Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(8):2209-2223. doi:10.7150/thno.30726 This issue Cite
Research Paper
Myeloid cell-derived LL-37 promotes lung cancer growth by activating Wnt/β-catenin signaling
Ping Ji1*, Yongxin Zhou2*, Yibao Yang1*, Junlu Wu1, Hao Zhou3, Wenqiang Quan1, Junjun Sun1, Yiwen Yao1, Anquan Shang1, Chenzheng Gu1, Bingjie Zeng1, Jenni Firrman4, Weidong Xiao5, Robert Bals6, Zujun Sun1, Dong Li1 ![]()
1. Department of Clinical Laboratory, Shanghai Tongji Hospital, Tongji University School of Medicine, Shanghai 200065, China.
2. Department of Thoracic-cardiovascular Surgery, Tongji hospital,Tongji University School of Medicine, Shanghai 200065, China.
3. Department of Pharmacy, Putuo People's Hospital, Shanghai 200060, China.
4. Dairy and Functional Foods Research Unit, Eastern Regional Research Center, Agriculture Research Service, United States Department of Agriculture, Wyndmoor, PA 19038, USA.
5. Sol Sherry Thrombosis Research Center, Temple University, Philadelphia, PA 19140, USA.
6. Department of Internal Medicine V-Pulmonology, Allergology, Respiratory Intensive Care Medicine, Saarland University Hospital, Homburg 66424, Germany.
*The authors contributed equally to this work.
Received 2018-10-16; Accepted 2019-3-28; Published 2019-4-12
Abstract

Rationale: Antimicrobial peptides, such as cathelicidin LL-37/hCAP-18, are important effectors of the innate immune system with direct antibacterial activity. In addition, LL-37 is involved in the regulation of tumor cell growth. However, the molecular mechanisms underlying the functions of LL-37 in promoting lung cancer are not fully understood.
Methods: The expression of LL-37 in the tissues and sera of patients with non-small cell lung cancer was determined through immunohistological, immunofluorescence analysis, and enzyme-linked immunosorbent assay. The animal model of wild-type and Cramp knockout mice was employed to evaluate the tumorigenic effect of LL-37 in non-small cell lung cancer. The mechanism of LL-37 involving in the promotion of lung tumor growth was evaluated via microarray analyses, recombinant protein treatment approaches in vitro, tumor immunohistochemical assays, and intervention studies in vivo.
Results: LL-37 produced by myeloid cells was frequently upregulated in primary human lung cancer tissues. Moreover, its expression level correlated with poor clinical outcome. LL-37 activated Wnt/β-catenin signaling by inducing the phosphorylation of protein kinase B and subsequent phosphorylation of glycogen synthase kinase 3β mediated by the toll-like receptor-4 expressed in lung tumor cells. LL-37 treatment of tumor cells also decreased the levels of Axin2. In contrast, it elevated those of an RNA-binding protein (tristetraprolin), which may be involved in the mechanism through which LL-37 induces activation of Wnt/β-catenin.
Conclusion: LL-37 may be a critical molecular link between tumor-supportive immune cells and tumors, facilitating the progression of lung cancer.
Keywords: LL-37, lung cancer, tumor microenvironment, Wnt/β-catenin
Introduction
Solid tumors comprise malignant cells, as well as numerous non-malignant types of cells (e.g., immune cells). Understanding the interactions between tumor cells and immune cells is pivotal in designing and developing novel immune therapeutics against cancer [1]. The tumor microenvironment (TME) contains many resident cell types (e.g., adipocytes and fibroblasts), and is also populated by migratory myeloid cells (e.g., macrophages, neutrophils, and mast cells) [2]. Emerging evidence indicates that myeloid cells affect the progression of cancer by directly interacting with tumor cells. Moreover, they indirectly affect disease progression by enabling a tumor stroma that promotes cancer growth [3, 4]. In a previous study using a murine lung cancer model, we provided evidence that LL-37 secreted by macrophages promotes lung tumor growth through further recruitment of inflammatory cells [5]. Cathelicidin represents an essential component of innate immunity. Together with defensins and other antimicrobial peptides (AMPs), it provides the first line of defense against a variety of pathogens [6, 7]. The human LL-37 AMP gene, CAMP, codes for the protein hCAP-18 and is the only known cathelicidin isolated from human cells [8, 9] consisting of three domains: a signal peptide, a cathelin domain, and an LL-37 peptide. Cathelicidin possesses antibacterial, antifungal, and antiviral functions [10, 11], as well as chemotactic and immunostimulatory/modulatory effects [12]. Furthermore, increasing evidence suggests that LL-37 promotes the proliferation of cancer. Under normal conditions, LL-37 plays a role in tumor growth [13]. However, since LL-37 is involved in proliferation, migration, and angiogenesis [14], uncontrolled expression of LL-37 may also lead to dysregulation of these processes and promotion of cancer [15]. In lung cancers, upregulation of LL-37 expression led to tumor promotion instead of tumor surveillance, rendering LL-37 a growth factor for this type of tumor [16]. Consistent with this, we found that knockdown of LL-37 expression decreased tumor proliferation and recruitment of inflammatory cells in a murine lung cancer model [5]. Other studies suggested that LL-37 may activate epidermal growth factor receptor and the downstream Ras and mitogen-activated protein kinase pathways, resulting in proliferation, angiogenesis, suppression of apoptosis, invasion, and metastasis [16, 17]. Monocytes and macrophages are an important source of LL-37. We have previously shown that versican V1 derived from tumor cells enhances the expression of hCAP18/LL-37 in macrophages through activation of the toll-like receptor-2 (TLR2) and subsequent vitamin D-dependent mechanisms to promote the progression of ovarian cancer [18]. In prostate tumors, overexpressed LL-37 initially chemo-attracts immature myeloid progenitors to the TME and mediates differentiation and polarization of early myeloid progenitors into pro-tumorigenic type2 (M2) macrophages to drive the progression of prostate cancer [19]. In addition, LL-37 may (re)direct macrophage differentiation toward macrophages with a proinflammatory profile [20]. However, the molecular mechanisms underlying the functions of LL-37 in promoting lung cancer are not fully understood.
Aberrant activation of the Wnt signaling pathway is a major trait of multiple types of tumors, including lung cancer. In the absence of a Wnt signal (off-state), the APC/Axin/Gsk3β complex phosphorylates β-catenin for ubiquitin-mediated degradation [21]. In the presence of Wnt stimulation (on-state), Wnt ligands bind to their membrane receptors, resulting in dissociation of β-catenin from the 'destruction complex'. Consequently, stabilized cytosolic β-catenin translocates to the cell nucleus to form the β-catenin-LEF/TCF complex. This process induces the transcription of various downstream genes implicated in tumorigenesis [22]. Genetic mutations in the core components of Wnt/β-catenin signaling lead to the initiation of intestinal tumorigenesis [23]. However, according to previous studies, β-catenin gene mutations are uncommon in non-small cell lung cancer (NSCLC) [24, 25]. Therefore, it is likely that other mechanisms contribute to the overactivation of Wnt signaling in NSCLC.
Materials and Methods
Animals and cells
Cramp knockout (Cramp-/-) mice previously described [5] were kindly provided by Prof. Richard L. Gallo (University of California, San Diego, CA, USA). All mice were in C57BL/6 background. All experiments involving animals were approved by the Tongji Hospital of Tongji University Ethics Committee on the Use and Care of Animals, Shanghai, China. The human lung adenocarcinoma cell line A549 and mouse LLC cells were obtained from the American Type Culture Collection, and cultured in Dulbecco's modified Eagle's medium (DMEM) (Invitrogen, Carlsbad, USA), supplemented with 10% fetal calf serum (Invitrogen, Carlsbad, USA), 100 U/ml penicillin, and 100 U/ml streptomycin (PAA Laboratories GmbH).
Human lung samples
Human tissue specimens were retrospectively obtained from lung biopsies or lung resections, fixed in buffered formalin, and embedded in paraffin using standard methods. Corresponding clinical data were obtained from medical records and de-identified. The study was approved by the institutional review board and research and development committees of the Tongji Hospital of Tongji University, Shanghai, China.
Histologic and immunohistochemical analyses
Immunohistochemistry was performed as previously described [5]. The following primary antibodies were used for immunohistochemical analysis: mouse anti-Ki-67 (Abcam, Cambridge, USA), rabbit anti-CD68 (Abbiotec, San Diego, CA, USA), mouse anti-CD68 (Dako, Glostrup, Denmark), anti-CD66b rabbit (Cell Signaling Technology, Danvers, USA), rabbit anti-hCAP18/LL-37 (Santa Cruz, CA, USA), rabbit anti-TLR2 (Santa Cruz, CA, USA), rabbit anti-TLR4 (Santa Cruz, CA, USA), and mouse anti-unphosphorylated β-catenin (Millipore, Billerica, USA). Secondary antibody incubation and staining were performed using the EnVision®+ System-HRP (DAB) kit (Dako, Glostrup, Denmark), according to instructions provided by the manufacturer. TUNEL staining was performed using the DeadEnd Colorimetric TUNEL System kit (Promega, Madison, USA).
