Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Methods
Abbreviations
Supplementary Material
Acknowledgements
References
Author biography
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(12):3485-3500. doi:10.7150/thno.32033 This issue Cite
Research Paper
Molecularly annotation of mouse avatar models derived from patients with colorectal cancer liver metastasis
Jingyuan Wang1*, Baocai Xing2*, Wei Liu2, Jian Li1, Xicheng Wang1, Juan Li2, Jing Yang1, Congcong Ji1, Zhongwu Li3, Bin Dong3, Jing Gao1 ![]() , Lin Shen1
, Lin Shen1 ![]()
1. Key laboratory of Carcinogenesis and Translational Research (Ministry of Education/Beijing), Department of Gastrointestinal Oncology, Peking University Cancer Hospital and Institute, 52 Fucheng Road, Haidian District, Beijing 100142, China.
2. Key Laboratory of Carcinogenesis and Translational Research (Ministry of Education), Hepatopancreatobiliary Surgery Department I, Peking University Cancer Hospital and Institute, 52 Fucheng Road, Haidian District, Beijing 100142, China.
3. Department of Pathology, Key Laboratory of Carcinogenesis and Translational Research (Ministry of Education/Beijing), Peking University Cancer Hospital and Institute, 52 Fucheng Road, Haidian District, Beijing 100142, China.
*These authors contributed equally to this work.
Received 2018-12-5; Accepted 2019-4-22; Published 2019-5-25
Abstract

Background: Liver is the most common metastatic site in advanced colorectal cancer. Most patients with colorectal cancer liver metastasis (CRLM) do not benefit from current treatment. Patient-derived xenografts (PDXs) with defined molecular signatures are attractive models for preclinical studies.
Methods: Successfully established PDXs were evaluated to elucidate their fidelity of patients' biologic characteristics (pathologic, genetic and protein properties, together with chemosensitivity). The genomic variations of PDXs were analyzed by next-generation sequencing to explore the underlying molecular mechanism of metastasis and potential therapeutic targets.
Results: CRLM (N=73) showed a significantly higher successful PDX establishment rate than primary specimens (N=26; 76.7% vs. 57.7%). CRLM PDXs recapitulated the pathologic, genetic and protein properties of parental tumors, as well as chemosensitivity. Frequent altered genes in PDXs showed high consistency compared to patients' genomic alterations and were enriched in MAPK, ErbB, cell cycle, focal adhesion pathways for CRLM PDXs, whereas primary tumor-derived PDXs only exhibited genomic variations involving ErbB and cell cycle. The genetic alterations showed high concordance between paired PDXs from primary and metastatic tissues, except for recurrent gene mutations (ARID1A, CDK8, ETV1, STAT5B and WNK3) and common copy number gains in chromosomes 20q (e.g., SRC/AURKA). Several potential drug targets such as KRAS, HER2, and FGFR2 were validated using corresponding inhibitors. Additionally, PDX models could also be used in screening efficient regimens for patients with no druggable alterations.
Conclusion: This study has successfully established and validated a large panel of molecularly annotated platforms from patients with CRLM for preclinical studies.
Keywords: Patient-derived xenograft, colorectal cancer liver metastasis, molecular signature, therapeutic target
Introduction
Liver is the most common metastatic site in advanced colorectal cancer (CRC). Surgical resection is currently the effective treatment for patients with colorectal cancer liver metastasis (CRLM), with five-year overall survival (OS) rates of 35% to 60% [1], and chemotherapy could likely improve patient selection for operation as well as postoperative survival. With improvements in understanding CRC molecular subtypes, drug therapy paradigms have shifted from “one gene, one drug” to “multi-molecular, multi-drug”. However, due to the extensive molecular and functional heterogeneity of CRLM, the overall progress of novel cytotoxic chemotherapies and targeted drugs has been more modest than expected [2]. Therefore, the elucidation of the underlying metastatic mechanisms and novel therapeutic targets for personal cancer therapy is imperative.
Appropriate animal models are of vital importance for preclinical cancer studies. Compared to cell line-derived xenografts, patient-derived xenograft (PDX) models, also known as Avatar mouse models, have been shown to be more clinically relevant preclinical models due to the high-fidelity of their biological characteristics (molecular, genetic, and histopathologic heterogeneity) with their corresponding parental tumors [3-5]. The introduction of PDX models together with major advances in next-generation sequencing has enabled investigators to identify novel, rare, targetable alterations to design more rational drugs, as well as explore and reverse possible resistance to current drugs [6, 7]. In addition, they provide opportunities to further develop and validate personalized approaches for the treatment of CRLM.
Previous studies have established a large biobank of PDX models from CRLM and identified HER2 as an effective therapeutic target in Cetuximab-resistant CRLM [6]. However, the established PDX models were limited to tumor samples from Caucasians or lacked clear clinical details and molecular characteristics. It was reported that the incidence of rectal cancer among all the CRCs in China is higher than that in Europe and North America, and the age of rectal cancer in China is younger than that in western countries [8]. Also, for gene mutations, China has lower non-exon 2 KRAS, BRAF and PIK3CA mutations incidence than western countries, indicating there is a difference of mutational spectra between Caucasians and Chinese Han population [8, 9]. Hence, there is a need to establish a large panel of PDX models from patients diagnosed as CRLM with comprehensive clinical and molecular characteristics based on the Chinese Han population, which might have the different characteristic, compared to PDX models from western populations.
This study was designed to focus on the establishment and characterization of pathological and molecular features of PDX models. Underlying liver metastasis mechanisms were explored by comparing the genomic alterations between PDXs from CRLM and corresponding primary specimens. Furthermore, we also validated potential therapeutic targets and explored novel drug therapies guided by genotyping or expression profiling, leading to potential implications for precision medicine.
Results
Establishment of PDX models and parameters related to in vivo tumor formation
A total of 93 patients with their tumor specimens from colorectal primary tumors (CRPT, N=13), CRLM (N=67), and paired specimens (N=13) from both primary tumors (PT) and liver metastases (LM) were included in this study. Sixteen PDX models from CRPT and sixty-four PDX models from CRLM were successfully established at P1 respectively (Figure 1A). Along with serial passage, the latency period was continuously shorter (P <0.0001; Figure 1B) with the transplantation rate increasing from 61.5% to 100% in CRPT and from 87.7% to 100% in CRLM (Figure 1A). Notably, 13 patients harboring primary tumors and corresponding liver metastases, but only five pairs were successfully established (Cases 19, 38, 40, 52, and 56). The overall transplantation rate of CRLM (56/73, 76.7%) was higher than that of CRPT (15/26, 57.7%) (Figure 1C, P <0.001; Table S1). Higher transplantation rate of CRLM allowed us to exclude any strong bias towards selection of more aggressive cases in our set of xenografts. After the fourth generation, the PDX models became stable without further changes in model formation and thus used in the subsequent study.
Establishment of PDX models from patients with CRLM. (A) A flow diagram described the steps taken to establish a stable PDX bank from patients with CRLM. (B) Latency period of CRLM PDX models was shorter with increasing passage. (C) The overall transplantation rates of CRLM at P3 were compared according to the sources of CRC tumor samples. (D) The relationship between the latency period and clinicopathological characteristics. Line and error bars represent median ± inter-quartile range. CRLM, Colorectal Cancer Liver Metastases; PT, Primary Tumor; LM, Liver Metastases; TRG, Tumor Regression Grading; Chemo, Chemotherapy; Meta, Metachronous; Syn, synchronous; WT, Wild Type; MT, Mutation; NS, Not Significant; * p < 0.05; ** p < 0.01; *** p < 0.001; ****p<0.0001 by one-way ANOVA, unpaired two-tailed t-test or chi-squared test.
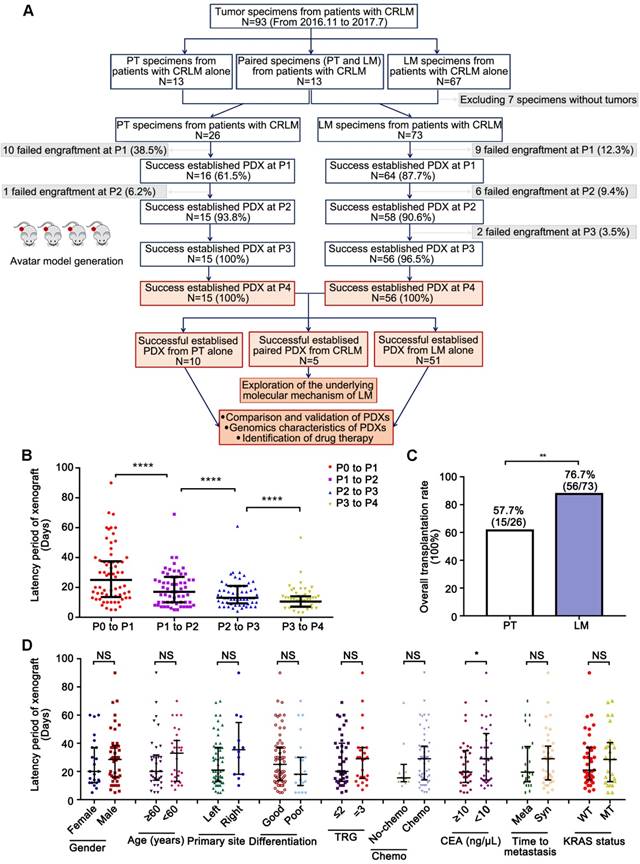
To determine the clinicopathologic parameters related to successful in vivo tumor formation, PDX model establishment rates were calculated and compared according to various patient characteristics (Table 1). No clinicopathologic features led to significant differences in the establishment results of CRLM PDX models. In addition, no differences were observed between latency period and characteristics except for the level of CEA (Figure 1D, Table 1). Samples with CEA≥10 ng/μL (24.71 ± 16.34 days) had shorter latency period than that with CEA<10 ng/μL (34.58 ± 20.33 days).
