Impact Factor
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Conclusion
Experimental Section
Supplementary Material
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(6):1809-1824. doi:10.7150/thno.27313 This issue Cite
Research Paper
Water-Mediated Nanostructures for Enhanced MRI: Impact of Water Dynamics on Relaxometric Properties of Gd-DTPA
Franca De Sarno1,2*, Alfonso Maria Ponsiglione1,2*, Maria Russo1,2, Anna Maria Grimaldi3, Ernesto Forte3, Paolo Antonio Netti1,2,4, Enza Torino1,2,4 ![]()
1. Department of Chemical, Materials Engineering & Industrial Production, University of Naples Federico II, Piazzale Tecchio 80, 80125 Naples, Italy.
2. Center for Advanced Biomaterials for Health Care, CABHC, Istituto Italiano di Tecnologia, IIT@CRIB, Largo Barsanti e Matteucci 53, 80125 Naples, Italy.
3. IRCCS SDN, Via E. Gianturco 113, 80143 Naples, Italy.
4. Interdisciplinary Research Center on Biomaterials, CRIB, Piazzale Tecchio 80, 80125 Naples, Italy.
*Equal Contribution
Received 2018-5-17; Accepted 2019-1-19; Published 2019-2-28
Abstract

Recently, rational design of a new class of contrast agents (CAs), based on biopolymers (hydrogels), have received considerable attention in Magnetic Resonance Imaging (MRI) diagnostic field. Several strategies have been adopted to improve relaxivity without chemical modification of the commercial CAs, however, understanding the MRI enhancement mechanism remains a challenge.
Methods: A multidisciplinary approach is used to highlight the basic principles ruling biopolymer-CA interactions in the perspective of their influence on the relaxometric properties of the CA. Changes in polymer conformation and thermodynamic interactions of CAs and polymers in aqueous solutions are detected by isothermal titration calorimetric (ITC) measurements and later, these interactions are investigated at the molecular level using NMR to better understand the involved phenomena. Water molecular dynamics of these systems is also studied using Differential Scanning Calorimetry (DSC). To observe relaxometric properties variations, we have monitored the MRI enhancement of the examined structures over all the experiments. The study of polymer-CA solutions reveals that thermodynamic interactions between biopolymers and CAs could be used to improve MRI Gd-based CA efficiency. High-Pressure Homogenization is used to obtain nanoparticles.
Results: The effect of the hydration of the hydrogel structure on the relaxometric properties, called Hydrodenticity and its application to the nanomedicine field, is exploited. The explanation of this concept takes place through several key aspects underlying biopolymer-CA's interactions mediated by the water. In addition, Hydrodenticity is applied to develop Gadolinium-based polymer nanovectors with size around 200 nm with improved MRI relaxation time (10-times).
Conclusions: The experimental results indicate that the entrapment of metal chelates in hydrogel nanostructures offers a versatile platform for developing different high performing CAs for disease diagnosis.
Keywords: nanoparticles, MRI, hydrogels, hydrodenticity, contrast agents
Introduction
Magnetic Resonance Imaging (MRI) is a promising technology in biomedical research and clinical diagnosis and provides high spatial resolution without the use of ionizing radiation [1]. Contrast Agents (CAs) are metal ions injected prior to MRI scanning in the human body to enhance the signal intensity and improve the contrast between healthy and pathological tissues [2]. Among them, the most extensively used CAs in the clinical practice are paramagnetic gadolinium (Gd) chelates [3, 4]. Despite their widespread use, these Gd-based CAs are limited by low sensitivity [5, 6]; therefore, a large amount of these paramagnetic agents needs to be used to obtain an appropriate diagnostic contrast [7]. In many cases, the exposure to Gd-based MRI CAs in patients with compromised renal function is associated with Nephrogenic Systemic Fibrosis (NSF) [8], a systemic disease that can lead to death [9]. Moreover, recent studies in humans conducted by McDonald, Kanda and coworkers [10-12] have revealed that these compounds are retained in some tissues (i.e. kidneys, bone, skin and brain) also in healthy subjects.
In this framework, the opportunity to develop a safer and more effective probe for MRI [13] starting by clinical approved CAs is a significant and valuable challenge [14-16].
It is well known that MRI CAs are classified based on their relaxivity, which represents the rate of change in longitudinal (r1) or transverse (r2) relaxation times of the water protons per mM concentration of metal ions. The relaxivity determines the enhancement of the image contrast and it can be influenced by many factors, such as molecular motion, size, rigidity and possible binding between Gd-chelates and other macromolecules [2, 4, 13].
The Solomon-Bloembergen-Morgan (SBM) [17] theory explains the principles of the relaxation enhancement and describes some variables, called characteristic times, which can be manipulated to produce changes in the relaxivity of a Gd-chelate. Among these parameters, the water exchange rates, the hydration number and the rotational correlation time of the metal chelate play a key role in the design of high relaxivity CAs [18].
As reported by Port et al. [19], rigidification of Gd-based CAs would be favorable to an increase in the relaxivity of the metal chelate since the presence of the ligand around the Gd ion induces shortening of the residence lifetime of the inner-sphere water molecules (τM) [20]. Also, they hypothesized that the presence of a rigid coordination cage of a chelate should limit its intramolecular conformational motions, which distorts the ligand field at the metal centre due to solvent molecules collisions, thus influencing the electronic relaxation times (τS1 and τS2) [19]. To assess the rigidification strategy, Port synthetized a constrained derivative of Gd-PCTA12, Gd-cyclo-PCTA12, in which one ethylene bridge connecting two nitrogen atoms of the triamine block is replaced by a cyclohexylene bridge, and the impact of rigidification was studied by comparing the physicochemical and relaxometric properties of both gadolinium MRI contrast agents, Gd-PCTA12 and Gd-cyclo- PCTA12.
Other experimental approaches studied by Decuzzi et al. [21, 22] proved that geometrical confinement could limit the mobility of water molecules and thereby enhance the relaxation response of Gd-based CAs without their chemical modification. In particular, they observed that nanometric pores of silica microparticles increase the rotational correlation time (τR) of Gd-DTPA (inner-sphere effect), which cannot tumble freely being adsorbed on the walls of the 100 nm pores. At the same time, it also increases the diffusion correlation time (τD) for water molecules (outer-sphere effect), which are geometrically confined and forced to interact longer with Gd-DTPA adsorbed to the inner pore surface [22]. Through the confinement strategy, a poor increment of the relaxivity can be obtained without modifying the chemical structure of the CA.
As advancement of the geometrical confinement [23], Courant et al. [24] and Callewaert et al. [25], showed that biocompatible hydrophilic hydrogels can be exploited to produce high water content nanoparticles (NPs) encapsulating the metal chelate. Inside the hydrogel, which creates a favorable aqueous environment for Gd-based CAs [26, 27], the rotational motion of the encapsulated CA (Gd-DOTP, Gd-DOTA and Gd-DTPA) is restricted and its magnetic properties are amplified.
In recent studies [14, 15, 28-30], the use of polymers to develop safer, more efficient and smart MRI CAs has significantly increased. Biopolymers, and particularly polysaccharides, have received considerable attention because the most of them are non-toxic and have various derivable groups (e.g. hydroxyl, carboxyl and amino groups) allowing easy chemical modifications or reaction with functional molecules [31-33]. Natural-occurring polysaccharides, like chitosan, alginate, heparin and hyaluronic acid, offer a suitable platform to produce nanoparticles due to their biodegradability, biocompatibility and ease in molecular modification and formation processes, such as by ionic or covalent crosslinking, ion-complex or self-assembly [33]. These properties not only make polysaccharides very promising materials for drug delivery but also proved to be useful in designing nanostructures for enhanced MRI [24, 34-37].
In our recently published works [5, 38], we have initially analysed the impact that hydrophilic biopolymer networks have on the relaxivity of Gd-based CAs and explained the role of water in the interaction between polymers and metal chelates. This concept, called “Hydrodenticity”, has been the subject of further investigations as reported by Russo et al. [39]. In a former work published by Russo and co-worker [6], crosslinked Hyaluronic Acid NanoParticles (cHANPs) containing a Gd-chelate (Gd-DTPA), are synthesized through a microfluidic platform that allows a high degree of control over particle synthesis, enabling the production of monodisperse particles as small as 35 nm for MRI applications. The relaxivity (r1) achieved with the cHANPs is 12-times higher than Gd-DTPA. Within cHANPs, the properties of Hydrodenticity can be modulated to obtain the desired mesh size, crosslink density, hydrophilicity and loading capability, as reported by Russo et al. [39, 40]. Moreover, they proved that an increase of the crosslinking degree of biopolymer can induce the enhancement of relaxivity by restricting molecular tumbling while maintaining the switching property [41] and allowing easy access of water throughout the structure, which is a key feature in MRI CAs. The possibility to adopt a unique platform to tune the hydrogel structural parameters and, consequently, increase the relaxivity of a metal chelate without any chemical modification, could have a great impact on the clinical outcome. In fact, thanks to their improved relaxometric properties, cHANPs could ensure a brighter contrast with a lower amount of metal chelate, thus enabling the potential reduction of the administration dosage as approved for clinical use.
In a further work [42], we reported an efficient way to produce Hybrid Core-Shell (HyCoS) NPs composed of a Chitosan core and a shell of Hyaluronic Acid (HA) with improved relaxometric properties (up to 5-times than the commercial CA). Subsequently, the same nanosystem is used to develop a new nanoprobe for simultaneous Positron Emission Tomography (PET)/MRI acquisitions as reported in our more recent publication [43].
Based on the above-reported works, it has been finally demonstrated that the polymer architecture affects some characteristic parameters of the metal chelate and tunes its relaxometric properties [24, 39, 44]. Moreover, it is clear that crosslinked biopolymers can have a significant role in overcoming the limitations of clinically relevant CAs without their chemical modification and as a compound in the design of advanced nanostructures with improved safety profile and switchable relaxometric properties. Indeed, it is known that the functional features as well as the swelling behaviour of hydrogels are influenced by the hydration degree, which can be likely modulated by changing the chemical composition of the system [45-47].
