Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results and Discussion
Conclusion
Experimental Section
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(9):2505-2525. doi:10.7150/thno.33280 This issue Cite
Research Paper
“Navigate-dock-activate” anti-tumor strategy: Tumor micromilieu charge-switchable, hierarchically activated nanoplatform with ultrarapid tumor-tropic accumulation for trackable photothermal/chemotherapy
Kondareddy Cherukula1, Saji Uthaman2, In-Kyu Park1 ![]()
1. Department of Biomedical Sciences, Chonnam National University Medical School, Hwasun, Jeollanam-do, 58128, Republic of Korea.
2. Department of Polymer Science and Engineering, Chungnam National University, 99 Daehak-ro, Yuseong-gu, Daejeon 34134, Republic of Korea
Received 2019-1-18; Accepted 2019-2-19; Published 2019-4-13
Abstract

The delivery of therapeutics into tumors remains a challenge in nanoparticle-mediated drug delivery. However, effective therapies such as photothermal therapy (PTT) are limited by quick systemic clearance and non-specific biodistribution. Anti-tumor strategies tailored to accommodate both tumor accumulation/retention and cellular internalization under a single platform would be a promising strategy. This work demonstrates a hierarchical activating strategy that would exhibit enhanced circulation and rapid tumor-tropism as well as facilitate tumor penetration, followed by tumor-specific drug release to realize trackable photothermal/chemotherapy.
Methods: We engineered a lithocholic acid-conjugated disulfide-linked polyethyleneimine micelle (LAPMi) loaded with paclitaxel (LAPMi-PTX, L), followed by the electrostatic adsorption of indocyanine green (ICG, I) on LAPMI-PTX and subsequently coated them with thermosensitive DPPC and DSPE-PEG-NH2 lipids (L), producing Lipid/ICG/LAPMi-PTX (LIL-PTX) nanoparticles (NPs). The characteristics of NPs, including physicochemical characterization, photothermal & pH responsiveness, cell uptake, tumor spheroid penetration, anti-tumor efficacy and hierarchical activation of LIL-PTX NPs were investigated in vitro and in vivo by using CT26 cell line. The anti-metastatic potential of LIL-PTX NPs were demonstrated using 4T1 orthotopic tumor model.
Results: The NPs synthesized possessed charge switchability in the mildly acidic pH, and were laser- and pH-responsive. Dual stimuli-responsive nature of LIL-PTX NPs improved the disposition of therapeutics to the tumor, reflected by enhanced intracellular uptake, tumor spheroid penetration and in vitro cytotoxicity studies. LIL-PTX NPs readily switched its surface charge from neutral to positive upon reaching the tumor milieu, thus resulting in rapid tumor tropism and accumulation. Under near-infrared laser irradiation, the thermosensitive lipids on LIL-PTX NPs were deshielded, and the tumor-penetrating LAPMi-PTX was subsequently exposed to the tumor milieu, thus resulting in enhanced intracellular internalization. Next, LAPMi-PTX evaded the endo-lysosomes, thereby releasing the PTX through the degradation of LAPMi mediated by intracellular GSH in the tumor. LIL-PTX NPs significantly improved the therapy by eradicating primary tumors completely and suppressing their subsequent lung metastasis.
Conclusion: The improved therapeutic index is due to enhanced passive targeting by rapid tumor-tropic accumulation and tumor penetration by laser-driven exposure of LAPMi, thereby improving the therapeutic delivery for image-guided photothermal/chemotherapy.
Keywords: photothermal therapy, stimuli-responsive, charge switchable, NIR laser, hierarchical targeting systems
Introduction
Photothermal therapy (PTT) is defined as localized heating in the tumor region via a near-infrared (NIR) laser to generate local heat that is potentially cytotoxic to neighboring cells at the photosensitizer location [1, 2]. Although PTT is considered to be an efficient anti-cancer treatment strategy, it is difficult to eradicate all the tumor cells, owing to the heterogeneous distribution of local heat in the tumor and surrounding healthy tissue, which often results in tumor recurrence and metastasis [3, 4].In addition, PTT is also limited by its penetration depth, since the tumor margin beyond the laser penetration initiates further tumor growth due to insufficient heat distribution [5]. Recent studies on cancer cells revealed that intracellular heating triggered more therapeutic efficacy than extracellular heating [6, 7]. Additionally, it has been suggested that nanosystems targeting intracellular organelles, such as the nucleus [8] and the mitochondria [9], achieved tumor-specific PTT with negligible adjacent cell damage. However, the materials explored thus far have been mostly metallic or inorganic in nature, which is of great concern regarding the biosafety and off-target toxicity [10]. Recently, PTT also demonstrated remarkable synergistic therapeutic outcomes in conjunction with chemotherapy [11, 12]. To execute the combination of PTT and chemotherapy, it is necessary to program a suitable stimuli-responsive nanocarrier with high drug loading capacity and the spatiotemporal release of the loaded drug from the nanocarrier upon endogenous/exogenous stimuli. Therefore, the precise design of a nano drug delivery system should enhance both tumor tissue accumulation and intracellular internalization. Until now, very few nanoparticulate systems have achieved these functionalities.
Longer circulation of nanoparticles is of prime importance for the successful delivery of therapeutics. Increasing evidence suggests that neutrally charged, PEGylated nanocarriers less than 100 nm in diameter have a high tumor uptake, thereby providing a long circulation time and a low RES uptake [13]. However, the tumor retention of these nanoparticles is mainly limited by the backflow into the blood circulation, the poor interaction with tumor cells and a low intracellular uptake [14, 15] . In addition, PEGylated nanoparticles still witness the RES uptake in the liver and spleen after systemic circulation, thus resulting in low tumor targeting efficiency [14]. In this context, using charge-switchable nanoparticles to improve tumor cell interactions, physiological stability, and blood circulation is an attractive strategy [16]. Despite stronger electrostatic interactions with the target cell surface, extravasation in the tumor and deep penetration of nanoparticles into its necrotic core have also limited the therapeutic efficacy of nanoparticles [17]. To address these challenges, one potential approach is to design a tailor-made hierarchically activated nanoplatform with a camouflage that would readily disassemble upon exogenous/endogenous triggers while maintaining the advantages of a long circulation time, enhanced tumor cell interactions and deep tumor penetration of nanoparticles. The delivered nanoplatform would dissociate into tumor-penetrating nanoparticles upon an external stimulus, such as near-infrared light, and release the therapeutics into the tumor cytoplasm initiated by an endogenous trigger that is characteristic of cancer cells.
We previously reported a tumor-penetrating polymeric nanomicelle comprising a lithocholic acid (LCA)-conjugated disulfide-linked polyethyleneimine (ssPEI) micelle (LAPMi) [18]. The PEI moiety in LAPMi aids in high internalization, tumor accumulation and endosomolytic properties [19-21]. In addition, disulfide linkages that are introduced in LAPMi allowed for the tumor cytoplasm-specific destabilization of nanoparticles and facilitated the release of cargos, owing to a higher level of GSH in cancer cells [22]. LCA, which is a naturally occurring bile acid, has been utilized as a tumor-penetrating agent, drug permeation enhancer and drug penetrator [23,24]. Thus, LAPMi, improved the tumor penetration and redox-responsive properties and was also proven to be an efficient drug delivery carrier targeting the tumor.
In this work, we utilized a 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy (polyethylene glycol)-2000] amine (DSPE-PEG-NH2) and dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) lipids as an outer coating layer on LAPMi. DPPC is a zwitterionic lipid with a gel-to-fluid phase transition temperature of 41°C [25], which could impart the thermosensitive property to the nanoconstruct and trigger instant drug release upon PTT laser irradiation. The PEG component of DSPE-PEG-NH2 will ensure systemic circulation and reticuloendothelial system (RES) escape [26]. The amino end group of the lipid will aid in accumulation at the tumor milieu by changing its surface charge to positive upon protonation in the tumor extracellular environment [27-29]. A two-step approach was utilized to formulate the nanoconstruct: first, positively charged LAPMi-loaded paclitaxel (PTX) (LAPMi-PTX) was electrostatically adsorbed with negatively charged indocyanine green (ICG) (ICG/LAPMi-PTX) for responsiveness to external NIR laser irradiation, thermal and fluorescence imaging properties; second, a lipid outer corona layer comprising a temperature-responsive lipid with protonatable NH2 and PEG moieties was coated onto ICG/LAPMi-PTX, forming the lipid/ICG/LAPMi-PTX (LIL-PTX) nanoparticles (NPs) with tumor tropism/accumulation and enhanced systemic circulation properties.
The possible mechanism of the hierarchically activated nanoplatform is illustrated in Figure 1. 1) Once accumulated in the tumor milieu by an enhanced permeation and retention (EPR) effect, the surface charge of LIL-PTX NPs switches to positive from amine protonation in the mild acidic environment (pH 6.8) [28-30] of the tumor, which further enhances the tumor homing of LIL-PTX NPs. 2) The tumor-delivered LIL-PTX NPs was irradiated with the NIR laser to disassociate the thermosensitive lipid corona through deshielding of the lipid layer by photothermal ICG activation, thus exposing the tumor-penetrating LAPMi-PTX to the tumor microenvironment. 3) The LAPMi-PTX would further facilitate the penetration into the tumor interstitium and intracellular trafficking, owing to its tumor penetration properties and positive surface charge. 4) Following the endocytotic pathway, the LAPMi-PTX would escape the endo/lysosome and become further disassembled due to the breakdown of disulfide linkage in the LAPMi-PTX mediated by the GSH that is present in cancer cells, thereby releasing PTX for tumor apoptosis. The results from this research further emphasized the critical roles of the tumor microenvironment in designing the nanoparticulate system, especially for tumor accumulation and penetration; more importantly, it created a new paradigm for improving passive targeting and their subsequent targeted therapeutic delivery in cancer cells.
Schematic illustration of the hierarchical targeting strategy based on LIL-PTX NPs. Physiologically stable LIL-PTX NPs accumulates preferentially in the tumor milieu via both the EPR effect and the positive surface charge switch due to the charge conversion over the surface amino groups on LIL-PTX NPs. Upon laser irradiation, thermosensitive lipids will be removed through the laser-activated photothermal property of ICG, thereby exposing the tumor-penetrating LAPMi-PTX into the tumor micromilieu. Furthermore, LAPMi-PTX experiences enhanced tumor penetration and subsequent cellular internalization mediated by LCA and PEI components. Next, the PEI moiety of LAPMi-PTX aids in endo-osmolysis and witnesses the PTX drug release for killing cancer cells upon exposure to intracellular GSH.
Results and Discussion
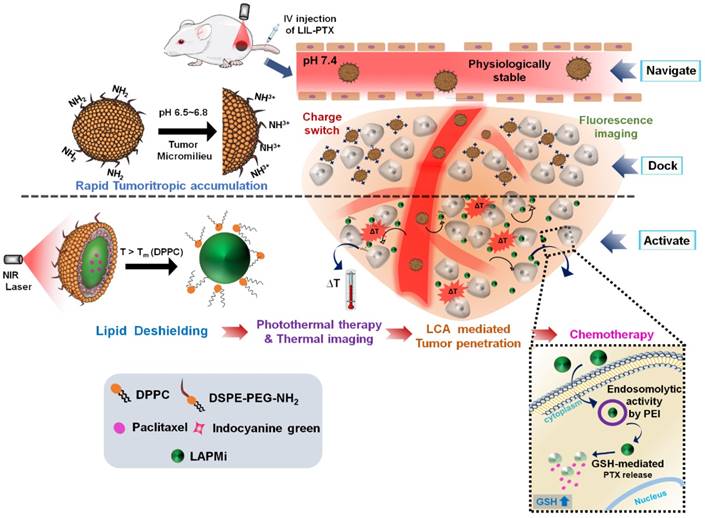
Preparation and Characterization of LIL-PTX NPs
LAPMi and paclitaxel containing LAPMi (LAPMi-PTX) was synthesized by following our previous work [18]. The as-synthesized LAPMi was characterized with proton nuclear magnetic resonance (1H NMR) (Figure S1A). The characteristic peaks of LCA appearing between δ: 0.6-2.3 & 3.8 ppm and characteristic peaks of ssPEI appearing between δ: 2.66-3.5 ppm verified the formation of LAPMi [18]. The physicochemical characterization of LAPMi was published elsewhere [18]. The number average molecular weight of the disulfide-crosslinked polyethyleneimine (ssPEI) was calculated by 1H NMR analysis and estimated to be 9,974 Da. In addition, we have performed MALDI-TOF analysis of ssPEI and (tris (2-carboxyethyl) phosphine) (TCEP)-treated LAPMi. As shown in Figure S1B-C, the spectrum was characteristic of a PEI polymer with repeating units differed by 43.04 Da corresponding to [CH2CH2NH] confirming the presence of PEI [31]. The molecular masses obtained from MALDI-TOF were around 921 Da and 2,100 Da for ssPEI and TCEP-treated LAPMi, respectively.
