2 expression was simultaneously knocked down (Figure 7C, D), confirming that SCARNA10 interacts with PRC2 to regulate the expression of α-SMA and Smad2/3. Additionally, to address whether SCARNA10 promotes the transcription of these target genes through inhibiting the binding of PRC2 to the promoters, ChIP analysis was performed in LX-2 and AML12 cells infected with lenti-SCARNA10-shRNA or control virus. The data demonstrated that knockdown of SCARNA10 increases the binding of SUZ12, EZH2 and H3K27me3 level across the promoters of target genes, including α-SMA, Col1α1, Col1α2, TGFβ, MMP2, SMAD2, SMAD3, KLF6 and SCARNA10 itself (Figure 7E, Figure S14B, C and Figure S15A-C). However, we observed that knockdown of SCARNA10 increases the binding of SUZ12 but not EZH2 and H3K27me3 levels across the TIMP1 and CTGF promoters in LX-2 cells, indicating that SCARNA10 may regulate TIMP1 and CTGF expression in other ways. Additionally, ChIP results showed that TGFβ decreases the binding of SUZ12 across the SCARNA10 promoters (Figure S16A, B). Taken together, our data demonstrated that SCARNA10 interacts with PRC2 and blocks the binding of PRC2 to the promoters of the target genes.
SCARNA10 interacts with PRC2 and releases PRC2-repressed pro-fibrogenic genes expression. (A, B) qRT-PCR detection of SCARNA10, MALAT1 and ACTIN retrieved by SUZ12- or EZH2-specific antibody compared with IgG in the RIP assay with LX-2 cells. (C, D) The protein level of α-SMA and Smad2/3 in SCARNA10 down-regulated LX-2 (C) and AML12 (D) cells simultaneously transfected with siEZH2 or siSUZ12 was determined by western blot. GAPDH was used as an internal control. (E) LX-2 cells were infected with lenti-shSCARNA10 or shRNA-control, and ChIP analyses were performed on indicated genes promoter regions using anti-SUZ12 antibody. Enrichment was shown relative to input. The data are expressed as the mean ± SD for at least triplicate experiments, *p< 0.05.
Discussion
Recent improvements in genome-wide surveys have revealed that less than 2% of the human genome encodes proteins and the majority of the human genome transcripts are non-coding RNAs, which participate in modulating biological processes through regulating gene expression at the transcriptional and posttranscriptional levels according to the cellular location [19]. In this study, we provided evidence for a functional role of Scarna10 in murine and human liver fibrosis. Based on our findings, we reported that Scarna10 promotes liver fibrosis both in vitro and in vivo through inducing HCs apoptosis and HSCs activation. Furthermore, our results demonstrated that Scarna10 functions as a novel positive regulator of TGFβ signaling in liver fibrogenesis by inhibiting the binding of PRC2 to the promoter of the genes involved in TGFβ pathway, thus promoting the transcription of these genes. In addition, we also found that the expression of SCARNA10 is increased in the serum and liver tissue from patients with advanced hepatic fibrosis as well as in liver tissues of mice developing liver fibrosis, and the level of SCARNA10 is associated with the stage of liver fibrosis. All these data supported our conclusion that SCARNA10 is not only a potential diagnosis marker, but also a possible therapeutic target against liver fibrosis.
