Impact Factor
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Experimental Methods
Results and Discussion
Conclusions
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(6):1752-1763. doi:10.7150/thno.30977 This issue Cite
Research Paper
Convection-Enhanced Delivery of a Virus-Like Nanotherapeutic Agent with Dual-Modal Imaging for Besiegement and Eradication of Brain Tumors
Hao-Han Pang1,2*, Pin-Yuan Chen3,4,5*, Kuo-Chen Wei5,6, Chiun-Wei Huang7, Yow-Ling Shiue2, Chiung-Yin Huang5,6 ![]() , Hung-Wei Yang1
, Hung-Wei Yang1 ![]()
1. Institute of Medical Science and Technology, National Sun Yat-sen University, 70 Lienhai Rd., Kaohsiung 80424, Taiwan
2. Institute of Biomedical Sciences, National Sun Yat-sen University, 70 Lienhai Rd., Kaohsiung 80424, Taiwan
3. Department of Neurosurgery, Chang Gung Memorial Hospital, Keelung, 222 Maijin Rd., Keelung 20401, Taiwan
4. Community Medicine Research Center and Laboratory Animal Center, Chang Gung Memorial Hospital, Keelung, 222 Maijin Rd., Keelung 20401, Taiwan
5. School of Medicine, Chang Gung University, 259 Wenhua 1st Rd., Guishan Dist., Taoyuan 33302, Taiwan
6. Department of Neurosurgery, Chang Gung Memorial Hospital, Linkou, 5 Fuxing St., Guishan Dist., Taoyuan 33305, Taiwan
7. Center for Advanced Molecular Imaging and Translation, Chang Gung Memorial Hospital, Linkou, 5 Fuxing St., Guishan Dist., Taoyuan 33305, Taiwan
*H. H. Pang and P. Y. Chen contributed equally to this work.
Received 2018-10-26; Accepted 2019-1-6; Published 2019-2-28
Abstract

Convection-enhanced delivery (CED) is a promising technique for infusing a therapeutic agent directly into the brain, bypassing the blood-brain barrier (BBB) with a pressure gradient to increase drug concentration specifically around the brain tumor, thereby enhancing tumor inhibition and limiting the systemic toxicity of chemotherapeutic agents. Herein, we developed a dual-imaging monitored virus-like nanotherapeutic agent as an ideal CED infusate, which can be delivered to specifically besiege and eradicate brain tumors.
Methods: We report one-pot fabrication of green-fluorescence virus-like particles (gVLPs) in Escherichia coli (E. coli) for epirubicin (EPI) loading, cell-penetrating peptide (CPP) modification, and 68Ga-DOTA labeling to form a positron emission tomography (PET)-fluorescence dual-imaging monitored virus-like nanotherapeutic agent (68Ga-DOTA labeled EPI@CPP-gVLPs) combined with CED for brain tumor therapy and image tracking. The drug delivery, cytotoxicity, cell uptake, biodistribution, PET-fluorescence imaging and anti-tumor efficacy of the 68Ga-DOTA labeled EPI@CPP-gVLPs were investigated in vitro and in vivo by using U87-MG glioma cell line and U87-MG tumor model.
Results: The 68Ga-DOTA-labeled EPI@CPP-gVLPs showed excellent serum stability as an ideal CED infusate (30-40 nm in size), and can be disassembled through proteolytic degradation of the coat protein shell to enable drug release and clearance to minimize long-term accumulation. The present results indicated that 68Ga-DOTA-labeled EPI@CPP-gVLPs can provide a sufficiently high drug payload (39.2 wt% for EPI) and excellent detectability through fluorescence and PET imaging to accurately represent drug distribution during CED infusion. In vivo delivery of the 68Ga-DOTA-labeled EPI@CPP-gVLPs through CED demonstrated that the median survival was prolonged to over 50 days when the mice received two administrations (once per week) compared with the control group (median survival: 26 days).
Conclusion: The results clearly indicated that a combination of 68Ga-DOTA-labeled EPI@CPP-gVLPs and CED can serve as a flexible and powerful synergistic treatment in brain tumors without evidence of systemic toxicity.