ELISA assay for LL-37
Samples of serum obtained from patients or mice were analyzed using hCAP18/LL-37 or CRAMP ELISA (USCN life science, Houston, TX, USA), respectively, according to the instructions provided by the manufacturer.
Preparation of cell total protein extract and western blotting
Initially, 10 mg of microdissected tumor tissue were homogenized in 500 ml of cell lysis buffer (Cell Signaling Technology, Danvers, USA) using a rotor-stator homogenizer. Western blotting analysis was performed as previously described [5]. Briefly, 30 mg of total protein extract were loaded on 10% SDS polyacrylamide gels, subjected to electrophoresis, and blotted onto Hybond-C Extra membranes. The cell supernatants were initially concentrated to 1/10 of their original volume through vacuum centrifugation, and subsequently subjected to electrophoresis in 4-12% NU/PAGE gradient gels (Invitrogen, Carlsbad, USA). The primary antibodies used for the western blotting analysis were as follows: mouse anti-cyclin D1 (Cell Signaling Technology, Danvers, USA), rabbit anti-c-myc (Cell Signaling Technology, Danvers, USA), rabbit anti-total β-catenin (Cell Signaling Technology, Danvers, USA), rabbit anti-Gsk3β (pSer9) (Cell Signaling Technology, Danvers, USA), rabbit anti-pan Gsk3β (Cell Signaling Technology, Danvers, USA), rabbit anti-protein kinase B (Akt) (pSer473) (Cell Signaling Technology, Danvers, USA), rabbit anti-pan Akt (Cell Signaling Technology, Danvers, USA), mouse anti-unphosphorylated β-catenin (Millipore, Billerica, USA), and mouse anti-β-actin (Sigma, St. Louis, USA). Notably, HRP-conjugated goat anti-rabbit (Santa Cruz, CA, USA) or rabbit anti-mouse (Dako, Glostrup, Denmark) was used as the secondary antibody.
Gene set enrichment analysis (GSEA)
A GSEA for LL-37 was performed using Pearson correlation coefficient with 1,000 permutations against the kegg pathway geneset compilation. The GSEA was performed using the GSEA 2.0.9 software (http://www.broadinstitute.org/gsea/).
RNA isolation and real-time polymerase chain reaction (PCR)
Total RNA was isolated from microdissected tumors and noncancerous lung tissues or cells, according to the instructions provided by the manufacturer, using an RNeasy plus mini kit (Qiagen, Duesseldorf, Germany). Real-time PCR reaction mixtures have been previously described [5]. Briefly, cDNA was synthesized through reverse transcription reaction using the First Strand cDNA synthesis kit (Invitrogen, Carlsbad, USA). Real-time PCR was performed using the qPCR SYBR Green Mix (Bio-Rad, Hercules, USA) on an AB 7300 Real time PCR system. The sequences of primers used in this analysis are listed in Supplementary Table 1. The specificity of RT-PCR was controlled using 'no reverse transcription' controls and melting curve analysis. Quantitative PCR results were obtained using the ΔΔCT (cycle threshold) method. Data were normalized to the levels of β-actin in each sample.
TOPflash reporter assay
The TOPflash/FOPflash luciferase reporter system (Genomeditech, Shanghai, China) measured β‐catenin-driven TCF/LEF transcriptional activation. A549 cells were transfected with the TOPflash construct at a concentration of 1 μg/ml per well in a 12-well plate using the Lipofectamine 2000 transfection reagent (Life Technologies, Gaithersburg, USA). For the normalization of transfection efficiency, cells were co-transfected with a pCMV-Renilla luciferase vector (Genomeditech, Shanghai, China) at a concentration of 100 ng/ml. Transfection complexes were removed after 2 h, and cells were stimulated with LL-37 in DMEM supplemented with 0.5% fetal calf serum for 24 h. Cell lysates were prepared, and luciferase activity was evaluated using a dual luciferase reporter system in accordance with the protocol provided by the manufacturer (Promega, Madison, USA). The activity of firefly luciferase was normalized to that of Renilla luciferase and expressed as the relative fold change.
Isolation of lung macrophages and neutrophil
Mouse macrophages and neutrophils were purified from individual lung tissue samples. Whole lung tissues were minced into a fine slurry and digested using 1 mg/mL collagenase A (Roche Applied Science, Basel, Switzerland) in DMEM media containing 10% fetal calf serum and 2 units/mL of DNAse (Roche Applied Science, Basel, Switzerland) for 1 h at 37℃ with constant shaking (85 rpm). The resultant digest was passed ×10 through an 18-gauge needle and filtered using a 40-µm cell strainer. The single-cell suspension of lung tissues was plated, and macrophages were isolated via plastic adherence for 1 h at 37℃. The extraction of neutrophils from free floating cells was performed using the Mouse neutrophil Isolation Kit (Solarbio, Beijing, China).
Statistical analysis
The data are presented as means ± standard deviation or ± standard error of the mean as indicated. Differences in means were analyzed using Student's t-test and one-way analysis of variance. Kaplan-Meier survival curves were analyzed using the log-rank test. A P<0.05 denoted statistical significance.
Results
LL-37 is overexpressed in the human lung cancer stroma, and its level in the serum correlates with poor prognosis
We compared the expression levels of LL-37 in 45 adjacent normal lung tissues and 86 NSCLC tissues (i.e., 47 adenocarcinomas and 39 squamous cell carcinomas) through immunohistochemistry. We detected significantly higher levels of LL-37 in lung cancer tissues versus those observed in adjacent normal lung tissues (Figure 1A). Additionally, contrary to previous reports for other carcinomas [26, 27], LL-37 was mostly restricted to tumor-infiltrating immune cells (i.e., macrophages and neutrophils) with limited or no expression in tumor cells per se. Immunohistological and immunofluorescence staining experiments using macrophage and neutrophil markers CD68 and CD66b, respectively, demonstrated that these types of cells were enriched in tumor tissues compared with those reported in adjacent normal lung tissue controls, and were the predominant site of LL-37 expression (Figure 1 A and B). Subsequently, we inoculated wild-type (WT) C57BL/6 mice with Lewis lung carcinoma (LLC) cells to further determine the expression of LL-37 in lung tumors. Macrophages and neutrophils were isolated from lung tissues of tumor-bearing mice and controls (Figure S1A). We observed an increased secretion of LL-37 by macrophages and neutrophils in tumor-bearing mice compared with that measured in control mice (Figure S1B). Similarly, induction of the LL-37 gene was observed in macrophages and neutrophils obtained from tumor-bearing mice (Figure S1C). These results were consistent with those obtained through immunohistochemical analysis of tumor tissues obtained from patients diagnosed with NSCLC.
LL-37 is overexpressed in the human lung cancer stroma, and its level in the serum correlates with poor prognosis. (A) Immunohistological analysis of the expression of LL-37, CD68 (macrophage marker), and CD66b (neutrophil marker) in normal lung tissues (n = 35) and NSCLC tissues (adenocarcinoma [n = 47] and squamous cell carcinoma [n = 39]). Scale bar, 100 μm. (B) Immunofluorescence analysis of the expression of LL-37, CD68, and CD66b in NSCLC tissues. Scale bar, 50 μm. (C) The levels of LL-37 in the sera of healthy donors and patients diagnosed with NSCLC. (D) The levels of LL-37 in the sera of patients with lung cancer prior to and after surgery. (E) Kaplan-Meier analysis of the 5-year RFS (left panel) and OS (right panel) of patients with NSCLC. Patients were stratified according to their expression of LL-37 as follows: high (i.e., greater than the median, n = 69) versus low (greater than or equal to the median, n = 70).
