Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Conclusion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(12):3410-3424. doi:10.7150/thno.32615 This issue Cite
Research Paper
O-GlcNAc modification of Sox2 regulates self-renewal in pancreatic cancer by promoting its stability
Nikita S Sharma1, Vineet K Gupta1, Patricia Dauer2, Kousik Kesh1, Roey Hadad1, Bhuwan Giri1, Anjali Chandra5, Vikas Dudeja1,4, Chad Slawson3, Santanu Banerjee1,4, Selwyn M Vickers6, Ashok Saluja1,4, Sulagna Banerjee1,4 ![]()
1. Department of Surgery, University of Miami, Miami, FL.
2. Department of Pharmacology, University of Minnesota, Minneapolis Minnesota.
3. University of Kansas Medical Center, Kansas City, KS.
4. Sylvester Comprehensive Cancer Center, Miami, FL.
5. Department of Psychology, Harvard University.
6. School of Medicine Dean's Office, University of Alabama at Birmingham.
Received 2018-12-27; Accepted 2019-4-24; Published 2019-5-24
Abstract

Pancreatic adenocarcinoma (PDAC) claims more than 90% of the patients diagnosed with the disease owing to its aggressive biology that is manifested by high rate of tumor recurrence. Aberrant upregulation in the transcriptional activity of proteins involved in self-renewal like Sox2, Oct4 and Nanog is instrumental in these recurrence phenomena. In cancer, Sox2 is aberrantly “turned-on” leading to activation of downstream genes those results in relapse of the tumor. Molecular mechanisms that regulate the activity of Sox2 in PDAC are not known. In the current study, we have studied the how glycosylation of Sox2 by O-GlcNAc transferase (OGT) can affect its transcriptional activity and thus regulate self-renewal in cancer.
Methods: RNA-Seq analysis of CRISPR-OGTi PDAC cells indicated a deregulation of differentiation and self-renewal pathways in PDAC. Pancreatic tumor burden following inhibition of OGT in vivo was done by using small molecule inhibitor, OSMI, on subcutaneous implantation of PDAC cells. Sox2 activity assay was performed by Dual Luciferase Reporter Assay kit.
Results: Our study shows for the first time that in PDAC, glycosylation of Sox2 by OGT stabilizes it in the nucleus. Site directed mutagenesis of this site (S246A) prevents this modification. We further show that inhibition of OGT delayed initiation of pancreatic tumors by inhibition of Sox2. We also show that targeting OGT in vivo with a small molecule-inhibitor OSMI, results in decreased tumor burden in PDAC.
Conclusion: Understanding this mechanism of SOX2 regulation by its glycosylation is expected to pave the way for development of novel therapy that has the potential to eradicate the cells responsible for tumor-recurrence.
Keywords: OGT, O-GlcNAc, Sox2, Pancreatic cancer, metabolism, self-renewal
Introduction
Pancreatic cancer is currently the third leading cause of cancer-related deaths in United States with over 53,000 cases per year and a 5-year survival of 7% (http://www.cancer.gov). This is primarily due to a high rate of recurrence of the tumor as a result of upregulated self-renewal genes following surgical resection or chemotherapy. Self-renewal in any cell is regulated by a concerted action of the “Yamanaka factors” SOX2, OCT4, Myc and KLF4 [1]. These transcription factors are robustly regulated by a number of interacting signaling pathways as well as cellular metabolism [2]. Alterations in tumor metabolism and/or bioenergetics pathways in cancer have recently emerged as a new cancer hallmark [3]. Cancer cells undergo drastic metabolic reprogramming that not only results in the classic Warburg Effect producing a large amount of lactate, but also in channeling a large amount of metabolites through biosynthetic pathways that support rapid growth and proliferation [4]. Among these, the hexosamine biosynthesis pathway (HBP) has emerged as a critical node that is instrumental in the regulation of a number of pro-oncogenic pathways [5-7]. This pathway, often considered as a “nutrient-sensing” pathway, is a shunt pathway of glycolysis, uses fructose-6- and glutamine to produce glucosamine-6-phosphate. About 2-3% of the total cellular glucose is channelized via HBP to culminate in synthesis of the nucleic acid sugar UDP-GlcNAc. UDP-GlcNAc is one of the major substrates for both N-linked glycosylation as well as O-GlcNAcylation of proteins. Since the HBP is dependent on the availability of glucose, slight increases in glucose uptake results in significantly more O-GlcNAcylated modifications of many proteins [8-10].
Protein O-GlcNAcylation is mediated via the activity of the enzyme O-GlcNAc transferase (OGT). A large number of proteins, particularly transcription factors are modified by OGT. This modification affects subcellular localization, protein-protein interaction or DNA binding ability of these proteins [11]. A number of studies have been focused on how O-GlcNAc modification of pro-survival transcription factors may regulate tumor progression [11]. Even though the O-GlcNAc modifications of transcription factors are well-known, the regulation of self-renewal genes in cancer by O-GlcNAc modification is a relatively understudied area. Recent reports have shown that SOX2 and OCT4, the key transcription factors that regulate self-renewal in embryonic stem cells and induced pluripotent stem cells are modified by O-GlcNAc [12]. In a recent study by Myers et al, O-GlcNAc modification of SOX2 regulates its interaction with PARP, which in turn led to differentiation [12]. However, there is no report on how O-GlcNAc modification of SOX2 regulates the self-renewal properties in a tumor cell.
Hexosamine biosynthesis pathway, and particularly OGT expression is upregulated in pancreatic cancer [6, 13, 14]. Studies from our laboratory have shown that O-GlcNAc modification of Sp1 regulates its nuclear translocation and thus its activity [5]. We have also shown that O-GlcNAc modification of β-catenin in pancreatic cancer regulates the Wnt signaling pathway [15]. In the current study, we have evaluated the role of O-GlcNAc modification on SOX2, the key transcription factor regulating self-renewal in cancer cells. Our results show that SOX2 is modified by O-GlcNAc transferase. O-GlcNAcylation of SOX2 stabilizes the protein in the nucleus and thus regulates its transcriptional activity. Further, mutating the Sox2 glycosylation site (S246A) prevents its O-GlcNAc modification and enhances its degradation. Inhibition of OGT in pancreatic cancer cells leads to a decrease in tumor initiation both in vivo and clonogenicity in vitro. This is the first report describing the O-GlcNAc modification of SOX2 in cancer cells and its role in regulation of self-renewal.
Materials and Methods
Cell Culture
S2-VP10, and L3.6pL (a generous gift from Dr. Masato Yamamoto, University of Minnesota) cells were grown in RPMI 1640 - Gibco) and DMEM without NEAA containing 10% Fetal Bovine Serum (FBS) and 1% Penicillin/Streptomycin respectively. SU86.86 (ATCC) cell line was also cultured in RPMI 1640 (Gibco) with 10% FBS and 1% Penicillin/Streptomycin. Human pancreatic stellate cells (ATCC, Manassas, VA, USA) were cultured in stellate cell medium (ScienCell, 5301) with stellate cell growth supplements provided by the manufacturer (SteCGS, Cat. No. 5352, P/S, Cat. No. 0503) ScienCell, Carlsbad, CA, USA. All cell lines were routinely tested for mycoplasma contamination and STR profiles (ATCC).
Transcriptome deep sequencing and analysis
Aliquots of RNA were derived from the qRT-PCR samples. 3 independent passages of OGTi and the respective sham shcells were analyzed. The RNA was quality tested using a Bioanalyzer 2100 (Agilent Technologies, CA, USA). cDNA was created by reverse transcription of oligo-dT purified polyadenylated RNA and fragmented, blunt-ended, and then ligated to barcoded adaptors. Then, the library was size selected, and the selection process was validated and quantified by capillary electrophoresis and qPCR, respectively. Samples were load on the HiSeq 2500 (Illumina Inc., CA, USA) to generate around 34 million paired-end 50bp reads for each sample.
Raw sequence data was processed through PartekFlow RNAseq pipeline (Partek Inc., St. Louis, MO) as follows- A pre-alignment QA/QC was performed and bases with Q>30 were retained for analysis. Bowtie2 was used to filter out non-human DNA, mtDNA and rDNA from the samples and STAR 2.5.3 aligner was used to map the high-quality reads to Hg19 human genome assembly. Aligned reads were quantified for differential abundance among samples using (a) Partek E/M annotation model and (b) Cufflinks algorithm using the Hg19- Ensemble transcripts release 75.
Site Directed Mutagenesis
For site directed mutagenesis, pDONR223_SOX2_WT obtained from Addgene (plasmid # 82233). SOX2 was modified at S246 to Alanine and T256 to Alanine using QC Lightning Multi SITE DIRECTED MUTAGENESIS kit from Agilent according to manufacturer's instruction. Clones were confirmed by sequencing [16].
Sox2 DNA binding assay
The DNA binding assay for Sox2 was done by using Sox2 transcription factor activity assay kit (RayBiotech). This assay uses a dsDNA coated plate with canonical SOX2 binding sequences to semi-quantitatively detect active SOX2 in lysates or nuclear extracts. Assay was performed using total cell lysates and nuclear lysates according to manufacturer's protocol.
Immunofluorescence and immuno-histochemistry
Tissues were deparaffanized by heating it at 56οC overnight and then hydrated by treating it with xylene (15 mins, two times) ,100% ethanol,90% ethanol, 70% ethanol (2 times) 5 mins each. The slides were then steamed with a pH 6 reveal decloaker (BIOCARE Medical, Concord, CA, USA) for antigen retrieval, blocked in Dako serum blocker (Agilent technologies, Santa Clara, CA, USA). Primary antibody was added overnight. Slides were washed 3X in PBS, secondary antibodies (Alexaflour) were diluted in SNIPER (BIOCARE Medical) and slides were stained for 30 mins at room temperature. Slides were washed again 3X in PBS and mounted using Prolong Gold anti-fade with DAPI (Molecular Probes, ThermoFisher Scientific, Weston, FL, USA). Slides were dried overnight and imaged by fluorescent microscope. SOX2 antibody was purchased from SIGMA-ALDRICH, St Louis, MO, USA (SAB2701974), OCT 4 (D7O5Z) and NANOG (1E6C4) antibody was purchased from CELL SIGNALLING TECHNOLOGY, Danvers, MA, USA. Antibody against OGT and O GlcNAc were purchased from SIGMA-ALDRICH (SAB2101676) and Millipore, Burlington, MA, USA (Clone CTD 110.6), respectively. Images were acquired at 20X and 60X.
For tissue microarrays (TMA) slides were obtained from UsBiomax. The slides were processed as above and immunohistochemistry was performed. Antigen retrieval of OGT was done at pH 6.0. Endogenous peroxidases were blocked with a 3% hydrogen peroxide solution. Tissues were then blocked with Dako protein block, and incubated with primary antibody overnight. Slides were stained with anti-OGT (Sigma) at 1:200 dilutions. Slides were washed with PBS, incubated with secondary anti-rabbit antibody, conjugated to horseradish peroxidase, for 30 minutes. Slides were washed again with PBS. Diaminobenzidine Peroxidase Substrate Kit (Vector Laboratories) was then added to slides. Primary antibody was omitted for negative controls. Slides were mounted with permount. Images were obtained on a Leica DM6B with a 20x objective. The TMAs were quantitated independently and blindly by 3 research personnel in the lab using an arbitrary intensity scale (0= no signal, 5= highest signal as seen in positive control). The 10-20 fields per section were quantitated by each personnel.
Generation of OGT-Knockout, Sox2 overexpression and mutant Cell line
Human OGT gene knockout kit was obtained from Origene, Rockville, MD, USA (KN206822). S2-VP10 cells were plated at a density of 80,000 in a 6 -well plate. One day after plating the cells were transfected with gRNA1 and gRNA2 along with donor vector. The cells were then cultured according to the manufacturer's protocols. After 8 passages puromycin pressure at a concentration of 0.5μg/ml was applied. The colonies were allowed to grow and the cells were then single-cell sorted by FACS in a 96-well plate. The select clones were allowed to grow and then characterized by mRNA expression and western blot.
For generating S246A and T256A cells, similar protocol was followed after site directed mutagenesis described above. Mutants were transfected both in S2VP10 (Sox2 S246A mut) and MIAPACA2 (MIA-Sox2-S246A-mut) pancreatic cancer cell lines. For Sox2 overexpression, SOX2 (Myc-DDK-tagged) plasmid was obtained from Origene (CAT#: RC200757). Both S2VP10 (Sox2 OE) as well as MIA-PACA2 (MIA-Sox2-OE) were stably transfected to generate Sox2 overexpressing cell lines.
Proximal Ligation Assay
S2VP10 and L3.6pL cells were plated at a density of 50,000 cell per well in a chamber slide. Cells were treated with OSMI for 24 hours. After 24 hour's media was removed, the cells were washed 3 times with PBS and fixed with 2% paraformaldehyde. After fixing the cells, they were stained according to the manufacturer's protocol (Duo link, DU 092101, SIGMA) and the images were acquired at 20X magnification.
In vivo Studies
Female athymic nude mice between the ages of 4-6 weeks were used for in vivo experiments and were purchased from the Jackson laboratory, Bar Harbour, ME, USA. For subcutaneous experiments 500,000 cells were implanted in the right flank of the mice. Corning® Matrigel® Growth Factor Reduced (GFR) Basement Membrane Matrix, purchased from Corning, inc, Corning NY, USA and 1X PBS at a ratio of 1:1 were used as a suspension medium for the cells. Tumors were allowed to reach a size of 100mm3 and then treatment was started. OSMI (SML1621 SIGMA) was given at a dose of 10mg/kg/day in DMSO for 3 weeks. At the end of 3 week's mice were sacrificed and tissues were collected.
For tumor initiation studies, OGT knockout (OGTi-S2VP10) cells were injected at a limiting dilution from 500,000 cells per mice to 500 cells in the right flank of female athymic nude mice. For tumor initiation studies using Sox2 mutant cells, 500 MIA-Sox2, MIA-empty vector control or MIA-Sox2-S246Amut cells were implanted in subcutaneously in athymic nude mice. The mice were monitored daily and the appearance of tumor was noted. All the experiments were performed according to the IACUC protocols as approved by the University of Miami.
RNA Isolation and cDNA synthesis
RNA was isolated using the Trizol method of isolation and cDNA was made using the High Capacity cDNA Reverse Transcription Kit. 2 μgm of RNA was used per reaction. RT-qPCR was performed in the Roche Light Cycle 480. Primers (SOX2 PPH02471A, OCT4 PPH66786A, NANOG PPH17032E-200, OGT PPH19166A, OGA PPH01061A) were purchased from Qiagen, Valencia, CA, USA.
Immunoprecipitation with DDK-tag
S2VP10 cells were plated at a density of 100,000 cells per well in a 6 well plate. The cells were then transfected with a Myc-DDK tagged-Human SRY plasmid (pCMV6-Entry) for 24 hours. After 24 hours cells were scrapped and protein was isolated. 150μg of protein was then used for immunoprecipitation with 15μl of Anti-DDK monoclonal antibody. The antibody-protein mixture was allowed to mix via rotating overnight at 4ºC. 30μl of Protein A/G was added to the antibody-protein mix and was incubated via rotating at 4ºC for 2 hours. After 2 hours, the samples were centrifuged at 5000rpm for 5 mins and the flow through was collected. This was repeated 3 times. After the final centrifugation, the beads were resuspended in 50μl of RIPA buffer. The samples were then run on a 4-20% SDS PAGE gel, transferred on a nitrocellulose membrane and blocked with 5% milk for 1 hour. O GlcNAc antibody was added at a dilution of 1:1000 in BSA and the blot was incubated overnight at 4ºC. The blot was washed 3X with TBST, anti-mouse secondary was added, washed again 3X with TBST and the blots were developed.
CHX Assay for protein Stability
For CHX assay, S2VP10 SC and OGTi cells were plated at a density of 500,000 in a 6cm dish for 24 hours. After 24 hours Cycloheximide was added at a dose of 300 μg/ml. Cells were scrapped and protein was isolated after 0, 30, 60 and 120 minutes of treatment. 50μg of protein was used to perform a western blot.
Sox2 Dual Luciferase Reporter Assay
Sox2 dual luciferase reporter assay kit was purchased from Qiagen. S2VP10 and L3.6pL cells were plated at a density of 0.8X106 cells per well in a 24 well plate. Cells were transfected with the SOX2 reporter plasmid the next day using Attractene as a transfection reagent. Transfection was done according to manufacturer's instruction. Negative and positive controls were kept according the kit manual. 24h after the transfection with the reporter plasmid, the cells were treated with OSMI and/ or MG132 at a dose of 50µM and 1µM respectively for another 24 hours respectively. 24 hours after treatment the cells were lysed using 1X passive lysis buffer and the luminescence was analyzed using a spectrophotometer using the Promega Dual Luciferase Assay System according to manufacturer's instruction.
The dual luciferase report kit for self-renewal was purchased from Qiagen. This is a multiplexed assay that can simultaneously test from transcriptional activity of 10 different transcription factors involved in self-renewal. Effect of OGT inhibition on the activity of these transcriptions was performed according to the manufacturer's instruction.
Sox2 ELISA
For estimating the amount of SOX2 protein in our cell lysates, we performed a Sandwich ELISA using the human SOX2 ELISA kit from My Biosource, San Diego, CA, USA (MBS2513016). Cells were plated in a 10 cm dish and allowed to attach and grow for 24 hours. After 24 hours of plating cells were treated with OSMI and MG1332 alone or in combination at a dose of 50 µM and 1µM for 24 hours respectively. After 24 hours of treatment cells were lysed using RIPA lysis buffer and the protein was estimated using BCA protein estimation method. Equal amount of protein was loaded for the assay and the assay was performed according to the manufacturer's protocol.
Nuclear and Cytosolic fractionation
For nuclear and cytosolic fractionation, cells were harvested once confluent. NE-PER nuclear and cytoplasmic fractionation kit (ThermoScientific) was used according to manufacturer's instruction to separate the two cellular compartments.
Ethics Statement
All procedures were performed according to protocols approved by the University of Miami Institutional Animal Care and Use Committee (IACUC).
Statistical Analysis
Values are expressed as the mean +/- SEM. All in vitro experiments were performed at least three times. The significance between any two samples was analyzed by t-test, values of p<0.05 were considered statistically significant.
Results
OGT expression correlates with the aggressiveness of the disease
OGT expression and O-GlcNAc modifications are elevated in pancreatic cancer [17]. Previously published results from our laboratory confirm this [5]. To further investigate this phenomenon in patient tumor tissues, we performed immunohistochemistry on a de-identified human pancreatic tumor tissue microarray. Our results showed that the expression of OGT increases with the aggressiveness of the disease. OGT expression was significantly higher in the T3N0M0 stage of the pancreatic tumor compared to the adjacent normal or the T2N0M0 stage (Figure 1A, B). The TNM staging of the TMA is shown in Supplementary Figure 1A, B. To study whether this was also demonstrated in the patient databases, we performed an analysis of the OGT expression and correlated it with patient survival in the TCGA database at www.cbioportal.org. Our results showed a negative correlation between the expression of OGT and patient survival and advanced histologic grade (Supplementary Figure 1C, D).
OGT expression in pancreatic cancer. Tumor tissue microarray from patient samples showed that OGT expression coincided with the aggressive stages of the disease (A). The quantitation of the histology confirmed this observation (B). In KrasG12DTP53R172H Pdx-cre or KPC model for pancreatic cancer, OGT (green) and O-GlcNAc (Red) expression increased as the tumor progressed. Representative immunofluorescence from 1 month old mice and mice with full tumor and the quantitation of O-GlcNAc and OGT staining (C). Tissues for KPC animals were stained for OGA as well. OGA expression was also high in full tumor tissues compared to the histologically normal pancreas at 1 month (D). As seen with OGT, immunofluorescence of Sox2 staining (E) and Sox2 glycosylation observed with proximal ligation assay (F) was observed to be higher in full tumors in KPC animals. * indicated statistical significance when p<0.05
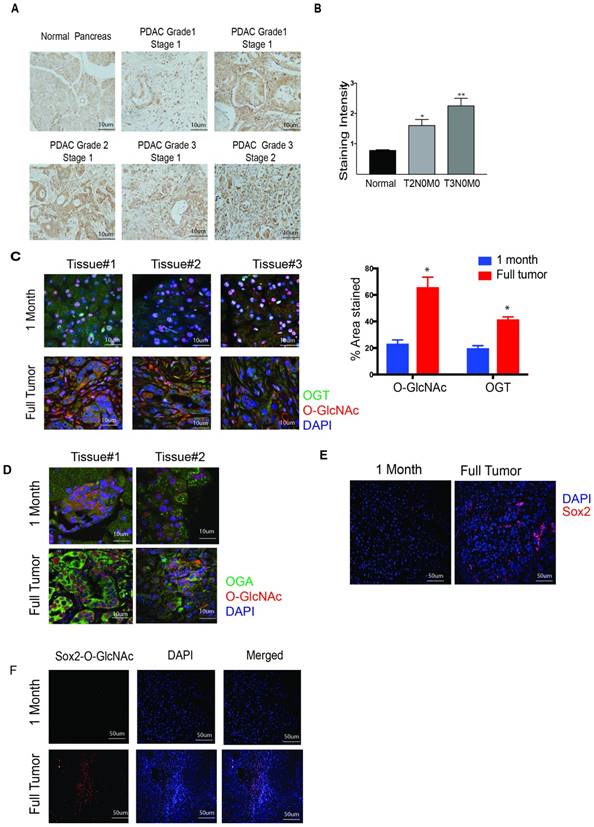
To evaluate whether O-GlcNAcylation of proteins is increased as the pancreatic tumor progresses, we first compared the expression of OGT in the pancreas of 1-month old KrasG12D, TP53 R172H Pdx-Cre (KPC) mice and 6-8-month-old KPC mice with a fully developed tumor. Our results showed that expression of OGT as well as O-GlcNAc modification was significantly less in the tissue of 1-month-old mice (lacking tumor) compared to that in a 6-8-month-old KPC mice (that has tumor) as can be seen from the quantitation of staining (Figure 1C). It is well known that the glycosyltransferase OGT and glycosidase OGA work on in partnership. We next evaluated the expression level of OGA in pancreatic tissues of KPC mice as tumors progressed. Like OGT, OGA was also overexpressed in the full tumor tissue while its expression was less in the early stages of the pancreatic tumor development (Figure 1D). Similarly, Sox2 was also found to be overexpressed in full tumor in the KPC animals while its expression was low in the pancreas of 1-month old KPC mice (Figure 1E). To study if the increased expression of OGT/OGA and Sox2 also corresponded to O-GlcNAc modification of Sox2, we next performed a proximal ligation assay with anti-Sox2 and Anti-O-GlcNAc antibody. Our results showed that in a fully developed pancreatic tumor, O-GlcNAc modification of Sox2 was increased as well (Figure 1F).
Our data corroborated with previous findings that OGT and OGA are overexpressed in pancreatic cancer cell line (Supplementary Figure 1 E-H) [7]. Since S2VP10 and L3.6 PL cell lines had the highest expression of OGT among those tested, we used these in the current study.
Transcriptomic analysis of OGT knockout cell line
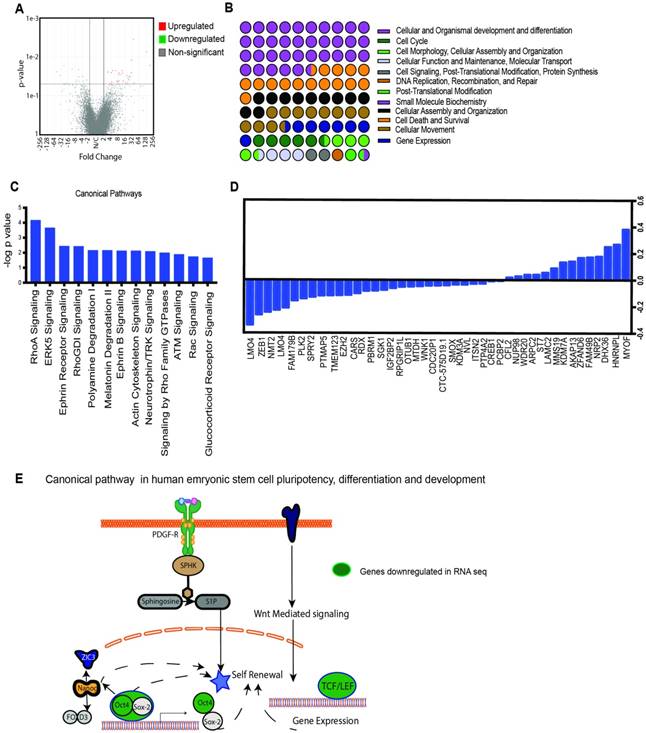
OGT is responsible for modification of a number of critical pathways that actively affect oncogenic signaling in cancer. To study the major pathways that are downregulated upon inhibition OGT, we performed a RNA-seq analysis on OGTi cells. Raw sequence data was processed through PartekFlow RNAseq pipeline (Partek Inc., St. Louis, MO) as follows- A pre-alignment QA/QC was performed and bases with Q>30 were retained for analysis. Bowtie2 was used to filter out non-human DNA, mtDNA and rDNA from the samples and STAR 2.5.3 aligner was used to map the high-quality reads to Hg19 human genome assembly. Aligned reads were quantified for differential abundance among samples using (a) Partek E/M annotation model and (b) Cufflinks algorithm using the Hg19- Ensemble transcripts release 75. Out of 13,401 identified transcripts, 43 were found to be significantly different among the two study groups. (Figure 2A). The Gene Ontology showed that the majority of the deregulated genes belonged to the category of embryogenesis, organogenesis and differentiation, cell survival and regulation of gene transcription (Figure 2B). To further analyze the pathways that are deregulated upon inhibition of OGT, we conducted pathway enrichment analysis and found that most significantly deregulated genes belonged to pathways were those involved in development and differentiation (Figure 2 C, D). Among these, the human embryonic stem cell and pluripotency network was observed to be present. This network encompassed the downstream signaling stemming from PDGF-PDGFR and Wnt and culminated in regulation of genes that were involved in self-renewal pathways (Figure 2E). One of the major transcription factors that regulate self-renewal is Sox2. Our analysis of the genes deregulated in the OGTi group showed that several of them were Sox2 target genes (Supplementary Table 1).
Transcriptomics analysis of OGTi cells: Volcano plot showing significantly deregulated genes (A) GO of the biological processes that are altered as a result of OGT inhibition (B). Canonical pathways altered by OGT inhibition as analyzed by the IPA software (C). List of 43 deregulated genes upon OGT inhibition (D). Schematic of embryonic stem cell pathways that are deregulated upon OGT inhibition (E).
Down-regulation of OGT leads to a decrease in self-renewal in pancreatic cancer
Tumor aggressiveness is typically characterized by their ability to recur after resection or therapy. This phenomenon is characterized by the “turning-on” of self-renewal genes like SOX2, OCT4 and Nanog as well as by estimating their “tumor initiation” frequency [18]. Lower tumor initiation frequency leads to delayed recurrence of tumors and decreased self-renewal ability in a tumor cell. To study the effect of OGT down-regulation on this, we used an OGT-CRISPR plasmid that stably knocked-out the expression of OGT in highly aggressive S2VP10 pancreatic cancer cells (OGTi). These cells showed decreased total O-GlcNAc level (Supplementary Figure 2A) as well as decreased expression of OGT (Supplementary Figure 2 B, C). When implanted in limiting dilution (500 cells) in athymic nude mice, they showed decreased tumor initiation suggesting a delay in tumor recurrence (Figure 3A). Further, upon comparing the tumor initiation frequency in the control and the OGTi animals we observed that the tumor initiation frequency was much lower in the OGTi group (Figure 3B). This indicated that OGT was instrumental in tumor initiation and self-renewal of pancreatic cancer cells.
Downregulation of OGT expression leads to a decrease in self-renewal in pancreatic cancer: CRISPR knockout of OGT in pancreatic cancer cell line S2VP10 showed delayed tumor initiation compared to scrambled control. Each incidence of detectable tumor initiation was plotted as a 1 in the Kaplan Meir Curve (A). Tumor initiation frequency was calculated for scrambled control and OGTi cells. OGTi cells had significantly low tumor initiation frequency compared to control (B). In S2VP10 and L3.6pL cells (C) inhibition of OGT by small molecule inhibitor OSMI decreased the colony forming units compared to untreated indicating that OGT was instrumental in determining clonogenicity. CRISPR knockout of OGT in S2VP10 cells (OGTi) also showed decreased colony formation (D) compared to scrambled control. Representative pictures are shown in (E). Transcriptional activity of Sox2 was decreased upon inhibition of OGT with an inhibitor, OSMI (F) and with siRNA (G) and in OGTi cells (H) with dual luciferase reporter assay for either self-renewal (S/R) or Sox2 transcription factor binding sites in the promoter of the luciferase gene. Consistent with this, DNA binding of Sox2 was decreased in OGTi cells (I) and when L3.6PL cells were treated with OSMI (J). * indicated statistical significance when p<0.05.
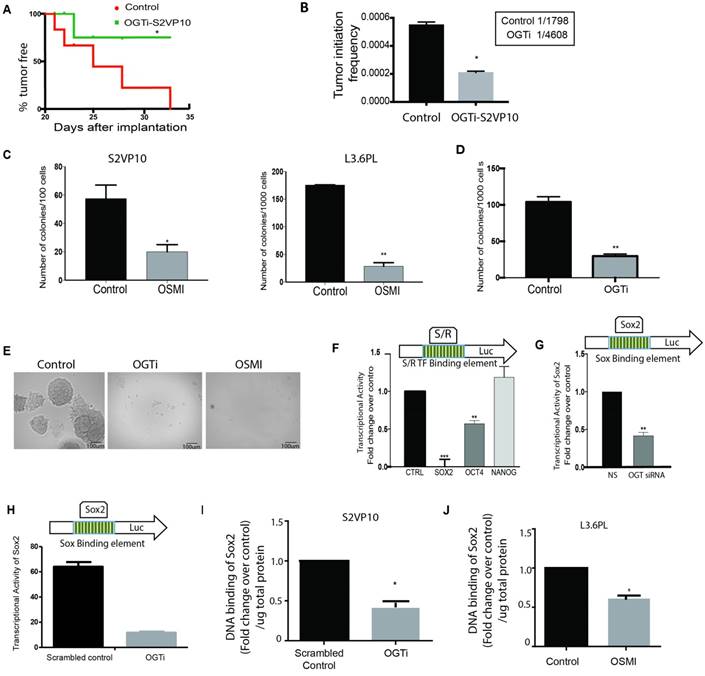
Since OGT inhibition resulted in a delay in tumor initiation, a decrease in tumor initiation frequency, and a number of deregulated genes in the OGTi group were Sox2 targets (Supplementary Table 1), we next studied the effect of this inhibition on self-renewal genes namely, SOX2, OCT4 and NANOG. Our results showed that OGT silencing (using siRNA) did not change the expression of core self-renewal transcription factors SOX2 or OCT 4 in S2VP10 cells while modestly changing its expression in L3.6pL cells (Supplementary Figure 2 D). However, a methyl-cellulose based colony forming unit assay on S2-VP10 and L3.6pL cells showed that treatment with OSMI (a small molecule inhibitor of OGT) decreased the colonies in S2VP10 and L3.6pL cells (Figure 3C) compared to untreated cells. Since small molecule inhibitors are notorious for their off-target effects, we next performed this assay using the OGTi-S2VP10 cells. The colony forming ability of these cells was also significantly decreased (Figure 3D). Representative pictures can be seen in Figure 3E. Since colony-forming ability is a measure of the cells self-renewal potential, we next performed a luciferase based transcriptional activity assay on the transcription factors regulating self-renewal. In this assay, luciferase was the reporter gene under the control of the promoter region containing the binding sites for self-renewal genes like Sox2, Oct4 etc. Binding of these self-renewal transcription factors to the promoter region would activate the transcription of luciferase, which as a result metabolizes the substrate luciferin to generate a measurable luminescent product. Pancreatic cancer cells S2VP10, were treated with small molecule inhibitor OSMI and used for this luciferase based stem cell reporter array. Our results showed that among the transcription factors involved in self-renewal SOX2 transcriptional activity was the most significantly down-regulated (Figure 3F). We validated this using siOGT on S2VP10 cells (Figure 3G) and on our OGTi-S2VP10 cells (Figure 3H). Our results showed that SOX2 transcriptional activity was affected significantly in cells in which OGT was inhibited either by OSMI or by siRNA compared to the control. Our results thus indicated that OGT is required for SOX2 transcriptional activity. It is well known that O-GlcNAc modification of a protein can affect its activity in a number of ways. In embryonic cells, modification of SOX2 facilitates its interaction with PARP and promotes differentiation [12]. Since our results showed that upon inhibition of OGT, SOX2 transcriptional activity was inhibited, we hypothesized that this could be because OGT regulates DNA binding ability of SOX2. To determine this, we studied if inhibition of OGT affected the DNA binding ability of SOX2. Our results indicated that the DNA binding ability of SOX2 is inhibited in S2VP10-OGTi cells (Figure 3I) as well as in L3.6PL cells when treated with OSMI (Figure 3J).
Transcriptional regulation of self-renewal in pancreatic cancer is dependent on O-GlcNAc modification of Sox2
Since our results indicated that OGT inhibition might affect self-renewal properties of pancreatic cancer cells via inhibition of SOX2 transcriptional activity and DNA binding, we next studied the glycosylation of SOX2. SOX2 is modified by OGT in embryonic stem cells. This modification of SOX2 is known to play a role in its protein-protein interaction in those cells [12]. To determine if SOX2 is modified by O-GlcNAc in pancreatic cancer cells, we overexpressed DDK tagged Sox2 (pCMV6-entrySox2-Myc-DDK) performed an immunoprecipitation with anti-DDK antibody and probed it with anti-O-GlcNAc antibody. SOX2 was seen to be glycosylated by O-GlcNAc in this experiment. Similar result was obtained when endogenous Sox2 was immunoprecipitated from S2VP10 cells using Anti-Sox2 antibody (Figure 4A). We next confirmed this, by Proximal Ligation Assay (PLA) on S2-VP10 cells. In this assay, a positive fluorescence is emitted only when Sox2 is glycosylated. Our results showed that SOX2 is indeed modified by O-GlcNAc (Figure 4B). Proximal ligation Assay was also done of L3.6PL cells. Our results are in Supplementary Figure 2E. This was further confirmed by mass spectrometry (Supplementary Data 1).
Transcriptional regulation of self-renewal in pancreatic cancer is dependent on O-GlcNAc modification of Sox2. Immunoprecipitation with anti-DDK antibody followed by western blotting with Anti-O-GlcNAc Ab in pCMV6-Entry Sox2 Myc-DDK overexpressing plasmid showed that Sox2 was modified by addition of an O-GlcNAc moiety. Similar results were observed when endogenous Sox2 was immunoprecipitated with anti-Sox2 antibody and immunoblotted with anti-O-GlcNAc (A). Proximal ligation assay with Anti-O-GlcNAc ab and Anti-Sox2 Ab confirmed this modification (B). Glycosylation site of Sox2 was mutated (S246A) and overexpressed in MIA-PACA2 cells with very low endogenous expression of Sox2. Upon immunoprecipitation with Anti-Sox2 antibody and immunoblotting with anti-O-GlcNAc antibody confirmed loss of glycosylation on this protein (C). Total Sox2 protein expression was decreased in L3.6PL cells as well as in S2VP10 cells (D). Colony forming assay was done as a functional assay for self-renewal. Silencing Sox2 showed decreased colonies in control as expected (red bar); overexpression of Sox2 in control increased colony formation (Green bar) in control while had no effect on OGTi cells (black bar). However, when OGT and Sox2 were overexpressed in OGTi cells, a robust increase in colony formation was seen, indicating a full rescue (E). When MIA-PACA2, MIA Sox2-OE and MIA-Sox2-S246A mut cells were implanted subcutaneously in athymic nude mice, a delay in tumor initiation was observed in MIA-PACA2 and MIASox2-S246Amut groups while in MIA-Sox2-OE all animals formed tumors by 21 days (F). Representative pictures of animals can be observed in (G) * indicated statistical significance when p<0.05.
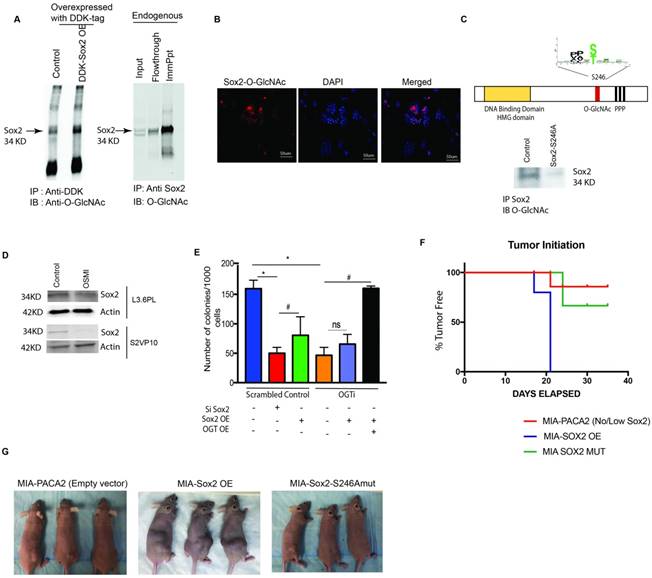
We next identified the site for O-GlcNAc modification on Sox2 in pancreatic cancer cells by using the software OGTsite (http://csb.cse.yzu.edu.tw/OGTSite/) as well as YinOYang1.2 (http://www.cbs.dtu.dk/services/YinOYang/). The results showed that S246 and T256 had the highest probability of getting modified by O-GlcNAc addition. To study if the modification of Sox2 is disrupted by mutating the site we next performed a site directed mutagenesis of Ser 246 and Thr 256 to Ala in Sox2 overexpressing plasmids (with DDK-Tag) obtained from Addgene. The plasmid was then transfected to generate a stable Sox2-S246A mutant S2-VP10 cells (Sox2 mut) as well as in MIA-PACA2, a cell line that lacks endogenous Sox2. We observed that S246A mutation in Sox2 gene resulted in loss of O-GlcNAc modification of SOX2 as observed via immunoprecipitation with the DDK-tag (Figure 4C), confirming that Ser246 in SOX2 indeed gets modified by O GlcNAcylation. Mutation of Thr 256 to Ala did not affect the stability of Sox2 (Supplementary Figure 2F). Interestingly, when glycosylation was inhibited by small molecule inhibitor OSMI, decreased protein level was observed in the western blot in S2VP10 and L3.6PL cell lines (Figure 4D, Supplementary Figure 3A).
We next performed a loss- and gain- of function assay on the OGTi clones. A limiting dilution colony formation assay was performed in which Sox2 was silenced and overexpressed in control pancreatic cancer cells (S2VP10). Sox-2 silencing showed a significant decrease in colony formation, while the Sox2 overexpression slightly increased number of colonies. However, in OGTi cells, less number of colonies were formed as expected (as lack of glycosylation was degrading the Sox2), but a Sox2 overexpression did not increase the CFUs (since the cells still lacked OGT, and thus could not glycosylate overexpressed Sox2 required for clonogenicity). This was only seen when Sox2 was simultaneously overexpressed with OGT in the OGTi cells. This further confirmed that the presence of Sox2 (without glycosylation) was not enough for self-renewal (Figure 4E). Similar study on an additional clone is shown in Supplementary Figure 3B.OGT overexpression and inhibition was confirmed at the RNA and protein level (Supplementary Figure 3C, D). Further, when we overexpressed Sox2 as well as Sox2S246A in MIA-PACA2 cells (cell line that lacks endogenous Sox2) and implanted these cells in athymic nude mice, there was 100% tumor initiation in the Sox2 overexpressing cells by day 21 following implantation, while in MIA-PACA2 control cells and MIA-PACA2 Sox2-S246A had 80% animals where tumors did not initiate (Figure 4 F, G). This further confirmed that glycosylated Sox2 was required for tumor initiation.
O-GlcNAc modification of SOX2 is required for its stability in pancreatic cancer cells
Since Sox2 protein levels were decreased upon inhibition of OGT, we next validated the decrease in Sox2 expression level in pancreatic cancer cells following inhibition of OGT in OGTi using IF (Supplementary Figure 3E). Since our observation (in Supplementary Figure 2C) indicated that SOX2 mRNA remained unaltered, we concluded that inhibition of OGT was affecting the stability of SOX2 protein. It is well known that SOX2 acts as a molecular rheostat for a cancer cell and its expression needs to be tightly regulated. Thus, the degradation machinery for SOX2 needs to be fairly robust in a cell [18]. To study stability of Sox2 in pancreatic cancer cells, we next performed a CHX assay to quantify the half-life of Sox2. Control and OGTi cells were treated with cycloheximide to block the synthesis of new protein for up to 2h and the expression of Sox2 was determined in pancreatic cancer cells by immunoblotting. Additionally, S2VP10 cells were treated with OSMI and cycloheximide and followed for 2h. Sox2 protein was degraded to 1/3rd its levels within 60 min. However, in the OGTi cells where OGT is knocked out by CRISPR constitutively, there was no expression of Sox2 even at time 0. Similar results were observed upon treatment with OSMI, where protein levels of Sox2 were decreased compared to control within 30 min. (Figure 5A). To study if Sox2 glycosylation was responsible for this, we next performed a CHX assay on our stably overexpressed Sox2-S246A mutant in pancreatic cancer cells. In these cells, S246 of Sox2 is mutated to an Ala-residue and is not glycosylated. Our results showed that in the mutant, where Sox2 was not glycosylated, the protein levels were lower compared to the control similar to that seen in OGTi cells (Figure 5B). To evaluate if the degradation of Sox2 upon inhibition of O-GlcNAc modification can be “rescued” by a proteasome inhibitor, we next treated S2VP10 cells with OSMI alone and OSMI with MG-132, a proteasome inhibitor. We observed a significant rescue in the protein levels of SOX2 when the cells were treated with the proteasome inhibitor MG132 (Figure 5C) thus demonstrating that O-GlcNAcylation of SOX2 is important for its stability. This was also quantitatively confirmed by an ELISA (Figure 5D). Further, treatment with MG132 in the presence of OSMI also showed a rescue in the transcriptional activity of SOX2 (Figure 5E). Additionally, our studies showed that mutation of the O-GlcNAc modification site (S246A), that resulted in a decrease in the SOX2 protein levels (Figure 5F), also showed decreased DNA binding by Sox2 (Figure 5G), which would lead to decreased Sox2 DNA binding activity (Figure 5G). We also observed that upon inhibition of OGT (by treatment with OSMI), there was an inhibition of nuclear translocation which was reverted upon treating with O-GlcNAcase inhibitor Thiamet G (Figure 5H). Similar lack of nuclear localization was observed in OGTi cells (Supplementary Figure 3F). When we fractionated the cytoplasmic and nuclear compartments in the Sox2 overexpressing cell line, we observed that Sox2 primarily localized in the nucleus in the overexpressed cell line. However, when the glycosylation site was mutated, there was an overall decrease in the total Sox2 levels both in the nucleus as well as the cytoplasm (Figure 5I).
O-GlcNAc modification of SOX2 is required for its stability in pancreatic cancer cells. Cycloheximide chase assay was performed to check for protein stability. In S2VP10 cells, endogenous Sox2 levels were decreased in 60 minutes while in OGTi cells there was minimal expression of Sox2. In OSMI treated cells Sox2 started degrading by 30 min (A). Similar to that observed in OGTi cells, Sox2-S246A mutant S2VP10 cells showed a decreased Sox2 expression even at 0h indicating that lack of glycosylation in these cells affected its stability (B). Upon inhibiting the proteasome with MG132, in OSMI treated S2VP10 cells, Sox2 expression was rescued as the degradation machinery was blocked as observed in western blot (C) and independently confirmed by Sox2 ELISA (D). Treatment with MG132 in the presence of OSMI also rescued Sox2 transcriptional activity as seen by a luciferase based reporter assay (E). Transfection of MIA-PACA2 cells with a SOX2 S246A mutant plasmid lead to a decrease in the protein levels of SOX2 (F) as well as in its DNA binding to promoter of Sox2 target genes (G). Treatment with OSMI (50uM) also resulted in inhibition of nuclear translocation of Sox2 while inhibition of OGA (the glycosidase that mediates removal of O-GlcNAc from a protein) using Thiamet-G (TMG), resulted in increased nuclear translocation compared to untreated cells (H). Similarly, in the Sox2 overexpressing MIA-PACA2, there was increased Sox2 in the nuclear fraction while in the S246A mutant, there was very little Sox2 in the nucleus (I) as observed after fractionating the cellular compartments. * indicated statistical significance when p<0.05.
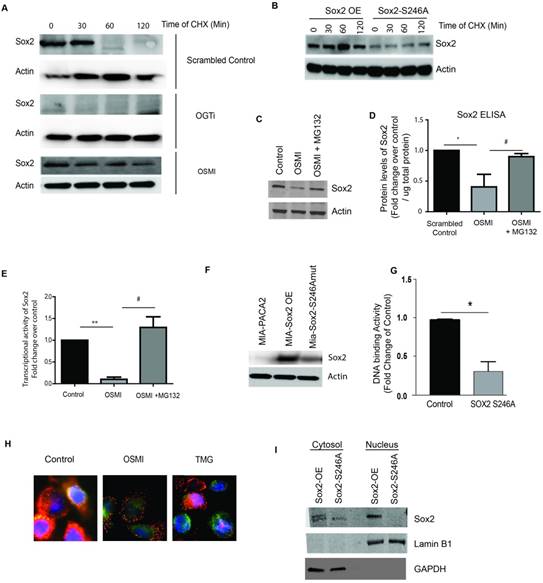
This indicated that the glycosylation of Sox2 was primarily involved in its stability in both nucleus as well as cytoplasm and was not playing an overall role in translocation.
Inhibition of OGT in vivo decreases tumor burden
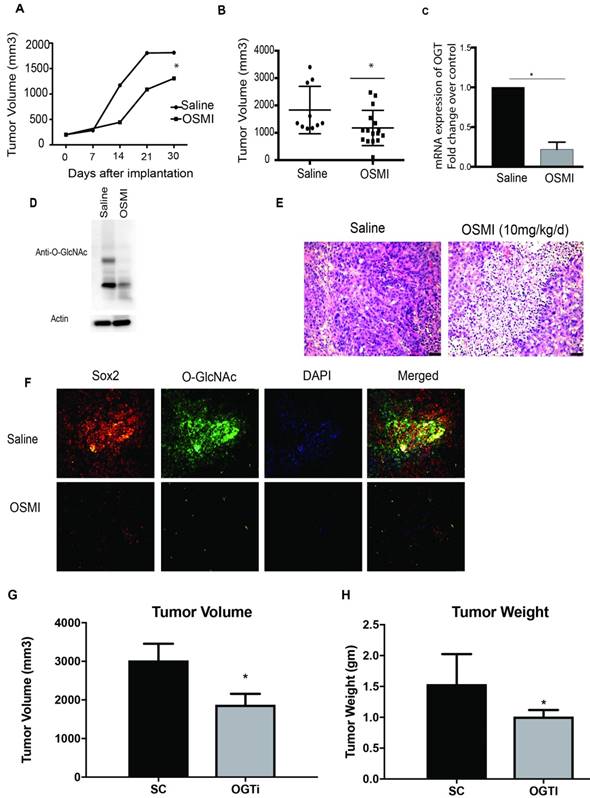
To evaluate the efficacy of OGT downregulation in vivo, we implanted S2VP10 cells subcutaneously in athymic nude mice. OSMI was administered at a dose of 10mg/kg/day for 30 days. Consistent with our in vitro data treatment with OSMI caused a delay in tumor progression (Figure 6A). Further, a significant decrease in tumor volume was observed upon inhibition of OGT (Figure 6 B). The tumors showed less OGT expression (Figure 6C) as well as less O-GlcNAc (Figure 6D), along with decreased cellularity in the H&E staining (Figure 6E). Further, as expected, there was less O-GlcNAc modification on SOX2 (Figure 6F). Upon analysis, the tumors showed a decrease in the OCT4 and NANOG expression when treated with OSMI (Supplementary Figure 4). Since OSMI being a small molecule can have other non-tumor autonomous response, we further did a xenograft of OGTi cells in the pancreas of the athymic nude mice. Our studies showed that OGTi was as efficient in decreasing tumor volume as OSMI in these animals (Figure 6 G, H).
Inhibition of OGT in vivo decreases tumor burden. Treatment with OGT inhibitor OSMI delayed tumor progression (A) and decreased tumor volume (B). Treatment with OSMI decreased mRNA expression of OGT in the tumors (C) and total O-GlcNAcylation (D). Tumors treated with OSMI also showed decreased cellularity as seen by H&E stains in immunohistochemistry (E). Sox2 protein levels and O-GlcNAcyltion was also decreased upon treatment with OSMI (F). In orthotopic implantation of OGTi cells in athymic nude mice showed a decreased tumor volume (G) and weight (H) when compared to control. * indicated statistical significance when p<0.05.
Discussion
O-GlcNAc transferase or OGT is an enzyme that transfers a single N-Acetyl glucosamine to serine or threonine residues of many proteins. This modification can “flag” the protein for phosphorylation [19], determine its interaction with other proteins or regulate its nuclear translocation [11, 20]. The role of O-GlcNAc modification on key oncogenic transcription factors has gained importance over the last decade [11, 21]. A number of proteins like Myc, Sp1 and NF-kB which are instrumental in promoting proliferation in cancer are known to be modified by O-GlcNAc [5, 7]. Previous results from our laboratory have shown that O-GlcNAc modification of Sp1 regulates its nuclear translocation and thus its activity [5]. Similarly, recent studies have shown that O-GlcNAc modification of the YAP component of the Hippo Pathway disrupts its interaction with LATS1 (an upstream kinase) thereby preventing its phosphorylation and up-regulating its transcriptional activity [22]. In gastric cancer, O-GlcNAc modification stabilizes FOXM1 protein by decreasing its phosphorylation by GSK-3β [23] .
The role of O-GlcNAc modifications in regulating stemness and self-renewal is gaining importance in cancer research [24, 25]. A recent study in embryonic stem cells has shown that Sox2, a key transcription factor for self-renewal is modified by O-GlcNAc, which in turn facilitates its interaction with PARP to regulate differentiation [12]. However, there are very few studies on whether this modification is responsible for initiation and regulation of self-renewal in cancer. Our current study shows that inhibition of OGT by a CRISPR results in a decreased tumor initiation frequency and delayed tumor initiation in animals (Figure 1). Tumor initiation is regulated by SOX2, OCT4 and NANOG transcription factors. These transcription factors act in a concerted manner to regulate genes that control “stemness” and differentiation and thus are responsible for recurrence of tumor. Our study shows that in pancreatic cancer, where there is a high incidence of tumor recurrence, SOX2 was getting modified by O-GlcNAc (Figure 4). O-GlcNAcylation of SOX2 regulates its transcriptional activity. Further, upon inhibition of OGT, Sox2 transcriptional activity was profoundly downregulated compared to other transcription factors responsible for self-renewal (Figure 3F). When OGT is inhibited either by siRNA, or the small molecule inhibitor OSMI, there is a distinct decrease in colony formation units (Figure 3). Additionally, his was rescued only when OGT was overexpressed in these cells along with Sox2 suggesting that it was the modification that was responsible for the colony formation (Figure 4E).
Our results also showed that O-GlcNAc modification was decreasing SOX2 transcriptional activity by affecting its stability and preventing its nuclear localization (Figure 5). This was an interesting observation as it had been reported earlier that in embryonic stem cells, the acetylation Sox2 regulates its nuclear translocation [26, 27]. However, our studies clearly indicated that upon inhibition of OGT, there is an inhibition of nuclear translocation when O-GlcNAc modification of SOX2 is abrogated. Conversely, treatment of pancreatic cancer cells with Thiamet-G, an inhibitor of O-GlcNAcse (an enzyme that facilitates removal of glycosylation) reverted this and promoted nuclear translocation. is beyond the scope of the current manuscript.
The stability of SOX2 has been implicated to play a role in its function. Sumoylation of SOX2 at Lysine 247 is instrumental in inhibition of DNA binding by SOX2 [28]. Our results showed that O-GlcNAc modification of SOX2 also regulates its stability (Figure 5). In a number of other cancers, O-GlcNAc modification is known to regulate protein stability. In prostate cancer, O-GlcNAc modification of Bm1-1 modulates its stability and promotes oncogenic function [29, 30]. However, its role in stability of SOX2 protein in pancreatic cancer cells has not been studied before. Our studies thus show for the first time that O-GlcNAc modification in pancreatic cancer may be instrumental in regulating its self-renewal properties by maintaining the stability of Sox2 and regulating its nuclear translocation.
Conclusion
Self-renewal in a tumor is considered to be one of the hallmarks of the cancer stem cells or tumor initiating cells that are responsible for tumor recurrence, chemoresistance and metastasis. Understanding the molecular switch that can potentially turn-on the self-renewal pathway (by activating Sox2), is thus a significant area of research. In this context, our study highlights that the O-GlcNAc transferase dependent SOX2 glycosylation has a profound effect on the transcriptional activity of SOX2 as this modification regulates its nuclear translocation as well as protein stability. Understanding this mechanism of “turning-on” Sox2 by its glycosylation is thus expected to pave the way for development of novel therapy that can not only decrease tumor progression but also eradicate the “roots” of cancer or the “cancer stem cells”.
Abbreviations
O-GlcNAc: O-N-acetylglucosamine; OGT: O-GlcNAc transferase; OSMI: OGT with a small molecule inhibitor; CHX: Cycloheximide; PDAC: Pancreatic Ductal Adenocarcinoma.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This study was funded by NIH grants R01-CA170946 and R01-CA124723 (to AKS); NIH grant R01-CA184274 (to SB); Katherine and Robert Goodale foundation support (to AKS), Minneamrita Therapeutics LLC (to AKS). The authors would also like to acknowledge Oncogenomics Core at the Sylvester Cancer Center, University of Miami for their help with the RNA-seq transcriptomics study. The authors would like to acknowledge Dr. David Robbins for his valuable comments on the manuscript.
Availability of data and material
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request. Additionally, RNA seq data is deposited in NCBI (GEO Accession number: GSE114472).
Authors' contributions
NS, VKG, PD, KK, RH, BG, AC performed experiments and generated data; CS, NS, VKG, SB, SB analyzed and interpreted the data; NS; AC, VD, Selwyn M Vickers, AS, Sulagna Banerjee revised and edited the manuscript; Santanu Banerjee analyzed the RNA seq and the proteomics data, NS and Sulagna B conceptualized the study, AS, SB provided funding for the study and supported it.
Competing Interests
University of Minnesota has a patent for Minnelide, which has been licensed to Minneamrita Therapeutics, LLC. AKS is the co-founder and the Chief Scientific Officer of this company. SB is a consultant with Minneamrita Therapeutics LLC and this relationship is managed by University of Miami. The remaining authors declare no conflict of interest.
References
1. Olariu V, Lovkvist C, Sneppen K. Nanog, Oct4 and Tet1 interplay in establishing pluripotency. Sci Rep. 2016;6:25438
2. Enomoto K, Watanabe-Susaki K, Kowno M, Takada H, Intoh A, Yamanaka Y. et al. Identification of novel proteins differentially expressed in pluripotent embryonic stem cells and differentiated cells. J Med Invest. 2015;62:130-6
3. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646-74
4. DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7:11-20
5. Banerjee S, Sangwan V, McGinn O, Chugh R, Dudeja V, Vickers SM. et al. Triptolide-induced cell death in pancreatic cancer is mediated by O-GlcNAc modification of transcription factor Sp1. J Biol Chem. 2013;288:33927-38
6. Guillaumond F, Leca J, Olivares O, Lavaut MN, Vidal N, Berthezene P. et al. Strengthened glycolysis under hypoxia supports tumor symbiosis and hexosamine biosynthesis in pancreatic adenocarcinoma. Proc Natl Acad Sci U S A. 2013;110:3919-24
7. Ma Z, Vocadlo DJ, Vosseller K. Hyper-O-GlcNAcylation is anti-apoptotic and maintains constitutive NF-kappaB activity in pancreatic cancer cells. J Biol Chem. 2013;288:15121-30
8. Ferrer CM, Sodi VL, Reginato MJ. O-GlcNAcylation in Cancer Biology: Linking Metabolism and Signaling. J Mol Biol. 2016;428:3282-94
9. Ferrer CM, Reginato MJ. Sweet connections: O-GlcNAcylation links cancer cell metabolism and survival. Mol Cell Oncol. 2015;2:e961809
10. Ferrer CM, Lynch TP, Sodi VL, Falcone JN, Schwab LP, Peacock DL. et al. O-GlcNAcylation regulates cancer metabolism and survival stress signaling via regulation of the HIF-1 pathway. Mol Cell. 2014;54:820-31
11. Ozcan S, Andrali SS, Cantrell JE. Modulation of transcription factor function by O-GlcNAc modification. Biochim Biophys Acta. 2010;1799:353-64
12. Myers SA, Peddada S, Chatterjee N, Friedrich T, Tomoda K, Krings G. et al. SOX2 O-GlcNAcylation alters its protein-protein interactions and genomic occupancy to modulate gene expression in pluripotent cells. Elife. 2016;5:e10647
13. Chen R, Lai LA, Sullivan Y, Wong M, Wang L, Riddell J. et al. Disrupting glutamine metabolic pathways to sensitize gemcitabine-resistant pancreatic cancer. Sci Rep. 2017;7:7950
14. Yang C, Peng P, Li L, Shao M, Zhao J, Wang L. et al. High expression of GFAT1 predicts poor prognosis in patients with pancreatic cancer. Sci Rep. 2016;6:39044
15. Garg B, Giri B, Majumder K, Dudeja V, Banerjee S, Saluja A. Modulation of post-translational modifications in beta-catenin and LRP6 inhibits Wnt signaling pathway in pancreatic cancer. Cancer Lett. 2017;388:64-72
16. Kim E, Ilic N, Shrestha Y, Zou L, Kamburov A, Zhu C. et al. Systematic Functional Interrogation of Rare Cancer Variants Identifies Oncogenic Alleles. Cancer Discov. 2016;6:714-26
17. Dong X, Li Y, Chang P, Tang H, Hess KR, Abbruzzese JL. et al. Glucose metabolism gene variants modulate the risk of pancreatic cancer. Cancer Prev Res (Phila). 2011;4:758-66
18. Wuebben EL, Rizzino A. The dark side of SOX2: cancer - a comprehensive overview. Oncotarget. 2017;8:44917-43
19. Hart GW, Slawson C, Ramirez-Correa G, Lagerlof O. Cross talk between O-GlcNAcylation and phosphorylation: roles in signaling, transcription, and chronic disease. Annu Rev Biochem. 2011;80:825-58
20. Eustice M, Bond MR, Hanover JA. O-GlcNAc cycling and the regulation of nucleocytoplasmic dynamics. Biochem Soc Trans. 2017;45:427-36
21. Hart GW. Three Decades of Research on O-GlcNAcylation - A Major Nutrient Sensor That Regulates Signaling, Transcription and Cellular Metabolism. Front Endocrinol (Lausanne). 2014;5:183
22. Peng C, Zhu Y, Zhang W, Liao Q, Chen Y, Zhao X. et al. Regulation of the Hippo-YAP Pathway by Glucose Sensor O-GlcNAcylation. Mol Cell. 2017;68:591-604 e5
23. Inoue Y, Moriwaki K, Ueda Y, Takeuchi T, Higuchi K, Asahi M. Elevated O-GlcNAcylation stabilizes FOXM1 by its reduced degradation through GSK-3beta inactivation in a human gastric carcinoma cell line, MKN45 cells. Biochem Biophys Res Commun. 2017
24. Sharma NS, Saluja AK, Banerjee S. "Nutrient-sensing" and self-renewal: O-GlcNAc in a new role. J Bioenerg Biomembr. 2017
25. Swamy M, Pathak S, Grzes KM, Damerow S, Sinclair LV, van Aalten DM. et al. Glucose and glutamine fuel protein O-GlcNAcylation to control T cell self-renewal and malignancy. Nat Immunol. 2016;17:712-20
26. Baltus GA, Kowalski MP, Zhai H, Tutter AV, Quinn D, Wall D. et al. Acetylation of sox2 induces its nuclear export in embryonic stem cells. Stem Cells. 2009;27:2175-84
27. Etchegaray JP, Chavez L, Huang Y, Ross KN, Choi J, Martinez-Pastor B. et al. The histone deacetylase SIRT6 controls embryonic stem cell fate via TET-mediated production of 5-hydroxymethylcytosine. Nat Cell Biol. 2015;17:545-57
28. Tsuruzoe S, Ishihara K, Uchimura Y, Watanabe S, Sekita Y, Aoto T. et al. Inhibition of DNA binding of Sox2 by the SUMO conjugation. Biochem Biophys Res Commun. 2006;351:920-6
29. Li Y, Wang L, Liu J, Zhang P, An M, Han C. et al. O-GlcNAcylation modulates Bmi-1 protein stability and potential oncogenic function in prostate cancer. Oncogene. 2017;36:6293-305
30. Yang X, Qian K. Protein O-GlcNAcylation: emerging mechanisms and functions. Nat Rev Mol Cell Biol. 2017;18:452-65
Author contact
![]() Corresponding author: 1550 NW 10th Ave. Papanicolau Building, Suite 109B, Miami, FL 33136, Sulagna.Banerjeemiami.edu (305)-243-8242
Corresponding author: 1550 NW 10th Ave. Papanicolau Building, Suite 109B, Miami, FL 33136, Sulagna.Banerjeemiami.edu (305)-243-8242