Patient characteristics, transplantation rate, and latency period of CRLM1 PDX models.
| Characteristics | No. of patients (%) | Latency period (days) | Transplantation rate (%)2 | ||
|---|---|---|---|---|---|
| Patient | 73 (100%) | 28.6±18.8 | 87.7% (64/73) | ||
| Gender | P=0.399 | P=0.467 | |||
| Male | 49 (67.12%) | 27.00±17.68 | 85.7% (42/49) | ||
| Female | 24 (32.88%) | 31.26±19.73 | 91.67% (22/24) | ||
| Age (years) | P=0.283 | P=0.395 | |||
| < 60 | 34 (46.58%) | 32.45±18.12 | 91.2% (31/34) | ||
| >= 60 | 39 (53.42%) | 27.30±19.77 | 84.6% (33/39) | ||
| Tumor site3 | P=0.150 | P=0.394 | |||
| Right side | 11 (15.07%) | 37.54±24.16 | 100% (11/11) | ||
| Left side | 60 (84.93%) | 27.96±24.16 | 85.4% (53/62) | ||
| Differentiation4 | P=0.127 | P=0.211 | |||
| good | 52 (71.23%) | 24.40±18.63 | 95.2% (44/52) | ||
| poor | 21 (28.77%) | 32.25±18.89 | 84.6% (20/21) | ||
| TRG5 | P=0.751 | P=0.223 | |||
| I | 12 (16.44%) | 26.11±16.20 | 91.67% (9/9) | ||
| II | 23 (31.51%) | 29.37±18.62 | 91.30% (30/33) | ||
| III | 36 (49.31%) | 31.64±20.83 | 91.67% (25/31) | ||
| Prior therapy | P=0.201 | P=0.723 | |||
| Neoadjuvant therapy | 62 (84.93%) | 31.11±19.01 | 87.10% (54/62) | ||
| No neoadjuvant therapy | 11 (15.07%) | 22.70 ±18.36 | 90.90% (10/11) | ||
| Time to metastasis6 | P=0.694 | P=0.086 | |||
| synchronous | 46 (63.01%) | 30.58±18.71 | 96.3% (38/46) | ||
| metachronous | 27 (36.99%) | 28.65±19.78 | 82.6% (26/27) | ||
| Kras status | P=0.907 | P=0.288 | |||
| Wild type | 45 (61.64%) | 29.33±20.40 | 84.44% (38/45) | ||
| Mutation | 28 (38.36%) | 29.92±20.01 | 92.86% (26/28) | ||
| CEA (ng/uL) | P=0.037 * | P=0.395 | |||
| <10 | 39 (53.42%) | 34.58±20.33 | 91.43% (32/35) | ||
| ≥10 | 34 (46.58%) | 24.71±16.34 | 91.67% (33/36) |
1CRLM: Colorectal cancer liver metastases.
2Transplation rates were calculated based on the successful establishment of P1.
3Left side of colorectum involved left colon and rectum.
4Good including well-differentiated and moderately differentiated adenocarcinoma, Poor including poor-differentiated adenocarcinoma and mucinous adenocarcinoma.
5TRG: Tumor Regression Grading.
6Synchronous liver metastasis was defined as liver metastatic lesions diagnosed before or within 6 months of the primary CRC diagnosis. All others were considered metachronous Liver metastasis.
*A two-sided P < 0.05 was considered statistically significant. All values were represented as the mean ± S.D. P was calculated by chi-square test, unpaired two-tailed t-test or one-way analysis of variance separately.
Preservation of the parental CRLMs' biologic characteristics and chemosensitivity in PDX models
The utility of PDXs as a model system for CRLM depends on the precise reflection of the parental tumors' pathologic and molecular characteristics. Parental CRLM and corresponding xenograft tumors were determined whether the engraftment of CRLM tissues in NOD/SCID (non-obese diabetic/severe combined immunodeficient) mice maintained the key features of the parental tumors. Pathologic comparison revealed a high degree of similarity in differentiation status between the xenografts and corresponding parental tumors, including intestinal type adenocarcinoma (Cases 05 and 45) and mucinous adenocarcinoma (Cases 03 and 18) (Figure 2A). Even though replacement of the initial human stroma by its murine counterpart occurred gradually after tumor implantation, indicated by IHC staining of human vimentin (Figure S1A), as well as flow cytometric analysis of human CD44 by culturing cancer associated fibroblast (CAF) in vitro (Figure S1B), protein expression of CRLM critical markers such as EGFR, HER2, MET, and pan-cytokeratin (CK, human cancer cell marker) was reproduced in the xenograft tumors and maintained during passage (Figure 2B). Besides, the histopathological features and leukocyte markers (human CD45, CD20, CD3) of all the established PDX models were evaluated at regular intervals to exclude lymphoma transformation, illustrated by case 24 and 86 (Figure S1C). Although the genomic profiles (gene copy number alterations and mutations) of some genes were variable during serial passage, the majority of genes remained stable and showed high consistence between the parental and corresponding xenograft tumors in nine PDX models (Figure 2C, Table S2). In particular, the KRAS mutation status of xenografts was identical to that of parental tumors (concordance rate: 100%), validated by Sanger sequencing (Figure S1D).
Fidelity was maintained to their corresponding patient tumors during the passage of CRLM PDXs regards to the pathological, protein and genetic properties, as well as chemosensitivity. (A) Representative histology of paired patient-PDX tumors. PDX models retained the histopathologic characteristics of original samples with different morphologic features including moderately differentiated phenotype (Case 05), poor-differentiated (Case 45) and mucinous adenocarcinoma (Cases 03 and 18). (B) PDX models demonstrated IHC marker expression patterns (including HER2, EGFR, MET, and CK) were preserved during passage. Positive staining was counted from five randomly selected areas in each slide at × 400 magnification. Scale bars = 100 μm. (C) The genetic alterations were compared among serial passages. For most cases, the genetic properties were conserved in PDX models, compared to corresponding parental tumors. The Venn diagram below demonstrated the number of variations during passage. (D) Chemosensitivity of PDX models was consistent to corresponding patients. Five PDX models of CRLM were evaluated for chemosensitivity compared to corresponding patients. Tumor volumes and proportion of tumor growth inhibition were expressed as means ± SD. The anti-tumor activity are depicted by %TGI (tumor growth inhibition). %TGI = (1-ΔT/ΔC) × 100%, (ΔT = Tumor volume change of the drug-treated group, ΔC = Tumor volume change of the control group on the final day of the study). (E) All of five PDX models had comparable therapeutic responses with patients including two progressive diseases, two stable diseases, and one partial response.
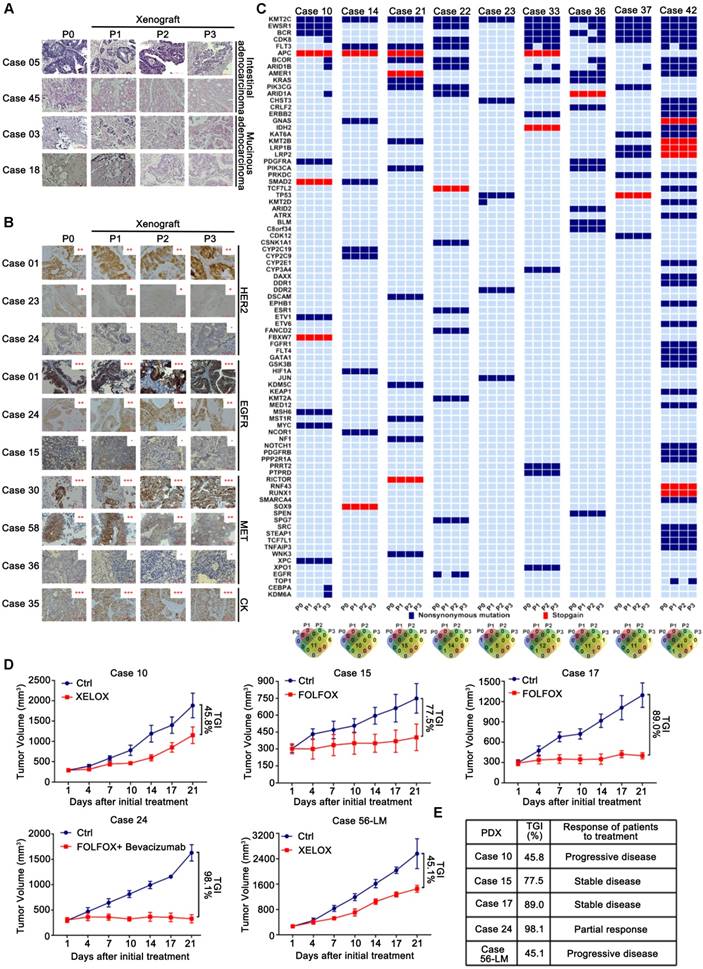
One of the most important elements to evaluate the PDX models is the therapeutic response compared to that of corresponding patients. Five PDX models were randomly used in this study to compare the chemosensitivity in patients treated with neoadjuvant chemotherapy regimens. All the PDX models could mimic patient responses, predicting two progressive diseases, two stable diseases, and one partial response (Figures 2D), suggesting that PDX models had comparable therapeutic activities relative to the corresponding patients (Figure 2E). Overall, these results implied that the PDX models well replicated the biological features of original patient tumors, thereby indicating that the models were good resources for investigating the molecular features of CRLM and responses to drug therapy.
