Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(12):3459-3475. doi:10.7150/thno.33292 This issue Cite
Research Paper
RUNX2 overexpression and PTEN haploinsufficiency cooperate to promote CXCR7 expression and cellular trafficking, AKT hyperactivation and prostate tumorigenesis
Yang Bai1,2*, Yinhui Yang1,2*, Yuqian Yan2, Jian Zhong2, Alexandra M. Blee2, Yunqian Pan2, Tao Ma2, R. Jeffrey Karnes3, Rafael Jimenez5, Wanhai Xu1 ![]() , Haojie Huang2,3,4
, Haojie Huang2,3,4 ![]()
1. Heilongjiang Key Laboratory of Scientific Research in Urology and Department of Urology, the Fourth Hospital of Harbin Medical University, Harbin, Heilongjiang Province, 150081, China.
2. Department of Biochemistry and Molecular Biology, Mayo Clinic College of Medicine and Science, Rochester, MN 55905, USA;
3. Department of Urology, Mayo Clinic College of Medicine and Science, Rochester, MN 55905, USA;
4. Mayo Clinic Cancer Center, Mayo Clinic College of Medicine and Science, Rochester, MN 55905, USA;
5. Department of Laboratory Medicine and Pathology, Mayo Clinic College of Medicine and Science, Rochester, MN 55905, USA.
*These authors contributed equally.
Received 2019-1-18; Accepted 2019-5-6; Published 2019-5-24
Abstract

Rationale: The overall success rate of prostate cancer (PCa) diagnosis and therapy has been improved over the years. However, genomic and phenotypic heterogeneity remains a major challenge for effective detection and treatment of PCa. Efforts to better classify PCa into functional subtypes and elucidate the molecular mechanisms underlying prostate tumorigenesis and therapy resistance are warranted for further improvement of PCa outcomes.
Methods: We generated Cre+;Runx2-cTg;Ptenp/+ (Runx2-Pten double mutant) mice by crossbreeding Cre+;Runx2-cTg males with Pten conditional (Ptenp/p) females. By using Hematoxylin and Eosin (H&E) staining, SMA and Masson's Trichrome staining, we investigated the effect of PTEN haploinsufficiency in combination with Runx2 overexpression on prostate tumorigenesis. Moreover, we employed immunohistochemistry (IHC) to stain Ki67 for cell proliferation, cleaved caspase 3 for apoptosis and AKT phosphorylation for signaling pathway in prostate tissues. Chromatin immunoprecipitation coupled quantitative PCR (ChIP-qPCR), reverse transcription coupled quantitative PCR (RT-qPCR), western blot (WB) analyses and immunofluorescence (IF) were conducted to determine the underlying mechanism by which RUNX2 regulates CXCR7 and AKT phosphorylation in PCa cells.
Results: We demonstrated that mice with prostate-specific Pten heterozygous deletion and Runx2 overexpression developed high-grade prostatic intraepithelial neoplasia (HGPIN) and cancerous lesions at age younger than one year, with concomitant high level expression of Akt phosphorylation and the chemokine receptor Cxcr7 in malignant glands. RUNX2 overexpression induced CXCR7 transcription and membrane location and AKT phosphorylation in PTEN-deficient human PCa cell lines. Increased expression of RUNX2 also promoted growth of PCa cells and this effect was largely mediated by CXCR7. CXCR7 expression also positively correlated with AKT phosphorylation in PCa patient specimens.
Conclusions: Our results reveal a previously unidentified cooperative role of RUNX2 overexpression and PTEN haploinsufficiency in prostate tumorigenesis, suggesting that the defined RUNX2-CXCR7-AKT axis can be a viable target for effective treatment of PCa.
Keywords: RUNX2, PTEN, CXCR7, tumorigenesis, prostate cancer
Introduction
Prostate cancer (PCa) is the second leading cause of cancer death among American men[1]. Despite the high incidence and mortality rate, there is an incomplete understanding of the full repertoire of clinically relevant molecular alterations responsible for prostate oncogenesis and progression. This remains a critical barrier that slows progress toward improving treatment in a significant subset of PCa patients.
Complete or partial loss of the tumor suppressor PTEN frequently occurs in localized and advanced/castration-resistant PCa [2-4], indicating that PTEN deficiency plays an important role in both PCa development and progression. PTEN primarily functions as a lipid phosphatase that specifically catalyzes dephosphorylation of the 3' phosphate of the inositol ring in phosphatidylinositol (3,4,5)-trisphosphate (PtdIns (3,4,5) P3 or PIP3) [5]. In agreement of this finding, homozygous loss of the Pten gene results in hyperactivation of Akt and thereby promotes formation of HGPIN and PCa in mice. However, Pten heterozygous deletion alone is insufficient to induce prostate tumorigenesis in mice only until very advanced ages [6, 7], suggesting that cooperating oncogenic lesions are required. Thus, it is both clinically and biologically significant to study how partial loss of PTEN works cooperatively with other genetic lesion(s) to promote PCa initiation and progression.
Runt-related transcription factor 2 (RUNX2) (also known as core-binding factor subunit alpha-1 (CBF-alpha-1)) plays a crucial role in regulation of cell proliferation in osteoblasts and endothelial cells. RUNX2 is also implicated in human PCa [8, 9] and RUNX2 overexpression has been linked to upregulation of matrix metalloproteinases, secreted bone resorbing factors, and PCa cell metastasis to bone [9-12]. We have demonstrated previously that elevated level of RUNX2 protein correlates with decreased expression of PTEN protein in a cohort of human PCa specimens [13], and others have shown that Runx2 is upregulated in prostate tumors in Pten knockout mice [14]. However, whether concomitant RUNX2 overexpression and PTEN deficiency play a causal role in prostate tumorigenesis and PCa progression remains elusive.
In this study, we generated a novel Runx2 conditional transgenic mouse model (Runx2-cTg). Characterization of this model revealed that Runx2 expression in combination with Pten heterozygous deletion (Pb-Cre+;Runx2-cTg;Ptenp/+) in the prostate leads to increased cell proliferation and formation of high-grade PIN and cancerous lesions. We further showed that the Akt kinase is highly phosphorylated in prostate tumors in Pb-Cre+;Runx2-cTg;Ptenp/+ mice, but not in nonmalignant prostate tissues in Pb-Cre+;Ptenp/+ mice. The concomitant Runx2 overexpression and Pten deficiency induce upregulation of the chemokine receptor Cxcr7, which is required for Akt hyperphosphorylation in prostate tumors in Runx2-Pten double mutant mice. We further showed that CXCR7 expression correlates with phosphorylated AKT in PCa specimens in patients and that CXCR7 is crucial for RUNX2-mediated growth of PTEN-deficient PCa cells.
Materials and Methods
Plasmids, antibodies, and reagents
CMV-driven mammalian expression vector for HA-tagged mouse Runx2 was described previously [13]. To generate a Runx2 transgene vector, HA-Runx2 was subcloned into a conditional vector, which was kindly provided by David Largaespada from the University of Minnesota (Minneapolis, MN). In this plasmid, HA-Runx2 was inserted after the LoxP-Stop-LoxP cassette and therefore it was termed as pLSL-HA-Runx2. pCMV-Cre plasmid was kindly provided by Jan van Deursen from Mayo Clinic (Rochester, MN). Antibodies used for western blot analysis were: anti-RUNX2 (12556S, Cell Signaling Technology); anti-ERK2 (sc1647, Santa Cruz Biotechnology); anti-HA 1.1 (Covance); anti-PTEN (9559L, Cell Signaling Technology); anti-pS473 AKT (9271S, Cell Signaling Technology); anti-AKT (9272S, Cell Signaling Technology); anti-pS473 AKT (4060L, Cell Signaling Technology); anti-CXCR7 (ab38089, Abcam); anti-P70 (9202S, Cell Signaling Technology); anti-p-P70 (9205S, Cell Signaling Technology); anti-rabbit or anti-mouse secondary antibodies (GE Healthcare UK Limited). Antibodies used for IHC were: anti-AR (ab108341, Abcam); anti-Ki-67 (ab15580, Abcam); anti-pS473 AKT (4060L, Cell Signaling Technology); anti-pS235/236-S6 (2211S, Cell Signaling Technology); anti-CXCR7 (MAB42273, R&D); anti-PTEN (9559L, Cell Signaling Technology); anti-RUNX2 (12556S, Cell Signaling Technology); biotinylated anti-rabbit or anti-mouse (BA-1000, BA-9200, Vector Lab).
Cell lines, cell culture and transfection
LNCaP, PC-3, LAPC-4 and 293T cells were purchased from ATCC (Manassas, VA). LNCaP, PC-3 cells were cultured in RPMI 1640 medium (Corning cellgro) supplemented with 10% fetal bovine serum (FBS) (Thermo Fisher Scientific). LAPC-4 cells were cultured in Iscove's modified Dulbecco's medium (Corning cellgro) supplemented with 15% FBS (Thermo Fisher Scientific). 293T cells for lentiviral packaging were cultured in Dulbecco's modified Eagle's medium (Corning cellgro) supplemented with 10% FBS (Thermo Fisher Scientific). Transfections were performed using Lipofectatmine2000 (Thermo Fisher Scientific), following manufacturer's instructions. 75-90% transfection efficiencies were achieved.
Clonogenic assays
The procedure was performed as described previously [15, 16]. Briefly, an appropriate number of cells for different drug treatments were plated onto 6-well plates. At the following day, cells were treated with 0.1% BSA or CXCL12 (200 ng/ml, Cat#300-28A, PeproTech). Around 10 days later, colonies were fixed with acetic acid:methanol (1:7) for 60 min and stained with (0.5% w/v) crystal violet for 1 h, followed by a gentle wash with running tap water. Colonies with more than 50 cells were counted. The linear regression was applied to generate the survival curve.
