Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results and Discussion
Conclusions
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(7):1980-1992. doi:10.7150/thno.31073 This issue Cite
Research Paper
Hyaluronic Acid Oligosaccharides Improve Myocardial Function Reconstruction and Angiogenesis against Myocardial Infarction by Regulation of Macrophages
Ning Wang1*, Chao Liu1*, Xinxin Wang1, Tao He1, Lu Li1, Xiuqi Liang1, Li Wang1, Linjiang Song2, Yuquan Wei1, Qinjie Wu1 ![]() , Changyang Gong1
, Changyang Gong1 ![]()
1. State Key Laboratory of Biotherapy and Cancer Center, West China Hospital, Sichuan University, Chengdu, 610041, P. R. China
2. School of Medical and Life Sciences, Chengdu University of Traditional Chinese Medicine, Chengdu, 611137, P. R. China
*These authors contributed equally to this work.
Received 2018-10-30; Accepted 2019-2-11; Published 2019-3-16
Abstract

Myocardial infarction (MI) is identified as one of the major causes of mortality and disability worldwide. For severe myocardial infarction, even advanced forms of clinical intervention often lead to unsatisfactory therapeutic results. Thus, alternative strategies for MI treatment are still desirable. Previously studies reported the capacity of degradative fragment of h-HA (high molecular weight hyaluronic acid), hyaluronan oligosaccharides (<10 disaccharides units, o-HA), for wound healing by influence on angiogenesis, inspiring us to study its potential for myocardial functional recovery against MI. However, there are few reports about o-HA in MI therapy.
Methods: In our study, we synthesized o-HA with 6~10 disaccharides (4-5 kDa) by enzymatic degradation and investigated its therapeutic effects on MI.
Results: We found that o-HA could reduce infarct size and apoptosis in MI region, also promote myocardial angiogenesis and myocardial function reconstruction in MI mouse model. Furthermore, our results also indicated that o-HA in cardiac improved polarization of M2 type macrophage, removed the inflammatory response caused by neutrophil for accelerating myocardial function reconstruction in vivo. The transcriptomic analyses revealed that o-HA could activate expression of chemokines Ccl2 and Cxcl5 for promoting macrophage polarization and stimulate MAPK and JAK/STAT signaling pathway for compensatory response of myocardial function.
Conclusion: Collectively, our results suggested o-HA with 6~10 disaccharides might be a potential agent for reconstruction of cardiac function against MI.
Keywords: hyaluronic acid oligosaccharides, myocardial infarction, M2 macrophage, neutrophil, LV remodeling, angiogenesis
Introduction
Myocardial infarction (MI) is one of the major causes of mortality and disability worldwide [1, 2]. MI appears when a coronary artery becomes blocked, inducing insufficient blood supply to myocardium of infarcted area. It will reduce oxygen distribution, input of nutrient and output of metabolic waste. The oxygen downstream caused apoptosis and necrosis of cardiomyocytes, which is leading to cascade damage amplification in left ventricular (LV) [3, 4]. Consequently, the function of LV decreased accompanied with hypertrophy [5].
Previous studies convinced that myocardial regeneration was limited by irreversible and inability of cardiac tissues after cardiac injuries [6, 7]. So far, the golden standard for treatment of myocardial injury is heart transplantation [8]. However, some challenges of heart transplantation exist, including a huge insufficiency of organ donors, easy to cause immune rejection and prolongation of hospitalization time [9]. The other treatment including pharmacological inhibition of G-protein coupled receptors and interventional therapy could clear vascular plaques or lead to vasodilation and restore blood flow for damaged cardiac muscles [10-12]. While, these approaches often impact an unexpected burst of reactive oxygen species (ROS), which may increase the risks of “ischemia-reperfusion injury” [13-15]. Therefore, there is a pressing need to develop new strategies for MI treatment.
The repair mechanism of MI is a complex progress, with different kinds of changes in cellular and extracellular components [16]. During the early stage of post-MI, the myocardial cell death takes a limited few hours to develop along with chronic inflammatory reaction [17]. Therein, inflammation component is involved and is got into “double-edged sword” for damage repair. Inflammatory reactions exist in the early stage of myocardial injury, mainly composing of neutrophils and mononuclear phagocyte system, which mainly applied to remove the damaged cardiomyocytes [18, 19]. However, the lack of blood supply generates persistent apoptosis and necrosis of LV cardiomyocytes, which could trigger inflammatory corpuscle participating continuously, which may hamper the process of myocardial repair [20]. Wherein, monocyte could convert to macrophage, subsequently, being devised to two kinds of activation patterns which displayed variously immunological functions: classical (M1 type) and alternative (M2 type) with different cell makers [21, 22]. Porcheray indicated that macrophage activation occurs initially through the M1 pathway, and then change to the M2 pathway, which the transition was invertible. The homeostasis of macrophages to switch from one activation state to another would affect tissue healing during ischemic injury [23].
The macromolecules existing in the extracellular matrix are essential for wound healing. Among them, hyaluronic acid (HA) was widely studied biological macromolecule that had the potential to promote damage repair. HA is one major component of tissue ECM, and is a non-protein extracellular molecule, negatively charged polymer with repeated units of β (1, 4)-D glucuronic acid-β (1, 3) N-acetyl-D-glucosamine [24, 25]. Researchers found that HA has a size-depended effect. The bioactivity of high molecule weight (h-HA) greater than 103 kDa gets the most interesting finding of antibacterial, anti-inflammatory and lubricant properties [26]. The partial degradative fragment of h-HA, hyaluronan oligosaccharides (< 10 disaccharides units, o-HA) has influence on angiogenesis, endothelial cell migration and proliferation for wound healing [27, 28]. However, the therapeutic effects of o-HA on MI by intravenous injection remained few reports, and the mechanism has not been elucidated.
In our study, we synthesized o-HA with 6~10 disaccharides (4-5 kDa) by enzymatic degradation. Then, therapeutic effects of o-HA on MI were investigated in detail, including infarct size, biomarkers in blood, apoptosis in MI region, and angiogenesis and cell proliferation around the damaged myocardium. In addition, M2 type macrophage polarization and neutrophil removal were also studied in MI mouse model. Furthermore, the mechanism of o-HA-induced myocardial function reconstruction was elucidated. Stimulation of macrophages migration, polarization, and secretion of growth cytokines by o-HA in vitro were explored. Furthermore, the transcriptomic analyses were also employed to study o-HA effects on macrophages.
Methods
Materials and animals
Hyaluronic acid (HA, ~1000 kDa) was purchased from Shandong Freda Biochem Co., Ltd. (Shandong, China). Bovine testicular hyaluronidase and Trichloroacetic aced (TCA) were purchased from Sigma-Aldrich (shanghai, China). All the organic chemical used in this work were analytical reagent (AR) grade. CD31 and Ki-67 immunohistochemistry antibody were purchased from Abcam (Cambridge, MA). Anti-CD45, anti-CD11b, and anti-Gr1 were provided by Cell Signal Technology (Boston, USA) for flow cytometry. Anti-CD206 was provided by Abcam (Cambridge, UK). Horseradish peroxidase (HRP)-labeled goat anti-rabbit or anti-mouse secondary antibodies were purchased from BD biosciences (New York, USA).