Identification of variants and pathway enrichment in PDX models from patients with CRLM
To discover the unique molecular profile of each PDX model, a panel of 483 genes (Table S3) was sequenced for all the PDX models. Of these, the nuclear accumulation rates of β-catenin indicating the activication of Wnt signaling pathway, were 48.2% (27/56) for CRLM PDXs, and 40% (6/15) for CRPT PDXs (Figure S2). Only one (1.8%, 1/56) of the CRLM PDXs displayed high-grade microsatellite instability and two showed HER2 positive status (3.6%, 2/56). Only one of the CRPT PDXs harbored BRAF mutation (6.7%, 1/15). The top 20 mutated genes in the 56 PDX models from CRLM and 15 PDX models from CRPT, and the 30 frequently mutated genes from other reports were shown in Figures 3A. The top altered genes, which included KMT2C, ARID1A/B, BCR, FANCD2, and ZNF703, have so far rarely been reported in mCRC. Furthermore, a substantial amount of well-known driver genes alterations, including APC, ERBB2, FBXW7, KRAS, PIK3CA, and SRC were identified, and were remarkably consistent with the MSK data from CRLM (N=313, Table S4) and CRPT (N=111, Table S5) (Figures S3A-B). Many of these well-known alterations might be potential targets, which will be analyzed in subsequent studies.
Identification of variants and pathway enrichment in PDX models from patients with CRLM. (A) Genomic alterations were analyzed with different clinicopathological features in PDX models from CRLM (Left) and CRPT (Right) respectively. Red, stopgain; Dark blue, nonsynonymous SNV; Maroon, Amplification; Dark khaki, inframeshift indel; Green, frameshift indel. (B) Several relevant pathways, including ErbB, MAPK, focal adhesion, adherens junction, cell cycle, Wnt and VEGF pathways in CRLM PDXs (Left), were found to be enriched by KEGG analysis, whereas only ErbB, cell cycle signaling pathways were enriched in the CRPT PDXs (Right). * p < 0.05. (C) Details for molecular alterations involved in the KEGG analysis were showed in CRLM (Left) and CRPT (Right) PDX models respectively. Blue, missense mutation; Pale green, indel; Orange, amplification. Copy number ≥ 5 was considered as amplification.
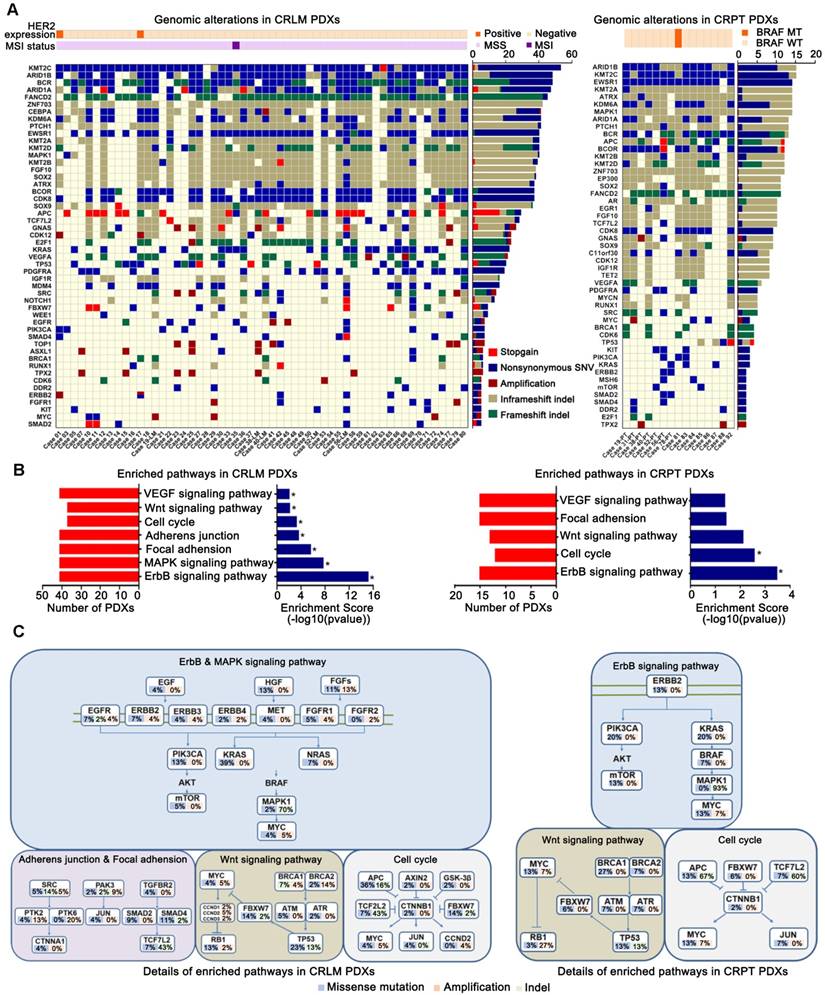
In this study, genes altered in more than two PDX models were selected for KEGG pathway analysis. Several significantly altered pathways, including ErbB, MAPK, focal adhesion, adherens junction, cell cycle, Wnt and VEGF signaling pathways were enriched in CRLM PDXs, whereas only ErbB and cell cycle signaling pathways were enriched in CRPT PDXs (Figure 3B). The detailed molecular alterations involved in the corresponding pathways were shown in Figures 3C. Therefore, PDXs could be used for drug development and mechanism research based on the unique genomic profiles.
Application of PDX models from patients with CRLM for the revelation of underlying molecular metastasis mechanism
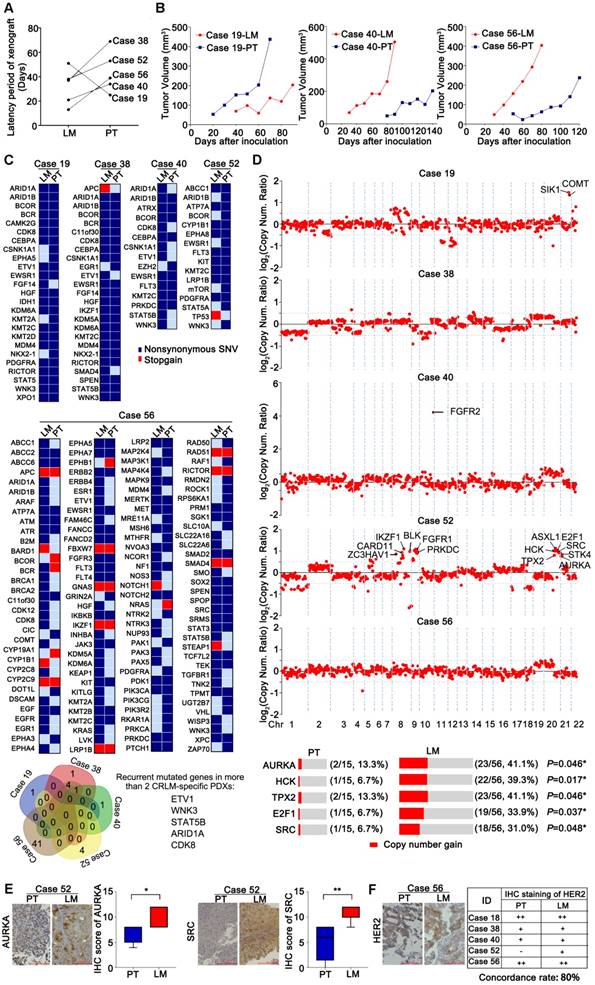
Five pairs of PDX models from CRPT and CRLM were successfully established. As shown in Figures 4A-B, the latency period of xenografts in CRLM was much shorter than that in CRPT, except for Case 19. To explore the underlying metastasis mechanism, these paired PDX models were sequenced. Consistent with previous studies [10], the overall similarity in terms of mutation status and copy number between paired PDXs from CRLM and CRPT was high (Figures 4C and S3C). However, several mutations (mutated in two more CRLM-specific PDXs: ETV1, ARID1A, WNK3, STAT5B, and CDK8) and common copy number alterations (chromosomes 8q and 20q) in the metastases were predicted to be deleterious, which might contribute to the initiation and progression of distant metastasis (Figures 4C-D). Notably, the copy number of FGFR2 in Case 40-LM was particularly higher than that in corresponding PDXs from CRPT, suggesting its potential role in metastatic process. Besides, all established PDXs were used as validation set, which showed that the copy number gain (CNG) rate of several indicated genes in CRLM PDXs was higher than that of CRPT PDXs (AURKA, 41.1% vs. 13.3%; E2F1, 33.9% vs. 6.7%; HCK, 39.3% vs. 6.7%; SRC, 31.0% vs. 6.7%; TPX2, 41.1% vs. 13.3%; P<0.05) (Figure 4D). Interestingly, these genes were all located on chromosomes 20q, suggesting that chromosomes 20q alterations might be associated with liver metastasis of colorectal cancer, which should be further confirmed in patient tumor samples. Meanwhile, the expression of ARUKA and SRC in CRLM were higher than that in corresponding CRPT PDXs (Figures 4E), validated by IHC detection. Additionally, consistent with previous study [11], HER2 expression between the paired PDXs showed high consistency (Figure 4F).