Generation of mutant mice
All animal study was approved by the Mayo Clinic Institutional Animal Care and Use Committee (IACUC). All mice were housed in standard conditions with a 12 hour light/12 hour dark cycle and access to food and water ad libitum. Runx2 conditional transgenic (Runx2-cTg) mice (FVB) were generated by Mayo Clinic core facility. Probasin (Pb)-driven Cre4 recombinase transgenic mice (C57BL/DBA2), originally generated in the laboratory of Dr. Pradip Roy-Burnam at University of Southern California, Los Angeles, CA [17], were acquired from the National Cancer Institute (NCI) Mouse Repository. Pten Loxp/Loxp conditional mice (129/Balb/c), originally generated in the laboratory of Dr. Hong Wu at University of California, Los Angeles, CA [18], were acquired from Jackson Laboratory (004597). Littermates with similar genetic backgrounds were used as controls throughout the study. PCR primers for genotyping of wild-type and conditional alleles of Pten, transgenic HA-Runx2, and Cre are listed in Supplementary Table S1.
RNA interference (RNAi)
Cells were transfected with control siRNA (siN05815122147, RIBOBIO), siRUNX2 pool (M-012665-01, Thermo Fisher Scientific Dharmacon), or siPTEN (M-003023-02, Sigma) following manufacturer's instructions. Control shRNA or CXCR7-specific shCXCR7 (NM_020311, Sigma) was transfected into 293T cells. Cells were treated with 1:100 sodium pyruvate 24 h after transfection. After another 24 h, media containing packaged virus was filtered with 0.45 μm filters and added to target cells. Polybrene was added to a final concentration of 8 μg/ml. Puromycin selection was performed to remove any cells without successful lentiviral transduction.
Western blot
Approximately 80 μg of protein from cell lysates or homogenized mouse prostate tissue was denatured in sample buffer (Thermo Fisher Scientific) with added protease inhibitor cocktail (Sigma-Aldrich), subjected to SDS-polyacrylamide gel electrophoresis (Bio-Rad) and transferred to nitrocellulose membranes (Thermo Fisher Scientific). Membranes were blocked in 1X TBST with 5% milk for 1 h at room temperature and immunoblotted with indicated primary antibodies at 4°C overnight. Membranes were incubated at room temperature for 1 h with horseradish peroxidase-conjugated secondary antibodies. Blots were visualized by SuperSignal West Pico Luminol Enhancer Solution (Thermo Fisher Scientific).
Reverse transcription quantitative polymerase chain reaction (RT-qPCR)
Total RNA was isolated from cells in culture or from mouse prostate tissues using Trizol. cDNA was synthesized using a GoScript kit (Promega). Two-step real-time polymerase chain reaction (PCR) was performed using the SYBR Green Mix (Bio-Rad) and C1000 Touch Thermal Cycler, CFX96 Real-Time System (Bio-Rad) according to manufacturer's instructions. Primers were used at a final concentration of 500 nM. GAPDH was used as an internal control. Primers for RT-qPCR are listed in Supplementary Table S1.
MTS assay for cell proliferation
Cell growth was measured by absorbance at 490 nm using a MTS assay kit (Promega) according to manufacturer's instructions. PC-3 cells were plated in 96-well plates with approximately 800 cells per well. At indicated time points, CellTiter 96R Aqueous One Solution Reagent was added to a final concentration of 10% (V/V). Absorbance was measured after 60 min incubation at 37ºC.
Hematoxylin and Eosin (H&E) staining and histological analysis
4 μm thick sections were cut from formalin-fixed paraffin-embedded (FFPE) tissues. Sides were de-paraffinized with xylene and rehydrated through a graded series of alcohol. Slides were incubated in Hematoxylin (Sigma-Aldrich) for 10 min before alcohol washing and incubation in 1% Eosin (Richard-Allan Scientific) for 90 seconds. After further washes with graded alcohol and a final wash in xylene to dehydrate slides, stained tissue was covered with cytoseal glue and coverslips. Presence of PIN lesions was scored based on previously published criteria [19], where mouse PIN I and II were classified as low-grade PIN (LGPIN) and mouse PIN III and IV were classified as high-grade PIN (HGPIN).
Immunohistochemistry (IHC)
4 μm thick sections were cut from FFPE tissues. Antigen retrieval and immunostaining was performed as described previously [20]. For quantitative analysis, a staining index (SI) for five randomly selected microscopic fields (400X) was calculated: SI = staining intensity × staining percentage and averaged to obtain the mean. Staining intensity scores ranged from 0-3 (0 = no staining, 1 = low staining, 2= medium staining, 3 = strong staining).
Masson's trichrome staining
Four-micrometer thick sections were cut from FFPE tissues and stained with Masson's trichrome statin kit (KTMTR, American MasterTech) by followed the manufacturer's instructions. Briefly, the sections were deparaffinized with xylene and rehydrated through a series of ethanol (100% > 95% > 80% > 70%) and running tap water. The sections were incubated with Bouin solution overnight at room temperature. After a thorough rinse with running tap water, the slides were stained with Weigert's hematoxylin for 5 mins and rinsed with tap water, and followed by incubation with Biebrich Scarlet-Acid Fuchsin solution for 15 minutes. The slides were incubated with phosphomolybdic-phosphotungstic acid solution for 10 to 15 mins and immediately stained with Aniline blue for 10 mins. After a wash with tap water, the slides were incubated with Acetic acid for 5 mins, and dehydrated with a series of ethanol (70% > 95% > 100%) and followed by 3 changes of xylene.
Human PCa tissue specimens
Human PCa tissue microarray (TMA) was purchased from US Biomax, Inc (Rockville, MD). Samples were de-identified to all individuals involved in this study. TMA specimens were used for antigen retrieval and immunohistochemistry as described under IHC. Primary antibodies used were anti-CXCR7 (MAB42273, R&D) and anti-pS473 AKT (9271S, Cell Signaling Technology).
Statistical Analysis
All experiments were conducted with three or more replicates unless noted otherwise. Statistical analyses were performed by Student's t-test unless noted otherwise. Values with P < 0.05 are considered statistically significant.
Results
Generation of Runx2 conditional transgenic (Runx2-cTg) mice
It has been shown previously that homozygous deletion of Pten substantially increased Runx2 protein expression in the mouse prostate [14]. In agreement with this observation, analysis of our RNA sequencing (RNA-seq) data demonstrated that Runx2 mRNA expression was largely upregulated in Pten homozygously deleted mouse prostate tumors compared to normal prostate tissues (Figure S1A, left). We further confirmed that it was the case at the protein level (Figure S1A, right), indicating that complete loss of PTEN induces RUNX2 expression in prostatic cells. The Cancer Genome Atlas (TCGA) PCa dataset shows that among tumors with PTEN defects, approximately 50% of them exhibit partial loss of PTEN (Figure S1B). We therefore examined the impact of PTEN partial loss on RUNX2 expression in mice. Different from the effect of homozygous deletion, heterozygous deletion of Pten failed to increase Runx2 mRNA and protein expression in the mouse prostate (Figure 1A, S1A, S1C), arguing that the Pten gene dose is critical in controlling Runx2 gene expression in mouse prostatic cells. Notably, we have shown previously that overexpression of RUNX2 protein co-occurs with partial loss of PTEN protein in PCa specimens in patients [13]. The data obtained from both human and mouse PCa indicate that while partial loss of PTEN expression is not necessarily able to induce RUNX2 overexpression in PCa, it appears that both events occur concomitantly in malignant prostatic cells. Therefore, we were very interested to employ genetically engineered mouse (GEM) models to determine whether RUNX2 overexpression in combination with partial loss of PTEN plays a causal role in prostate tumorigenesis and progression.
Generation of Runx2 conditional transgenic (Runx2-cTg) mice. (A) Western blot showing Runx2 levels in “wild-type” (Cre-;Ptenp/+) or Pten heterozygous (Cre+;Ptenp/+) murine prostate tissues. Erk2 was used as a loading control. (B) Schematic of HA-Runx2 conditional transgenic construct pLSL-HA-Runx2. (C) Western blot for HA-tagged Runx2 in LNCaP cells transfected with CMV-Runx2 (positive control) or the pLSL-HA-Runx2 plasmid in combination with or without CMV-Cre. (D) Agarose gel shows the genotyping results of two independent transgenic founder Runx2-cTg mice. (E), Western blot showing Runx2 levels in “wild-type” (Cre-;Runx2-cTg) and Runx2 transgenic (Cre+;Runx2-cTg) murine prostate tissues.