C57BL/6 male mice (5-6 weeks) were used for modeling MI. The animals were purchased from Beijing HFK Bioscience Co., Ltd., which were acclimatized at a controlled temperature of 20-22 oC, and a relative humidity of 50-60% under 12 h light-dark condition. All animal procedures were approved by the Institutional Animal Care and Treatment Committee of Sichuan University (Chengdu, P.R. China).
Preparation and characterization of o-HA
o-HA (6~10 disaccharides, 4-5 kDa) was prepared by hyaluronidase degradation methods [29, 30]. One hundred milligram of h-HA was dissolved in 50 mL of 0.1 M sodium acetate buffer (pH 5.4) containing 0.15 M NaCl, and then digested by 20000 units of bovine testicular hyaluronidase (Sigma) incubating at 37 oC. At regular intervals (2, 4, 6, 8, and 24 h), 10 mL aliquots were removed and the reaction was terminated by the addition of 1 mL of 100% Trichloroacetic acid (TCA, Sigma). The precipitate was eliminated by centrifugation and the supernatants from different time was mixed. The aliquots were dialyzed against distilled water with dialysis membrane (MWCO, 1000 Da) for 72 h at 4 oC. The internal solution was dialyzed against distilled water with dialysis membrane (MWCO, 5000 Da) for another 72 h. At last, the external phase was collected, followed by lyophilized. Lyophilized o-HA was preserved at 4 oC.
The o-HA samples were further characterized by Gel Permeation Chromatography (GPC) to determine macromolecular weight and weight distribution. The samples were dissolved in chloroform and cast on KBr plate, and the Fourier Transforms Infrared Spectroscopy (FTIR) spectra were kept. Nuclear Magnetic Resonance Analysis (1H NMR) was used to characterize chemical composition.
MI surgery
6-8 weeks male mice (C57) were used. MI was induced by permanent ligation of the left anterior descending coronary artery (LAD) as described previously [31-33]. The animals were anaesthetized by ketamine (100 mg/kg) and xylazine (10 mg/kg) with intraperitoneal administration. The mice were ventilated with air using small-animal respirator with a tidal volume of 0.1 mL and a respiratory rate of 110 strokes per minute by tracheotomy. The chest was opened, LV (left ventricle) was visualized and a left intercostal thoracotomy was performed. The pericardium was removed, and ligation of the LAD was achieved with 5-0 silk suture ligatures about 1 mm distal from the tip of the left atrium. Successful coronary blocking of the LAD was visualized by significant color change of the ischemic region. The chest was closed with 4-0 silk suture. After that, the post-MI mice were ventilated continuously for 30 min at 37 oC until their lung re-expanding again. Sham-operated group underwent the same procedure without occlusion of the LAD. NS group stands for the post-MI mice group which was treated with normal saline as control.
The post-MI mice were randomly divided into four groups (Sham, NS, h-HA and o-HA treated groups, n=8), and intravenous injection every other day. The dose of h-HA and o-HA was 5 mg/kg per mouse. The Sham group was used as negative control.
Biomarker release of post-MI after o-HA treatment
The cardiac troponin-I (cTn I) and creatine kinase-MB (CK-MB) in serum are specific biomarker for reflecting the myocardial necrosis level in post-MI [34, 35]. For detection the recovery effect of o-HA on myocardial cell in ischemic area, the post-MI mice were drawn blood at 6 h, 8 h, 12 h, 24 h, 48 h, 72 h, 96 h, 120 h, 144 h and 168 h. The serum samples of different groups (n=6) were analyzed quantitatively by the high-sensitivity ELISA (enzyme linked immunosorbent assay) Kit (CUSABIO, USA) for cTn I analysis. At 28 d, the CK-MB in serum was detected by blood biochemical analysis for myocardial cell repair.
Histology, morphometry and histochemistry for hearts
We evaluated the heart function by echocardiograph. We assessed in vivo cardiac function in total of 24 animals (n=4 in normal mice group, n=6 in sham, NS, h-HA and o-HA groups respectively) at 4 weeks after MI by echocardiographic imaging system (Vevo 770, VisualSonic, Toronto, Canada).
Histological evidence of MI with necrosis was detected by histopathological techniques. The mice were euthanized at 28 d, and the hearts were collected for further examination (n=8).
Hearts (n=4) were cut into 3 mm sections from apex to base, then hatched in 1.5% triphenyl-terazolium-chloride (TTC, Sigma) in PBS (pH 7.4) for 20 min at 37 oC followed by formalin fixation. The infarction area emerged yellow-white, and the normal myocardium was red. Each heart section was recorded by digital camera (Sony, Tokyo, Japan), and the infarct size was measured with ImageJ software (National Institutes of Health, Bethesda, MD).
The hearts (n=4) were fixed in 4% paraformaldehyde, embedded in paraffin, and sectioned for H&E, Masson trichrome histopathology analysis. The photographs were taken using a CCD camera (Zeiss Axioskop2, Germany). The infarction size was defined as a percentage of the sum of the epicardial and endocardial infarct circumference divided by the sum of the total LV epicardial and endocardial circumferences [36]. LV anterior wall thickness was expressed as previous study [36].
We evaluated the heart function by echocardiograph. We assessed in vivo cardiac function in total of 24 animals (n=4 in normal mice group, n=6 in sham, NS, h-HA and o-HA groups respectively) at 4 weeks after MI by echocardiographic imaging system (Vevo 770, VisualSonic, Toronto, Canada).
After treatment, mice hearts were fixed in 4% paraformaldehyde, embedded in paraffin, and sectioned. A commercially available TUNEL (TdT-mediated dUTP Nick-End Labeling) kit (Vazyme Biotech co., Ltd) was used to detect apoptotic cells in the heart tissues. This analysis was performed following the manufacturer's protocol. The samples were then observed using a fluorescence microscope (× 200). The apoptotic index in per group was calculated in four equal-sized fields of LV.
The angiogenesis in the ischemia sites were stained by epithelial cell marker CD31 (goat anti-mouse CD31 polyclonal antibody, 1 : 100) (Abcam, Cambridge, MA) with the labeled streptavidin-biotin method. The samples were observed using microscope (× 200). The number of microvessel per group was calculated to determine the microvessels density in four randomly fields of LV region.
The proliferation of endothelial cells in LV was stained with Ki-67, (Abcam, Cambridge, MA) (1 : 150) and biotin-horseradish peroxidase labeled secondary antibody (BD Biosciences Pharmingen) using the labeled streptavidin-biotin method. The images were captured by microscopy. In LV, the Ki-67 labeling index was counted as the number of Ki-67+ cells/total number of cells in four randomly regions per group.