Genomic alterations were compared between five pairs of PDX models from CRLM and CRPT, respectively. (A) The difference of latency period of xenografts between CRPT and CRLM PDXs. Four of five CRLM PDX models showed a faster growth than the corresponding CRPT PDXs, except for Case 19. (B) Representative growth curve of three paired PDXs (Cases 19, 40, and 56). (C) Numerous mutated genes were identified and compared in five paired PDX models. Several recurrent mutated genes in two more CRLM-specific PDXs were identified by Venn diagram in the bottom. Red, stopgain; Dark blue, nonsynonymous SNV. (D) Copy number alterations were also compared, displayed by log2 (copy number ratio). Values more than 0.5 was assumed to be different between primary tumors and corresponding liver metastases. Several copy number alterations of genes indicated in C1 were selected for the validation in a larger biobank of PDX models from CRLM/PT. Copy number ≥ 3 was considered as gain. (E) The expression of AURKA and SRC were further validated at protein level by IHC staining. The expression of AURKA and SRC in PDXs from CRLM were higher than that of PDXs from corresponding primary tumors. (F) HER2 expression was also evaluated in five paired PDX from CRLM. The table demonstrated an 80% overlap in HER2 expression. * p < 0.05; ** p < 0.01 by one-way ANOVA or chi-squared test.
In order to further explore the role of AURKA and SRC indicated by the comparison of paired PDXs from CRPT and CRLM, we selected high expression of AURKA cell lines (SW480 and SW620) for the knockdown of AURKA and low expression of AURKA cell line (RKO) for exogenous AURKA overexpression (Figure S4A-B). Migratory and invasive potential was dramatically impaired by AURKA knockdown compared to control groups, as measured by the Transwell assays, while migratory and invasive potential was increased by AURKA overexpression (Figure S4C). To explore the mechanism of AURKA in the promotion of cell migration and invasion, molecules involved in epithelial-mesenchymal transition (EMT) and Wnt signaling pathway were assessed by western-blotting. Our results demonstrated that the knockdown of AURKA in SW620 and SW480 cell lines lead to higher expression of the epithelial marker E-cadherin and lower expression of the mesenchymal markers vimentin, N-cadherin and β-catenin, as well as the inactivacation of wnt signaling pathway, as indicated by the increased expression of Axin1 and decreased expression of c-Myc, TCF1/TCF7, and phosphorylated GSK-3β (Figure S4D). In contrast, upregulated expression of AURKA in RKO had the opposite effects (Figure S4D). Also, the inhibition of AURKA decreased metastasis ability of SW620 in vivo, by intraperitoneally injectinig SW620/NC and SW620/sh-AURKA cell lines respectively (Figure S4E). It was reported that AURKA interacted with SRC to promote motility of fibroblasts [12]. Thus, we wondered whether AURKA mediated the oncogenic role of SRC in CRC. After downregulating the expression of AURKA in the SW620/SRC cells (Figure S4F), we found the inhibition of AURKA eliminated the SRC enhanced migration and invasion (Figure S4G), by inactivating SRC-induced Wnt signaling pathway and suppressing the SRC induced EMT (Figure S4H). These results suggested AURKA could serve as a mediator of SRC to promote the migration and invasion of CRC cell lines by via inducing EMT and activating wnt signaling pathway (Figure S4I).
Application of PDX models from patients with CRLM for preclinical evaluation of potential targets
To validate that the PDX models are reliable for preclinical studies based on the molecular features, we evaluated the efficiency of anti-epidermal growth factor receptor (EGFR) monoclonal antibodies (mAb) Cetuximab, a FDA approved targeted drug for mCRC, in PDX models (Figures 5A). Our results confirmed the inefficacy of Cetuximab in mCRC with common KRAS mutations (G12V, G13D), PIK3CA (E545D, T1025A), or HER2 amplification (Figure 5B), which is highly consistent with the findings of large retrospective and prospective trials [13-15]. These results suggested that our PDX models were valuable for the identification of other candidate targets.
Validation of targets and discovery of novel drugs in CRLM PDX models for preclinical studies. (A1) The efficacy of anti-EGFR monoclonal antibodies Cetuximab on five PDX models (N=5/group). (B) Comparison of the efficacy of Cetuximab among five PDX models with genetic alterations of KRAS, PIK3CA and HER2. PDX models with KRAS mutations, PIK3CA mutations or HER2 amplification showed primary resistance to Cetuximab, which agreed with the findings of large retrospective and prospective trials. (C) The efficacy of RC48 and Herceptin on four PDX models (N=5/group). (D) Representative examples of xenograft tumors with HER2 copy number variations and expression, assessed by NGS (Left) and IHC (Right). RC48 and Herceptin exhibited stronger tumor suppressive activity in PDX models with HER2 amplification than those with HER2 un-amplification. (E) The efficacy of AZD4547 in four PDX models (N=5/group). (F) Representative examples of xenograft tumors with FGFR2 copy number variations and expression, as assessed by NGS (Left) and IHC (Right). AZD4547 exhibited selective antitumor activity in the PDX models with FGFR2 amplification and high expression. The antitumor activity was depicted by %TGI. TGI = (1-ΔT/ΔC) × 100%, (ΔT = Tumor volume change of the drug-treated group, ΔC = Tumor volume change in the control group on the final day of the study). Tumor volumes and proportion of tumor growth inhibition were expressed as means ± SD. Positive staining of IHC was counted from five randomly selected areas in each slide at × 400 magnification. Scale bars = 100 μm.
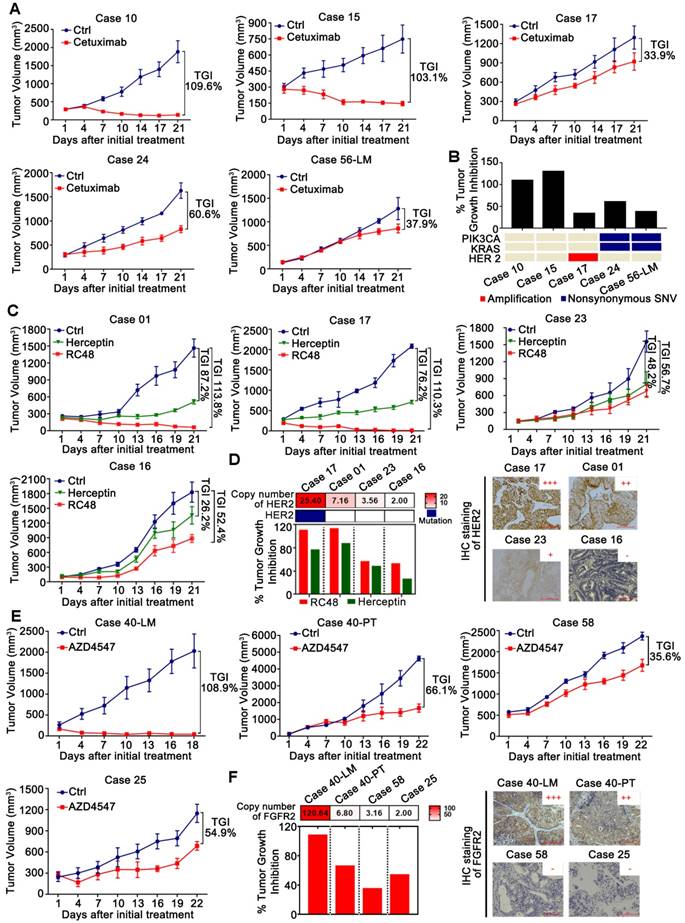
Based on the above results, several potential targets were selected for further investigation, including HER2 and FGFR2. HER2 has now emerged as an important target in HER2-amplified mCRC using the combination of trastuzumab (also called Herceptin) and lapatinib (HERACLES) [16] or pertuzumab (MyPathway) [17]. RC48, a novel antibody-drug conjugate that targets HER2, was explored in this study. We found that RC48 exerted stronger tumor suppressive activity in PDX models with HER2 amplification than those with HER2 non-amplification (Figures 5C-D) by inactivating the PI3K/AKT/mTOR and MAPK pathways (Figure S5B). In addition, RC48 was more efficient than Herceptin in mCRC regardless of the HER2 amplification status, suggesting that RC48 is an alternative choice to Herceptin in mCRC. Besides, KRAS mutation (G12V) in Case 16 and PDGFRA (S478P), which leads to the activation of downstream signaling pathways (such as MAPK pathway), might potentially be the mechanism underlying the resistance to anti-HER2 therapy (Table 2).