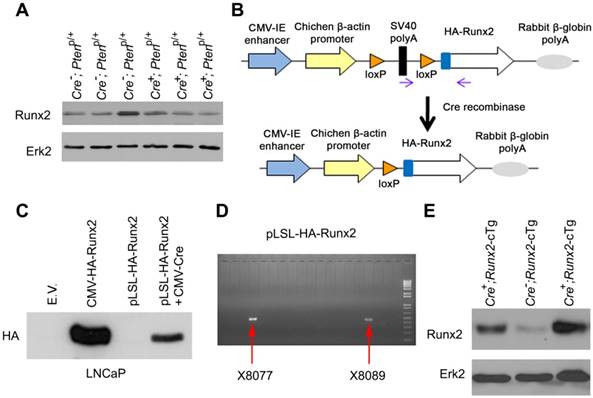
The promoter of mouse Pb gene is often used to generate prostate-specific Cre recombinase transgenic mice [17, 21]. Given that Pten deletion inhibits Pb gene expression by downregulating AR protein in the mouse prostate [22-24], it is possible that Pten deletion may impair Pb promoter-mediated transgene expression in the mouse prostate. To avoid this potential problem, we cloned HA-tagged Runx2 into a conditional expression vector where a LoxP-Stop-LoxP cassette is located between the promoter and the transcription start site of HA-Runx2 transgene (termed pLSL-HA-Runx2) (Figure 1B). pLSL-HA-Runx2 vector was transfected with or without CMV-Cre recombinase expression vector into AR-expressing LNCaP PCa cells. We demonstrated that HA-Runx2 protein was detected only in LNCaP cells co-transfected with CMV-Cre, but not in Cre non-transfected LNCaP cells (Figure 1C), indicating that the pLSL-HA-Runx2 vector worked as expected. This plasmid was linearized and utilized to generate Runx2-cTg mice, and two founder mice were obtained (Figure 1D). Cre-positive and -negative Runx2 transgenic mice were generated by crossbreeding Runx2-cTg mice with Pb-driven Cre recombinase transgenic mice (Pb-Cre4) as reported previously [17]. Western blot analysis showed that Runx2 protein level was much higher in Cre-positive prostate tissues compared to Cre-negative counterparts and Pten heterozygous deletion prostates (Figure 1E, S1A). These data indicate that we have successfully generated a prostate-specific Runx2-cTg murine model. To our knowledge, it represents the first of the kind which allows for more detailed investigation of the role of RUNX2 overexpression in PCa tumorigenesis and progression.
Runx2 overexpression and Pten heterozygous deletion cooperate to induce HGPIN and cancerous lesions in mice
To determine whether transgenic expression of RUNX2 promotes prostate tumorigenesis and whether overexpression of RUNX2 cooperates with partial loss of PTEN to enhance PCa progression, prostate-specific Cre transgenic males (Pb-Cre4) [17] were crossbred with Runx2-cTg females to generate prostate-specific HA-Runx2-expressing mice (Cre+;Runx2-cTg). We further cross bred Cre+;Runx2-cTg males with Pten conditional (Ptenp/p) females to establish four cohorts of mice: 1) Cre-;Runx2-cTg;Ptenp/+ (“wild-type” littermate control), 2) Cre+;Runx2-cTg (Runx2 transgenic alone), 3) Cre+;Ptenp/+ (Pten heterozygous alone), and 4) Cre+;Runx2-cTg; Ptenp/+ (Runx2-Pten double mutant). Firstly, we have verified the overexpression of RUNX2 in the prostate tissues from both Cre+;Runx2-cTg (Runx2 transgenic alone) and Cre+;Runx2-cTg;Ptenp/+ (Runx2-Pten double mutant) by using prostate tissues from Cre+;Ptenp/p mice as a positive control (Figure S1A, right). H&E staining analyses of mouse prostates demonstrated that Runx2 transgenic mice exhibited no evidence of LGPIN (mouse PIN I and II) or HGPIN (mouse PIN III and IV) [25], precursors of prostate cancer at 4 months of age (Figure 2A, S2A). Consistent with previous reports [6, 7], no HGPIN was detected in the lobes of the prostate from Pten heterozygous mice at 4 months of age, including anterior prostate (AP), ventral prostate (VP), and dorsolateral prostate (DLP) (Figure 2A, S2A). However, LGPIN was detected in approximately 10% of the Pten heterozygous mice at 4 months of age (Figure 2A, S2A).
Runx2 overexpression and Pten heterozygous deletion cooperate to induce HGPIN and cancerous lesions in mice. (A) Quantitative data for 4-month-old mice from each genotype (n = 10/group) with LGPIN, HGPIN and/or cancerous lesions (HGPIN/Ca) or no lesion (nonmalignant). (B) H&E staining of DLP and VP from mice with indicated genotypes (n = 10 mice/genotype) at 8 months of age. The high magnification image is corresponding to the framed area in low magnification image in each genotype. Scale bars are indicated in the images. (C) Quantitative analysis of LGPIN, HGPIN and/or cancerous lesion (HGPIN/Ca) or no lesion (nonmalignant) in 8-month-old mice with indicated genotypes (n = 10 mice/genotype). (D) H&E, IHC for smooth muscle actin (SMA) and Masson's Trichrome staining (pointed by red asterisks respectively) in the prostate of mice with five different genotypes (n = 5 mice/genotype) at age of 8 months. Scale bars are indicated in the images.
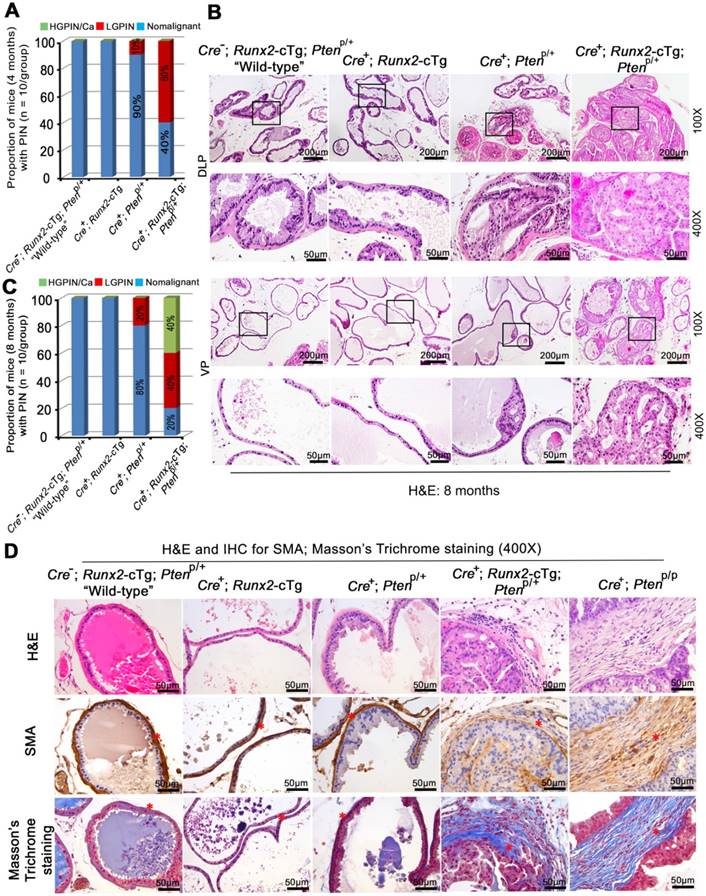
Notably, a much higher percentage of Runx2-Pten mice displayed LGPIN at 4 months compared to Pten heterozygous mice (Figure 2A, S2A). To determine whether tumors in Runx2-Pten mice continue to progress with aging, we examined prostate histology at an extended time-point, 8 months. In additional to LGPIN, focal HGPIN was also detected in the prostates of Runx2-Pten mice at this age (Figure 2B, 2C, S2B). In contrast, no neoplastic changes were observed in the prostate of “wild-type” littermate controls at the same age (Figure 2B, 2C, S2B). Together, these data demonstrate that Pten heterozygous deletion and Runx2 overexpression cooperate to induce HGPIN and cancerous lesions in the mouse prostate within 8 months, while Pten heterozygosity or Runx2 transgenic expression alone do not promote HGPIN to the same extent. This novel observation supports the notion that co-occurrence of reduced expression of PTEN protein and RUNX2 protein overexpression in patient samples is functionally critical in driving prostate tumorigenesis.
We also performed smooth muscle actin (SMA) IHC and detected a highly condensed layer of SMA-positive stroma in the prostate of “wild-type”, Runx2-cTg and Cre+;Ptenp/+ knockout mice (Figure 2D, column 1-3), which is recognized as a natural barrier against the invasion of tumor cells into the adjacent stromal tissue. In contrast, we observed much loosened layers of SMA-positive stroma surrounding malignant epithelial cells in the prostate of Cre+;Runx2-cTg;Ptenp/+ double mutant mice (Figure 2D, column 4), an observation similar to that in prostate tumors in Pten homozygous knockout mice (Cre+;Ptenp/p) (Figure 2D, column 5). Additionally, invasive adenocarcinoma often induces a desmoplastic response, in which collagenous material is diffusely distributed in the surrounding stromal tissue. We performed Masson's trichrome staining that marks keratin and muscle fibers in red and collagen in blue. We detected a relatively condensed layer of collagen surrounding the stroma in the prostate of “wild-type”, Runx2-cTg and Cre+;Ptenp/+ mice (Figure 2D, column 1-3). On the contrary, we found the existence of multiple layers of collagen (blue) integrated with layers of small muscle fibers (pink) in the prostate of Cre+;Runx2-cTg;Ptenp/+ mice (Figure 2D, column 4). The result was also similar to the observation in Pten homozygous knockout (Cre+;Ptenp/p) prostate tumors (Figure 2D, column 5). Thus, similar to the effect of Pten homozygous deletion, Runx2-cTg in combination with Pten heterozygous deletion can induce tumor microenvironment alterations such as extracellular matrix remodeling and aberrant collagen synthesis and deposit, an aggressive phenotype similarly seen in other genetically engineered mouse models such as Pten homozygous deletion mice.