Toxicity assessment
In order to assess the possible side effects in the o-HA-treated mice, all mice were observed after intravenously administration every other day for 28 d, including the general conditions (the activity, skin, hair, behavior, energy and other clinical signs). Before being euthanized, their blood was acquired for complete blood count (CBC) test and blood chemistry profile analysis. Meanwhile, the major organs (liver, spleen, lung and kidney) were fixed in 4% paraformaldehyde with H&E staining for further histopathology analysis.
Flow cytometry analysis of the myocardial microenvironment
The above results showed that o-HA could encourage MI repair. Furthermore, how o-HA played an important role in regulating the microenvironment of the myocardium attracted our attentions. Therefore, we meant to detect if there were some changes in myocardial microenvironment after o-HA treatment in vivo. The LV region of heart samples were separated into infarct and non-infarct area after being treated at different time points (24 h, 48 h, 72 h, 7 d, 14 d and 21 d), and then be prepared to be single-cell suspension following standard procedure. After stained by CD206 and Gr1 (all the flow cytometry antibodies, which were provided by CST, USA), the cells in infarcted area were analyzed using a flow cytometer (Arial III, BD Biosciences).
Wound healing, transwell and transwell invasion assay in vitro
By studying the function of o-HA on the cardiac microenvironment, the results above indicated that the MI recovery of o-HA mainly based on increased macrophages, improving M2 polarization, and inhibiting excessive inflammation. Then, we made further research for the effect of o-HA on macrophages in vitro. The peritoneal macrophages were extracted from C57 mice as previously reported [20]. Briefly, the mice were anaesthetized and decapitated, and 15 mL serum and antibiotics-free DMEM was injected into intraperitoneal. After 15 min, the medium was sucked out by 20 mL syringe. The medium was centrifuged at 2000 rpm for 5 min. The macrophages were washed for three times with serum and antibiotics-free DMEM and counted for further usage.
The wound healing test was performed on peritoneal macrophages as described previously. In briefly, the cell suspension (2.0 × 106 cells/well) were seeded in 12-well plates and cultured for 2 h in fresh DMEM medium containing 10% serum. After removing the suspended cells, the adherent macrophages were culture for another 20 h before migration test. The macrophages were scraped by sterile 0.1 mL pipette tips and fresh 700 μL of serum and antibiotics-free DMEM containing different concentration of o-HA (250 μg/mL, and 500 μg/mL) and h-HA (500 μg/mL) were added into each well, respectively. After incubation for 24 h, cells were fixed and imaged using a microscope (Zeiss, Germany) and the percentage of migrated macrophages was counted.
Transwell assay was conducted by Transwell plate (8 μm gap size, Millipore) as previously described with some modifications. The peritoneal macrophages were extracted according to the above method. Subsequently, a total of 2 × 106 cells in 100 μL serum-free medium were added in the upper chamber. Then, 400 μL of serum-free medium containing different concentration of o-HA (250 μg/mL, and 500 μg/mL) and h-HA (500 μg/mL) was added to the bottom chamber. Macrophages allowed to migrate for 24 h. Non-migrated cells in the upper chamber were removed. The migrated cells were fixed in methanol and stained with 0.5% crystal violet. Migrated cells in five randomly selected regions were counted and taken pictures under microscope.
Transwell invasion assay was performed according to previous studies with some modifications. The peritoneal macrophages were extracted according to the above method. In brief, the upper surface of the Transwell plate was coated with serum-free medium diluted Matrigel (1: 1, 50 μL/well, BD Biosciences). After Matrigel polymerizetion, the bottom chambers were filled with 400 μL 10% FBS-DMEM medium and 100 μL secrum-free medium containing 2 × 106 cells was seeded to the top chambers. The same agents as above were added to top chambers and cells allowed to migrate for 24 h. The following cell process were similar with the above method.
The promote-polarization ability of o-HA was evaluated in vitro with flow FCM. The peritoneal macrophages were extracted according to the above method. 5 × 106 cells were seeded in 6-well plates and cultured for 2 h. after being removed of the suspended cells, the adherent-macrophages were cultured for another 20 h with 10% FBS-DMEM medium. Then the macrophages were incubated with o-HA (0, 250 and 500 μg/mL) and h-HA (500 μg/mL) for 48 h. after the incubation, the macrophages were stained with CD206 antibody (M2 type macrophage specific biomaker) and the samples were analyzed by FCM.
Analysis of macrophage polarization by o-HA in vitro
Order to determining whether o-HA could strengthen the secretion of VEGF in M2 type macrophages. Western blots assay was conducted on different concentration of o-HA (0, 250 and 500 μg/mL) and h-HA (500 μg/mL) treated-macrophages. Briefly, after treated for 48 h, the total protein was extracted from the macrophages by a buffer containing 1% triton X-100, 1% deoxycholate and proteinase inhibitor cocktail (Sigma). After centrifugation for 15 min at 4 oC, the protein supernatant was collected and its concentration was measured by BCA protein assay kit. Total protein samples were loaded and separated by 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and incubated with antibodies against VGEF (Abcam) at 4 oC overnight and HRP-conjugated secondary antibodies at 37 oC for 1 h. After the incubation, the blots were reacted with chemiluminescent substrates visualized using a chemiluminescent (ECL) detection system.
Transcriptomic analyses
The peritoneal macrophages were extracted from the above procedure. Then, the macrophages were incubated with o-HA and h-HA for 48 h. After the incubation, the total RNA of macrophages was extracted. RNA-seq libraries were prepared using the Illumina HiSeq according to the manufacturer's instructions. Through screening comparison, H-cluster analysis was used to analyze the expression of differential genes and functional enrichment was studied by KEGG analysis.
Statistical analysis
Data are presented as mean ± SD. Paired t test or ANOVA was performed as indicated to determine statistical differences. The difference was considered significant for P values of less than 0.05, and the statistical analyses were carried out using GraphPad Prism software.
Results and Discussion
Preparation and Characterization of o-HA
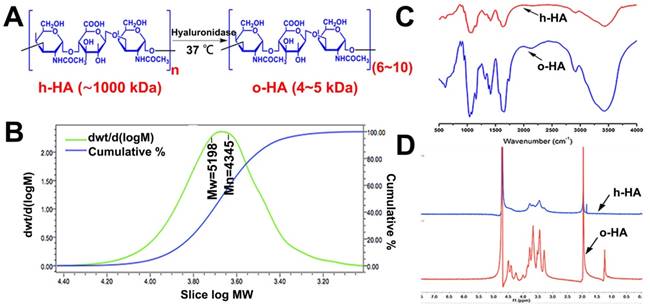
We prepared o-HA (6~10 disaccharides, 4-5 kDa) by hyaluronidase mediating degradation of high molecule weight HA (h-HA, ~1000 kDa). The synthesis scheme of o-HA was shown in Figure 1 and Figure 2 A. GPC spectrum indicated that the Mn was 4345, and the polymer dispersity index (PDI) of o-HA was 1.52 (Figure 2B). The FTIR and 1H NMR were applied to confirm the chemical structure of o-HA, which displayed the structural consistency of o-HA and h-HA (Figure 2C, D). The above data demonstrated that o-HA obtained by enzymatic degradation was prepared successfully.
