Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Materials and methods
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(12):3541-3554. doi:10.7150/thno.32908 This issue Cite
Research Paper
Polo-Like Kinase 1 phosphorylates and stabilizes KLF4 to promote tumorigenesis in nasopharyngeal carcinoma
Jia Mai1*, Zhuo-Yan Zhong1*, Gui-Fang Guo1,2*, Xiu-Xing Chen1,2, Yan-Qun Xiang1,3, Xuan Li1, Hai-Liang Zhang1, Yu-Hong Chen1, Xue-Lian Xu1, Rui-Yan Wu1, Yan Yu1, Zhi-Ling Li1, Xiao-Dan Peng1, Yun Huang1, Li-Huan Zhou1, Gong-Kan Feng1, Xiang Guo1,3, Rong Deng1 ![]() , Xiao-Feng Zhu1
, Xiao-Feng Zhu1 ![]()
1. State Key Laboratory of Oncology in South China, Collaborative Innovation Center for Cancer Medicine, Guangdong Key Laboratory of Nasopharyngeal Carcinoma Diagnosis and Therapy, Sun Yat-sen University Cancer Center, Guangzhou, China
2. Department of VIP Inpatient, Sun Yat-sen University Cancer Center, Guangzhou, China
3. Department of Nasopharyngeal Carcinoma, Sun Yat-sen University Cancer Center, Guangzhou, China
*Equally contributing first authors
Received 2019-1-7; Accepted 2019-4-24; Published 2019-5-26
Abstract

Rationale: Advanced nasopharyngeal carcinoma (NPC) is an aggressive disease with no targeted therapies and poor outcomes. New innovative targets are urgently needed. KLF4 has been extensively studied in the context of tumors, and current data suggest that it can act as either a tissue-specific tumor-inhibiting or a tumor-promoting gene. Here, we found that KLF4 played as a tumor-promoting gene in NPC, and could be mediated by PLK1.
Methods: Tissue immunohistochemistry (IHC) assay was performed to identify the role of KLF4 in NPC. Global gene expression experiments were performed to explore the molecular mechanisms underlying KLF4-dependent tumorigenesis. Small-molecule kinase inhibitor screening was performed to identify potential upstream kinases of KLF4. The pharmacologic activity of polo-like kinase inhibitor volasertib (BI6727) in vitro and in vivo was determined.
Result: Our investigation showed that high expression of KLF4 was correlated with poor prognosis in NPC. Moreover, genome-wide profiling revealed that KLF4 directly activated oncogenic programmes, including gene sets associated with KRAS, VEGF, and MYC signalling. We further found that inhibition of polo-like kinase 1 could downregulate the expression of KLF4 and that PLK1 directly phosphorylated KLF4 at Ser234. Notably, phosphorylation of KLF4 by PLK1 caused the recruitment and binding of the E3 ligase TRAF6, which resulted in KLF4 K32 K63-linked ubiquitination and stabilization. Moreover, KLF4 could enhance TRAF6 expression at the transcriptional level, thus initiating a KLF4-TRAF6 feed-forward loop. Treatment with the PLK1 inhibitor volasertib (BI6727) significantly inhibited tumor growth in nude mice.
Conclusion: Our study unveiled a new PLK1-TRAF6-KLF4 feed-forward loop. The resulting increase in KLF4 ubiquitination leads to stabilization and upregulation of KLF4, which leads to tumorigenesis in NPC. These results expand our understanding of the role of KLF4 in NPC and validate PLK1 inhibitors as potential therapeutic agents for NPC, especially cancer patients with KLF4 overexpression.
Keywords: PLK1, KLF4, nasopharyngeal carcinoma, K63-linked ubiquitination
Introduction
Nasopharyngeal carcinoma (NPC) is an aggressive head and neck cancer characterized by a high rate of local invasion and early distant metastasis [1]. It has a particularly high prevalence in South China. Due to the intrinsic invasiveness and asymptomatic nature of the disease, the majority of NPC patients are diagnosed with advanced disease (60-70% of cases) with poor outcomes. Thus far, there is no effective targeted therapy for advanced NPC.
Krüppel-like factor 4 (KLF4) is a complex transcription factor that contains a highly conserved C-terminal DNA-binding domain with three zinc fingers [2]. Under physiological conditions, upon binding to specific sequences, including CACCC boxes and GC boxes, KLF4 can exert multiple functions by regulating many cellular processes, such as cell proliferation, development, apoptosis, and homeostasis [3]. KLF4 regulates embryonic stem cell self-renewal [4], and together with OCT4, SOX2, and C-MYC, it can also reprogram somatic cells into induced pluripotent stem cells [5].
KLF4 has been extensively studied in the context of tumors, and current data suggest that it can act as either a tissue-specific tumor-inhibiting or a tumor-promoting gene, with the underlying mechanism remaining unclear [6]. KLF4 has been reported to inhibit tumorigenesis and/or malignant properties in a variety of contexts, including neuroblastoma, leukaemia, and pancreatic, lung, and colon cancer [7]. Conversely, adverse impacts on clinical outcomes and/or protumorigenic effects in functional assays have been reported for KLF4 in breast, skin, and other cancers. The experimental data are rather controversial, and there is no general agreement about the role of KLF4 in nasopharyngeal carcinoma.
Polo-like kinase 1 (PLK1) is a serine/threonine protein kinase that is widely recognized as an oncogene that drives cellular proliferation by promoting mitosis and cytokinesis [8, 9]. There are the 5 polo-like kinase (PLK) family members, PLK1 to PLK5. PLK1, the best-studied member of the family, is frequently used as a tumor marker [8]. PLK1 promotes cell-cycle progression by regulating multiple steps during mitosis [9]. Moreover, non-mitotic roles of PLK1 have been suggested, including regulation of cancer cell invasiveness and protection against apoptosis [10]. It has been reported that PLK1 can regulate MYC stabilization and activate a feed-forward circuit promoting tumor cell survival [11], but it is not yet clear how elevated levels of PLK1 reprogram cells to promote cancer progression.
In this study, we found that high expression of KLF4 was correlated with poor prognosis in NPC. We found that Polo-like kinase 1 (PLK1) could bind the repression domain of KLF4 and directly phosphorylated KLF4 at Ser234. Phosphorylation of KLF4 by PLK1 induced the recruitment and binding of the E3 ligase TRAF6 to KLF4, which resulted in KLF4 K63 ubiquitination and stabilization. Volasertib, a small molecular PLK1 inhibitor, caused a decrease in KLF4 protein levels and exhibited potent antitumor activity in vivo. These results indicate that PLK1 inhibitors are potential therapeutic agents for NPC that function by inhibiting KLF4.
Results
KLF4 is functionally overexpressed in human nasopharyngeal carcinoma
Krüppel-like factor 4 (KLF4) is an important regulator of cell fate decisions, including the DNA damage response, inflammation, apoptosis and stem cell renewal. KLF4 has been reported to have tumor-suppressive properties in gastrointestinal, lung, pancreatic and oesophageal cancer, while it acts as an oncogenic factor in breast and squamous cell carcinoma [12, 13]. To investigate the role of KLF4 in NPC, we examined the expression of KLF4 in human NPC (n=152) with a tissue immunohistochemistry (IHC) assay. The tissues were divided into groups with low (score 0-4) and high (score 5-9) levels of KLF4 according to their immunostaining scores (Figure 1A). Strikingly, high KLF4 expression was significantly associated with poor outcome in NPC patients (Figure 1B). We subsequently analysed the correlations between KLF4 expression levels and the clinical characteristics of the NPC patients. In the training cohort, no significant correlations between KLF4 expression levels and patient age, sex, and so on (Table S1). To determine whether the expression level of KLF4 was an independent prognostic factor, multivariate cox regression analyses were used. In the training cohort, the KLF4 expression level was identified as an independent prognostic factor for local relapse-free survival (LRFS) (Table S2). Moreover, 8 samples from NPC and areas adjacent to carcinoma tissues were removed, and Western blotting analysis was employed to test the expression levels of KLF4. We found that KLF4 expression in seven tumor tissues was much higher than that in the tissue adjacent to the carcinoma (Figure 1C).