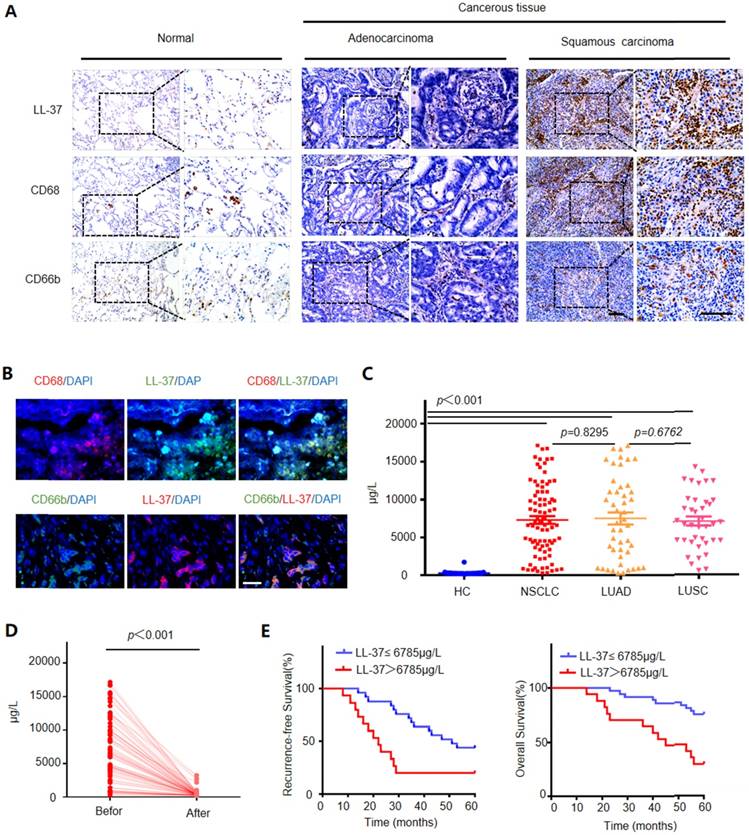
We subsequently investigated the levels of LL-37 in the sera of healthy donors and patients diagnosed with NSCLC. The concentration of LL-37 in the sera of healthy controls was 275.6±30.6 μg/L. However, the level of LL-37 in the serum of NSCLC patients was significantly increased (7,312±532.3 μg/L). There were no significant differences in the levels of LL-37 between patients with adenocarcinoma and those with squamous cell carcinoma (7,498±799.5 μg/L vs. 7,065±597.5 μg/L, respectively) (Figure 1C). We subsequently investigated whether the higher level of LL-37 required the presence of the tumors, by measuring the levels of LL-37 in the serum prior to and after surgery. Indeed, 1 month after surgery the levels of LL-37 in the serum were significantly decreased (Figure 1D), suggesting that this peptide may be associated with the presence of a tumor.
Based on these results, we further investigated the prognostic role of the levels of LL-37 in peripheral blood. A total of 86 patients with lung cancer, who had received curative resection, were divided into the following two groups: 1) those with LL-37 levels above the median level (6,785 μg/L) in peripheral serum at the time of diagnosis; and 2) those with LL-37 levels below the median level. As shown in Figure 1E, patients in the former group (i.e., LL-37 levels >6,785 μg/L) exhibited worse recurrence-free survival and overall survival compared with patients in the latter group (i.e., LL-37 levels ≤6,785 μg/L). This result suggested that elevated levels of LL-37 in the blood are negatively correlated with survival in patients with lung cancer.
Knockdown of LL-37 in non-tumor cells inhibits metastatic growth of lung cancer in vivo
The levels of LL-37 were consistently higher in NSCLC. Therefore, we examined the impact of LL-37 deficiency on the development of lung tumors and its role as a tumor promoter. In mice, the gene Cnlp is homologous to the human gene CAMP, coding for the LL-37-related antimicrobial peptide (Cramp) [28]. Therefore, we injected WT and Cramp-/- mice with 1×105 LLC cells via tail vein injection. After inoculation, Cramp-/- mice exhibited markedly reduced mortality versus WT mice (Figure 2a) and decreased tumor multiplicity (Figure 2B and C), further demonstrating the significant role of LL-37 in the growth of lung tumors.
Knockdown of LL-37 in non-tumorous cells inhibits metastatic growth of lung cancer in vivo. (A) Kaplan-Meier survival analysis of WT and Cramp-/- mice injected with 1×105 LLC cells via the tail vein (P<0.001, log-rank test for statistical analysis; (B) Lungs of WT and Cramp-/- mice 21 days after inoculation with 1×105 LLC cells. Lung appearance and histology (n = 5 for each group) (H&E stain; lower panel). Scale bar, 200 μm. (C) Number of tumor nodules detectable on the surface of the lung (n = 5 for each group). (D) Immunohistochemical analysis of the expression of PCNA, Ki-67, and cyclin D1 in WT and Cramp-/- mice injected with 1×105 LLC cells via the tail vein. Scale bar, 50 μm (n = 5 for each group). (E) Immunoblotting analysis in isolated tumors from WT and Cramp-/- mice injected with 1×105 LLC cells via the tail vein. (F) Apoptotic cells were identified using TUNEL staining. Scale bar, 100 μm (n = 5 for each group).
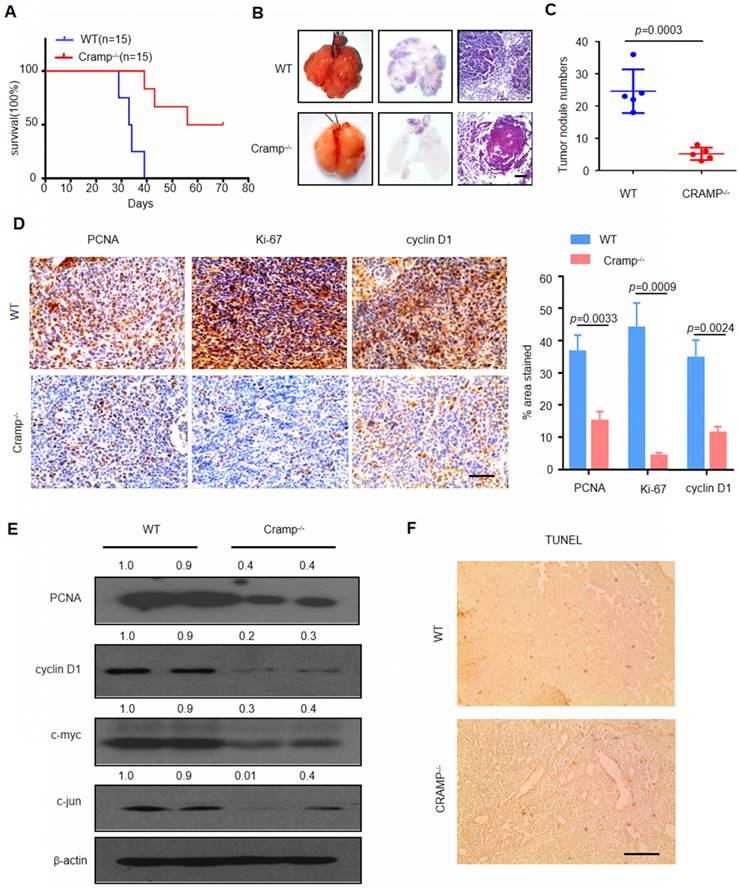
Subsequently, we investigated whether the LL-37-induced changes in lung tumor burden were due to altered cell proliferation or apoptosis. We analyzed the proliferation of tumor cells in WT and Cramp-/- mice (Figure 2D). Immunohistochemical analysis revealed a marked decrease in key cell cycle proteins and proliferation markers, such as proliferating cell nuclear antigen (PCNA), Ki-67, and cyclin D1, in tumor cells injected in Cramp-/- mice. These results were further confirmed through western blotting analysis and extended to c-jun and c-myc (Figure 2E). In contrast, we did not observe a significant difference in the rate of cellular apoptosis between WT and Cramp-/- mice (Figure 2F). Therefore, LL-37 plays an important role in tumor growth by affecting the expression of proliferative genes.