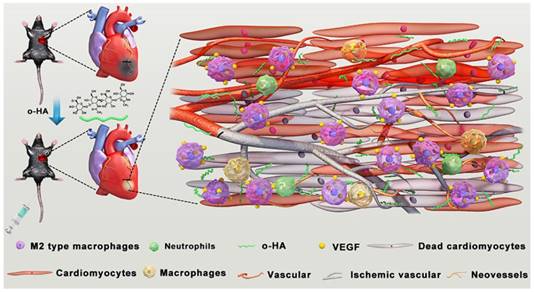
Schematic illustration of preparation, post-MI treatment and LV repair mechanism of o-HA.
Synthesis and characterization of o-HA. A) Synthesis of o-HA. B, C, D) GPC, 1H NMR, and FTIR spectrum of o-HA.
o-HA reduced infarct size against MI
To evaluate in vivo therapeutic efficiency of o-HA against MI, we applied o-HA into the cardiac function deficient mice. 28 d after treatment, morphological and histological analysis of heart tissues were used to assess the effects of o-HA. As shown in Figure 3A and B, the hearts of mice were stained by TTC, in which the infarct area of LV performed yellow-white and the living myocardium appeared red. Therefore, we could determine the effects of o-HA against MI by calculating the size of infarct area. o-HA-treated group showed less yellow-white area than NS and h-HA-treated groups.
Histological analysis of hearts harvested 28 d after MI. A) infarcted region assessed based on TTC staining of heart sections, the infarcted field was white. B) photograph of hearts causing MI after treated by four groups. (n=8). C) Images from four-treated groups showing morphological changes with H&E staining of heart paraffin sections. D) Images from four-treated groups showing myocardial fibrosis with MT staining of heart paraffin sections. E) % LV infarct area of different groups. F) The relative thickness of LV (%) in different groups.
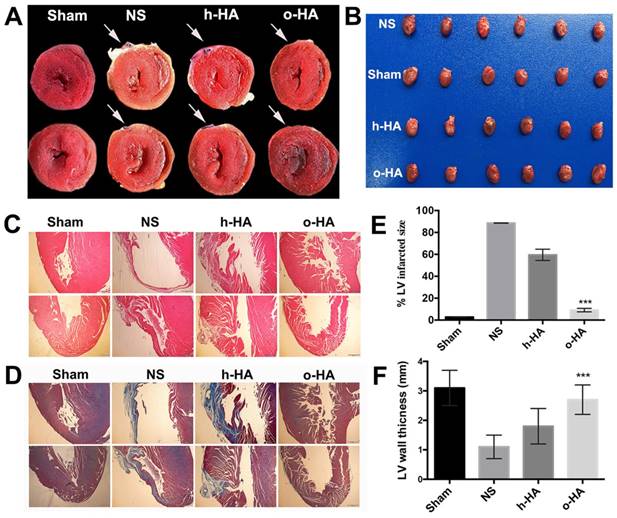
Heart tissues from mice in each group were collected and stained with haematoxylin and eosin (H&E) and Masson trichrome (MT). Fewer inflammatory cell of LV (compared to NS and h-HA group) could be observed in o-HA group (Figure 3C and Figure S1A).
Simultaneously, MT staining was consistent with the results of H&E. We observed that less collagen deposition in LV after o-HA treatment by the size of blue region (compared to NS and h-HA group). It indicated a striking decreasing level of scar tissue (Figure 3D and Figure S1B). The LV infarct size (%) were 100% in NS group, 59.4 ± 4.3% in h-HA group and 10.5 ± 1.2% in o-HA group, respectively (Figure 3E). The infarct size was significantly reduced ~10-fold in o-HA treatment group than NS group (P < 0.01). The wall thickness of LV was 1.1 ± 0.4 mm in NS group, 1.8 ± 0.6 mm in h-HA group and 2.7 ± 0.5 mm in o-HA group (Figure 3F). The relative thickness of LV was increased ~2.7-fold in o-HA group than NS from statistical analysis (P < 0.01). The Sham group was used as negative control (3.1 ± 0.6 mm).
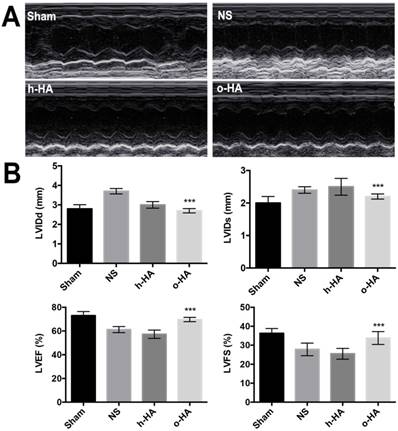
As shown in Figure 4, the echocardiography was used as noninvasive method to measure cardiac function 4 weeks after MI. There were significant differences between o-HA group and NS group (***P< 0.001). While, o-HA group had no statistical difference with Sham group.
showed cardiac function measured by echocardiography at 28 d after surgery. A) Representative echocardiograph from the various groups. B) LV internal diameters of diastole (LVIDd) and systole (LVIDs) were measured at the view of short axis from M mode. LV ejection fraction (LVEF) and fractional shortening (LVFS) was calculated using VisualSonic analysis software (n=6 animals/group). ***P< 0.001 vs NS.
Toxicity evaluation of o-HA in vivo
Subsequently, in vivo toxicity evaluation of o-HA was performed. The blood of mice in each group was collected for CBC test and blood chemistry profile analysis (Figure S2). Then, the major organs were collected and fixed in 4% paraformaldehyde for H&E staining analysis (Figure S3). The results of CBC test and blood chemistry profile analysis indicated that all the indicators were in normal range and there were no significant changes in different groups. In addition, the histological analysis of liver, spleen, lung and kidney revealed there were no histopathologic changes and inflammatory cell infiltration compared with sham group.
Biomarkers revealed decline of myocardial necrosis
Owing to the absolutely cardiac tissue specificity, cTn I and CK-MB were used as the main biomarkers for the diagnosis of myocardial necrosis [37]. The concentration changes of the both cTn I and CK-MB could reflect the severity of myocardial injury which had a temporal correlation [38]. The concentration of cTn I in NS and h-HA group rose within 24 h (598.2 ± 37.1 and 489.5 ± 8.2 pg/mL at 24 h, respectively), whereafter, declined in a 28 d-period. However, in the o-HA treatment group, the expression of cTn I in the serum at 24 h was significantly lower (378.4 ± 12.5 Pg/mL, P<0.001) than that in the NS group and lasted with a lower concentration for 28 d (Figure 5A).
Release of cTn I and CK-MB after MI. A) Values of cTn I in blood at various time point were displayed. cTn I released with early rising, reaching peak, and decreasing over several days. The o-HA group with smaller cTn I peak. B) Concentration of CK-MB in post-MI mice after being treated for 28 d.