Liver fibrosis is a complicated pathological process, driven at the cellular level by activation of quiescent HSCs and characterized by the sustained induction of a fibrotic gene program [2]. Development of liver fibrosis is orchestrated by a network of fibrogenic and inflammatory signaling pathways, including TGFβ, Wnt, Notch, and Hedgehog pathways [1, 3, 4]. Within these, the TGFβ signaling pathway is probably the most prominent direct inducer of collagen transcription. Thus, researchers are trying to block the TGFβ signal in order to suppress liver fibrosis. In addition, KLF6, which was initially characterized as an immediate-early gene in hepatic fibrosis, has emerged as an alternative candidate for therapeutic interventions because it could directly up-regulate the pro-fibrotic genes such as TGFβ, TGFβRI, TGFβRII and collagen I [7]. Here, we demonstrated that lentivirus-mediated knockdown of SCARNA10 only slightly inhibits TGFβ-induced Smad2/3 phosphorylation; however, both the mRNA and protein levels of Smad2, Smad3, TGFβ and KLF6 were decreased in SCARNA10 down-regulated HSCs and HCs. On the other hand, over-expression of SCARNA10 increases the expression of Smad2, Smad3, TGFβ and KLF6. These findings were also confirmed in mouse models. Taken together, these results showed that SCARNA10 aggravates liver fibrosis mainly through promoting the transcription of the genes involved in TGFβ pathway.
Recently, increasing evidence has indicated that a significant number of lncRNAs involved in transcription regulation often function through binding with chromatin modifying enzymes to promote epigenetic activation or silencing of gene expression [20, 35, 37]. It has been reported that twenty percent of all human lncRNAs physically associate with the PRC2 which consists of three core components: EED, SUZ12 and one of the two histone H3K27 methyltransferases, EZH1 or EZH2 [38]. Biochemically, PRC2 uses either EZH2 or EZH1 as the catalytic subunit, and other core components such as EED and SUZ12, to help the propagation of H3K27 trimethylation (H3K27me3) marks by allosteric activation of PRC2. H3K27me3 strongly associates with transcriptional repression [38]. However, in mammals, it is not clear how sequence-specific recruitment of the PRC2 occurs across the genome. In the current study, we performed RIP assay to pull down endogenous RNAs associated with SUZ12 or EZH2, and the results showed that SCARNA10 physically interacts with PRC2. However, our result revealed that over-expression of SCARNA10 promotes the transcription of Smad2, Smad3, TGFβ, α-SMA, Collagens and KLF6. Further study revealed that knockdown of SCARNA10 increases PRC2 binding and the H3K27me3 modification across the promoters of target genes, suggesting that SCARNA10 may promote the expression of these genes by physically binding with PRC2 and masking its DNA binding sites. SCARNA10's mechanism of action is reminiscent similarly to lncRNA Gas5 or Lethe that titrates glucocorticoid receptor or NF-κB transcription factors away from their cognate binding sites, respectively [39, 40]. Taken together, our data demonstrated for the first time that SCARNA10 interacts with PRC2 and inhibits the binding of PRC2 to the promoter of the target genes.
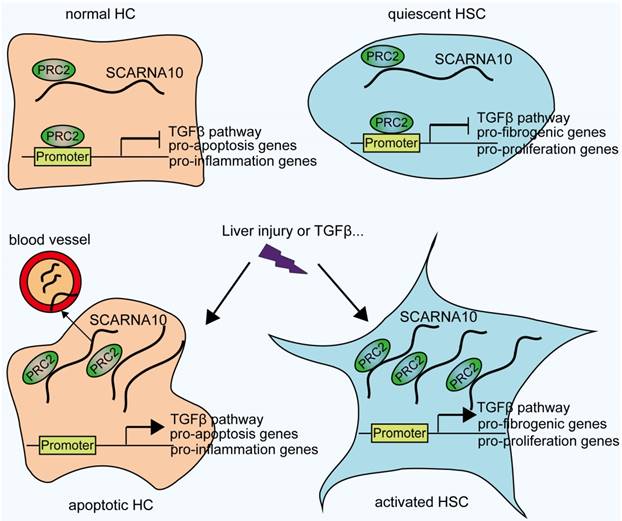
In conclusion, our findings identified SCARNA10 as a novel regulator of TGFβ signaling in liver fibrosis. Increased SCARNA10 expression in apoptotic HCs, activated HSCs and fibrotic liver tissues promotes the progression of liver fibrosis through inhibiting the binding of PRC2 to the promoters of the genes involved in TGFβ pathway, suggesting this lncRNA may be a possible therapeutic target against liver fibrosis. More importantly, the results of patient data indicated that SCARNA10 may prove to be an accurate biomarker for the fibrotic condition and these studies provided a new avenue to be explored for research of translational medicine.