Here, we aim to highlight the basic principles ruling biopolymer-CA interactions in the perspective of their influence on the relaxometric properties of the CA by adopting a multidisciplinary experimental approach. HA [26] is used as a model polymer because of its biocompatibility and high hydrophilicity. We characterize, physically and chemically, the interactions between hydrophilic biopolymers and Gd-based CAs. In this theoretical framework, the peculiar effect of Hydrodenticity on the polymer conformation and the formation of the stable water compartments responsible for the enhancement of the MRI signal is introduced and discussed. Finally, we use the acquired knowledge about polymer-CA systems to apply the concept of Hydrodenticity to the design of Gd-based polymer NPs with enhanced relaxometric properties.
Results
Changes in polymer conformation induced by a Gd-based contrast agent
The polymer conformation can be modified by the affinity with the solvent solution [48, 49]. Furthermore, the addition of a solute can still induce a change in the polymer conformation. In our previous work, we proved that the relaxivity of CAs can be modulated combining them with macromolecules or polymers [5]. Therefore, the understanding of the interaction between polymers and CAs in aqueous solution could be critical to tune the relaxometric properties of CAs. We aim to show how the presence of the Gd-DTPA in the aqueous solution can influence the behaviour of the polymer matrix and, on the other side, how these adjustments of the polymer conformation can govern the characteristic correlation times of the Gd-DTPA [5, 6, 39].
To investigate thermodynamic interactions between polymer and contrast agent, HA and Gd-DTPA respectively, are selected to be tested by Isothermal Titration Calorimetry (ITC). We aim to take advantage of the molecular interactions that are accompanied by some level of heat exchange between the interacting system and its surrounding medium; indeed, these interactions can be evaluated, at constant temperature, through the ITC. Basic principles of this technique have been widely discussed elsewhere [50, 51].
Titration experiments are conducted injecting a solution of Gd-DTPA in the ITC cell containing the polymer solution. Different HA concentrations, ranging from 0.3 to 0.7% w/v, are tested and more representative results are reported in Figure 1 (peaks above the baseline represent exothermic phenomena while peaks below the baseline represent endothermic phenomena). It is clear that significant enthalpy variations are obtained in the titration experiments (Figure 1A-C), which induces changes in the polymer chains' conformation as explained below. Since Figure 1A-C show ITC thermograms varying the HA concentration in the sample cell, a wide range of Gd-DTPA/HA molar ratios is examined and the relative energetic contribution and enthalpy values are calculated by integrating peaks of the experimental curves and are reported in Figure 1D. Simple dilution of Gd-DTPA in water (Figure S1) exhibits only small constant exothermic peaks over the whole experiment.
Titration curves of Gd-DTPA into aqueous polymer solutions at 25 °C. Calorimetric traces (heat flow against time) for (A) 0.3% w/v HA, (B) 0.5% w/v HA and (C) 0.7% w/v HA. In (D) it is reported the normalized enthalpy vs Gd-DTPA/HA molar ratio for Gd-DTPA in 0.3% w/v HA (red circles), in 0.5% w/v HA (blue triangles) and in 0.7% w/v HA (magenta diamonds). The curves were shifted vertically for clarity; y-offset were set at 2 (red circles), 0 (blue triangles) and -10 (magenta diamonds).
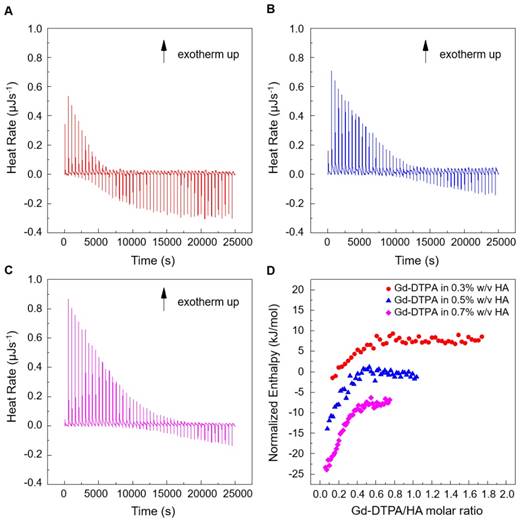
In Figure 1, it is worth noting that the energetic contribution decreases as the Gd-DTPA/HA molar ratio increases; thus, the higher is the concentration of HA in the sample cell, the higher is the Gd-DTPA concentration needed to observe endothermic peaks. It can also be noted that the endothermic contribution exceeds the exothermic one at the recurrent Gd-DTPA/HA ratio approximatively equal to 0.5 through all the experiments at different HA concentrations in the sample cell. It means that a specific energetic contribution is needed to induce the adjustment of the polymer conformation. Then, when the Gd-DTPA/HA molar ratio equals 0.5, the endothermic peaks start slightly increasing until reaching a plateau, which corresponds to the thermodynamic equilibrium established within the ternary system (polymer-CA-water). The measured energetic variation reflects the conformational changes of polymer chains due to the presence of the CA in solution and leads to the formation of stable sub-domains in which a balanced exchange of water molecules occurs between the polymer, the CA and the bulk.
This is confirmed by the relation between Gd-DTPA/HA molar ratio and enthalpy values showed in Figure 1D. Indeed, at low Gd-DTPA/HA ratio, the enthalpy term is negative, as expected for polyelectrolytes in water such as HA, which generally dissolve easily in aqueous media and behaves like a long, more or less randomly mobile chain and its conformation is governed by electrostatic forces [52]. Indeed, it is well-known that, at low polymer concentrations (dilute solution regime), the intrachain electrostatic interactions and interactions with the surrounding polyelectrolyte chain ions determine the chain conformations since the polyelectrolyte chains are separated from each other by average distances larger than their size [52, 53]. As Gd-DTPA/HA ratio increases the enthalpy increases too, meaning that a change in conformation is occurring due to the presence of a ionic compound (Gd-DTPA). Indeed, apart from the solvent type (e.g., water), the polymer conformation can be modified by the concentration of added co-solutes. In particular, in the presence of ionic compounds, a screening of the electrostatic repulsion between the charged monomers occurs at low salt concentration form a cloud surrounding the chain and the polymer collapses from an extended coil to a more compact conformation as the salt is added [52, 54, 55].
At Gd-DTPA/HA = 0.5, the enthalpy becomes constant, meaning that an equilibrium is reached. The attainment of this thermodynamic equilibrium derives from a water- mediated interaction occurring between HA and Gd-DTPA. As both hydrophilic components, HA and Gd-DTPA interact with the water by forming hydrogen bonds and by coordinating water molecules. This competitive behaviour generates a measurable heat that reflects the change in polymer chains conformation and the exchange of bound water molecules with the bulk, thereby, bringing the system to a more stable configuration. As also observed in other metal-polymer systems [56, 57], in the presence of small amounts of metal-chelate compounds, a change of the polymer structure occurs due to the weak macromolecule-metal interactions, which favors the formation of a new hierarchy in the structural organization of the polymer as compared with metal-free system.
In our previous paper [39], we preliminary showed how this new structural organization is able to affect the relaxometric properties of the system, as an effect of the new concept of Hydrodenticity, which will be further explained in the following paragraphs.
NMR study of DPTA interactions and water mobility in polymer solution
Since previous ITC measurements reported by Gouin et al. [58] have shown no binding energy between free Gd3+ ions and HA, we hypothesize that polymer conformational changes are mainly induced by the presence of the chelating macromolecule, DTPA. NMR spectroscopy is used to confirm this hypothesis. NMR spectra are calculated for HA and DTPA solutions.
Considering the spectra of the only DTPA and HA (Figure 2A-B), whose characteristic peaks are circled in blue and red respectively, the observations of DTPA/HA solutions at different molar ratios are reported (Figure 2C-I). The molar ratio ranges from 2 to 100 and is obtained by decreasing the HA concentration from 150 to 10 µM.
(A) 1H NMR spectrum of DTPA; (B) 1H NMR spectrum of HA; (C-I) 1H NMR spectra of DTPA/HA solutions at different molar ratios, from DTPA/HA = 2 to DTPA/HA = 100. Characteristic peaks of DTPA and HA are highlighted in blue and red respectively.
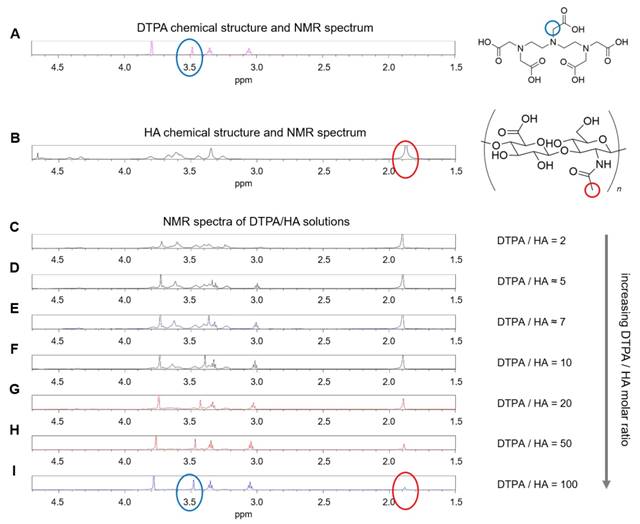
In Figure 2, it can be observed that the characteristic DTPA peak at 3.50 ppm (s, 2 H, CH2-COOH) is influenced by the presence of HA in solution. In fact, it seems to shift and reduce its intensity far more than the other peaks by increasing the HA concentration. As an example, the shift is evident by comparing Figure 2I, where the DTPA peak is highlighted in blue, with Figure 2C, where the signal is dramatically reduced. It results that an interaction between the two components of the system exists and generates changes in the NMR spectrum of the solution. This can be due to electrostatic repulsion between the two anionic macromolecules, HA and DTPA.
Through NMR-DOSY, instead, we investigate how the presence of both HA and Gd-DTPA can affect the mobility of water molecules.
Figure 3 shows the normalized time-dependent self-diffusion coefficient of water in both polymer solutions (Figure 3A) and polymer-CA solutions (Figure 3B). For short diffusion delays, the measured self-diffusion coefficient D is nearly equal to the free self-diffusion coefficient D0 of water at 25°C (2.5·10-9 m2/s), since the molecules travel over a short distance and only a few of them feel the surrounding macromolecules. As the diffusion time increases, more water molecules go through these restrictions and the self-diffusion coefficient reaches a plateau value.