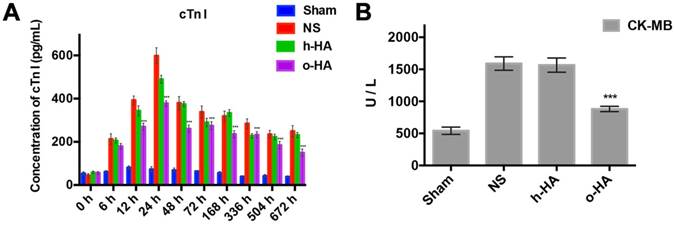
The data showed that CK-MB concentration was lower in o-HA-treated group (879.3 ± 14.2 U/L) compared to NS group (1583.5 ± 32.6 U/L, P<0.001). The Sham group was used as negative control (507.2 ± 21.6 U/L), and h-HA group also had higher CK-MB concentration (1465.6 ± 42.7 U/L, P<0.001) (Figure 5B). It suggested that o-HA treatment could reduce the degree of injury and necrosis of myocardial tissues in post-MI mouse.
Apoptosis analysis in myocardium
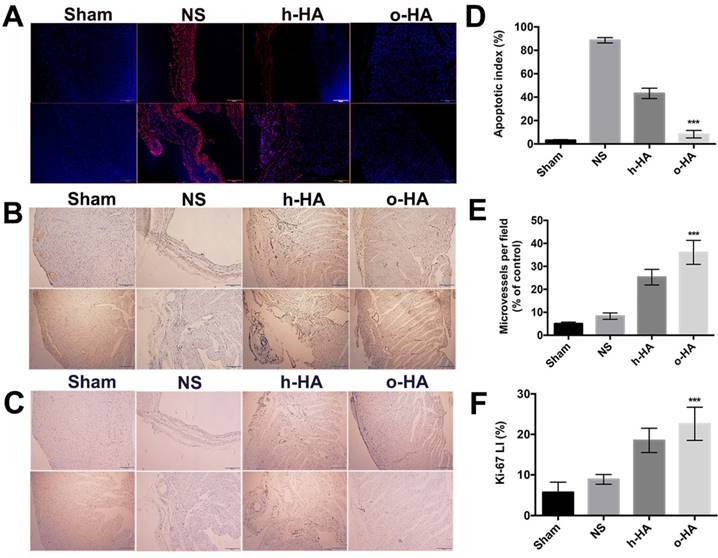
Apoptosis in LV was then investigated by immunofluorescent TUNEL assay. More TUNEL+ signals (red) were found within LV region in NS and h-HA groups than o-HA group (Figure 6A). The apoptotic index (%) in o-HA group was 8.3 ± 2.4%, versus NS (88.6 ± 3.2%, P<0.001) group and h-HA (43.2 ± 3.5%, P<0.001) group. The Sham group was used as negative control (3.2 ± 0.5%) (Figure 6D). The results showed that NS and h-HA group had more apoptotic cardiac myocytes and bigger infarct size than o-HA group in LV (n=4). It is also indicated that o-HA could attenuate apoptosis, reduce the infarction area and play a protective role in cardiomyocytes in LV.
IHC analysis of expression of A) TUNEL, B) CD31 and C) Ki-67 in heart sections of different group after treated for 28 d.
o-HA promoted angiogenesis and cell proliferation
Above data showed that o-HA could attenuate myocardial necrosis and reduce infarction area after MI, and then we investigated whether o-HA could promote cell proliferation. First, we set out to determine whether o-HA had beneficial activity in myocardium angiogenesis. The LV region of four groups were experimented by immunohistochemical CD31 staining. The neovascularization density of o-HA group (36.1 ± 4.8) significantly increased compared with NS (8.3 ± 1.4, P<0.001) and h-HA (25.4 ± 3.4, P< 0.001) group. The Sham group was used as negative control (5.4 ± 0.7) (Figure 6B, E and Figure S4A). The increased counts of blood vessels were noted in the injury regions after o-HA treatment (n=4). The angiogenesis effect of o-HA may improve the myocardial outcome after MI.
Next, Ki-67 immunohistochemical staining was used to analysis the effect of o-HA on cell proliferation ability in vivo. Remarkably, more Ki-67+ survival cells (brown) likewise exhibited in infarct heart tissues in o-HA group compared to NS and h-HA group (Figure 6C and Figure S4B). The difference of Ki67 LI (%) was readily apparent on inspection of heart: the Ki67 LI (%) in o-HA group was 22.6 ± 4.1%, versus NS group (8.9 ± 1.3%, P<0.001) and h-HA group (18.5 ± 2.7%, P<0.001). The Sham group was used as negative control (12.4 ± 2.5%) (Figure 6F). The results demonstrated a significant contribution to the cell proliferation in the cardiac function deficient mice with o-HA treatment, which provided function reconstruction for traumatic myocardium.
These observations suggested a robust capability of o-HA to improve angiogenesis which furnish a compensatory recover the conditions of myocardial cells in the MI regions. Meanwhile, o-HA could be beneficial to cell proliferation which leaded to improvement of MI [38].
Myocardial microenvironment analysis
Subsequently, we aimed to figure out the in vivo mechanisms which could interpret the effect of o-HA on MI repair. In the microenvironment of injured-myocardium, many kinds of immune cells and stem cell populations could often be observed. The different cell populations had time-depended behavior. The influence of o-HA on several of these cell lines at different time (1 d, 3 d, 7 d, 14 d, 21 d) were analysis by flow cytometry (FCM) and the results were displayed in Figure 7 (n=4).
FCM analysis in infarcted area microenvironment at 24 h, 72 h, 7 d, 14 d and 21 d. A, C) M2 type macrophages distribution after treatment. B, D) neutrophils inhibition.
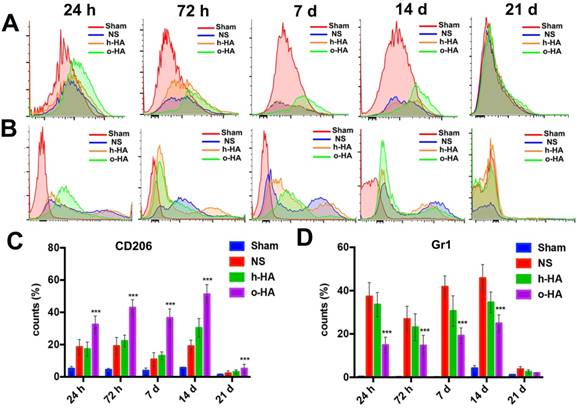
Among macrophage populations, M2 type macrophage could promote injury repair. We used FCM to analysis the levels of macrophage polarization in LV after o-HA treatment. Compared with NS (1 d: 18.5 ± 4.6%, 3 d: 19.1 ± 5.4%, 7 d: 10.8 ± 4.2%, 14 d: 19 ± 3.8%) (P<0.01) and h-HA (1 d: 17.2 ± 4.4%, 3 d: 22.3 ± 3.6%, 7 d: 13.1 ± 2.4%, 14 d: 30.4 ± 5.8%) group (P<0.01), o-HA group contained a greater number of M2 type macrophage (1 d: 32.4 ± 5.3%, 3 d: 42.9 ± 5%, 7 d: 36.6 ± 5.5%, 14 d: 53.2 ± 5.1%) which indicated that o-HA could promote recruitment of M2 type macrophages. However, the M2 type macrophage decreased at 21 d in all the groups (NS: 2.2 ± 1.3%, h-HA: 3.2 ± 1.2%, o-HA: 5.7 ± 1.4%). The Sham group was used as negative control (1 d: 5.1 ± 1.3%, 3 d: 4.4 ± 0.9%, 7 d: 3.8 ± 1.4%, 14 d: 5.4 ± 0.4%, 21 d: 1.3 ± 0.5%) (Figure 7A, C and Figure S5).