We used ICG, an FDA-approved imaging agent for photothermal therapy (PTT), which has been exploited extensively due to its biocompatibility [1, 32-34]. At first, the negatively charged ICG was electrostatically adsorbed onto positively charged LAPMi-PTX in an acidic aqueous medium at different weight ratios (Figure 2A). The surface charge of LAPMi-PTX gradually reduced with increasing concentrations of ICG; for instance, from approximately 28 ± 0.8 mV at the ICG/LAPMi-PTX weight ratio of 0.25 to approximately 14 ± 2 mV at the ratio of 1 and approximately 6 ± 0.7 mV at the ratio of 2 (Figure 2B). In addition, the hydrodynamic size was subsequently increased upon ICG adsorption from 161 ± 2 nm at a ratio of 0.25 to 212 ± 9 nm at a ratio of 2, which clearly showed the increasing amount of ICG adsorption onto LAPMi-PTX. Moreover, it was found that the ICG/LAPMi-PTX ratio of 2 has an adsorption efficiency of 87.5 %, which was found by calculating the ICG absorbance from the unadsorbed samples that were collected postfiltration.
Synthesis and physicochemical characterization of LIL-PTX NPs. (A) Synthesis scheme of LIL-PTX NPs by thin film hydration and the extrusion method. (B) Hydrodynamic size and zeta potential changes upon ICG adsorption onto LAPMi-PTX. (C) Hydrodynamic size and zeta potential changes upon lipids coating onto ICG/LAPMi-PTX. (D) Field-emission transmission electron microscopy (FE-TEM) of LIL-PTX NPs (i, ii) and a uranyl acetate negatively stained FE-TEM image of LIL-PTX NPs (iii,iv).
Next, the fusion of DPPC and DSPE-PEG-NH2 (molar ratio of 4:1) lipids with an ICG/LAPMi-PTX core was carried out by the thin film hydration and extrusion technique to form lipid/ICG/LAPMi-PTX (LIL-PTX) NPs. The lipids were dried into a thin film and then were rehydrated in the presence of aqueous ICG/LAPMi-PTX. As an amphiphilic molecule, ICG could interact with both the hydrophilic LAPMi-PTX and the hydrophobic acyl chains of lipids. In addition, there exists a noncovalent interaction between ICG and lipid chains, presumably due to an increase in Van Der Waals forces and hydrophobic force interactions by the acyls of lipid chains [11, 35] thereby forming a stable fusion of lipids with a LAPMi-PTX core. We observed that the concentrations of lipid and polymer affect the hydrodynamic size and surface charge of the formulation. Figure 2C exhibits that, with an increase in the lipid:ICG/LAPMi-PTX weight ratio, there was a slight reduction in the hydrodynamic size, for instance, from approximately 212 ± 11 nm at a ratio of 0.5 and approximately 187 ± 9.9 nm at a ratio of 2.5 to approximately 183 ± 4.8 nm at a ratio of 3.5, thus confirming the fusion of the lipid shell over the polymer core. In addition, the zeta potential measurements showed a large variation with the concentration change in the components, for instance, approximately 15.2 ± 0.9 mV at a ratio of 0.25, approximately 7.9 ± 0.6 mV at a ratio of 2.5, and approximately 9.9 ± 0.7 mV at a ratio of 3.5. It was observed that the increase in the lipid concentration reduced the zeta potential, owing to the increasing concentrations of neutrally charged DPPC. We chose the optimized LIL-PTX NPs formulation at a ratio of 2.5 (Figures S1D-E) for further studies because of its neutral charge (7.9 ± 0.6 mV) and optimal size (187 ± 9.9 nm), with poly dispersity index (PDI) of 0.044 for enhanced circulation.
It was evidenced by several studies that the ideal nanoparticle size for escaping mononuclear phagocytosis is in the range of 70-200 nm [36, 37]. Thus, it is an important prerequisite for engineering the nanoparticle surface with an optimal size and zeta potential to prevent phagocytosis of the nanoparticles and therefore to ensure longer systemic circulation. Field-emission transmission electron microscopy (FE-TEM) imaging of LIL-PTX NPs revealed a size of 81.4 ± 9 nm (Figures 2D i-ii). Disparity between FE-TEM and the hydrodynamic size can be attributed to the hydrodynamic volume and thick hydration layer formed by the PEG segment of lipids [38]. In addition, the FE-TEM image of prepared LIL-PTX NPs negatively stained with uranyl acetate demonstrated a uniform lipid layer over the LAPMi-PTX core (Figures 2D iii-iv). The as-prepared LIL-PTX NPs showed good dispersion and solubility (Figure S2A). In addition, Figure S2B suggests that bare ICG tends to aggregate and precipitate in PBS, whereas LIL-PTX NPs showed enhanced solubility and stability in all the tested media (DW, PBS and culture media). Also, stability was studies as means of hydrodynamic size changes for 5 days (Figure S2C). The result suggested that LIL-PTX NPs bearing PEG shield did not witness any appreciable size changes in all tested buffer and serum conditions.
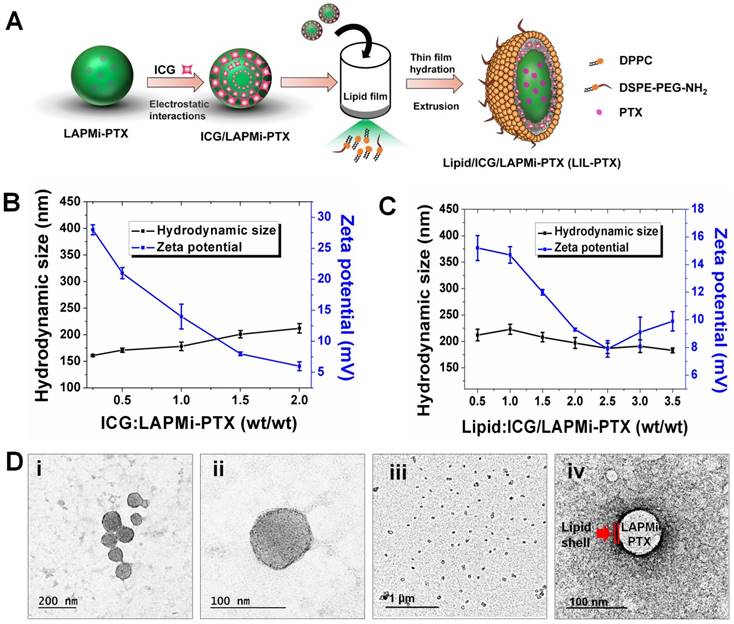
The optical properties of LIL-PTX NPs were evaluated to confirm the presence of ICG. As Figure 3A-B shows, both the absorbance and fluorescence spectra revealed a redshift of 27 nm and 7 nm, respectively, compared to free ICG, which confirms the presence of ICG aggregates between the lipid chains. This shift towards the longer wavelengths contributes to the higher heating efficiency than the bare ICG, as observed by other research groups [39, 40]. Since the photothermal heating efficiency of the ICG is crucial for PTT, the temperature changes after laser irradiation (808 nm, 2 W/cm2, 10 min) were recorded with the thermal camera. As shown in Figure 3C, ICG entrapped in the LIL-PTX NPs showed a superior heating elevation within a few seconds of irradiation. LIL-PTX NPs exhibited a redshift closer to 808 nm, and this is the primary reason for heat enhancement, as was observed by other researchers [39].
UV-VIS absorbance (A) and fluorescence spectrum (B) of ICG and LIL-PTX NPs. (C) Thermal images of the LIL-PTX NPs (50 µg/mL) subjected to NIR laser irradiation (808 nm, 2 W/cm2). (D) Thermal elevation of LIL-PTX NPs, ICG and DW subjected to NIR laser irradiation (808 nm, 2 W/cm2) recorded at different time points. (E) Scheme demonstrating the LIL-PTX NPs protonation at tumor acidic pH 6.5. (F) Zeta potential variations of LIL-PTX NPs at different pH levels.
Moreover, LIL-PTX NPs exhibited an enhanced temperature elevation compared to the ICG and PBS controls (Figure 3D). This indicates that ICG molecules bound between LAPMi-PTX and lipids possess the light-to-heat conversion ability. The bare ICG-containing solution exposed to irradiation underwent photodegradation; thus, the temperature dropped quickly, whereas the ICG in LIL-PTX NPs experienced lesser photodegradation and hence enhanced temperature elevation. This demonstrates the efficacy of LIL-PTX NPs as a PTT carrier. In addition, we observed the time-dependent NIR absorbance degradation of bare ICG compared to that of LIL-PTX NPs (Figure S3A-B) upon laser irradiation. These results suggest that LIL-PTX NPs can be an efficient carrier for thermal therapies and enhances both the stability and heating efficiency. Similarly, the fluorescence spectrum of ICG and LIL-PTX NPs after laser irradiation was evaluated. Furthermore, Figure S4A-B shows the time-dependent photobleaching effect in the bare ICG compared to that of LIL-PTX NPs upon laser irradiation. This, in turn, demonstrated the efficacy of LIL-PTX NPs as an efficient PTT and trackable nanosystem.
Next, we investigated the surface charge variations of LIL-PTX NPs at different pH levels. Charge switchable groups, such as amine and histidine, undergo protonation in the acidic pH of the tumor milieu [41]. The amine group of the LIL-PTX NPs underwent protonation at mild acidic conditions and turned the surface charge to a positive scale, as shown in Figure 3E-F. For instance, the surface charge switched from 6.6 ± 1.6 mV at pH 7.4 to 21.3 ± 1.85 mV at an acidic pH of 6.5 (Figure S5). This sudden switch to a positive charge aids in the increased recruitment and retention of nanoparticles inside the tumor region. In addition, the neutral charge at physiological pH will aid in minimal interaction with the serum proteins [42].