(A) Normalized time dependent water self-diffusion coefficient in 0.1% w/v HA (squares), 1% w/v HA (triangles), 2% w/v HA (flipped triangles), 3% w/v HA (diamonds). (B) Normalized time dependent water self-diffusion coefficient in 1% w/v HA (triangles), 5 μM Gd-DTPA in 1% w/v HA (flipped triangles) and 30 μM Gd-DTPA in 1% w/v HA (stars).
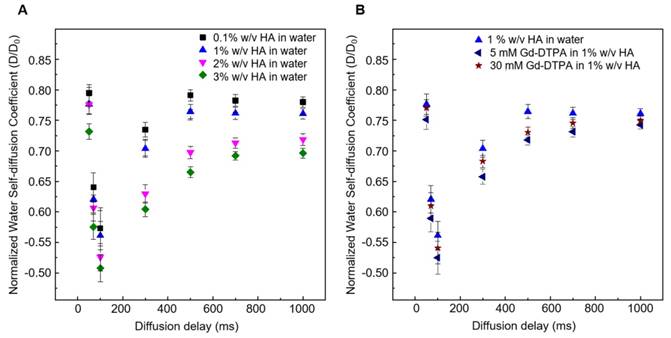
We can hypothesize that the presence of Gd-DTPA competes with those HA-molecular sites beared by water molecules and that are responsible for polymer hydration and hydrogel formation. As highlighted with ITC results, the polymer conformation can be modified by the presence of Gd-DTPA, which could interplay with the water molecules and with the formation of hydrogen bonding. NMR-DOSY measurements are carried out to assess these hypothesized changes in water mobility. It can be observed that, in the case of the ternary system, the diffusivity of water beyond decreases, as expected for solvent molecules within polymer matrices or in confined environments [59, 60], suggesting that the polymer-CA combination affects the water mobility more than the polymer itself.
Figure 3A clearly shows that water diffusion behaviour is affected by the polymer concentration. In particular, the diffusion coefficient decreases at increasing polymer concentration. Besides, Figure 3B shows the additional contribution of the CA to the water mobility. In fact, the presence of Gd-DTPA, even at relatively low concentrations (5 - 30 µM), can further reduce the value of the water self-diffusion coefficient for both short and long diffusion times.
It is worth noting that low Gd-DTPA concentrations are chosen (Figure 3B) because Gd-DTPA is highly paramagnetic and it can interfere with NMR measurements [61, 62], while the HA concentrations (0.1 - 3% w/v) are slightly higher than those used in the ITC experiments to highlight and make more evident the differences in diffusion behaviour between samples. In particular, as illustrated in Figure 3B, a fixed polymer concentration of 1% w/v is selected to show the effect of CA on the diffusion of water molecules.
A data comparison between ITC and NMR spectra confirms the hypothesized fundamental properties behind the concept of Hydrodenticity: the ability of Gd-DTPA to induce changes in polymer conformation and in water mobility.
Water dynamics within hydrated polymer matrix containing Gd-DTPA
To analyse further the role of water mobility in the Hydrodenticity, a study of the dynamics and behaviour of water molecules is needed. Within hydrated polymer matrices (hydrogels), containing metal chelates, water molecules mediate polymer-CA interactions and, therefore, play a dual role: on the one hand, the amount of absorbed water [63, 64] and its interaction with the hydrogel structure affects the chain motion of the hydrophilic polymer; on the other, the mobility of water molecules in the polymer matrix is responsible for the relaxometric properties of the CA.
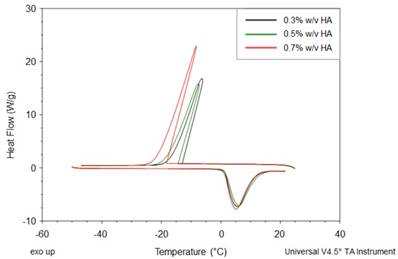
We investigate the water dynamics in water-HA systems, with and without Gd-DTPA, using the Differential Scanning Calorimetry (DSC). We focus on the thermal effects that the polymer (Figure 4) and the CA (Table 1) have on the water dynamics. According to the literature, indeed, the crystallization of water changes with the polymer concentration and with the hydration degree [46].
DSC thermograms of HA at different concentrations (0.3, 0.5 and 0.7% w/v).
Melting (Tm) and crystallization (Tc) temperatures for free Gd-DTPA in water and HA solutions with and without CA.
| Sample | Concentration | Tc (°C) | ΔH (J/g) | Tm (°C) | ΔH (J/g) |
|---|---|---|---|---|---|
| Gd-DTPA | 60 µM 80 µM 100 µM 140 µM | -7.47 -8.36 -8.00 -9.27 | 218.7 245.0 239.7 238.6 | 5.37 6.52 5.36 5.17 | 249.0 255.0 266.0 269.9 |
| HA | 0.3 % w/v 0.5 % w/v 0.7 % w/v | -6.40 -7.72 -8.31 | 243.0 248.4 239.2 | 5.62 6.05 5.21 | 260.8 253.3 270.0 |
| HA + Gd-DTPA | 0.3 % w/v + 60 µM 0.5 % w/v + 100 µM 0.7 % w/v + 140 µM | -9.73 -7.88 -6.24 | 229.1 244.9 254.5 | 7.94 6.47 5.02 | 255.1 273.0 259.0 |
In Figure 4, thermograms of water-polymer systems at different HA concentrations (0.3 - 0.7% w/v) are displayed. We can observe that, during the cooling phase, the crystallization peaks shift to lower temperatures and lower enthalpy values. As expected, the enthalpy, given as the peak area, reaches its maximum value at the highest HA concentration (0.7% w/v).
Table 1 shows a comparison of melting (Tm) and crystallization (Tc) temperatures between HA solutions with and without Gd-DTPA (concentration range: 60 - 200 µM). It can be noted that the transition properties remained unaffected in the presence of the CA, suggesting that the influence of the polymer on the thermal behaviour of water is predominant with respect to the CA at the selected concentration.
Relaxation times, rates and relaxivity of the polymer matrix
The existence of a water-mediated interaction between Gd-DTPA and HA, observed through ITC, NMR and DSC, and the effect of the polymer conformation on the characteristic correlation time of the metal chelate could explain the boosting of the relaxivity in the studied systems.
Relaxometric properties are investigated using time-domain relaxometry on two different systems: non-crosslinked and crosslinked polymer matrix (0.5% w/v HA) containing Gd-DTPA.
In the latter case, rheological and chemical- physical properties of the polysaccharide can be modulated by changing the crosslinker (DVS) concentration, as known as crosslinking density. In fact, thanks to the presence of hydrophilic groups in the skeleton of HA, the hydrogel is able to uptake a large amount of water. Under these conditions, water is in an abnormal aggregate state that influences the relaxivity of hydrated Gd-DTPA.
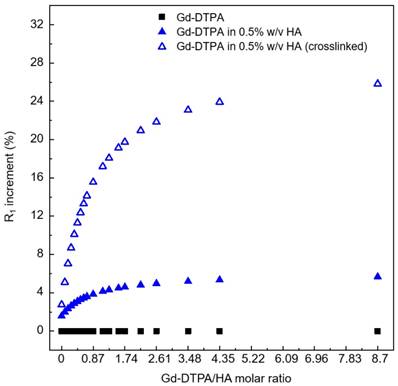
Figure 5 shows the results of relaxometric measurements for the hydrogel system (0.5% w/v HA) studied by loading different concentration of Gd-DTPA. The hydrogel system is analysed and compared to the free Gd-DTPA solution. In particular, we display the increment in the percentage of the paramagnetic relaxation as a function of Gd-DTPA/ HA ratio.
Increment in longitudinal relaxation rate, R1, at different Gd-DTPA/HA ratio (from 0 to 9) for: free Gd-DTPA in water (black squares); Gd-DTPA in 0.5% w/v HA solution (blue filled triangles); Gd-DTPA in 0.5% w/v HA crosslinked with DVS (blue empty triangles). The R1 increment is calculated in percentage with respect to the corresponding R1 of Gd-DTPA in water. A fast increment in R1 is observed until a 2.5 Gd-DTPA/HA ratio (Gd-DTPA concentration equal to 300 µM). For higher ratios (i.e. higher Gd-DTPA concentrations), the R1 increment reaches a plateau.
The increment in percentage of the longitudinal relaxation rate (R1) has been calculated as follows (Equation 1):
(1)
Details on relaxation times and rates values are reported in Table S1 of the Supporting Information.
We find that the stronger is the interaction between Gd-DTPA and HA, the better is the MRI enhancement. Moreover, the crosslinked system is much more efficient than the non-crosslinked one. Indeed, in the crosslinked system, since the enhancement reaches a plateau at Gd-DTPA/HA molar ratio equal to 2.5 (i.e. at 300 µM of Gd-DTPA in 0.5% w/v HA water solution), it is not necessary to overload the system with Gd-DTPA in order to achieve higher relaxation. It is worth noting that, for both studies, with and without crosslinker, the Gd-DTPA concentration of 200 μM seems to represent a threshold for the maximum effect of the hydrogel on the Gd-DTPA relaxation mechanism.
Furthermore, transversal relaxation times (T2) and rates (R2) have also been measured and reported in Table S1 of the Supporting Information.
Figure 6 displays a schematic representation of the hydrogel network formation, even in the presence of the crosslinking agent, and its influence on the polymer conformation.
Formation mechanism affects hydrogel network structure: schematic representation of: formation of a hydrogel of hyaluronic acid in water (bottom); complex HA-Gd-DTPA structure (middle); crosslinked hydrogel network containing the contrast agent (top).
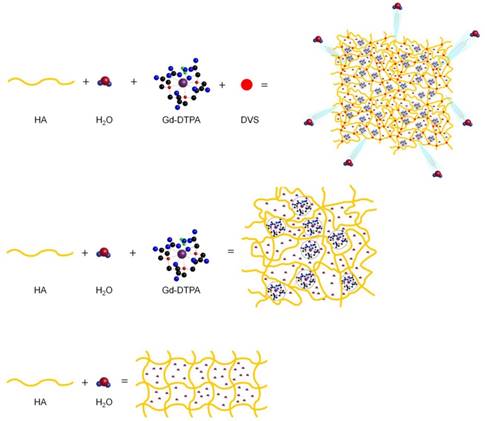
We hypothesize that the boosting of Gd-DTPA relaxivity in a hydrogel matrix is due to a proper complexation between the polymer and the CA in solution, mediated by the water and further amplified by the addition of a crosslinker. It is confirmed that the reached equilibrium among osmotic pressure and elastodynamic forces of the polymer meshes and hydration degree of the CA in the matrix are able to tune finely the relaxometric properties of the metal chelates in the ternary system. The overall ensemble of these phenomena is defined as Hydrodenticity [39].