LL-37 is required for activation of Wnt/β-catenin signaling
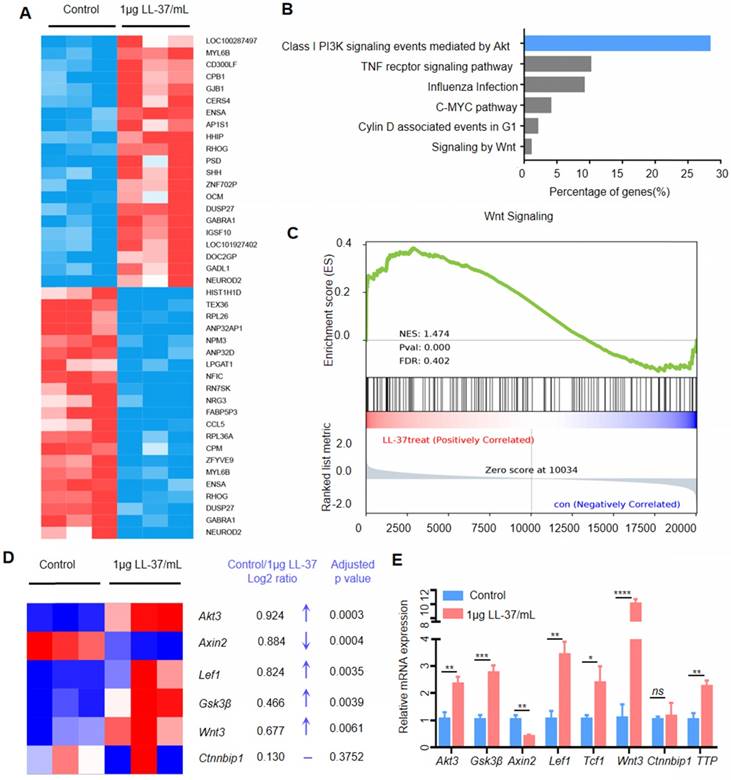
We performed RNA sequencing of A549 cells treated with LL-37 to identify differentially expressed genes (Figure 3A) and signaling pathways influenced by LL-37. An ingenuity pathway analysis revealed that the Class I PI3K signaling event mediated by Akt was the most upregulated pathway in LL-37-treated A549 cells (Figure 3B). Activation of the Akt pathway is a common feature in human cancers, leading to increased cell survival, growth, and proliferation [29]. Activated Akt phosphorylates numerous downstream targets, including forkhead box O3 and proline-rich Akt substrate of 40 kDa [30, 31]. Moreover, Akt phosphorylates and inactivates the glycogen synthase kinase Gsk3β at Serine 9. Gsk3β functions as a negative regulator of β-catenin stabilization. Therefore, Akt phosphorylation of Gsk3β may lead to stabilization of β-catenin and accumulation of nuclear β-catenin [32]. Numerous studies suggested that Wnt signaling cross-talks with the PI3K/Akt pathway [33, 34]. In this respect, the GSEA of RNA-sequencing data indicated that LL-37 also increased the expression of Wnt-responsive genes (Figure 3C). In agreement with the results of the GSEA, a heatmap depiction showed that treatment with LL-37 increased the expression of Wnt-responsive genes, including Akt3, LEF1, Gsk3β, and Wnt3. However, it decreased the expression of Axin2 (Figure 3D). Quantitative reverse-transcription-PCR (qRT-PCR) validation of RNA-sequencing in A549 cells treated with LL-37 for 4 h showed similar results (Figure 3E), suggesting that LL-37 is involved in the activation of Akt-Wnt-β-catenin signaling in NSCLC.
LL-37 is required for the activation of Wnt/β-catenin signaling. (A) Heatmap of differentially expressed genes between A549 and LL-37-treated A549 cells (2logFC cutoff value < -0.5 or > 0.5, P<0.05, FDR < 5%). (B) Ingenuity pathway analysis of genes differentially represented in RNA-seq data from LL-37-treated A549 cells (n = 3). Top canonical pathways are ranked according to the percentage of genes (%). (C) GSEA plot showing that LL-37-treated A549 cells were positively correlated with Wnt-activated gene signatures and inversely correlated with Wnt-activated gene signatures in the WNT_SIGNALING dataset (n = 3). (D) Heatmap representation of upregulated genes in LL-37 treated A549 cells relative to A549 cells (n = 3) associated with 'Wnt-responsive genes, including Log2FC and an adjusted P value. (E) RT-qPCR analysis of the expression levels of Akt3, Gsk3β, Axin2, LEF1, TCF1, Wnt3, Ctnnbip1 and TTP, in LL-37-treated A549 cells versus control cells. A two-tailed Student's t-test was used for statistical analysis (*P<0.05, **P<0.01, ***P<0.001, ****P<0.001).
We analyzed the phosphorylation of β-catenin in lung tumor cells of WT mice and Cramp-/- mice after inoculation with LLC cells to examine whether LL-37 is required for the activation of Wnt/β-catenin in LLC tumors. We found strong staining of unphosphorylated β-catenin in the tumor cells of WT mice. In contrast, decreased staining was noted in Cramp-/- mice (Figure 4A). Downregulation of the β-catenin pathway was further confirmed via immunoblotting analysis, showing lower levels of unphosphorylated β-catenin in Cramp-/- mice tumors. This finding indicated a potential role of the β-catenin pathway during the development of lung tumors (Figure 4B).
LL-37 activates Wnt/β-catenin signaling to promote growth of lung tumors. (A) Immunohistochemical analysis of the expression of unphosphorylated β-catenin in WT and Cramp-/- mice injected with 1×105 LLC cells via the tail vein. Scale bar, 100 μm. (B) Immunoblotting analysis in isolated tumors from WT and Cramp-/- mice injected with 1×105 LLC cells via the tail vein. (C) Immunohistochemical analysis of the expression of unphosphorylated β-catenin and LL-37 in serial sections of NSCLC tumor specimens. Scale bar, 100 μm. (D) Immunofluorescence staining shows the subcellular localization of unphosphorylated β-catenin and actin (phalloidin) in A549 cells. Scale bar, 20 μm. (E) Western blotting analysis of unphosphorylated β-catenin, total β-catenin, phosphorylated Gsk3β, pan Gsk3β, phosphorylated Akt, pan Akt, Cyclin D1, c-myc, TTP, and β-actin in A549 cells after stimulation with LL-37 for indicated time periods.
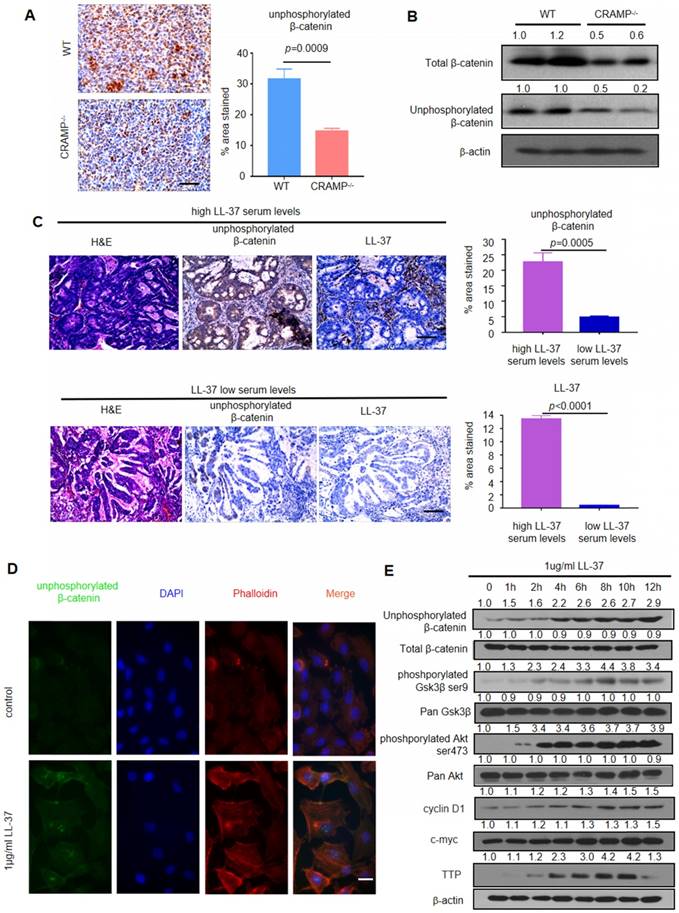
We further examined the expression of unphosphorylated β-catenin through immunohistochemical staining on two sets of human NSCLC tissue arrays containing high (>6,785 μg/L) (n=60) and low (≤6,785 μg/L) (n=19) levels of LL-37 in the serum. We found that the former group exhibited significantly higher levels of unphosphorylated β-catenin versus the latter group (Figure 4C). These data indicated that the role of LL-37 is conserved between humans and mice, and that Wnt/β-catenin signaling plays a critical role in the LL-37-induced proliferation of lung cancer cells in vivo.
LL-37 activates Wnt/β-catenin signaling to promote growth of lung tumors
We analyzed the β-catenin pathway of LL-37-treated lung cancer cell line A549 to further investigate the molecular mechanisms mediating the effects of LL-37 on tumor cell physiology and development. Treatment with LL-37 caused accumulation of unphosphorylated β-catenin in the nuclei of A549 cells versus untreated cells, exhibiting diffuse localization with a small fraction of unphosphorylated β-catenin in the nuclei (Figure 4D). A dual-luciferase reporter assay was employed to determine whether the canonical Wnt signaling pathway was regulated by LL-37. We found that the activity of the TCF/LEF-reporter was significantly increased in A549 cells treated with LL-37 for 24 h. (Figure S2). It is established that the GSK3β and Akt proteins regulate Wnt/β-catenin signaling [35]. The levels of GSK3β phosphorylation on Ser9 and Akt phosphorylation on Ser473 were significantly increased in A549 cells treated with LL-37 for 4 h. The accumulation of unphosphorylated β-catenin was associated with the phosphorylation of GSK3β and Akt (Figure 4E). Tristetraprolin (TTP) downmodulates the induction of Axin2 by increasing the rate of mRNA decay [36]. In the present study, we found that LL-37 significantly stimulated the expression of TTP protein in A549 cells observed for 12 h (Figure 4E).