The alterations of RTK/RAS/RAF/PI3K/AKT and WNT signaling pathways of twelve PDX models.
| Gene | Case 10 | Case 15 | Case 24 | Case 56-LM | Case 17 | Case 01 | Case 23 | Case 16 | Case 40-LM | Case 40-PT | Case 58 | Case 25 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EGFR | FS del | p.D368N | CNV 4.22 | p.A1013V | ||||||||
| ERBB2 | p.A293T | CNV 25.44 | CNV 7.16 | CNV 3.56 | ||||||||
| ERBB3 | p.S310F | CNV 3.4 | ||||||||||
| ERBB4 | p.S1078A | |||||||||||
| FGF10 | NF insert | NF insert | NF insert | NF insert | NF insert | NF insert | NF inert | |||||
| FGF14 | CNV 4.14 | CNV 3.38 | CNV 4.38 | NF insert | p.D33G | |||||||
| FGFR2 | CNV 120.64 | CNV 6.8 | CNV 3.16 | |||||||||
| FGFR1 | CNV 4.14 | |||||||||||
| KRAS | p.G12V | p.G13D | p.G12V | p.G13D | ||||||||
| PDGFRA | p.S478P | p.S478P | ||||||||||
| PIK3CA | p.E545D | p.T1025A | p.Q546E | |||||||||
| APC | p.Q978X | p.Q1367X | FS indel | p.E1577X | p.E1322X | |||||||
| CCND2 | CNV 10.78 | CNV 3.44 | ||||||||||
| FBXW7 | p.R393X | p.R658X | p.R465C | |||||||||
| JUN | p.P220R | AMP 4.62 | AMP 4.02 | |||||||||
| MYC | p.H374R | CNV 4.64 | CNV 4.18 | |||||||||
| TCLF7L2 | NF indel | p.E17G | NF indel | NF indel | NF indel |
Notably, the FGFR2 copy number of Case 40-LM was markedly higher than that of the corresponding primary tumor Case 40-PT (CN: 120.64 vs. 6.80), which impelled us to evaluate the efficiency of FGFR2 inhibitor AZD4547 in CRLM PDX models. Four PDX models with different copy numbers of FGFR2 were selected for AZD4547 treatment. We found that AZD4547 exerted selective antitumor activity (Figures 5E-F) by inactivating both the PI3K/AKT/mTOR and MAPK pathways in the PDX models with FGFR2 amplification and high expression (Figure S5C). Interestingly, even though the copy number of Case 58 was higher than that of case 25, Case 58 showed poorer response to AZD4547 than Case 25, possibly due to KRAS mutation (G13D) in Case 58 and FGFR1 copy number gains in Case 25 (Table 2).
Furthermore, PDX models could also be used to test empirically potential active drugs for individualized patient treatment when there were no druggable alterations identified. This was well illustrated by Case 21. Several FDA approved drugs were used to evaluate the anti-tumor activities in this case. As shown in Figure S5D, we found that Irinotecan plus Capecitabine, or triple combination (Capecitabine, bevacizumab plus Oxaliplatin, or plus Irinotecan) resulted in significant tumor suppression with tumor growth inhibition (TGI) of 92%, 102%, and 99%, respectively.
Discussion
In past decades, although targeted therapy based on the mutational status of RAS/RAF could prolong survival time for CRC, mCRC (mainly CRLM) remains, for the most part, incurable. It is time to integrate novel technologies for biomarker discovery and advance to a 'multi-molecular, multi-drug' paradigm for precision medicine [18]. PDX models are of great relevance in this process and have been established from various tumors, including CRLM, which were mostly from Caucasian [6, 19]. Given the difference survival and clinicopathologic features between races, the establishment of PDX models from patients with CRLM in the Chinese Han population is imperative. Therefore, large panels of PDX models for CRLM in the Chinese Han population were successfully established to explore the underlying metastasis mechanisms, evaluate the efficacy of new anticancer drugs, therapeutic combinations, and identify biomarkers for sensitivity.
CRLM specimens displayed a better tumor take rate in NOD/SCID mice (76.7%, 56/73) compared to primary tumors (57.7%, 15/26), gastric cancers (34.1%, 63/185) and esophageal squamous carcinoma (13.3%, 25/188) [3, 4]. However, the take rate of our results was a bit lower than that of Andrea's group [6], possibly due to epidemiological and molecular characteristics among populations. No major bias with respect to the clinical characteristics was introduced by grafting. In addition, elevated serum CEA levels were observed, indicating higher aggressive behavior, which was negatively correlated with the latency period of PDXs. Consistent with previous reports [3, 4], the latency period was shorter with any additional serial passage, accompanied by higher transplantation rates.
As already described for other PDXs, histologic and molecular fidelity of the model to the original patient sample was conserved across multiple serial passages [3-5]. All of these are key advantages of these models with respect to the cell line-derived xenografts. Compared to other studies (the rates of lymphoma transformation: 2.3% to 38%) [20, 21], the lower rate of lymphoma transformation in our study for PDX derived from CRLM could be explained by several facts as followed: First, fewer periportal tissue-resident lymphocytes were present in colorectal liver metastasis than that in primary colorectal cancer [22, 23]. Second, the degree of immunodeficiency may also play a role in lymphomation [24]. Most studies which has the high rate of lymphomation, used NSG mice, the most immunodeficient mouse strains up to now. NSG mice are more susceptible to EBV infection than NOD/SCID mice. Third, PDX models we detected were limited to passage one to four. Rare B-lymphocytes present in the early-generation PDX may be difficult to be discovered by IHC detection, and these cells might extensively proliferate in subsequent PDX generations in vivo [25]. Besides, although the tumor microenvironment could be reprogrammed with the substitution by murine stroma component gradually, Arnaud et al have shown that murine stromal cells could adopt a human-like metabolomic profile in the PDX models and conserve metabolomic identity of original tumors for at least four generations in both primary sites and liver metastases of CRC [26]. Besides, PDXs from primary sites of CRC could also conserve CMS-specific features, including metabolic and mesenchymal subtypes [27, 28]. The high frequency of gene mutations related to the epigenetic modifications, including KMT2A/C/D, ARID1A/B, BCR, FANCD2, ZNF703, CEBPA, and KAM6A, at passage 3 and the minor difference of genetic alterations between PDXs and corresponding patients, were possibly due to the amplification of minor cell populations in primary tumors, the replacement of human stroma by murine cells, very low allele frequency of the mutations in the patient samples with higher frequency of mutations in the PDXs or the continued evolving mutational process during passage, paralleled with the evolution of tumors in the patients [29-31]. Whether the small amount of drift matter functionally was still controversial. Ben Davis et al revealed CNVs could influence the response of the PDX to targeted therapeutics instead of chemotherapy [32], while Gao et al's study showed the effects on drug responses of models was minimal in the early passage [33]. Possible reasons responsible for the different impacts on the drug response lie over concerns with the Avatar approach including the application of early- and late-passage tumors from PDXs and integrative analysis of multilayer omics data (DNA methylation, gene expression, mutation, proteomics, metabolomics, etc.), which may provide greater predictive power in assessing the therapeutic responses of PDXs compared to the corresponding primary tumor. Particularly, unlike the results of Uri Ben, no significantly rapid accumulation of copy number variations during passaging was observed in this study. Additionally, the loss of immune cells after a few passages limited us to explore the drugs targeting immune system. Thus, all these limitations need to be taken into consideration by researchers when analyzing the effect of drugs on tumor environment in the PDX models. Also, one of our future goals is to establish humanized immune mice.
Given that our PDX models could recapitulate the broad histopathological and molecular spectra of tumors from patients with CRLM, they were really informative biobanks that might be used to explore the potential metastasis mechanism. Based on our data, we found that the copy numbers of genes located in chromosomes 20q such as AURKA, E2F1, HCK, SRC, and TPX2 seemed to be subject to gain in CRLM PDXs compared to that of CRPT PDXs. The TPX2/AURKA axis, as co-regulators on the MYC pathway, could drive colon tumorigenesis and metastasis [34]. Additionally, SRC and HCK, which belong to the SRC family of kinases, are associated with tumor progression in various cancers. Besides, the SRC inhibitor, dasatinib, could also reduce tumoral mass and decrease the metastatic dissemination of tumoral cells [35], suggesting its potential therapeutic value for patients with CRLM. Our results further revealed that inhibition of AURKA eliminated the SRC enhanced migration and invasion. The role of recurrent mutations involving genes associated with CRLM has not been investigated except for that of ARID1A, which was associated with poor prognosis in gastrointestinal cancers [36]. However, all these need to be validated further in a larger population and at the mechanistical level.
Some evidence showed that PDX models from other tumors responded similarly to pharmacological agents when compared to their matched patients' tumors[3-4], which was also confirmed in our study. Moreover, we also validated those identical to clinical observations, KRAS (G12V, G13D) or PIK3CA (E545D, T1025A) mutant xenografts were resistant to EGFR blockade, as well as cases involving HER2 amplification [13-15, 37]. These findings indicated that genomics-characterized PDXs models could also enable systematic investigation on the efficiency and mechanisms of drug response or resistance, and the identification of potential predictive biomarkers for novel drugs, thereby contributing to the design of early-phase clinical trials.
To fully utilize PDX models for translational research, we annotated each PDX model molecularly. Dysfunction of the Wnt signaling pathway resulting in the nuclear accumulation of β-catenin is important in colorectal carcinogenesis. The nuclear accumulation rates of β-catenin in both CRLM and CRPT PDXs were similar to reported results in patients' tumor tissues [38, 39]. HER2 expression and microsatellite instability-high (MSI-H) status were also roughly similar to that in patients with CRLM [40, 41]. Several important pathways, including the MAPK, ErbB, cell cycle, focal adhesion, and adherence junction were enriched in our study. These prompted us to explore candidate targets that were involved in these pathways.