Schematic diagram illustrates the role of lnc-SCARNA10 in the progression of liver fibrosis.
Abbreviations
α-SMA: alpha-smooth muscle actin; BDL: bile duct ligation; CCl4: carbon tetrachloride; CTGF: connective tissue growth factor; ECM: extracellular matrix; GFAP: glial fibrillary acidic protein; HCs: hepatocytes; HSCs: hepatic stellate cells; LncRNAs: long non-coding RNAs; ORFs: open reading frames; PDGF: platelet-derived growth factor; PRC2: Polycomb Repressive Complex 2; TGFβ: transforming growth factor-β; TIMPs: tissue inhibitor of metalloproteinases.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No.81670558; 81870429; 81800542), the National 13th 5-year Plan for Hepatitis Research (No.2017ZX10203201-007), the Natural Science Foundation of Tianjin (grant numbers 18JCZDJC99000 and 18JCQNJC81500), and the Science & Technology Development Fund of Tianjin Education Commission for Higher Education (No.2017KJ221).
Author contributions
K.Z., T.H., and W.H. conceived and designed the studies. Y.H., Z.H., Z.Z., and K.Z. performed the majority of the experiments. S.S., L.Z., Q.Y., Y.Z., J.W., X.H., and T.C. collected the clinical samples. K.Z., Y.H., Z.Y., W.H., and T.H. analyzed the data. K.Z., T.H., and W.H. wrote the manuscript. All authors critically reviewed and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Lee YA, Wallace MC, Friedman SL. Pathobiology of liver fibrosis: a translational success story. Gut. 2015;64:830-41
2. Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209-18
3. Trautwein C, Friedman SL, Schuppan D, Pinzani M. Hepatic fibrosis: Concept to treatment. J Hepatol. 2015;62:S15-24
4. Seki E, Schwabe RF. Hepatic inflammation and fibrosis: functional links and key pathways. Hepatology. 2015;61:1066-79
5. Herrmann J, Gressner AM, Weiskirchen R. Immortal hepatic stellate cell lines: useful tools to study hepatic stellate cell biology and function? J Cell Mol Med. 2007;11:704-22
6. Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev. 2008;88:125-72
7. Tu X, Zhang H, Zhang J, Zhao S, Zheng X, Zhang Z. et al. MicroRNA-101 suppresses liver fibrosis by targeting the TGFbeta signalling pathway. J Pathol. 2014;234:46-59
8. Al-Mossawi MH, Chen L, Fang H, Ridley A, de Wit J, Yager N. et al. Unique transcriptome signatures and GM-CSF expression in lymphocytes from patients with spondyloarthritis. Nat Commun. 2017;8:1510
9. Ulitsky I, Bartel DP. lincRNAs: genomics, evolution, and mechanisms. Cell. 2013;154:26-46
10. Krol J, Krol I, Alvarez CP, Fiscella M, Hierlemann A, Roska B. et al. A network comprising short and long noncoding RNAs and RNA helicase controls mouse retina architecture. Nat Commun. 2015;6:7305
11. Song Y, Liu C, Liu X, Trottier J, Beaudoin M, Zhang L. et al. H19 promotes cholestatic liver fibrosis by preventing ZEB1-mediated inhibition of epithelial cell adhesion molecule. Hepatology. 2017;66:1183-96
12. Mondal T, Subhash S, Vaid R, Enroth S, Uday S, Reinius B. et al. MEG3 long noncoding RNA regulates the TGF-beta pathway genes through formation of RNA-DNA triplex structures. Nat Commun. 2015;6:7743
13. Mei YP, Liao JP, Shen J, Yu L, Liu BL, Liu L. et al. Small nucleolar RNA 42 acts as an oncogene in lung tumorigenesis. Oncogene. 2012;31:2794-804