TLRs - required for innate immune responses - plays a significant role in inflammation, regulation of immune cells, survival, and proliferation [37, 38]. In addition, TLRs have been associated with the activation of Wnt/β-catenin signaling [39-42]. We assayed the expression of TLR2 and TLR4 - closely related to lung cancer growth - to examine the contribution of TLRs to LL-37-induced activation of Wnt/β-catenin signaling [43]. We performed an analysis of patients with adenocarcinoma and squamous cell carcinoma using data available in The Cancer Genome Atlas database. This analysis did not identify a significant correlation between the expression of LL-37 and that of TLR2 or TLR4 (Figure S3A and B). In addition, immunoblotting and qRT-PCR did not reveal statistically significant differences in the expression of TLR2 or TLR4 following incubation of A549 cells with LL-37 (Figure S3C and D). Furthermore, there was no LL-37-induced increase in the expression of TLR2 or TLR4 in lungs obtained from WT and Cramp-/- mice (Figure S4 and S5). Furthermore, A549 cells were treated with the TLR2 or TLR4 inhibitor to further investigate whether TLRs are required for the activation of Wnt/β-catenin in tumor cells. The results showed that the LL-37-induced accumulation of unphosphorylated β-catenin was dependent on TLR4, but not on TLR2 (Figure S6A and C). Moreover, TLR4 was required for optimal LL-37-induced phosphorylation of Akt and Gsk3β, as well as expression of LEF1 and TCF1 (Figure S6A and Figure B). Thus, the LL-37 induced activation of Wnt/β-catenin in lung cancer cells requires functional TLR4.
The β-catenin pathway plays a critical role in the stimulation of lung tumor growth by LL-37
We treated mice with LL-37 (5 μg or 10 μg) or phosphate-buffered saline every 2 days after 1-week of inoculation with 2×105 LLC cells, to test whether LL-37 is continuously required for the stimulation of lung tumor growth. Tumors were analyzed 21 days after injection of LLC cells (Figure 5). We found that continuous treatment with LL-37 resulted in a significant increase in tumor size and multiplicity (Figure 5A and B). Immunoblotting analysis of tumor lysates obtained from LL-37-treated mice revealed the upregulated expression of gene products targeted by β-catenin, including c-Myc, cyclin D1, and c-jun. These results indicated that LL-37 may affect the formation and growth of tumors (Figure 5C). In addition, treatment with LL-37 enhanced the expression of unphosphorylated β-catenin in lung tumors and importantly, elevated the levels of LL-37 in the serum (Figure 5D). This observation was consistent with previous results based on human serum.
The β-catenin pathway plays a critical role in the stimulation of lung tumor growth by LL-37. C57BL/6 mice were inoculated with 2×105 LLC cells via the tail vein. One week after inoculation, C57BL/6 mice were treated with XAV939 (200 μg) or LL-37 (5 μg or 10 μg) every 2 days for 3 weeks. (A) Macroscopic and microscopic pathology (hematoxylin and eosin staining) of lungs in mice with indicated treatments (n = 5 for each group). (B) Number of tumor nodules detectable on the surface of the lung with indicated treatments at the experimental endpoint (n = 5 for each group). (C) Immunoblotting analysis of PCNA, cyclin D1, c-myc, c-jun, and β-actin in tumor lysates from C57 BL/6 mice inoculated with 2×105 LLC cells with indicated treatments. (D) The levels of LL-37 in the sera of mice with indicated treatments.
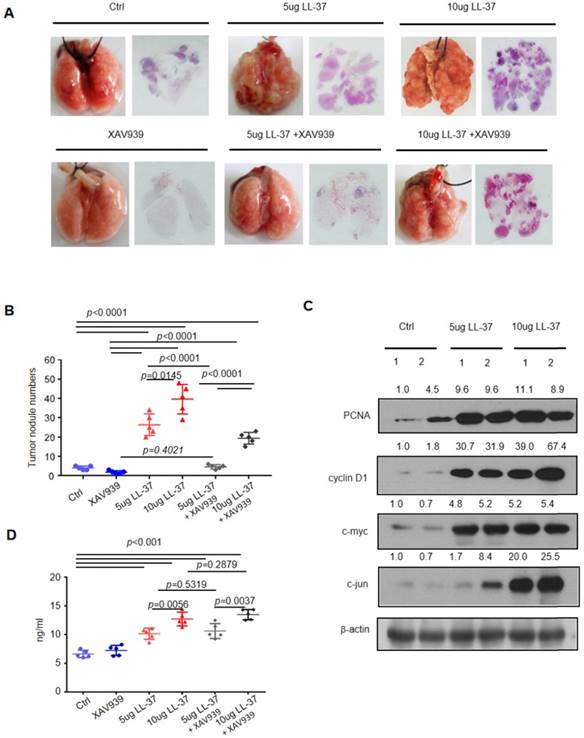
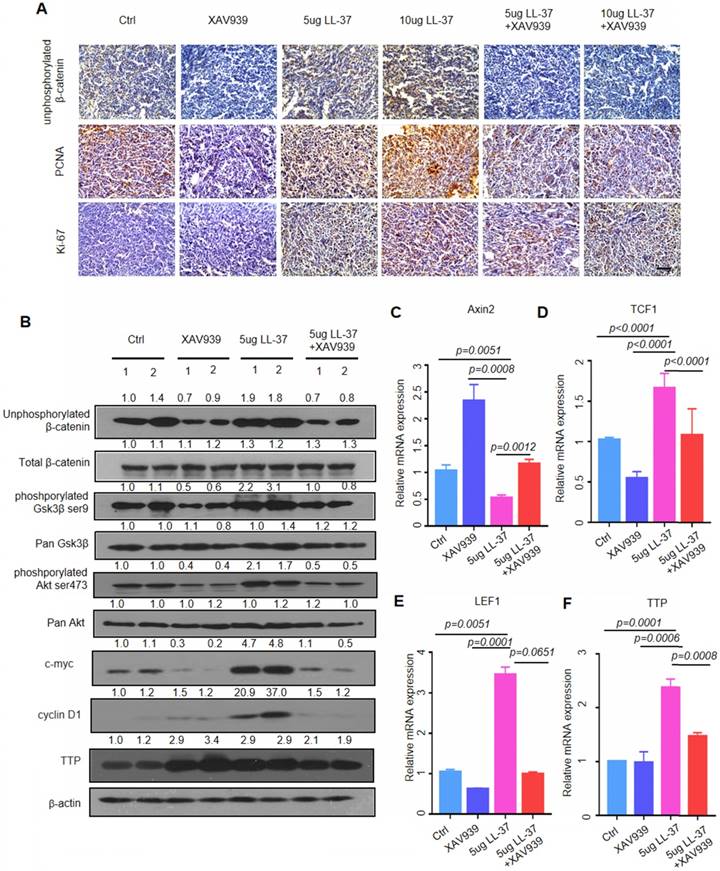
In addition, we tested whether inhibition of β-catenin may block the action of LL-37, to determine the importance of β-catenin in the observed effects of LL-37 in vivo. XAV939 - a tankyrase inhibitor - selectively inhibits β-catenin-mediated transcription by stabilizing Axin, a component of the β-catenin destruction complex [44]. Combined treatment with XAV939 (200 μg) and LL-37 (5 μg or 10 μg) in the LLC tumor model in C57BL/6 mice significantly reduced the number of lung tumor nodules compared with LL-37 monotherapy (Figure 5A and B). Moreover, we analyzed cell proliferation in tumors treated with XAV939 or combined XAV939/LL-37. The combined treatment resulted in a significant decrease in staining with PCNA-specific and Ki-67-specific antibodies versus LL-37 monotherapy (Figure 6A). Immunoblotting analysis of tumor lysates revealed that the accumulation of unphosphorylated β-catenin was associated with the phosphorylation of Gsk3β and Akt (Figure 6B). Notably, the expression of TTP and cell cycle-related proteins were also increased in LL-37-treated mice (Figure 6B). The qRT-PCR analysis indicated that treatment with XAV939 resulted in the upregulation of Axin2 mRNA in tumor cells. However, treatment with LL-37 suppressed the expression of Axin2 (Figure 6C). The TCF/LEF family of transcription factors are the major mediators of Wnt signaling and their expression is upregulated by stabilized β-catenin [45]. As shown in Figure 6D and E, the expression of TCF1 and LEF1 was significantly increased in tumor lysates of LL-37-treated mice. In contrast, this effect was blocked by treatment with XAV939. We also found that LL-37 significantly stimulated the expression of TTP mRNA in vivo (Figure 6F). The results of the animal model were consistent with those of the in vitro cell model, providing mechanistic insight and suggesting that LL-37 mediates growth of lung cancer via activation of Wnt/β-catenin.