Case study: production of polymer particles based on Hydrodenticity
Recent recommendations from Food and Drug Administration (FDA) and European Medicine Agency (EMA) about the Gd deposition in the brain and other tissues have highlighted the importance to design polymer biocompatible NPs with enhanced relaxivity without chemical modification of the clinical relevant CAs [11, 65]. Thus, crosslinked NPs formed by HA, a biodegradable, biocompatible, non-toxic, non-immunogenic and non-inflammatory linear polysaccharide [66], could represent a successful candidate among nanovectors for MRI applications [67]. Indeed, in the last decades, it is undisputed the growing research interest toward the therapeutic action of HA and in developing new diagnostic tools based on this polymer [67]. In this work, starting from the above-presented results, we aim to apply the Hydrodencity in the design of biocompatible hydrogel nanostructures to obtain improved relaxometric properties. We propose a concrete example of the concept of Hydrodenticity applied to the production of crosslinked HA NPs for MRI, loaded with Gd-DTPA. An emulsion-based method is used to obtain stable W/O nanoemulsions as templates.
Study of emulsion stability
Stable W/O emulsions are prepared by stirring appropriate amounts of the oil phase (Mineral oil) and an aqueous phase containing different concentrations of Span-80 (S80) or S80 with Tween-85 (S80/T85). The pH, ranging from 12 to 14 is adjusted by adding appropriate amounts of NaOH from a stock solution (0.2 M). Further details are reported in the Materials and Method Section. As expected, in the absence of any surfactant, W/O emulsions prepared in the same conditions split very rapidly in two phases due to their unfavourable thermo-dynamic state. Visual comparison, turbidimetry and backscattering are successfully used to study emulsion stability (Figure S2 and S3) [68]. Comparing emulsions obtained at different W/O ratio, 10:90 and 20:80, but at the same concentration of surfactant, the stability is more extended for emulsions with lower water content. In particular, a formulation 10/90 W/O volume ratio containing S80 (1% w/v) and T85 (0.5 % w/v) resulted the more stable. However, even though the stability of the emulsion is crucial to reduce polydispersity, an alkaline environment (addition of NaOH) is necessary for the crosslinking reaction to take place. Indeed, Balazs and Leshchiner [69] showed that the crosslinking reaction starts shortly after addition of DVS (5 - 10 min) and, that, 1 hour is sufficient for the completion of the reaction [70, 71]. By these requirements, to conduct the experimental campaign, we select the formulation with S80 (1% w/v) and NaOH (0.2 M) as the optimal trade-off to obtain an emulsion stable for at least 3 hours (Figure S2 and S3), enough for the DVS to react.
Preparation of DVS-crosslinked nanoparticles with and without CA
The exploitation of the best process conditions to design biocompatible nanostructures based on Hydrodenticity and control their relaxation parameters for MRI application is reported. In particular, the effect of the homogenization, HA concentration and the role of the crosslinking reaction is analysed. Different experimental parameters and conditions are tested and details are reported in the Materials and Methods section. A preliminary mixing is performed at 5000 or 7000 rpm for 10 min, by keeping constant the temperature at 25 °C. A 5000 rpm speed is preferred to avoid and uncontrolled increase of the temperature.
After the homogenization, a crosslinking reaction is performed at high pH values (12 - 14) and creates sulfonyl bis-ethyl linkages between the hydroxyl groups of HA [72]. This crosslinking method has the advantage of occurring at room temperature, which limits the degradation of HA in alkaline solutions. Even though the starting material DVS is highly reactive and toxic, the biocompatibility of the HA-DVS hydrogels is confirmed by histological analysis [73].
In our protocol, a study of the modalities of injection of the crosslinking agent at different steps of the homogenization process has shown that only when DVS is added after the homogenization step spherical NPs are obtained. On the contrary, when the addition of the crosslinker is performed at any time point during the homogenization phase, a shear stress behavior of the polymer phase, interfering with the formation of those particles, is observed (Figure S4).
The best experimental condition for production of crosslinked NPs is reached at 0.045% v/v DVS (Table S2).
Based on these results and using the same process conditions, loaded NPs are obtained by adding the CA in the water phase of the emulsion. Among several FDA approved CAs, we have chosen to encapsulate a Gd chelated, Gd-DTPA (9.13 mM).
Purification and characterization of HA-NPs
Ultracentrifugation (UC) and dialysis are performed to purify HA NPs. Dynamic Light Scattering (DLS) measurements are made on aqueous dilute NP suspension (1:10). The smaller NPs' size without CA (217.57 ± 34.65 nm) is obtained at 0.25% w/v of HA solution. At higher polymer concentration (0.5% w/v) particle size is higher (401.67± 77,65 nm), while the formulation with 0.1% w/v HA shows a reverse phenomenon with larger particles (760.15 ± 86 nm), probably due to less stability of the nuclei that tend to coalesce. When Gd-DTPA is added to the process, the particle size at HA 0.25% w/v is slightly increased (258.77 ± 15.65 nm) for the same process conditions. After purification, NPs are investigated by electron microscopy techniques (SEM and TEM). The morphology of the NPs observed revealed that the particles are spherical in shape and monodisperse (Figure S5).
Loading Capability (LC) and Encapsulation Efficiency (EE) is determined through ICP-MS by comparing the theoretical amount initially used to prepare the particles and the Gd encapsulated in the system after ultracentrifugation. The higher encapsulation results for 0.25 % w/v HA (1:2 w/w HA/Gd- DTPA ratio). Results show that probably ionic nature of Gd-DTPA impacts on its encapsulation. In addition, HA NPs result stable up to 6 months or more, at 4°C and the release of Gd-DTPA from HA NPs is tested at physiological condition (up to 48 hr), because it is expecting that the clearance is shorter than 48 hr.
The zeta potential value of the 0.25% HA-NPs, with and without CA, indicate that they had a negatively charged surface (-37.4 ± 1.34 mV and -31.8 ± 0.88 mV, respectively), due to the carboxylic group of HA. IR measurements were also performed (Figure S6).
Relaxivity studies
T1 and T2 measurements at 60 MHz (1.5 Tesla) and 120 MHz (3 Tesla): Relaxivity and relaxation times are measured for both unloaded and loaded NPs and compared with free Gd-DTPA solution. Measurements are performed on a 120 MHz (3 Tesla) MRI system and on a 60 MHz (1.5 T) benchtop relaxometer.
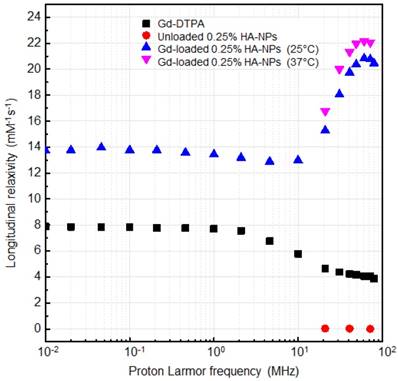
Relaxivity results obtained at 120 MHz are presented on a per millimolar Gd basis in Figure 8 and show a maximum r1 of 33.3 s-1mM-1 (i.e. 10 times higher compared to free Gd-DTPA). Even though all the proposed formulations of Gd-DTPA-loaded HA nanostructures show an increase of the r1 signal, as reported in Figure 7A, the highest boosting of the relaxivity is provided by the NPs obtained using the formulation at 0.25% HA and 1:2 w/w HA/Gd-DTPA (Figure 7A-C).
NMRD profiles showing relaxivity of Gd-DTPA in water (squares), unloaded 0.25% HA-NPs (circles), Gd-loaded 0.25% HA-NPs at 25°C (triangles) and 37°C (flipped triangles).
(A) Relaxivity values r1 determined at magnetic field strengths of 3T for different set of HA-NPs with respect to free Gd-DTPA in water. (B) Longitudinal relaxation rate (1/T1) versus Gd-DTPA concentration for free Gd-DTPA in water and for HA-NPs at different polymer concentrations loaded with Gd-DTPA. (C) T1-weighted images of Gd-DTPA, unloaded HA-NPs (used as control) and HA-NPs at different polymer concentrations loaded with Gd-DTPA. All samples are imaged at 3T, 25°C, using standard spin echo (SE) sequence. (D) Distribution of longitudinal relaxation times of (T1) of 200 μM Gd-DTPA in water (squares), unloaded 0.25% HA-NPs (circles) and 0.25% HA-NPs loaded with 200 μM Gd-DTPA (triangles).
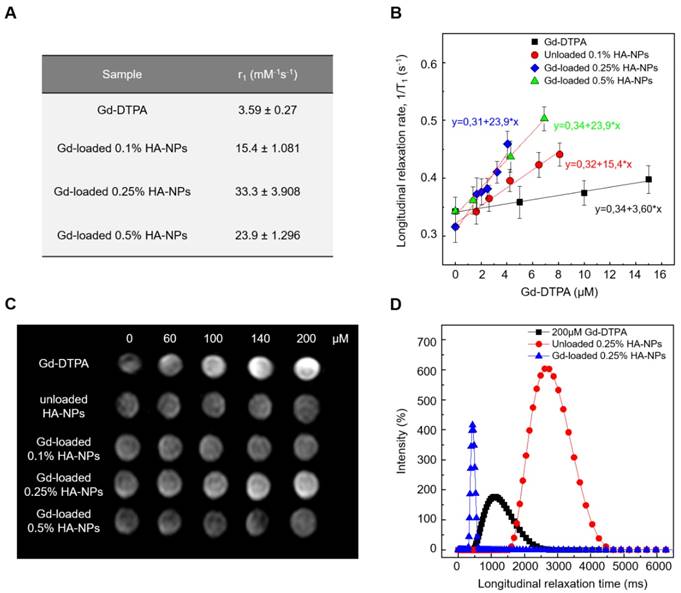
T1 relaxation time distributions at 37°C and 60 MHz are investigated for loaded and unloaded NPs (HA at 0.25% w/v) as well as for free Gd-DTPA solution (Figure 7D). Compared to the 200 µM free Gd-DTPA solution, which shows a broad distribution around 1000 ms, NPs loaded with 200 µM Gd-DTPA exhibit an excellent T1 distribution with a sharp peak centered below 500 ms. Gd concentration within loaded NPs was determined through Inductively Coupled Plasma Mass Spectrometry (ICP-MS).