Stimuli Responsive Disassembly of LIL-PTX NPs
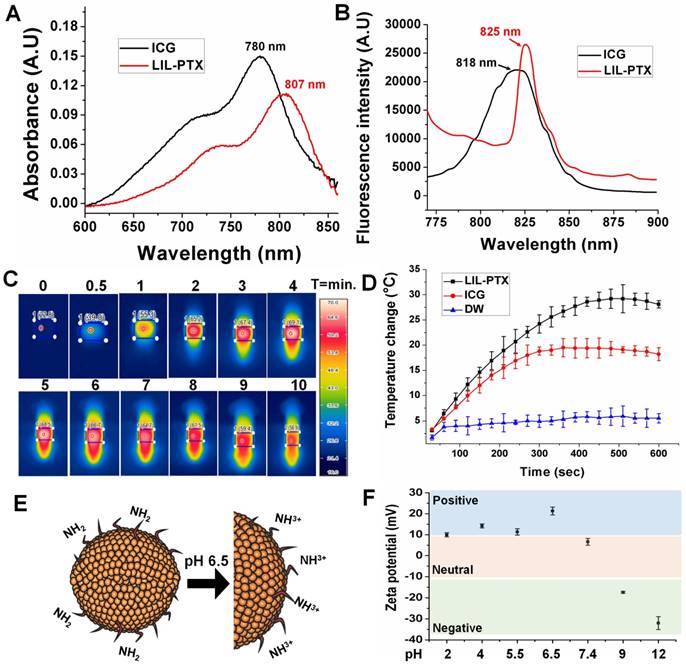
We investigated the hierarchical disassembly of the LIL-PTX NPs, which would determine the fate of LIL-PTX NPs in vivo conditions by FE-TEM and dynamic light scattering (DLS) (Figure 4A-D & Figure S6). LIL-PTX NPs incubated with DMEM media at pH 6.5 were irradiated with an NIR laser (808 nm, 2 W/cm2, 10 min). Upon laser irradiation, thermosensitive DPPC lipids of the LIL-PTX NPs quickly experienced a phase change from the solid gel phase to the liquid crystalline phase after a temperature increase above the melting phase temperature (41.4 °C) [43], thereby creating a higher permeability of lipids and the subsequent disassembly of lipid corona. This further exposes the hydrophilic LAPMi-PTX to the medium, as visualized by the FE-TEM image (Figure 4A & Figure S6A). This result showed a fairly undisturbed LAPMi-PTX after laser irradiation in the media, showing distinct size distribution in DLS analysis (Figure 4D). When the laser-irradiated nanoparticles were further exposed to the intracellular glutathione concentration (10 mM) of cancer cells, the LAPMi-PTX structure became destabilized and released the PTX drugs (Figure 4A & Figure S6B), which is similar to the result of our previous work [18].
FE-TEM images of (A-i) LIL-PTX NPs (A-ii) LIL-PTX NPs irradiated with a laser (808 nm, 2 W/cm2, 10 min) in the DMEM medium (pH 6.5). (A-iii) Laser-irradiated nanoparticles further incubated with GSH (10 mM). (B) Release profiles of the PTX from LIL-PTX NPs formulation, laser irradiation (808 nm, 2 W/cm2, 10 min) and combined laser and GSH (10 mM) incubation at acidic pH 6.5. (C) The UV-VIS absorbance graph of various formulations by laser irradiation (808 nm, 2 W/cm2, 10 min) and combined laser and GSH (10 mM) incubation at acidic pH 6.5. (D) DLS analysis of the LIL-PTX NPs irradiated with the laser (808 nm, 2 W/cm2, 10 min) at acidic pH 6.5.
PTX drug loading and encapsulation efficiency in LAPMi were estimated to be 26.6 % and 90.1 %, respectively, from our previous report [18]. The PTX release from LIL-PTX NPs was investigated in laser irradiated (LIL-PTX + Laser), as well as in the combination of laser and GSH (LIL-PTX + Laser + GSH) at acidic pH 6.5, along with the control (LIL-PTX). As shown in Figure 4B, the drug release from LIL-PTX NPs was primarily inhibited, owing to the presence of double barriers for the drug leakage in the form of disulfide linkages and a lipid shell. For instance, the drug release accounted for 14 % at 2 h, 19 % at 8 h, 22 % at 24 h, and 31 % at 144 h time points, which clearly demonstrates that LIL, as a nanodrug carrier, inhibited drug leakage, which is of prime importance for preventing systemic toxicity. In contrast, the LIL-PTX + Laser group showed a slightly increased drug release pattern compared to the control group, presumably due to the nanoparticle destabilization by laser irradiation. For instance, the drug releases were 18 % at 2 h, 26 % at 8 h, 36 % at 24 h, and 64 % at 144 h time points when the LIL-PTX NPs was irradiated with a laser in PBS (pH 6.5). This result demonstrated that powerful laser irradiation alone was not sufficient to trigger the release of drugs from the LIL-PTX NPs. In contrast, further incubation of GSH (10 mM) with the laser-irradiated samples displayed a remarkable enhancement in the drug release due to the cleavage of the disulfide linkages on LAPMi-PTX, which acted as barriers to the drug release. For instance, the GSH presence triggered an initial burst of 47 % at 2 h, 69 % at 8 h, 89 % at 24 h, and 98 % at 144 h time points. This result clearly indicated that the hierarchical trigger to deliver drugs is an efficient strategy to avoid toxicity and to deliver the drugs completely from the carrier. In addition, coumarin-6 (Cou) as a model hydrophobic drug was encapsulated in LIL (LIL-Cou), and its release was checked upon irradiation by UV-VIS spectroscopy (Figure 4C). Laser irradiation of LIL-Cou NPs made the peak of coumarin-6 visible at 480 nm, which otherwise was not shown in the nonirradiated (LIL-Cou) sample. This further demonstrated that dual stimuli accelerated drug release from the carrier.
Tumoral pH- Specific Cell Uptake and LIL-PTX -Mediated Hierarchical Targeting
We next investigated the intracellular uptake of LIL NPs in the CT26 cells. To confirm the effect of weak acidic pH on intracellular trafficking, the pH in the culture media was adjusted to 6.5, and the cellular uptake was evaluated after treatment with LIL NPs. As shown in Figure 5A, the cellular treatment of LIL NPs at pH 7.4 demonstrated a weak ICG fluorescence at 2 h postincubation. In contrast, LIL NPs treatment at tumoral pH 6.5 (Figure 5B) exhibited significantly enhanced fluorescence intensity of the ICG in the CT26 cells. Moreover, flow cytometry analysis also revealed a significant LIL-PTX NPs uptake in CT26 cells at pH 6.5 (Figure 5C & Figure S7). This result strongly suggests that the loading of therapeutics in LIL NPs significantly promoted the intracellular uptake under the mild acidic conditions of the tumor milieu, which was presumably mediated by the positive charge switch.
Confocal microscopy images of CT26 cells treated with the LIL NPs at pH 7.4 (A) and pH 6.5 (B) Scale bar indicates 10 µm. (C) Flow cytometry histogram of CT26 cells treated with the LIL NPs at either pH 7.4 or pH 6.5. (D) Schematic illustration demonstrating the effect of laser- and GSH-triggered release of coumarin-6 from LIL-Cou NPs. (E) Confocal microscopy images of CT26 cells treated with LIL-Cou NPs at pH 6.5. Scale bar indicates 20 µm. (F) Flow cytometry histograms of the CT26 cells treated with LIL-Cou NPs under various conditions. (G) Mean fluorescence intensity (MFI) of the flow cytometric profiles of the CT26 cells treated with LIL-Cou NPs under various conditions (*p < 0.05 and **p < 0.01).
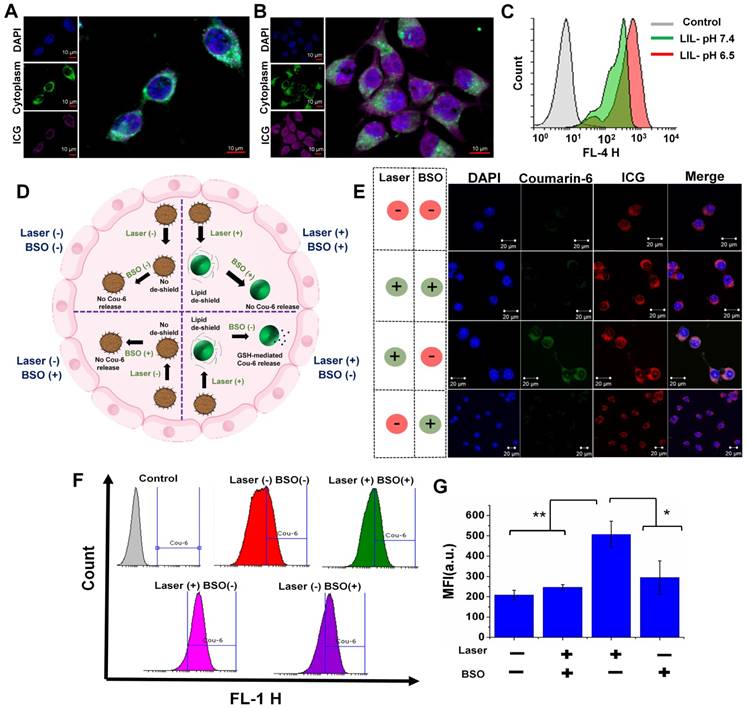
A hierarchical targeting strategy was adopted in our study, in which both exogenous (laser) and endogenous (GSH) triggers were utilized to disassemble the nanoplatform and elicit the efficient therapeutic strategy. A confocal microscope was utilized to demonstrate the dual responsive nature of LIL NPs. Figure 5D shows the schematic of the experimental design to evaluate the responsiveness of LIL NPs against GSH and laser light. We used L-buthionine sulfoximine (BSO), which is an intracellular glutathione inhibitor, to validate the redox responsive nature. In addition, we used LIL-Cou NPs to evaluate the drug release response according to the stimulus. As depicted in Figure 5E, the confocal microscopy images of the laser (-)/BSO (-) group demonstrated that, with the absence of both GSH and laser stimuli, the drug release from the LIL-Cou NPs was minimal; thus, there was a very faint green signal in the images. Next, the laser (+)/BSO (+) group demonstrated that, despite the laser irradiation destabilizing lipid chains on LIL-Cou NPs, coumarin-6 release from the LIL-Cou NPs was inhibited, owing to their strong disulfide chains on LAPMi. In contrast, when the laser light stimulus was complemented with the GSH presence as shown in the laser (+)/BSO (-) group, an intense green fluorescence was prominent in the confocal imaging.
Interestingly, even in the presence of GSH, the coumarin-6 release could not be witnessed without the laser irradiation, as in the laser (-)/BSO (+) group, because there was a minimal green signal in the cells. This result indicated that the presence of both stimuli (laser & GSH) was required to release the drugs inside the cytoplasm. In addition, the flow cytometry analysis (Figure 5F-G) demonstrated a significant coumarin-6 release upon the combined application of both stimuli (laser & GSH). In addition, the hierarchical targeting strategy allowed for spatiotemporal control of the therapeutics, which could be very effective in achieving a high therapeutic index with minimal off-target toxicity.
In vitro Cytotoxicity Evaluation
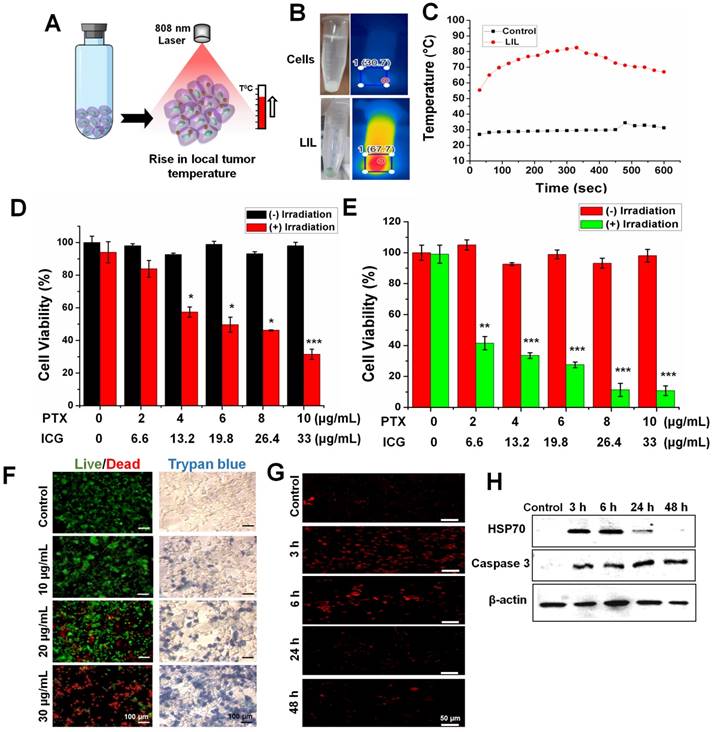
Encouraged by the effective PTT efficacy in the nanoparticle solution, we investigated the thermal conversion efficiency in the CT26 cells at pH 6.5. LIL-PTX NPs (30 µg/mL) internalized CT26 cells were collected in a tube after multiple washes to remove extracellular-located nanoparticles. The cell pellet was subjected to laser irradiation to check the thermal elevation (Figure 6A). As shown in Figure 6B-C, the temperature rise was significant compared to nontreated control cells and attained a maximum temperature of ~68 °C for a 10-min laser irradiation, which is primarily because of the presence of high intracellular concentrations of LIL-PTX NPs. These findings confirmed that LIL-PTX-mediated photothermal therapy induced a temperature rise in the intracellular environment, which is one of important reasons to trigger the desired cancer cell death.