KLF4 Is Functionally Overexpressed in nasopharyngeal cancer. (A) Immunohistochemical staining for KLF4 in nasopharyngeal cancer tissue (n=152). Score = 0-4 (low) and score = 5-9 (high) indicate KLF4 levels in representative tumor tissues. (B) Kaplan-meier analysis of local relapse-free survival (LRFS) in a set of 152 nasopharyngeal cancer patients (n=152) according to KLF4 expression, **P<0.01, log rank tests.(C) Immunoblot analysis of KLF4 in the nasopharyngeal carcinoma (T, tumor) and adjacent to carcinoma tissues (N, normal tissue). (D) Protein levels of KLF4 with or without KLF4 shRNA knockdown. S26 cells were infected with the control shRNA or validated shRNAs targeting KLF4 virus to build a stable cell line. (E and F) Colony formation assay using S26 shNC, S26 shKLF4-1 and S26 shKLF4-2 cells for 10 days. Crystal violet was used to stain the formed colonies (E). The colony numbers were calculated as mean ± SD (n=3). ***p<0.001, student's t-test (F). (G and H) The migratory ability of S26 shNC, S26 shKLF4-1 and S26 shKLF4-2 cells was assayed using an uncoated transwell assay. Crystal violet was used to stain the cells (G). The migratory cell numbers were calculated as mean ± SD (n=3). ***p<0.001, student's t-test (H). (I-K) Tumor volumes at the indicated dates (I) and tumor images (J) as well as tumor weight at day 17 (K) of S26 xenografts of shNC and shKLF4. (n=8 for each group).
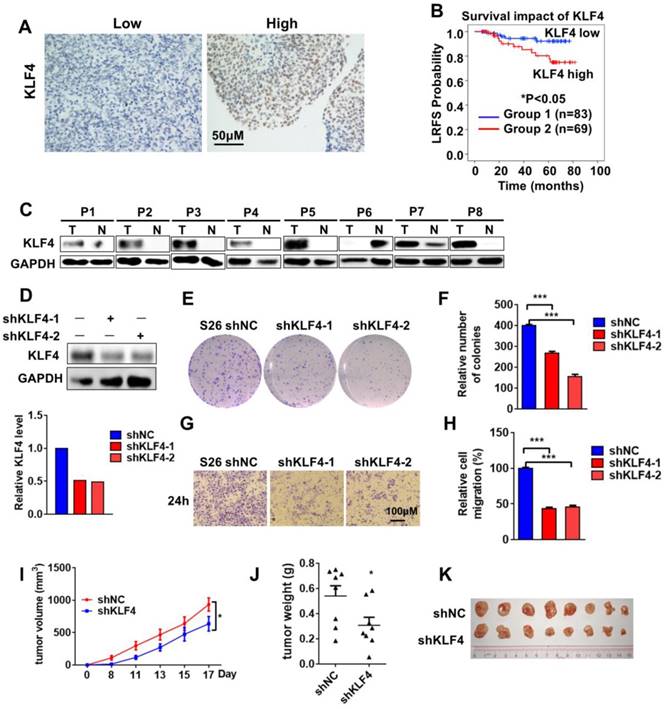
To evaluate the impact of KLF4 on NPC, KLF4 knockdown in the NPC cell line S26 was performed with two different small hairpin RNAs (shRNAs) (Figure 1D). The colony-forming ability of S26 cells was greatly decreased (Figure 1E-F) following KLF4 knockdown, accompanied by a decrease in cell proliferation (Figure S1A) and cell migratory ability (Figure 1G-H and Figure S1B). All these results indicated that KLF4 deficiency decreased NPC malignant behaviour in vitro. Next, in vivo malignant behaviour was determined in a xenograft mouse model. As expected, the absence of KLF4 dramatically decreased tumorigenesis (Figure 1I-K).
KLF4 knockdown affects gene expression profiling in NPC cells
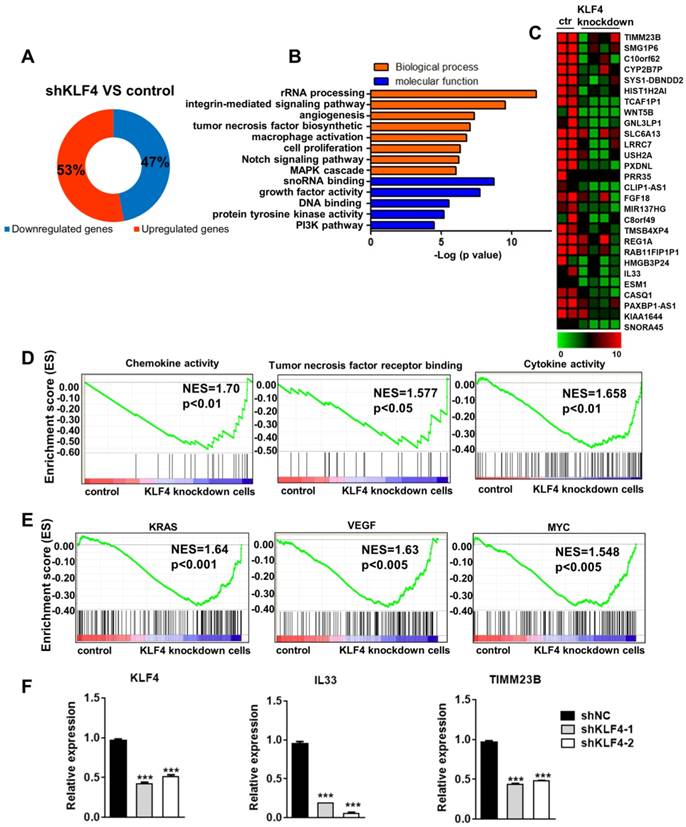
Although KLF4 and some of its downstream targets have been well studied, especially in gastrointestinal and pancreatic cancer, it remains unclear why elevated KLF4 protein levels enhance malignant transformation in NPC [14, 15]. To explore the molecular mechanisms underlying KLF4-dependent tumorigenesis, we performed global gene expression experiments in control and KLF4 knockdown cells. We found that 53% of genes were upregulated and 47% of genes were downregulated (Figure 2A) by KLF4 knockdown. As KLF4 functions as a transcription activator, we chose the downregulated genes for further analysis. As expected, gene ontology analysis showed that these genes were mainly associated with proliferation and survival processes, such as angiogenesis, cell proliferation, the Notch signalling pathway, growth factor activity, and the PI3K pathway (Figure 2B). Consistently, GSEA revealed a marked downregulation of proliferation and survival signatures, such as chemokine activity, tumor necrosis factor receptor binding, and cytokine activity, in KLF4 knockdown cells (Figure 2D). In addition, we identified inactivation of tumorigenic pathways, including gene sets associated with KRAS, VEGF, and MYC signalling, in KLF4 knockdown cells (Figure 2E). We then analysed the gene expression changes using a 2-fold cut-off in KLF4 knockdown NPC cells compared to control NPC cells. A heat map shows the most highly changed genes (Figure 2C). Q-PCR was used to confirm the changes in gene expression (Figure 2F).These results suggest that aberrantly high KLF4 expression can promote tumorigenic proliferation of cancer cells by controlling oncogenic pathways.
KLF4 depletion downregulated oncogenic genes expression. (A) Proportions of up- and downregulated genes targeted by KLF4. (B) GO analysis of the downregulated genes in KLF4-deficient tumor cells. (C) Heatmap showed the genes most differentially expressed in KLF4-deficient tumor cells. (D) GSEA analysis of KLF4-deficient tumor cells for top differentially regulated gene sets in KEGG pathway. (E) GSEA analysis of KLF4-deficient tumor cells for top differentially regulated gene sets in Oncogene pathway. (F) Real-time qPCR of specific genes in control or KLF4- deficient tumor cells. Expression was normalized to the housekeeping gene GAPDH.