Loaded NPs perform far better even compared to the unloaded ones, whose distribution appears to be broad and centered around 2800 ms.
It is worth mentioning that, compared to T1 distribution for bulk water (3600 ms), unloaded NPs' distribution shows that a slight contribution to the longitudinal relaxivity is ascribable to the crosslinked polymer nanostructure, which is able by itself to tune the water mobility for a non-nanostructured material. The contribution, therefore, to the overall relaxivity is further enhanced in the ternary system, thanks to the water-mediated interaction between the polymer and metal chelate.
Modeling of NMR dispersion: NMRD profile
The NMRD profiles as a function of the static magnetic field of the aqueous solutions of Gd-DTPA and loaded and unloaded NPs (Figure 8) are set up to establish the effects caused by Hydrodenticity functionalities on the parameters that determine the observed relaxivities. The longitudinal relaxation rates are recorded at 37°C as a function of resonance frequency and according to NP Gd-loading obtained by ICP-MS. The NMRD experimental curve for free Gd-DTPA shows a plateau in longitudinal relaxivity at low fields and significantly decreases as the applied magnetic field increases starting from 1 MHz. Conversely, longitudinal relaxivity (r1) for loaded NPs (HA at 0.25% w/v) is characterized by the presence of a low-field plateau and a gradual increase starting from 10 MHz, reaching a “dispersion peak” between 60 and 70 MHz. The same peak and trend in the high field region (20 - 70 MHz) is observed for both at 25°C and 37°C. As a control, unloaded NPs do not exhibit increase in relaxivity in this field region, confirming that the nanohydrogel structure containing Gd-DTPA contributes to slowing the chelator's tumbling motion and allows water exchange thanks to its hydrophilic properties, as hypothesized in the concept of Hydrodenticity.
Discussion
Theory of the Gado-Mesh formation
This work reports the use of polymer hydrogels for boosting the relaxivity of clinical relevant CAs.
Results show that a spontaneous complexation exists between HA and Gd-DTPA due to thermodynamic interactions as demonstrated by both calorimetric and NMR measurements. It is already known that, in an aqueous polymer solution, solvent affinity can alter polymer conformation by inducing local solvation of the structure that influences the number of available conformations of the polymer chains. Moreover, the introduction of another soluble ionic component, such as Gd-DTPA, in the system can induce further changes in polymer conformation, detectable via diverse thermodynamic approaches [74-77]. Based on our findings, we prove for the first time that these conformational changes, induced by the metal chelate, contribute to an increase in CA's relaxivity.
To tune the MRI enhancement, we exploit the versatile structural characteristics of HA. In fact, the high porosity of the hydrogel, which shows a mesh-like structure, is controlled by varying the density of covalent crosslinking [78, 79]. Moreover, the presence of negatively charged groups and the degree of crosslinking influence water adsorption capacity and, thus, the relaxometric properties of the ternary system [45].
Therefore, the presence of the hydrogel matrix can significantly amplify the magnetic properties of the encapsulated Gd-DTPA, suggesting a strong outer-sphere and second-sphere contribution to the relaxivity.
Based on these observations, we hypothesize that increased relaxivity is mainly related to the creation of water domains or clusters (water compartments) around the CA within the polymer matrix. In fact, biopolymer systems contain intermolecular cavities that can be considered as molecular nano-domains in which various self-assembly processes can be implemented in principle [80]. The formation of peculiar structures within these cavities can be associated with thermodynamic transitions and it is a characteristic of many metallopolymer systems [81, 82].
The sub-nanostructures, here defined “Gado- Meshes”, are generated from a three-way interaction between HA, Gd-DTPA and water. The entrapment of the CA inside the hydrophilic matrix of HA results in a reduction of the rotational tumbling rate, due to an increase of the effective viscosity of the aqueous solution into the hydrogel matrix. At the same time, multiple CA-water interaction pathways occur between the exchangeable protons of the water molecules coordinated by the CA and the other water molecules freely moving within the hydrogel mesh or bonded to the polymer chains [22, 44, 83].
Our “Gado-mesh” consists of highly relaxing Gd-water compartments spontaneously generated within the hydrogel matrix by the combination of these multiple physico-chemical interactions. The so created nanostructure is composed of different water layers departing from the polymer chains that surround the Gd-molecules. It is known, in fact, that hydrated polysaccharides, such as HA, are characterized by the presence of multiple water layers, contiguous regions of variable water density within a polyelectrolyte solution [84], differing in their physical properties depending on the distance from the polysaccharide chain [85]. The hydration process of HA generates: the “bound water layer”, which is the water fractions closely associated with the polymer matrix; the “unbound water layer”, made by the water molecules which are not directly interacting with the polymer; and the “free water layer”, which resembles dynamics of pure water. In addition, the developed polymer network can be dependent not only on the water layers organization but also on other solute species, such as Gd-DTPA, altering the bound water layer with non-negligible effect on the HA conformation and dynamic [86]. In the “Gado-mesh”, Gd-DTPA has a competitive behaviour with the respect of HA, similar to the cation shielding of the HA due to the presence of salts [87], and interposes itself between the water molecules around the HA, altering the bound water layer and generating water compartments with high MRI enhancing properties. The “Gado-mesh” influences the τR, τD and τM times through the action of the Hydrodenticity, whose effect is magnified by the crosslinking. Hydrodenticity, hence, refers to the status of the hydrated Gd-DTPA with the coordination water subjected to osmotic pressure deriving from elastodynamics equilibrium of swollen gels [39, 88-93].
We hypothesize that the attainment of this equilibrium is reached when the standard energetic stability of the meshes is compromised by the presence of the Gd-DTPA and evolves to a new spontaneous equilibrium involving the formation of nanocompartments, so-called “Gado-Meshes”, in which water is in an abnormal aggregate state that influences the relaxivity. Water molecules in the hydrogel matrix that are subjected to the effect of Hydrodenticity are able to change their water dynamics and can mediate the hydrogel conformation and the physical and relaxometric properties of the metal chelate.
Conclusion
From a biomedical point of view, the possibility to tune relaxometric properties of CAs by controlling hydrogel structural parameters can pave the way to new advancements in the design of nanovectors for diagnosis and therapy. This work proves that a new generation of more efficient nanoparticle-based CAs can be developed by exploiting the affinity between CAs and biopolymers. It can be done using biocompatible and clinical relevant CAs without their chemical modification as approved in the clinical practice. Furthermore, the size of the resulting NPs is in a range that makes them suitable for delivery to cells and tissues and a further increase in relaxivity can be potentially achieved by tuning the system to the most efficient structure by choosing the correct biopolymer-CA combination and optimizing concentration and crosslinking degree of the structure.
Experimental Section
Materials
All chemicals used are of analytical reagent grade quality and are employed as received. Sorbitan monooleate (Span® 80) (S80), Polyoxyethylenesorbitan trioleate (Tween® 85) (T85), Mineral oil (light oil, 0.8 gr/cm at 25°C), Divinyl sulfone (DVS, 118.15 Da), Diethylenetriaminepentaacetic acid gadolinium(III) dihydrogen salt hydrate (Gd-DTPA, 547.57 Da), Sodium hydroxide pellets (NaOH), Acetone and Ethanol are purchased from Sigma Aldrich Chemical (Italy). Sodium Hyaluronate, with an average molecular weight of 850 kDa (purity 99%; Hyasis® 850P) and 42 kDa, is respectively supplied by Novozymes Biopharma and Bohus Biotech (Sweden) as dry powder and used without purification.
Magnevist® (Bracco Imaging, Italy), a contrast agent commercially available, is used in this study. The water is purified by distillation, deionization, and reserve osmosis (Milli-Q Plus) and systematically used for sample preparation, purification and analysis. All experiments are repeated in triplicate and conducted at room temperature, 25°C.
Isothermal Titration Calorimetry
Titration experiments are performed by using a Nano ITC Low Volume calorimeter (TA Instruments). CA and polymer are prepared in double-distilled filtered water without any additives. The sample cell (700 µL) and the syringe (50 µL) are filled with aqueous solutions of HA and Gd-DTPA respectively. Syringe Gd-DTPA concentration is fixed at 1.5 mM, while different HA concentrations in the sample cell are tested, ranging from 0.3 to 0.7% w/v. The measurements are performed at 25 °C and at fixed stirring rate of 200 rpm. Fifty Injections, each of 1 μL of Gd-DTPA, are delivered in intervals of 500 s. The concentration of polymer is expressed as the mass of the repeat unit (unit mol/L).
Data analysis and processing to provide ITC and enthalpy change (ΔH) profiles is carried out using the NanoAnalyze (TA instruments) and the OriginPro software.
NMR Spectroscopy
1H NMR spectra are recorded at 25 °C with Varian Agilent NMR spectrometer operating at 600 MHz to observe chemical interactions between polymer and chelating agent (DTPA). The NMR samples consisted of water solution of HA-DTPA at different molar ratios (HA/DTPA ranging from 0 to 0.5), with 10% v/v D2O.
Diffusion-ordered NMR Spectroscopy (DOSY) is also performed and the z-gradient strengths (Gz) is varied in 20 steps from 500 to 32500 G/cm (maximum strength). The gradient pulse duration (δ) and the diffusion delay (Δ) are kept constant, 2 ms for δ and ranging from 7 to 1000 ms for Δ. After Fourier transformation and baseline correction, DOSY spectra are processed and analysed using Varian software VNMRJ (Varian by Agilent Technologies, Italy) in order to obtain the values of water self-diffusion coefficient.
Differential Scanning Calorimetry
For all measurements the HA/water solution (Mw = 42 kDa) is used. The aqueous solutions are prepared in a concentration range of polymer 0.3-0.7% w/v. Next, Gd-DTPA is added as CA at different molar ratio HA/Gd-DTPA (from 1:0.25 to 1:3) and stirred for 12 h. The hydrated polymer samples, with and without CA, are sealed at room temperature in a Tzero hermetic pans prior to analysis. DSC measurements are performed in a TA Instruments' Q20TM calorimeter on samples between 5 and 10 mg. The samples are cooled down from 25°C to -50°C followed by heating scan up to 25°C. The same heating and cooling rate are 10°C/min for all runs. Samples are tested in triplicate to ensure reproducibility. For DSC and ITC measurements, we used low molecular weight HA (42 kDa) to highlight better the energetic contributions of different components without exceeding the maximum scale of the instruments.
