Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(6):1794-1808. doi:10.7150/thno.31841 This issue Cite
Research Paper
H2Se Induces Reductive Stress in HepG2 Cells and Activates Cell Autophagy by Regulating the Redox of HMGB1 Protein under Hypoxia
Xiaohong Pan1,2, Xiaoxiao Song1, Cheng Wang2, Tingting Cheng2, Dongrui Luan1, Kehua Xu1 ![]() , Bo Tang1
, Bo Tang1 ![]()
1. College of Chemistry, Chemical Engineering and Materials Science, Key Laboratory of Molecular and Nano Probes, Ministry of Education, Collaborative Innovation Center of Functionalized Probes for Chemical Imaging in Universities of Shandong, Shandong Normal University, Jinan 250014, P. R. China.
2. Department of Pharmaceutical Sciences, Binzhou Medical University, Shandong, Yantai 264003, P. R. China.
Received 2018-11-28; Accepted 2019-1-17; Published 2019-2-28
Abstract

Rationale: Selenium has been shown to have chemotherapeutic effects against cancer. However, the anti-cancer mechanism of selenium is not fully understood, and the role of hydrogen selenide (H2Se), which is a common metabolite of dietary selenium compounds, has not been elucidated due to the lack of detection methods. In this study, we revealed a new anti-cancer mechanism of selenite with the help of a H2Se fluorescent probe.
Methods: HepG2 cells were cultured under a simulated tumor hypoxic microenvironment. The H2Se and H2O2 levels were detected by fluorescent probes in living cells and in mice. Autophagic and apoptotic proteins were detected by Western blotting. The redox of HMGB1 protein were analyzed by non-reducing sodium dodecyl sulfate polyacrylamide gel electrophoresis.
Results: After pharmacological doses of Na2SeO3 treatment of HepG2 cells under hypoxic conditions, high levels of H2Se were produced before cell death. The H2Se accumulation resulted in reductive stress instead of oxidative stress, which was induced by Na2SeO3 treatment under normoxic conditions. Furthermore, H2Se targeted the HMGB1 protein and induced cell autophagy. H2Se could interrupt the disulfide bond in HMGB1 and promote its secretion. The reduced HMGB1 outside the cells stimulated cell autophagy by inhibiting the Akt/mTOR axis. Here, autophagy played a dual role, i.e., mild autophagy inhibited apoptosis, while excessive autophagy led to autophagy-associated cell death.
Conclusions: These results show that H2Se plays a key role during HepG2 cell death induced by selenite. Our findings reveal a new anti-cancer mechanism of selenite and provide a new research area for selenium studies.
Introduction
H2Se is a common metabolite of dietary selenium compounds (selenite, SeMet, MeSeCys and CysSeSeCys)1, 2. Dietary selenium compounds significantly differ in their metabolic pathways and their abilities to produce various selenium metabolites, but their metabolic pathways intersect at a common metabolite, which has widely been identified as hydrogen selenide (H2Se)1. H2Se is a highly reducible selenide with very high volatility and reactivity that cannot be directly detected in cell and animal models. Selenium compounds may have a potential use in the prevention and treatment of cancers3. However, the role of H2Se in selenium compound treatments for cancers has not been elucidated due to the lack of detection methods. In our previous study, we developed a specific fluorescent probe for the real-time monitoring of H2Se in living cells and in vivo4 (Fig. 1A). This probe can rapidly respond to H2Se with high selectivity and sensitivity. In this study, we reveal an important effect of H2Se during selenium treatment for cancer with the help of this fluorescent probe.
H2Se fluorescent probe and the cytotoxicity of Na2SeO3 in HepG2 cells. (A) Probe used to detect H2Se. (B, C, D and E) HepG2 cells were treated with different concentrations of Na2SeO3 for 24 h under hypoxic conditions (1% O2) or normoxic conditions (20% O2). Cell viabilities were determined by an MTT assay (B). Cell death was measured by LDH release assay (C) and Typan blue staining analysis (E). Morphological changes were observed under an inverted microscope (Olympus, Japan) (D). (*p< 0.05, **p< 0.01, ***p< 0.001, t test).
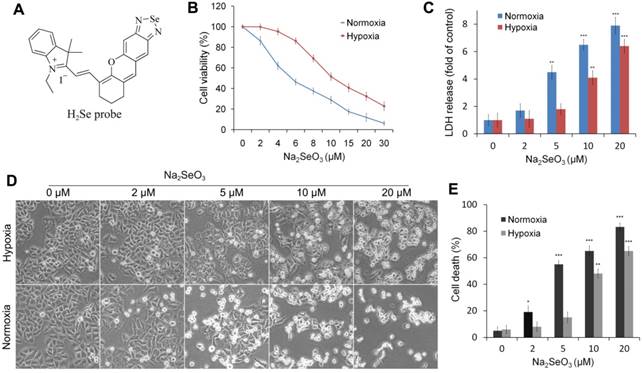
Sodium selenite (Na2SeO3) is the first dietary selenium compound shown to produce H2Se during metabolism and is considered to have cancer treatment properties1. Therefore, we chose Na2SeO3 as the supplier of H2Se in this study. Na2SeO3 has anti-tumor effects, but the mechanisms are very complex and not fully understood. Previous studies have attributed the anti-cancer mechanism of Na2SeO3 to oxidative stress5-8. Selenite is reduced to H2Se by glutathione reductase (GR); then, H2Se can rapidly react with O2 to form elementary selenium and superoxide anion radicals (O2.-), leading to DNA strand breaks and apoptosis in cancer cells9, 10 or resulting in a decrease in the mitochondrial membrane potential and release of cytochrome c into the cytosol, which then leads to cell apoptosis11,12. Although the anti-cancer mechanism of selenite has been under investigation for two decades, there are still inconsistencies between in vitro studies and clinical outcomes. The main reason for these differences is that the tumor microenvironment is very complicated, and most in vitro studies do not fully consider the influence of the tumor microenvironment on the experimental results.
Hypoxia, which refers to low levels of O2, is a well-known feature of the microenvironment of solid tumors. It has been estimated that 50 to 60% of solid tumors contain regions of hypoxia due to the increased tumor size, abnormal growth of the tumor vasculature, and reduced oxygen concentration in the blood13-15. The intratumoral O2 levels in many solid tumors ranges between 5.3 and 14 mmHg (0.7-1.8%)16. In hepatocellular carcinoma (HCC), most regions inside the tumor have O2 values within the range of 0-10 mmHg (0-1.32%)13,17. Clinical studies have shown that intratumoral hypoxia is closely related to the effect of chemotherapy. However, previous studies have overlooked this problem in most cases and tested cancer cells cultured in a normoxic environment in vitro, which provides sufficient O2 levels to produce reactive oxygen species (ROS). Therefore, we question whether H2Se produced by selenium metabolism could completely react with oxygen to produce ROS under hypoxic conditions.
Here, we chose human hepatocellular carcinoma HepG2 cells to reveal the important role of H2Se produced by Na2SeO3 under hypoxia (1% O2). Once HepG2 cells were treated with pharmacological doses of Na2SeO3 under hypoxic conditions, high levels of H2Se were produced, but no obvious increase in ROS was observed. The H2Se accumulation resulted in reductive stress in cells and a reduction in HMGB1 protein. The reduced HMGB1 was secreted and promoted cell autophagy, eventually leading to autophagy-associated cell death via the inhibition of Akt/mTOR phosphorylation.
Materials and Methods
Cell culture
Hepatocellular carcinoma HepG2 cells were obtained from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China) and cultured in DMEM supplemented with 10% FBS at 37 °C in a humidified atmosphere of 95% air (20% O2) and 5% CO2. For simulating the tumor microenvironment, cells were cultured in a mixture containing 1% O2, 94% N2 and 5% CO2 using Bugbox M (Ruskinn, England).
Reagents and antibodies
Sodium selenite (Na2SeO3) and 3-(4,5-dimethyl- 2-thiazolyl)-2,5-diphnyl-2H-tetrazolium bromide (MT T) were purchased from Sigma-Aldrich (St. Louis, MO, USA). NADP/NADPH Assay Kit (ab 65349) was obtained from Abcam (Cambridge, MA, USA). GSH/ GSSG Assay Kit, total Superoxide Dismutase Assay Kit and Catalase Assay Kit were from Beyotime (Shanghai, China). Recombinant human high-mobility group protein B1 (HMGB1) was from ProSpec- Tany (Israel). Anti-LC3 (16 kDa, 14 kDa), anti-p62 (62 kDa), anti-cleaved caspase 9 (35 kDa), anti-cleaved caspase3 (17 kDa), anti-AKT (60 kDa), anti-p-AKT (ser473) (60 kDa), anti-p-AKT (thr308) (60 kDa), anti-mTOR (289 kDa), anti-p-mTOR (ser2448) (289 kDa) and anti-Histone H3 (17 kDa) antibodies were from Cell Signaling Technology (Danvers, MA, USA). Anti-superoxide dismutase 1 (18 kDa), anti-catalase (60 kDa), anti-HMGB1 (25 kDa), anti-caspase9 (46 kDa), anti-caspase3 (35 kDa) antibodies and the secondary antibody were from Abcam (Cambridge, MA, USA).