LL-37 is required for the activation of Wnt/β-catenin signaling. (A) Immunohistochemical analysis of the expression of unphosphorylated β-catenin, PCNA, and Ki67 in C57BL/6 mice inoculated with 2×105 LLC cells with indicated treatments. Scale bar, 50 μm. (B) Immunoblotting analysis of unphosphorylated β-catenin, total β-catenin, phosphorylated Gsk3β, pan Gsk3β, phosphorylated Akt, pan Akt, cyclin D1, c-myc, TTP, and β-actin in tumor lysates from C57BL/6 mice inoculated with 2×105 LLC cells with indicated treatments. (C)(D)(E)(F) qRT-PCR analysis of the relative mRNA levels of Axin2 (C), LEF1 (D), TCF1 (E), and TTP (F) in tumor lysates from C57BL/6 mice inoculated with 2×105 LLC cells with indicated treatments.
Discussion
The main finding of the present study is that LL-37 expressed by myeloid and non-tumorous cells is an important regulator of the development and growth of lung tumors. We observed a marked increase in the expression of LL-37 in cancerous lung tissues obtained from NSCLC patients. Macrophages and neutrophils infiltrated the tumor area and secreted LL-37. Importantly, the concentration of LL-37 in the serum correlated with worse recurrence-free survival and overall survival in NSCLC patients. The peptide exerted a direct tumor-promoting effect by activating Wnt/β-catenin signaling. This conclusion was supported by our observations that the levels of LL-37 in the serum were negatively associated with the survival of NSCLC patients. Our data suggest that LL-37 may be a potential prognostic biomarker and serve as a therapeutic target.
Previous studies performed by our research group and other investigators have suggested that the expression of LL-37 is increased in a variety of cancers, including ovarian [18], lung, colon [46], breast [47], and melanoma [48]. However, gastric cancers produce lower amounts of LL-37, which inhibit tumor growth in this type of cancer. This evidence indicates that the actions of LL-37 are tissue-specific [49]. Previous studies reported low expression of LL-37 in colon cancer, with possible antitumor activity. However, our previous research showed that LL-37 was strongly expressed and secreted by infiltrating macrophages in tumor stroma, significantly contributing to the progression of colon cancer [50].
LL-37-deficient mice developed fewer and smaller tumors versus WT mice. Besides its importance during early tumor promotion, LL-37 affects tumor growth during all stages of lung cancer. LL-37 modulates cellular immune responses by increasing the production of chemokines, particularly interleukin 8 (IL-8) [51]. LL-37 directly induces the transcription of IL-8 and regulates the inflammatory process in various inflammatory lung diseases [52]. IL-8 is a potent attractant of myeloid cells, facilitating the recruitment and survival of macrophages and neutrophils capable of producing proinflammatory factors [53]. These immune cells regulate tumor growth by producing cytokines and chemokines that act in both the autocrine and paracrine manner. In the present study, we found that fewer myeloid cells (macrophages, neutrophils) were recruited in LL-37-deficient tumors. This finding was consistent with the role of LL-37 in the migration of leukocytes.
We previously reported that inhibition of nuclear factor-κB (NF-κB) activation in myeloid cells, especially the lack of RelA/p65 in the myeloid lineage, inhibited the proliferation of lung tumor cells in a mouse model of lung cancer and resulted in decreased expression of LL-37 [54]. This observation indicated that the expression of this peptide is strongly correlated with NF-κB signaling. However, ablation of RelA/p65 in myeloid cells - affecting the induction of LL-37 - does not significantly alter the recruitment of myeloid cells [54]. Thus, in addition to controlling the expression of LL-37, NF-κB may also be involved in the production of chemotactic factors. Sequestosome 1 (SQSTM1/p62), an autophagic adaptor protein whose expression is dependent on NF-κB, is one such candidate. NF-κB exerts its anti-inflammatory activity by inducing delayed accumulation of the autophagy receptor p62/SQSTM1, suppressing the production of TNF-α and IL-1β. TNF-α and IL-1β induce the production of chemokines and expression of chemokine-receptors in tumor and stromal cells [55]. By binding to diverse receptors, LL-37 activates four signaling pathways (i.e., purinergic receptor P2X7 [56], epidermal growth factor receptor [17], insulin-like growth factor-1 receptor [57], and Wnt/β-catenin) to increase growth of tumor cells. Our results suggest that, among these signaling pathways, β-catenin is a critical LL-37 effector in the growth of lung tumors. Unphosphorylated β-catenin is highly expressed in tumor cells of mice and patients with NSCLC.
Wnt/β-catenin signaling is activated in various types of cancer, including colon cancer [58] and hepatocellular carcinoma [59]. However, the mechanisms involved in its activation remain obscure. Wnt/β-catenin induces the expression of genes important for cell proliferation (i.e., cyclin D1 and c-myc [23, 60]) and migration (i.e., MMP-7 and CD44 [61, 62]). In the present study, we demonstrated that activation of Wnt/β-catenin signaling is an important contributor to the tumor-promoting activity of LL-37, leading to enhanced proliferation of lung tumor cells. Gsk3β is a major component, regulating the activation of Wnt/β-catenin signaling by constitutively phosphorylating β-catenin. This leads to its degradation, consequently blocking β-catenin-mediated gene transcription. Our current results showed that LL-37 is responsible for the phosphorylation of several Wnt/β-catenin signaling proteins, including β-catenin, Akt, and Gsk3β (Figure 7).
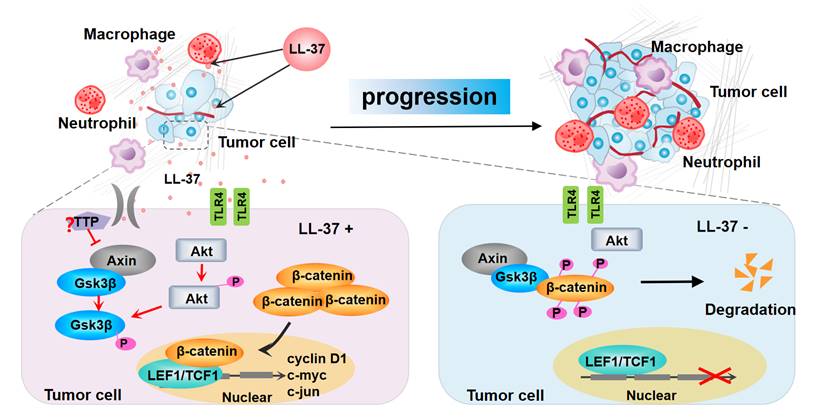
Model for the progression of LL-37-mediated lung cancer in NSCLC. LL-37 expressed by myeloid cells is an important regulator of the development and growth of lung tumors. Myeloid cell-secreted LL-37 directly targets TLR4, leading to the activation of Wnt/β-catenin signaling in NSCLC cells. LL-37 activates Wnt/β-catenin signaling through the induction of Akt and subsequent phosphorylation of Gsk3β, resulting in the stabilization and nuclear translocation of β-catenin. This permits the accumulation of β-catenin in the cytoplasm. Subsequently, β-catenin translocates to the nucleus to bind with TCF1 and LEF1, thereby initiating the transcription of multiple factors promoting cell growth. P, phosphorylation.