Contemporary, removal the over-reaction events of neutrophils could be better for injured-myocardium repair [18,19]. We used FCM to analysis the ability of o-HA to eliminate the excessive inflammation. As shown in Figure 7B and D, o-HA may maintain normal inflammatory response at early injury stage (1 d: 15.1 ± 3.3%, 3 d: 16.0 ± 4.3%) for scavenging of the necrotic cells and reduce the excessive inflammatory response as time prolong (7 d: 19.2 ± 3.2%, 14 d: 25.2 ± 3.3%, 21 d:1.9 ± 0.2%). While, NS (1 d: 38.1 ± 5.5%, 3 d: 27.2 ± 4.3%, 7 d: 42.0 ± 5.6%, 14 d: 47.5 ± 6.2%, 21 d: 4.0 ± 1.1%) (P<0.01) and h-HA (1 d: 33.6 ± 5.6%, 3 d: 23.0 ± 5.3%, 7 d: 31.1 ± 4.7%, 14 d: 35.3 ± 4.3%, 21 d: 3.1 ± 0.7%) group (P<0.01) kept a higher percentage of neutrophils which could lead to excessive and persistent inflammation. The Sham group was used as negative control (1 d: 0.3 ± 0.1%, 3 d: 0.2 ± 0.2%, 7 d: 0.14 ± 0.1%, 14 d: 4.1 ± 1.2%, 21 d: 1.1 ± 0.2%).
In vivo myocardial microenvironment analysis showed o-HA could increase the number of macrophages in LV region, reflecting that o-HA had the recruitment function of macrophages, and could promote M2 macrophage polarization, thereby increasing cytokine secretion. In addition, o-HA could also suppress chronic inflammatory reactions that accelerated compensatory response of myocardial function.
o-HA induced migration and invasion of macrophage
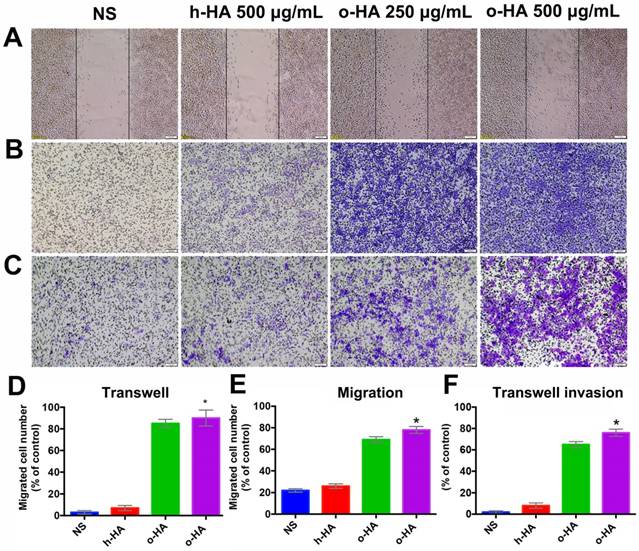
To further investigate mechanisms that underlie the favorable activity of o-HA on myocardial regeneration, we then investigated the effect of o-HA on migration and invasion of macrophage. Wound-healing and transwell migration assays were performed using mice peritoneal macrophage. o-HA enhanced migration of macrophages in dose-dependent manners. Comparing with NS (22.4 ± 2.2%, P<0.001) and h-HA (26.3 ± 1.3%, P<0.001) group, o-HA with concentration of 250 μg/mL (67.6 ± 3.3%) and 500 μg/mL (91.4 ± 2.5%) could enhance macrophage migration (Figure 8A, D). Consistent results were obtained in transwell migration assay, o-HA (250 μg/mL: 81.3 ± 5.8%, 500 μg/mL: 94.5 ± 7.4%) showed stronger enhancement on macrophage migration compared with h-HA (7.2 ± 2.1%, P<0.001) and NS (3.1 ± 1.1%, P<0.001) (Figure 8B, E). In transwell invasion assay, macrophages exhibited dramatically invasion capability in the presence of o-HA (250 μg/mL: 32.3 ± 4.5%, 500 μg/mL: 76.5 ± 3.7%) compared with h-HA (8.4 ± 2.4%, P<0.001) and NS (0%, P<0.001) (Figure 8C, F). Altogether, our results showed that o-HA increased migration and invasion of macrophages.
Migration and invasion of peritoneal macrophages by o-HA. A) and D) promotion of o-HA on macrophages in “Wound healing” analysis. B) and E) Transwell migration of macrophages triggered by o-HA compared with NS and h-HA. C) and F) invasion of macrophage induced by o-HA in Transwell assay.
Macrophage polarization assay in vitro
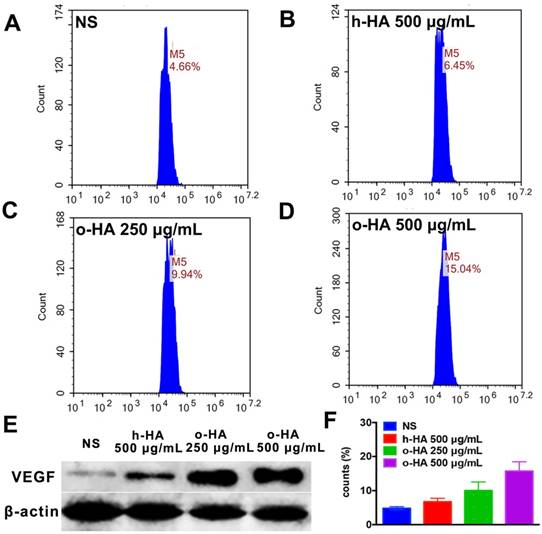
We also evaluated the polarization-inducing ability of o-HA on macrophages. o-HA induced higher level of polarization of M2 type macrophage (15.7 ± 2.7%) than h-HA (10.0 ± 2.5%, P<0.001) and NS (4.8 ± 0.5%, P<0.001) group (Figure 9A to D and F). The expression of VEGF (vascular endothelial growth factor) was also upregulated in the o-HA groups compared with NS and h-HA group (Figure 9E). In summary, the results suggested that o-HA, not only being a promotor-agent for polarization of M2 type macrophage, but also up-regulating VEGF secretion.