Cell viability assay and LDH release assay
The cytotoxicity of Na2SeO3 to HepG2 cells under normoxic (20% O2) and hypoxic (1% O2) conditions was evaluated by MTT assays. Briefly, HepG2 cells were seeded in 96-well flat-bottom microtiter plates at a density of 20,000 cells/mL with 100 μL per well and incubated for 24 h at 37 °C. Then, cells were exposed to different concentrations of Na2SeO3 under normoxic and hypoxic conditions for 24 h. 20 μL MTT solution (5 mg/mL) was added to each well and incubated at 37 °C for 4 h. The supernatant was discarded, and 100 μL DMSO was added to each well to dissolve the crystals. Absorbance was recorded at a wavelength of 490 nm with a microtiter plate reader (Bio-Tek ELX800, Winooski, VT, USA). LDH release assay was measured according to LDH cytotoxicity assay kit (Beyotime Biotechnology, China) according to the manufacturer's instructions. At least three replicates were carried out for each treatment.
Fluorescence imaging in living cells
The fluorescence of H2O2 and H2Se was detected by confocal microscopy. HepG2 cells were grown on 15 mm glass-bottom culture dishes. After treatment with Na2SeO3, the cells were incubated with 10 μM H2O2 probe or 10 μM H2Se probe in FBS-free DMEM medium at 37 °C for 15 min and then washed three times with PBS buffer. Then, the cells were imaged immediately using a confocal microscope (×40) with 532 nm excitation and 600-700 nm collection for H2O2 and 633 nm excitation and 650-750 nm collection for H2Se.
Tumor model preparation
All animal experiments were carried out according to the Principles of Laboratory Animal Care (People's Republic of China) and the Guidelines of the Animal Investigation Committee, and approved by the local Animal Care and Use Committee. Eight-week-old Kunmin mice were purchased from the Shanghai SLAC Laboratory Animal Co., Ltd. 1×107 hepatocellular carcinoma H22 cells derived from mice were injected into the enterocoelia of one mouse, and ascites were formed after 5-7 days, which were then used after three passages. Eight-week-old mice received a subcutaneous injection of H22 ascites tumor cells into the axillary lateral subcutaneous of their left forelimbs at a density of 1×107 cells/mL with 100 μL per mice. After 5 days, these mice were given different concentrations of Na2SeO3 through oral administration for different times. Tumor volume was measured using digital calipers and calculated as: volume = 0.5 × (length×width2) 18.
Fluorescence imaging in vivo
After administration of 0-10 mg/kg of Na2SeO3 via oral administration for 10 days, the mice were subcutaneously injected with an H2O2 probe or H2Se probe and incubated for 30 min. The fluorescence images were obtained using an in vivo imaging system (IVIS) with 532 nm excitation and 600-700 nm collection for H2O2, and 633 nm excitation and 650-750 nm collection for H2Se.
Measurement of SOD and CAT activities
Tumor-bearing mice were treated with 0-10 mg/kg of Na2SeO3 through oral administration for 10 days. The tumor tissues were harvested and homogenized on ice. Total SOD and CAT activities were measured using a Total Superoxide Dismutase Assay Kit with NBT (Beyotime Biotechnology) and a Catalase Assay Kit (Beyotime Biotechnology), respectively. The assays were performed according to the instructions provided by the manufacturer.
NADPH and GSH detection
Tumor-bearing mice were treated with different concentrations of Na2SeO3 through oral administration for 10 days. The tumor tissues (approximately 50 mg) were then harvested and washed with cold PBS. The tissues were homogenized on ice using a Dounce homogenizer (30-50 passages) with 500 μL of NADP/NADPH extraction buffer. The samples were transferred to a tube and centrifuged at 14,000 rpm at 4 °C for 5 min. Then, the extracted NADP/NADPH supernatant was transferred into a 10 kD Spin Column (ab 93349) and centrifuged at 10,000×g for 20 min at 4 °C to remove the enzymes contained in tissues that can consume NADPH rapidly. Then, the samples were assayed according to the instructions of the NADP/NADPH Assay Kit (colorimetric) (ab 65349). Samples preparation for GSH detection was similar to the above, and the samples were assayed according to the instructions of the GSH/GSSG Assay Kit (Beyotime Biotechnology).
HMGB1 ELISA assays
HepG2 cells were exposed to 10 μM Na2SeO3 under hypoxic conditions or 5 μM Na2SeO3 under normoxic conditions for different time (0-24 h). After treatment, the supernatants of each well were harvested and analyzed for HMGB1 levels using an HMGB1 ELISA kit (Shino-TestCorporation, Kanagawa, Japan) according to the manufacturers' instructions. Simultaneously, the cells of each well were also collected and lysed to test the intracellular HMGB1 levels by Western blot analysis.
Western blot analysis
After treatment, HepG2 cells were harvested and lysed using 100 μL of lysis buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, EDTA, Complete protease inhibitor) for 15 min at 4 °C. The lysates were centrifuged at 15,000 rpm for 15 min at 4 °C. The supernatants were transferred to a new tube and quantified by BCA Assay Kit (P0009) (Beyotime, China). An equal amount of protein was separated by SDS-PAGE (12%) and transferred to PVDF membranes. After the membrane was blocked with 5% non-fat dried milk for 2 h, it was incubated with the desired primary antibodies overnight at 4 °C. Then, the membrane was incubated with HRP-conjugated IgG secondary antibodies for 2 h. Chemiluminescence was detected using the Western Blotting Detection System (ImageQuant LAS 500). For quantification of equal loading, the membrane was reprobed with Histone H3 antibody.
Transmission electron microscopy (TEM)
HepG2 cells were exposed to Na2SeO3 for different time (0-24 h) under hypoxic and normoxic conditions. After treatment, cells were harvested and fixed according to our previous report19. The data were then analyzed by TEM.
H2Se interrupts the disulfide bond in HMGB1
Exogenous H2Se was prepared according to our previous report4. Briefly, Al2Se3 reacted with H2O in an N2 atmosphere for 30 min, then, the aqueous solution containing H2Se was used for the experiment. HMGB1 proteins were dissolved in sterile 18 MΩ-cm H2O not less than 100 µg/ml, which can then be further diluted to other aqueous solutions. To examine whether H2Se can interrupt the disulfide bond in HMGB1, 10 μg protein samples were incubated with sodium dodecyl sulfate (SDS) sample buffer ( 4% SDS, 40% Glycerol, 0.032% Bromophenol blue, 40 mM Tris-HCl PH 8.0 ) containing 2 mM H2Se or 200 mM DTT for 30 min, respectively; the mixture was then separated by non-reducing SDS-PAGE (12% gel). The gel was run at 90 V for 2 h followed by staining with Coomassie blue. The image was acquired using a gel imaging system (ImageQuant LAS 500).
Statistical analysis
Data were analyzed using GraphPad Prism 5.0 software. Student t test (two-tailed) was used for single comparisons. All data are presented as mean ± SEM, data were considered statistically significant when p < 0.05, where *: p<0.05; **: p<0.01; ***: p<0.001.
Results
Na2SeO3 has different cytotoxicity under hypoxic conditions and normoxic conditions
To investigate the different effects of Na2SeO3 under hypoxia and normoxia, we evaluated the cell viability of HepG2 cells treated with Na2SeO3 under hypoxic conditions (1% O2) and normoxic conditions (20% O2), respectively. After the HepG2 cells were treated with different concentrations of Na2SeO3 for 24 h, the semi-inhibitory concentration (IC50) of Na2SeO3 under the hypoxic conditions was approximately 10 μM. However, under the normoxic conditions, the IC50 was 5 μM (Fig. 1B) (the IC50 concentration was used in the subsequent experiments). The cell death assay and morphological observations revealed that after the treatment with 5 μM of Na2SeO3 for 24 h, about half of the cells cultured under the normoxic conditions died, while the cells cultured under the hypoxic conditions displayed minimal cell death. When the concentration of Na2SeO3 was increased to 10 μM, most cells under the normoxic conditions died, while only approximately half of the cells under the hypoxic conditions died (Fig. 1C, D and E). Remarkably, numerous vacuoles appeared inside the undead cells under the hypoxic conditions (Fig. 1D). These results suggest that the anti-cancer mechanism of Na2SeO3 under hypoxia may differ from that under normoxia.