Runx2 is critical for neoplastic prostatic cell proliferation in vitro and in mice
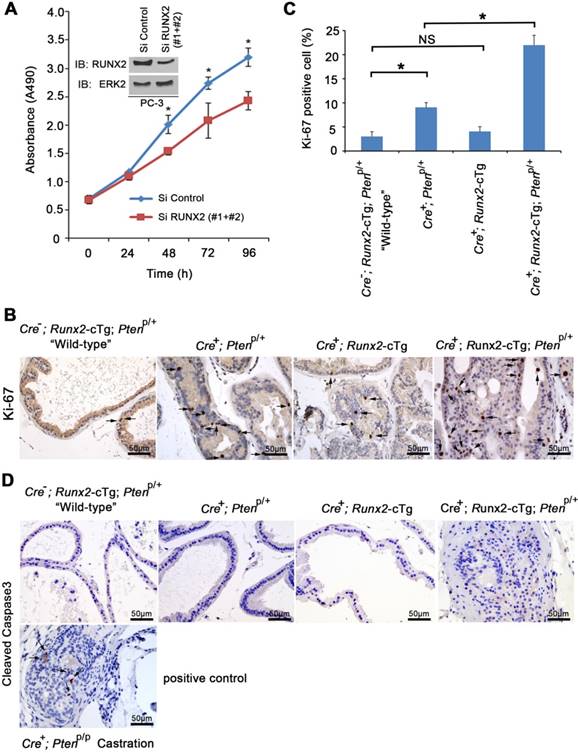
To better understand molecular mechanisms driving HGPIN formation in Runx2-Pten mice at 8 months of age, we first examined cell proliferation. RUNX2 is known to regulate endothelial cell cycle progression [26]. In addition, we and others have demonstrated that RUNX2 is highly expressed in the rapidly proliferative, PTEN-negative PC-3 human PCa cell line [9, 13]. Not surprisingly, siRNA-mediated knockdown of endogenous RUNX2 decreased PC-3 cell proliferation compared to those transfected with control siRNAs (Figure 3A). To further explore cell proliferation in the mouse model, IHC for Ki-67, a cell proliferation marker, was performed on mouse prostate tissues (Figure 3B). The number of Ki-67-positive cells was significantly increased in the prostates of Pten heterozygous mice compared to “wild-type” littermate controls, and this effect was further enhanced by concomitant Runx2 overexpression in Runx2-Pten mice (Figure 3B, 3C). In agreement with the histological characterization (Figure 2), Runx2 overexpression alone in Runx2 transgenic mice did not significantly increase Ki-67-positive cells. Thus, in addition to the established role of Runx2 in PCa cell invasion and migration [9, 13], our data highlights that Runx2 plays an important role in proliferation of both PTEN-negative PCa cells in culture and in the mouse prostate with reduced expression of Pten. IHC analysis of expression of cleaved caspase 3, a marker of apoptosis showed no overt alteration in the number of apoptotic cells among all groups of mice examined in 8 months of age (Figure 3D). As a positive control, positive staining of cleaved caspase 3 was detected in Pten homozygous knockout prostate tumors in castrated mice (Figure 3D), which is consistent with the previous report [18]. Together, these data support the notion that cell proliferation is a major contributor to prostate tumorigenesis induced by partial loss of PTEN and RUNX2 overexpression.
Runx2 regulates proliferation of neoplastic prostatic cells in vitro and in mice. (A) MTS assay measuring proliferation of PC-3 cells transfected with control or RUNX2-specific siRNAs at different time points. Inset, western blot showing the effectiveness of RUNX2 knockdown. ERK2 was used as a loading control. (B) IHC of proliferation marker Ki-67 in prostate tissues collected from mice with the indicated genotypes at age of 8 months. Arrows indicate Ki-67 positive cells. (C) Quantification of Ki-67 positive cells in each indicated mouse genotype. Data are shown as mean ± SD (n = 6 mice/genotype). * P < 0.01, NS, no significance. (D) IHC analysis of cleaved Caspase 3 expression in the prostate of mice with the indicated genotypes at age of 8 months (n = 6 mice/genotype). Cleaved Caspase 3 IHC in PCa tissues from castrated prostate-specific Pten homozygous deletion mice was included as positive control. Photos are representatives of results from 6 mice in each group (n = 6).
Akt is hyperphosphorylated in neoplastic lesions in Runx2-Pten double mutant mice
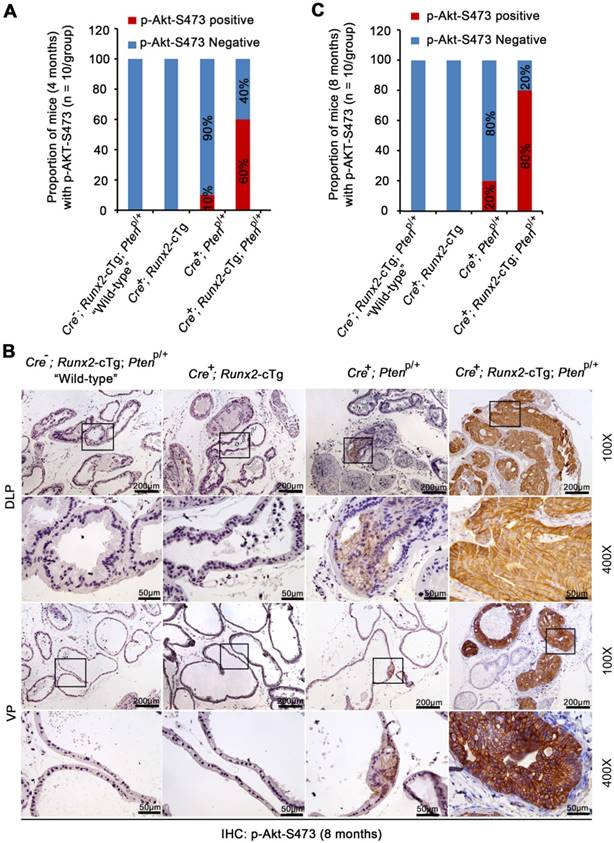
Akt activation is crucial for Pten homozygous deletion-induced prostate tumorigenesis in mice [18]. Consistent with previous reports [27], focal modest Akt phosphorylation (e.g. serine 473 (p-Akt-473)) was detected in a very limited number of prostate acini in Pten heterozygous (Cre+;PtenP/+) mice at 4 or 8 months of age (Figure 4A-C, S3A-B). In contrast, Akt was hyperphosphorylated (indicated by strong plasma membrane staining) in HGPIN in all types of glands (DLP, VP and AP) of Runx2-Pten mice at both 4 and 8 months of age (Figure 4A-C, S3A-B). S6 ribosomal protein is one of the downstream targets of the AKT signaling and S6 phosphorylation is often used as a surrogate for AKT signaling activation [28]. Similar to AKT phosphorylation, only modest level of S6 phosphorylation was observed in minimal areas of the prostate from Pten heterozygous mice at 8 months of age (Figure S4A). However, S6 phosphorylation straining was much stronger in HGPIN from Runx2-Pten mice at the same age (Figure S4A); suggesting hyperphosphorylated AKT is functionally active in prostate tumors in double mutant mice.
Akt is hyperphosphorylated in neoplastic lesions in Runx2-Pten mice. (A) Quantitative data of Akt S473 phosphorylation (p-Akt-S473) IHC in the prostate of 4-month-old mice with the indicated genotypes (n = 10 mice/genotype). (B) Representative images of IHC for p-Akt-S473 in DLP and VP of 8 month-old mice with the indicated genotypes (n = 10 mice/genotype). The high magnification image (400X) is corresponding to the framed area in low magnification image (100X) in each genotype. Scale bars are indicated in the images. Scale bars are shown in the images. (C) Quantitative data of p-Akt-S473 IHC in the prostate of 8-month-old mice with the indicated genotypes (n = 10 mice/genotype).
Akt hyperphosphorylation in prostate tumors in Runx2-Pten mice was unlikely due to complete loss of Pten protein in tumor cells, because moderate expression of Pten protein (indicated by both cytoplasmic and nuclear staining) was detected, although the level was lower compared to that in the prostate of “wild-type” littermates (Figure S4B). To ensure there was no large internal deletion in the remaining allele of the Pten gene in prostatic tissues from Runx2-Pten mice, genomic DNA was isolated from the microdissected FFPE prostate tissues for PCR amplification for each exon (Figure S5A). There was no difference in size of PCR products amplified from prostate tissues of the “wild-type” littermate controls, nonmalignant tissues (p-Akt-473 negative) and HGPIN (p-Akt-473 positive) of Runx2-Pten mice (Figure S5B). To rule out the possibility that the hyperphosphorylation of Akt was due to mutations in the undeleted allele of Pten gene, at least in the phosphatase domain, RNA samples were isolated from microdissected prostate tissues of the “wild-type” littermate controls, nonmalignant tissues (p-Akt-473 negative) and HGPIN (p-Akt-473 positive) from Runx2-Pten mice, and RT-PCR product containing exon 5 (encoding the phosphatase domain) (Figure 5C, upper panel) was extracted for Sanger sequencing. The sequencing results indicated that there was no mutation in p-Akt-473 positive HGPIN samples from Runx2-Pten mice (Figure S5C, lower panel). Thus, AKT hyperactivation in HGPIN of Runx2-Pten double mice is unlikely caused by the genomic alterations in the remaining allele of the Pten gene.
Co-regulation of the CXCR7 gene by RUNX2 and PTEN in PCa. (A) UCSC genome browser screenshots showing signal profiles of RUNX2 ChIP-seq (reported previously [32]) in the ACKR3 (CXCR7) gene locus in C4-2B cells. H3K4me1 and H3K4me3 ChIP-seq data were acquired from LNCaP cells as reported previously [52]. P, promoter, NC, negative control region, E, enhancer. (B, C) PC-3 cells were transfected with empty vector (EV) or HA-Runx2 for 24 h for western blot (B) and ChIP-qPCR analysis of RUNX2 binding at the CXCR7 gene promotor and enhancers (C). ERK2 was used as a loading control. All data are shown as mean values ± SD (n = 3). * P < 0.05 comparing RUNX2 binding in EV transfected cells. (D-G) LAPC-4 cells were transfected with EV and HA-Runx2 expression vector in combination with control or PTEN-specific siRNAs (siPTEN) for 48 h followed by ChIP-qPCR analysis of RUNX2 binding at the CXCR7 gene promoter and enhancers (D), RT-qPCR assessment of CXCR7 mRNA expression (E), and western blot analysis of effectiveness of murine Runx2 transfection and PTEN knockdown on expression of p-AKT-473 (F) and CXCR7 protein level in LAPC-4 cells. Quantified data (G) was resulted from the normalization by ERK2 western blot intensity.