Previous research reported that LL-37 affects both the surface and intracellular expression of TLR. However, almost all research studies investigating LL-37 and TLRs were performed using immune cells. In mast cells, it has been shown that LL-37 may enhance the expression of TLR2, TLR4, and TLR9 on the surface, and TLR3, TLR5, and TLR7 in the interior of the cell [63]. Moreover, LL-37 binds to agonists of multiple TLRs in human monocytic cells and macrophages [64, 65], B lymphocytes and plasmacytoid dendritic cells [66], and DC cells [67]. However, it has not been determined whether binding of LL-37 to the TLR agonists may differentially affect signaling in cancer cells. Furthermore, although Wnt signaling activity has been linked to TLR signaling activity [68-70], the currently available evidence remains inconclusive. Further studies are warranted to investigate the link between TLR signaling and Wnt signaling in lung cancer cells. Additionally, there is no evidence linking LL-37 with TLR/Wnt signaling in tumor cells. Our results illustrated that, in lung tumor cells, the expression of TLR4 was not significantly altered following treatment with LL-37. However, the activity of Wnt/β-catenin and expression of related genes were greatly reduced by the TLR4 inhibitor TAK-242. This finding indicated that TLR4 mediates the activation of Wnt/β-catenin by LL-37 in tumor cells. The regulation of Wnt/β-catenin activity is complex and involves cross-talk with other factors. In a previous study, we demonstrated that LL-37 also activates the PI3K/Akt signaling pathway by binding to its specific receptor FPRL1 [71]. Moreover, we reported that the induction of Axin2 mRNA - a component of the β-catenin destruction complex inducing the phosphorylation and subsequent ubiquitin-mediated degradation of β-catenin - was significantly decreased by LL-37. It appears that TTP is a RNA-binding protein, which enhances mRNA decay by binding to AU-rich elements located in the 3'-untranslated region of the Axin2 transcript [72]. Our results showed that the expression of TTP was significantly enhanced by LL-37. This finding indicated that a possible mechanism through which LL-37 induces Axin2 mRNA decay may involve the production of TTP.
In summary, the present results establish a link between LL-37 and Wnt/β-catenin signaling in lung cancer. Moreover, they provide a rationale for the use of LL-37 blockers and Wnt/β-catenin inhibitors in the treatment and prevention of lung cancer.
Abbreviations
NSCLC: non-small cell lung cancer; Akt: protein kinase B; Gsk3β: glycogen synthase kinase 3β; TLR4: toll-like receptor 4; TLR2: toll-like receptor 2; TTP: tristetraprolin; TME: tumor microenvironment; AMPs: antimicrobial peptides; EGFR: epidermal growth factor receptor; MAPK: mitogen-activated protein kinase; LEF1: lymphoid enhancer factor 1; TCF1: transcription factor 1; LLC: Lewis lung carcinoma; PCNA: proliferating cell nuclear antigen; PI3K: phosphatidylinositol 3 kinase; GSEA: gene set enrichment analysis; qRT-PCR: Quantitative reverse transcription-polymerase chain reaction; RNA-Seq: RNA sequencing; TUNEL: terminal deoxynucleotidyl transferase dUTP nick end labeling; H&E: hematoxylin-eosin staining; NF-κB: nuclear factor-κB; SQSTM1/p62: Sequestosome 1; IGF-1R: insulin-like growth factor-1 receptor; MMP-7: Matrix metalloprotein 7.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This research was supported by the National Natural Science Foundation of China (81472179, 81873975), the Excellent Academic Leader Training Program of Shanghai Health System (2018BR31), Three-Year Action Plan for Promoting Construction of Public Health (2015-2017) (15GWZK0301), the Fundamental Research Funds for the Central Universities (22120170071), and the Clinical Research and Cultivation Project of Shanghai Tongji Hospital [ITJ(ZD)1803]. This research was also supported by the Innovation Group Project of Shanghai Municipal Health Commission (2019CXJQ03). We would like to thank Dr. Ming Guan for the helpful discussion.
Author contributions
D.L. and P.J. designed the study. Z.S and R.B. oversaw the study. Y. Z. provided clinical samples. P.J., Y.Z., and Y.Y. performed the majority of the experiments and analyzed the data. J.W. and H.Z. performed a number of the animal experiments. J.S., Y.W.Y., A.S., C.G., and B.Z. provided research assistance. J.F. reviewed and edited the article. W.X. takes responsibility for the accuracy of the data analysis. D.L., P.J., and Z.S wrote the article.
Competing Interests
The authors have declared that no competing interest exists.
References
1. De Henau O, Rausch M, Winkler D. et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kγ in myeloid cells. Nature. 2016;539:443-7
2. Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. 2013;14:1014-22
3. Engblom C, Pfirschke C, Pittet MJ. The role of myeloid cells in cancer therapies. Nat Rev Cancer. 2016;16:447-62
4. Croci DO, Zacarías Fluck MF, Rico MJ. et al. Dynamic cross-talk between tumor and immune cells in orchestrating the immunosuppressive network at the tumor microenvironment. Cancer Immunol Immunother. 2007;56:1687-700
5. Li D, Beisswenger C, Herr C. et al. Expression of the antimicrobial peptide cathelicidin in myeloid cells is required for lung tumor growth. Oncogene. 2014;33:2709-16
6. Lehrer RI, Ganz T. Cathelicidins: a family of endogenous antimicrobial peptides. Curr Opin Hematol. 2002;9:18-22
7. Agerberth B, Charo J, Werr J. et al. The human antimicrobial and chemotactic peptides LL-37 and alpha-defensins are expressed by specific lymphocyte and monocyte populations. Blood. 2000;96:3086-93
8. Bals R, Wilson JM. Cathelicidins-a family of multifunctional antimicrobial peptides. Cell Mol Life Sci. 2003;60:711-20
9. Larrick JW, Hirata M, Balint RF. et al. Human CAP18: a novel antimicrobial lipopolysaccharide-binding protein. Infect Immun. 1995;63:1291-7
10. Bals R, Wang X, Zasloff M. et al. The peptide antibiotic LL-37/hCAP-18 is expressed in epithelia of the human lung where it has broad antimicrobial activity at the airway surface. Proc Natl Acad Sci U S A. 1998;95:9541-6
11. Barlow PG, Svoboda P, Mackellar A. et al. Antiviral activity and increased host defense against influenza infection elicited by the human cathelicidin LL-37. PLoS One. 2011;6:e25333
12. Piktel E, Niemirowicz K, Wnorowska U. et al. The Role of Cathelicidin LL-37 in Cancer Development. Arch Immunol Ther Exp (Warsz). 2016;64:33-46
13. Kim JE, Kim HJ, Choi JM. et al. The antimicrobial peptide human cationic antimicrobial protein-18/cathelicidin LL-37 as a putative growth factor for malignant melanoma. Br J Dermatol. 2010;163:959-67
14. Koczulla R, von Degenfeld G, Kupatt C. et al. An angiogenic role for the human peptide antibiotic LL-37/hCAP-18. J Clin Invest. 2003;111:1665-72
15. Jia J, Zheng Y, Wang W. et al. Antimicrobial peptide LL-37 promotes YB-1 expression, and the viability, migration and invasion of malignant melanoma cells. Mol Med Rep. 2017;15:240-8
16. von Haussen J, Koczulla R, Shaykhiev R. et al. The host defence peptide LL-37/hCAP-18 is a growth factor for lung cancer cells. Lung Cancer. 2008;59:12-23
17. Tjabringa GS, Aarbiou J, Ninaber DK. et al. The antimicrobial peptide LL-37 activates innate immunity at the airway epithelial surface by transactivation of the epidermal growth factor receptor. J Immunol. 2003;171:6690-6
18. Li D, Wang X, Wu JL. et al. Tumor-produced versican V1 enhances hCAP18/LL-37 expression in macrophages through activation of TLR2 and vitamin D3 signaling to promote ovarian cancer progression in vitro. PLoS One. 2013;8:e56616
19. Cha HR, Lee JH, Hensel JA. et al. Prostate cancer-derived cathelicidin-related antimicrobial peptide facilitates macrophage differentiation and polarization of immature myeloid progenitors to protumorigenic macrophages. Prostate. 2016;76:624-36
20. van der Does AM, Beekhuizen H, Ravensbergen B. et al. LL-37 directs macrophage differentiation toward macrophages with a proinflammatory signature. J Immunol. 2010;185:1442-9
21. Waltzer L, Bienz M. The control of beta-catenin and TCF during embryonic development and cancer. Cancer Metastasis Rev. 1999;18:231-46