Na2SeO3 does not induce oxidative stress under hypoxic conditions
Several previous studies have shown that Na2SeO3 can induce cell apoptosis through oxidative stress5,7. Thus, we assessed whether oxidative stress was induced by Na2SeO3 in the process of cell death under the hypoxic conditions. In this experiment, a previously described H2O2 probe20,21 (Fig. S1) was employed because of its high selectivity and sensitivity. HepG2 cells were exposed to 10 μM Na2SeO3 for 0-24 h or 0-10 μM Na2SeO3 for 6 h under hypoxic conditions, and then, the cells were incubated with the H2O2 probe for 15 min. Negligible H2O2 intracellular background fluorescence was observed under the hypoxic conditions (Fig. 2A, B). Conversely, under the normoxic conditions, the fluorescence intensity increased after the cells were treated with 0-10 μM Na2SeO3 for 6 h (Fig. 2C). These results confirm that Na2SeO3 exhibits a pro-oxidant function under normoxic conditions. However, under the hypoxic conditions, no oxidative stress was involved in the process of Na2SeO3-mediated HepG2 cell death. To further confirm these findings, we conducted a similar experiment in vivo. Tumor-bearing mice were orally treated with different concentrations of Na2SeO3 for 10 days. Then, the mice were subcutaneously injected with 10 μM of the H2O2 probe, and fluorescence images were obtained using an in vivo imaging system after 30 min. The fluorescence signals of H2O2 were not enhanced in the Na2SeO3 treatment groups (Fig. 2D). These findings support the in vitro results obtained using the HepG2 cells showing that no oxidative stress was involved in the Na2SeO3-induced cell death.
Detection of oxidative stress. (A) HepG2 cells were exposed to 10 μM Na2SeO3 for 0-24 h under hypoxic conditions (1% O2) and then incubated with 10 μM of the H2O2 probe for 15 min before the fluorescence images were obtained using confocal microscopy. (B and C) HepG2 cells were exposed to 0-10 μM Na2SeO3 for 6 h under hypoxic conditions (1% O2) or normoxic conditions (20% O2), and then, the cells were incubated with 10 μM of the H2O2 probe for 15 min before the fluorescence images were obtained using confocal microscopy. The fluorescence intensity of Figure A, B and C was quantified based on the results of the relative fluorescence intensity of per cell in the scanned area. (D) Tumor-bearing mice were treated with 0-10 mg/kg Na2SeO3 for 10 days, and then, 10 μM H2O2 probe was subcutaneously injected into the tumor for 30 min before the fluorescence was analyzed using an in vivo imaging system. (E, F and G) After tumor-bearing mice were treated with 0-10 mg/kg Na2SeO3 for 10 days, the tumor tissues were harvested and lysed for CAT and SOD protein expression detection by western-blot assay with Histone H3 as an internal reference for protein standardization (E), and for CAT and SOD activity detection (F and G). The scale bar in all fluorescence images of cells is 50 μm.
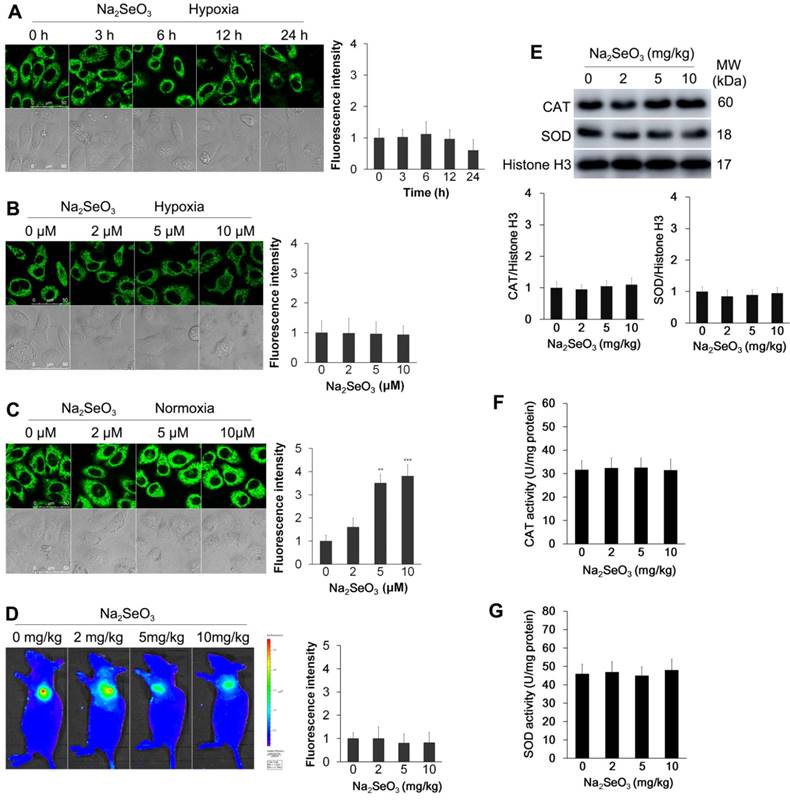
Furthermore, two antioxidant enzymes, superoxide dismutase (SOD) and catalase (CAT), were detected. Tumor-bearing mice were orally treated with different concentrations of Na2SeO3 for 10 days. The tumor tissues were obtained to detect the protein expression and enzyme activity of SOD and CAT. Immunoblot analysis revealed that the Na2SeO3 treatment did not cause changes in the expression of SOD and CAT (Fig. 2E). Consistent with these observations, the enzyme activities of SOD and CAT also showed minimal changes after the mice were treated with different concentrations of Na2SeO3 (Fig. 2F and G).
Na2SeO3 induces reductive stress by the accumulation of H2Se
Na2SeO3 can be reduced to H2Se by GSH and other reduction systems1. In our previous work, we reported the synthesis of a novel small-molecule fluorescent probe (NIR-H2Se) for the detection of H2Se4. NIR-H2Se can rapidly respond to H2Se with a high selectivity and has been successfully used for detecting endogenous H2Se both in vitro and in vivo. In this study, we used the probe to observe the H2Se levels produced by Na2SeO3 metabolism during cell death. HepG2 cells were exposed to 0-10 μM Na2SeO3 for 12 h or 10 μM Na2SeO3 for 0-24 h under hypoxic conditions. After treatment, the cells were loaded with 10 μM NIR-H2Se for 15 min. The results indicate that the HepG2 cells treated with 0-10 μM Na2SeO3 had a concentration-dependent increase in fluorescence, and a nearly 4-fold increase in the fluorescence intensity was observed in the 10 μM group compared with that in the control group (Fig. 3A). Additionally, after the HepG2 cells were treated with 10 μM Na2SeO3 for 0-12 h, the fluorescence intensity was also enhanced in a time-dependent manner. However, when the treatment was extended to 24 h, the fluorescence intensity decreased due to the cell was going to die (Fig. 3B). Cell death analysis results showed that approximately half of the HepG2 cells died after 10 μM Na2SeO3 treatment for 24 h (Fig. 3C and D). These results indicate that H2Se is produced before cell death. In contrast, after the HepG2 cells were exposed to 0-10 μM Na2SeO3 for 12 h under the normoxic conditions, the cells exhibited much lower fluorescence compared with that under the hypoxic conditions (Fig. 3E), which may be due to the rapid oxidation of H2Se in the aerobic environment9.
Detection of reductive stress. (A) HepG2 cells were exposed to 0-10 μM Na2SeO3 for 12 h under hypoxic conditions (1% O2) and then incubated with 10 μM of the H2Se probe for 15 min before the fluorescence images were obtained using confocal microscopy. (B) HepG2 cells were exposed to 10 μM Na2SeO3 for 0-24 h under hypoxic conditions (1% O2), and then, the H2Se probe was added using the method described in (A). The fluorescence images were obtained using confocal microscopy. The fluorescence intensity of Figure A and B was quantified based on the results of the relative fluorescence intensity of per cell in the scanned area. (C and D) HepG2 cells were exposed to 10 μM Na2SeO3 for 0-24 h under hypoxic conditions (1% O2), the cytotoxicity of Na2SeO3 was observed under an inverted microscope (C), then, the cells were collected and stained by trypan blue (D). (E) HepG2 cells were exposed to 0-10 μM Na2SeO3 for 12 h under normoxic conditions (20% O2), and then, the H2Se probe was added using the method described in (A). The fluorescence images were obtained using confocal microscopy and the fluorescence intensity was quantified as described in (A and B). (F) Tumor-bearing mice were treated with 0-10 mg/kg Na2SeO3 for 10 days, and then, 10 μM NIR-H2Se probe was subcutaneously injected into the tumor for 30 min before the fluorescence was analyzed using an in vivo imaging system. (G and H) Tumor-bearing mice were treated with 0-10 mg/kg Na2SeO3 for 10 days, and then, the tumor tissues were harvested for the NADPH/NADPHt and GSH/GSHt ratio detection. (I) Tumor-bearing mice were treated with 0-10 mg/kg Na2SeO3 for 10 days, then, the tumor tissues were taken out and photographed. (J) While the tumor-bearing mice were treated with 0-10 mg/kg Na2SeO3 for different days, the tumor volumes were measured using digital calipers. (*p< 0.05, **p< 0.01, ***p< 0.001, t test). The scale bar in all fluorescence images of cells is 50 μm.