14. Zhong F, Zhou N, Wu K, Guo Y, Tan W, Zhang H. et al. A SnoRNA-derived piRNA interacts with human interleukin-4 pre-mRNA and induces its decay in nuclear exosomes. Nucleic Acids Res. 2015;43:10474-91
15. Ogawa T, Enomoto M, Fujii H, Sekiya Y, Yoshizato K, Ikeda K. et al. MicroRNA-221/222 upregulation indicates the activation of stellate cells and the progression of liver fibrosis. Gut. 2012;61:1600-9
16. Roderburg C, Luedde M, Vargas Cardenas D, Vucur M, Mollnow T, Zimmermann HW. et al. miR-133a mediates TGF-beta-dependent derepression of collagen synthesis in hepatic stellate cells during liver fibrosis. J Hepatol. 2013;58:736-42
17. Hu J, Chen C, Liu Q, Liu B, Song C, Zhu S. et al. The role of the miR-31/FIH1 pathway in TGF-beta-induced liver fibrosis. Clin Sci. 2015;129:305-17
18. Wang P, Luo ML, Song E, Zhou Z, Ma T, Wang J. et al. Long noncoding RNA lnc-TSI inhibits renal fibrogenesis by negatively regulating the TGF-beta/Smad3 pathway. Sci Transl Med. 2018:10
19. Zhang K, Shi ZM, Chang YN, Hu ZM, Qi HX, Hong W. The ways of action of long non-coding RNAs in cytoplasm and nucleus. Gene. 2014;547:1-9
20. Matera AG, Terns RM, Terns MP. Non-coding RNAs: lessons from the small nuclear and small nucleolar RNAs. Nat Rev Mol Cell Biol. 2007;8:209-20
21. Prensner JR, Chinnaiyan AM. The emergence of lncRNAs in cancer biology. Cancer Discov. 2011;1:391-407
22. Roderburg C, Urban GW, Bettermann K, Vucur M, Zimmermann H, Schmidt S. et al. Micro-RNA profiling reveals a role for miR-29 in human and murine liver fibrosis. Hepatology. 2011;53:209-18
23. Ichigozaki Y, Fukushima S, Jinnin M, Miyashita A, Nakahara S, Tokuzumi A. et al. Serum long non-coding RNA, snoRNA host gene 5 level as a new tumor marker of malignant melanoma. Exp Dermatol. 2016;25:67-9
24. Schwarzenbach H. Circulating nucleic acids as biomarkers in breast cancer. Breast Cancer Res. 2013;15:211
25. Dhahbi JM. Circulating small noncoding RNAs as biomarkers of aging. Ageing Res Rev. 2014;17:86-98
26. Qi P, Zhou XY, Du X. Circulating long non-coding RNAs in cancer: current status and future perspectives. Mol Cancer. 2016;15:39
27. He Y, Lin J, Kong D, Huang M, Xu C, Kim TK. et al. Current State of Circulating MicroRNAs as Cancer Biomarkers. Clin Chem. 2015;61:1138-55
28. Bedossa P, Poynard T. An algorithm for the grading of activity in chronic hepatitis C. The METAVIR Cooperative Study Group. Hepatology. 1996;24:289-93
29. Suh YG, Kim JK, Byun JS, Yi HS, Lee YS, Eun HS. et al. CD11b(+) Gr1(+) bone marrow cells ameliorate liver fibrosis by producing interleukin-10 in mice. Hepatology. 2012;56:1902-12
30. Maschmeyer P, Flach M, Winau F. Seven steps to stellate cells. J Vis Exp. 2011;51:pii 2710
31. Klattenhoff CA, Scheuermann JC, Surface LE, Bradley RK, Fields PA, Steinhauser ML. et al. Braveheart, a long noncoding RNA required for cardiovascular lineage commitment. Cell. 2013;152:570-83
32. Yang F, Zhang L, Huo XS, Yuan JH, Xu D, Yuan SX. et al. Long noncoding RNA high expression in hepatocellular carcinoma facilitates tumor growth through enhancer of zeste homolog 2 in humans. Hepatology. 2011;54:1679-89