Patient selection has been proven critical in the development of HER2-targeted agents in CRC. An early effort to recruit patients with HER2-amplified CRC for treatment with the HER2 mAb Herceptin in combination with the standard-of-care chemotherapy irinotecan was halted owing to the low prevalence of the alteration, despite of promising antitumor activity in the biomarker-positive population [42]. The amplification rate of HER2 was about 3.7% in Chinese Han population [43], which was involved in Cetuximab-resistance. More recently, a dual HER2-targeted regimen, Herceptin in combination with the tyrosine kinase inhibitor lapatinib, was relatively successful, with substantial clinical activity in advanced CRC [16]. RC48, a novel antibody-drug conjugate by targeting HER2, which exerted significant tumor suppressor activity in HER2-amplified gastric cancer [44], was also explored in this study. As a single agent, RC48 had a higher tumor inhibition of CLRM compared to Herceptin, especially in HER2-amplified PDX models, inspiring us to launch a new clinical trial of RC48 in patients with CRLM in the near future. Apart from the amplification, activating mutations of HER2 are also important. Targeting the activating mutation of HER2 was really efficient in CRC via Herceptin plus tyrosine kinase inhibitors [45], which also need to be explored in our PDX models continuously using RC-48. All the results provide a novel therapy for patients with CRLM. Except for the exploration of drug targets, PDX models could also be informative platforms for the elucidation of case-specific drug resistance mechanism. Based on our study of anti-HER2 therapy, we found KRAS or PDGFRA mutation might be responsible for RC48 resistance.
FGFR2 amplification is rare in mCRC [46]. Surprisingly, we detected one pair of primary tumors and corresponding liver metastases with FGFR2 amplification. FGFR2 pathway activation is required for driving growth in NCI-716 CRC cells [47]. Recent research demonstrated that the FGFR2 inhibitor AZD4547 could exert significant tumor inhibition in FGFR2-amplified PDX models of other solid tumors [48, 49] and in early clinical trials [50, 51]. Despite the rare frequency of FGFR2 amplification in mCRC, the exploration of this target is essential for this case. AZD4547 showed selective anti-tumor activity by inactivating the pathways of PI3K/AKT/mTOR and MAPK against FGFR2-amplified PDX models. These results indicated that PDX models could guide the treatment in the case with rare alteration, inspired by the experience of drug therapy in other cancers. Besides, KRAS mutation (G12V) might be the negative predictive biomarker of AZD4547, whereas FGFR1 copy number gains might be the positive predictive biomarker based on the results of Cases 58 and 25, which need to be further explored due to the limited samples.
It was reported that the aberrant activation of Wnt signaling might be a primary cause and a resistance mechanism in MAPK pathway-activated colorectal cancers [52, 53]. However, our results suggested that Wnt signaling pathway might not play the important role in the resistance to Cetuximab, Herceptin, RC48 and AZD4547 in our study (Table 2 and Figure S5A-C). In consideration of the limited number of available PDX models and the complex relationship between Wnt and MAPK signaling pathway dependent on the specific cellular context [54], more PDX models need to be used to further explore that whether the status of Wnt and other signaling pathways plays the role in the efficacy of Cetuximab, anti-HER2 therapy (Herceptin and RC48), and anti-FGFR2 (AZD4547) therapy.
Usually, genomic analysis provides minimal insights or failed targeting pathways, and more conventional drugs and combinations could also be appropriately applied based on the results of PDX models [55], as exemplified by Case 21 in this study. This further proved that drug screening strategy using a large number of PDX models may be used pre-clinically to assess responses to candidate treatment. This model developed the concept of a combination between mouse hospital and co-clinical trial project and could enable repositioning and/or repurposing of previously approved drugs.
In conclusion, we have successfully established and validated a large panel of molecularly annotated CRLM platforms for the elucidation of underlying metastatic mechanisms, preclinical evaluation of novel therapeutics and biomarkers discovery, and integrating genomic sequencing into cancer treatment decision-making. While limitations still exist, these models would be further optimized in future studies.
Methods
Patients and tumor samples
Surgical specimens and clinical records were obtained from 93 patients with CRLM (primary tumors, N=26; liver metastases, N=80) at the Department of Hepato-Pancreato-Biliary Surgery I of Peking University Cancer Hospital from November 2016 to July 2017. The samples of seven patients, confirmed with no tumor cells from pathology, were excluded. All patients gave their written informed consent for their tumor samples to be used for research. This study was approved by the medical ethics committee of Peking University Cancer Hospital and conducted in accordance with the approved guidelines.
Establishment of PDX models
NOD/SCID mice were from Beijing HFK Bio-Technology Co., Ltd. Fresh surgical specimens (P0 = passage zero) were obtained directly from the operating room, dissected carefully, and implanted subcutaneously into one flank of a six-week-old NOD/SCID mice as previously described [3, 4]. Briefly, once tumors reached ~750 mm3 or the animal developed sickness, tumor fragments were harvested and re-inoculated into mice for passage. Each model derived from the individual patient was passaged up to four generations (referred to P1-P4). Successful transplantations of PDXs were defined as the stable passaging to P3. Animals in which implanted tumors failed to grow at six months were euthanized and defined as non-PDX forming. All procedures were performed under sterile conditions at Peking University Cancer Hospital specified-pathogens free facility and carried out in accordance with the Guide for the Care and Use of Laboratory Animals of the NIH.
Hematoxylin and eosin (H&E) staining and Immunohistochemistry (IHC) staining
Formalin-fixed paraffin-embedded (FFPE) samples were processed according to conventional experimental protocols. Serial 4-μm-thick sections from each FFPE block were processed for H&E staining according to the manufacturer's instructions with an H&E staining kit (C0105, Beyotime, China). IHC was performed as previously described [56] using the corresponding primary antibodies (primary antibodies were shown in supplementary methods). H&E staining and IHC staining were reviewed and scored according to the criteria reported previously [56] by two independent pathologists who were blinded to this study. Tumor Regression Grading (TRG) was graded as followed: TRG 0: No regression; TRG I: Dominant tumor mass with obvious fibrosis and/or vasculopathy; TRG II: Predominantly fibrosis with scattered tumor cells; TRG III: Only scattered tumor cells in the space of fibrosis with/without acellular mucin; TRG IV: Complete regression. The samples with TRG IV (N=7), were excluded.
Targeted next-generation sequencing and data analysis
Genomic DNA was extracted from the PDXs using a QIAamp DNA Mini Kit (QIAGEN Ltd., Crawley, UK) according to the manufacturer's instructions. The capture-based library was generated from 500 ng of DNA from each sample using a KAPA Hyper Prep Kit (Kapa Biosystems, Boston, MA, USA), followed by Agilent's SureSelectXT Target Enrichment System (Agilent Technologies, Santa Clara, CA, USA). Library quality was assessed using Agilent 2100 Bioanalyzed on-chip electrophoresis (Agilent Technologies, Inc.), and the library was quantified with an Agilent QPCR NGS Library Quantification Kit (Agilent Technologies, Inc.), and sequenced on an Illumina HiSeq 2000 system (Illumina, San Diego, CA, USA). The details of gene variant calling were shown in Supplementary Methods, and detailed data were shown in the Table S7.
Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis of the genes identified in more than two PDX models were conducted using the Database for Annotation, Visualization, and Integrated Discovery Bioinformatics Resources 6.8 (DAVID; http://david.abcc.ncifcrf.gov). Genetic alterations from the Memorial Sloan Kettering (MSK) Cancer cohort were publicly available through the cBioPortal for Cancer Genomics (http://cbioportal.org/msk-impact). A genomic landscape analysis of CRLM across the PDX models and MSK data was also conducted in cBioportal.
In vivo animal experiments
In this study, in vivo experiments were performed to evaluate the chemosensitivity, as well as the antitumor activity of several targeted drugs, including: RC-48 targeting HER2, AZD4547 targeting FGFR2, Cetuximab, and Herceptin.
Tumors were subcutaneously implanted into NOD/SCID mice, and when the tumors reached a size of 150-200 mm3, tumor-bearing mice were randomized into different groups (N=5/group) as required (Details of drug information were shown in supplementary methods). Tumor size and mouse weight were measured every three days, and tumor volume was calculated using the following formula: Volume = (Length × Width2)/2, where Length and Width were the long and short diameter of tumor. TGI was calculated using the following formula: TGI = (1- ΔT/ΔC) × 100% (ΔT = tumor volume change of the drug-treated group, ΔC = tumor volume change of the control group on the final day of the study). Twenty-four hours after the last treatment, mice were euthanized followed by immediately tumor dissection and photograph. According to previous reports, the PDX models were classified as high-responder with TGIs > 60% and poor-responders with TGIs < 30% [7].
Statistical analysis
Statistical analysis was performed with the SPSS 20.0 software (SPSS) or Graphpad Prism version 7.0 (Graphpad software). The relationships between clinicopathological characteristics and transplantation rate or latency period of xenografts were analyzed using the chi-square test, unpaired two tailed t-test or one-way ANOVA. The differences of alteration rates in different subgroups were evaluated by the chi-square test. For the in vivo study, tumor growth between two groups was compared using repeated-measured analysis of variance. A two-sided p<0.05 was considered statistically significant.