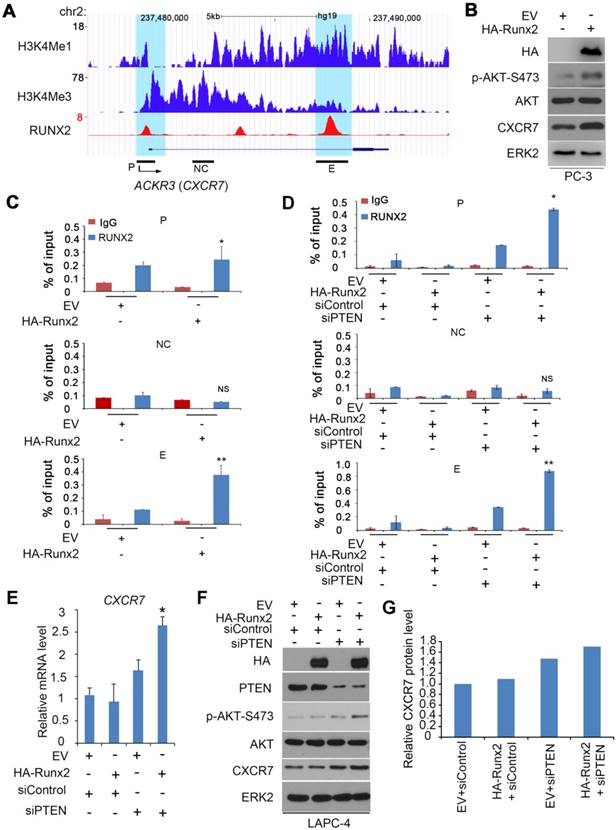
The chemokine receptor CXCR7 is co-regulated by RUNX2 and PTEN in PCa cells
To define the molecular mechanism by which concomitant RUNX2 overexpression and PTEN partial loss induces AKT hyperphosphorylation and activation, we examined expression of the p85 and p110 subunits of PI3K, two upstream activators of AKT as well as mTOR, which activates AKT through the mTOR complex 2 (mTORC2). However, expression of these genes was not affected by ectopic expression of RUNX2 in combination with PTEN siRNAs in PTEN-positive LAPC-4 PCa cell line (data not shown), suggesting that RUNX2 overexpression may promote AKT activation through noncanonical pathway(s).
CXCR7 (encoded by ACKR3 gene) is an atypical G protein-coupled receptor of chemokines such as CXCL12/SDF-1 and it primarily signals through β-arrestin, but not G proteins [29]. CXCR7 expression is upregulated during PCa progression [30]. Intriguingly, increased expression of CXCR7 induces AKT phosphorylation in PCa cells [30], and this effect is likely mediated by CXCR7-dependent recruitment of the adaptor protein β-arrestin which scaffolds AKT to growth factor receptor, although the exact underlying mechanism remains largely unclear [29, 31].
Both CXCR7 and its ligand SDF1 can be upregulated by RUNX2 overexpression in prostate cancer C4-2B cells [32]. Through in-depth analysis of the RUNX2 chromatin immunoprecipitation-sequencing (ChIP-seq) data reported by that study [32], we observed two major putative RUNX2 binding peaks, one in the promoter (marked by high level of H3K4me3, a promoter histone marker) and the other in the enhancer (indicated by high level of H3K4me1, an enhancer histone marker) located in the first intron of CXCR7 gene (Figure 5A). RUNX2 binding at these two sites, but not a non-specific site was further confirmed by ChIP-qPCR in PTEN-negative PC-3 cells without transfection of HA-Runx2 (Figure 5B, 5C). RUNX2 binding at both promoter and enhancer of CXCR7 gene was largely enhanced by ectopic expression of HA-Runx2 (Figure 5B, 5C). In agreement with these findings, overexpression of RUNX2 not only substantially induced CXCR7 protein expression, but also increased AKT phosphorylation in PC-3 cells (Figure 5B). Different from the results in PTEN-deficient PC-3, however, transient overexpression of HA-Runx2 alone had little or no effect on RUNX2 binding at CXCR7 gene promoter and enhancer as well as CXCR7 mRNA and protein expression in PTEN-proficient LAPC-4 cells (Figure 5D-F). These findings imply that PTEN dose is a key factor for RUNX2-mediated expression of CXCR7 in PCa cells. To test this hypothesis, we examined the effect of RUNX2 overexpression on CXCR7 expression in the presence or absence of PTEN knockdown. Only concomitant RUNX2 overexpression and PTEN knockdown, but not RUNX2 overexpression or PTEN knockdown alone, significantly increased RUNX2 binding in the promoter and enhancer of CXCR7 gene (Figure 5D), and the same was true for CXCR7 mRNA and protein expression in LAPC-4 cells (Figure 5E-G). Accordingly, only RUNX2 overexpression in combination with PTEN knockdown substantially increased AKT phosphorylation in PTEN-positive LAPC-4 cells (Figure 5F). These data indicate that PTEN deficiency is required for RUNX2 overexpression-mediated upregulation of CXCR7 expression in PCa cells.
CXCR7 expression correlates with AKT phosphorylation in PCa from mice and patients and is required for RUNX2-mediated AKT activation in PTEN-deficient cells
The findings in cultured PCa cell lines (Figure 5) prompted us to determine whether there is any correlation between CXCR7 expression and AKT phosphorylation (p-AKT) in HGPIN from Runx2-Pten double mutant mice and patients. IHC analysis with sequential tissue sections showed that little or no expression of Cxcr7 and p-Akt was detected in the non-malignant prostate glands in “wild-type”, Runx2-cTg and Cre+;Ptenp/+ knockout mice (Figure 6A, S6A, S6B). IHC staining of Cxcr7 and p-Akt in HGPIN in Runx2-Pten double mutant mice clearly showed the plasma membrane staining of both Cxcr7 and p-Akt and demonstrated that Cxcr7 protein staining positively correlates with hyperphosphorylated Akt in double mutant mice (Figure 6A, S6A, S6B). In contrast, little or no expression of Cxcr7 was detected in HGPIN in Cre+;Ptenp/p (Runx2 wild-type) mice although p-Akt was highly expressed (Figure 6A, S6A, S6B). This data is consistent with the finding that Runx2 protein level was lower in HGPIN of Cre+;Ptenp/p mice compared to double mutant mice (Figure S1A, right), further supporting the importance of the Runx2 protein level in regulation of Cxcr7 expression in PTEN-deficient background. Two groups have shown most recently that CXCR7 is a repression target of AR and increased expression of CXCR7 contributes to castration resistance in prostate cancer [33, 34]. The expression of AR protein was comparable in the prostate of the four groups of mice we studied (Figure S6C), suggesting that increased expression of CXCR7 in the cell membrane in the prostate tumors of Runx2 transgenic-Pten+/- mice was less likely mediated by AR function.
CXCR7 expression correlates with AKT phosphorylation in PCa in Runx2-Pten mice and patient PCa specimens and CXCR7 is required for Runx2-induced PCa cell growth. (A) H&E and IHC for Cxcr7 and S473 phosphorylated Akt (p-Akt-S473) in the prostate tissues of mice with five different genotypes at age of 8 months. Scale bars are indicated in the images. Photos are representatives of results from 3 mice in each group (n = 3). (B-D) IHC for CXCR7 and p-AKT-S473 in PCa specimens in a cohort of 55 patients. Representative CXCR7 and p-AKT-S473 IHC images from two cases of PCa are shown in (B). Heatmap is utilized to summarize IHC of CXCR7 and p-AKT-S473 protein expression in all cases of PCa analyzed (C). Spearman correlation analysis exhibits a positive correlation between CXCR7 and p-AKT-S473 expression in this cohort of patients and the correlation is statistically significant (D). (E-G) PC-3 cells were infected with lentivirus for empty vector or HA-Runx2 in combination with control shRNA (shC) and CXCR7-specific shRNAs (shCXCR7). At 48 h after infection, cells were harvested for western blot analysis of indicated proteins (E) and colony formation assays with representatives of colonies shown in (F) and quantification data shown in (G). Data represent mean values ± SD (n = 3). * P < 0.05; **, P < 0.001.