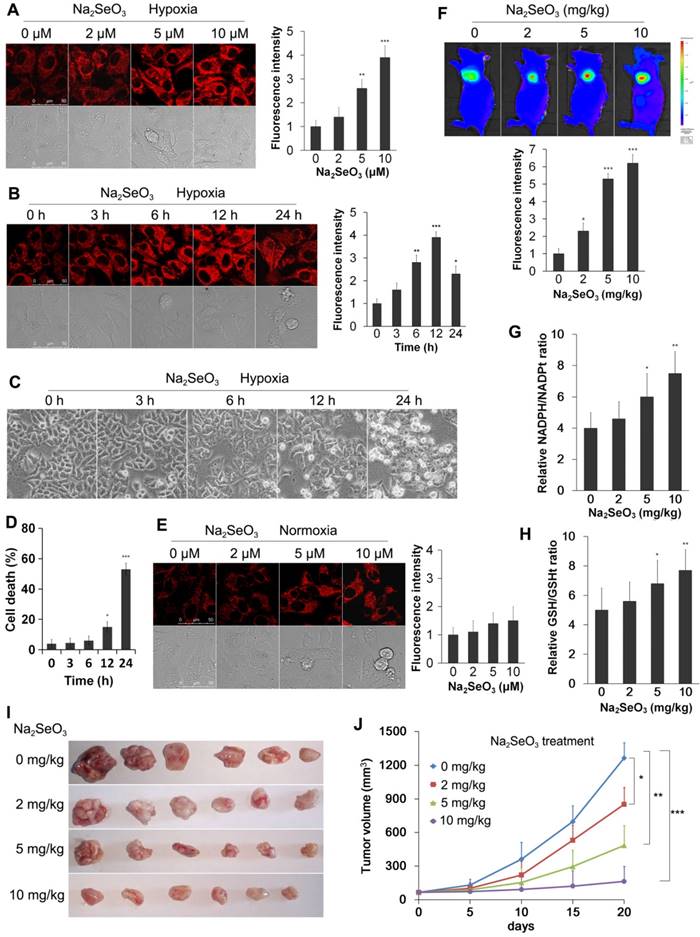
Further support for above phenomenon under the hypoxic conditions was provided by the in vivo experiment. Tumor-bearing mice were treated with different concentrations of Na2SeO3 via oral administration for 10 days. Then, the mice received a subcutaneous administration of the NIR-H2Se probe (10 μM) into the tumor. The probe was incubated for 30 min before the fluorescence images were obtained using an in vivo imaging system. As a result, the fluorescence signal of the probe was substantially increased in the Na2SeO3 treatment groups (Fig. 3F). Furthermore, we tested the NADPH levels and GSH levels in the tumor tissues in parallel mouse experiments. The NADPH levels were significantly increased in a dose-dependent manner in the Na2SeO3 treatment groups (Fig. 3G). Consistent with the NADPH results, the GSH levels were also increased in the Na2SeO3 treatment groups (Fig. 3H). H2Se, NADPH and GSH are three highly reactive reducible molecules, and excess NADPH and/or GSH is a marker of reductive stress22-24. Several studies have reported that reductive stress can also cause body damage and lead to cell death22,25. Our tumor volume measurement experiments indicated that Na2SeO3 could significantly inhibit tumor growth in a dose- and time-dependent manner (Fig. 3I and 3J). Therefore, here, we propose that the cell death induced by Na2SeO3 under the hypoxic conditions in vitro and the liver cancer growth inhibition in vivo may be attributed to reductive stress caused by the accumulation of H2Se.
H2Se targets the redox of the HMGB1 protein and activates cell autophagy
Our data indicate that H2Se may play a key role in the cell death caused by Na2SeO3. Subsequently, we elucidated the anticancer mechanism of H2Se. Because H2Se is a highly reactive and reducible molecule, we first considered its effect on redox proteins. High-mobility group protein B1 (HMGB1) is a redox-sensitive protein containing three cysteines (Cys23, 45, and 106), and the two Cys23-Cys45 residues can form an intramolecular disulfide bond26. The activity of HMGB1 strongly depends on its redox state27. In addition, the HMGB1 protein is both a nuclear factor and a secreted protein associated with cell survival and death28. In the nucleus, HMGB1 acts as a non-histone architectural chromatin-binding factor that bends DNA and promotes protein assembly in specific DNA targets29, 30. HMGB1 is also a secreted protein that can be secreted from cells in response to damage or stress and functions as an extracellular signaling molecule in cell survival/death pathways31, 32. Therefore, we hypothesize that HMGB1 is a target of H2Se.
Subsequently, we examined the intracellular and extracellular HMGB1 levels under hypoxic conditions and normoxic conditions. HepG2 cells were treated with IC50 concentrations of Na2SeO3 for 0-24 h under hypoxic conditions and normoxic conditions, respectively. The cells were then harvested and lysed to assess the intracellular HMGB1 protein levels. The data showed that the intracellular HMGB1 protein levels were down-regulated significantly in the Na2SeO3 treatment group in a time-dependent manner under the hypoxic conditions, while there was less reduction under the normoxic conditions (Fig. 4A). Simultaneously, the culture supernatants were also collected, and the extracellular HMGB1 levels were analyzed by ELISA. We found that the extracellular HMGB1 levels increased obviously in a time-dependent manner under the hypoxic conditions. In comparison, under the normoxic conditions, the HMGB1 levels also showed an increasing trend over time but were lower than those observed under the hypoxic conditions (Fig. 4B). These results indicate that intracellular HMGB1 is released outside the cell after HepG2 cells are treated with Na2SeO3 under both hypoxic and normoxic conditions; however, the released amount under the hypoxic conditions is higher than that under the normoxic conditions.
HMGB1 protein analysis and cell autophagy detection. (A and B) HepG2 cells were treated with 10 μM Na2SeO3 under hypoxic conditions (1% O2) or 5 μM Na2SeO3 under normoxic conditions (20% O2) for 0-24 h, respectively; then, the cells were collected to detect the intracellular HMGB1 levels by western blot assay (A), and the culture supernatants were collected to test the extracellular HMGB1 levels by ELISA (B) (*p< 0.01, **p< 0.01, t test). (C) The cell treatment methods are the same as those described in (A). After treatment, the cells were harvested and lysed to detect the LC3, p62, clCaspase 9 and clCaspase 3 levels by western-blot analysis with Histone H3 as an internal reference. (D) HepG2 cells were treated with 10 μM Na2SeO3 under hypoxia or 5 μM Na2SeO3 under normoxia for 6-24 h, then the formation of autophagosomes and autolysosomes was assessed by transmission electron microscopy. Red arrow indicating autophagosome or autolysosome. (E) Under hypoxic conditions, HepG2 cells were incubated with the autophagy specific inhibitor CQ (10 μM) for 3 h or 3-MA (5 mM) for 1 h, followed by treatment with 10 μM Na2SeO3 for 24 h. After treatment, the cells were collected to detect the LC3 and p62 levels by western-blot analysis. (F) Under hypoxic conditions, HepG2 cells were treated with 10 μg/mL reduced HMGB1 or 10 μg/mL oxidized HMGB1 for 24 h, respectively; then, the cells were harvested and lysed to detect the LC3, p62, clCaspase 9 and clCaspase 3 levels by western-blot analysis. “R” represents reduced HMGB1, and “O” represents oxidized HMGB1.