Time-Domain Relaxometry
The spin-lattice relaxation times (T1) are measured in a Bruker Minispec (mq 60) bench-top relaxometer operating at 60 MHz for protons (magnetic field strength: 1.41 T). Measurements are taken at 37°C, and before NMR measurements, the tube is placed into the NMR probe for about 15 min for thermal equilibration. Experiments are made using water solutions of Gd-DTPA (from 0 to 0.1 mM) and HA (0.3, 0.5 and 0.7% w/v) crosslinked with DVS (DVS/HA weight percentage ratio equal to 1:8). T1 values are determined by both saturation (SR) and inversion recovery (IR) pulse sequences. The relaxation recovery curves are fitted using a multi-exponential model. Relaxivities, r1, are calculated from the slope of the regression line of 1/T1 [s-1] versus concentration [mM] with a least-squares method.
Emulsion Preparation
The emulsions are prepared at different water to oil (W/O) ratio (10/90 and 20/80 v/v). Mineral oil is used as oil phase (or continuous phase, PC) and W/O emulsions are made by varying the concentration of surfactants for the PC and water phase (or dispersed phase, PD) and the concentration of NaOH (from 0 to 0.2 M) for the PD in order to obtain emulsion systems. In particular, non-ionic surfactants, Span-80 (S80) and Tween-85 (T85), are used to prepare mixtures with a range from 4.3 to 7.65 of HLB values. Depending on the initial HLB to be used, mixtures of S80 and T85 are pre-dissolved in the appropriate S80/T85 mass ratios (from 50/50 to 75/25) in PC and PD, respectively. PD containing T85 and NaOH, is added dropwise to PC and W/O emulsions are prepared using a high-shear homogenizer (Silverson L5M-A, Silverson Machines Ltd, Waterside, UK). Homogenization of the Emulsion is performed from 5000 to 7000 rpm for 10 min at room temperature (25°C).
Temporal Emulsion Stability Determination
The stability of emulsions is evaluated, at regular time intervals, by visual observation, measuring the height of the phase separated by creaming in centimeters as a function of the time. In addition, optical characterization of emulsion stability made is using a Turbiscan (Turbiscan LabThermo) by static multiple light scattering (MLS), sending a light beam from an electroluminescent diode (λ=880 nm) through a cylindrical glass cell containing the sample. The emulsion sample without dilution is placed in a cylindrical glass cell and two synchronous optical sensors receive the light transmitted through the sample (180° from the incident light) and the light backscattered by the droplets in the sample (45° from the incident light). The optical reading head scans the height of the sample in the cell (about 40 mm), by acquiring transmission and backscattering data every 40 μm. Transmitted and backscattered light are monitored as a function of time and cell height for 24 hours at an interval of 30 min at 25°C.
Preparation of DVS-Crosslinked Nanoparticles
Based on these preliminary results, PD/PC ratio in all samples is set at 10/90 v/v. In particular, for the preparation of cross-linked NPs, HA powder (Mw = 850 kDa) is dissolved at different concentrations (from 0.1 to 0.5% w/v) under alkaline condition (NaOH ranging from 0 to 0.2 M) by vigorous stirring at room temperature for 4 hours until a homogenous solution is obtained. Mineral oil and S80 (from 0.5 to 2% w/v) are separately mixed by stirring. PD is added drop-wise in the PC without stirring and all the components are completely mixed by homogenization at various times (5-15 minutes) and speeds (5000 - 7000 rpm). Then, the cross-linking agent (DVS) is added to the final emulsion (40 ml), which is kept in agitation on a laboratory tube rotator for 24 hours to obtain a homogeneous DVS distribution in the PD. To test DVS activity, various conditions of crosslinking reaction are explored: (1) at different DVS concentrations (from 0.01 to 0.5% v/v); (2) at three starting times of reaction (beginning, during and post homogenization) and (3) at different temperatures (4 and 25°C).
The best experimental conditions for production of crosslinked HA-NPs are reported in Table S2 of the Supporting Information.
Loading of HA NPs with Contrast Agents
After identifying the protocol to obtain NPs, Gd-DTPA is chosen as CA and mixed in the PD before homogenization. Gd-loaded HA NPs (HA-Gd NPs) are prepared using different HA/CA mass ratios (1:1, 1:2 and 1:5). DVS is added post homogenization to the batch at room temperature using the same procedure reported above.
Collection of the Nanoparticles
Recovery of the NPs and their separation from W/O emulsion system is made using dialysis and/or ultracentrifugation. For dialysis method, the obtained emulsion is placed in a pre-washed cellulose membrane tubing (Spectra/Por® Dialysis Tubing, cut-off of 25 kDa). Organic impurities (Mineral oil and S80) are removed dialyzing first against solvents as acetone and/or ethanol, and gradually against water. Dialyzing solutions are changed at regular time intervals. In the case of ultracentrifugation, 1 ml of the emulsion is added to 5 ml of ethanol and mixed for 2 hours. Then, this mix is centrifuged with an ultracentrifuge (Beckman-Coulter OPTIMA MAX-XP) at 55000 rpm for 20 min at 15°C. The resulting pellet is washed twice and resuspended in MilliQ water. The second step of ultracentrifugation (70000 rpm, 10 min, 15°C) is applied to the pellet in order to obtain purified NPs.
Characterization of the Nanoparticles
To determine the size distribution of NPs, dynamic light scattering (DLS) is performed using a Zetasizer S-90 1000 HS (Malvern Instruments, UK).
All samples are diluted (1:10) with deionized water to prevent the effects of multiple scattering. The measurement temperature is set at 25°C. The morphology and size of NPs are investigated using ULTRA PLUS field emission Scanning Electron microscope (FE-SEM Carl Zeiss, Oberkochen, Germany) and a Transmission Electron microscopy (TEM, TECNAI). In the first case, the samples are coated with gold (7 nm).
Determination Of Gadolinium Loading by ICP-MS
The quantitative determination of loaded Gd in HA NPs is assessed by ICP-MS (NexION 350, Perkin Elmer) without any previous digestion processes. For all examinations, purified NP suspensions are used. The non-encapsulated Gd-complexes are separated from the NPs by high-speed centrifugation (55000 rpm, 20 min, 15 °C).
MRI Testing
To explore the potential of Gd-loaded HA NPs as MRI contrast agent, MRI in vitro test is performed at two different magnetic fields, 1.5 T and 3 T MR (Philips Achieva) using Sense Head 8 coil. The T1-weighted MR images of HA NPs, unloaded and loaded with Gd-DTPA at different concentrations using an inversion recovery sequence are measured with the following parameters: TR = 2500 ms; TE = 12 ms; TI = 50, 100, 200, 400, 800, 1100, 1800 ms; FOV= 180x146 mm; slice thickness = 4 mm, acquisition matrix = 360x292.
The signal intensity of the samples is measured on the obtained T1-weighted MR images and compared to Gd-DTPA.
NMR Dispersion Measurements
The proton 1/T1 NMRD profiles are measured using a fast-field-cycling Stelar SmarTracer relaxometer over a continuum of magnetic field strengths from 0.00024 to 0.25 T (which correspond to 0.01 - 10 MHz proton Larmor frequencies). The uncertainty of these measurements is less than 1%. Additional data points in the range 15 - 70 MHz are obtained using a Stelar Relaxometer and a Bruker WP80 NMR electromagnet adapted to variable-field measurements (15 - 80 MHz proton Larmor frequency).
Fourier Transform Infrared Spectroscopy
Fourier transform infrared spectra (FT-IR) were collected from Nicolett 6700 FT-IR spectrometer (Thermo Scientific). All the IR spectra of the specimen were collected at 0.09 cm-1 resolution with 2 min interval. Empty HA NPs and Gd-DTPA- Lodaed HA NPs were analyzed to observe the interaction. Spectra were recorded and analyzed for signal assignation.
Supplementary Material
Supplementary figures and tables.
Abbreviations
CA: contrast agent; cHANPs: crosslinked hyaluronic acid nanoparticles; DOSY: diffusion-ordered nmr spectroscopy; DSC: differential scanning calorimetry; DVS: divinyl sulphone; EE: encapsulation efficacy; EMA: european medicines agency; FDA: food and drug administration; Gd: gadolinium; Gd-DTPA: diethylenetriaminepentaacetic acid gadolinium(iii) dihydrogen salt hydrate; HA: hyaluronic acid; HLB: hydrophilic-lipophilic balance; HyCoS: hybrid core-shell; ICP-MS: inductively coupled plasma mass spectrometry; IR: inversion recovery; ITC: isothermal titration calorimetry; LC: loading capability; MLS: multiple light scattering; MRI: magnetic resonance imaging; NaOH: sodium hydroxyde pellets; NMR: nuclear magnetic resonance; NMRD: nuclear magnetic relaxation dispersion; NPs: nanoparticles; NSF: nephrogenic systemic fibrosis; PC: continuous phase; PD: dispersed phase; PET: positron emission tomography; S80: sorbitan monooleate (span® 80); SBM: solomon-bloembergen-morgan; SEM: scanning electron microscopy; SR: saturation recovery; T85: polyoxyethylenesorbitan trioleate (tween® 85); TEM: transmission electron microscopy; W/O: water in oil.