(A) Schematic illustration showing the PTT-induced heat in the in vitro cell culture. (B) Thermal images of heat generated in the CT26 cells treated with LIL-PTX NPs (30 µg/mL of ICG) for 2 h incubation at pH 6.5 and subsequently irradiated with an NIR laser (808 nm, 2 W/cm2, 10 min). (C) Thermal elevation of the in vitro PTT. (D) Cell viability of CT26 cells at pH 7.4 incubated with LIL-PTX NPs for 24 h, either with or without laser irradiation (808 nm, 2 W/cm2, 10 min) (*p < 0.05 and ***p < 0.001). (E) Cell viability of the CT26 cells at pH 6.5 incubated with LIL-PTX NPs for 24 h, either with or without irradiation (808 nm, 2 W/cm2, 10 min) (**p < 0.01 and *** p < 0.001). (F) Live/dead assay and trypan blue assay of the CT26 cells incubated with LIL-PTX NPs for 24 h, either with or without laser irradiation (808 nm, 2 W/cm2, 10 min). Green color indicates live cells and red color indicates dead cells. Scale bar indicates 100 µm. (G) Immunofluorescence imaging of HSP70 (Scale bar indicates 50 µm) and (H) Western blot analysis of CT26 cells incubated with LIL-PTX NPs for 24 h, either with or without laser irradiation (808 nm, 2 W/cm2, 10 min).
Since the stimuli responsiveness and in vitro PTT efficacy was superior with LIL-PTX NPs, we next evaluated the synergistic therapeutic efficacy in the mouse colon carcinoma cells (CT26). At first, we tested the effectiveness of monotherapy, i.e., chemotherapy or photothermal therapy alone. Figure S8A shows that the ICG alone did not elicit the cytotoxicity up to 25 µg/mL concentrations even with the laser irradiations. Despite the enhanced intracellular trafficking of LIL NPs, PTT therapy alone could not kill the cells completely, as depicted in Figure S8B. Similarly, we checked the cytotoxicity effects of LIL-PTX NPs. As depicted in Figure S9A-B, both PTX and LIL-PTX NPs did not elicit potential toxicity to the CT26 cells. These results indicated that, despite an efficient intracellular uptake, LIL-PTX NPs could not elicit potential toxicity without laser irradiation. This is particularly important for the systemic applications to prevent off-target toxicity.
LIL NPs was formulated to elicit the synergistic effects of both PTT and PTX-mediated chemotherapy. To study the synergistic therapeutic efficacy, the viability of CT26 cells was treated with LIL-PTX NPs at different concentrations and at pH 7.4 and pH 6.5. As shown in Figure 6D, CT26 cells incubated with LIL-PTX NPs at pH 7.4 showed concentration-dependent toxic effects, retaining viability of approximately 30~35 % even at higher concentrations of PTX and ICG. This result indicated that the lower intracellular uptake of LIL-PTX NPs at the physiological pH 7.4 reduced cytotoxicity. In contrast, the viability of the cells treated at pH 6.5 was significantly reduced and displayed a strong thermal ablation and chemotherapeutic effect in a concentration-dependent manner. Importantly, the equivalent concentrations of PTX and ICG treated at pH 6.5 showed enhanced cytotoxicity than those at pH 7.4 due to the facilitated intracellular trafficking of the LIL-PTX NPs, as shown in Figure 6E. Notably, the IC50 values at pH 7.4 & pH 6.5 were 4.34 µg/mL and 2.14 µg/mL respectively (Figure S10A). This result was primarily ascribed to the superior light-activated hierarchical targeting strategy exhibited by LIL-PTX NPs in the mild acidic condition of cancer cells. In the absence of laser irradiation, LIL-PTX NPs did not show toxicity at pH 7.4 and pH 6.5.
GSH-responsive cytotoxicity in in vitro conditions was evaluated by pretreating the CT26 cells with the BSO, followed by laser irradiation (808 nm, 2 W/cm2, 10 min). Figure S10B shows that the BSO pretreatment significantly inhibited the cytotoxicity compared to the untreated control, even at the higher concentrations of PTX. These results strongly indicated that intracellular GSH could be utilized as a potential trigger to release the drugs. This result also proposed the applicability of LIL-PTX NPs as a stable, biocompatible and on-demand drug delivery vehicle. Furthermore, the anti-tumor activity was qualitatively witnessed by live/dead staining and trypan blue assay (Figure 6F) after laser irradiation (808 nm, 2 W/cm2, 10 min). The results demonstrated the enhanced cell killing in a concentration-dependent manner, which was evidenced by the bright red color and blue color of the live/dead assay and the trypan blue assay, respectively. Heat-induced cellular damage is resisted by heat shock proteins (HSPs). HSP70 directly inhibits cellular apoptosis by preventing caspase activation [44]. Hence, we investigated the effect of HSP70 on anti-tumor activity. Figure 6G depicted the immunofluorescence imaging of HSP70 after laser irradiation (808 nm, 2 W/cm2, 10 min) in CT26 cells treated with the LIL-PTX NPs. The results suggested that HSP70 levels increased immediately after the irradiation and remained activated up to 6 h postirradiation, followed by a decrease thereafter.
Similarly, a western blot analysis of the cell samples postirradiation was analyzed (Figure 6H & Figure S11A). Similar to immunofluorescence imaging, laser irradiation causes cancer cells to undergo a compensatory stage, wherein HSP70 levels increase to protect the cells from laser-induced heat stress [45]. However, after 6 h postirradiation, the irradiated cells led to a decompensatory stage with irreversible cell damage and characteristic downregulation of the HSP70 expression. These results verified the potential PTT effect by LIL-PTX NPs since most of the PTT efficacies have been compromised with the overexpression of HSP, even after the treatment of HSP inhibitors [45, 46]. Caspase proteins play crucial roles in cell apoptosis. Caspase-3 plays a crucial role in the early stages of apoptosis and eventually degrades the substrates in the cytoplasm and nuclei, thus leading to apoptosis [47]. As shown in Figure 6H & Figure S11B, the expression of the caspase-3 protein was upregulated upon laser irradiation. Downregulation of the HSP70 protein complemented with an increased caspase-3 expression accelerated the LIL-PTX-mediated cytotoxicity.
Tumoral pH-Based Enhanced Penetration in Multicellular Tumor Spheroid
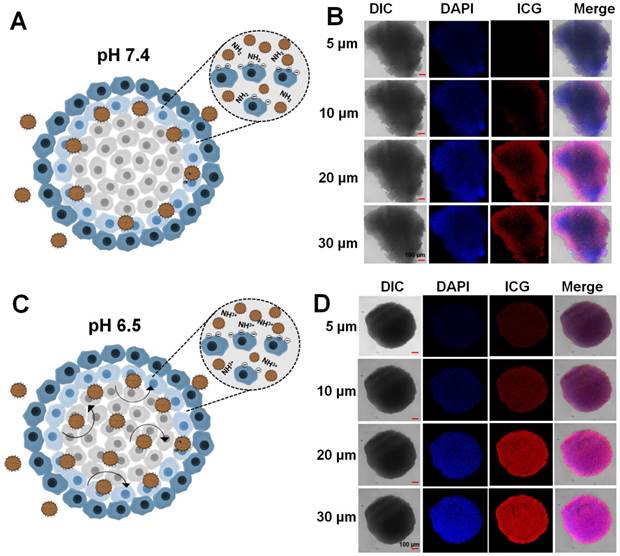
A multicellular tumor spheroid (MCTS) mimics the solid tumor and in vivo complex tumor microenvironment [48]. An MCTS based on CT26 cells was prepared and tested for the tumor penetration efficacy of LIL-PTX NPs. As depicted in Figure 7A-B, LIL-PTX NPs penetrated into the outer regions of the tumor spheroid at pH 7.4. However, LIL-PTX NPs incubated at tumoral pH 6.5 witnessed bright red fluorescence throughout and in deeper parts of the tumor spheroid, as shown in Figure 7C-D. This deep spheroid penetration of LIL-PTX NPs could be attributed to the charge transition of LIL-PTX NPs into a positive charge in the acidic tumor milieu. In addition, the positively charged lipids have a higher tumor internalization effect than the neutrally charged lipids [49] . In contrast, ICG/LAPMi-PTX, a formulation without lipid coating witnessed the accumulation in the outer sphere of the spheroid (Figure S12). This is possibly due to the charge repulsions of the ICG deposited on the surface of nanoparticles with a negative charge of the cell surface. Thus, this result demonstrated that lipid coating enhanced the tumor penetration and internalization properties. In addition, we tested the live/dead assay in the MCTS model (Figure S13). Similar to fluorescence imaging in the MCTS model (Figure 7), LIL-PTX NPs was distributed throughout the deeper regions, and displayed an enhanced cell death at pH 6.5, compared with those of either the treatments of LIL-PTX NPs or the ICG/LAPMi-PTX sample at pH 7.4. Taken together, these results suggested that lipid coating significantly enhanced tumor penetration and cell death.
Multicellular tumor spheroid of CT26 cells incubated with the LIL-PTX for 4 h at pH 7.4 (A, B) and pH 6.5 (C, D). Scale bar corresponds to 100 µm.
LIL-PTX-Mediated Ultrarapid Tumor-tropic Accumulation and in vivo Photothermal Conversion Ability
Encouraged by the overwhelming in vitro results for anti-tumor activity, we tested our formulations in the in vivo tumor model, which was developed by subcutaneous CT26 cell injection. We evaluated the biodistribution studies for LIL-PTX NPs in the subcutaneous tumor model (Figure 8A & Figure S14). For this, free ICG (1.5 mg/kg), ICG/LAPMi-PTX (1.5 mg/kg of ICG) and LIL-PTX NPs (1.5 mg/kg of ICG) were injected intravenously and monitored by the FOBI imaging system. The robust fluorescence intensity of ICG can be seen in the tumor region as early as 1 h postinjection of LIL-PTX NPs. The fluorescence intensity in the tumor region peaked at 12 h postinjection (Figure 8C) and substantially declined by 120 h. In addition, ex vivo imaging of the organs collected at different time points postinjection were analyzed (Figure 8B). The results demonstrated that a bright fluorescence intensity in the tumor region was evidenced along with the other organs, such as the lungs and kidneys, at 1 h. At the 12 h and 24 h time points, fluorescence in the tumor region was remarkably sustained by simultaneously clearing it from the organs, which can be characterized by the dimmer fluorescence.
(A) Time-dependent biodistribution of LIL-PTX NPs (1.5 mg/kg of ICG) injected via the tail vein in CT26 tumor-bearing mice. (B) Ex vivo images of individual organs collected from LIL-PTX NPs -injected mice at different time points postinjection (K: kidney, Lu: lung, Li: liver, S: spleen, I: intestine, T: tumor, H: heart). (C) ICG fluorescence quantification from the operator-defined region of interest at the tumor region. (D) Fluorescence images captured from live imaging of LIL-PTX NPs (1.5 mg/kg of ICG)-injected mice. (Refer to supporting information for the movie clip). (E) Ex vivo fluorescence intensity of various organs collected from LIL-PTX NPs-injected mice at different times postinjection. (F) Thermal images and (G) Thermal profiles of CT26 tumor-bearing mice with laser irradiation (808 nm, 2 W/cm2, 10 min) following the tail-vein injection of PBS, ICG, ICG/LAPMi-PTX and LIL-PTX NPs (**p<0.01).