PLK1 Enhances KLF4 Protein Stability
The results above show that KLF4 plays an important role in NPC. Therefore, we were interested in determining how KLF4 is regulated. Based on recent studies indicating that post-translational modifications exist in OCT4 and SOX2 [16], we hypothesized that KLF4 expression might be regulated by post-translational modification, specifically phosphorylation and ubiquitination. To identify potential upstream kinases of KLF4, we performed small-molecule kinase inhibitor screening using nasopharyngeal cancer cells. A total of 56 kinase inhibitors targeting PI3K signalling, mTOR, Wnt signalling, Aurora Kinase, EGFR, and MEK were used for screening (Table S3). We found that the polo-like kinase inhibitor BI6727 could markedly decrease KLF4 expression (Figure 3A and S2A-B). Furthermore, S18 cells were treated with the selective PLK inhibitor BI6727 at different concentrations. As expected, BI6727 dramatically decreased KLF4 protein levels but not mRNA levels in a concentration-dependent manner in NPC cell lines (Figure 3B-3C, Figure S2C). Another PLK inhibitor, BI2536, was also and showed similar results (Figure 3D, Figure S2D), and do not depend on the cell cycle stage (Figure S2H-2I).
KLF4 is a newly identified substrate of PLK1. (A) Heatmap showed changes of KLF4 protein level in treated by different kinase inhibitors by western blot analysis. Calculate gray value with image J. (B) S18 cells were treated with BI6727 for 24H and cell lysates analyzed for the level of KLF4. (C) S26, CNE1, CNE2 and HONE1 cells were treated with BI6727 for 24H and cell lysates analyzed for the level of KLF4. (D) S18 and S26 cells were treated with BI2536 for 24H and cell lysates analyzed for the level of KLF4. (E) PLK1 depletion by specific siRNA in S26 cells. PLK1 and KLF4 protein levels were analyzed by immunoblot, with GAPDH as a loading control. (F-G) PLK1 and KLF4 protein levels were analyzed by immunoblot, with GAPDH as a loading control in 293T (F) and S26 (G) cells. (H) Relative KLF4 mRNA levels were quantitated by real-time qPCR. Data shown represent the means (±SEM) of triplicates. (I) MG132 rescued KLF4 loss resulted from CHX treated. (J-k) MG132 rescued KLF4 loss resulted from PLK1 inhibition. (L) PLK1 can upregulate KLF4 ubiquitination. FLAG-KLF4 and HA-ubiquitin were transfected into 293T cells together with HIS-PLK1 or vector. Protein extracts were immunoprecipitated (IP) using anti-FLAG antibody. (M) FLAG-KLF4 and HA-ubiquitin were transfected into 293T cells together with si-PLK1 or negative control. Protein extracts were immunoprecipitated (IP) using anti-FLAG antibody. (N) FLAG-KLF4 and HA-ubiquitin were transfected into 293T cells together with BI6727 or DMSO. Protein extracts were immunoprecipitated (IP) using anti-FLAG antibody.
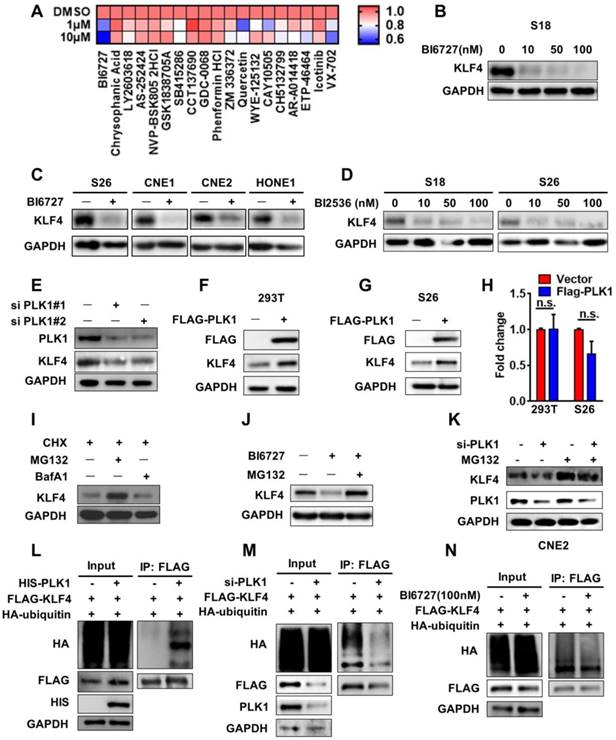
To evaluate the impact of PLK1 on KLF4 expression, we performed PLK1 knockdown within siRNAs in nasopharyngeal carcinoma CNE2 cells. Genetic depletion of PLK1 led to a marked decrease in KLF4 protein abundance (Figure 3E). We then sought to understand how PLK1 regulates KLF4 expression. Ectopic expression of PLK1 in S26 cells and 293T cells did not lead to upregulation of KLF4 mRNA levels but markedly increased its protein abundance (Figure 3F-H), and it depended on the PLK1 kinase activity (Figure S2J), suggesting post-translational regulation of KLF4 by PLK1. To validate this hypothesis, we co-treated S18 cells with cyclohexamide (CHX) and MG132 (a proteasome inhibitor) or bafilomycin A1 (BafA1, a lysosome inhibitor) and found that the KLF4 protein levels were restored by co-treatment with MG132 (Figure 3I). The results indicated that KLF4 protein stability was mediated through the ubiquitin-proteasomal protein degradation pathway. In support of this notion, administration of BI6727 in S18 cells consistently decreased endogenous KLF4 protein levels, which was reversed by addition of MG132 (Figure 3J, Figure 3K). To determine whether PLK1 affects KLF4 protein turnover, we measured KLF4 protein half-life in the presence of BI6727, siPLK1 and FLAG-PLK1. As shown in Figure S2E-2G, depletion or inhibition of PLK1 led to enhanced KLF4 protein turnover while PLK1 overexpress decreased KLF4 protein turnover.
To elucidate the mechanism of stabilization of KLF4, a ubiquitination assay was performed to test whether PLK1 affects the ubiquitination of KLF4 in vivo. We co-transfected either empty vector or His-tagged PLK1 with Flag-tagged KLF4 and HA-tagged ubiquitin into 293T cells. We found that KLF4 ubiquitination was markedly increased under PLK1 overexpression (Figure 3L). Overexpression of PLK1 could increase KLF4 ubiquitination, whereas knockdown of PLK1 with PLK1 siRNA led to a decrease in KLF4 ubiquitination (Figure 3M). In addition, the PLK inhibitor BI6727 also downregulated the ubiquitination of KLF4 (Figure 3N). These results confirmed that PLK1 was involved in KLF4 protein stability in a post-translational modification-dependent manner.
KLF4 interacts with PLK1 in nasopharyngeal cancer cells
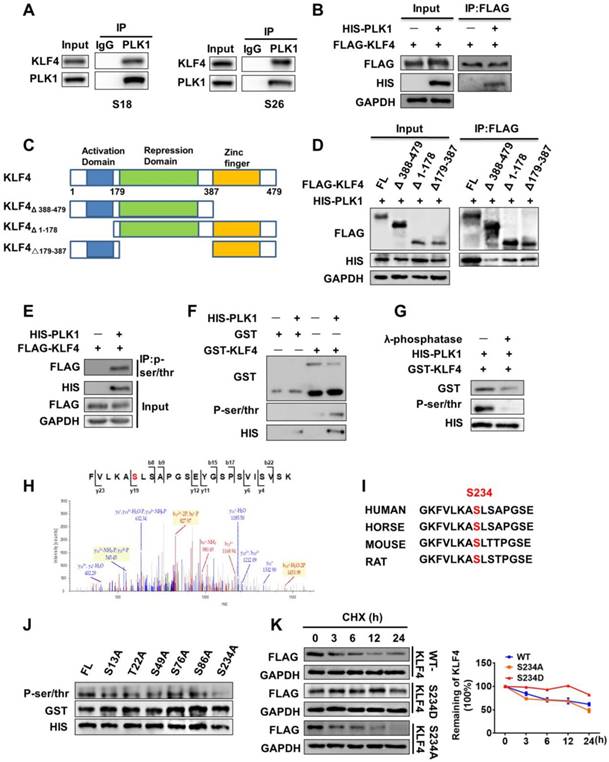
To understand exactly how PLK1 regulates KLF4 protein stability, we examined whether PLK1 can interact with KLF4 and performed endogenous co-immunoprecipitation in S18 and S26 cells. Immunoprecipitation with PLK1 antibody showed that endogenous KLF4 was present in PLK1 immunoprecipitates (Figure 4A). Co-immunoprecipitation in 293T cells also revealed that KLF4 could interact with PLK1 (Figure 4B). To identify the region or regions of KLF4 responsible for interaction with PLK1, several Flag-tagged KLF4 deletion mutants were generated (Figure 4C). Using a co-IP assay, we found that deletion of C-terminus (aa Δ 388-479) abolished the KLF4 and PLK1 interaction, whereas deletion of the N-terminus (aa Δ 1-178) or the middle portion (aa △179-387) had little effect on the KLF4 and PLK1 interaction (Figure 4D). This result indicates that the C-terminus (aa 387-479) of KLF4 mediated the interaction with PLK1.