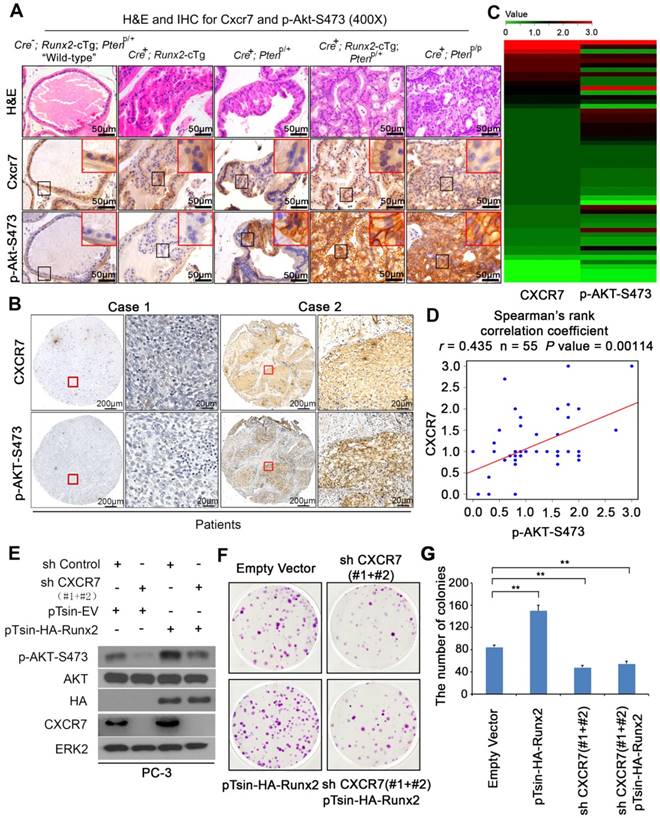
We also performed IHC for CXCR7 and p-AKT-S473 on a tissue microarray (TMA) containing PCa specimens from a cohort of 55 patients. IHC staining was evaluated by measuring both staining intensity and percentage of positive cells. Representative IHC images displaying low/lost or high staining of CXCR7 and p-AKT-S473 are shown in Figure 6B. Further analysis revealed a positive correlation between CXCR7 expression and AKT S473 phosphorylation in these patients (Spearman correlation r = 0.435, P = 0.00114) (Figure 6C-D).
We next examined whether CXCR7 plays a causal role in upregulation of AKT phosphorylation induced by RUNX2 overexpression and PTEN loss in PCa cells. We overexpressed RUNX2 in combination with or without CXCR7 knockdown in PC-3 cells which are PTEN-null, but express high level of endogenous RUNX2 [13]. Knockdown of endogenous CXCR7 largely decreased AKT S473 phosphorylation in PC-3 cells (Figure 6E). Importantly, RUNX2 expression-induced elevation of AKT S473 phosphorylation was abolished by CXCR7 knockdown (Figure 6E). Clonogenic assays demonstrated that RUNX2 overexpression largely increased anchorage-independent growth PC-3 cells and this effect was completely abolished by co-knockdown of CXCR7 (Figure 6F, 6G). Together, these data suggest that RUNX2 overexpression and PTEN loss cooperatively induce CXCR7 expression, which in turn promotes AKT hyperphosphorylation and contributes to prostate tumorigenesis.
To explore the role of co-occurrence of RUNX2 overexpression and PTEN insufficiency in induction of CXCR7 expression and AKT hyperphosphorylation in human PCa samples, we performed meta-analysis of the TCGA data. To our surprise, we found that PTEN deletion status had no correlation with RUNX2 mRNA expression in the cohort of TCGA patients (Figure S7A). The same was true for mRNA expression of CXCR7 and other two RUNX2 target genes BGLAP (encoding osteocalcin) and CXCL8 (encoding IL8) (Figure S7B-D). These data indicate that at mRNA level, expression of RUNX2 and its downstream genes does not correlate with PTEN deletion status in patient samples, at least in the TCGA dataset. We have shown previously that low expression of PTEN protein correlated with high level of RUNX2 protein in a cohort of human prostate cancer [13] and that RUNX2 protein can be targeted for proteasomal degradation in osteoblasts mediated by HECT domain-based E3 ubiquitin ligases such as Smurf1 and Smurf2 and cullin1-based E3 ubiquitin ligases such as Fbw7 and Skp2 [35-37]. We therefore examined whether RUNX2 is a potential proteasomal degradation protein in PCa cells. We demonstrated that treatment of PC-3 cells with two proteasome inhibitors MG132 and bortezomib invariably increased RUNX2 protein level, but no change at mRNA level (Figure S7E, S7F). Together, while our data showed that expression of RUNX2 and its downstream genes does not correlate with PTEN deletion status at mRNA level in patient samples, our new findings indicate that RUNX2 protein is a proteasomal target in PCa, suggesting that the relationship between PTEN loss and RUNX2 elevation in PCa is very complex. Future detailed investigation of their relationship and the impact on the expression of their downstream target genes in human PCa is warranted.
RUNX2 enhances localization of CXCR7 in the plasma membrane
Previous studies have shown that ligand binding triggers internalization of CXCR7 and it can recycle back to the cell surface after internalization despite the continuous presence of ligands [38]. Given that the plasma membrane localization of CXCR7 is important for its receptor functions such as interaction with β-arrestin and AKT activation, we sought to determine whether RUNX2 expression affects endocytic recycling of CXCR7. Beside its expression in the plasma membrane, CXCR7 was also substantially localized in recycling endosomes where it co-localized with TFRC and RAB11, two markers that define the endocytic recycling process [39] (Figure 7A, 7B). Accordingly, we demonstrated that treatment of cells with the lysosomal inhibitor concanamycin A (ConA) enhanced the intensity of CXCR7 localization in the cytosolic compartment (Figure 7C). These data confirmed the recycling feature of CXCR7. Importantly, immunofluorescent cytochemistry (IFC) analysis demonstrated that ectopic expression of RUNX2 increased CXCR7 localization in the plasma membrane (Figure 7D). We further examined the effect of the CXCR7 ligand CXCL12 on phosphorylation of AKT and its downstream signaling and colony formation in PCa cells. We demonstrated that CXCL12 stimulation increased AKT phosphorylation and its downstream effector P70 in PC-3 cells (Figure 7E). Accordingly, colony formation assays demonstrated that CXCL12 treatment increased PC-3 cell proliferation (Figure 7F). These new data further support the notation that increased expression and/or activation of CXCR7 plays an important role in regulation of AKT signaling and prostate oncogenesis.
RUNX2 overexpression facilitates CXCR7 endocytic recycling. (A-B), Co-localization of immunostaining of CXCR7 and two endocytic recycling markers TFRC (A) and RAB11 (B) in LNCaP cells. (C) LNCaP cells were transiently transfected with HA-Runx2 and treated with vehicle (DMSO) and the lysosome inhibitor concanamycin A (ConA) for 16 h. The cells were fixed and immunostained for CXCR7. (D) Overexpression of Runx2 enhanced the distribution of CXCR7 on the plasma membrane as shown by IFC. (E-F) PC-3 cells were treated with vehicle (0.1% BSA) or CXCL12 (200 ng/ml). After 48 h treatment, cells were harvested for western blot analysis of indicated proteins (E) and colony formation assays with representatives of colonies and quantification data shown in (F). *, p85 form of S6 kinase. Data represent mean values ± SD (n = 3). P value was indicated in the image. (G) A hypothetical model wherein RUNX2 overexpression in combination of PTEN lose (PTEN+/- or PTEN-/-) elicits a feed forward signaling loop that leads to increased expression and cellular trafficking of CXCR7, hyperphosphorylation of AKT, and prostate tumorigenesis and progression.
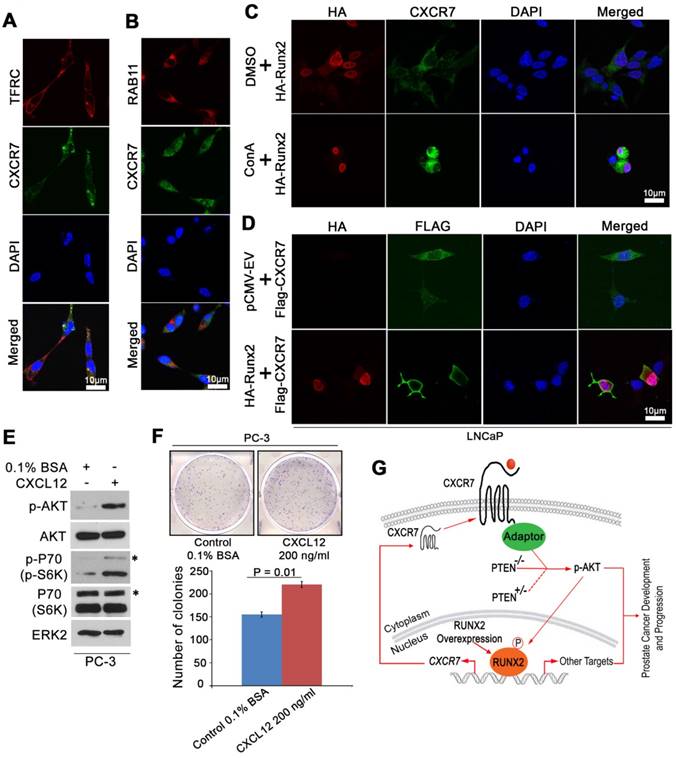
These findings and those described above suggest that CXCR7 is not a canonical binding target of RUNX2 in PTEN-proficient cells, but partial loss of PTEN or RUNX2 overexpression permits RUNX2 to bind to and promote the transcription of CXCR7 and its membrane relocation, ultimately leading to hyperactivation of AKT and development and progression of prostate cancer (Figure 7G).
Discussion
It is increasingly clear that PCa is a heterogeneous disease both at the genetic and clinical levels. Extensive characterization of large patient datasets reveals many distinct genetic subtypes that might explain the variable success of PCa therapies [40-42]. A key barrier to overcoming disease relapse is the clarification of PCa etiologies and understanding of the molecular mechanisms that are distinct to each tumor subtype. Partial loss of PTEN frequently occurs in PCa in patients. However, previous studies have shown that Pten heterozygous deletion alone is insufficient for timely prostate tumorigenesis in mice [6, 7, 43]. Similarly, RUNX2 has long been implicated in PCa progression [9, 13, 44, 45], but its precise role in prostate tumorigenesis in vivo is unclear.