Polarization of peritoneal macrophages with o-HA in vitro. A, F) NS. B, F) h-HA, 500 μg/mL. C, F) o-HA, 250 μg/mL. D, F) o-HA, 500 μg/mL. E) VEGF expression by western blots assay of M2 macrophage.
Transcriptomic analyses of o-HA on heart function
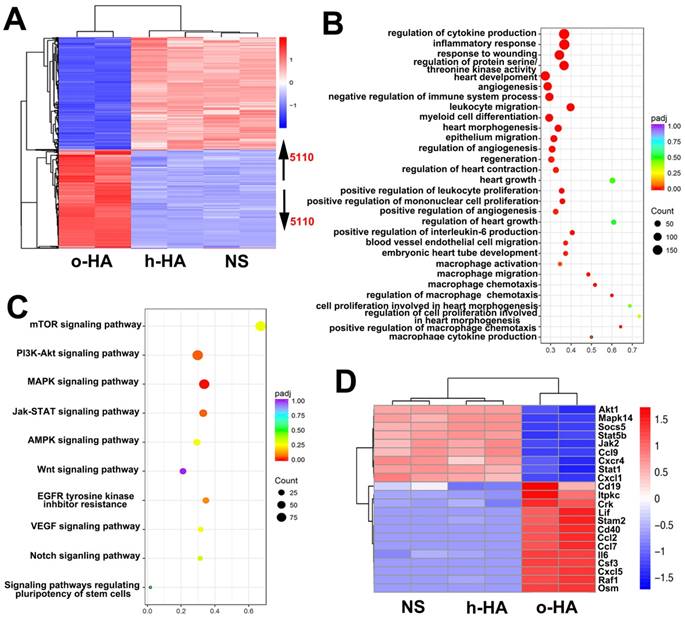
To gain insight into the molecular mechanism of the enhanced heart function activity of o-HA, we leveraged transcriptomics data to study o-HA effects on macrophages. In Figure 10A, the expression profiles of o-HA, h-HA and NS samples were clearly separated and 5110 genes were found to be differentially expressed (false discovery rate < 0.05) between the o-HA and NS groups. Subsequently, to determine molecular pathways associated with o-HA, we performed KEGG analysis for biological function and signal pathway (Figure 10B and C). The analysis revealed significant enrichment for function of myocardial repair and related signal pathways of macrophage action [39-41]. Furthermore, well-characterized genes in the above signaling pathways were found to be upregulated in the transcriptomic data upon treatment of o-HA (Figure 10D). The transcriptomic analyses revealed that o-HA could activate expression of chemokines (Ccl2, Cxcl5, ect.) for promoting macrophage polarization and stimulate MAPK and JAK/STAT signaling pathway for myocardial function reconstruction. Consistent with the functional studies above, these findings suggested that o-HA may activate macrophages for cardiac function recovery.
(A) Heatmap depiction of differentially expressed genes among o-HA, h-HA and NS groups treated macrophages from two independent experiments. (B, C) KEGG analysis of all differentially function and signaling pathways enrichment between o-HA and NS treated macrophages. (D) Heatmap depiction of differentially expressed genes in signaling pathways among o-HA, h-HA and NS groups treated macrophages from two independent experiments.
Conclusions
In this study, we have synthesized o-HA (4 ~ 5 kDa) by enzymatic degradation and assigned it for the treatment of MI. The results indicated that o-HA could effectively reduce infracted area, concomitant reduce cardiomyocyte apoptosis, and increase myocardial angiogenesis in the ischemic area. But treatment with h-HA had only minor therapy effects. Additionally, we identified the crucial role of o-HA in regular ischemic myocardial microenvironment. Moreover, our study provided further evidence that o-HA could suppress neutrophil over-reaction caused chronic inflammatory response. We found that o-HA increased the recruitment of macrophages and promoted the polarization of M2 macrophages. The transcriptomic analyses revealed that o-HA could activate expression of chemokines such as Ccl2 and Cxcl5 for promoting macrophage polarization and stimulate MAPK and JAK/STAT signaling pathway for myocardial function reconstruction. In summary, our results suggested o-HA with 6~10 disaccharides might be a potential agent for reconstruction of cardiac function against MI.
Abbreviations
MI: myocardial infarction; h-HA: high molecular weight hyaluronic acid; o-HA: hyaluronan oligosaccharides; LV: left ventricular; ROS: reactive oxygen species; HA: hyaluronic acid; TCA: trichloroacetic aced; GPC: Gel Permeation Chromatography; FTIR: Fourier Transforms Infrared Spectroscopy; 1H NMR: Nuclear Magnetic Resonance Analysis; LAD: left anterior descending coronary artery; cTn I: cardiac troponin-I; CK-MB: creatine kinase-MB; ELISA: enzyme linked immunosorbent assay; TTC: triphenyl-terazolium-chloride; CBC: complete blood count; SDS-PAGE: sodium dodecyl sulfate-polyacrylamide gel electrophoresis; ECL: chemiluminescent detection system; H&E: haematoxylin and eosin; MT: masson trichrome; TUNEL: TdT-mediated dUTP Nick-End Labeling; FCM: flow cytometry.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This work was financially supported by National Natural Science Foundation of China (No. 81822025), National Program for Support of Top-notch Young Professionals (W02070141) and 1·3·5 project for disciplines of excellence, West China Hospital, Sichuan University.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Dutta P, Courties G, Wei Y, Leuschner F, Gorbatov R, Robbins CS. et al. Myocardial infarction accelerates atherosclerosis. Nature. 2012;487:325-9
2. Correction to. Heart Disease and Stroke Statistics-2017 Update: A Report From the American Heart Association. Circulation. 2017;136:e196
3. Lindsey ML. MMP induction and inhibition in myocardial infarction. Heart Fail Rev. 2004;9:7-19
4. Pfeffer MA, Braunwald E. Ventricular remodeling after myocardial infarction. Experimental observations and clinical implications. Circulation. 1990;81:1161-72
5. Lambert JM, Lopez EF, Lindsey ML. Macrophage roles following myocardial infarction. Int J Cardiol. 2008;130:147-58
6. Curtis MW, Russell B. Cardiac tissue engineering. J Cardiovasc Nurs. 2009;24:87-92
7. Radhakrishnan J, Krishnan UM, Sethuraman S. Hydrogel based injectable scaffolds for cardiac tissue regeneration. Biotechnol Adv. 2014;32:449-61
8. Hasan A, Khattab A, Islam MA, Hweij KA, Zeitouny J, Waters R. et al. Injectable Hydrogels for Cardiac Tissue Repair after Myocardial Infarction. Adv Sci (Weinh). 2015;2:1500122
9. Halade GV, Kain V, Ingle KA, Prabhu SD. Interaction of 12/15-lipoxygenase with fatty acids alters the leukocyte kinetics leading to improved postmyocardial infarction healing. Am J Physiol Heart Circ Physiol. 2017;313:H89-102
10. Schumacher SM, Gao E, Zhu W, Chen X, Chuprun JK, Feldman AM. et al. Paroxetine-mediated GRK2 inhibition reverses cardiac dysfunction and remodeling after myocardial infarction. Sci Transl Med. 2015;7:277ra31