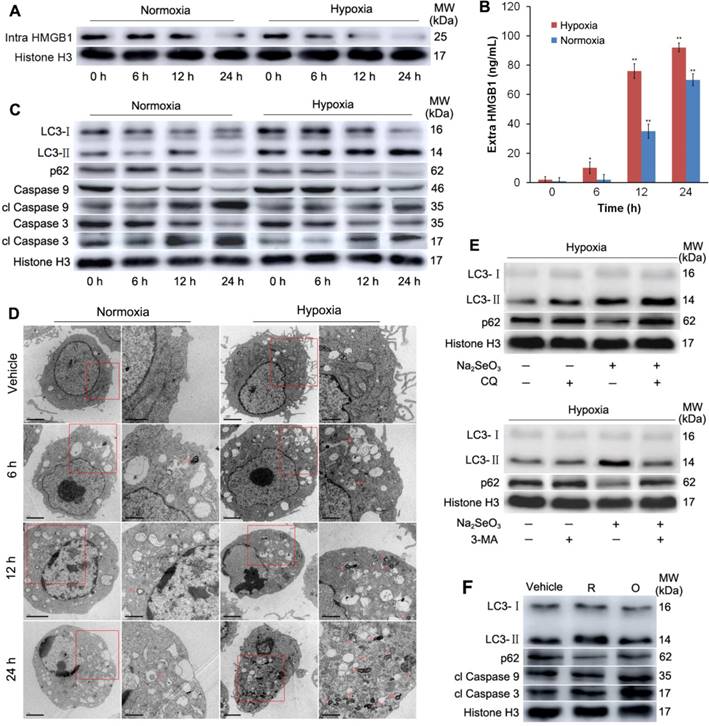
The activity of the secreted/released HMGB1 depends on its redox state26,27. Previous reports have shown that reduced exogenous HMGB1 can increase autophagy and that oxidized HMGB1 increases apoptosis33-35. Therefore, we investigated whether autophagy was induced after HepG2 cells were treated with Na2SeO3 under hypoxic conditions. HepG2 cells were exposed to IC50 concentrations (10μM) of Na2SeO3 for 0-24 h under hypoxic conditions. Simultaneously, the same treatment was applied under normoxic conditions for comparison. After treatment, the cells were harvested and lysed. Then, the LC3 and p62 proteins, a pair of autophagy markers, were assessed. Immunoblot analysis revealed that the Na2SeO3 treatment resulted in a drastic conversion of LC3 I/II and down-regulation of p62 in a time-dependent manner during the process of cell death. In contrast, the characteristic autophagy-related events were observed less under the normoxic conditions (Fig. 4C, Fig. S2A and B). The transmission electron microscopy results showed that the Na2SeO3 treatment caused the accumulation of autophagosomes and/or autolysosomes in a time-dependent manner under hypoxic conditions, comparatively, there were only mild autophagy appeared under normoxic conditions (Fig. 4D). Moreover, chloroquine (CQ) and 3-methyladenine (3-MA), which are two autophagy inhibitors, inhibited the autophagy process successfully (Fig. 4E), indicating that an autophagic flux occurred under the hypoxic conditions. At the same time, two apoptosis-related markers, i.e., caspase-9 and caspase-3, were also detected. The Na2SeO3 treatment for 12-24 h significantly increased the levels of cleaved caspase-9 and cleaved caspase-3 under the normoxic conditions. However, under the hypoxic conditions, the Na2SeO3 treatment resulted in a mild increase in cleaved caspase-9 and cleaved caspase-3 (Fig. 4C, Fig. S2C and D). These data indicate that Na2SeO3 mainly induces cell apoptosis under normoxic conditions but activates cell autophagy under hypoxic conditions, suggesting that under hypoxic conditions, the released HMGB1 may exist in the reduced form, while under normoxic conditions, it should exist in the oxidized form.
To further identify the redox state of the released HMGB1 under hypoxic and normoxic conditions, we treated HepG2 cells with recombinant HMGB1 proteins. A portion of the protein was subjected to dithiothreitol (DTT), which is a well-known thiol-based protein disulfide reducing agent, to obtain reduced HMGB1, and another portion was exposed to H2O2 to obtain oxidized HMGB1. As shown in Fig. 4F, under hypoxia, the treatment with reduced HMGB1 induced LC3-II formation and reduced the expression of p62. There was no significant change in the expression of cl Caspase9 and cl Caspase3. Conversely, oxidized HMGB1 led to obviously increased levels of cl Caspase9 and cl Caspase3, but the LC3-II and p62 levels did not significantly change. These results indicate that reduced HMGB1 promotes autophagy, while oxidized HMGB1 promotes apoptosis in HepG2 cells, which is consistent with previous studies33,34.
According to the above results, we believe that the released HMGB1 after Na2SeO3 treatment under hypoxia should be in the reduced form, and the reduction of HMGB1 may be attributed to the strong reduction of H2Se. To determine whether H2Se could interrupt the Cys23-Cys45 disulfide bond in HMGB1, an in vitro simulation system experiment was performed. Because the redox states of proteins can be monitored by mobility shift 36-38, we tested whether H2Se could interrupt the disulfide bond in HMGB1 using non-reducing sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). In this experiment, we used the strong reducing agent DTT for comparison. Similar to the DTT-treated group, the protein incubated with H2Se showed an electrophoretic mobility that was smaller than that in the group without H2Se (Fig. 5A). These results demonstrate that H2Se could interrupt the disulfide bond contained in HMGB1. To further prove that H2Se can transform the disulfide bond to the mercapto groups in proteins, we tested the lysozyme, which contains four disulfide bonds, by an ESSI-MS analysis. After adding H2Se to the lysozyme, all disulfide bonds were interrupted and formed eight mercapto groups (Fig. S3). Thus, we speculated that the secreted HMGB1 protein under hypoxic conditions in the above experiment (Fig. 4B) should exist in its reduced form and that the reduction in HMGB1 is attributed to the accumulation of H2Se produced by Na2SeO3. Then, the reduced HMGB1 promotes autophagy.
H2Se interrupts the disulfide bonds in HMGB1 and induces cell autophagy. (A) Non-reducing SDS-PAGE indicated the reduction in HMGB1. HMGB1 (10 μg/lane) with or without H2Se and DTT was loaded on a 12% gel. Lanes marked with “O” represents oxidized HMGB1 in the absence of H2Se and DTT. Lanes marked with “R” represents reduced HMGB1 by H2Se or DTT. (B) oxDTT can clear H2Se. HepG2 cells were incubated with 10 μM Na2SeO3 and 100 μM oxDTT for 12 h under hypoxic conditions. After treatment, the cells were loaded with 10 μM H2Se probe for 15 min and fluorescence images were obtained using confocal microscopy. Scale bar = 50 μm. The fluorescence intensity was quantified based on the results of the relative fluorescence intensity of per cell in the scanned area. (**p< 0.01, ***p< 0.001, t test). (C) H2Se induced cell autophagy. HepG2 cells were treated using the method described in (B), and then, the cells were harvested and lysed to detect the LC3 levels by western-blot analysis.
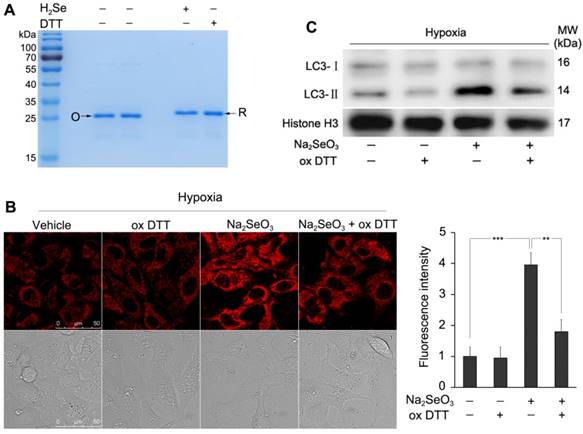
To further determine the relationship between H2Se and autophagy, we removed H2Se with oxidized DTT (oxDTT) and observed the change in autophagy. HepG2 cells were incubated with Na2SeO3 (10 μM) and oxDTT (100 μM) for 12 h under hypoxic conditions. After the treatment, a fraction of the cells was loaded with 10 μM NIR-H2Se for 15 min and submitted to a fluorescence analysis. As shown in Fig. 5B, the enhanced fluorescence intensity of H2Se induced by Na2SeO3 was markedly decreased by oxDTT, indicating that oxDTT can clear H2Se. Another fraction of the cells was collected for a western-blot analysis. The results showed that the Na2SeO3-induced increase in the protein expression of LC3-II was obviously reversed by oxDTT (Fig. 5C). These results further indicate that H2Se induces autophagy in HepG2 cells under the hypoxic conditions.