In this study, we generated a Runx2 conditional transgenic Runx2-cTg mouse model. While our results showed that prostate-specific overexpression of Runx2 alone was insufficient to promote prostate tumorigenesis in mice even at 8 months of age, Runx2 overexpression in combination with Pten heterozygosity caused HGPIN and cancerous lesions within 8 months. To our knowledge, this is the first report that identifies a causal role for RUNX2 overexpression and PTEN haploinsufficiency in prostate tumorigenesis. These findings significantly enhance our understanding of PCa etiology, and generation of this unique mouse model may lead to identification of new molecular targets and therapeutic strategies to improve the clinical outcome of PCa patients with alterations in PTEN and RUNX2.
It is well established that Pten homozygous deletion results in Akt hyperphosphorylation and prostate tumorigenesis in mice [18]. In contrast, Pten heterozygous deletion not only rarely drastically induces AKT phosphorylation, but also fails to promote timely tumor formation in the mouse prostate [6, 27], further highlighting the significance of AKT activation in prostate tumorigenesis. In support of this notion, we found Akt was highly phosphorylated in HGPIN and cancerous prostate tissues in Runx2-Pten mice, but not in the counterparts in Runx2 transgenic or Pten heterozygous deletion mice. Our findings imply the presence of a major mechanism by which RUNX2 overexpression works synergistically with partial loss of PTEN to promote AKT hyperphosphorylation in PCa. Whether our findings in PCa can be applied to other cancer types such as glioblastomas, pancreatic or endometrial cancers warrants further investigation.
In an effort to elucidate the underlying mechanism underlying AKT hyperactivation in RUNX2 and PTEN deregulated PCa cells, we first found that AKT hyperactivation in these cells is likely mediated through noncanonical pathway(s). CXCR7 is an atypical G protein-coupled receptor, signaling of which is primarily mediated through the adaptor protein β-arrestin, but not G proteins [29]. In support of this notion, it has been shown that CXCR7 increases cell viability by inducing AKT phosphorylation and this effect is likely mediated through β-arrestin [29, 31]. By analyzing RUNX2 transcriptome in PTEN-deficient C4-2B PCa cells [46], we noticed that CXCR7 is one of the genes upregulated by RUNX2 and this observation is further supported by the meta-analysis of RUNX2 ChIP-seq data [32] showing that there are two major putative RUNX2 binding peaks in the CXCR7 gene locus. Furthermore, we showed that ectopic expression of RUNX2 not only increased RUNX2 protein binding at the CXCR7 gene locus, but also largely increased AKT phosphorylation in another PTEN-null PC-3 PCa cell line. To our surprise, we demonstrated that RUNX2 expression alone failed to do so in PTEN-positive LAPC-4, but in combination with PTEN knockdown RUNX2 overexpression synergistically enhanced its binding at CXCR7 gene locus and increased CXCR7 mRNA and protein expression as well as AKT phosphorylation. Indeed, it has been shown previously that RUNX2 is a direct substrate of AKT and AKT phosphorylation of the runt homology domain (RHD) increases the DNA binding ability of RUNX2 and enhances expression of its downstream genes such as MMP9 and MMP13 [47]. Thus, it is possible that similar to the induction of MMP9 and MMP13, activation of AKT due to PTEN loss can increase RUNX2-mediated transcription of CXCR7 through RUNX2 phosphorylation (Figure 7G). Importantly, this hypothesis is fully supported by our finding that overexpression of RUNX2 alone had little or no induction of CXCR7 in PTEN-positive LAPC-4, but RUNX2 induction of CXCR7 was largely enhanced by PTEN silencing. Moreover, this working model also provides a plausible explanation as to why overexpression of RUNX2 alone is sufficient to upregulate CXCR7 in PTEN-negative prostate cancer cells such as C4-2B and PC-3 as demonstrated in this study and a previous report [32].
CXCR7, as a receptor of chemokines, has been reported to be involved in membrane internalization to facilitate the uptake of chemokines in cancer cells [48]. In this study, we confirmed CXCR7 involvement in endocytic recycling in PCa cells. Importantly, we found that overexpression of RUNX2 potentially accelerated this process. However, the underlying mechanism is unknown. Since the overexpression of RUNX2 might trigger downstream targets that are involved in endocytic recycling, it warrants further investigation to identity such effectors that mediate CXCR7 membrane distribution. Nevertheless, our study uncovers a feed forward loop that promotes Runx2 activation, CXCR7 transcription and membrane relocation, and AKT hyperactivation (Figure 7G). The discovery of this “vicious cycle” is consistent with the previous report that AKT phosphorylates RUNX2 DNA binding domain and enhances RUNX2 binding on its consensus DNA binding element [49].
One possible mediator of the CXCR7-induced hyperphosphorylation of AKT could be the scaffold protein β-arrestin-2, which has been implicated in CXCR7-mediated AKT phosphorylation [29-31]. However, more studies are required to confirm this hypothesis as well as to identify other possible mediators. The novel Runx2-cTg mouse model will be an essential resource to answer these questions, as well as to investigate therapy responsiveness in the context of Pten heterozygosity and Runx2 overexpression, and additional combinations of frequent PCa mutations. This study and those that follow are essential to contribute to a more detailed understanding of many PCa subtypes that are distinct both at the genetic and the therapeutic levels. It is worth noting that CXCR7-specific small molecule inhibitors have been developed and its anti-cancer efficacy has been demonstrated in brain tumors and castration-resistant PCa [50, 51]. These inhibitors could be exploited for chemoprevention and/or effective treatment of the subtype of PCa with RUNX2 overexpression and PTEN heterozygosity.
Abbreviations
PCa: prostate cancer; HGPIN: high-grade prostatic intraepithelial neoplasia; LGPIN: low-grade prostatic intraepithelial neoplasia; RUNX2: Runt-related transcription factor 2; Runx2-cTg: murine Runx2 conditional transgenic mouse model; IACUC: Institutional Animal Care and Use Committee; NCI: National Cancer Institute; RNAi: RNA interference; RT-qPCR: Reverse transcription quantitative polymerase chain reaction; H&E: Hematoxylin and Eosin; FFPE: formalin-fixed paraffin-embedded; IHC: Immunohistochemistry; SI: staining index; TMA: tissue microarray; TCGA: The Cancer Genome Atlas; GEM: genetically engineered mouse; Pb: probasin; AP: anterior prostate; VP: ventral prostate; DLP: dorsolateral prostate; SMA: smooth muscle actin; ChIP-seq: chromatin immunoprecipitation-sequencing; ConA: concanamycin A; RHD: runt homology domain.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This work was supported in part by grants from NIH (CA134514, CA130908, CA193239, and CA203849 to H. Huang), Natural Science Foundation of China (No. 2017YFC0908001, 81611130070 and 81771898 to W. Xu), and Natural Science Foundation of China (No. 2017YFC0908004, 81802534, HYDSYWTS201903 to Y. Yang).