PLK1 enhanced KLF4 stability through regulation of its phosphorylation. (A) Endogenous interaction between KLF4 with PLK1. Lysates from S18 or S26 cells were subjected to immunoprecipitation using anti-PLK1 antibody, and the co-precipitation KLF4 was detected by western blot (WB) using anti-KLF4 antibody. (B) 293T cells were transfected with FLAG-KLF4, HIS-PLK1 and as indicated. (C) The illustration of KLF4 deletion constructs. (D) Coimmunoprecipitation and immunoassay of extracts of 293T cells transfected with HIS-PLK1 together with FLAG-KLF4 (FL), FLAG-KLF4 (Δ 388-479), FLAG-KLF4 (Δ 1-178), or FLAG-KLF4 (Δ179-387). (E) KLF4 is phosphorylated by PLK1 in vivo. FLAG-KLF4 was transfected into 293T cells together with HIS-PLK1 or vector. Protein extracts were immunoprecipitated (IP) using anti-P-ser/thr antibody. (F) KLF4 is phosphorylated by PLK1 in vitro. The GST-KLF4 fusion protein was analyzed by in vitro kinase assay with ATP and active PLK1. (G) The GST-KLF4 fusion protein were incubated with or without λ-phosphatase and analyzed by in vitro kinase assay with ATP and active PLK1. (H) Mass spectrometry analysis of KLF4 proteins after incubation with recombinant PLK1 in a kinase reaction. The data showed that the serine residue corresponding to Ser234 (indicated in red in peptide sequence) was phosphorylated. y, product ion number from C terminus of the peptide; b, product ion numbered from N terminus of the peptide. (I) The predicted PLK1 phosphorylation sites in KLF4 are highly conserved during evolution. Putative PLK1 phosphorylation sites are indicated with red. (J) In vitro kinase assay showing that bacterially purified recombinant S234A KLF4 has the most significant effect on phosphorylation levels. (K) KLF4 Ser234 phosphorylation Mimicking mutation enhanced KLF4 Stability.
PLK1 enhances KLF4 Stability through Regulation of Its Phosphorylation at Ser234
To examine whether PLK1 directly phosphorylates KLF4, we co-transfected either empty vector or His-tagged PLK1 with Flag-tagged KLF4 in 293T cells. We then performed a co-immunoprecipitation (IP) assay using a P-Ser/Thr antibody, and KLF4 was present in immunoprecipitates when the cells were co-transfected with His-tagged PLK1 but not empty vector (Figure 4E, Figure S2K). Next, we constructed a glutathione S-transferase (GST)-fusion bacterial expression vector and purified the GST-KLF4 protein from BL21 E. coli. The protein was subjected to an in vitro kinase assay with ATP and commercially available active PLK1. The results demonstrated that PLK1 phosphorylated KLF4 (Figure 4F). To further confirm this finding, λ-phosphatase (PPase), a phosphatase capable of removing phosphates from serine, threonine, and tyrosine residues, was added to the in vitro kinase assay system, and the signal of KLF4 phosphorylation disappeared (Figure 4G).
To determine the specific site at which KLF4 is phosphorylated by PLK1, we used GPS (Group-based Prediction System) software to predict the possible sites [17]. The results showed that KLF4 contains 33 putative sites that can be phosphorylated by PLK1 (Table S4). Then, mass spectrometry analysis was performed after the in vitro kinase assay. The results showed that there were 12 potential sites of KLF4 that could be phosphorylated by PLK1 (Table S5). We constructed six mutants (S13A, T22A, S49A, S76A, S86A, and S234A) of GST-KLF4, as shown in the results. An in vitro kinase assay with PLK1 indicated that the KLF4 S234A mutation abolished most of the phosphorylation signal compared to wild-type KLF4 (Figure 4J).
We analysed the motif containing Ser234 in KLF4 from residue 227 to residue 241 (GKFVLKAS★LSAPGSE) and found that this sequence was highly evolutionarily conserved and followed the sequence pattern of PLK1 phosphorylation substrates (Figure 4H-I). To verify that Ser234 of KLF4 was a target amino acid for PLK1, we constructed an active mutant of Ser234 (S234D). As expected, KLF4 S234D stabilized KLF4 (Figure 4K). Overall, these data demonstrate that polo-like kinase 1 can mediate phosphorylation of KLF4 at Ser234 and promote KLF4 stability.
PLK1 promotes K63-Linked ubiquitination of KLF4
Protein ubiquitination is an important post-translational modification that regulates protein fate. Although ubiquitination often results in protein degradation, certain types of ubiquitination, such as K63-linked ubiquitination, often regulate signalling activation and protein stability [18]. Therefore, we investigated whether KLF4 underwent K63-linked ubiquitination and found that this was the case (Figure 5A). We co-transfected Flag-tagged KLF4 with wild-type ubiquitin, a K63 ubiquitin mutant (only able to form K63-linked chains) or a K48 ubiquitin mutant (only able to form K48-linked chains) in 293T cells. We found that KLF4 can undergo both types of ubiquitination (Figure 5B). In addition, overexpression of PLK1 increased KLF4 K63-linked chain ubiquitination but not K48-linked chain ubiquitination (Figure S3A-S3B), whereas PLK1 knockdown with PLK1 siRNAs led to a decrease in KLF4 K63-linked chain ubiquitination but not K48-linked chain ubiquitination (Figure 5C and Figure S3C). These results further confirm that PLK1 can phosphorylate KLF4 and then affect the K63-linked ubiquitination levels of KLF4.
PLK1 recruits TRAF6 to induce K63-linked ubiquitination of KLF4. (A) Protein extracts from S18 and S26 cells were immunoprecipitated using anti-KLF4 antibody or with IgG as a negative control and analyzed by Western blotting using anti-Lys63 specific ubiquitin and anti-KLF4 antibodies. (B) 293T cells were co-transfected with FLAG- KLF4 alone or in combination with wile type ubiquitin or its mutant (K48R or K63R) HA-tagged constructs. Protein extracts were immunoprecipitated using an anti-FLAG beads. (C) FLAG-KLF4 and HA-K63-UB were transfected into 293T cells together with PLK1 siRNA or control. Protein extracts were immunoprecipitated (IP) using anti-FLAG beads. (D) 293T cells were co-transfected with HA-K63-UB alone or in combination with wile type FLAG-KLF4 or its mutant (K32R, K232R or K252R). Protein extracts were immunoprecipitated using an anti-FLAG beads. (E) 293T cells were transfected with FLAG-KLF4 and HA-TRAF6 as indicated. Protein extracts were immunoprecipitated using an anti-FLAG beads. (F-G) TRAF6 depletion by specific siRNA in S26 and CNE2 cells. TRAF6 and KLF4 protein levels were analyzed by immunoblot, with GAPDH as a loading control. (H) 293T cells were transfected with FLAG-KLF4 and HIS-PLK1 as indicated. Protein extracts were immunoprecipitated using an anti-FLAG beads. (I) 293T cells were transfected with FLAG-KLF4, HA-K63-UB and si-TRAF6 as indicated. (J) 293T cells were transfected with FLAG-KLF4, HA-K63-UB, HIS-PLK1 and V5-TRAF6 as indicated. (K) 293T cells were co-transfected with HA-TRAF6 alone or in combination with wile type FLAG-KLF4 or its mutant (S234D, S234A). Protein extracts were immunoprecipitated using anti-FLAG beads. (L-M) PLK1 and KLF4 protein levels were analyzed by immunoblot, with GAPDH as a loading control in S26 (G) and 293T (F) cells. (N) Real-time qPCR analysis of TRAF6 expression upon KLF4 shRNA knockdown.