11. Ban K, Park HJ, Kim S, Andukuri A, Cho KW, Hwang JW. et al. Cell therapy with embryonic stem cell-derived cardiomyocytes encapsulated in injectable nanomatrix gel enhances cell engraftment and promotes cardiac repair. ACS Nano. 2014;8:10815-25
12. Binder A, Ali A, Chawla R, Aziz HA, Abbate A, Jovin IS. Myocardial protection from ischemia-reperfusion injury post coronary revascularization. Expert Rev Cardiovasc Ther. 2015;13:1045-57
13. Braunwald E, Kloner RA. Myocardial reperfusion: a double-edged sword? J Clin Invest. 1985;76:1713-9
14. Cho DI, Kim MR, Jeong HY, Jeong HC, Jeong MH, Yoon SH. et al. Mesenchymal stem cells reciprocally regulate the M1/M2 balance in mouse bone marrow-derived macrophages. Exp Mol Med. 2014;46:e70
15. Kempf T, Zarbock A, Widera C, Butz S, Stadtmann A, Rossaint J. et al. GDF-15 is an inhibitor of leukocyte integrin activation required for survival after myocardial infarction in mice. Nat Med. 2011;17:581-8
16. Takahashi K, Ito Y, Morikawa M, Kobune M, Huang J, Tsukamoto M. et al. Adenoviral-delivered angiopoietin-1 reduces the infarction and attenuates the progression of cardiac dysfunction in the rat model of acute myocardial infarction. Mol Ther. 2003;8:584-92
17. Vorob'eva EA. Structural changes in the myocardium during intermediate variants of chronic ischemic heart disease. Vrach Delo. 1983;7:80-2
18. Zhang GW, Gu TX, Guan XY, Sun XJ, Qi X, Li XY. et al. HGF and IGF-1 promote protective effects of allogeneic BMSC transplantation in rabbit model of acute myocardial infarction. Cell Prolif. 2015;48:661-70
19. Zhao ZQ, Corvera JS, Halkos ME, Kerendi F, Wang NP, Guyton RA. et al. Inhibition of myocardial injury by ischemic postconditioning during reperfusion: comparison with ischemic preconditioning. Am J Physiol Heart Circ Physiol. 2003;285:H579-88
20. Hilgendorf I, Gerhardt LM, Tan TC, Winter C, Holderried TA, Chousterman BG. et al. Ly-6Chigh monocytes depend on Nr4a1 to balance both inflammatory and reparative phases in the infarcted myocardium. Circ Res. 2014;114:1611-22
21. Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K. Development of monocytes, macrophages, and dendritic cells. Science. 2010;327:656-61
22. Epelman S, Liu PP, Mann DL. Role of innate and adaptive immune mechanisms in cardiac injury and repair. Nat Rev Immunol. 2015;15:117-29
23. Porcheray F, Viaud S, Rimaniol AC, Leone C, Samah B, Dereuddre-Bosquet N. et al. Macrophage activation switching: an asset for the resolution of inflammation. Clin Exp Immunol. 2005;142:481-9
24. Mahoney DJ, Aplin RT, Calabro A, Hascall VC, Day AJ. Novel methods for the preparation and characterization of hyaluronan oligosaccharides of defined length. Glycobiology. 2001;11:1025-33
25. Day AJ. The structure and regulation of hyaluronan-binding proteins. Biochem Soc Trans. 1999;27:115-21
26. Chen CH, Wang SS, Wei EI, Chu TY, Hsieh PC. Hyaluronan enhances bone marrow cell therapy for myocardial repair after infarction. Mol Ther. 2013;21:670-9
27. Jiang D, Liang J, Noble PW. Hyaluronan in tissue injury and repair. Annu Rev Cell Dev Biol. 2007;23:435-61
28. Thygesen K, Alpert JS, Jaffe AS, Simoons ML, Chaitman BR, White HD. et al. Third universal definition of myocardial infarction. Nat Rev Cardiol. 2012;9:620-33
29. West DC, Hampson IN, Arnold F, Kumar S. Angiogenesis induced by degradation products of hyaluronic acid. Science. 1985;228:1324-6
30. Gao F, Cao M, Yang C, He Y, Liu Y. Preparation and characterization of hyaluronan oligosaccharides for angiogenesis study. J Biomed Mater Res B Appl Biomater. 2006;78:385-92
31. Chi NH, Yang MC, Chung TW, Chen JY, Chou NK, Wang SS. Cardiac repair achieved by bone marrow mesenchymal stem cells/silk fibroin/hyaluronic acid patches in a rat of myocardial infarction model. Biomaterials. 2012;33:5541-51
32. Park J, Kim B, Han J, Oh J, Park S, Ryu S. et al. Graphene oxide flakes as a cellular adhesive: prevention of reactive oxygen species mediated death of implanted cells for cardiac repair. ACS Nano. 2015;9:4987-99
33. Swirski FK, Nahrendorf M. Imaging macrophage development and fate in atherosclerosis and myocardial infarction. Immunol Cell Biol. 2013;91:297-303
34. Giannitsis E, Katus HA. Cardiac troponin level elevations not related to acute coronary syndromes. Nat Rev Cardiol. 2013;10:623-34
35. Bertinchant JP, Robert E, Polge A, Marty-Double C, Fabbro-Peray P, Poirey S. et al. Comparison of the diagnostic value of cardiac troponin I and T determinations for detecting early myocardial damage and the relationship with histological findings after isoprenaline-induced cardiac injury in rats. Clin Chim Acta. 2000;298:13-28
36. Yoshizumi T, Zhu Y, Jiang H, D'Amore A, Sakaguchi H, Tchao J. et al. Timing effect of intramyocardial hydrogel injection for positively impacting left ventricular remodeling after myocardial infarction. Biomaterials. 2016;83:182-93
37. Nagajothi N, Trivedi A. Biomarkers in acute cardiac disease. J Am Coll Cardiol. 2006;48:2358
38. Kawaguchi M, Takahashi M, Hata T, Kashima Y, Usui F, Morimoto H. et al. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation. 2011;123:594-604
39. Kretzschmar K, Post Y, Bannier-Hélaouët M, Mattiotti A, Drost J, Basak O. et al. Profiling proliferative cells and their progeny in damaged murine hearts. Proceedings of the National Academy of Sciences. 2018;115:E12245-54
40. Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3:23-35
41. Imhof BA, Aurrand-Lions M. Adhesion mechanisms regulating the migration of monocytes. Nat Rev Immunol. 2004;4:432-44
42. Weninger W, Biro M, Jain R. Leukocyte migration in the interstitial space of non-lymphoid organs. Nat Rev Immunol. 2014;14:232-46
Author contact
![]() Corresponding authors: C Gong and Q Wu. E-mail: chygong14com, cellwqjcom.
Corresponding authors: C Gong and Q Wu. E-mail: chygong14com, cellwqjcom.