Authors' Contributions
H.H. conceived the study. Y.B., Y.Y., Y.Yan, J.Z., Y.P., T.M. generated mouse models, performed experiments, and analyzed the data. R.J.K. and R.J. supervised and performed patient tissue collection and histological and IHC data analysis. H.H., W.X., A.M.B., Y.Y. and Y.B. wrote the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69:7-34
2. Wang SI, Parsons R, Ittmann M. Homozygous deletion of the PTEN tumor suppressor gene in a subset of prostate adenocarcinomas. Clin Cancer Res. 1998;4:811-5
3. Suzuki H, Freije D, Nusskern DR, Okami K, Cairns P, Sidransky D. et al. Interfocal heterogeneity of PTEN/MMAC1 gene alterations in multiple metastatic prostate cancer tissues. Cancer Res. 1998;58:204-9
4. Cairns P, Okami K, Halachmi S, Halachmi N, Esteller M, Herman JG. et al. Frequent inactivation of PTEN/MMAC1 in primary prostate cancer. Cancer Res. 1997;57:4997-5000
5. Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998;273:13375-8
6. Di Cristofano A, De Acetis M, Koff A, Cordon-Cardo C, Pandolfi PP. Pten and p27KIP1 cooperate in prostate cancer tumor suppression in the mouse. Nat Genet. 2001;27:222-4
7. King JC, Xu J, Wongvipat J, Hieronymus H, Carver BS, Leung DH. et al. Cooperativity of TMPRSS2-ERG with PI3-kinase pathway activation in prostate oncogenesis. Nat Genet. 2009;41:524-6
8. Pratap J, Lian JB, Javed A, Barnes GL, van Wijnen AJ, Stein JL. et al. Regulatory roles of Runx2 in metastatic tumor and cancer cell interactions with bone. Cancer Metastasis Rev. 2006;25:589-600
9. Akech J, Wixted JJ, Bedard K, van der Deen M, Hussain S, Guise TA. et al. Runx2 association with progression of prostate cancer in patients: mechanisms mediating bone osteolysis and osteoblastic metastatic lesions. Oncogene. 2010;29:811-21
10. Baniwal SK, Khalid O, Gabet Y, Shah RR, Purcell DJ, Mav D. et al. Runx2 transcriptome of prostate cancer cells: insights into invasiveness and bone metastasis. Mol Cancer. 2010;9:258
11. Guo ZJ, Yang L, Qian F, Wang YX, Yu X, Ji CD. et al. Transcription factor RUNX2 up-regulates chemokine receptor CXCR4 to promote invasive and metastatic potentials of human gastric cancer. Oncotarget. 2016;7:20999-1012
12. Ito Y, Bae SC, Chuang LS. The RUNX family: developmental regulators in cancer. Nat Rev Cancer. 2015;15:81-95
13. Zhang H, Pan Y, Zheng L, Choe C, Lindgren B, Jensen ED. et al. FOXO1 inhibits Runx2 transcriptional activity and prostate cancer cell migration and invasion. Cancer Res. 2011;71:3257-67
14. Lim M, Zhong C, Yang S, Bell AM, Cohen MB, Roy-Burman P. Runx2 regulates survivin expression in prostate cancer cells. Lab Invest. 2010;90:222-33
15. Franken NA, Rodermond HM, Stap J, Haveman J, van Bree C. Clonogenic assay of cells in vitro. Nat Protoc. 2006;1:2315-9
16. Yan Y, An J, Yang Y, Wu D, Bai Y, Cao W. et al. Dual inhibition of AKT-mTOR and AR signaling by targeting HDAC3 in PTEN- or SPOP-mutated prostate cancer. EMBO Mol Med. 2018:10
17. Wu X, Wu J, Huang J, Powell WC, Zhang J, Matusik RJ. et al. Generation of a prostate epithelial cell-specific Cre transgenic mouse model for tissue-specific gene ablation. Mech Dev. 2001;101:61-9
18. Wang S, Gao J, Lei Q, Rozengurt N, Pritchard C, Jiao J. et al. Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell. 2003;4:209-21
19. Park JH, Walls JE, Galvez JJ, Kim M, Abate-Shen C, Shen MM. et al. Prostatic intraepithelial neoplasia in genetically engineered mice. Am J Pathol. 2002;161:727-35
20. Yemelyanova A, Vang R, Kshirsagar M, Lu D, Marks MA, Shih Ie M. et al. Immunohistochemical staining patterns of p53 can serve as a surrogate marker for TP53 mutations in ovarian carcinoma: an immunohistochemical and nucleotide sequencing analysis. Mod Pathol. 2011;24:1248-53
21. Stanbrough M, Leav I, Kwan PW, Bubley GJ, Balk SP. Prostatic intraepithelial neoplasia in mice expressing an androgen receptor transgene in prostate epithelium. Proc Natl Acad Sci U S A. 2001;98:10823-8
22. Lei Q, Jiao J, Xin L, Chang CJ, Wang S, Gao J. et al. NKX3.1 stabilizes p53, inhibits AKT activation, and blocks prostate cancer initiation caused by PTEN loss. Cancer Cell. 2006;9:367-78
23. Magee JA, Abdulkadir SA, Milbrandt J. Haploinsufficiency at the Nkx3.1 locus. A paradigm for stochastic, dosage-sensitive gene regulation during tumor initiation. Cancer Cell. 2003;3:273-83
24. Jiao J, Wang S, Qiao R, Vivanco I, Watson PA, Sawyers CL. et al. Murine cell lines derived from Pten null prostate cancer show the critical role of PTEN in hormone refractory prostate cancer development. Cancer Res. 2007;67:6083-91
25. Park JH, Walls JE, Galvez JJ, Kim M, Abate-Shen C, Shen MM. et al. Prostatic intraepithelial neoplasia in genetically engineered mice. Am J Pathol. 2002;161:727-35
26. Qiao M, Shapiro P, Fosbrink M, Rus H, Kumar R, Passaniti A. Cell cycle-dependent phosphorylation of the RUNX2 transcription factor by cdc2 regulates endothelial cell proliferation. J Biol Chem. 2006;281:7118-28
27. Kim MJ, Cardiff RD, Desai N, Banach-Petrosky WA, Parsons R, Shen MM. et al. Cooperativity of Nkx3.1 and Pten loss of function in a mouse model of prostate carcinogenesis. Proc Natl Acad Sci U S A. 2002;99:2884-9
28. Majumder PK, Febbo PG, Bikoff R, Berger R, Xue Q, McMahon LM. et al. mTOR inhibition reverses Akt-dependent prostate intraepithelial neoplasia through regulation of apoptotic and HIF-1-dependent pathways. Nat Med. 2004;10:594-601
29. Rajagopal S, Kim J, Ahn S, Craig S, Lam CM, Gerard NP. et al. Beta-arrestin- but not G protein-mediated signaling by the "decoy" receptor CXCR7. Proc Natl Acad Sci U S A. 2010;107:628-32
30. Wang J, Shiozawa Y, Wang J, Wang Y, Jung Y, Pienta KJ. et al. The role of CXCR7/RDC1 as a chemokine receptor for CXCL12/SDF-1 in prostate cancer. J Biol Chem. 2008;283:4283-94
31. Luan B, Zhao J, Wu H, Duan B, Shu G, Wang X. et al. Deficiency of a beta-arrestin-2 signal complex contributes to insulin resistance. Nature. 2009;457:1146-9
32. Little GH, Noushmehr H, Baniwal SK, Berman BP, Coetzee GA, Frenkel B. Genome-wide Runx2 occupancy in prostate cancer cells suggests a role in regulating secretion. Nucleic Acids Res. 2012;40:3538-47
33. Li S, Fong KW, Gritsina G, Zhang A, Zhao JC, Kim J. et al. Activation of MAPK signaling by CXCR7 leads to enzalutamide resistance in prostate cancer. Cancer Res. 2019
34. Rafiei S, Gui B, Wu J, Liu XS, Kibel AS, Jia L. Targeting the MIF/CXCR7/AKT Signaling Pathway in Castration-Resistant Prostate Cancer. Mol Cancer Res. 2019;17:263-76
35. Kaneki H, Guo R, Chen D, Yao Z, Schwarz EM, Zhang YE. et al. Tumor necrosis factor promotes Runx2 degradation through up-regulation of Smurf1 and Smurf2 in osteoblasts. J Biol Chem. 2006;281:4326-33
36. Kumar Y, Kapoor I, Khan K, Thacker G, Khan MP, Shukla N. et al. E3 Ubiquitin Ligase Fbw7 Negatively Regulates Osteoblast Differentiation by Targeting Runx2 for Degradation. J Biol Chem. 2015;290:30975-87
37. Thacker G, Kumar Y, Khan MP, Shukla N, Kapoor I, Kanaujiya JK. et al. Skp2 inhibits osteogenesis by promoting ubiquitin-proteasome degradation of Runx2. Biochim Biophys Acta. 2016;1863:510-9
38. Canals M, Scholten DJ, de Munnik S, Han MK, Smit MJ, Leurs R. Ubiquitination of CXCR7 controls receptor trafficking. PLoS One. 2012;7:e34192
39. Grant BD, Donaldson JG. Pathways and mechanisms of endocytic recycling. Nat Rev Mol Cell Biol. 2009;10:597-608
40. Barbieri CE, Baca SC, Lawrence MS, Demichelis F, Blattner M, Theurillat J-P. et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet. 2012;44:685-9
41. Cancer Genome Atlas Research Network. Electronic address scmo, Cancer Genome Atlas Research N. The Molecular Taxonomy of Primary Prostate Cancer. Cell. 2015;163:1011-25
42. Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM. et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161:1215-28
43. Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS. et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18:11-22
44. Yang Y, Bai Y, He Y, Zhao Y, Chen J, Ma L. et al. PTEN Loss Promotes Intratumoral Androgen Synthesis and Tumor Microenvironment Remodeling via Aberrant Activation of RUNX2 in Castration-Resistant Prostate Cancer. Clin Cancer Res. 2018;24:834-46
45. Ge C, Zhao G, Li Y, Li H, Zhao X, Pannone G. et al. Role of Runx2 phosphorylation in prostate cancer and association with metastatic disease. Oncogene. 2016;35:366-76
46. Baniwal SK, Khalid O, Gabet Y, Shah RR, Purcell DJ, Mav D. et al. Runx2 transcriptome of prostate cancer cells: insights into invasiveness and bone metastasis. Mol Cancer. 2010;9:258
47. Pande S, Browne G, Padmanabhan S, Zaidi SK, Lian JB, van Wijnen AJ. et al. Oncogenic cooperation between PI3K/Akt signaling and transcription factor Runx2 promotes the invasive properties of metastatic breast cancer cells. J Cell Physiol. 2013;228:1784-92
48. Luker KE, Steele JM, Mihalko LA, Ray P, Luker GD. Constitutive and chemokine-dependent internalization and recycling of CXCR7 in breast cancer cells to degrade chemokine ligands. Oncogene. 2010;29:4599-610
49. Pande S, Browne G, Padmanabhan S, Zaidi SK, Lian JB, van Wijnen AJ. et al. Oncogenic cooperation between PI3K/Akt signaling and transcription factor Runx2 promotes the invasive properties of metastatic breast cancer cells. J Cell Physiol. 2013;228:1784-92
50. Walters MJ, Ebsworth K, Berahovich RD, Penfold ME, Liu SC, Al Omran R. et al. Inhibition of CXCR7 extends survival following irradiation of brain tumours in mice and rats. Br J Cancer. 2014;110:1179-88
51. Luo Y, Azad AK, Karanika S, Basourakos SP, Zuo X, Wang J. et al. Enzalutamide and CXCR7 inhibitor combination treatment suppresses cell growth and angiogenic signaling in castration-resistant prostate cancer models. Int J Cancer. 2018;142:2163-74
52. Wang D, Garcia-Bassets I, Benner C, Li W, Su X, Zhou Y. et al. Reprogramming transcription by distinct classes of enhancers functionally defined by eRNA. Nature. 2011;474:390-4
Author contact
![]() Corresponding author: Haojie Huang (huang.haojieedu, +15072931311); Wanhai Xu (xuwanhaicom, +8613313602566).
Corresponding author: Haojie Huang (huang.haojieedu, +15072931311); Wanhai Xu (xuwanhaicom, +8613313602566).