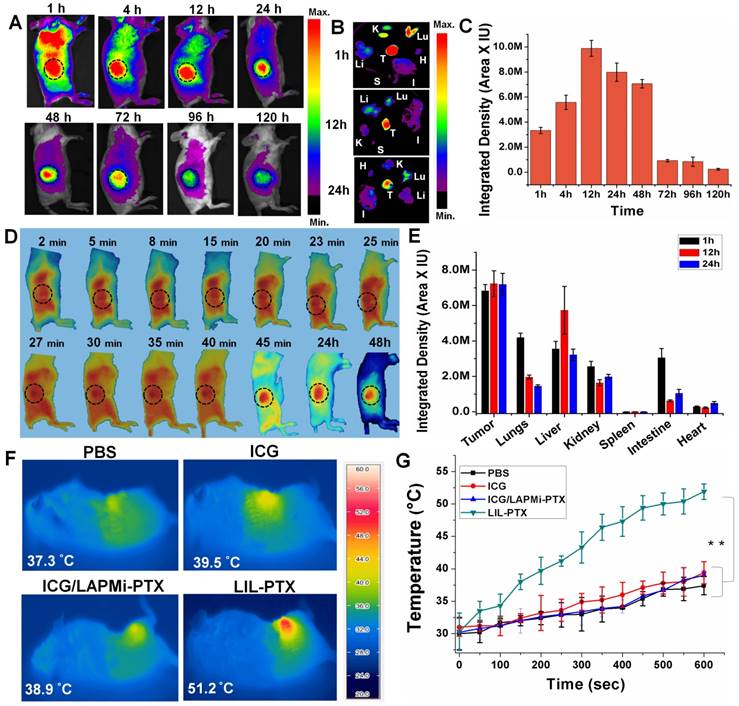
In contrast, ICG- and ICG/LAPMi-PTX-treated groups evidenced minimal tumor uptake, and the fluorescence quickly disappeared after 12 h of injection (Figure S14A). Moreover, ex vivo imaging from organs collected after 24 h confirmed that the fluorescence intensity in the tumor was negligible compared to that of other organs in these groups (Figure S14B-C). Importantly, LIL-PTX NPs uptake in other major sites, such as the lungs, liver and kidneys, was subsequently cleared; at the same time, the abundant tumor uptake was maintained (Figure 8E). Remarkably, LIL-PTX NPs were able to escape from the major RES organs, such as the liver and spleen, over a 24 h period. The liver and spleen are considered to remove the foreign entities rapidly from circulation, and avoiding the liver and spleen in developing nanoparticle-mediated therapies remains a major challenge. This improved circulation of LIL-PTX NPs can be ascribed to the PEGylated lipid layer on the nanoparticle surface complemented with a tumor-milieu charge switch compared with the ICG and non-lipid coated formulation (ICG/LAPMi-PTX). Furthermore, these results demonstrated that the lipid layer improved ICG protection during circulation and tumor homing, as lipids can protect the cargo from shear forces and complement mediated lysis during blood circulation [50].
The tumor-homing properties of LIL-PTX NPs were studied in a detailed manner by a live imaging system (Figure 8D). Once injected into the system, LIL-PTX NPs fluorescence rapidly increased in the tumor region (which was indicated by a black dotted circle). The tumor tropism of LIL-PTX NPs increased in a time-dependent manner. For instance, 27 min postinjection, the LIL-PTX NPs fluorescence was evidenced to be higher than that in all other organs. Remarkably, 45 min postinjection, LIL-PTX NPs accumulation was seen to be maximum in the tumor region with simultaneous clearance from other major organs. We hypothesized that this rapid tumor accumulation property of LIL-PTX NPs minimized the interaction of LIL-PTX NPs with the RES organs and other immune components, wherein the foreign particles are recognized and subjected to quick systemic clearance. Thus, the superior performance of LIL-PTX NPs in tumor targeting can be ascribed to their physicochemical parameters during circulation, such as sub-100 nm size and neutral surface charge. Furthermore, the positive charge switch of LIL-PTX NPs aided in the enhanced tumor retention and interaction with tumor cells, thereby preventing the backflow or clearance of therapeutics.
To investigate the in vivo applications of LIL-PTX NPs, pharmacokinetic parameters were evaluated. After an intravenous administration of LIL-PTX NPs (10 mg/kg of PTX), PTX concentrations in plasma and major organs including tumor were analyzed (Figure S15A-B). As shown in Figure S15A, PTX was rapidly distributed and was detectable even after 48 h post-administration. Moreover, half-life (t1/2) of LIL-PTX NPs was found to be 10.76 h and area under curve (AUC) estimated to be 39.66 µg.h/mL, which imply the longer circulation property of LIL-PTX NPs in the blood (Table S1). Also, clearance time (CL) was found to be 0.117 L/h. Figure S15B suggests that PTX distribution in tumor was largely enhanced in a time-dependent manner. Results indicate that PTX concentration in tumor was increased upto 24 h, and decreased thereafter. These results are in accordance with the fluorescence imaging analysis, where higher tumor accumulations of NPs were achieved at 24h post-injection. This enhanced PTX distribution in the tumor is attributable to the hierarchical activation strategy, through which drug leakage and non-specific distribution could be avoided. Also, PTX concentrations were detectable in the initial 4h time point in other major organs, and then eventually cleared from the system, emphasizing the non-toxic nature of LIL-PTX NPs.
To evaluate the thermal elevation capacity of LIL NPs in the in vivo conditions, LIL-PTX NPs was intravenously injected into tumor-bearing mice at a dose of 1.5 mg/kg of ICG, followed by laser irradiation (808 nm, 2 W/cm2, 10 min) 24 h postinjection. The tumor surface temperature was captured by an infrared thermal camera (Figure 8F). Figure 8G shows that the LIL-PTX NPs-treated group witnessed a temperature rise very quickly upon laser irradiation, whereas other groups demonstrated negligible hyperthermia. After 10 min of laser irradiation, a thermal rise in the tumor region of the PBS-, ICG-, ICG/LAPMi-PTX- and LIL-PTX NPs-treated groups were recorded as 37.4 ±1.4 °C, 39.4 ±1.7 °C, 39 ±0.6 °C and 51.9±1.2 °C, respectively. The temperature rise in the LIL-PTX NPs-treated group was above the threshold temperature of irreversible tissue damage [51]. Distinctly, this effective tumor hyperthermia was attributable to the significant amount of LIL-PTX NPs homing to the tumor region; thus, thermal enhancement upon laser irradiation was observed.
Enhanced Anti-Tumor Therapy in Subcutaneous Tumor Model
To demonstrate synergistic anti-cancer efficacy by using a hierarchical targeting strategy, LIL-PTX NPs was administered intravenously into CT26 tumor-bearing mice at a dose of 1.5 mg/kg of ICG and 10 mg/kg of PTX and laser irradiated (Figure 9A). The results demonstrated that the group of tumor mice treated with PBS + PTT underwent rapid tumor growth, which suggested that laser irradiation alone cannot affect tumor inhibition (Figure 9B, 9C). Tumor growth inhibition was not observed in the free ICG +PTT group as well. The chemotherapy group (LAPMi-PTX) showed negligible tumor inhibition, as the tumor targeting ability of LAPMi-PTX was minimal. Interestingly, the PTT alone group (LIL+PTT) showed a tumor inhibition immediately after laser irradiation, which was ascribed to the tumor-homing ability of LIL NPs (Figure 9C). However, tumor regrowth was evidenced 5 days postirradiation. This result indicated that the heat generated in PTT alone is not sufficient to treat intractable solid tumors.
(A) Schematic illustration explaining the tumor treatment strategy of CT26 tumor-bearing mice with LIL-PTX NPs. (B) Digital images of CT26 tumor-bearing mice depicted the tumor growth/inhibition, followed by intravenous injection of various groups after laser irradiation (808 nm, 2 W/cm2, 10 min) with observation for 15 days post-treatment. (C) Tumor volume changes following PTT/chemotherapy (***p<0.001). (D) Immunohistochemistry analysis of the tumor tissues collected from various groups 15 days post-treatment. H&E assay indicate the tissue morphology (Scale bar indicate 100 µm). TUNEL assay indicates the apoptotic cells (green signal) and Ki67 assay demonstrate the tumor cell proliferation (red signal) (Scale bar indicate 50 µm). Cytokine levels in serum from mice collected at 2 h, 24 h and 48 h post-treatment of LIL-PTX NPs: (E) TNF-alpha;(F) IL-6.
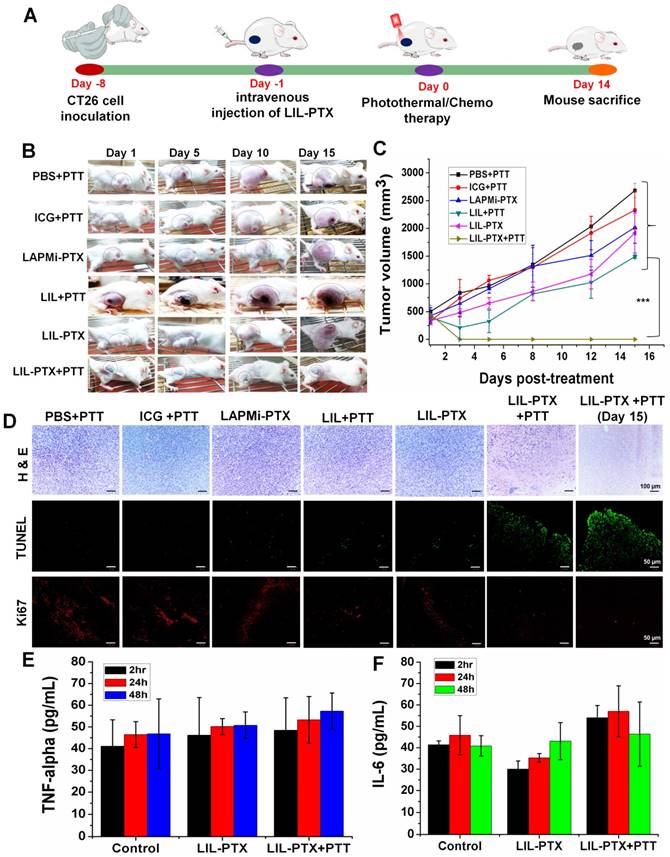
Moreover, a recent study found that the tumor margin beyond laser penetration evidenced tumor recurrence after laser irradiation [5]. Thus, this result suggested that PTT therapy alone could not demonstrate efficient tumor inhibition, owing to its limitation in laser penetration and heterogeneous heat distribution in the tumor region. Despite the efficient tumor accumulation and therapeutic dose of PTX, tumors treated with LIL-PTX NPs did not show any tumor inhibition throughout the treatment period. Without laser irradiation, LIL-PTX NPs were not destabilized to elicit anti-tumor activity. In addition, this result reaffirms the physiological stability of LIL-PTX NPs and its role in preventing systemic toxicity in healthy tissues. Notably, a single dose of LIL-PTX NPs plus laser irradiation was observed to result in complete tumor ablation without any recurrence, thus leaving the tumor region with scars, which reduced the post-treatment period. Representative images of dissected tumors after the treatment period suggested that synergistic PTT/chemotherapy caused efficient tumor ablation without tumor recurrence compared to that of other groups (Figure S16A).
Furthermore, the changes in body weight in mice in the LIL-PTX+PTT-treated group were not significant throughout the treatment course, which suggested that the PTT/chemotherapy had no systemic side effects (Figure S16B). In contrast, other groups showed an increase in body weight during the treatment period, presumably due to the tumor burden. Additionally, regenerated skin around the scars was observed in the LIL-PTX+PTT group, which suggested superior anti-tumor efficacy. H&E staining was used to evaluate the therapeutic efficacy. As shown in Figure 9D, no obvious tissue damage or necrosis was found in the control groups. In contrast, as expected, the tumor tissue underwent significant necrosis and tissue damage in the LIL-PTX+PTT group, which was characterized by damaged cellular structures and large voids.