H2Se-mediated autophagy can inhibit apoptosis and result in autophagy-associated cell death
To investigate the role of autophagy in selenite- induced cell death, HepG2 cells were incubated with the autophagy specific inhibitor CQ (10 μM) for 3 h, followed by treatment with Na2SeO3 under hypoxic conditions. After treatment, the cells were collected and submitted to a western-blot analysis and MTT assay. The results showed that CQ blocked the progression of autophagy by blocking the fusion of autophagosomes and lysosomes to upregulate LC3-II expression (Fig. 4E). Furthermore, MTT assay revealed that compared with the Na2SeO3 treatment alone, the CQ-pretreatment enhanced the inhibitory effect of Na2SeO3 on cell viability when HepG2 cells were treated with 10 μM Na2SeO3 for 6-12 h. In contrast, when the Na2SeO3 treatment time was extended to 24-48 h, the CQ-pretreatment weakened the effect of Na2SeO3 (Fig. 6A). Similarly, after treating the HepG2 cells with 5-7.5 μM Na2SeO3 for 24 h, the CQ-pretreatment boosted the cell growth inhibition of Na2SeO3. However, as the concentration of Na2SeO3 increased to 10-20μM, the CQ-pretreatment attenuated the inhibitory effect of Na2SeO3 (Fig. 6B). To observe the relationship between autophagy and apoptosis induced by selenite, we also tested the activity of caspases. After the HepG2 cells were treated with 10 μM Na2SeO3 for 12 h, the CQ-pretreatment significantly enhanced the activity of caspase 3 and caspase 9 compared with that following the Na2SeO3 treatment alone (Fig. 6C and D). However, the CQ-pretreatment only caused a mild increase in caspase 3 and caspase 9 activities when Na2SeO3 treatment time was extended to 24 h (Fig. 6C and D). These observations indicate that the autophagy induced by H2Se plays a dual role. Mild autophagy can inhibit cell apoptosis, and autophagy plays a cytoprotective role. However, excessive autophagy leads to autophagy-associated cell death, and, autophagy and apoptosis simultaneously promote cell death.
Detection of apoptosis after the inhibition of autophagy and an Akt/mTOR axis analysis. (A and B) HepG2 cells were pretreated with 10 μM CQ for 3 h followed by treatment with 10 μM Na2SeO3 for 0-48 h or 0-20 μM Na2SeO3 for 24 h under hypoxic conditions, then the cell viabilities were determined by an MTT assay. (*p< 0.05, t test). (C and D) HepG2 cells were pretreated with 10 μM CQ for 3 h followed by treatment with 10 μM Na2SeO3 for 0-24 h under hypoxic conditions; then, the cells were harvested for the caspase 9 and caspase 3 activity detection (*p< 0.05, t test). (E) HepG2 cells were treated with 10 μM Na2SeO3 for 0-24 h under hypoxic conditions; then, the cells were harvested and lysed to detect p-AKT and p-mTOR levels by western-blot analysis with Histone H3 as an internal reference.
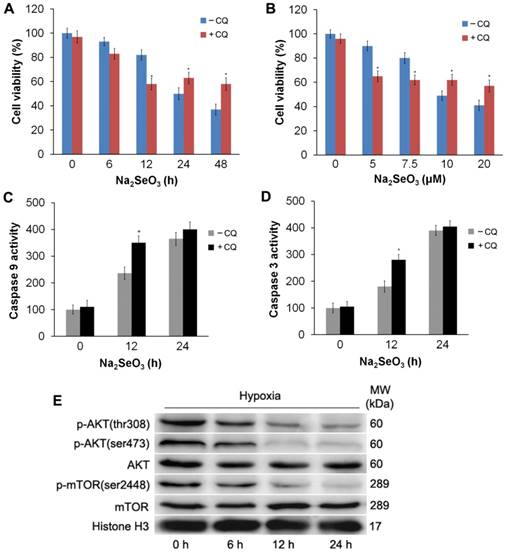
H2Se activates autophagy by inhibiting Akt/mTOR phosphorylation in HepG2 cells
Subsequently, we examined the upstream targets of autophagy signaling to further illuminate the mechanism of H2Se-induced autophagy. It has been demonstrated that blocking the Akt/mTOR axis stimulates autophagy39. Therefore, we examined whether the Akt/mTOR pathway was inhibited in selenite-treated liver cancer cells. HepG2 cells were treated with 10 μM Na2SeO3 for 0-24 h under hypoxic conditions. The western blot analysis showed that the phosphorylation of Akt (Ser473 and Thr308) was attenuated by Na2SeO3 in a time-dependent manner, while the total protein levels of Akt remained unchanged compared to those in the control (Fig. 6E). Moreover, Na2SeO3 suppressed the phosphorylation of mTOR (Ser2448) without affecting its total protein expression (Fig. 6E). These results suggest that the Akt/mTOR signaling pathway is inhibited by Na2SeO3.
Discussion
Selenium is an essential trace element involved in different physiological functions in the human body and has both cancer preventive and cancer treatment properties. Selenium plays a possible role in cancer prevention largely because of its antioxidant properties40. As a component of antioxidant selenoproteins (e.g., glutathione peroxidase), selenium can have antioxidant effects by reducing the formation of free radicals and reactive oxygen species1, 41. Most importantly for this study, selenium is considered to have cancer treatment properties. Previous studies have shown that the cytotoxicity and anti-cancer activity of selenium are mainly attributed to its prooxidant action, including the oxidation of protein thiols and ROS generation1,7. The generation of ROS by selenium has been well-documented by in vitro studies, but the ultimate source of ROS generation and the mechanism responsible for exerting selenium's proapoptotic effects are uncertain1. Notably, previous studies have tested cancer cells cultured in a normoxic environment in vitro, which provides sufficient O2 levels. However, the solid tumor microenvironment is known to be hypoxic (low levels of O2)42,43. Thus, whether selenium promotes cell death by producing ROS in a hypoxic environment is unclear.
In this study, we investigated the anti-tumor mechanism of selenite from the perspective of selenium metabolism under simulated tumor microenvironment (hypoxia) conditions. The results showed that treating liver cancer cells with pharmacological concentrations of Na2SeO3 did not cause an increase in ROS and did not induce changes in the activity of antioxidant enzymes in vitro and in vivo, indicating that Na2SeO3 does not induce oxidative stress under hypoxic conditions. However, under the normoxic conditions, Na2SeO3 resulted in oxidative stress, which is consistent with another report that showed Na2SeO3 leads to cell apoptosis due to increases in ROS5. More importantly, under hypoxic conditions, pharmacological concentrations of Na2SeO3 induced the accumulation of H2Se in vitro and in vivo. H2Se is a common intermediate metabolite of dietary selenium compounds and has high reducibility. In the metabolism of selenite, selenite is first reduced by GSH to produce selenodiglutathione (GSSeSG), which can then be reduced by NADPH and glutathione reductase (GR) to glutathione selenenylsulfide (GSSeH). Finally, H2Se (hydrogen selenide ion (HSe-) at a physiological pH) is produced in a system containing GSH, NADPH and GR44-46. Previously, the role of H2Se in the anti-tumor effect of selenium has not been elucidated due to the lack of detection methods and its high volatility and reactivity. Here, we explored the important role of H2Se with the help of a fluorescent probe4 which can image H2Se specifically.
Reductive stress is the opposite of oxidative stress, and is defined as an excessive amount of reducing equivalents in the forms of NAD(P)H and/or GSH23,24. Similar to oxidative stress, reductive stress also represents a disturbance in the redox state that is harmful to biological systems. An excess of reducing equivalents can prevent growth factor-mediated signalling, promote mitochondrial dysfunction, decrease cell survival, and increase apoptosis47. Our experiments showed that a large amount of H2Se was produced before cell death after Na2SeO3 treatment of liver cancer cells under hypoxic conditions; simultaneously, there was also an increase in the NADPH and GSH levels. According to these results, we propose that pharmacological concentrations of selenite metabolism can cause reductive stress in cancer cells, which may be mainly due to the production and accumulation of H2Se.
The above results suggest that H2Se may play a key role in the cell death caused by Na2SeO3. Because H2Se is a highly reactive and reducible molecule, we first considered its effect on HMGB1 protein because HMGB1 is a redox-sensitive protein and its activity strongly depends on its redox state. HMGB1 contains three cysteines encoded at positions 23, 45, and 106. The two Cys23-Cys45 residues of HMGB1can rapidly form an intramolecular disulfide bond at the standard redox potential (237 mV)36. The cellular glutathione redox system is not enough to keep HMGB1 completely reduced in cells. Thioredoxin can reduce oxidized HMGB1, but the reaction is much slower than other reducing reactions mediated by thioredoxin. The low efficiency of HMGB1 with the thioredoxin reaction along with the protein stabilization by the Cys23-Cys45 disulfide bond may cause the HMGB1 protein to exist in an oxidized form in cells under physiological conditions and oxidative stress26,48. (Fig. S4). Our above results show that the accumulation of H2Se produced by Na2SeO3 metabolism under hypoxic conditions provides a strong reducing environment that may lead to the reduction of HMGB1 in cells. The in vitro simulation system experiment indicated that H2Se could interrupt the disulfide bond contained in HMGB1 and transform the disulfide bond to mercapto groups.