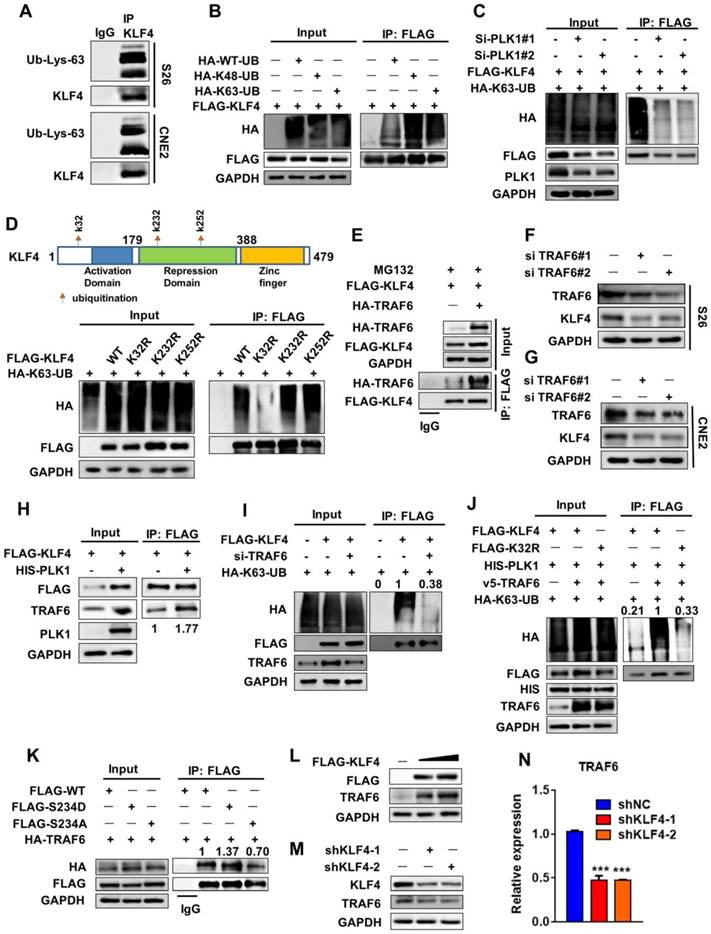
To determine the specific K63-linked chain ubiquitination site of KLF4, we referred to two bioinformatics databases, UbPred (http://www.ubpred.org/) and NetChop (http://www.cbs.dtu.dk/services/NetChop/), to predict the putative lysine sites responsible for polyubiquitination of KLF4 [19]. We found that K32, K232 and K252 were the sites with the highest confidence. Then, corresponding KLF4 mutants bearing a single lysine (K)-to-arginine (R) substitution at every potential ubiquitination site were generated. We found that K63-linked chain ubiquitination was attenuated in the K32R KLF4 mutant (Figure 5D). These results suggested that K32 could be a critical lysine residue for KLF4 K63-linked chain ubiquitination.
PLK1 Recruits TRAF6 to induce K63-Linked Ubiquitination of KLF4
We next attempted to identify the E3 ligase that could mediate this modification of KLF4 [20]. The E3 ligase TRAF6 always mediates K63 ubiquitination [21, 22]. 293T cells were co-transfected with KLF4 and TRAF6 or vector. Interestingly, TRAF6 not only bound to KLF4 but also enhanced the stability of KLF4 (Figure 5E and Figure S3D-S3E). Furthermore, TRAF6 siRNA-treated cells exhibited a decrease in KLF4 protein levels and stability (Figure 5F-G and Figure S3F). Moreover, PLK1 overexpression strengthened the capability of TRAF6 to interact with KLF4 (Figure 5H). Overexpression of PLK1 caused an increase in TRAF6, and PLK1 siRNA-treated cells exhibited a decrease in TRAF6 protein levels (Figure 5H and Figure S3G). These results show that TRAF6 can also be regulated by PLK1.
Finally, when TRAF6 was downregulated, the capability of KLF4 to form a K63-linked ubiquitination chain was strongly affected (Figure 5I). And, the capability of KLF4 to form a K63-linked ubiquitination chain was reduced under K32R and S234A mutant (Figure 5J, Figure S3H). In addition, the active mutant of Ser234 (S234D) had a stronger ability to interact with TRAF6 than the wild-type sequence (Figure 5K). These findings demonstrate that both TRAF6 and PLK1 are required for KLF4 stability.
Interestingly, we noticed that the TRAF6 protein was expressed at higher levels upon KLF4 transfection (Figure 5L). Knocking down KLF4 with shRNA led to a decrease in TRAF6 protein (Figure 5M). A Q-PCR assay showed that depletion of KLF4 expression caused a decrease in TRAF6 mRNA levels (Figure 5N). A luciferase reporter assay was then used to further identify that TRAF6 is the direct downstream target of KLF4. As shown in Figure S3I the promoter activity of TRAF6 was significantly increased upon KLF4 overexpression. Taken together, these findings demonstrate that KLF4 regulates TRAF6 expression at the transcriptional level. Thus, KLF4 and TRAF6 exhibit positive feedback regulation; TRAF6 promotes K63-linked ubiquitination of KLF4 and then increases KLF4 protein stability, and KLF4 enhances TRAF6 expression at the transcriptional level and further promotes KLF4 expression.
Increases in KLF4 mediated by PLK1 function in oncogenesis in NPC
PLK1 can phosphorylate KLF4 and promote its stability. Next, we sought to explore the physiological roles of KLF4 phosphorylation by polo-like kinase 1 in NPC. Stable cell lines were first established as shown in Figure 6A. Ectopic expression of PLK1 significantly enhanced the protein levels of KLF4. In this analysis, we found that ectopic expression of PLK1 promoted cell proliferation and migration, supporting a possible oncogenic role for KLF4. More notably, depletion of endogenous KLF4 by short hairpin RNA (shRNA) suppressed cell proliferation and migration (Figure 6B-C and Figure S4A). These results indicate that KLF4 can present oncogenic function. Furthermore, overexpression of PLK1 greatly increased the colony-forming ability of S26 cells, and simultaneous knockdown of KLF4 could largely reverse the tumorigenic effect of PLK1 overexpression (Figure S4B-C). These in vitro results suggested a functional interplay between PLK1 and KLF4. To test this in vivo, we established xenografts using stable cell lines in nude mice. The results showed that PLK1 inhibition suppressed tumorigenesis (Figure 6D-F).
Targeting PLK1 inhibited KLF4 expression and tumor growth in vivo. (A) S26 cells were stable transfected with FLAG-PLK1 with or without KLF4 shRNA knockdown as indicated. KLF4, PLK1 and GAPDH were analysed by Western blotting. (B and C) The migratory ability of cell lines as indicated was assayed using an uncoated transwell assay. Crystal violet was used to stain the cells (B). The migratory cell numbers were calculated as mean ± SD (n=3). ***p<0.001, student's t-test (C). (D-F) Tumor volumes at the indicated dates (D) and tumor weight at day 17 (E) as well as tumor images (F) of S26 xenografts as indicated. (n=10 for each group). (G) Immunohistochemical analyses of 152 specimens from nasopharyngeal cancer patients using anti-PLK1 and anti-KLF4 antibodies were performed. Representative images of IHC staining of tumors form two human nasopharyngeal cancer patients are presented. (H) Kaplan-meier analysis of local relapse-free survival (LRFS) in a set of 152 nasopharyngeal cancer patients (n=152) according to PLK1 expression, **P<0.01, log rank tests. (I) Correlation study of KLF4 and PLK1 expression in the asopharyngeal cancer consisting of 152 samples. (J) Schematic modeling the treatment of the mouse. (K-M) Representative tumors from S18 allografts mice treated with BI6727, 10mg/kg/day. Tumor volumes and tumor weight shows in figure. (n=7 for each group). (N) S18 xenografts were taken off from the mice, and then extract the protein for western blot analysis. The expression of KLF4 was shown (C, control; T, treated). (O) Model of Polo-Like Kinase 1 triggers a feed-forward loop between KLF4 and TRAF6.