Likewise, anti-tumor efficacy was also demonstrated by Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) and Ki67 assays. The results indicated that the LIL-PTX+PTT group demonstrated high apoptosis and reduced tumor cell proliferation, as indicated by the Ki67 antigen, compared with that of other control groups. In addition, we observed splenomegaly in all the control groups (Figures S17A-B). However, the spleen in the LIL-PTX+PTT-treated group did not demonstrate any enlargement to indicate the non-inflammatory treatment efficacy of the LIL-PTX NPs formulation. Major organs, such as the lung, heart, liver, kidney and spleen, were collected and analyzed by H&E assay (Figure S18), and no visible signs of potential toxic effects were indicated for any of the groups.
We also investigated the potent immunological response post-PTT/chemotherapy by LIL-PTX NPs. Changes in the serum concentrations of inflammatory cytokines, such as TNF-ɑ and IL-6, were measured after 2 h, 24 h and 48 h post-PTT. As shown in Figure 9E, 9F, changes in the levels of these cytokines before and after PTT were insignificant. This result was consistent with a previous report [52], wherein non-adjuvant-like PTT agents such as ICG, CuS nanoparticles, and graphene oxide, showed a minimal immunologic response after laser irradiation. Cancer therapies such as PTT can trigger inflammatory responses upon laser irradiation, which has a clear association with tumor regeneration and therapy resistance. Thus, recent studies focused on mitigating PTT-associated inflammation by using anti-inflammatory drugs, thereby preventing further tumor recurrence [53].
Biodistribution, Anti-Tumor Activity and Metastasis Inhibition in Orthotopic Tumor Model
By observing superior anti-tumor efficacy in the CT26 tumor model, it would be interesting to determine similar anti-tumor efficacies in the orthotopic tumor model and to evaluate their inhibitory potential in controlling subsequent lung metastasis. For this purpose, we developed an orthotopic breast cancer model by injecting 4T1 tumor cells in the mammary pad (Figure 10A). Similar to the subcutaneous model, we evaluated the biodistribution and tumor-homing abilities of LIL-PTX NPs. An intravenous injection of LIL-PTX NPs (1.5 mg/kg of ICG) into 4T1 orthotopic tumor-bearing mice indicated that LIL-PTX NPs exhibited rapid tumor tropic accumulation in the orthotopic breast tumors (Figure 10B). LIL- PTX NPs fluorescence can be visualized in the tumor region as early as 30 min, and it peaked 16 h postinjection (Figure 10C). Superior tumor tropism of LIL-PTX NPs in the breast tumors consolidates the targeting efficacy of the LIL-PTX NPs in orthotopic tumors as well. Next, the tumor temperature elevations were measured after laser irradiation (808 nm, 2 W/cm2, 10 min) with a thermal camera. As shown in Figure S19A-B, the LIL-PTX-treated group demonstrated a temperature rise of 52.6 ± 1.1 °C after 10 min of laser irradiation, which was sufficient to ablate the malignant cancer cells. Anti-tumor efficacy was evaluated in the 4T1 orthotopic tumor-bearing mice, followed by laser irradiation (808 nm, 2 W/cm2, 10 min) and treatment with similar dosages of ICG and PTX in the CT26 tumor model. Figure 10D & Figure S20 indicated that the LIL-PTX+PTT-treated group significantly inhibited the tumor growth compared to other groups (PBS+PTT, LIL-PTX) and elicited synergistic photothermal/chemotherapy post-laser irradiation.
(A) Schematic illustrating the photothermal/chemotherapy treatment strategy of the 4T1 orthotopic tumor. (B) Time-dependent biodistribution analysis of 4T1 orthotopic tumor-bearing mice, followed by an intravenous injection of LIL-PTX NPs (1.5 mg/kg of ICG). (C) Fluorescence imaging of 4T1 orthotopic tumor-bearing mice depicting the localization of LIL-PTX NPs in the tumor region recorded 16 h postinjection. (D) Tumor volume changes over a treatment period of 2 weeks after laser irradiation (808 nm, 2 W/cm2, 10 min) (***p<0.001). (E) H&E stained sections of tumor tissues collected from various groups at a post-treatment period of 14 days. Scale bar indicates 100 µm. (F) H&E stained sections of lungs collected from various groups at a post-treatment period of 14 days. Black arrow indicates the tumor cell region. The 4X on the scale bar indicates 1000 µm, and the 40X scale corresponds to 200 µm. (G) The tumor cell percentage area quantified as a fraction of the blue signal from the lungs of various groups. An analysis was performed using ImageJ software (**p<0.01 and ***p<0.001).
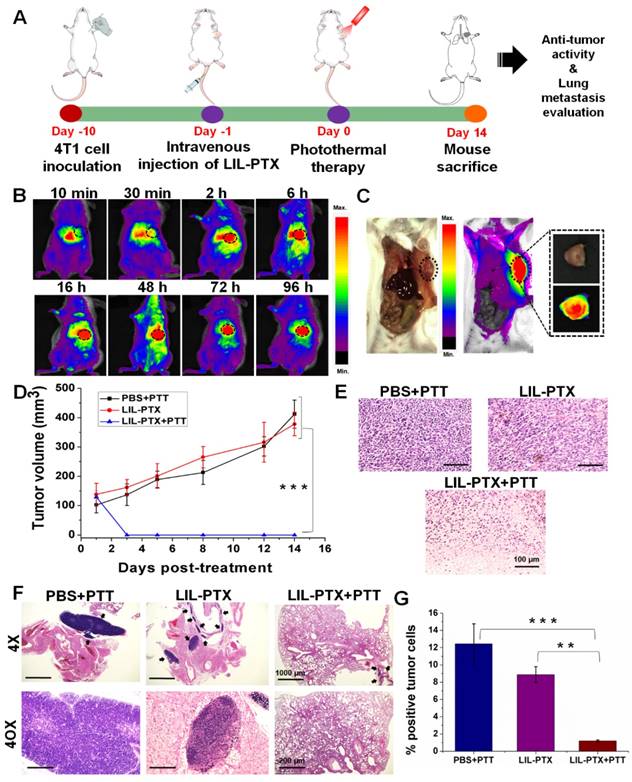
In addition, the therapeutic efficacy was evaluated by H&E-stained tumor tissue (Figure 10E). The results indicated complete tumor cell damage accompanied by large necrotic nuclei and large voids compared with that of other control groups. 4T1 inoculation in the mammary fat pad tends to metastasize to the lungs. Therefore, the anti-metastatic effect was analyzed by the H&E assay of the lung sections of the 4T1 orthotopic breast tumor mice from all groups (Figure 10F). H&E sections showed that the prevalence of tumor cells in the lung sections was significantly lowered in the LIL-PTX+PTT group compared with that of the other groups. Furthermore, an ImageJ analysis of the lung sections quantified for tumor cell percentage (%) revealed that the lung tumor burden was reduced by 6-fold and 4-fold compared with that of the PBS+PTT and LIL-PTX+PTT groups, respectively (Figure 10G). Thus, these results demonstrated that our hierarchical targeting strategy of therapeutics successfully ablated the primary tumor along with lung metastasis.
Conclusion
In this study, we prepared a novel formulation to precisely deliver therapeutics to tumors in an ultrarapid manner for combinatorial photothermal/chemotherapy. Our approach to the hierarchical delivery of therapeutics through LIL-PTX NPs formulation possesses the following advantages: (1) prolonged systemic circulation of LIL-PTX NPs and remarkable stability in the physiological conditions of encapsulated ICG protected by the outer lipid layer, thus leading to an enhanced heating effect; (2) excellent responsiveness to both the laser and intracellular GSH, as well as a demonstrated hierarchical disassembly of therapeutics upon exogenous and endogenous triggers; (3) rapid tumor tropic accumulation of LIL-PTX NPs while clearing that from the other organs to deliver therapeutics specifically to tumors; (4) enhanced tumor penetration of LIL-PTX NPs; and (5) cancer-specific activatable drug release. Anti-tumor studies conducted in subcutaneous and orthotopic tumor models manifested the enhanced therapeutic efficiency that is characterized by complete tumor ablation, without any recurrence or metastasis. The immunohistochemistry studies on tumor sections also reaffirmed the antitumor efficacy that was demonstrated by enhanced apoptosis and inhibited tumor cell proliferation. Thus, the LIL-PTX NPs-mediated hierarchical activating strategy significantly improved the image-guided combinatorial photothermal/chemotherapy and proposed a paradigm shift in the clinical translation for anti-tumor therapy.
Experimental Section
Materials
1,2-distearoyl-sn-glycero-3 phosphoethanolamine-N-[methoxy (polyethylene glycol) - 2000] amine (DSPE-PEG-NH2) and dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) were purchased from Avanti polar lipid (AL, USA). ICG was purchased from BioActs (Korea). Branched polyethyleneimine (1800 Da) was purchased from Polysciences Inc. (USA). Methanol and dimethyl sulfoxide (DMSO) were obtained from Merck (Germany). 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide hydrochloride (EDC), L-glutathione reduced (GSH), N-hydroxysuccinimide (NHS), lithocholic acid and L-buthionine sulfoximine (BSO) were purchased from Sigma (St. Louis, MO). 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4sulfophenyl)- tetrazolium (MTS) reagent was obtained from Promega (Madison, WI, USA). Propidium iodide (PI), fluorescein diacetate (FDA) and Dulbecco's modified Eagle's medium (DMEM) Ellman's reagent were obtained from Thermo Scientific (Waltham, MA, USA). All other chemicals were obtained from commercial suppliers unless otherwise specified.
Synthesis of LIL-PTX NPs
ssPEI, LAPMi & LAPMi-PTX synthesis was carried out following our previous report [18], with some minor modifications. For LIL NPs synthesis, ICG was adsorbed onto LAPMi-PTX in an acidic aqueous solution at different ratios and was incubated for 1 h at 37°C. Next, ICG adsorbed LAPMi-PTX (ICG/LAPMi-PTX) were collected with a 3 kDa centrifugal filter (Merck, NJ, USA) at 10000 rpm for 30 min and were washed with DW. The amount of adsorbed ICG was determined by measuring the absorbance of ICG from the filtrate. Next, DSPE-PEG-NH2 and DPPC at a molar ratio of 4:1 was mixed in chloroform and made into a thin film. The thin film was hydrated with the as-prepared ICG/LAPMi-PTX at 60°C for 15 min and was sonicated for 10 min. The sonicated samples were extruded through a mini extruder (Avanti polar lipid, AL, USA) using a polycarbonate filter of 200 nm in 11 cycles. The resultant sample lipid/ICG/LAPMi-PTX (LIL-PTX NPs) was dialyzed against DW (MWCO=10 kDa) for 12 h at 4°C, and then lyophilized. LIL NPs was synthesized in a similar manner using LAPMi as the core material.
Characterization of NPs
The nanoparticle size (DLS analysis) and zeta potential were measured using a Zetasizer Nano Z instrument (Malvern Instruments, Malvern, UK). Proton nuclear magnetic resonnce (1H NMR) was analyzed by NMR spectrophotometry (500 Hz; Bruker, MA) in D2O. The molecular weight distribution of polymers is analyzed by Bruker Ultraflex MALDI TOF/TOF (IL, USA). The morphology of the nanoparticles was confirmed using a field emission transmission electron microscope (FE-TEM). LIL-PTX NPs (0.5 mg/mL in DMEM, pH 7.4), was irradiated (808 nm, 2 W/cm2, 10 min), and further treated with 10 mM GSH in DMEM (pH 6.5) for 24 h. Negative staining for lipids in LIL-PTX NPs was analyzed by uranyl acetate. These samples were analyzed by FE-TEM (JEM-2100F, USA) and DLS. The UV absorbance was analyzed by a UV-VIS spectrophotometer (UV-2700, Shimadzu, USA). The fluorescence spectrum was recorded by a Tecan Spark Multimode plate reader (Mannedorf, Switzerland). The thermal elevation of the LIL-PTX NPs (30 µg/mL ICG), ICG (30 µg/mL) was irradiated with an 808 nm laser for 10 min and was imaged by a thermal camera (Avio IR camera/Thermometer, Japan).