Keywords: virus-like particles (VLPs), nanomedicine, dual-modal imaging, convection-enhanced delivery (CED), brain tumor
Introduction
Among all astroglial tumors, glioblastoma multiforme (GBM) is known to be the most lethal and aggressive primary malignant tumor [1-3]. The median survival of GBM patients is less than 15 months, and less than 5% of patients with GBM survive longer than 3 years. At present, chemotherapy and radiotherapy are the most common adjuvant therapy methods for GBM after surgery. To date, only two standard drugs are used in clinical chemotherapy, temozolomide (TMZ) and bis-chloroethyl nitrosourea (BCNU) [4-6]. The failure of chemotherapy is partly attributed to the functional blood-brain barrier (BBB), which prevents effective delivery of sufficient quantities of drugs to the brain [7-9]. Thus, GBM tumors present unique challenges in the local treatment of brain tumors; it is crucial to devise strategies to increase drug availability in tumor tissues as well as to obtain the desired therapeutic responses in manners that do not rely on the circulatory system.
Convection-enhanced delivery (CED) uses a direct infusion technique that relies on pressure-driven bulk flow within central nervous system (CNS); it is one of the most promising delivery systems for enhancing therapeutic agents and reducing systemic toxicity [10]. The bulk flow is created by a small pressure gradient from a pump that pushes solute through a catheter targeted within the CNS that provides much greater volumes of drug distribution than are achievable through diffusion [11]. Compared with traditional convection delivery, CED produces higher concentrations of therapeutic agents, longer infusion times, and larger distribution volumes at the infusion site, resulting in minimal systemic toxicity [12]. Relevant studies have shown that drug delivery through direct CED injection provides intraparenchymal concentrations that are 100-fold greater than those through intranasal delivery, and 1000- to 10,000-fold greater than those through intravenous delivery [13, 14]. Furthermore, CED has been clinically tested as a safe delivery method for brain tumor therapy agents as well as for the treatment of brain tumors and neurodegenerative disease [15, 16].
Epirubicin (EPI) is an anthracycline that has been considered one of the most potent antiglioblastoma drugs approved by the US Food and Drug Administration (US FDA) from the NHI Clinical Collection library [17]. However, it results in harsh side‐effects and cardiotoxicity that drastically reduce quality of life. The severity of EPI's toxicity can be significantly reduced by encapsulating it in nanoparticles (NPs); however, only a few liposomal and protein‐based NPs have been approved for cancer drug delivery despite numerous attempts [18]. Liposomes have been used to encapsulate a multitude of therapeutics and prolong their half-life systemically, and can potentially reduce unwanted early drug-tissue interaction, allowing for greater volumes of distribution and reduced tissue clearance rates [19]. However, they are limited by high production cost, complicated preparation procedure, particle instability, and spontaneous membrane fusion with off‐target cells [20]. Polymer‐based NPs offer similar advantages in that they can be conjugated to numerous chemotherapies, but they suffer from structural heterogeneity, particle instability, slow and nonuniform drug release, and potential immunogenicity [21]. Magnetic nanoparticles, such as iron oxide (15-80 nm), can also be delivered via CED, loaded with bioactive molecules, and utilized as magnetic resonance (MR) imaging contrast agents; however, they are limited by difficult degradation and clearance in the brain [22]. While many permutations are being investigated in animal models, no particular vehicle has been proven to be reliably superior, and few have been tested in clinical trials. An alternate method of drug delivery to NPs are virus-like particles (VLPs), which are extremely promising vaccine candidates because of their native-like and noninfective properties [23, 24]. VLPs originate from several types of virus but only the capsid proteins (or coat proteins) are expressed and form noninfectious empty viral particles via self-assembly [25]. To date, the most commonly used method is in vitro assembly of VLPs from isolated subunits to encapsulate small molecules, chemotherapeutic agents, and RNAs with therapeutic potential in the presence of a reducing agent; however, the procedure is too complicated, time-consuming, and easily contaminated [26, 27].