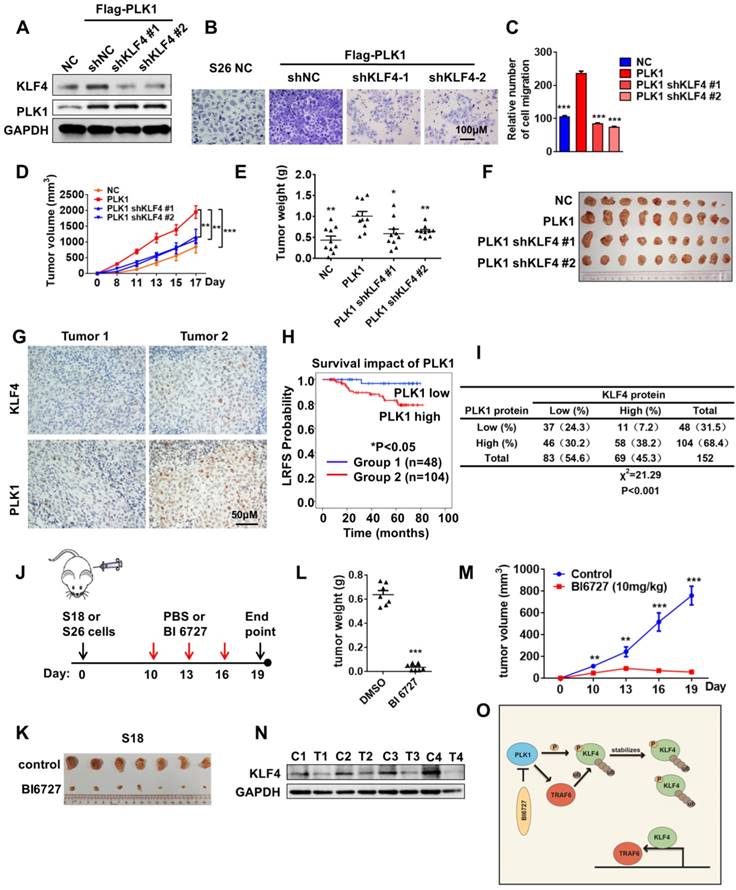
We then performed immunohistochemical analysis to evaluate the potential association between PLK1 and KLF4 in 152 human NPC specimens using anti-PLK1 and anti-KLF4 antibodies (Figure 6G). As expected, high PLK1 expression was significantly associated with poor outcome in nasopharyngeal cancer patients (Figure 6H). We observed a positive correlation between PLK1 and KLF4 (Figure 6I; Χ2= 21.29, P<0.001). Altogether, our results demonstrated that KLF4 was positively correlated with PLK1, and the expression levels of KLF4 and PLK1 were upregulated in nasopharyngeal cancer patients with poor prognoses.
Polo-Like Kinase 1 Inhibitor is an Effective Therapeutic agent for Nasopharyngeal Cancer
The data above have demonstrated the biological consequences of crosstalk regulation of KLF4 by ubiquitylation and protein phosphorylation in the maintenance of protein stability as well as tumor progression. To determine whether targeting the PLK1-KLF4-TRAF6 axis can be used for NPC treatment, we examined the effect of BI6727, a small molecular inhibitor of PLK1, on NPC cells. The results of a transwell assay showed that the migration of NPC cells was significantly inhibited by BI6727 (Figure S4D-E). To test whether a polo-like kinase inhibitor would inhibit tumor cell growth in vivo, we evaluated the effects of BI6727 in S18 and S26 xenograft tumors. Mice were treated with 10 mg/kg BI6727 every three days until tumors in the PBS-treated group reached the allowed maximum tumor size (Figure 6J). Tumor growth was tremendously inhibited by BI6727 (Figure 6K-M and Figure S4F-H). Then, protein was extracted from the xenografts for Western blot experiments. Interestingly, inhibition of PLK1 with BI6727 was correlated with extreme expression of KLF4 in S18 and S26 xenografts (Figure 6N and Figure S4I), suggesting that polo-like kinase 1 inhibitor can play a potential therapeutic role in KLF4-driven nasopharyngeal cancer. To evaluate the relationship between KLF4 and PLK1 inhibitors, KLF4-WT, S234D and K32R mutant stable cell lines were established at the base of shKLF4 S26 cell line, and the cells were treated with PLK1 inhibitor BI6727 in a MTT assay. The result shows that KLF4 S234D and K32R mutants do not have the same sensitivity to BI6727 as KLF4-WT, as S234D and K32R mutation partially overcomes the PLK1 inhibition (Figure S4J-4K). We have identified a pharmacogenetic interaction between BI6727 and the oncogene KLF4, suggesting that KLF4 status may serve as a genetic predictor of the efficacy of PLK1 inhibitors.
Discussion
In this study, we revealed that KLF4 is upregulated in nasopharyngeal cancer and is correlated with poor prognoses. Our study demonstrated for the first time that PLK1 can directly interact with KLF4 and phosphorylate Ser234, alter the affinity of KLF4 for the E3 ligase TRAF6, and promote the K63-linked ubiquitination of KLF4 K32. In addition, KLF4 can enhance TRAF6 expression at the transcriptional level and further promote KLF4 expression. These enhance KLF4 stability and therefore increase the oncogenic potential of KLF4 in NPC (Figure 6O).
Expression of KLF4 can be regulated at both the transcriptional and post-transcriptional levels [23]. Previous studies have shown that hypermethylation of CPG islands in the promoter of KLF4 and methylation of histones modulates its activity in cancer [24]. ERK1 and ERK2 have been shown to negatively regulate KLF4 activity by phosphorylating serine residue 132 [25]. Acetylation of KLF4 at lysine residues 225 and 229 mediated by the P300/CBP complex inhibits the ability of KLF4 to activate downstream targets [26]. The observations in this study suggest an additional regulatory layer orchestrating KLF4 function in cancer through the mechanism of protein K63-linked ubiquitination. In colon and hepatocellular cancer, high expression of KLF4 can induce transcription of a series of cyclin-dependent kinase inhibitors, such as p21, p27 and p57, causing it to function as a tumor suppressor [27]. In breast cancer, high expression of KLF4 can inhibit P53-dependent cell apoptosis, causing it to function as a protumorigenic factor [28]. Here, we showed that knockdown of KLF4 in NPC downregulated KRAS, VEGF, and MYC signalling and that KLF4 thus functioned as a protumorigenic factor. These results suggest that aberrantly high KLF4 expression can promote the tumorigenesis of cancer cells, which is associated with activation of oncogenic pathways.
Regarding the role of K63-linked ubiquitination in protein stability, we show here that this modification is a crucial factor in KLF4 protein stability and is mediated by TRAF6. TRAF6-dependent K63-linked ubiquitination of ULK1 has been shown to be essential for ULK1 activation and stability [29]. TRAF6 ubiquitinates some protein kinases, such as TAK1 and AKT, leading to their subsequent activation [18]. Here, we revealed that TRAF6 interacted with KLF4, promoting its K63-linked ubiquitination and enhancing its stability. In addition, KLF4 could enhance TRAF6 expression at the transcriptional level, further promoting KLF4 protein stability. Furthermore, PLK1 overexpression increased TRAF6 protein levels. Thus, PLK1 enhanced the ability of TRAF6 to mediate substrate stability and promoted its own stability. These results implicate PLK1 as an essential regulator upstream of the feed-forward loop between KLF4 and TRAF6. Therefore, the use of specific PLK1 inhibitors could be an exciting approach for therapeutic purposes.
PLK1 is a promising therapeutic target. BI6727 (volasertib), a highly specific and potent PLK1 inhibitor, is under multiple clinical trials [30]. However, the trials performed to date suggest that BI6727 exhibits quite varied clinical efficacies and that only a fraction of patients respond well to single-agent BI6727, suggesting that optimization of patient populations is urgently needed [11]. We have identified a pharmacogenetic interaction between volasertib and the oncogene KLF4, suggesting that KLF4 may partially play roles in antitumor activity of PLK1 inhibitor.
Both PLK1 and KLF4 could deregulate cell growth and oncogenesis through multiple mechanisms. We assume that when both PLK1 and KLF4 are high expressed in the NPC, the PLK1-KLF4 axis may impact NPC tumorigenesis more potently. However, it needs to be further verified.
Materials and methods
Cell culture and reagents
S18, S26 and CNE2 cells were maintained in 1640 medium supplemented with 10% FBS. 293T cells were cultured in DMEM with 10% FBS.