Drug release tests
The paclitaxel (PTX) and coumarin-6 loading into LAPMi was carried out following our previous publication [18]. Two milliliters of LIL-PTX NPs (pH 7.4), LIL-PTX + Laser (pH 6.5) and LIL-PTX + Laser + GSH (pH 6.5) were loaded into dialysis bags (MWCO 3.5 kDa) and into a 48 mL PBS (pH 7.4) solution while stirring. Laser (808 nm, 2 W/cm2, 10 min) and GSH (10 mM) were used as the stimulus to trigger the release. Then, 1 mL of samples (n=3) was collected and replaced with 1 mL 1X PBS buffer (pH 7.4). HPLC (Shimadzu HPLC system, Kyoto, Japan) was used to quantify the amount of PTX release.
Cell culture and multicellular tumor spheroid (MCTS) formation
The mouse colon carcinoma cell line CT26 was cultured in DMEM (10 % fetal bovine serum (FBS), 100 µg/mL streptomycin) in a humidified cell culture chamber (5 % CO2, 37°C). For the MCTS formation, CT26 cells were incubated in an agar-coated 96-well plate as reported earlier [54]. Briefly, 1 % (wt./vol.) agarose (SeaMatrix Agarose LE) was utilized to coat the 96-well plate, and the CT26 cell density was 50,000 cells/well. The DMEM culture media was replaced with fresh media every alternate day, and spheroids were collected at the end of day 10.
Cell uptake studies
CT26 cells were cultured in DMEM (10 % FBS) in an 8-well culture chamber (Lab-Tek, Thermo Fisher, USA) at pH 7.4 or pH 6.5 followed by the addition of LIL NPs in DMEM solutions for 4 h. Then, cells were washed with PBS and fixed with 4 % paraformaldehyde followed by counterstaining with ProLong DAPI (Thermo Fisher Scientific, USA) and CytoFlamma 496 (BioActs, Korea). For stimuli- responsive imaging, BSO (20 µM) was pretreated for 30 min followed by PBS wash and incubated with LIL-Cou NPs (30 µg/mL) in DMEM solutions (pH 6.5) for 4 h. Then, cells were subjected to NIR laser (808 nm, 2 W/cm2, 10 min) and were washed with PBS, fixed with 4 % PFA and counterstained with DAPI. Nanoparticle uptake and stimuli responsive imaging were analyzed by confocal laser scanning microscopy (Zeiss LSM510, Germany) and a FACSCalibur flow cytometer (Becton Dickinson, USA).
In vitro studies
Cell viability was evaluated on CT26 cells (3 × 104 cells/well) with DMEM and was incubated overnight. Next, the cells were incubated with predetermined concentrations of ICG, PTX, LIL NPs and LIL-PTX NPs for 24 h. BSO (20 µM) was pretreated for 30 min prior to nanoparticle treatment. For laser treatment groups, the laser (808 nm, 2 W/cm2, 10 min) was irradiated, and the cells were replaced with fresh media followed by 24 h incubation. Thermal elevation in CT26 cells was performed by collecting the cell pellets post treatment with LIL-PTX NPs, followed by laser irradiation (808 nm, 2 W/cm2, 10 min). The temperature rise was recorded by a thermal camer (Avio IR camera/Thermometer, Japan). For the cytotoxicity evaluation, MTS (20 µL) reagent was incubated for 3 h, and the absorbance was recorded using a Spark 10 M multimode microplate reader (Tecan Trading AG, Switzerland) at 490 nm. The live/dead assay was carried out on LIL-PTX-treated cells with a laser (808 nm, 2 W/cm2, 10 min) by FDA and PI treatments according to the manufacturer's protocol and were imaged by a ZOE fluorescent cell imager (Bio-Rad, USA). The trypan blue assay was performed by adding 100 µL trypan blue (0.1 wt %), followed by PBS wash and was visualized by using a ZOE fluorescent cell imager (Bio-Rad, USA).
Western blot analysis and immunofluorescence
CT26 cells were treated with LIL-PTX NPs (33 µg/mL ICG, 10 µg/mL PTX) for 4 h, followed by a laser treatment (808 nm, 2 W/cm2, 10 min) and were incubated for another 24 h. Then, the cells were prepared for Western blot analysis by using a procedure that was reported earlier [55]. HSP70 (ThermoFisher Scientific, USA), caspase-3 and β-actin (Santa Cruz Biotechnology, Santa Cruz, CA) antibodies were used according to the manufacturer's protocol and were detected by SmartChemi (Korea Lab Tech, Korea). For immunofluorescence imaging of HSP70, the cells were formalin fixed and permeabilized with Triton X-100 (0.1 %) for 10 min at RT and were further blocked with 1 % BSA for 1 h at room temperature. Then, the cells were incubated with an HSP70 antibody (1:50 dilution) for 1 h at RT and were further incubated with the goat anti-mouse IgG Flamma 594 (BioActs, Korea) for 30 min at RT. Slides were stained with ProLong DAPI and were visualized with a ZOE fluorescent cell imager (Bio-Rad, USA).
Penetration and anti-tumor studies in MCTS
MCTS penetration was visualized by confocal microscopy. For this, spheroids were treated with the ICG/LAPMi-PTX (30 µg/mL ICG) and LIL-PTX NPs (30 µg/mL ICG) in DMEM solutions of pH 7.4 and pH 6.5 for 4 h. Spheroids were further washed with PBS and fixed with 4 % PFA, counterstained with DAPI, placed in a cell dish containing agarose (1 %) and imaged by confocal microscope. For the live/dead assay, MCTS were treated with ICG/LAPMi-PTX (33 µg/mL ICG, 10 µg/mL PTX) and LIL-PTX NPs (33 µg/mL ICG, 10 µg/mL PTX) at pH 7.4 and pH 6.5 for 4 h. Laser-irradiated (808 nm, 2 W/cm2, 10 min) MCTS were incubated for 24 h with fresh media. The next day, MCTS were stained with FDA and PI, followed by imaging via a ZOE fluorescent cell imager (Bio-Rad, USA).
Biodistribution, in vivo anti-tumor studies & treatment plan in a subcutaneous tumor model
Animal experiments were conducted according to the regulations of the Ethics Committee of Chonnam National University Medical School and Chonnam National University Hwasun Hospital, South Korea (CNU IACUC-H-2018-59). 4-week-old BALB/c female mice were injected with CT26 cells (1x 106) subcutaneously in the right flank, and tumors were allowed to grow for 10 days. For the biodistribution analysis, ICG, ICG/LAPMi-PTX and LIL-PTX NPs at concentrations of 1.5 mg/kg ICG were injected intravenously, and optical imaging was performed with a fluorescence-labeled organism bio-imaging instrument (FOBI, NEO science, Gyeonggi, Korea). At predetermined time points, the tumors and major organs were dissected from the mice and were analyzed using the FOBI in vivo imaging system. Live imaging of LIL-PTX-injected mice was recorded by the FLARE imaging system. For biodistribution, mice were randomly distributed into the following groups: free ICG (0.5 mg/kg), ICG/LAPMi-PTX (0.5 mg/kg ICG) and LIL-PTX NPs (0.5 mg/kg ICG) (n=5 for each group). For the in vivo anti-tumor treatment, the groups were PBS, ICG+PTT (1.5 mg/kg ICG), LAPMi-PTX (10 mg/kg PTX) and ICG/LAPMi-PTX (1.5 mg/kg ICG & 10 mg/kg PTX), LIL+PTT (1.5 mg/kg ICG), LIL-PTX (1.5 mg/kg ICG & 10 mg/kg PTX), LIL-PTX+PTT (1.5 mg/kg ICG & 10 mg/kg PTX), (n=5). A near-infrared laser (808 nm, 2 W/cm2) was irradiated after 24 h onto tumor regions for 10 min. The rise in the tumor surface temperature was recorded by a thermal camera (Avio IR camera/Thermometer, Japan). Mouse blood was collected at predetermined time points, and an ELISA analysis of TNF-ɑ and IL-6 (eBioscience Inc., CA, USA) from the serum was conducted according to the manufacturer's protocol. The tumor volume and mouse weight were recorded over a treatment period of 15 days.
Pharmacokinetics of LIL-PTX
PTX concentrations in plasma and major tissues including tumors were obtained using HPLC method. Samples were homogenized with saline and subsequntly mixed with acetonitrile for plasma protein precipitation. The sample mixture was vortexed briefly and centrifuged at 4 oC for 20 min, then the sample was analyzed using HPLC system (Shimadzu HPLC system, Kyoto, Japan).
Biodistribution and antitumor studies in an orthotopic breast tumor model
4T1 cells at a density of 5X105 cells were injected directly into the mammary fat pad of female Balb/C mice. Tumors grew 10 days post implantation and LIL-PTX (1.5 mg/kg ICG) was injected via the tail vein. The nanoparticle distribution as a means of the ICG fluorescence was recorded by a FOBI in vivo imaging system. For anti-tumor studies, PBS, LIL-PTX (1.5 mg/kg ICG & 10 mg/kg PTX) were intravenously injected (n=5). After 24 h post treatment of LIL-PTX+PTT, the laser was irradiated (808 nm, 2 W/cm2) onto tumor regions for 10 min. The rise in the tumor surface temperature was recorded by a thermal camera (Avio IR camera/Thermometer, Japan). The tumor volume and mouse weight were recorded over a treatment period of 15 days.
Histology, immunohistochemistry and immunofluorescence analysis
All major organs, including tumors, were fixed with 10 % formalin, and the tissue sections were embedded in paraffin. The tissue sections were dewaxed with xylene and were hydrated with alcohols from 100 %, 95 % and 75 % ethanol and DW; then, the tissues were stained with hematoxylin and eosin (H&E). The TUNEL assay was analyzed according to the manufacturer's instructions (DeadEndTM Fluorometric TUNEL system, Promega, USA), and the images were recorded by a ZOE fluorescent cell imager (Bio-Rad, USA). For Ki67 assay, tumor sections (6 µm) were cryosectioned and fixed in ice-cold acetone, washed with PBS, and probed with an anti-mouse Ki67 antibody (1:50 dilution; Abcam, USA), according to the manufacturer's protocol.
Abbreviations
NIR: near-infrared; LCA: lithocholic acid; ssPEI: disulfide-linked polyethyleneimine; PTX: paclitaxel; 1H-NMR: proton nuclear magnetic resonance; FE-TEM: Field emission transmission electron microscopy; DLS: Dynamic light scattering; BSO: L-buthionine sulfoximine; DSPE-PEG-NH2: 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] amine; DPPC: dipalmitoyl-sn-glycero-3-phosphocholine; RES: Reticuloendothelial system; PTX: paclitaxel; ICG: indocyanine green; EPR: enhanced permeation and retention; Cou: coumarin-6; HSP: heat shock proteins; MCTS: multicellular tumor spheroid; TUNEL: Terminal deoxynucleotidyl transferase dUTP nick end labeling.
Supplementary Material
Supplementary figures and table.
Supplementary movie.