Acknowledgements
Franca De Sarno and Alfonso Maria Ponsiglione contributed equally to this work. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Mansfield P. Snapshot magnetic resonance imaging (Nobel lecture). Angew Chem Int Ed Engl. 2004;43:5456-64
2. Caravan P. Strategies for increasing the sensitivity of gadolinium based MRI contrast agents. Chem Soc Rev. 2006;35:512-23
3. Shokrollahi H. Contrast agents for MRI. Mater. Sci Eng C Mater Biol Appl. 2013;33:4485-97
4. Zhang W, Liu L, Chen H, Hu K, Delahunty I, Gao S. et al. Surface impact on nanoparticle-based magnetic resonance imaging contrast agents. Theranostics. 2018;8:2521-48
5. Ponsiglione AM, Russo M, Netti PA, Torino E. Impact of biopolymer matrices on relaxometric properties of contrast agents. Interface Focus. 2016:6 (6)
6. Russo M, Bevilacqua P, Netti PA, Torino E. A Microfluidic Platform to design crosslinked Hyaluronic Acid Nanoparticles (cHANPs) for enhanced MRI. Sci Rep. 2016:6 (37906)
7. Caravan P. Protein-targeted gadolinium-based magnetic resonance imaging (MRI) contrast agents: design and mechanism of action. Acc Chem Res. 2009;42:851-62
8. Klemm PJ, Floyd WC, Smiles DE, Fréchet JMJ, Raymond KN. Improving T1 and T2 magnetic resonance imaging (MRI) contrast agents through the conjugation of an esteramide dendrimer to high water coordination Gd(III) hydroxypyridinone (HOPO) complexes. Contrast Media Mol Imaging. 2012;7:95-9
9. Thomsen HS, Morcos SK, Almén T, Bellin M-F, Bertolotto M, Bongartz G. et al. Nephrogenic systemic fibrosis and gadolinium-based contrast media: updated ESUR Contrast Medium Safety Committee guidelines. Eur Radiol. 2013;23:307-18
10. Kanda T, Ishii K, Kawaguchi H, Kitajima K, Takenaka D. High signal intensity in the dentate nucleus and globus pallidus on unenhanced T1-weighted MR images: relationship with increasing cumulative dose of a gadolinium-based contrast material. Radiology. 2014;270:834-41
11. McDonald RJ, McDonald JS, Kallmes DF, Jentoft ME, Murray DL, Thielen KR. et al. Intracranial Gadolinium Deposition after Contrast-enhanced MR Imaging. Radiology. 2015;275:772-82
12. Rogosnitzky M, Branch S. Gadolinium-based contrast agent toxicity: a review of known and proposed mechanisms. Biometals. 2016;29:365-76
13. Huang J, Zhong X, Wang L, Yang L, Mao H. Improving the Magnetic Resonance Imaging Contrast and Detection Methods with Engineered Magnetic Nanoparticles. Theranostics. 2012;2:86-102
14. Guo C, Sun L, Cai H, Duan Z, Zhang S, Gong Q. et al. Gadolinium-Labeled Biodegradable Dendron-Hyaluronic Acid Hybrid and Its Subsequent Application as a Safe and Efficient Magnetic Resonance Imaging Contrast Agent. ACS Appl Mater Interfaces. 2017;9:23508-19
15. Luo Q, Xiao X, Dai X, Duan Z, Pan D, Zhu H. et al. Cross-Linked and Biodegradable Polymeric System as a Safe Magnetic Resonance Imaging Contrast Agent. ACS Appl Mater Interfaces. 2018;10:1575-88
16. Corti M, Lascialfari A, Marinone M, Masotti A, Micotti E, Orsini F. et al. Magnetic and relaxometric properties of polyethylenimine-coated superparamagnetic MRI contrast agents. J Magn Magn Mater. 2008:320 (14)
17. Bloembergen N, Morgan LO. Proton Relaxation Times in Paramagnetic Solutions. Effects of Electron Spin Relaxation. J Chem Phys. 1961;34:842-50
18. Li Y, Beija M, Laurent S, Elst L vander, Muller RN, Duong HTT. et al. Macromolecular Ligands for Gadolinium MRI Contrast Agents. Macromolecules. 2012;45:4196-204
19. Port M, Raynal I, Vander Elst L, Muller RN, Dioury F, Ferroud C. et al. Impact of rigidification on relaxometric properties of a tricyclic tetraazatriacetic gadolinium chelate. Contrast Media Mol Imaging. 2006;1:121-7
20. Jászberényi Z, Sour A, Tóth É, Benmelouka M, Merbach AE. Fine-tuning water exchange on GdIII poly(amino carboxylates) by modulation of steric crowding. Dalton Trans. 2005;0:2713-9
21. Gizzatov A, Stigliano C, Ananta JS, Sethi R, Xu R, Guven A. et al. Geometrical confinement of Gd(DOTA) molecules within mesoporous silicon nanoconstructs for MR imaging of cancer. Cancer Lett. 2014;352:97-101
22. Sethi R, Ananta JS, Karmonik C, Zhong M, Fung SH, Liu X. et al. Enhanced relaxivity of Gd3+-based contrast agents geometrically confined within porous nanoconstructs. Contrast Media Mol Imaging. 2012;7:501-508
23. Kuang Y, Cao Y, Liu M, Zu G, Zhang Y, Zhang Y. et al. Geometrical Confinement of Gadolinium Oxide Nanoparticles in Poly(ethylene glycol)/Arginylglycylaspartic Acid-Modified Mesoporous Carbon Nanospheres as an Enhanced T1 Magnetic Resonance Imaging Contrast Agent. ACS Appl Mater Interfaces. 2018;10:26099-107
24. Courant T, Roullin VG, Cadiou C, Callewaert M, Andry MC, Portefaix C. et al. Hydrogels Incorporating GdDOTA: Towards Highly Efficient Dual T1/T2 MRI Contrast Agents. Angew Chem Int Ed. 2012;51:9119-22
25. Callewaert M, Roullin VG, Cadiou C, Millart E, Gulik LV, Andry MC. et al. Tuning the composition of biocompatible Gd nanohydrogels to achieve hypersensitive dual T1/T2 MRI contrast agents. J Mater Chem B. 2014;2:6397-405
26. Necas J, Bartosikova L, Brauner P, Kolar J. Hyaluronic acid (hyaluronan): a review. Vet Med. 2008;53:397-411
27. Zhu W, Artemov D. Biocompatible blood pool MRI contrast agents based on hyaluronan. Contrast Media Mol Imaging. 2011;6:61-8
28. Zhu L, Yang Y, Farquhar K, Wang J, Tian C, Ranville J. et al. Surface Modification of Gd Nanoparticles with pH-Responsive Block Copolymers for Use As Smart MRI Contrast Agents. ACS Appl Mater Interfaces. 2016;8:5040-50
29. Sun L, Li X, Wei X, Luo Q, Guan P, Wu M. et al. Stimuli-Responsive Biodegradable Hyperbranched Polymer-Gadolinium Conjugates as Efficient and Biocompatible Nanoscale Magnetic Resonance Imaging Contrast Agents. ACS Appl Mater Interfaces. 2016;8:10499-512
30. Wang S, Zhou Z, Yu G, Lu N, Liu Y, Dai Y. et al. Gadolinium Metallofullerene-Polypyrrole Nanoparticles for Activatable Dual-Modal Imaging-Guided Photothermal Therapy. ACS Appl Mater Interfaces. 2018;10:28382-9
31. Huang Y, Hu H, Li R-Q, Yu B, Xu F-J. Versatile Types of MRI-Visible Cationic Nanoparticles Involving Pullulan Polysaccharides for Multifunctional Gene Carriers. ACS Appl Mater Interfaces. 2016;8:3919-27
32. Swierczewska M, Han HS, Kim K, Park JH, Lee S. Polysaccharide-based Nanoparticles for Theranostic Nanomedicine. Adv Drug Deliv Rev. 2016;99:70-84
33. Nitta SK, Numata K. Biopolymer-based nanoparticles for drug/gene delivery and tissue engineering. Int J Mol Sci. 2013;14:1629-54
34. Almalik A, Karimi S, Ouasti S, Donno R, Wandrey C, Day PJ. et al. Hyaluronic acid (HA) presentation as a tool to modulate and control the receptor-mediated uptake of HA-coated nanoparticles. Biomaterials. 2013;34:5369-80
35. Almalik A, Benabdelkamel H, Masood A, Alanazi IO, Alradwan I, Majrashi MA. et al. Hyaluronic Acid Coated Chitosan Nanoparticles Reduced the Immunogenicity of the Formed Protein Corona. Sci Rep. 2017;7:10542
36. Almalik A, Alradwan I. et al. Cellular responses of hyaluronic acid-coated chitosan nanoparticles. Toxicol Res. 2018;7:942-50
37. Lallana E, Donno R, Magrì D, Barker K, Nazir Z, Treacher K. et al. Microfluidic-assisted nanoprecipitation of (PEGylated) poly (d,l-lactic acid-co-caprolactone): Effect of macromolecular and microfluidic parameters on particle size and paclitaxel encapsulation. Int J Pharm. 2018;548:530-9
38. Russo M, Bevilacqua P, Netti PA, Torino E. Commentary on “A Microfluidic Platform to Design Crosslinked Hyaluronic Acid Nanoparticles (cHANPs) for Enhanced MRI”. Mol Imaging. 2017;16:1-3
39. Russo M, Ponsiglione AM, Forte E, Netti PA, Torino E. Hydrodenticity to enhance relaxivity of gadolinium-DTPA within crosslinked hyaluronic acid nanoparticles. Nanomed. 2017;12:2199-210
40. Russo M, Grimaldi AM, Bevilacqua P, Tammaro O, Netti PA, Torino E. PEGylated crosslinked hyaluronic acid nanoparticles designed through a microfluidic platform for nanomedicine. Nanomed. 2017;12:2211-22
41. Okada S, Mizukami S, Kikuchi K. Switchable MRI contrast agents based on morphological changes of pH-responsive polymers. Bioorg Med Chem. 2012;20:769-74
42. Vecchione D, Grimaldi AM, Forte E, Bevilacqua P, Netti PA, Torino E. Hybrid Core-Shell (HyCoS) Nanoparticles produced by Complex Coacervation for Multimodal Applications. Sci Rep. 2017;7:45121
43. Vecchione D, Aiello M, Cavaliere C, Nicolai E, Netti PA, Torino E. Hybrid core shell nanoparticles entrapping Gd-DTPA and 18F-FDG for simultaneous PET/MRI acquisitions. Nanomed. 2017;12:2223-31
44. Courant T, Roullin GV, Cadiou C, Callewaert M, Andry MC, Portefaix C. et al. Biocompatible nanoparticles and gadolinium complexes for MRI applications. Comptes Rendus Chim. 2013;16:531-9
45. Pasqui D, De Cagna M, Barbucci R, Pasqui D, De Cagna M, Barbucci R. Polysaccharide-Based Hydrogels: The Key Role of Water in Affecting Mechanical Properties. Polymers. 2012;4:1517-34