HMGB1 protein is both a nuclear factor and a secreted protein. When HMGB1 is released outside the cell, it can function as an extracellular signaling molecule in cell survival/death pathways32. Our results showed that the intracellular HMGB1 is released outside the cell when HepG2 cells were treated with Na2SeO3 under hypoxic conditions. Comparatively, under normoxic conditions, HMGB1 was also released outside the cell; however, the released amount was lower than that under hypoxic conditions. Interestingly, the activity of secreted HMGB1 strongly depends on its redox state. Reduced exogenous HMGB1 can increase autophagy and oxidized HMGB1can increase apoptosis33. Our present data showed that the Na2SeO3 treatment induced cell autophagy under hypoxic conditions, but mainly activated cell apoptosis under normoxic conditions, suggesting that the released HMGB1 may exist in its reduced form under hypoxic conditions, while under normoxic conditions, it should exist in the oxidized form. Under hypoxic conditions, the reduction of HMGB1 should be attributed to the accumulation of H2Se, while the oxidized form of HMGB1 under normoxic conditions should be caused by oxidative stress.
The relationship between H2Se and autophagy was detected using oxidized DTT, which can clear H2Se. The results showed that autophagy was obviously blocked after H2Se was removed, indicating that H2Se induces cell autophagy. Combined with the above experimental results, we speculate that H2Se induces autophagy by reducing the HMGB1 protein and causing its release from the intracellular to the extracellular space. Subsequently, the autophagy signaling pathway detection results displayed that Na2SeO3 could suppress the phosphorylation of Akt and mTOR, indicating that H2Se activates autophagy by inhibiting the Akt/mTOR pathway in HepG2 cells.
In tumor therapy, autophagy and apoptosis are intricately related. In general, autophagy may impair stress-induced apoptosis, and apoptotic caspases breakdown autophagy-related proteins to suppress autophagy49,50. However, sometimes, autophagy-associated proteins may help apoptosis, and excessive autophagy degrades essential cellular components to induce autophagic cell death (ACD)51. In our study, we found that H2Se induced both autophagy and apoptosis in HepG2 cells treated with pharmacological concentrations of Na2SeO3 under hypoxic conditions. However, the degree of apoptosis under the hypoxic conditions was significantly lower than that under the normoxic conditions. Interestingly, two phenomena appeared after autophagy was inhibited by CQ under the hypoxic conditions. If the inhibition occured during the early stages of autophagy, cell apoptosis was accelerated. However, if autophagy was advanced, the inhibition of autophagy delayed the rate of cell death. These results indicate that mild autophagy plays a cytoprotective role and inhibits cell apoptosis, but excessive autophagy leads to autophagy- associated cell death under hypoxic conditions.
In addition, the cytotoxicity test results showed that Na2SeO3 had different cytotoxicity under hypoxic conditions and normoxic conditions. The IC50 concentration of Na2SeO3 under the hypoxic conditions was twice that the under normoxic conditions. This difference may be attributed to the fact that selenite induces apoptosis by oxidative stress under normoxic conditions; whereas, under hypoxic conditions, H2Se activates autophagy, which initially delays cell death but ultimately leads to autophagy-associated cell death. This autophagy-associated cell death is a slow cell death process compared to apoptosis.
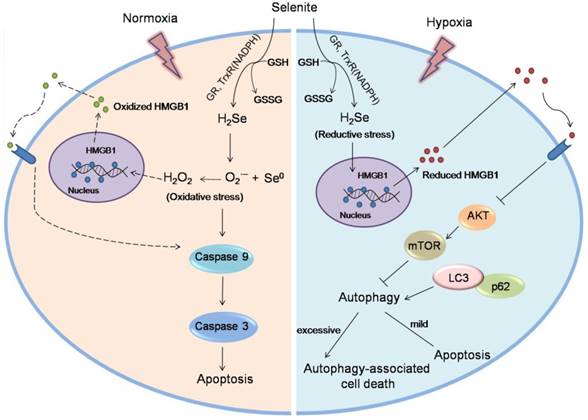
Taken together, this work reveals a new anti-tumor mechanism for selenium. Under hypoxic conditions, H2Se produced by selenite metabolism gradually accumulates and induces reductive stress in cancer cells. Furthermore, H2Se causes the reduction of the HMGB1 protein, which is secreted and promotes cell autophagy. Here, autophagy plays a dual role, i.e., mild autophagy inhibits apoptosis, while excessive autophagy leads to autophagy-associated cell death. In contrast, in a normoxic environment, H2Se may rapidly react with O2 to form ROS, then leading to caspase-dependent apoptosis via oxidative stress 6,11(Fig. 7). Thus, we propose that in vitro studies investigating the treatment of hypoxic tumors with reducing drugs should be performed under the corresponding hypoxic conditions because different oxygen conditions may lead to widely different conclusions and mislead the clinical use of these drugs. Our findings also suggest that appropriate oxygen supplementation during selenite treatment for hypoxic tumors may improve the cancer therapeutic effect.
Schematic of the mechanism of selenite under hypoxic and normoxic conditions. In tumor cells, selenite metabolism produces H2Se in a system containing GSH, NADPH and glutathione reductase (GR). Under hypoxic conditions, the H2Se gradually accumulates and induces reductive stress. Furthermore, H2Se causes the reduction of the HMGB1 protein, which is secreted and promotes cell autophagy by inhibiting the Akt/mTOR axis. Here, autophagy plays a dual role, i.e., mild autophagy inhibits apoptosis, while excessive autophagy leads to autophagy-associated cell death. In contrast, under hypoxic conditions, H2Se may rapidly react with O2 to form ROS, then leading to caspase-dependent apoptosis via oxidative stress. The ROS may cause the oxidation of the HMGB1 protein, which is secreted and promotes cell apoptosis.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This work was supported by National Natural Science Foundation of China (21575081, 21535004, 21775091 and 91753111), the Shandong Provincial Natural Science Foundation of China (ZR2014JL055) and the Key Research and Development Program of Shandong Province (2018YFJH0502).