Plasmids
pCDNA3.1B-Flag-KLF4, pCDNA3.1B-His-PLK1, pCDNA3.1B-HA-ub, pGEX6p1-KLF4, pCDH-Flag-PLK1 and other mutation plasmids were generated using the ClonExpress II One Step Cloning Kit (Vazyme Biotech Co., Ltd., Nanjing, China c112) according to the manufacturer's instructions. For transfection, cells were plated to form a 60-80% confluent culture and transfected using branched polyethylenimine (PEI, Sigma-Aldrich 480727).
Lentiviral infection
pLKO.1-shKLF4-shRNA or pCDH-flag-PLK1 was co-transfected with pCMV-VSVG and pCMV-△8.2 into 293T cells using Lipofectamine 2000. The packaged lentiviral particles were collected, mixed with polybrene and added into the target cells. Stable cell lines were generated by culturing the cells in medium containing the antibiotic neomycin (20 µg/ml) or puromycin (1 µg/ml).
RNA interference
The target sequences for siRNAs against the human PLK1 and TRAF6 genes were as follows:
PLK1 no. 1: 5'-CGAGGUGCUGAGCAAGAAATT-3'
PLK1 no. 2: 5'- UCUUUACUUCUGGCUAUAUTT-3'
PLK1 no. 3: 5'- UCAUCUUGUGCCCACUGAUTT-3'
TRAF6 no. 1: 5'-GGGUACAAUACGCCUUACATT-3' [31]
TRAF6 no. 2: 5'-CGAAGAGAUAAUGGAUGCCTT-3'
The target sequences for shRNAs against the human KLF4 gene were as follows:
KLF4 no. 1: 5'-CAGAATTGGACCCGGTGTA-3'
KLF4 no. 2: 5'-GGCAAAACCTACACAAAGA-3'
Immunoblots and immunoprecipitation
Cells were harvested and lysed in whole-cell lysate buffer containing 20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% NP-40, 1% sodium deoxycholate, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, and 1 μg/ml leupeptin, (RIPA buffer, CST, #9806) supplemented with protease inhibitor (1% PMSF). The protein concentrations of the lysates were measured on a spectrophotometer using a Thermo Fisher BCA Protein Assay Kit (23227). The same amount of each whole-cell lysate was resolved by SDS-PAGE and immunoblotted with the indicated antibodies. For immunoprecipitation, 1,000 μg of lysate was incubated with the indicated antibody (1-2 μg) overnight at 4°C followed by incubation for 1H with Protein A /G Sepharose beads (Thermo Fisher 20421). The immunoprecipitates were washed five times with 1×PBS before being resolved by SDS-PAGE and immunoblotted with the indicated antibodies. Quantification of immunoblot band intensity was performed with Image J software.
Antibodies and chemicals
All antibodies were used at a 1:1,000 dilution in 1% BSA for Western blots. An anti-KLF4 antibody (12173S), an anti-DYKDDDDK tag antibody (2368S), an anti-TRAF6 antibody (8028S), an anti-His tag antibody (12698S), an anti-HA tag antibody (3724S) and an anti-PLK1 antibody (4513S) were purchased from CST. An anti-Flag antibody was purchased from Sigma-Aldrich (F1804). An anti-Gapdh antibody was purchased from Vazyme (L/N307041). Kinase inhibitors were purchased from Selleckchem.
Real-time RT-PCR analyses
Total RNA was extracted using a HiPure Universal RNA Mini Kit (Magen R4130-03), and reverse transcription was performed using a PrimeScript™ RT Reagent Kit with gDNA Eraser (Takara RR047D). Real-time PCR was performed using ChamQ SYBR qPCR Green Master Mix (Vazyme Biotech Co., Ltd., Nanjing, China Q311-03). The data are shown as the mean ± s.d. for three independent experiments.
In vivo ubiquitination assays
293T cells were transfected with an HA-tagged ubiquitin plasmid and other plasmids (flag-klf4 and/or his-plk1). Twenty-four to 48 h after transfection, the cells were lysed in denaturing buffer (1% SDS, 50 mM Tris pH 8.0, 10 mM EDTA). After incubation at 4°C for 10 min, the lysate was sonicated and diluted tenfold with buffer (0.01% SDS, 1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris pH 8.0, 167 mM NaCl) and sonicated again. Then, the cells were incubated with anti-Flag-conjugated agarose beads (Sigma-Aldrich, mouse antibody) for 2H at 4°C. The immunoprecipitates were washed five times with 1×PBS before being resolved by SDS-PAGE and immunoblotted with the indicated antibodies.
Migration assays
For cell migration assays, 3-5×104 cells were plated in a 24-well plate chamber insert (Falcon 353504) with serum-free medium on top of the insert and DMEM containing 10% FBS added to the bottom of the plate. The cells were incubated for 24-48 h, washed with PBS once and then fixed with 4% paraformaldehyde for 10 min. The cells were stained with 0.1% crystal violet blue for 30 min and then washed with double-distilled water (ddH2O). The cells on the top of the insert were scraped with a cotton swab. The positive-stained cells were examined under a microscope. The data are shown as the mean± s.d. from three independent experiments.
Cell proliferation assays
The indicated cell lines were seeded in 96-well plates (1000 cells/well) and cultured in 200 µl of 1640 medium containing 10% serum. Cell proliferation was measured every day using MTT (MP) according to the manufacturer's instructions. The data are shown as the mean± s.d. from three independent experiments.
Cell colony formation assay
S18 and S26 cells were counted, plated in triplicate at a density of 800 cells per well in 6-well plates, and cultured in DMEM (supplemented with 10% foetal bovine serum) for approximately 7 days. Then, the cells were washed twice with PBS and fixed in methanol for approximately 10 min. After two additional washes with PBS, the cells were dyed with crystal violet for 30 min. Then, the crystal violet was washed out, and the numbers of colonies were counted.
Immunohistochemistry
Nasopharyngeal cancer clinical samples were fixed in 4% paraformaldehyde (PFA), embedded in paraffin, sectioned and stained with haematoxylin and eosin. Immunohistochemical staining of the paraffin-embedded tumor tissues was performed using KLF4 (ab215036, Abcam) and PLK1 (ab17056, Abcam) primary antibodies and an ABC Elite immunoperoxidase kit according to the manufacturer's instructions.
Kinase assays
Full-length WT and mutant KLF4 fusion proteins were purified from BL21 E. coli that were transfected with pGEX6p1-WT-KLF4 or pGEX6p1-mutant-KLF4 using GST beads. Active PLK1 was purchased from Sino Biological (10676-H07B). The kinase reactions were performed in kinase buffer (10×) (9802, CST) with 500 mM ATP, 2 µg of soluble KLF4, and 100 ng of PLK1. The reaction was incubated at 30°C for 1 h and then resolved by SDS-PAGE followed by western blotting.
Mass spectrometry
GST-KLF4 fusion protein was excised from the SDS-PAGE gel after the in vitro kinase assay. The GST-KLF4 band was subjected to trypsin digestion and liquid chromatography coupled tandem mass spectrometry as described previously [17]. Protein and modification identification was performed with a database search, and peptide identifications were validated with PeptideProphet.
Mouse xenograft assays
The indicated cells at a density of 5×105 were suspended in 50 µl of 1640 medium, mixed with Matrigel (Corning, 354230; 1:1) and injected into the flanks of male nude mice. Tumor size was measured every 3 days with a calliper, and the tumor volume was determined with the formula πLW2/6, where L is the longest diameter and W is the shortest diameter. At the end of the studies, the mice were sacrificed, and in vivo solid tumors were dissected and weighed. For the BI6727 treatment assay, when the tumor volume reached 100-150 mm3, xenografted mice were randomized and then received an injection of vehicle or 10 mg/kg bodyweight BI6727 once every three days. Tumor volume and weight were measured as mentioned above.
Statistical analyses
Differences between two groups were assessed by Student's t-test. Student's t-test was used for all statistical analyses with GraphPad Prism 5.0 software.
Acknowledgements
This study was supported by the National Key R&D Program of China (2017YFC0908501), the Natural Science Foundation of China ( 81630079, 81772624, 81572605, 81803006, 81802789), the Natural Science Foundation of Guangdong Province (2017A030313481) , the Science and Technology Project of Guangzhou (201803010007), and the Fundamental Research Funds for the Central Universities of China (17ykjc25).