22. Hamada F. [Wnt signaling and cancer]. Kaibogaku Zasshi. 2009;84:111-2
23. Zhan T, Rindtorff N, Boutros M. Wnt signaling in cancer. Oncogene. 2017;36:1461-73
24. Ohgaki H, Kros JM, Okamoto Y. et al. APC mutations are infrequent but present in human lung cancer. Cancer Lett. 2004;207:197-203
25. Sunaga N, Kohno T, Kolligs FT. et al. Constitutive activation of the Wnt signaling pathway by CTNNB1 (beta-catenin) mutations in a subset of human lung adenocarcinoma. Genes Chromosomes Cancer. 2001;30:316-21
26. Sainz B Jr, Alcala S, Garcia E. et al. Microenvironmental hCAP-18/LL-37 promotes pancreatic ductal adenocarcinoma by activating its cancer stem cell compartment. Gut. 2015;64:1921-35
27. Coffelt SB, Waterman RS, Florez L. et al. Ovarian cancers overexpress the antimicrobial protein hCAP-18 and its derivative LL-37 increases ovarian cancer cell proliferation and invasion. Int J Cancer. 2008;122:1030-9
28. Nizet V, Ohtake T, Lauth X. et al. Innate antimicrobial peptide protects the skin from invasive bacterial infection. Nature. 2001;414:454-7
29. Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene. 2008;27:5497-510
30. Shaw RJ, Cantley LC. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature. 2006;441:424-30
31. Havel JJ, Li Z, Cheng D. et al. Nuclear PRAS40 couples the Akt/mTORC1 signaling axis to the RPL11-HDM2-p53 nucleolar stress response pathway. Oncogene. 2015;34:1487-98
32. Sharma M, Chuang WW, Sun Z. Phosphatidylinositol 3-kinase/Akt stimulates androgen pathway through GSK3beta inhibition and nuclear beta-catenin accumulation. J Biol Chem. 2002;277:30935-41
33. He XC, Yin T, Grindley JC. et al. PTEN-deficient intestinal stem cells initiate intestinal polyposis. Nat Genet. 2007;39:189-98
34. Macdonald BT, Semenov MV, He X. SnapShot: Wnt/beta-catenin signaling. Cell. 2007;131:1204
35. Stewart DJ. Wnt signaling pathway in non-small cell lung cancer. J Natl Cancer Inst. 2014;106:djt356
36. Gillespie J, Ross RL, Corinaldesi C. et al. Transforming Growth Factor β Activation Primes Canonical Wnt Signaling Through Down-Regulation of Axin-2. Arthritis Rheumatol. 2018;70:932-42
37. Kim S, Takahashi H, Lin WW. et al. Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature. 2009;457:102-6
38. Reuven EM, Fink A, Shai Y. Regulation of innate immune responses by transmembrane interactions: lessons from the TLR family. Biochim Biophys Acta. 2014;1838:1586-93
39. Martin M, Rehani K, Jope RS. et al. Toll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat Immunol. 2005;6:777-84
40. Schaale K, Brandenburg J, Kispert A. et al. Wnt6 is expressed in granulomatous lesions of Mycobacterium tuberculosis-infected mice and is involved in macrophage differentiation and proliferation. J Immunol. 2013;191:5182-95
41. Bansal K, Trinath J, Chakravortty D. et al. Pathogen-specific TLR2 protein activation programs macrophages to induce Wnt-beta-catenin signaling. J Biol Chem. 2011;286:37032-44
42. Royer PJ, Henrio K, Pain M. et al. TLR3 promotes MMP-9 production in primary human airway epithelial cells through Wnt/β-catenin signaling. Respir Res. 2017;18:208
43. Kim S, Takahashi H, Lin WW. et al. Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature. 2009;457:102-6
44. Huang SM, Mishina YM, Liu S. et al. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature. 2009;461:614-20
45. Hovanes K, Li TW, Munguia JE. et al. Beta-catenin-sensitive isoforms of lymphoid enhancer factor-1 are selectively expressed in colon cancer. Nat Genet. 2001;28:53-7
46. Ren SX, Cheng AS, To KF. et al. Host immune defense peptide LL-37 activates caspase-independent apoptosis and suppresses colon cancer. Cancer Res. 2012;72:6512-23
47. Weber G, Chamorro CI, Granath F. et al. Human antimicrobial protein hCAP18/LL-37 promotes a metastatic phenotype in breast cancer. Breast Cancer Res. 2009;11:R6
48. Dolkar T, Trinidad CM, Nelson KC. et al. Dermatologic toxicity from novel therapy using antimicrobial peptide LL-37 in melanoma: A detailed examination of the clinicopathologic features. J Cutan Pathol. 2018;45:539-44
49. Hase K, Murakami M, Iimura M. et al. Expression of LL-37 by human gastric epithelial cells as a potential host defense mechanism against Helicobacter pylori. Gastroenterology. 2003;125:1613-25
50. Li D, Liu W, Wang X. et al. Cathelicidin, an antimicrobial peptide produced by macrophages, promotes colon cancer by activating the Wnt/β-catenin pathway. Oncotarget. 2015;6:2939-50
51. Bucki R, Leszczyńska K, Namiot A. et al. Cathelicidin LL-37: a multitask antimicrobial peptide. Arch Immunol Ther Exp (Warsz). 2010;58:15-25
52. Zuyderduyn S, Ninaber DK, Hiemstra PS. et al. The antimicrobial peptide LL-37 enhances IL-8 release by human airway smooth muscle cells. J Allergy Clin Immunol. 2006;117:1328-35
53. Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow. Lancet. 2001;357:539-45
54. Li D, Beisswenger C, Herr C. et al. Myeloid cell RelA/p65 promotes lung cancer proliferation through Wnt/β-catenin signaling in murine and human tumor cells. Oncogene. 2014;33:1239-48
55. Mantovani A, Allavena P, Sica A. et al. Cancer-related inflammation. Nature. 2008;454:436-44
56. Montreekachon P, Chotjumlong P, Bolscher JG. et al. Involvement of P2X(7) purinergic receptor and MEK1/2 in interleukin-8 up-regulation by LL-37 in human gingival fibroblasts. J Periodontal Res. 2011;46:327-37
57. Girnita A, Zheng H, Grönberg A. et al. Identification of the cathelicidin peptide LL-37 as agonist for the type I insulin-like growth factor receptor. Oncogene. 2012;31:352-65
58. Voloshanenko O, Erdmann G, Dubash TD. et al. Wnt secretion is required to maintain high levels of Wnt activity in colon cancer cells. Nat Commun. 2013;4:2610
59. Capurro MI, Xiang YY, Lobe C. et al. Glypican-3 promotes the growth of hepatocellular carcinoma by stimulating canonical Wnt signaling. Cancer Res. 2005;65:6245-54
60. Moon RT, Kohn AD, De Ferrari GV. et al. WNT and beta-catenin signalling: diseases and therapies. Nat Rev Genet. 2004;5:691-701
61. Aman A, Piotrowski T. Wnt/beta-catenin and Fgf signaling control collective cell migration by restricting chemokine receptor expression. Dev Cell. 2008;15:749-61
62. Iwai S, Yonekawa A, Harada C. et al. Involvement of the Wnt-β-catenin pathway in invasion and migration of oral squamous carcinoma cells. Int J Oncol. 2010;37:1095-103
63. Agier J, Brzezinska-Blaszczyk E, Zelechowska P. et al. Cathelicidin LL-37 Affects Surface and Intracellular Toll-Like Receptor Expression in Tissue Mast Cells. J Immunol Res. 2018;2018:7357162
64. Mookherjee N, Brown KL, Bowdish DM. et al. Modulation of the TLR-mediated inflammatory response by the endogenous human host defense peptide LL-37. J Immunol. 2006;176:2455-64
65. Kahlenberg JM, Kaplan MJ. Little peptide, big effects: the role of LL-37 in inflammation and autoimmune disease. J Immunol. 2013;191:4895-901
66. Hurtado P, Peh CA. LL-37 promotes rapid sensing of CpG oligodeoxynucleotides by B lymphocytes and plasmacytoid dendritic cells. J Immunol. 2010;184:1425-35
67. Di Nardo A, Braff MH, Taylor KR. et al. Cathelicidin antimicrobial peptides block dendritic cell TLR4 activation and allergic contact sensitization. J Immunol. 2007;178:1829-34
68. Wahlin YB. Effects of chlorhexidine mouthrinse on oral health in patients with acute leukemia. Oral Surg Oral Med Oral Pathol. 1989;68:279-87
69. Martin M, Rehani K, Jope RS. et al. Toll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat Immunol. 2005;6:777-84
70. Bansal K, Trinath J, Chakravortty D. et al. Pathogen-specific TLR2 protein activation programs macrophages to induce Wnt-beta-catenin signaling. J Biol Chem. 2011;286:37032-44
71. Koczulla R, von Degenfeld G, Kupatt C. et al. An angiogenic role for the human peptide antibiotic LL-37/hCAP-18. J Clin Invest. 2003;111:1665-72
72. Gillespie J, Ross RL, Corinaldesi C. et al. Transforming Growth Factor β Activation Primes Canonical Wnt Signaling Through Down-Regulation of Axin-2. Arthritis Rheumatol. 2018;70:932-42
Author contact
![]() Corresponding authors: Zujun Sun, email: 14111010030edu.cn; and Dong Li, email: lidongedu.cn
Corresponding authors: Zujun Sun, email: 14111010030edu.cn; and Dong Li, email: lidongedu.cn