Author contributions
X.H.P. performed all the experiments with the support of C.W. and T.T.C. and wrote the manuscript. X.X.S. performed all analytical measurements, in vitro assays, and cell culture experiments. D.R.L. and F.P.K provided expertise and supervised animal experiments. Z.Z.C. provided expertise with LC-MS experiments. K.H.X. and B.T. designed experimental strategies.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Weekley CM, Harris HH. Which form is that? The importance of selenium speciation and metabolism in the prevention and treatment of disease. Chem Soc Rev. 2013;42:8870-8894
2. Brigelius-Flohé R. Selenium compounds and selenoproteins in cancer. Chem Biodivers. 2008;5:389-95
3. Hatfield DL, Tsuji PA, Carlson BA, Gladyshev VN. Selenium and selenocysteine: roles in cancer, health, and development. Trends Biochem Sci. 2014;39:112-20
4. Kong FP, Ge LH, Pan XH, Xu KH, Liu XJ, Tang B. A highly selective near-infrared fluorescent probe for imaging H2Se in living cells and in vivo. Chem Sci. 2016;7:1051-1056
5. Shen HM, Yang CF, Ong CN. Sodium selenite-induced oxidative stress and apoptosis in human hepatoma HepG2 cells. Int J Cancer. 1999;81:820-828
6. Shen HM, Yang CF, Ding WX, Liu J, Ong CN. Superoxide radical-initiated apoptotic signalling pathway in selenite-treated HepG(2) cells: mitochondria serve as the main target. Free Radic Biol Med. 2001;30:9-21
7. Luo H, Yang Y, Duan J, Wu P, Jiang Q, Xu C. PTEN-regulated AKT/FoxO3a/Bim signaling contributes to reactive oxygen species-mediated apoptosis in selenite-treated colorectal cancer cells. Cell Death Dis. 2013;4:e481
8. Chen P, Wang L, Li N, Liu Q, Ni J. Comparative proteomics analysis of sodium selenite-induced apoptosis in human prostate cancer cells. Metallomics. 2013;5:541-550
9. Spallholz JE. On the nature of selenium toxicity and carcinostatic activity. Free Radic Biol Med. 1994;17:45-64
10. Shen HM, Ding WX, Ong CN. Intracellular glutathione is a cofactor in methylseleninic acid-induced apoptotic cell death of human hepatoma HEPG(2) cells. Free Radic Biol Med. 2002;15:552-561
11. Xiang N, Zhao R, Zhong W. Sodium selenite induces apoptosis by generation of superoxide via the mitochondrial-dependent pathway in human prostate cancer cells. Cancer Chemother Pharmacol. 2009;63:351-362
12. Ma Q, Fang H, Shang W, Liu L, Xu Z, Ye T. et al. Superoxide flashes: early mitochondrial signals for oxidative stress-induced apoptosis. J Biol Chem. 2011;286:27573-81
13. Xiong XX, Qiu XY, Hu DX, Chen XQ. Advances in Hypoxia-Mediated Mechanisms in Hepatocellular Carcinoma. Mol Pharmacol. 2017;92:246-255
14. Tohme S, Yazdani HO, Liu Y, Loughran P, van der Windt DJ, Huang H, Simmons RL, Shiva S, Tai S, Tsung A. Hypoxia mediates mitochondrial biogenesis in hepatocellular carcinoma to promote tumor growth through HMGB1 and TLR9 interaction. Hepatology. 2017;66:182-197
15. Rankin EB, Giaccia AJ. Hypoxic control of metastasis. Science. 2016;352:175-80
16. Rey S, Schito L, Koritzinsky M, Wouters BG. Molecular targeting of hypoxia in radiotherapy. Adv Drug Deliv Rev. 2017;109:45-62
17. Liu XB, Cheng Q, Geng W, Ling CC, Liu Y, Ng KT. et al. Enhancement of cisplatin-based TACE by a hemoglobin-based oxygen carrier in an orthotopic rat HCC model. Artif Cells Nanomed Biotechnol. 2014;42:229-236
18. Henry WS, Laszewski T, Tsang T, Beca F, Beck AH, McAllister SS, Toker A. Aspirin Suppresses Growth in PI3K-Mutant Breast Cancer by Activating AMPK and Inhibiting mTORC1 Signaling. Cancer Res. 2017;77:790-801
19. Pan X, Liu D, Wang J, Zhang X, Yan M, Zhang D. et al. Peneciraistin C induces caspase-independent autophagic cell death through mitochondrial-derived reactive oxygen species production in lung cancer cells. Cancer Sci. 2013;104:1476-82
20. Karton-Lifshin N, Segal E, Omer L, Portnoy M, Satchi-Fainaro R, Shabat D. A unique paradigm for a Turn-ON near-infrared cyanine-based probe: noninvasive intravital optical imaging of hydrogen peroxide. J Am Chem Soc. 2011;133:10960-10965
21. Pan X, Wang X, Wang L, Xu K, Kong F, Tang B. Near-Infrared Fluorescence Probe for Monitoring the Metabolic Products of Vitamin C in HepG2 Cells under Normoxia and Hypoxia. Anal Chem. 2015;87:7092-7
22. Rajasekaran NS, Connell P, Christians ES, Yan LJ, Taylor RP, Orosz A. et al. Human alpha B-crystallin mutation causes oxido-reductive stress and protein aggregation cardiomyopathy in mice. Cell. 2007;130:427-439
23. Dimmeler S, Zeiher AM. A "reductionist" view of cardiomyopathy. Cell. 2007;130:401-402
24. Brewer AC, Mustafi SB, Murray TV, Rajasekaran NS, Benjamin IJ. Reductive stress linked to small HSPs, G6PD, and Nrf2 pathways in heart disease. Antioxid Redox Signal. 2013;18:1114-27
25. Zhang H, Limphong P, Pieper J, Liu Q, Rodesch CK, Christians E, Benjamin IJ. Glutathione-dependent reductive stress triggers mitochondrial oxidation and cytotoxicity. FASEB J. 2012;26:1442-51
26. Tang D, Kang R 3rd Zeh HJ, Lotze MT. High-mobility group box 1, oxidative stress, and disease. Antioxid Redox Signal. 2011;14:1315-1335
27. Janko C, Filipović M, Munoz LE, Schorn C, Schett G, Ivanović-Burmazović I, Herrmann M. Redox modulation of HMGB1-related signaling. Antioxid Redox Signal. 2014;20:1075-85
28. Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191-195
29. Bianchi ME, Beltrame M. Upwardly mobile proteins. Workshop: the role of HMG proteins in chromatin structure, gene expression and neoplasia. EMBO Rep. 2000;1:109-114
30. Thomas JO, Travers AA. HMG1 and 2, and related 'architectural' DNA-binding proteins. Trends Biochem Sci. 2001;26:167-174
31. Lotze MT, Tracey KJ. High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol. 2005;5:331-342
32. VanPatten S, Al-Abed Y. High Mobility Group Box-1 (HMGb1): Current Wisdom and Advancement as a Potential Drug Target. J Med Chem. 2018;61:5093-5107
33. Tang D, Kang R, Cheh CW, Livesey KM, Liang X, Schapiro NE. et al. HMGB1 release and redox regulates autophagy and apoptosis in cancer cells. Oncogene. 2010;29:5299-5310
34. Tang D, Loze MT, Zeh HJ, Kang R. The redox protein HMGB1 regulates cell death and survival in cancer treatment. Autophagy. 2010;6:1181-1183
35. Tang D, Kang R, Livesey KM, Cheh CW, Farkas A, Loughran P. et al. Endogenous HMGB1 regulates autophagy. J Cell Biol. 2010;190:881-92
36. Kienast A, Preuss M, Winkler M, Dick TP. Redox regulation of peptide receptivity of major histocompatibility complex class I molecules by ERp57 and tapasin. Nat Immunol. 2007;8:864-72
37. He H, Li X, Kong X, Zhang C, Hua Y, Chen Y. Effects of Disulfide Bond Reduction on the Conformation and Trypsin/Chymotrypsin Inhibitor Activity of Soybean Bowman-Birk Inhibitor. J Agric Food Chem. 2017;65:2461-2467
38. Tiwari A, Liba A, Sohn SH, Seetharaman SV, Bilsel O, Matthews CR. et al. Metal deficiency increases aberrant hydrophobicity of mutant superoxide dismutases that cause amyotrophic lateral sclerosis. J Biol Chem. 2009;284:27746-58
39. Dou Q, Chen HN, Wang K, Yuan K, Lei Y, Li K. et al. Ivermectin Induces Cytostatic Autophagy by Blocking the PAK1/Akt Axis in Breast Cancer. Cancer Res. 2016;76:4457-69
40. Allen NE, Travis RC, Appleby PN, Albanes D, Barnett MJ, Black A. et al. Selenium and Prostate Cancer: Analysis of Individual Participant Data From Fifteen Prospective Studies. J Natl Cancer Inst. 2016:108 (11)
41. Hauksdóttir HL, Webster TJ. Selenium and Iron Oxide Nanocomposites for Magnetically-Targeted Anti-Cancer Applications. J Biomed Nanotechnol. 2018;14:510-525
42. Denko NC. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat Rev Cancer. 2008;8:705-713
43. Harris AL. Hypoxia-a key regulatory factor in tumour growth. Nat Rev Cancer. 2002;2:38-47
44. Ganther HE. Selenotrisulfides. Formation by the reaction of thiols with selenious acid. Biochemistry. 1968;7:2898-905
45. Ganther HE. Reduction of the selenotrisulfide derivative of glutathione to a persulfide analog by glutathione reductase. Biochemistry. 1971;10:4089-98
46. Hsieh HS, Ganther HE. Acid-volatile selenium formation catalyzed by glutathione reductase. Biochemistry. 1975;14:1632-6
47. Handy DE, Loscalzo J. Responses to reductive stress in the cardiovascular system. Free Radic Biol Med. 2017;109:114-124
48. Sahu D, Debnath P, Takayama Y, Iwahara J. Redox properties of the A-domain of the HMGB1 protein. FEBS Lett. 2008;582:3973-3978
49. Liu PF, Tsai KL, Hsu CJ, Tsai WL, Cheng JS, Chang HW. et al. Drug repurposing screening identifies tioconazole as an ATG4 inhibitor that suppresses autophagy and sensitizes cancer cells to chemotherapy. Theranostics. 2018;8:830-845
50. Tang JY, Farooqi AA, Ou-Yang F, Hou MF, Huang HW, Wang HR. et al. Oxidative stress-modulating drugs have preferential anticancer effects - involving the regulation of apoptosis, DNA damage, endoplasmic reticulum stress, autophagy, metabolism, and migration. Semin Cancer Biol. 2018 [Epub ahead of print]
51. Ha JH, Noh HS, Shin IW, Hahm JR, Kim DR. Mitigation of H2O2-induced autophagic cell death by propofol in H9c2 cardiomyocytes. Cell Biol Toxicol. 2012;28:19-29
Author contact
![]() Corresponding authors: Bo Tang (tangbedu.cn) and Kehua Xu (xukehuaedu.cn.)
Corresponding authors: Bo Tang (tangbedu.cn) and Kehua Xu (xukehuaedu.cn.)