Supplementary Material
Supplementary figures and tables.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Li YY, Chung GT, Lui VW, To KF, Ma BB, Chow C. et al. Exome and genome sequencing of nasopharynx cancer identifies NF-kappaB pathway activating mutations. Nat Commun. 2017;8:14121
2. Hu J, Li H, Wu C, Zhao X, Liu C. The Prognostic Value of Decreased KLF4 in Digestive System Cancers: A Meta-Analysis from 17 Studies. Dis Markers. 2017;2017:3064246
3. Wei D, Kanai M, Huang S, Xie K. Emerging role of KLF4 in human gastrointestinal cancer. Carcinogenesis. 2006;27:23-31
4. Moon BS, Bai J, Cai M, Liu C, Shi J, Lu W. Kruppel-like factor 4-dependent Staufen1-mediated mRNA decay regulates cortical neurogenesis. Nat Commun. 2018;9:401
5. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663-76
6. Ferralli J, Chiquet-Ehrismann R, Degen M. KLF4alpha stimulates breast cancer cell proliferation by acting as a KLF4 antagonist. Oncotarget. 2016;7:45608-21
7. Chen HY, Lin YM, Chung HC, Lang YD, Lin CJ, Huang J. et al. miR-103/107 promote metastasis of colorectal cancer by targeting the metastasis suppressors DAPK and KLF4. Cancer Res. 2012;72:3631-41
8. Sanhaji M, Kreis NN, Zimmer B, Berg T, Louwen F, Yuan J. p53 is not directly relevant to the response of Polo-like kinase 1 inhibitors. Cell cycle. 2012;11:543-53
9. Kim J, Ishiguro K, Nambu A, Akiyoshi B, Yokobayashi S, Kagami A. et al. Meikin is a conserved regulator of meiosis-I-specific kinetochore function. Nature. 2015;517:466-71
10. Rizki A, Mott JD, Bissell MJ. Polo-like kinase 1 is involved in invasion through extracellular matrix. Cancer Res. 2007;67:11106-10
11. Xiao D, Yue M, Su H, Ren P, Jiang J, Li F. et al. Polo-like Kinase-1 Regulates Myc Stabilization and Activates a Feedforward Circuit Promoting Tumor Cell Survival. Mol Cell. 2016;64:493-506
12. Foster KW, Liu Z, Nail CD, Li X, Fitzgerald TJ, Bailey SK. et al. Induction of KLF4 in basal keratinocytes blocks the proliferation-differentiation switch and initiates squamous epithelial dysplasia. Oncogene. 2005;24:1491-500
13. Cittelly DM, Finlay-Schultz J, Howe EN, Spoelstra NS, Axlund SD, Hendricks P. et al. Progestin suppression of miR-29 potentiates dedifferentiation of breast cancer cells via KLF4. Oncogene. 2013;32:2555-64
14. Hu D, Gur M, Zhou Z, Gamper A, Hung MC, Fujita N. et al. Interplay between arginine methylation and ubiquitylation regulates KLF4-mediated genome stability and carcinogenesis. Nat Commun. 2015;6:8419
15. Tetreault MP, Yang Y, Katz JP. Kruppel-like factors in cancer. Nat Rev Cancer. 2013;13:701-13
16. Fang L, Zhang L, Wei W, Jin X, Wang P, Tong Y. et al. A methylation-phosphorylation switch determines Sox2 stability and function in ESC maintenance or differentiation. Mol Cell. 2014;55:537-51
17. Li X, Wu XQ, Deng R, Li DD, Tang J, Chen WD. et al. CaMKII-mediated Beclin 1 phosphorylation regulates autophagy that promotes degradation of Id and neuroblastoma cell differentiation. Nat Commun. 2017;8:1159
18. Yang WL, Wang J, Chan CH, Lee SW, Campos AD, Lamothe B. et al. The E3 ligase TRAF6 regulates Akt ubiquitination and activation. Science. 2009;325:1134-8
19. Lim KH, Kim SR, Ramakrishna S, Baek KH. Critical lysine residues of Klf4 required for protein stabilization and degradation. Biochem Biophys Res Commun. 2014;443:1206-10
20. Walsh MC, Lee J, Choi Y. Tumor necrosis factor receptor- associated factor 6 (TRAF6) regulation of development, function, and homeostasis of the immune system. Immunol Rev. 2015;266:72-92
21. Ohtake F, Saeki Y, Ishido S, Kanno J, Tanaka K. The K48-K63 Branched Ubiquitin Chain Regulates NF-kappaB Signaling. Mol Cell. 2016;64:251-66
22. Zhang X, Li CF, Zhang L, Wu CY, Han L, Jin G. et al. TRAF6 Restricts p53 Mitochondrial Translocation, Apoptosis, and Tumor Suppression. Mol Cell. 2016;64:803-14
23. Ghaleb AM, Yang VW. Kruppel-like factor 4 (KLF4): What we currently know. Gene. 2017;611:27-37
24. Yang WT, Zheng PS. Promoter hypermethylation of KLF4 inactivates its tumor suppressor function in cervical carcinogenesis. PLoS One. 2014;9:e88827
25. Kim MO, Kim SH, Cho YY, Nadas J, Jeong CH, Yao K. et al. ERK1 and ERK2 regulate embryonic stem cell self-renewal through phosphorylation of Klf4. Nat Struct Mol Biol. 2012;19:283-90
26. Evans PM, Zhang W, Chen X, Yang J, Bhakat KK, Liu C. Kruppel-like factor 4 is acetylated by p300 and regulates gene transcription via modulation of histone acetylation. J Biol Chem. 2007;282:33994-4002
27. Xie VK, Li Z, Yan Y, Jia Z, Zuo X, Ju Z. et al. DNA-Methyltransferase 1 Induces Dedifferentiation of Pancreatic Cancer Cells through Silencing of Kruppel-Like Factor 4 Expression. Clin Cancer Res. 2017;23:5585-97
28. Farrugia MK, Vanderbilt DB, Salkeni MA, Ruppert JM. Kruppel-like Pluripotency Factors as Modulators of Cancer Cell Therapeutic Responses. Cancer Res. 2016;76:1677-82
29. Nazio F, Strappazzon F, Antonioli M, Bielli P, Cianfanelli V, Bordi M. et al. mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nat Cell Biol. 2013;15:406-16
30. Gjertsen BT, Schoffski P. Discovery and development of the Polo-like kinase inhibitor volasertib in cancer therapy. Leukemia. 2015;29:11-9
31. Jiao L, Zhang HL, Li DD, Yang KL, Tang J, Li X. et al. Regulation of glycolytic metabolism by autophagy in liver cancer involves selective autophagic degradation of HK2 (hexokinase 2). Autophagy. 2018;14:671-84
Author contact
![]() Corresponding author: Xiao-Feng Zhu, State Key Laboratory of Oncology in South China, Collaborative Innovation Center for Cancer Medicine, Guangdong Key Laboratory of Nasopharyngeal Carcinoma Diagnosis and Therapy, Sun Yat-sen University Cancer Center, 651 Dongfeng Road East, Guangzhou, China, 510060. Phone: (8620) 8734-3149. E-mail: zhuxfengsysu.edu.cn; Rong Deng, State Key Laboratory of Oncology in South China, Collaborative Innovation Center for Cancer Medicine, Guangdong Key Laboratory of Nasopharyngeal Carcinoma Diagnosis and Therapy, Sun Yat-sen University Cancer Center, 651 Dongfeng Road East, Guangzhou, China, 510060. Phone: (8620) 8734-3170. E-mail: dengrongorg.cn
Corresponding author: Xiao-Feng Zhu, State Key Laboratory of Oncology in South China, Collaborative Innovation Center for Cancer Medicine, Guangdong Key Laboratory of Nasopharyngeal Carcinoma Diagnosis and Therapy, Sun Yat-sen University Cancer Center, 651 Dongfeng Road East, Guangzhou, China, 510060. Phone: (8620) 8734-3149. E-mail: zhuxfengsysu.edu.cn; Rong Deng, State Key Laboratory of Oncology in South China, Collaborative Innovation Center for Cancer Medicine, Guangdong Key Laboratory of Nasopharyngeal Carcinoma Diagnosis and Therapy, Sun Yat-sen University Cancer Center, 651 Dongfeng Road East, Guangzhou, China, 510060. Phone: (8620) 8734-3170. E-mail: dengrongorg.cn