Acknowledgements
This work was financially supported by Basic Science Research Program (No. 2016R1A2B4011184) and the Bio & Medical Technology Development Program (No. NRF-2017M3A9F5030940 and NRF-2017M3A9E2056374) through the National Research Foundation of Korea (NRF) funded by the Korean government, MSIP; and the Pioneer Research Center Program through the National Research Foundation of Korea funded by the Ministry of Science, ICT & Future Planning (2014M3C1A3053035). This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (No. 2018R1A5A2024181).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Yoon HJ, Lee HS, Lim JY, Park JH. Liposomal Indocyanine Green for Enhanced Photothermal Therapy. ACS Applied Materials and Interfaces. 2017;9:5683-91
2. Cherukula K, Manickavasagam Lekshmi K, Uthaman S, Cho K, Cho C-S, Park I-K. Multifunctional Inorganic Nanoparticles: Recent Progress in Thermal Therapy and Imaging. Nanomaterials. 2016;6:76 -
3. Zhou M, Zhao J, Tian M, Song S, Zhang R, Gupta S. et al. Radio-photothermal therapy mediated by a single compartment nanoplatform depletes tumor initiating cells and reduces lung metastasis in the orthotopic 4T1 breast tumor model. Nanoscale. 2015;7:19438-47
4. Jin R, Liu Z, Bai Y, Zhou Y, Gooding JJ, Chen X. Core-Satellite Mesoporous Silica-Gold Nanotheranostics for Biological Stimuli Triggered Multimodal Cancer Therapy. Advanced Functional Materials. 2018;28:1801961 -
5. Hirsch LR, Stafford RJ, Bankson JA, Sershen SR, Rivera B, Price RE. et al. Nanoshell-mediated near-infrared thermal therapy of tumors under magnetic resonance guidance. Proceedings of the National Academy of Sciences. 2003;100:13549-54
6. Zhou W, Liu X, Ji J. More efficient NIR photothermal therapeutic effect from intracellular heating modality than extracellular heating modality: An in vitro study. Journal of Nanoparticle Research. 2012:14
7. Creixell M, Bohórquez AC, Torres-Lugo M, Rinaldi C. EGFR-targeted magnetic nanoparticle heaters kill cancer cells without a perceptible temperature rise. ACS Nano. 2011;5:7124-9
8. Li N, Sun Q, Yu Z, Gao X, Pan W, Wan X. et al. Nuclear-Targeted Photothermal Therapy Prevents Cancer Recurrence with Near-Infrared Triggered Copper Sulfide Nanoparticles. ACS Nano. 2018;12:5197-206
9. Ma Z, Han K, Dai X, Han H. Precisely Striking Tumors without Adjacent Normal Tissue Damage via Mitochondria-Templated Accumulation. ACS Nano. 2018;12:6252-62
10. Luo Y, Huang L, Yang Y, Zhuang X, Hu S, Ju H. et al. A programmed nanoparticle with self-adapting for accurate cancer cell eradication and therapeutic self-reporting. Theranostics. 2017;7:1245-56
11. Lu Z, Huang FY, Cao R, Tan GH, Yi G, He N. et al. Intrinsic, Cancer Cell-Selective Toxicity of Organic Photothermal Nanoagent: A Simple Formulation for Combined Photothermal Chemotherapy of Cancer. ACS Appl Mater Interfaces. 2018;10:26028-38
12. Meng Z, Wei F, Wang R, Xia M, Chen Z, Wang H. et al. NIR-Laser-Switched in Vivo Smart Nanocapsules for Synergic Photothermal and Chemotherapy of Tumors. Advanced Materials. 2016;28:245-53
13. Alexis F, Pridgen E, Molnar LK, Farokhzad OC. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol Pharm. 2008;5:505-15
14. Yu M, Zheng J. Clearance Pathways and Tumor Targeting of Imaging Nanoparticles. ACS Nano. 2015;9:6655-74
15. Li SD, Huang L. Pharmacokinetics and biodistribution of nanoparticles. Mol Pharm. 2008;5:496-504
16. Chen X, Liu L, Jiang C. Charge-reversal nanoparticles: novel targeted drug delivery carriers. Acta Pharm Sin B. 2016;6:261-7
17. Mellor HR, Davies LA, Caspar H, Pringle CR, Hyde SC, Gill DR. et al. Optimising non-viral gene delivery in a tumour spheroid model. J Gene Med. 2006;8:1160-70
18. Cherukula K, Bae WK, Lee JH, Park IK. Programmed 'triple-mode' anti-tumor therapy: Improving peritoneal retention, tumor penetration and activatable drug release properties for effective inhibition of peritoneal carcinomatosis. Biomaterials. 2018;169:45-60
19. Suzuki H, Bae YH. Evaluation of drug penetration with cationic micelles and their penetration mechanism using an in vitro tumor model. Biomaterials. 2016;98:120-30
20. Shen T, Guan S, Gan Z, Zhang G, Yu Q. Polymeric Micelles with Uniform Surface Properties and Tunable Size and Charge: Positive Charges Improve Tumor Accumulation. Biomacromolecules. 2016;17:1801-10
21. Yan J, Su T, Cheng F, Cao J, Zhang H, He B. Multifunctional nanoparticles self-assembled from polyethylenimine-based graft polymers as efficient anticancer drug delivery. Colloids and Surfaces B: Biointerfaces. 2017;155:118-27
22. Cheng R, Feng F, Meng F, Deng C, Feijen J, Zhong Z. Glutathione-responsive nano-vehicles as a promising platform for targeted intracellular drug and gene delivery. Journal of Controlled Release. 2011;152:2-12
23. Atanackovic M, Posa M, Heinle H, Gojkovic-Bukarica L, Cvejic J. Solubilization of resveratrol in micellar solutions of different bile acids. Colloids Surf B Biointerfaces. 2009;72:148-54
24. Elnaggar YS. Multifaceted applications of bile salts in pharmacy: an emphasis on nanomedicine. Int J Nanomedicine. 2015;10:3955-71
25. Poudel BK, Gupta B, Ramasamy T, Thapa RK, Pathak S, Oh KT. et al. PEGylated thermosensitive lipid-coated hollow gold nanoshells for effective combinational chemo-photothermal therapy of pancreatic cancer. Colloids and Surfaces B: Biointerfaces. 2017;160:73-83
26. Paliwal SR, Paliwal R, Vyas SP. A review of mechanistic insight and application of pH-sensitive liposomes in drug delivery. Drug Deliv. 2015;22:231-42
27. Zhang X, Zhang K, Haag R. Multi-stage, charge conversional, stimuli-responsive nanogels for therapeutic protein delivery. Biomater Sci. 2015;3:1487-96
28. A. M. Taylor SM, Gupton SL. HHS Public Access. Lab Chip. 2015;2:2781-9
29. Russi AE, Brown MA. HHS Public Access. 2016; 165: 255-69.
30. Kato Y, Ozawa S, Miyamoto C, Maehata Y, Suzuki A, Maeda T. et al. Acidic extracellular microenvironment and cancer. Cancer Cell Int. 2013;13:89
31. Altuntas E, Knop K, Tauhardt L, Kempe K, Crecelius AC, Jager M. et al. Tandem mass spectrometry of poly(ethylene imine)s by electrospray ionization (ESI) and matrix-assisted laser desorption/ionization (MALDI). J Mass Spectrom. 2012;47:105-14
32. Sheng G, Chen Y, Han L, Huang Y, Liu X, Li L. et al. Encapsulation of indocyanine green into cell membrane capsules for photothermal cancer therapy. Acta Biomaterialia. 2016;43:251-61
33. Li W, Zhang H, Guo X, Wang Z, Kong F, Luo L. et al. Gold nanospheres-stabilized indocyanine green as a synchronous photodynamic-photothermal therapy platform that inhibits tumor growth and metastasis. ACS Applied Materials and Interfaces. 2017;9:3354-67
34. Ma Y, Tong S, Bao G, Gao C, Dai Z. Indocyanine green loaded SPIO nanoparticles with phospholipid-PEG coating for dual-modal imaging and photothermal therapy. Biomaterials. 2013;34:7706-14
35. Zheng X, Zhou F, Wu B, Chen WR, Xing D. Enhanced tumor treatment using biofunctional indocyanine green-containing nanostructure by intratumoral or intravenous injection. Mol Pharm. 2012;9:514-22
36. Storm G, Belliot SO, Daemen T, Lasic DD. Surface modification of nanoparticles to oppose uptake by the mononuclear phagocyte system. Advanced Drug Delivery Reviews. 1995;17:31-48
37. Gaumet M, Vargas A, Gurny R, Delie F. Nanoparticles for drug delivery: the need for precision in reporting particle size parameters. Eur J Pharm Biopharm. 2008;69:1-9
38. Ling K, Jiang H, Zhang Q. A colorimetric method for the molecular weight determination of polyethylene glycol using gold nanoparticles. Nanoscale Res Lett. 2013;8:538
39. Zheng X, Zhou F, Wu B, Chen WR, Xing D. Enhanced Tumor Treatment Using Biofunctional Indocyanine Green-Containing Nanostructure by Intratumoral or Intravenous Injection. Molecular Pharmaceutics. 2012;9:514-22
40. Kim TH, Chen Y, Mount CW, Gombotz WR, Li X, Pun SH. Evaluation of temperature-sensitive, indocyanine green-encapsulating micelles for noninvasive near-infrared tumor imaging. Pharm Res. 2010;27:1900-13
41. Fernandes C, Suares D, Yergeri MC. Tumor Microenvironment Targeted Nanotherapy. Front Pharmacol. 2018;9:1230
42. Crayton SH, Tsourkas A. pH-titratable superparamagnetic iron oxide for improved nanoparticle accumulation in acidic tumor microenvironments. ACS Nano. 2011;5:9592-601
43. Kneidl B, Peller M, Winter G, Lindner LH, Hossann M. Thermosensitive liposomal drug delivery systems: state of the art review. Int J Nanomedicine. 2014;9:4387-98
44. Mackey MA, Ali MRK, Austin LA, Near RD, El-Sayed MA. The Most Effective Gold Nanorod Size for Plasmonic Photothermal Therapy: Theory and In Vitro Experiments. The Journal of Physical Chemistry B. 2014;118:1319-26
45. Yang L, Tseng YT, Suo G, Chen L, Yu J, Chiu WJ. et al. Photothermal therapeutic response of cancer cells to aptamer-gold nanoparticle-hybridized graphene oxide under NIR illumination. ACS Appl Mater Interfaces. 2015;7:5097-106
46. Takayama S, Reed JC, Homma S. Heat-shock proteins as regulators of apoptosis. Oncogene. 2003;22:9041
47. Depraetere V, Golstein P. Dismantling in cell death: molecular mechanisms and relationship to caspase activation. Scand J Immunol. 1998;47:523-31
48. Wen Z, Liao Q, Hu Y, You L, Zhou L, Zhao Y. A spheroid-based 3-D culture model for pancreatic cancer drug testing, using the acid phosphatase assay. Brazilian Journal of Medical and Biological Research. 2013;46:634-42
49. Li YH, Wang J, Gao Y, Zhu JB, Wientjes MG, Au JLS. Relationships between Liposome Properties, Cell Membrane Binding, Intracellular Processing, and Intracellular Bioavailability. Aaps J. 2011;13:585-97
50. Liu XS, Situ A, Kang YA, Villabroza KR, Liao YP, Chang CH. et al. Irinotecan Delivery by Lipid-Coated Mesoporous Silica Nanoparticles Shows Improved Efficacy and Safety over Liposomes for Pancreatic Cancer. Acs Nano. 2016;10:2702-15
51. Peng CL, Shih YH, Lee PC, Hsieh TM, Luo TY, Shieh MJ. Multimodal image-guided photothermal therapy mediated by 188Re-labeled micelles containing a cyanine-type photosensitizer. ACS Nano. 2011;5:5594-607
52. Wang C, Xu L, Liang C, Xiang J, Peng R, Liu Z. Immunological responses triggered by photothermal therapy with carbon nanotubes in combination with anti-CTLA-4 therapy to inhibit cancer metastasis. Advanced Materials. 2014;26:8154-62
53. Dong Q, Wang X, Hu X, Xiao L, Zhang L, Song L. et al. Simultaneous Application of Photothermal Therapy and an Anti-inflammatory Prodrug using Pyrene-Aspirin-Loaded Gold Nanorod Graphitic Nanocapsules. Angewandte Chemie International Edition. 2018;57:177-81
54. Charoen KM, Fallica B, Colson YL, Zaman MH, Grinstaff MW. Embedded multicellular spheroids as a biomimetic 3D cancer model for evaluating drug and drug-device combinations. Biomaterials. 2014;35:2264-71
55. Yang WL, Nair DG, Makizumi R, Gallos G, Ye XM, Sharma RR. et al. Heat shock protein 70 is induced in mouse human colon tumor xenografts after sublethal radiofrequency ablation. Annals of Surgical Oncology. 2004;11:399-406
Author contact
![]() Corresponding author: In-Kyu Park, e-mail: pik96ac.kr
Corresponding author: In-Kyu Park, e-mail: pik96ac.kr