46. Panagopoulou A, Molina JV, Kyritsis A, Pradas MM, Lluch AV, Ferrer GG. et al. Glass Transition and Water Dynamics in Hyaluronic Acid Hydrogels. Food Biophys. 2013;8:192-202
47. Davies G-L, Kramberger I, Davis JJ. Environmentally responsive MRI contrast agents. Chem Commun. 2013;49:9704-21
48. Budkov YA, Kolesnikov AL, Georgi N, Kiselev MG. A flexible polymer chain in a critical solvent: Coil or globule? EPL Europhys Lett. 2015;109:36005
49. Flory PJ. Thermodynamics of High Polymer Solutions. J Chem Phys. 1942;10:51-61
50. Martinez JC, Murciano-Calles J, Cobos ES, Iglesias-Bexiga M, Ruiz-Sanz IL. Isothermal Titration Calorimetry: Thermodynamic Analysis of the Binding Thermograms of Molecular Recognition Events by Using Equilibrium Models. Application of Calorimetry in a Wide Context. Amal Ali Elkordy, IntechOpen. 2013
51. Kabiri M, Unsworth LD. Application of isothermal titration calorimetry for characterizing thermodynamic parameters of biomolecular interactions: peptide self-assembly and protein adsorption case studies. Biomacromolecules. 2014;15:3463-73
52. Carrillo J-MY, Dobrynin AV. Polyelectrolytes in Salt Solutions: Molecular Dynamics Simulations. Macromolecules. 2011;44:5798-816
53. Cesaro A, Paoletti S, Rizzo R, Benegas JC. Thermodynamics of polysaccharide polyelectrolytes: Enthalpy changes in the conformational transition of κ-carrageenan. Pure Appl Chem. 1994;66:461-467
54. Chudoba R, Heyda J, Dzubiella J. Tuning the Collapse Transition of Weakly Charged Polymers by Ion-Specific Screening and Adsorption. Soft matter. 2018;14:9631-9642
55. Horkay F, Hecht AM, Rochas C, Basser PJ, Geissler E. Anomalous small angle x-ray scattering determination of ion distribution around a polyelectrolyte biopolymer in salt solution. J Chem Phys. 2006;125:234904
56. Kozak N, Lobko E. Bottom-Up Nanostructured Segmented Polyurethanes with Immobilized in situ Transition and Rare-Earth Metal Chelate Compounds - Polymer Topology - Structure and Properties Relationship. Polyurethane. Fahmina Zafar and Eram Sharmin, IntechOpen. 2012
57. Kozak N, Nizelskii Y, Mnikh N, Shtompel V, Grischuk O. Formation of Nanostructures in Multi Component Systems Based on Organic Polymer and Coordination Metal Compound. Macromol Symp. 2006;243:247-60
58. Gouin S, Winnik FM. Quantitative assays of the amount of diethylenetriaminepentaacetic acid conjugated to water-soluble polymers using isothermal titration calorimetry and colorimetry. Bioconjug Chem. 2001;12:372-7
59. Ori G, Massobrio C, Pradel A, Ribes M, Coasne B. Structure and Dynamics of Ionic Liquids Confined in Amorphous Porous Chalcogenides. Langmuir. 2015;31:6742-51
60. Ori G, Villemot F, Viau L, Vioux A, Coasne B. Ionic liquid confined in silica nanopores: molecular dynamics in the isobaric-isothermal ensemble. Mol Phys. 2014;112:1350-61
61. Strain SM, Fesik SW, Armitage IM. Structure and metal-binding properties of lipopolysaccharides from heptoseless mutants of Escherichia coli studied by 13C and 31P nuclear magnetic resonance. J Biol Chem. 1983;258:13466-77
62. Prudêncio M, Rohovec J, Peters JA, Tocheva E, Boulanger MJ, Murphy MEP. et al. A caged lanthanide complex as a paramagnetic shift agent for protein NMR. Chem Weinh Bergstr Ger. 2004;10:3252-60
63. Yoshida H, Hatakeyama T, Hatakeyama H. Characterization of water in polysaccharide hydrogels by DSC. J Therm Anal. 1993;40:483-9
64. Yoshida H, Hatakeyama T, Hatakeyama H. Effect of Water on the Main Chain Motion of Polysaccharide Hydrogels. Viscoelasticity Biomater American Chemical Society. 1992;489:217-30
65. Boyken J, Frenzel T, Lohrke J, Jost G, Pietsch H. Gadolinium Accumulation in the Deep Cerebellar Nuclei and Globus Pallidus After Exposure to Linear but Not Macrocyclic Gadolinium-Based Contrast Agents in a Retrospective Pig Study With High Similarity to Clinical Conditions. Invest Radiol. 2018;53:278-85
66. Jin Y-J, Ubonvan T, Kim D-D. Hyaluronic Acid in Drug Delivery Systems. J Pharm Investig. 2010;40:33-43
67. Tripodo G, Trapani A, Torre ML, Giammona G, Trapani G, Mandracchia D. Hyaluronic acid and its derivatives in drug delivery and imaging: Recent advances and challenges. Eur J Pharm Biopharm. 2015;97:400-416
68. Mengual O, Meunier G, Cayré I, Puech K, Snabre P. TURBISCAN MA 2000: multiple light scattering measurement for concentrated emulsion and suspension instability analysis. Talanta. 1999;50:445-56
69. Balazs EA, Leshchiner A. Crosslinked gels of hyaluronic acid and products containing such gels, U.S. Patent 4,582,865 (1986).
70. Sahiner N, Jia X. One-Step Synthesis of Hyaluronic Acid-Based (Sub)micron Hydrogel Particles: Process Optimization and Preliminary Characterization. Turk. J Chem. 2008;32:397-410
71. Collins MN, Birkinshaw C. Investigation of the swelling behavior of crosslinked hyaluronic acid films and hydrogels produced using homogeneous reactions. J Appl Polym Sci. 2008;109:923-31
72. Quignard F, Di Renzo F, Guibal E. From natural polysaccharides to materials for catalysis, adsorption, and remediation. Top Curr Chem. 2010;294:165-97
73. Oh EJ, Kang S-W, Kim B-S, Jiang G, Cho IH, Hahn SK. Control of the molecular degradation of hyaluronic acid hydrogels for tissue augmentation. J Biomed Mater Res A. 2008;86:685-93
74. Diao Y, Whaley KE, Helgeson ME, Woldeyes MA, Doyle PS, Myerson AS. et al. Gel-induced selective crystallization of polymorphs. J Am Chem Soc. 2012;134:673-84
75. Shogbon CB, Brousseau J-L, Zhang H, Benicewicz BC, Akpalu YA. Determination of the Molecular Parameters and Studies of the Chain Conformation of Polybenzimidazole in DMAc/LiCl. Macromolecules. 2006;39:9409-18
76. Tao Z. and P.T. Cummings, Molecular dynamics simulation of inorganic ions in PEO aqueous solution. Mol Simul. 2007;33:1255-1260
77. Teraoka I. Polymer solutions: An introduction to physical properties. 2002;ISBN:978-0-471-38929-3.
78. Shetye SP, Godbole DA, Shilpa D, Gajare P. Hydrogels: Introduction, Preparation, Characterization and Applications. 2015:1:47-71.
79. Tondera C, Wieduwild R, Röder E, Werner C, Zhang Y, Pietzsch J. In Vivo Examination of an Injectable Hydrogel System Crosslinked by Peptide-Oligosaccharide Interaction in Immunocompetent Nude Mice. Adv Funct Mater. 2017;27:1605189
80. Mikhailov OV. Molecular nanotechnologies of gelatin-immobilization using macrocyclic metal chelates. Nano Rev. 2014;5:1
81. Riess G. Micellization of block copolymers. Prog Polym Sci. 2003;28:1107-70
82. Pomogailo PAD. Polvmer-Immobilised Clusters of (he Platinum Group Metals. Platinum Metals Rev. 1994:38 (2):60
83. Aime S, Frullano L, Crich SG. Compartmentalization of a Gadolinium Complex in the Apoferritin Cavity: A Route To Obtain High Relaxivity Contrast Agents for Magnetic Resonance Imaging. Angew Chem. 2002;114:1059-61
84. Vogler E. A, Structure and Reactivity of Water at Biomaterial Surfaces. Adv Colloid and Interface Sci. 1998:74 69-117
85. Průšová A, Šmejkalová D, Chytil M, Velebný V, Kučerík J. An alternative DSC approach to study hydration of hyaluronan. Carbohydr Polym. 2010;82:498-503
86. Ivanov D, Neamtu A. Molecular dynamics evaluation of hyaluronan interactions with dimethylsilanediol in aqueous solution. Rev Roum Chim. 2013:58 (2):229-238
87. Guillaumie F, Furrer P, Felt-Baeyens O, Fuhlendorff BL, Nymand S, Westh P. et al. Comparative studies of various hyaluronic acids produced by microbial fermentation for potential topical ophthalmic applications. J Biomed Mater Res A. 2010;92:1421-30
88. Velasco D, Tumarkin E, Kumacheva E. Microfluidic encapsulation of cells in polymer microgels. Small Weinh. Bergstr Ger. 2012;8:1633-42
89. Peppas NA, Huang Y, Torres-Lugo M, Ward JH, Zhang J. Physicochemical foundations and structural design of hydrogels in medicine and biology. Annu Rev Biomed Eng. 2000;2:9-29
90. Johnson DL. Elastodynamics of gels. J Chem Phys. 1982;77:1531-9
91. Ström A, Larsson A, Okay O. Preparation and physical properties of hyaluronic acid-based cryogels. J Appl Polym Sci. 2015:132 (29)
92. Utech S, Boccaccini AR. A review of hydrogel-based composites for biomedical applications: enhancement of hydrogel properties by addition of rigid inorganic fillers. J Mater Sci. 2016;51:271-310
93. Phinikaridou A, Andia ME, Protti A, Indermuehle A, Shah A, Smith A. et al. Noninvasive magnetic resonance imaging evaluation of endothelial permeability in murine atherosclerosis using an albumin-binding contrast agent. Circulation. 2012;126:707-19
Author contact
![]() Corresponding author: Enza Torino, PhD. Department of Chemical, Materials Engineering & Industrial Production, University of Naples Federico II, Piazzale Tecchio 80, 80125 Naples, Italy. Tel. +390817685990; email: enza.torinoit
Corresponding author: Enza Torino, PhD. Department of Chemical, Materials Engineering & Industrial Production, University of Naples Federico II, Piazzale Tecchio 80, 80125 Naples, Italy. Tel. +390817685990; email: enza.torinoit