Abbreviations
Patient-derived xenograft: PDX; Colorectal cancer liver metastasis: CRLM; Primary tumors: PT; Liver metastases: LM; Copy number gain: CNG; Epidermal growth factor receptor: EGFR; Monoclonal antibodies: mAb; Tumor growth inhibition: TGI; Nonobese diabetic/severe combined immunodeficient: NOD/SCID; Microsatellite instability-high: MSI-H; Kyoto Encyclopedia of Genes and Genomes: KEGG; Memorial Sloan Kettering: MSK.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This study was supported by the National Key Research and Development Program of China (No. 2017YFC1308900, 2017YFC0908400), and Beijing Municipal Administration of Hospital Clinical Medicine Development of Special Funding Support (ZYLX201701). We thank LetPub (www.letpub.com) for its linguistic assistance during the preparation of this manuscript.
Author contributions
L.S. and J.G. proposed the concepts and designed the experiments. J.W. and B.X. carried out the experiments and wrote the manuscript with help from all co-authors and all authors contributed to interpretation of the data. W.L., J.L., X.W., and J.L. provided patients' data. J.Y., C.J., Z.L., and B.D carried out partial experiments and performed the analysis.
Competing Interests
The authors have declared that no competing interest exists.
References
1. House MG, Ito H, Gonen M, Fong Y, Allen PJ, DeMatteo RP. et al. Survival after hepatic resection for metastatic colorectal cancer: trends in outcomes for 1,600 patients during two decades at a single institution. J Am Coll Surg. 2010;210:744-52
2. Segal NH, Saltz LB. Evolving treatment of advanced colon cancer. Annu Rev Med. 2009;60:207-19
3. Zhu Y, Tian T, Li Z, Tang Z, Wang L, Wu J. et al. Establishment and characterization of patient-derived tumor xenograft using gastroscopic biopsies in gastric cancer. Sci Rep. 2015;5:8542
4. Zou J, Liu Y, Wang J, Liu Z, Lu Z, Chen Z. et al. Establishment and genomic characterizations of patient-derived esophageal squamous cell carcinoma xenograft models using biopsies for treatment optimization. J Transl Med. 2018;16:15
5. Wang D, Pham NA, Tong J, Sakashita S, Allo G, Kim L. et al. Molecular heterogeneity of non-small cell lung carcinoma patient-derived xenografts closely reflect their primary tumors. Int J Cancer. 2017;140:662-73
6. Bertotti A, Migliardi G, Galimi F, Sassi F, Torti D, Isella C. et al. A molecularly annotated platform of patient-derived xenografts ("xenopatients") identifies HER2 as an effective therapeutic target in cetuximab-resistant colorectal cancer. Cancer Discov. 2011;1:508-23
7. Chen Z, Huang W, Tian T, Zang W, Wang J, Liu Z. et al. Characterization and validation of potential therapeutic targets based on the molecular signature of patient-derived xenografts in gastric cancer. J Hematol Oncol. 2018;11:20
8. Deng Y. Rectal Cancer in Asian vs. Western Countries: Why the Variation in Incidence? Curr Treat Options Oncol. 2017;18:64
9. Li L, Ma BB. Colorectal cancer in Chinese patients: current and emerging treatment options. Onco Targets Ther. 2014;7:1817-28
10. Vignot S, Lefebvre C, Frampton GM, Meurice G, Yelensky R, Palmer G. et al. Comparative analysis of primary tumour and matched metastases in colorectal cancer patients: evaluation of concordance between genomic and transcriptional profiles. Eur J Cancer. 2015;51:791-9
11. Lee WS, Park YH, Lee JN, Baek JH, Lee TH, Ha SY. Comparison of HER2 expression between primary colorectal cancer and their corresponding metastases. Cancer Med. 2014;3:674-80
12. Mahankali M, Henkels KM, Speranza F, Gomez-Cambronero J. A non-mitotic role for Aurora kinase A as a direct activator of cell migration upon interaction with PLD, FAK and Src. J Cell Sci. 2015;128:516-26
13. Lievre A, Bachet JB, Boige V, Cayre A, Le Corre D, Buc E. et al. KRAS mutations as an independent prognostic factor in patients with advanced colorectal cancer treated with cetuximab. J Clin Oncol. 2008;26:374-9
14. Sawada K, Nakamura Y, Yamanaka T, Kuboki Y, Yamaguchi D, Yuki S. et al. Prognostic and Predictive Value of HER2 Amplification in Patients With Metastatic Colorectal Cancer. Clin Colorectal Cancer. 2018;17:198-205
15. Xu JM, Wang Y, Wang YL, Wang Y, Liu T, Ni M. et al. PIK3CA Mutations Contribute to Acquired Cetuximab Resistance in Patients with Metastatic Colorectal Cancer. Clin Cancer Res. 2017;23:4602-16
16. Sartore-Bianchi A, Trusolino L, Martino C, Bencardino K, Lonardi S, Bergamo F. et al. Dual-targeted therapy with trastuzumab and lapatinib in treatment-refractory, KRAS codon 12/13 wild-type, HER2-positive metastatic colorectal cancer (HERACLES): a proof-of-concept, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016;17:738-46
17. Hainsworth JD, Meric-Bernstam F, Swanton C, Hurwitz H, Spigel DR, Sweeney C. et al. Targeted Therapy for Advanced Solid Tumors on the Basis of Molecular Profiles: Results From MyPathway, an Open-Label, Phase IIa Multiple Basket Study. J Clin Oncol. 2018;36:536-42
18. Dienstmann R, Vermeulen L, Guinney J, Kopetz S, Tejpar S, Tabernero J. Consensus molecular subtypes and the evolution of precision medicine in colorectal cancer. Nat Rev Cancer. 2017;17:79-92
19. Julien S, Merino-Trigo A, Lacroix L, Pocard M, Goere D, Mariani P. et al. Characterization of a large panel of patient-derived tumor xenografts representing the clinical heterogeneity of human colorectal cancer. Clin Cancer Res. 2012;18:5314-28
20. Zhang L, Liu Y, Wang X, Tang Z, Li S, Hu Y. et al. The extent of inflammatory infiltration in primary cancer tissues is associated with lymphomagenesis in immunodeficient mice. Sci Rep. 2015;5:9447
21. Collins AT, Lang SH. A systematic review of the validity of patient derived xenograft (PDX) models: the implications for translational research and personalised medicine. PeerJ. 2018;6:e5981
22. Hand F, Harmon C, Elliott LA, Caiazza F, Lavelle A, Maguire D. et al. Depleted polymorphonuclear leukocytes in human metastatic liver reflect an altered immune microenvironment associated with recurrent metastasis. Cancer Immunol Immunother. 2018;67:1041-52
23. Zou YF, Cai ZR, Chen YF, Rong YM, Wu XR, Yuan RX. et al. [Comparison of local immune microenvironment between liver-metastasis colorectal cancer and non-liver-metastasis colorectal cancer]. Zhonghua Wei Chang Wai Ke Za Zhi. 2013;16:547-51
24. Choi YY, Lee JE, Kim H, Sim MH, Kim KK, Lee G. et al. Establishment and characterisation of patient-derived xenografts as paraclinical models for gastric cancer. Sci Rep. 2016;6:22172
25. Dieter SM, Giessler KM, Kriegsmann M, Dubash TD, Mohrmann L, Schulz ER. et al. Patient-derived xenografts of gastrointestinal cancers are susceptible to rapid and delayed B-lymphoproliferation. Int J Cancer. 2017;140:1356-63
26. Blomme A, Van Simaeys G, Doumont G, Costanza B, Bellier J, Otaka Y. et al. Murine stroma adopts a human-like metabolic phenotype in the PDX model of colorectal cancer and liver metastases. Oncogene. 2018;37:1237-50
27. Linnekamp JF, Hooff SRV, Prasetyanti PR, Kandimalla R, Buikhuisen JY, Fessler E. et al. Consensus molecular subtypes of colorectal cancer are recapitulated in in vitro and in vivo models. Cell Death Differ. 2018;25:616-33
28. Sveen A, Bruun J, Eide PW, Eilertsen IA, Ramirez L, Murumagi A. et al. Colorectal Cancer Consensus Molecular Subtypes Translated to Preclinical Models Uncover Potentially Targetable Cancer Cell Dependencies. Clin Cancer Res. 2018;24:794-806
29. Ding L, Ellis MJ, Li S, Larson DE, Chen K, Wallis JW. et al. Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nature. 2010;464:999-1005
30. Bergamaschi A, Hjortland GO, Triulzi T, Sorlie T, Johnsen H, Ree AH. et al. Molecular profiling and characterization of luminal-like and basal-like in vivo breast cancer xenograft models. Mol Oncol. 2009;3:469-82
31. Gu Q, Zhang B, Sun H, Xu Q, Tan Y, Wang G. et al. Genomic characterization of a large panel of patient-derived hepatocellular carcinoma xenograft tumor models for preclinical development. Oncotarget. 2015;6:20160-76
32. Ben-David U, Ha G, Tseng YY, Greenwald NF, Oh C, Shih J. et al. Patient-derived xenografts undergo mouse-specific tumor evolution. Nat Genet. 2017;49:1567-75
33. Gao H, Korn JM, Ferretti S, Monahan JE, Wang Y, Singh M. et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat Med. 2015;21:1318-25
34. Takahashi Y, Sheridan P, Niida A, Sawada G, Uchi R, Mizuno H. et al. The AURKA/TPX2 axis drives colon tumorigenesis cooperatively with MYC. Ann Oncol. 2015;26:935-42