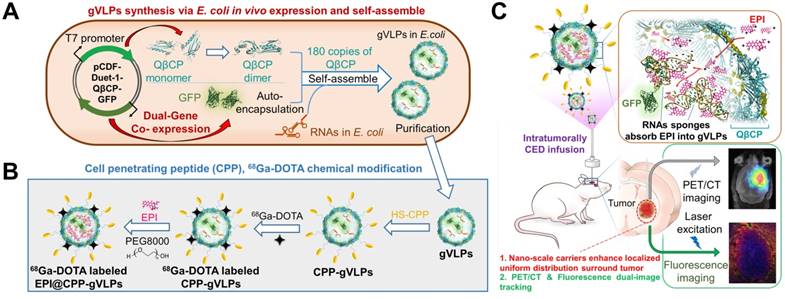
In light of this, we report in vivo one-pot fabrication of green-fluorescence VLPs (gVLPs) with EPI loading, cell-penetrating peptide (CPP) modification, and 68Ga-DOTA labeling, to form a dual-imaging monitored virus-like nanotherapeutic agent (EPI@CPP-gVLPs) as an ideal CED infusate, which can be delivered to specifically besiege and eradicate brain tumors through CED, reducing EPI systemic toxicity (Figure 1); thus, no purchasing of an expensive tracing agent is necessary, and the stability is high in terms of size in concentrated protein environments when compared with artificial drug delivery vectors.
(A) Bacteriophage QβCP, GFP coexpression system, and gVLPs complex self-assembly process in vivo. (B) Modification of the CPPs on gVLPs (CPP-gVLPs), 68Ga-DOTA labelling (68Ga-DOTA labelled CPP-gVLPs), and EPI loading in 68Ga-DOTA labelled CPP-gVLPs (68Ga-DOTA labelled EPI@CPP-gVLPs). (C) Schematic of 68Ga-DOTA labelled EPI@CPP-gVLPs infusion in a brain tumor by CED.
Experimental Methods
Vectors for Qβ-coat protein (QβCP) and green fluorescent protein (GFP) coexpression
DNA sequences of QβCP and GFP were cloned into pCDF-Duet-1 vector (Table S1) to form protein expression pCDF-Duet-GFP-QβCP for the coexpression of QβCP and GFP in E. coli. Vectors were transformed to E. coli cell line BL21 competent cells for protein coexpression to generate GFP-encapsulated virus-like particles (gVLPs). For gVLP production and purification, E. coli BL21 cells harboring the appropriate plasmids were grown in either LB broth or NZY solution supplemented with antibiotic (kanamycin or streptomycin) at 50 μg/mL, respectively. Starter culture was grown for 18 h at 37°C and used to inoculate 1 L of expression culture. One minimolar IPTG was performed as a protein expression reagent at an OD600 of 0.8-1.0 in culture solution (LB broth, BD, LOT: 244620, France) overnight at 37°C. The overnight culture was harvested by centrifugation at 6,500 g, resuspended in 20 mL of PBS buffer (pH = 7.4), and then lysed by sonication. The lysate was centrifuged for 30 min at 23,000 g, followed by precipitation with ammonium sulfate to obtain crude VLP-based samples (gVLPs). The crude VLP-based samples were resuspended in PBS buffer followed by 20% w:v PEG8000-NaCl precipitation to obtain VLPs. These VLPs were resuspended in 1 mL of PBS buffer and extracted with 1:1 n-butanol : chloroform. The VLP-based samples, from the aqueous layer, were purified by step sucrose gradient ultracentrifugation and then precipitated with 20% w:v PEG8000-NaCl solution and resuspended in 25 mL of PBS buffer, followed by exhaustive dialysis (SnakeSkin® Dialysis Tubing, 10,000 MWCO. Thermo, LOT: QD213952, USA) against PBS buffer (pH = 7.4) for 48 h. The obtained pure VLP-based samples were concentrated by protein concentrate filter tubes (Amicon Ultra-15 Centrifugal Filter Units; 100,000 MWCO; Merck Millipore, LOT: R6EA45140, Ireland). The final concentration of VLPs was assessed using a Pierce BCA Protein Assay kit (Thermo, LOT: PD202250, USA).
Cell-penetrating peptides (CPPs) modification on gVLPs
The gVLPs were modified with Cys-CPP to enhance cell uptake. The Cys-CPP (KYGRRRQRRKKRG-cys-SH) was conjugated on the surface of the gVLPs by sulfosuccinimidyl 4-(N-maleimidomethyl)cyclohexane-1carboxylate (sulfo-SMCC; Sigma-Aldrich, St. Louis, MO, USA) as a crosslinker. Briefly, 5 μl of sulfo-SMCC solution (10 mg/mL in DI-H2O) was mixed with 2 μM of gVLPs in 600 μL of PBS (pH = 7.4) for 30 min at 25°C in the dark, and then purified by filter column (Amicon Ultra-15 Centrifugal Filter Units; 100,000 MWCO; