Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2019; 9(10):2967-2983. doi:10.7150/thno.30562 This issue Cite
Research Paper
Lgr5-mediated p53 Repression through PDCD5 leads to doxorubicin resistance in Hepatocellular Carcinoma
Zhili Ma*, Deliang Guo*, Quanxiong Wang*, Pengpeng Liu, Yushao Xiao, Ping Wu, Yitao Wang, Baiyang Chen, Zhisu Liu ![]() , Quanyan Liu
, Quanyan Liu ![]()
Department of General Surgery, Research Center of Digestive Diseases, Zhongnan Hospital of Wuhan University, Wuhan 430071, P.R. China
*These authors contributed equally to this work
Received 2018-10-10; Accepted 2019-4-24; Published 2019-5-9
Abstract

The devastating prognosis of hepatocellular carcinoma (HCC) is partially attributed to chemotherapy resistance. Accumulating evidence suggests that the epithelial-mesenchymal transition (EMT) is a key driving force of carcinoma metastasis and chemoresistance in solid tumors. Leucine-rich repeat-containing G protein-coupled receptor 5 (Lgr5), as an EMT inducer, is involved in the potentiation of Wnt signaling in HCC. This study proposes uncovering the roles of Lgr5 in Doxorubicin (Dox) resistance of HCC to improve treatment efficacy for HCC.
Methods: We investigated the expression and significance of Lgr5 in HCC tissue and different cell lines. The effect of Lgr5 in EMT and Dox resistance was analyzed in HCC cells and implanted HCC tumor models. A two-hybrid analysis, using the Lgr5 gene as the bait and a HCC cDNA library, was used to screen targeted proteins that interact with Lgr5. The positive clones were identified by coimmunoprecipitation (Co-IP) and Glutathione-S-transferase (GST) pull-down. The impact of the interaction on Dox resistance was investigated by a series of assays in vitro and in vivo .
Result: We found that Lgr5 was upregulated and positively correlated with poor prognosis in HCC. Additionally, it functioned as a tumor promoter to increase cell migration and induce EMT in HCC cells and increase the resistance to Dox. We identified programmed cell death protein 5 (PDCD5) as a target gene of Lgr5 and we found that PDCD5 was responsible for Lgr5-mediated Dox resistance. Further analysis with Co-IP and GST pull-down assays showed that the N-terminal extracellular domain of Lgr5 could directly bind to PDCD5. Lgr5 induced p53 degradation by blocking the nuclear translocation of PDCD5 and leading to the loss of p53 stabilization. Lgr5 showed a protection against the inhibition of Dox on the growth of tumor subcutaneously injected. Moreover, Lgr5 suppressed Dox-induced apoptosis via the p53 pathway and attenuated the cytotoxicity of Dox to HCC.
Conclusion: Lgr5 induces the EMT and inhibits apoptosis, thus promoting chemoresistance by regulating the PDCD5/p53 signaling axis. Furthermore, Lgr5 may be a potential target gene for overcoming Dox resistance.
Keywords: Lgr5, PDCD5, hepatocellular carcinoma, resistance
Introduction
HCC is one of the leading causes of cancer-related death worldwide [1]. Surgical resection is the most common therapy for HCC, but hepatectomy cannot be performed in many patients with inappropriate indications [2]. Chemotherapy is the best option for patients who are ineligible for surgical treatment. However, HCC is often resistant to chemotherapeutic drugs, with only a few drugs eliciting a therapeutic effect in patients with HCC [3]. Doxorubicin (Dox) is a chemotherapeutic agent that inhibits the synthesis of DNA and RNA by binding to DNA-associated enzymes[4,5]. Although Dox is the cornerstone of chemotherapy for HCC, the effects of either Dox monotherapy or combination treatment with other chemotherapy drugs remain disappointing [6,7]. Therefore, explorations of the potential mechanism of Dox resistance are urgently needed and will provide researchers opportunities to identify or develop new therapeutic strategies for HCC.
The EMT is a developmentally conserved transdifferentiation process that plays a major role in cancer spread by inducing the formation of highly motile cancer cells [8,9]. Recently, accumulating evidence has revealed a close link between the EMT-like morphology of cancer cells and chemotherapy resistance [10]. EMT pathways facilitate the acquisition of stem cell properties and chemoresistance [11,12]. Studies on oxaliplatin-resistant HCC cells show that resistance can be reversed by suppressing the cell EMT [13]. Cisplatin chemoresistance in epithelial ovarian cancer is regulated by the Snail-induced EMT [14]. AMP-activated protein kinase family member 5 drives chemoresistance in HCC via the EMT [15]. Moreover, mounting evidence suggests that the EMT process is essential for tumor progression and induces chemotherapy resistance in many solid tumors, including colorectal cancer, pancreatic cancer and HCC [16-18]. All these studies provide strong confirmation that the EMT is a key driving force of carcinoma metastasis and chemoresistance in solid tumors.
Leucine-rich repeat-containing G protein-coupled receptor 5 (Lgr5) belongs to the family of G protein-coupled receptors, which may have specific ligands [19-20]. Lgr5 is a marker of adult stem cells and a potentiator of Wnt/R-spondin signaling in numerous stem cell contexts that has driven major advances in our understanding of stem cell biology during homeostasis, regeneration and cancer [21-25]. The expression of Lgr5 is significantly increased in subsets of colorectal, liver, pancreatic, stomach, and epithelial ovarian cancers [26-30]. Lgr5 is involved in the potentiation of Wnt signaling in these human cancers. It is widely accepted that amplification of Wnt/β-catenin signaling is a significant driving force in a subset of human cancers, including HCC. Recent research has shown that Lgr5 promotes the EMT in ovarian cancer and gastric cancer [31]. Ectopic expression of any EMT inducer is sufficient to repress CDH1 and other genes encoding for epithelial cell-to-cell adhesion proteins and EMT pathways also facilitate the acquisition of stem cell properties and chemoresistance. Therefore, it is reasonable to presume that Lgr5, as an EMT inducer, could induce Dox chemoresistance in HCC.
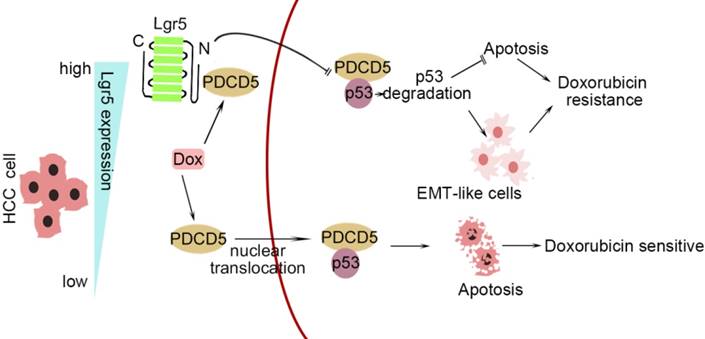
In this study, we investigated the correlation between upregulated Lgr5 expression in HCC tissues and a poor prognosis. Overexpression of Lgr5 resulted in EMT and Dox resistance, and these effects were ascribed to the inactivation of the PDCD5/p53 signaling pathway. Lgr5 also suppressed Dox-induced apoptosis via the p53 pathway in HCC cells. We suggest that Lgr5 is a novel biomarker for predicting the response to Dox-based chemotherapy (Scheme 1).
A graphical model for Lgr5-mediated Dox resistance in HCC cell lines.
Materials and methods
Ethical application
The protocols used in this study conformed to the ethical guidelines of the 1975 Declaration of Helsinki and were approved by the Human Subjects Committee of Zhongnan Hospital. Written informed consent was obtained from all the patients.
HCC specimens and patients
HCC specimens, including tumor tissue and corresponding adjacent nontumor liver tissue, were collected from HCC patients at the Zhongnan Hospital of Wuhan University (Hubei Province, China). Fresh samples for RNA extraction were placed in RNAlater (Invitrogen) and then stored at -80 ℃. The clinicopathologic data of the specimens from 100 HCC patients were obtained and are summarized in Table S1.
Cell lines and culture
The HCC cell lines HepG2, HepG2.215, HCCLM9, Huh7, and SK-Hep1 and the immortalized human hepatic cell line HL-7702 (L02) were purchased from the Cell Bank of the Type Culture Collection (CBTCC, Chinese Academy of Sciences, Shanghai, China) and cultured in DMEM (HyClone, USA) supplemented with 10% fetal bovine serum (Gibco, Carlsbad, CA, USA).
Immunofluorescence and immunohisto-chemistry
To identify the protein subcellular locations, cells grown on coverslips were washed with cold PBS and fixed with 4% paraformaldehyde for 15 min. Then, the cells were permeabilized with 0.5% Triton X-100 for 20 min followed by blocking with 5% goat serum (Gibco, USA) for 30 min. The cells were incubated with primary antibodies overnight at 4 ℃ and with secondary antibodies for 1 h. After washing 3 times, the cells were stained with DAPI (4'-6-Diamidino-2-phenylindole) (Thermo Fisher Scientific) and imaged with a confocal laser scanning microscope (Olympus FV1000). Paraffin-embedded sections of tissue were deparaffinized and rehydrated. After antigen retrieval, the slides were incubated with 3% H2O2 for 10 min and blocked with 5% corresponding serum for 1 h. The slides were incubated with primary antibodies and then incubated with HRP-conjugated secondary antibodies. The sections were stained with diaminobenzidine (DAB) and counted in five visual fields per slide under a microscope. The expression level of the protein in immunohistochemistry (ICH) were scored according to the extent of cell staining (the percentage of positive cells: ≤10%, 0; 11~50% : 2; 51~80% : 3; >80% : 4) and the intensity of staining cell (no staining: 0; slight staining: 1; moderate staining: 2; strong staining: 3). Then the score for the extent of cell staining was multiplied by the intensity of staining cell. Score of 0~3 was negative staining, 4~6 was weak staining, 7~9 was moderate staining and 10~12 was strong staining.The antibodies used are listed in Table S4.
Yeast two-hybrid screening
The full-length Lgr5 cDNA was cloned into pGBKT7 as bait and used to screen a human HCC cDNA library constructed in the prey plasmid pGADT7. The Y2H yeast strain transformed with bait was combined and incubated with the Y187 yeast strain transformed with prey plasmids. The mated cells were selected with four reporter genes. The yeast two-hybrid screens were performed according to the manufacturer's instructions (Clontech).
Plasmid constructions
For Co-IP, full-length Lgr5 with a C-terminal Flag tag and PDCD5 with a C-terminal Myc tag was cloned into pcDNA3.1(+). Deletion mutants of Lgr5, (1) L1:Lgr5Δ22-561, (2) L2:Lgr5Δ562-823 and (3) L3:Lgr5Δ624-907, were cloned into pcDNA3.1(+). For the GST pull-down assay, the full-length sequence of PDCD5 was cloned into pGEX-6P-1 (GE) containing a GST tag. Deletion mutants of Lgr5 (22-561) were cloned into pCold III for expression of the 6×His tag fusion protein. For Lgr5 knockdown, three shRNAs against Lgr5 were cloned into the psi-LVRH1GP vector. The vector and lentivirus were transfected according to the manufacturer's instructions, and the stable cells were selected by puromycin. The cDNA sequences of PDCD5, Lgr5 and its three deletion mutants and the Lgr5 shRNA sequence are listed in Table S6.
In vivo assay
For tumor growth and metastasis assay, ten 5-week-old male BALB/c nude mice were randomized into two groups (n=5), SK-Hep1 cells (1×106) stably transfected with Lgr5 or empty vector were injected into the tail veins for the establishments of pulmonary metastatic model. Mice were sacrificed 8 weeks post injection. The lung metastatic foci were detected by hematoxylin and eosin (H&E) staining.
For tumor drug resistance assay, 20 nude mice were randomized into four groups (n=5) including Lgr5, NC, Lgr5+Dox and NC+Dox. SK-Hep1 cells (5×105) stably transfected with Lgr5 or empty vector were subcutaneously injected into these nude mice. The Lgr5+Dox and NC+Dox groups were received Dox (5mg/kg body weight, iv) once every five days. The Lgr5 and NC groups were received saline as control. The growth of the implanted tumor was calculated by vernier caliper and the volume was calculated with the formula: V=ab2/2, where a is the maximum axis and b is the minimal axis. Mice were sacrificed 30 days after the first injection. Animals were purchased from the Central Laboratory of Animal Science, Wuhan University (Wuhan, China) and were maintained in a specific pathogen-free facility.
Western blot analysis
Total protein extracted from the HCC tissues and cells was measured by BCA protein assay (Beyotime, Shanghai, China). Equal amounts of protein were separated by SDS-polyacrylamide gel electrophoresis at 100 V for 2 h. Then, we transferred the protein from the gels to PVDF membranes (Millipore, USA). The membranes were incubated with appropriate dilutions of primary antibodies overnight at 4 ℃ after blocking with 5% bovine serum albumin in TBST. The membranes were incubated with secondary antibodies conjugated to HRP for 1 h at room temperature followed by treatment with Clarity Max™ Western ECL Substrate (Bio-Rad, USA). The antibodies are listed in Table S4.
Coimmunoprecipitation
To verify the interaction between Lgr5 and PDCD5 by co-IP, the overexpression plasmids Flag-Lgr5 and Myc-PDCD5 were cotransfected into HEK 293T cells. The cell lysates in IP lysis buffer (pH 7.4, 0.025 M Tris, 0.15 M NaCl, 0.001 M EDTA, 1% NP40, 5% glycerol) were incubated with anti-Flag and anti-Myc antibodies or IgG overnight at 4 ℃ with shaking. The immune complex solution was incubated with protein A/G magnetic beads for 1 h at room temperature with mixing and then washed to remove the unbound immune complex. The bound immune complex was dissociated from the beads with low-pH buffer for western blotting analysis (Pierce™ Classic Magnetic IP/Co-IP Kit, USA). The antibodies used in this study are listed in Table S4.
Quantitative real-time PCR
Total RNA was extracted from HCC tissue and cells using TRIzol reagent (Invitrogen) and reverse transcribed into cDNA using the PrimeScript™ RT Reagent Kit with gDNA Eraser (Takara). RT-qPCR was performed with SYBR Green mix. These experiments were performed in triplicate. The primers for each gene are listed in Table S5.
GST pull-down assay
The plasmids for GST-PDCD5 and His-Lgr522-561 were transfected into E. coli. The fusion proteins were prepared as described previously. Approximately 100 µg of GST and GST-PDCD5 fusion protein was immobilized in 50 µL of glutathione agarose and equilibrated before being incubated together at 4 ℃ for 60 min with gentle rocking motion. Approximately 100 µg of His-Lgr522-561 fusion protein was added to the immobilized GST-PDCD5 and GST after 3 washes with PBST. The two fusion proteins were incubated overnight at 4°C under gentle rotation. The bound proteins were eluted with elution buffer (10 mM glutathione in PBS, pH 8.0) and analyzed by immunoblotting.
Cell proliferation and cytotoxicity assay
The proliferation of HCC cells transfected with Lgr5, PDCD5 or shRNA-Lgr5 and the cell cytotoxicity following treatment with different concentrations of doxorubicin were assessed using a CCK8 assay (Dojundo, Japan). To evaluate cell proliferation, HCC cells were seeded in 96-well plates at a density of 104 cells/well in 100 µL of culture medium. CCK8 solution was added to each well, and the plate was incubated for 1 h at 37 ℃. Then, the optical density was measured at 450 nm. To evaluate the cell cytotoxicity, the cells were treated with Dox when they reached 70-80% confluence and tested by CCK8 assay. All experiments were performed three times.
Flow cytometric analysis
SK-Hep1 cells transfected with Lgr5 or PDCD5 or cotransfected with Lgr5 and PDCD5 were seeded into 6-well plates and cultured to 70-80% confluence. Then, the cells were not treated or were treated with doxorubicin (1 µg/mL) for 12 h and harvested followed by staining with an annexin V-FITC/PI staining kit according to the manufacturer's instructions (Sigma-Aldrich). The cells were then analyzed by flow cytometry to identify apoptosis (BD Biosciences). The experiments were repeated three times.
Cell colony formation assay, wound healing assay and invasion assay
For the cell colony formation assay, cells were seeded into a 6-well plate at a density of 103 cells per well and cultured for 2 weeks. The colonies were stained by crystal violet and counted. For the wound healing assay, cells were seeded into a 6-well plate at a density of 5×105 cells per well, and wounds were created with a 100 µL plastic pipette tip when the cells were 80- 90% confluent. The cells were cultured in serum-free medium, and the wound lines were measured after 12 or 24 h. For the invasion assay, Matrigel-coated chambers with 8 µm pores (BD Biosciences, Franklin Lakes, NY, USA) were used. Cells were seeded in the upper chamber at a density of 5×104 cells per well and cultured in medium with 0.1% bovine serum albumin for 24 h. The cells that invaded the membrane were counted after staining by crystal violet. All results were from at least three separate experiments.
Statistical analysis
The data are presented as the mean ± s.d. or as a percentage from at least three independent experiments. Χ2 tests, multivariate Cox regression analysis, Spearman's rank correlation coefficient analysis and Kaplan-Meier survival analysis were performed using IBM SPSS statistical software (IBM Corporation, USA). Student's t-test was used for comparisons in GraphPad (GraphPad Software Inc. USA). P values < 0.05 were considered statistically significant in all tests.
Results
Lgr5 expression is upregulated and negatively correlated with prognosis in HCC
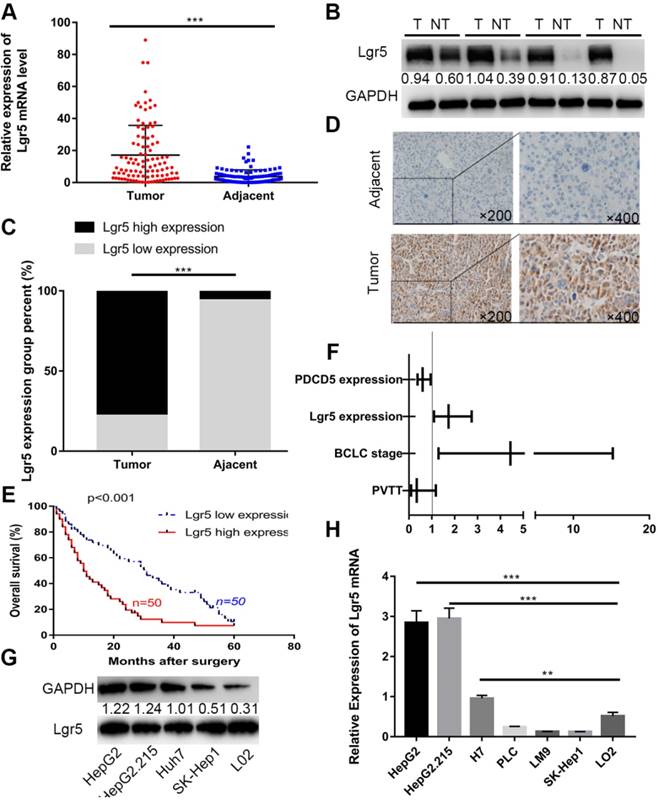
We first investigated the expression of Lgr5 in 100 HCC specimens between tumor and adjacent nontumor tissues using RT-qPCR and western blot analysis. The results showed that Lgr5 expression was significantly upregulated in tumor tissue compared with adjacent nontumor tissue (Figure 1A-B). We then confirmed these findings using IHC in these pairs of HCC specimens, which indicated that 78 (78%) of tumor tissue samples had high expression of Lgr5 (moderate and strong staining) that in tumor tissue, while 94 (94%) of nontumor tissue samples had a low expression of Lgr5 that in adjacent non-tumor tissue (Figure 1C-D). Furthermore, Kaplan-Meier survival curves revealed an obvious correlation between high Lgr5 expression and poor prognosis in patients with HCC (Figure 1E). Moreover, an analysis of the clinical characteristics of HCC revealed that Lgr5 expression was significantly correlated with tumor size (P=0.0093), tumor number (P=0.0253), BCLC stage (P=0.0005) and PVTT (P=0.0007) (Table S1). Multivariate analysis showed that high Lgr5 expression was an independent predictor of poor prognosis in patients with HCC (Figure 1F and Table S3). Finally, we investigated the levels of Lgr5 mRNA and protein in various HCC cell lines, including the HepG2, HepG2.215, PLC, LM9, Huh7 and SK-Hep1 cell lines and the immortalized normal human hepatic cell line L02. The results showed that Lgr5 mRNA and protein expression levels were increased in most HCC cell lines compared with the L02 cell line Figure 1G-H). Taken together, these data demonstrated that Lgr5 was upregulated in HCC tumor tissue and hepatoma cell lines and that its high expression indicated a negative prognosis in patients with HCC.
Lgr5 expression is upregulated and negatively correlated with prognosis in HCC. A. The expression of Lgr5 was different between tumor and adjacent nontumor tissue from 100 patients with HCC as determined by RT-qPCR. B. The protein levels of Lgr5 between tumor and adjacent nontumor tissue from 4 patients with HCC were detected by western blotting. The density ratio of protein to GAPDH calculated from the band density is shown. C. Semiquantitative data showed Lgr5 expression analyzed by IHC in HCC tissue compared with adjacent tissue. D. Representative IHC staining images of Lgr5 in HCC tissue as well as in adjacent nontumor tissue. Scale bars, 50 μm. E. Kaplan-Meier survival curve of the 100 HCC patients grouped according to the expression of Lgr5 in tumors. The median expression level was used as the cutoff. F. Forest plot of risk factors using multivariate Cox regression analysis. G. The expression of Lgr5 in HCC cell lines and a normal liver cell line (L02) was analyzed by western blotting (the ratio of molecule/GAPDH was indicated below). H. Expression of Lgr5 in different HCC cell lines and the normal human liver cell line (L02) as detected by qPCR. *p<0.05, **p<0.01, **p<0.001, t-test.
Lgr5 functions as a tumor promoter to increase cancer cell migration and induce an EMT-like phenotype in HCC cells
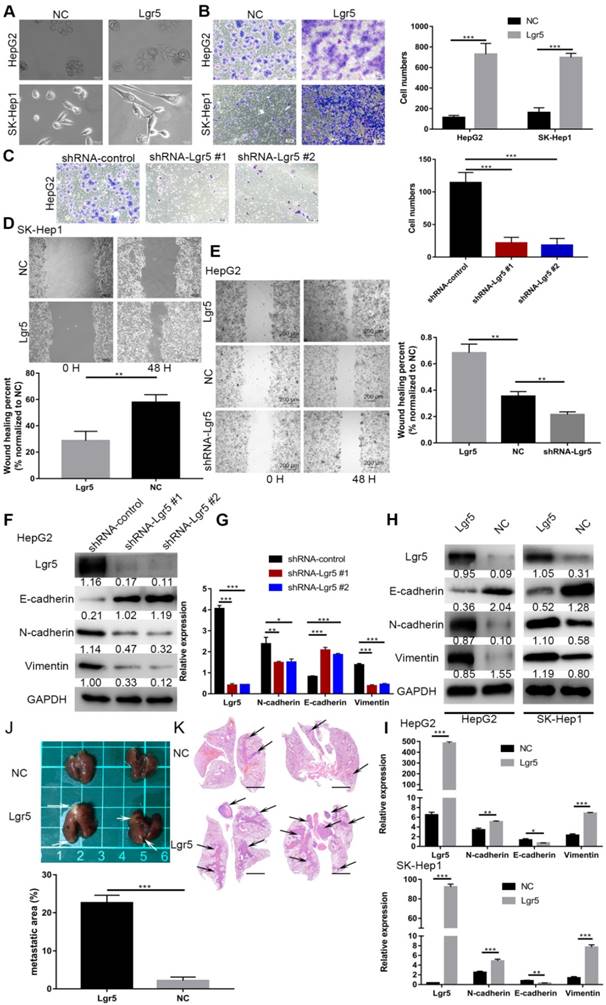
Considering the important role of Lgr5 in the development of HCC, we investigated its potential function as an oncogene or oncosuppressor. According to analysis by CCK8 assay, Lgr5 could significantly promote cell proliferation of SK-Hep1 cells in Lgr5 overexpression and knockdown experiments (Figure S1A). Furthermore, a colony formation assay confirmed that Lgr5 could stimulate cancer cell proliferation, and a quantitative apoptosis assay indicated that Lgr5 could inhibit hepatoma apoptosis (Figure 5D-F). When analyzing the correlation between Lgr5 expression and clinicopathological parameters, we found that Lgr5 was associated with PVTT in HCC patients, suggesting that Lgr5 affects the metastasis of cancer cells. We next detected the relationship between Lgr5 and the EMT, which is regarded as an initial event of neoplasm metastasis. Upon establishing stable Lgr5-overexpressing hepatoma cells, we observed morphological changes to a cell morphology resembling an EMT-like phenotype (Figure 2A). Transwell assays showed that overexpression of Lgr5 induced migration and invasion in HCC cells (Figure 2B), while knockdown of Lgr5 significantly inhibited the migration and invasion capacity of HCC cells (Figure 2C). Likewise, the wound-healing assay showed the same influence of Lgr5 on cell migration (Figure 2D-E). Furthermore, we detected the expression of E-cadherin, an epithelial marker, and N-cadherin and vimentin, which are mesenchymal markers. Western blotting and RT-PCR analysis showed that the levels of N-cadherin and vimentin were decreased but that the levels of E-cadherin were increased in HCC cells with stable knockdown of Lgr5 (Figure 2F-G). Moreover, we also observed that the expression of N-cadherin and vimentin was increased and that E-cadherin expression was decreased in Lgr5-overexpressing HCC cells (Figure 2H-I). Consistent with these in vitro assays, the pulmonary metastatic model showed a greater number of pulmonary metastatic nodules in the lung of mice injected with Lgr5 overexpression cells (Figure 2J). H&E staining of lung sections showed that Lgr5 overexpression increased the area of visible lung metastases (Figure 2K). All the data suggested that Lgr5 could induce an EMT-like phenotype and promote metastasis in HCC cells.
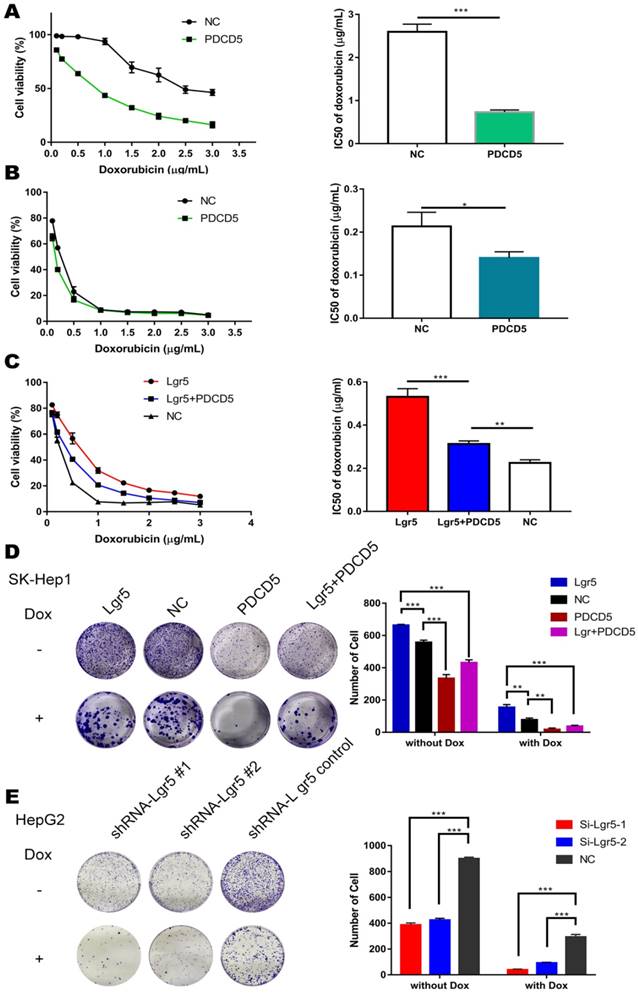
PDCD5 overexpression partially rescues Lgr5-mediated drug resistance. A, B. HepG2 and SK-Hep1 cells transfected with PDCD5 or empty vector as NC (negative controls) were treated with Dox at different concentrations for 48 h, and their viability was tested by CCK8 assay. The IC50 was calculated with a dose-response curve. C. SK-Hep1 cells transfected with Lgr5 or empty vector as NC (negative controls) or cotransfected with Lgr5 and PDCD5 were treated with Dox at different concentrations for 48 h, and their viability was tested by CCK8 assay. The IC50 was calculated with a dose-response curve. D, E. Colony formation assay in SK-Hep1 and HepG2 cells transfected with Lgr5, PDCD5 or shRNA-Lgr5. *p<0.05, **p<0.01, **p<0.001, t-test.
Lgr5 functions as a tumor promoter to increase cancer cell migration and induce an EMT-like phenotype in HCC cells. A. Morphology of HCC cells transfected with Lgr5 and shRNA-Lgr5. Scale bars, 20 µm. B,C. The migration capacity of HCC cells transfected with Lgr5 and shRNA-Lgr5 was analyzed by transwell assay. The quantitation of the data is shown. Scale bars, 50 µm. D,E. Wound-healing migration assays. The quantification of the open area is shown. Scale bars, 200 µm. F,H. The expression of protein associated with EMT in the cells with overexpression or knockdown of Lgr5 was detected by western blotting (the ratio of molecule/GAPDH was indicated below). G,I. The mRNA levels of E-cadherin, N-cadherin and vimentin in cells with overexpression or knockdown of Lgr5 were detected by qPCR. J. Representative image of the visible metastatic nodules in the mouse lungs of the SK-Hep1-Lgr5 group and the SK-Hep1-control group. K. Representative images of H&E stained lung section from the mice. Scale bars, 1000 µm. Percentage of metastatic area in lung tissue of mice injected with SK-Hep1-Lgr5 and SK-Hep1-control control is shown on the right. *p<0.05, **p<0.01, **p<0.001, t-test.
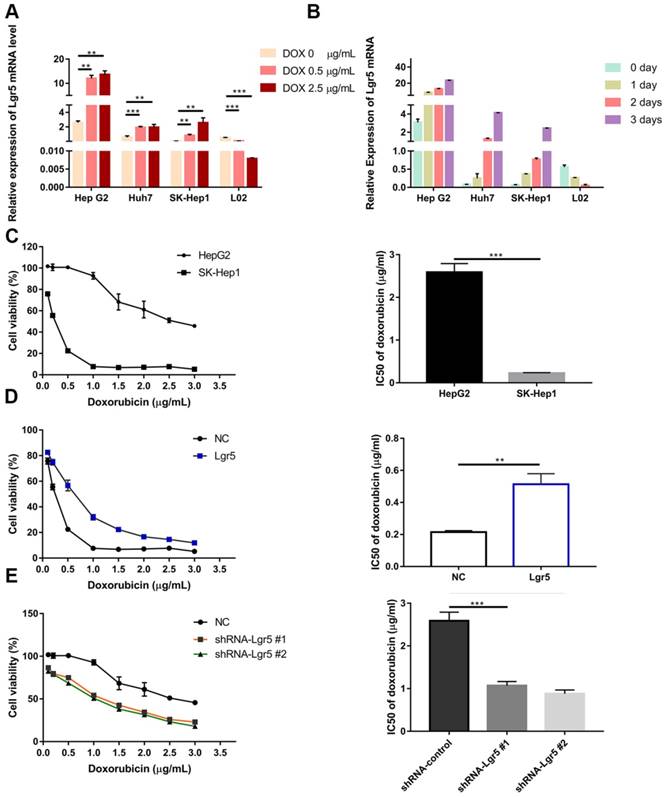
Lgr5 overexpression increases HCC cell resistance to Dox
Since EMT pathways facilitate the acquisition of stem cell properties and chemoresistance and since Lgr5 expression appears to promote the EMT-like phenotype, we investigated the relationship between Lgr5 and Dox efficacy in HCC cells. We found that cancer cell viability was positively associated with Lgr5 expression after treatment with Dox (1 µg/mL) in hepatoma cell lines (Figure S1B). We subsequently incubated HCC cell lines (HepG2, Huh7 and SK-Hep1) and the normal liver cell line L02 with different concentrations of Dox. The results showed that the expression of Lgr5 increased following treatment with increasing Dox concentrations in the different HCC cell lines. However, similar results were not found in L02 (the normal liver cell line) treated with Dox (Figure 3A). Then, we treated cells with Dox (1 µg/mL) and then detected the mRNA and protein levels of Lgr5 at different times. The results showed that Dox increased the expression of Lgr5 in HCC cell lines in a time-dependent manner but decreased Lgr5 expression in L02 cells (Figure 3B and Figure S1E). We further analyzed the effects of Lgr5 depletion or overexpression on Dox-treated cells through CCK8 assays. Strikingly, high Lgr5 expression levels effectively decreased the Dox sensitivity of HCC cells. The IC50 value for Dox in HepG2 cells was higher than that in SK-Hep1 cells because of the higher expression of Lgr5 in HepG2 cells compared with SK-Hep1 cells (Figure 3C). To examine whether the overexpression of Lgr5 enhances the resistance of HCC cells to Dox, we cultured SK-Hep1 cells transfected with Lgr5 with different concentrations of Dox and found that overexpression of Lgr5 could increase the IC50 value in SK-Hep1 cells (Figure 3D). Consistently, knockdown of Lgr5 could sensitize HepG2 cells to Dox (Figure 3E). These data suggested that the expression of Lgr5 has a significant correlation with Dox resistance.
Lgr5 overexpression increases HCC cell resistance to Dox. A. HCC cell lines and a normal liver cell line (L02) were treated with different concentrations of Dox for 24 h, and their expression of Lgr5 was detected by RT-qPCR. B. HCC cell lines and a normal liver cell line (L02) were treated with Dox (1 µg/mL), and their expression of Lgr5 was detected by qPCR once every three days. C. SK-Hep1 and HepG2 cells were treated with different concentrations of Dox for 48 h, and their cell viability was tested by CCK8 assay. The IC50 was calculated with a dose-response curve. D. SK-Hep1 cells transfected with Lgr5 or empty vector as NC (negative controls) were treated with Dox at different concentrations for 48 h, and their viability was tested by CCK8 assay. The IC50 was calculated with a dose-response curve. E. HepG2 cells transfected with shRNA-Lgr5 #1, shRNA-Lgr5 #2 or shRNA-control as NC were treated with Dox at different concentrations for 48 h, and their viability was tested by CCK8 assay. The IC50 was calculated with a dose-response curve. *p<0.05, **p<0.01, **p<0.001, t-test.
The N-terminal extracellular domain of Lgr5 can directly bind to PDCD5
To further understand how Lgr5 could influence drug resistance, we used a yeast two-hybrid assay with full-length Lgr5 as the bait and an HCC cDNA library as the prey. A total of 41 positive clones grown on the selective agar plate were identified to encode 30 different proteins by sequencing (Figure 4A). We tested these proteins and focused our attention on the programmed cell death protein 5 (PDCD5) as a putative mediator of Lgr5-dependent Dox resistance. Furthermore, we found that the expression of PDCD5 was downregulated in HCC tissue compared with the corresponding adjacent nontumor tissue, as detected by RT-qPCR and western blot analysis (Figure S2A-B). Consistently, IHC staining in 50 pairs of HCC specimens showed that 36 (72%) of the cases had lower expression of PDCD5 (negative or weak staining) in tumor tissue than in adjacent nontumor tissue, while 4 (8%) of cases had lower expression of PDCD5 in adjacent nontumor tissue than in tumor tissue (Figure S2C-D). Kaplan-Meier survival curves showed that elevated expression of PDCD5 in the tumor tissue of HCC patients was associated with improved overall survival (Figure S2E). The expression of PDCD5 was negatively correlated with tumor size (P=0.0278), BCLC stage (P=0.0088) and PVTT (P=0.0132) (Table S2). Multivariate Cox regression analysis suggested that PDCD5 is an independent predictor of overall survival (Figure 1F and Table S3).
The N-terminal extracellular domain of Lgr5 can directly bind to PDCD5. A. Yeast two-hybrid assay. Bait constructed with pGBKT7-Lgr5 was analyzed against prey constructed with human hepatocarcinoma tissue cDNA libraries. B. Co-IP of Lgr5 and PDCD5. Flag-Lgr5 and Myc-PDCD5 plasmids were transfected into HEK 293T cells. The precipitate was analyzed by western blotting with anti-Flag and anti-Myc antibodies. C. Interaction between endogenous Lgr5 and PDCD5 in cells. Cell lysates of HepG2 were prepared and used for Co-IP. The coimmunoprecipitates were analyzed by western blotting with anti-Lgr5 and anti-PDCD5 antibodies. D. Immunofluorescence was analyzed in HEK 293T cells transfected with Flag-Lgr5 and Myc-PDCD5 and detected with anti-Flag and anti-Myc antibodies. Fluorescence images were captured by confocal fluorescence microscopy. Scale bars, 20 µm. E. Flag-tagged full-length and deletion mutants (Lgr5, L1: Lgr5Δ22-561, L2: Lgr5Δ562-823, and L3: Lgr5Δ624-907) of the Lgr5 gene were cotransfected with Myc-PDCD5 in HEK 293T cells. Cell lysates were immunoprecipitated with an anti-Flag antibody, and the precipitates were analyzed with an anti-Myc antibody. F. His-Lgr5Δ22-561 and GST-PDCD5 proteins expressed in an E. coli system were detected by SDS-PAGE. G. Detection of His-Lgr5Δ22-561 bound to GST-PDCD5 or GST in a GST pull-down assay.
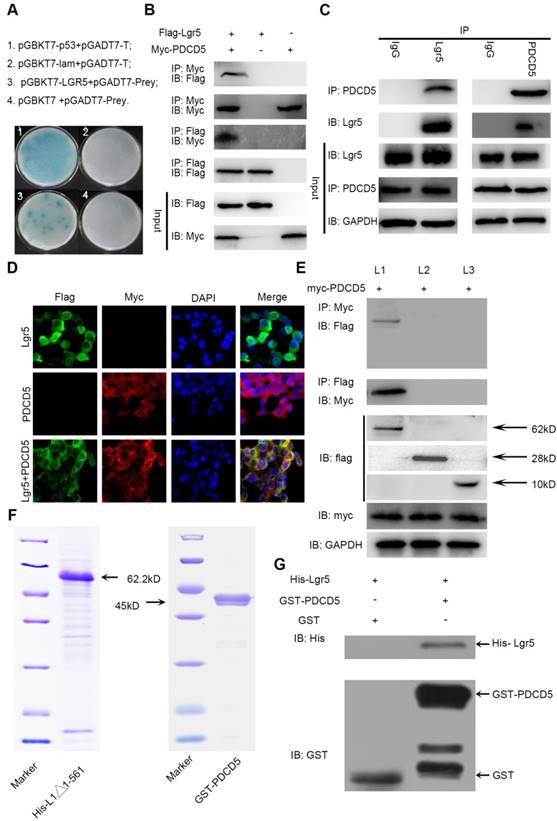
To confirm the interaction between Lgr5 and PDCD5, we performed immunoprecipitation in HEK 293T cells transfected with Flag-Lgr5 or Myc-PDCD5 or cotransfected with both plasmids. The western blotting images showed that the anti-Flag antibody could immunoprecipitate the Flag-Lgr5/Myc-PDCD5 complex in the cells cotransfected with Flag-Lgr5 and Myc-PDCD5, but the anti-Flag antibody could not immunoprecipitate Myc-PDCD5 in the cells transfected with only Flag-Lgr5. Additionally, a reciprocal immunoprecipitation showed that the anti-Myc antibody could immunoprecipitate Flag-Lgr5 (Figure 4B). To further determinate the endogenous interaction between Lgr5 and PDCD5, we chose the HepG2 cell line as a model for immunoprecipitation analysis because of its higher expression of Lgr5 compared with that in the other HCC cell lines. The anti-Lgr5 antibody could detect Lgr5 in the immune complex precipitated by anti-PDCD5 rather than the IgG antibodies. A reciprocal immunoprecipitation with anti-Lgr5 antibodies also confirmed the interaction between Lgr5 and PDCD5 (Figure 4C). Moreover, the interaction between Lgr5 and PDCD5 could be confirmed through immunofluorescence analysis by their colocation in HEK 293T cells transfected with both Flag-Lgr5 and Myc-PDCD5 (Figure 4D). To identify the interaction domain of Lgr5 binding to PDCD5, we carried out Co-IP with different domains of Lgr5. Since the Lgr5 structure can be divided into three parts, the extracellular region, the transmembrane region and the intracellular region [32], we established three truncated mutants of Lgr5: (1) L1:Lgr5Δ22-561, consisting of the N-terminal extracellular region; (2) L2:Lgr5Δ562-823, consisting of the transmembrane region, three extracellular loops and three intracellular loops; and (3) L3:Lgr5Δ624-907, consisting of the C-terminal intracellular region (Figure S3A). The three Lgr5 mutants were cotransfected into HEK 293T cells with Myc-PDCD5, and the cell lysates were coimmunoprecipitated with anti-Flag or anti-Myc antibodies. The results showed that the N-terminal extracellular domain included the binding domain of PDCD5 (Figure 4E). Next, to investigate whether PDCD5 directly binds to Lgr5, we performed GST pull-down assays with Lgr5Δ624-907 and PDCD5 in an E. coli system. The purified proteins were identified by SDS-PAGE and western blotting analysis (Figure 4F and Figure S3B-C). This assay showed that Lgr5Δ624-907 directly binds to GST-PDCD5, but not GST (Figure 4G). Altogether, these data suggested that the N-terminal extracellular domain of Lgr5 could directly bind to PDCD5.
PDCD5 overexpression partially rescues Lgr5-Mediated Drug Resistance
To address the idea that Lgr5-mediated Dox resistance is at least partially dependent on increases in PDCD5, we overexpressed PDCD5 in SK-Hep1 and HepG2 cells and analyzed cellular viability upon Dox treatment. The results showed that the IC50 of HCC cells transfected with PDCD5 was significantly lower than that of cells transfected with empty vector, suggesting that PDCD5 could enhance the cytotoxicity of Dox to HCC cells (Figure 5A-B). Then, we transfected PDCD5 into HCC cells stably overexpressing Lgr5 and observed that overexpression of PDCD5 could partly inhibit the enhancement of resistance to Dox mediated by Lgr5 (Figure 5C). A colony formation assay confirmed that Lgr5 and PDCD5 could impact cell proliferation and resistance to Dox (Figure 5D-E). These results suggested that cancer cells overexpressing PDCD5 become more sensitive to Dox-mediated cell death, thus confirming this pathway as a mechanism exploiting Lgr5-mediated drug resistance.
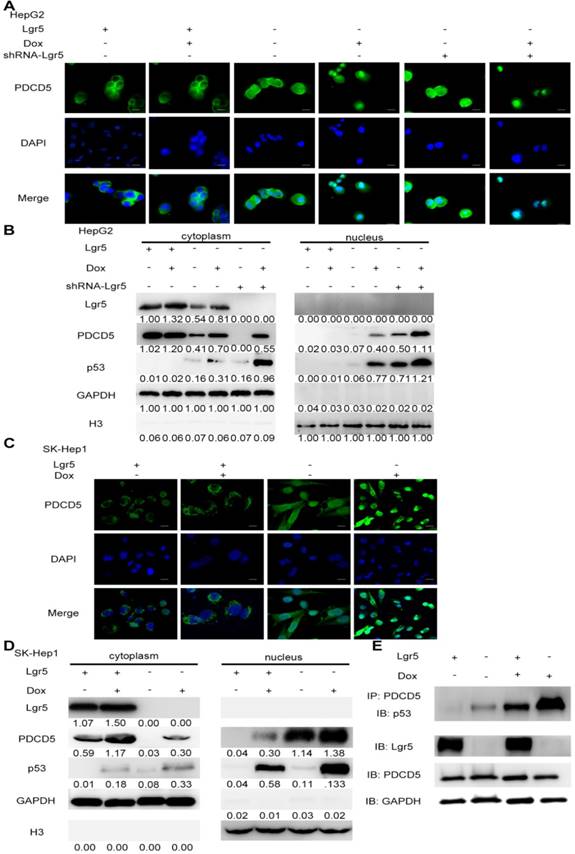
Lgr5 induces p53 degradation by blocking the nuclear translocation of PDCD5
To investigate the interaction between Lgr5 and PDCD5, we established overexpression plasmids for Lgr5 and PDCD5 and transfected them into SK-Hep1 and HepG2 cell lines. We found that overexpression of Lgr5 could not influence the expression of PDCD5 and that PDCD5 could not change the level of Lgr5 expression (Figure S4A-B). Owing to the high level of Lgr5 in the HepG2 cell line, we stably transfected shRNA-Lgr5 into HepG2 cells and found that the knockdown of Lgr5 could not change the expression of PDCD5 (Figure S4C-E). However, we found that Dox could promote PDCD5 mRNA and protein levels in HCC cells (Figure S1C-E).
PDCD5 can translocate to the nucleus in response to genotoxic stress and interact with P53 to regulate its dynamics in the DNA damage response [33-35]. It is well accepted that loss of p53 function is associated with drug resistance and cancer recurrence [36-38]. Therefore, it is reasonable to presume that Lgr5-mediated drug resistance may be attributed to its influence on the p53 pathway. To verify whether Lgr5 affects the translocation of PDCD5, we analyzed the subcellular location of PDCD5 in HCC cell lines transfected with shRNA-Lgr5 and an overexpression vector of Lgr5 by immunofluorescence assay. The results showed that PDCD5 could translocate from the cytosol to the nucleus in the SK-Hep1 and HepG2 cell lines treated with Dox but that Lgr5 could prevent this translocation regardless of whether HCC cells were treated with Dox (Figure 6A-D). Lgr5 knockdown induced PDCD5 nuclear translocation in these HCC cells (Figure 6A-B). We then confirmed that Lgr5 could block the nuclear translocation of PDCD5 in overexpression and knockdown experiments with western blotting analysis. We also found that Lgr5 could decrease p53 expression by blocking the nuclear translocation of PDCD5 (Figure 6A-D). To study the specific role of PDCD5 nuclear translocation in p53 expression, we performed immunoprecipitation with an anti-PDCD5 antibody. We found that Lgr5 overexpression could reduce the level of p53 precipitated by PDCD5 in HCC cells (Figure 6E), which suggested that the interaction between PDCD5 and p53 was weakened by Lgr5. These results demonstrated that Lgr5 could induce p53 degradation by trapping PDCD5 in the cytoplasm and preventing its interaction with p53.
Lgr5 induces p53 degradation by blocking the nuclear translocation of PDCD5. A,C. Subcellular colocalization of PDCD5. SK-Hep1 and HepG2 cells were transfected with Lgr5 for overexpression. shRNA-Lgr5 was transfected into HepG2 cells for knockdown. Immunofluorescence analysis in the cells treated as indicated shows the subcellular location of PDCD5. B,D. The cytoplasmic and nuclear extracts of protein from the cells treated as indicated were separated and prepared for western blotting with anti-PDCD5, anti-Lgr5 and anti-p53 antibodies (the ratio of molecule/GAPDH was indicated below). E. The interaction between PDCD5 and p53 was analyzed by immunoprecipitation and immunoblotting.
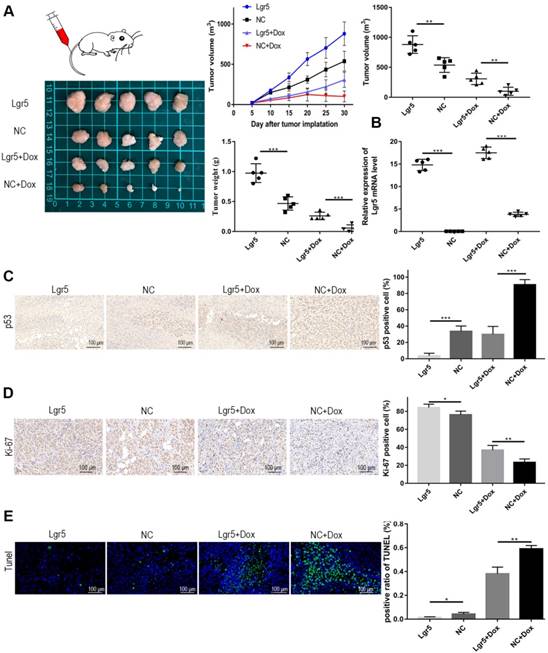
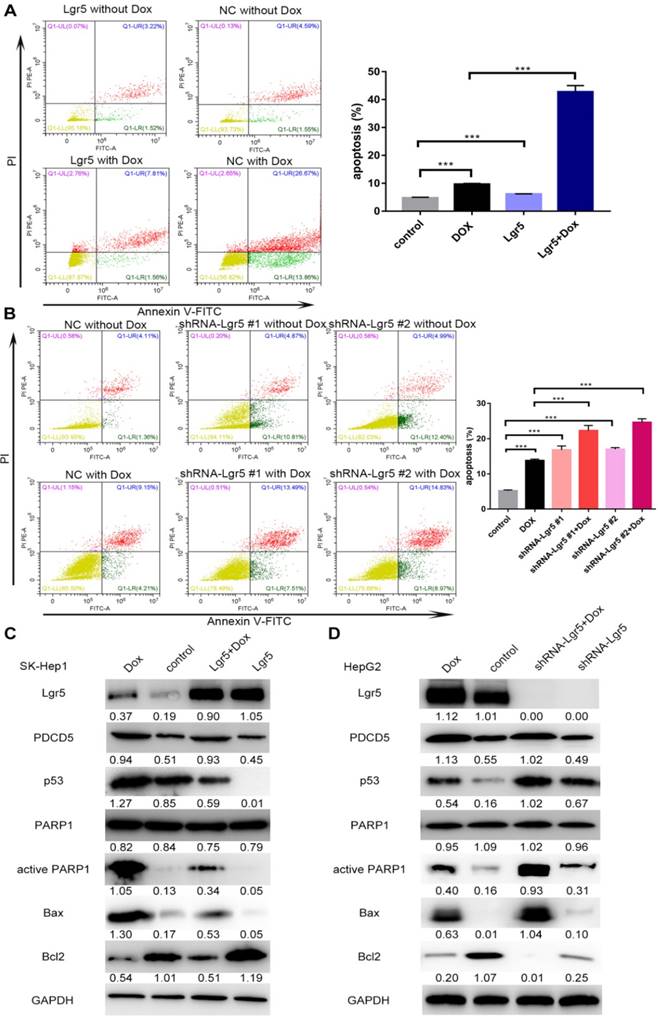
Lgr5 suppresses Dox-induced apoptosis via the p53 pathway in HCC cells
Modulation of apoptosis may influence resistance to chemotherapy and therefore affect the outcome of cancer treatment. The efficiency of Dox in apoptosis induction is partially dependent on p53 status in HCC. To further verify whether Lgr5 could suppress Dox-induced apotosis, we established a xenograft model by stably overexpressing Lgr5 in SK-Hep1 cells. As expected, the overexpression of Lgr5 could promote the tumor growth and Dox resistance in HCC (Figure 7A). The overexpression of Lgr5 was confirmed by qPCR (Figure 7B). And we noticed that Dox inhibited the expression of Ki-67, a cell proliferation marker, while Lgr5 enhanced the expression of Ki67 under treatment of Dox (Figure 7C). Lgr5 also reduced the level of p53 in the tumor that detected by ICH (Figure 7D). Furthermore, TUNEL assay showed that Lgr5 could inhibit the apoptosis induced by Dox in tumor model (Figure 7E).
Lgr5 promotes Dox resistance in HCC in vivo . A. Representative images of SK-Hep1-Lgr5 and SK-Hep1-control tumor growth in nude mice. The tumor weight and volume were shown (right). B. The expression of Lgr5 in the tumors detected by RT-qPCR. C,D. Immunohistochemical analysis of p53 and Ki-67 expression in tumors. Scale bar, 100 µm. E. TUNEL apoptosis assay analysis in tumors. Scale bar, 100 µm. *p<0.05, **p<0.01, **p<0.001, t-test.
In this study, we found that overexpression of PDCD5 enhanced the apoptosis induced by Dox (Figure S5A-B). We also detected the influence of Lgr5 on Dox-induced apoptosis in HCC cells using FACS analysis. The results showed that Dox could induce apoptosis when Lgr5 expression was reduced; however, the percentage of apoptotic cells, including those in early apoptosis (annexin V+/PI-) and late apoptosis (annexin V+/PI+), were significantly suppressed by Lgr5 in the SK-Hep1 cells treated with Dox (Figure 8A). Next, the results of a knockdown cell model using HepG2 cells with higher Lgr5 expression showed that knockdown of Lgr5 could increase the rate of apoptosis, which was beneficial for Dox-induced apoptosis (Figure 8B). To obtain further insight into the possible mechanism by which Lgr5 suppresses Dox-induced apoptosis, we investigated the effects of Lgr5 and Dox on apoptosis-related proteins with western blotting. We found that Dox upregulated the expression of cleaved PARP1 and p53 proteins and changed the expression of several apoptosis-related targets of p53; for example, Bax protein expression was increased and Bcl2 protein expression was decreased (Figure 8C-D). We also observed that Dox promoted Lgr5 and PDCD5 protein expression as previously indicated (Figure S1E). Furthermore, we found that Lgr5 could suppress Dox-induced p53 and Bax protein expression in SK-Hep1 and HepG2 cells in overexpression and knockdown experiments (Figure 8C-D). These results confirmed that Lgr5 suppresses Dox-induced apoptosis via the p53 pathway in HCC cells.
Lgr5 induces cell proliferation and inhibits Dox-induced apoptosis in HCC cells via interaction with PDCD5. A. SK-Hep1 cells were transfected with Lgr5. Flow cytometric analysis was used to detect apoptosis in cells treated with Dox (0 µg/mL or 1 µg/mL). B. HepG2 cells transfected with shRNA-Lgr5 #1, shRNA-Lgr5 #2 or shRNA-control were treated with Dox (0 µg/mL or 1 µg/mL), and their apoptosis was detected by flow cytometric analysis. C. SK-Hep1 cells transfected with Lgr5 were treated with Dox (1 µg/mL or 0 µg/mL) for 24 h. The cell lysates were analyzed by western blotting with antibodies against apoptosis-related proteins (the ratio of molecule/GAPDH was indicated below). D. HepG2 cells transfected with shRNA-Lgr5 #1, shRNA-Lgr5 #2 or shRNA-control were treated with Dox (1 µg/mL or 0 µg/mL) for 24 h. The western blot shows the expression of apoptosis-related proteins in the cells previously described. *p<0.05, **p<0.01, **p<0.001, t-test.
Discussion
Lgr5 is an oncoprotein associated with tumorigenesis and cancer progression [24]. Lgr5 potentiates Wnt/b-catenin signaling with R-spondin, Znrf3, and Rnf43 [39,40]. However, little is known about the molecular mechanisms involved in Lgr5 function in homeostatic stem cells and cancers. In this work, we observed that Lgr5 was upregulated in HCC tumor tissue and hepatoma cell lines, and its high expression indicated poor prognosis in patients with HCC. Clinical analysis showed that Lgr5 is positively related to multiple tumor numbers and PVTT, suggesting that Lgr5 is a critical factor for HCC metastasis. Next, upon investigating the potential function of Lgr5 as an oncogene, we found that Lgr5 could increase cancer cell migration and induce an EMT-like phenotype in HCC cells [41]. EMT-like cell phenotypes have been well established as being related to chemoresistance. It was reported that two Dox- resistant MCF-7 cell lines underwent EMT [42]. EMT is associated with diverse tumor including breast cancer and pancreatic cancer [43,44]. EMT in HCC caused by high expression of Lgr5 partly blame for a poor prognosis in HCC patient [27]. So we play attention to the effect of Lgr5 on drug resistance. Interestingly, we found that Lgr5 overexpression increased Dox resistance in HCC cells and that Dox induced Lgr5 expression in a concentration-dependent manner.
Dox is the cornerstone of chemotherapy for HCC; however, Dox resistance is an obstacle to successful treatment in patients with HCC. Dox induces apoptosis in human HCC cells via the p53 pathway. To elucidate the molecular mechanism of Dox resistance, we investigated the relationship between Lgr5 and p53 expression in HCC. We found that there is a negative correlation between Lgr5 and p53 expression in HCC tissues and that Lgr5 induces p53 degradation in HCC cell lines. PDCD5 was identified as a target gene; PDCD5 is a protein participating in apoptosis that is regarded as a tumor suppressor in various tumor types, including lung cancer, breast cancer and glioma [45-48]. PDCD5 can interact with and stabilize p53 to promote apoptosis under conditions of DNA damage [49,50]. We observed that PDCD5 is an independent prognostic factor in HCC patients and functions as an antioncogene. PDCD5 overexpression partially rescues Lgr5-mediated drug resistance. To further study the interaction between Lgr5 and PDCD5, we first established transfected HCC cell lines that overexpressed Lgr5 and PDCD5 and found that neither protein could improve the expression of the other at the transcriptional and posttranscriptional levels. Furthermore, we found that Lgr5 in the cytoplasm could interact with PDCD5 and prevent its translocation. Thus, PDCD5 could not translocate to the nucleus and stabilize p53 via binding to p53, suggesting that Lgr5 reduces the effective level of PDCD5 rather than the total level of this protein. High expression of Lgr5 could decrease the protein level of p53 and inhibit apoptosis in HCC cells treated with Dox or even in cells without Dox treatment. To further investigate the effect of Lgr5 on PDCD5, we overexpressed PDCD5 in HCC cells stably transfected with Lgr5 and found that their resistance to Dox was inhibited and that their apoptosis was induced, suggesting the antagonism of PDCD5 overexpression against Lgr5. These phenomena suggested that Lgr5 could block the nuclear translocation of PDCD5 through the binding of its N-terminal extracellular domain directly to PDCD5, thus inducing p53 degradation. This finding is supported by recent research showing that PDCD5 can translocate from the cytoplasm to the nucleus under Dox treatment [37]. We also found that Lgr5 suppresses Dox-induced apoptosis via the p53 pathway in HCC cells.
As shown in Scheme 1, we propose a model for a novel mechanism of Dox resistance in HCC cells that includes a pivotal role for Lgr5, which is directly activated by this chemotherapeutic drug. The induced Lgr5 acts as a direct negative regulator of PDCD5 by blocking the nuclear translocation of PDCD5 when the N-terminal extracellular domain of Lgr5 binds directly to PDCD5. The decreased PDCD5 levels in the nucleus induce the loss of p53 stabilization, leading to an EMT-like phenotype, inhibiting apoptosis and eventually causing resistance to Dox.
Taken together, our results reveal a novel molecular mechanism that underlies Dox resistance to HCC, providing insights into the players that take part in this process: Lgr5, PDCD5, and p53, all of which have the potential for pharmacological targeting. Moreover, it is worth noting that our findings provide the first evidence for a role of Lgr5 expression and function in the resistance to Dox in HCC cells. Based on our findings, we propose that Lgr5 may act as a vital predictive biomarker for clinical chemotherapy selection.
Abbreviations
HCC: hepatocellular carcinoma; Lgr5: leucine-rich repeat-containing G protein-coupled receptor 5; EMT: epithelial-mesenchymal transition; Dox: doxorubicin; Co-IP: coimmunoprecipitation; GST: Glutathione-S-transferase; PDCD5: programmed cell death protein 5; DAPI: 4'-6-Diamidino-2-phenylindole; DAB: diaminobenzidine; ICH: immunohisto-chemistry.
Supplementary Material
Supplementary figures and tables.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017;67:7-30
2. Kokudo N, Hasegawa K, Akahane M, Igaki H, Izumi N, Ichida T. et al. Evidence-based Clinical Practice Guidelines for Hepatocellular Carcinoma: The Japan Society of Hepatology 2013 update (3rd JSH-HCC Guidelines). Hepatol Res. 2015:45
3. EASL Clinical Practice Guidelines. Management of hepatocellular carcinoma. J Hepatol. 2018;69:182-236
4. Momparler RL, Karon M, Siegel SE, Avila F. Effect of adriamycin on DNA, RNA, and protein synthesis in cell-free systems and intact cells. Cancer Res. 1976;36:2891-5
5. Tacar O, Sriamornsak P, Dass CR. Doxorubicin: an update on anticancer molecular action, toxicity and novel drug delivery systems. J Pharm Pharmacol. 2013;65:157-70
6. Qin S, Bai Y, Lim HY, Thongprasert S, Chao Y, Fan J. et al. Randomized, multicenter, open-label study of oxaliplatin plus fluorouracil/leucovorin versus doxorubicin as palliative chemotherapy in patients with advanced hepatocellular carcinoma from Asia. J Clin Oncol. 2013;31:3501-8
7. Song MJ, Bae SH, Chun HJ, Choi JY, Yoon SK, Park JY. et al. A randomized study of cisplatin and 5-FU hepatic arterial infusion chemotherapy with or without adriamycin for advanced hepatocellular carcinoma. Cancer Chemother Pharmacol. 2015;75:739-46
8. Ding W, You H, Dang H, LeBlanc F, Galicia V, Lu SC. et al. Epithelial-to-mesenchymal transition of murine liver tumor cells promotes invasion. Hepatology. 2010;52:945-53
9. Acloque H, Adams MS, Fishwick K, Bronner-Fraser M, Nieto MA. Epithelial-mesenchymal transitions: the importance of changing cell state in development and disease. J Clin Invest. 2009;119:1438-49
10. Chen X, Lingala S, Khoobyari S, Nolta J, Zern MA, Wu J. Epithelial mesenchymal transition and hedgehog signaling activation are associated with chemoresistance and invasion of hepatoma subpopulations. J Hepatol. 2011;55:838-45
11. Dave B, Mittal V, Tan NM, Chang JC. Epithelial-mesenchymal transition, cancer stem cells and treatment resistance. Breast Cancer Res. 2012;14:202
12. Huang D, Duan H, Huang H, Tong X, Han Y, Ru G. et al. Cisplatin resistance in gastric cancer cells is associated with HER2 upregulation-induced epithelial-mesenchymal transition. Sci Rep. 2016;6:20502
13. Ren WW, Li DD, Chen X, Li XL, He YP, Guo LH. et al. MicroRNA-125b reverses oxaliplatin resistance in hepatocellular carcinoma by negatively regulating EVA1A mediated autophagy. Cell Death Dis. 2018;9:547
14. Cao L, Wan Q, Li F, Tang CE. MiR-363 inhibits cisplatin chemoresistance of epithelial ovarian cancer by regulating snail-induced epithelial-mesenchymal transition. Bmb Rep. 2018;51:456-61
15. Xu T, Zhang J, Chen W, Pan S, Zhi X, Wen L. et al. ARK5 promotes doxorubicin resistance in hepatocellular carcinoma via epithelial-mesenchymal transition. Cancer Lett. 2016;377:140-8
16. Sun L, Ke J, He Z, Chen Z, Huang Q, Ai W. et al. HES1 Promotes Colorectal Cancer Cell Resistance To 5-Fu by Inducing Of EMT and ABC Transporter Proteins. J Cancer. 2017;8:2802-8
17. Zheng X, Carstens JL, Kim J, Scheible M, Kaye J, Sugimoto H. et al. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature. 2015;527:525-30
18. Zhang Y, Lu Y, Zhang C, Huang D, Wu W, Zhang Y. et al. FSCN1 increases doxorubicin resistance in hepatocellular carcinoma through promotion of epithelial-mesenchymal transition. Int J Oncol. 2018;52(5):1455-1464
19. Venkatakrishnan AJ, Deupi X, Lebon G, Tate CG, Schertler GF, Babu MM. Molecular signatures of G-protein-coupled receptors. Nature. 2013;494:185-94
20. Hsu SY, Liang SG, Hsueh AJ. Characterization of two LGR genes homologous to gonadotropin and thyrotropin receptors with extracellular leucine-rich repeats and a G protein-coupled, seven-transmembrane region. Mol Endocrinol. 1998;12:1830-45
21. de Lau W, Barker N, Low TY, Koo BK, Li VS, Teunissen H. et al. Lgr5 homologues associate with Wnt receptors and mediate R-spondin signalling. Nature. 2011;476:293-7
22. Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M. et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. 2007;449:1003-7
23. Li XB, Yang G, Zhu L, Tang YL, Zhang C, Ju Z. et al. Gastric Lgr5(+) stem cells are the cellular origin of invasive intestinal-type gastric cancer in mice. Cell Res. 2016;26:838-49
24. Huch M, Dorrell C, Boj SF, van Es JH, Li VS, van de Wetering M. et al. In vitro expansion of single Lgr5+ liver stem cells induced by Wnt-driven regeneration. Nature. 2013;494:247-50
25. Ng A, Tan S, Singh G, Rizk P, Swathi Y, Tan TZ. et al. Lgr5 marks stem/progenitor cells in ovary and tubal epithelia. Nat Cell Biol. 2014;16:745-57
26. Jiang Y, Li W, He X, Zhang H, Jiang F, Chen Z. Lgr5 expression is a valuable prognostic factor for colorectal cancer: evidence from a meta-analysis. Bmc Cancer. 2016;16:12
27. Chen W, Fu Q, Fang F, Fang J, Zhang Q, Hong Y. Overexpression of leucine-rich repeat-containing G protein-coupled receptor 5 predicts poor prognosis in hepatocellular carcinoma. Saudi J Biol Sci. 2018;25:904-8
28. Mizuno N, Yatabe Y, Hara K, Hijioka S, Imaoka H, Shimizu Y. et al. Cytoplasmic expression of LGR5 in pancreatic adenocarcinoma. Front Physiol. 2013;4:269
29. Wang X, Wang X, Liu Y, Dong Y, Wang Y, Kassab MA. et al. LGR5 regulates gastric adenocarcinoma cell proliferation and invasion via activating Wnt signaling pathway. Oncogenesis. 2018;7:57
30. Liu W, Zhang J, Gan X, Shen F, Yang X, Du N. et al. LGR5 promotes epithelial ovarian cancer proliferation, metastasis, and epithelial-mesenchymal transition through the Notch1 signaling pathway. Cancer Med. 2018;7:3132-3142
31. Wang B, Chen Q, Cao Y, Ma X, Yin C, Jia Y. et al. LGR5 Is a Gastric Cancer Stem Cell Marker Associated with Stemness and the EMT Signature Genes NANOG, NANOGP8, PRRX1, TWIST1, and BMI1. Plos One. 2016;11:e168904
32. Kumar KK, Burgess AW, Gulbis JM. Structure and function of LGR5: an enigmatic G-protein coupled receptor marking stem cells. Protein Sci. 2014;23:551-65
33. Chen Y, Sun R, Han W, Zhang Y, Song Q, Di C. et al. Nuclear translocation of PDCD5 (TFAR19): an early signal for apoptosis? Febs Lett. 2001;509:191-6
34. Choi HK, Choi Y, Park ES, Park SY, Lee SH, Seo J. et al. Programmed cell death 5 mediates HDAC3 decay to promote genotoxic stress response. Nat Commun. 2015;6:7390
35. Li G, Ma D, Chen Y. Cellular functions of programmed cell death 5. Biochim Biophys Acta. 2016;1863:572-80
36. Xu Y, Wang S, Chan HF, Lu H, Lin Z, He C. et al. Dihydromyricetin Induces Apoptosis and Reverses Drug Resistance in Ovarian Cancer Cells by p53-mediated Downregulation of Survivin. Sci Rep. 2017;7:46060
37. Yang M, Li Y, Shen X, Ruan Y, Lu Y, Jin X. et al. CLDN6 promotes chemoresistance through GSTP1 in human breast cancer. J Exp Clin Cancer Res. 2017;36:157
38. Yang-Hartwich Y, Soteras MG, Lin ZP, Holmberg J, Sumi N, Craveiro V. et al. p53 protein aggregation promotes platinum resistance in ovarian cancer. Oncogene. 2015;34:3605-16
39. Yan KS, Janda CY, Chang J, Zheng G, Larkin KA, Luca VC. et al. Non-equivalence of Wnt and R-spondin ligands during Lgr5(+) intestinal stem-cell self-renewal. Nature. 2017;545:238-42
40. de Lau W, Peng WC, Gros P, Clevers H. The R-spondin/Lgr5/Rnf43 module: regulator of Wnt signal strength. Genes Dev. 2014;28:305-16
41. Liu J, Yu GZ, Cheng XK, Li XD, Zeng XT, Ren XQ. LGR5 promotes hepatocellular carcinoma metastasis through inducting epithelial-mesenchymal transition. Oncotarget. 2017;8:50896-903
42. Sommers CL, Heckford SE, Skerker JM, Worland P, Torri JA, Thompson EW. et al. Loss of epithelial markers and acquisition of vimentin expression in adriamycin- and vinblastine-resistant human breast cancer cell lines. Cancer Res. 1992;52:5190-7
43. Arumugam T, Ramachandran V, Fournier KF, Wang H, Marquis L, Abbruzzese JL. et al. Epithelial to mesenchymal transition contributes to drug resistance in pancreatic cancer. Cancer Res. 2009;69:5820-8
44. Huang J, Li H, Ren G. Epithelial-mesenchymal transition and drug resistance in breast cancer (Review). Int J Oncol. 2015;47:840-8
45. Liu H, Wang Y, Zhang Y, Song Q, Di C, Chen G. et al. TFAR19, a novel apoptosis-related gene cloned from human leukemia cell line TF-1, could enhance apoptosis of some tumor cells induced by growth factor withdrawal. Biochem Biophys Res Commun. 1999;254:203-10
46. Spinola M, Meyer P, Kammerer S, Falvella FS, Boettger MB, Hoyal CR. et al. Association of the PDCD5 locus with lung cancer risk and prognosis in smokers. J Clin Oncol. 2006;24:1672-8
47. Murshed F, Farhana L, Dawson MI, Fontana JA. NF-kappaB p65 recruited SHP regulates PDCD5-mediated apoptosis in cancer cells. Apoptosis. 2014;19:506-17
48. Li H, Zhang X, Song X, Zhu F, Wang Q, Guo C. et al. PDCD5 promotes cisplatin-induced apoptosis of glioma cells via activating mitochondrial apoptotic pathway. Cancer Biol Ther. 2012;13:822-30
49. Xu L, Hu J, Zhao Y, Hu J, Xiao J, Wang Y. et al. PDCD5 interacts with p53 and functions as a positive regulator in the p53 pathway. Apoptosis. 2012;17:1235-45
50. Bock FJ, Tanzer MC, Haschka MD, Krumschnabel G, Sohm B, Goetsch K. et al. The p53 binding protein PDCD5 is not rate-limiting in DNA damage induced cell death. Sci Rep. 2015;5:11268
Author contact
![]() Corresponding authors: Quanyan Liu. Tel.: +86-27-67812588; Fax: +86-27-8731935; E-mail: lqyedu.cn; Zhisu Liu. Tel.:+86-27-67812569; E-mail: spss2005com
Corresponding authors: Quanyan Liu. Tel.: +86-27-67812588; Fax: +86-27-8731935; E-mail: lqyedu.cn; Zhisu Liu. Tel.:+86-27-67812569; E-mail: spss2005com