35. Montero JC, Seoane S, Ocana A, Pandiella A. Inhibition of SRC family kinases and receptor tyrosine kinases by dasatinib: possible combinations in solid tumors. Clin Cancer Res. 2011;17:5546-52
36. Kim YS, Jeong H, Choi JW, Oh HE, Lee JH. Unique characteristics of ARID1A mutation and protein level in gastric and colorectal cancer: A meta-analysis. Saudi J Gastroenterol. 2017;23:268-74
37. Yonesaka K, Zejnullahu K, Okamoto I, Satoh T, Cappuzzo F, Souglakos J. et al. Activation of ERBB2 signaling causes resistance to the EGFR-directed therapeutic antibody cetuximab. Sci Transl Med. 2011;3:99ra86
38. Morikawa T, Kuchiba A, Yamauchi M, Meyerhardt JA, Shima K, Nosho K. et al. Association of CTNNB1 (beta-catenin) alterations, body mass index, and physical activity with survival in patients with colorectal cancer. JAMA. 2011;305:1685-94
39. Chen Z, He X, Jia M, Liu Y, Qu D, Wu D. et al. beta-catenin overexpression in the nucleus predicts progress disease and unfavourable survival in colorectal cancer: a meta-analysis. PLoS One. 2013;8:e63854
40. El-Deiry WS, Vijayvergia N, Xiu J, Scicchitano A, Lim B, Yee NS. et al. Molecular profiling of 6,892 colorectal cancer samples suggests different possible treatment options specific to metastatic sites. Cancer Biol Ther. 2015;16:1726-37
41. Fujiyoshi K, Yamamoto G, Takenoya T, Takahashi A, Arai Y, Yamada M. et al. Metastatic Pattern of Stage IV Colorectal Cancer with High-Frequency Microsatellite Instability as a Prognostic Factor. Anticancer Res. 2017;37:239-47
42. Ramanathan RK, Hwang JJ, Zmaboni WC, Sinicrope FA, Safran H, Wong MK. et al. Low overexpression of HER-2/PPPNeu in advanced colorectal cancer limits the usefulness of trastuzumab (herceptin (R)) and irinotecan as therapy. A phase II trial. Cancer Invest. 2004;22:858-65
43. Wang X, Sun Y, Gao J, Zheng J, Shen L. [Human epidermal growth factor receptor 2 expression in rectal cancer and its clinical implication]. Zhonghua Wei Chang Wai Ke Za Zhi. 2015;18:597-601
44. Li H, Yu C, Jiang J, Huang C, Yao X, Xu Q. et al. An anti-HER2 antibody conjugated with monomethyl auristatin E is highly effective in HER2-positive human gastric cancer. Cancer Biol Ther. 2016;17:346-54
45. Kavuri SM, Jain N, Galimi F, Cottino F, Leto SM, Migliardi G. et al. HER2 activating mutations are targets for colorectal cancer treatment. Cancer Discov. 2015;5:832-41
46. Carter JH, Cottrell CE, McNulty SN, Vigh-Conrad KA, Lamp S, Heusel JW. et al. FGFR2 amplification in colorectal adenocarcinoma. Cold Spring Harb Mol Case Stud. 2017;3:a001495
47. Mathur A, Ware C, Davis L, Gazdar A, Pan BS, Lutterbach B. FGFR2 is amplified in the NCI-H716 colorectal cancer cell line and is required for growth and survival. PLoS One. 2014;9:e98515
48. Delpuech O, Rooney C, Mooney L, Baker D, Shaw R, Dymond M. et al. Identification of Pharmacodynamic Transcript Biomarkers in Response to FGFR Inhibition by AZD4547. Mol Cancer Ther. 2016;15:2802-13
49. Jang J, Kim HK, Bang H, Kim ST, Kim SY, Park SH. et al. Antitumor Effect of AZD4547 in a Fibroblast Growth Factor Receptor 2-Amplified Gastric Cancer Patient-Derived Cell Model. Transl Oncol. 2017;10:469-75
50. Pearson A, Smyth E, Babina IS, Herrera-Abreu MT, Tarazona N, Peckitt C. et al. High-Level Clonal FGFR Amplification and Response to FGFR Inhibition in a Translational Clinical Trial. Cancer Discov. 2016;6:838-51
51. Van Cutsem E, Bang YJ, Mansoor W, Petty RD, Chao Y, Cunningham D. et al. A randomized, open-label study of the efficacy and safety of AZD4547 monotherapy versus paclitaxel for the treatment of advanced gastric adenocarcinoma with FGFR2 polysomy or gene amplification. Ann Oncol. 2017;28:1316-24
52. Chen G, Gao C, Gao X, Zhang DH, Kuan SF, Burns TF. et al. Wnt/beta-Catenin Pathway Activation Mediates Adaptive Resistance to BRAF Inhibition in Colorectal Cancer. Mol Cancer Ther. 2018;17:806-13
53. Ganesh S, Shui X, Craig KP, Koser ML, Chopda GR, Cyr WA. et al. beta-Catenin mRNA Silencing and MEK Inhibition Display Synergistic Efficacy in Preclinical Tumor Models. Mol Cancer Ther. 2018;17:544-53
54. Guardavaccaro D, Clevers H. Wnt/beta-catenin and MAPK signaling: allies and enemies in different battlefields. Sci Signal. 2012;5:pe15
55. Garralda E, Paz K, Lopez-Casas PP, Jones S, Katz A, Kann LM. et al. Integrated next-generation sequencing and avatar mouse models for personalized cancer treatment. Clin Cancer Res. 2014;20:2476-84
56. Wang J, Liu Z, Wang Z, Wang S, Chen Z, Li Z. et al. Targeting c-Myc: JQ1 as a promising option for c-Myc-amplified esophageal squamous cell carcinoma. Cancer Lett. 2018;419:64-74
Author biography
 Jingyuan Wang obtained her bachelor's degree from Nanjing Medical University in 2015. She is currently a Ph.D. student in Peking University under the supervision of Prof. Lin Shen. Her research is centered on the establishment and characterization of pathological and molecular features of PDX models.
Jingyuan Wang obtained her bachelor's degree from Nanjing Medical University in 2015. She is currently a Ph.D. student in Peking University under the supervision of Prof. Lin Shen. Her research is centered on the establishment and characterization of pathological and molecular features of PDX models.
 Baocai Xing is a professor at Hepatopancreatobiliary Surgery Department I, Peking University Cancer Hospital & Institute. He specialized in colorectal cancer with liver metastases. He has published many famous papers on famous journals, such as Cancer Cell, Nature, Nucleic Acids Research on the field of colorectal cancer metastasis and liver cancer.
Baocai Xing is a professor at Hepatopancreatobiliary Surgery Department I, Peking University Cancer Hospital & Institute. He specialized in colorectal cancer with liver metastases. He has published many famous papers on famous journals, such as Cancer Cell, Nature, Nucleic Acids Research on the field of colorectal cancer metastasis and liver cancer.
 Jing Gao is an associate professor at Department of GI Oncology, Peking University Cancer Hospital & Institute. She got her PhD's degree in Peking Union Medical College Hospital. She focuses on precision medicine of advanced gastric cancer, colorectal cancer with liver metastases, gastrointestinal stromal tumor and other gastrointestinal malignancies.
Jing Gao is an associate professor at Department of GI Oncology, Peking University Cancer Hospital & Institute. She got her PhD's degree in Peking Union Medical College Hospital. She focuses on precision medicine of advanced gastric cancer, colorectal cancer with liver metastases, gastrointestinal stromal tumor and other gastrointestinal malignancies.
 Lin Shen, professor and doctoral supervisor, is the deputy director of Beijing cancer hospital, the director of the department of gastrointestinal oncology and phase I clinical ward. Engaged in the precision drug treatment and multidisciplinary treatment, clinical trials and translational research of new drugs in gastrointestinal cancers. She formulated the Asia/China guidelines for the diagnosis and treatment of gastric cancer, the China guidelines for the diagnosis and treatment of colorectal cancer/GIST. All these guidelines or books became the criteria in clinical practice.
Lin Shen, professor and doctoral supervisor, is the deputy director of Beijing cancer hospital, the director of the department of gastrointestinal oncology and phase I clinical ward. Engaged in the precision drug treatment and multidisciplinary treatment, clinical trials and translational research of new drugs in gastrointestinal cancers. She formulated the Asia/China guidelines for the diagnosis and treatment of gastric cancer, the China guidelines for the diagnosis and treatment of colorectal cancer/GIST. All these guidelines or books became the criteria in clinical practice.
![]() Corresponding authors: Professor Lin Shen, 52 Fucheng Road, Haidian District, Beijing, 100142, China. Tel: +86-10-88196561. Email: shenlinedu.cn. Professor Jing Gao, 52 Fucheng Road, Haidian District, Beijing, 100142, China. Tel: +86-10-88196747. Email: gaojing_pumccom.
Corresponding authors: Professor Lin Shen, 52 Fucheng Road, Haidian District, Beijing, 100142, China. Tel: +86-10-88196561. Email: shenlinedu.cn. Professor Jing Gao, 52 Fucheng Road, Haidian District, Beijing, 100142, China. Tel: +86-10-88196747. Email: gaojing_pumccom.