33. Zhang K, Han X, Zhang Z, Zheng L, Hu Z, Yao Q. et al. The liver-enriched lnc-LFAR1 promotes liver fibrosis by activating TGFbeta and Notch pathways. Nat Commun. 2017;8:144
34. Cheng K, Yang N, Mahato RI. TGF-beta1 gene silencing for treating liver fibrosis. Mol Pharm. 2009;6:772-9
35. Kotake Y, Nakagawa T, Kitagawa K, Suzuki S, Liu N, Kitagawa M. et al. Long non-coding RNA ANRIL is required for the PRC2 recruitment to and silencing of p15(INK4B) tumor suppressor gene. Oncogene. 2011;30:1956-62
36. Sarma K, Cifuentes-Rojas C, Ergun A, Del Rosario A, Jeon Y, White F. et al. ATRX directs binding of PRC2 to Xist RNA and Polycomb targets. Cell. 2014;159:869-83
37. Liu GY, Zhao GN, Chen XF, Hao DL, Zhao X, Lv X. et al. The long noncoding RNA Gm15055 represses Hoxa gene expression by recruiting PRC2 to the gene cluster. Nucleic Acids Res. 2016;44:2613-27
38. Xu B, Konze KD, Jin J, Wang GG. Targeting EZH2 and PRC2 dependence as novel anticancer therapy. Exp Hematol. 2015;43:698-712
39. Rapicavoli NA, Qu K, Zhang J, Mikhail M, Laberge RM, Chang HY. A mammalian pseudogene lncRNA at the interface of inflammation and anti-inflammatory therapeutics. eLife. 2013;2:e00762
40. Kino T, Hurt DE, Ichijo T, Nader N, Chrousos GP. Noncoding RNA gas5 is a growth arrest- and starvation-associated repressor of the glucocorticoid receptor. Sci Signal. 2010;3:ra8
Author contact
![]() Corresponding author: Zhi Yao, Department of Immunology, Tianjin Key Laboratory of Cellular and Molecular Immunology, Key Laboratory of Immune Microenvironment and Disease of Ministry of Education, Tianjin Medical University, Tianjin, China. E-mail: yaozhiedu.cn, or Tao Han, The Third Central Clinical College of Tianjin Medical University, Department of Hepatology and Gastroenterology, Tianjin Third Central Hospital affiliated to Nankai University, Tianjin Key Laboratory of Artificial Cells, Artificial Cell Engineering Technology Research Center of Public Health Ministry, Tianjin, China. E-mail: hantaomdcom, or Wei Hong, Department of Histology and Embryology, Tianjin Key Laboratory of Cellular and Molecular Immunology, Key Laboratory of Immune Microenvironment and Disease of Ministry of Education, School of Basic Medical Sciences, Tianjin Medical University, Tianjin, China. E-mail: hongweiedu.cn.
Corresponding author: Zhi Yao, Department of Immunology, Tianjin Key Laboratory of Cellular and Molecular Immunology, Key Laboratory of Immune Microenvironment and Disease of Ministry of Education, Tianjin Medical University, Tianjin, China. E-mail: yaozhiedu.cn, or Tao Han, The Third Central Clinical College of Tianjin Medical University, Department of Hepatology and Gastroenterology, Tianjin Third Central Hospital affiliated to Nankai University, Tianjin Key Laboratory of Artificial Cells, Artificial Cell Engineering Technology Research Center of Public Health Ministry, Tianjin, China. E-mail: hantaomdcom, or Wei Hong, Department of Histology and Embryology, Tianjin Key Laboratory of Cellular and Molecular Immunology, Key Laboratory of Immune Microenvironment and Disease of Ministry of Education, School of Basic Medical Sciences, Tianjin Medical University, Tianjin, China. E-mail: hongweiedu.cn.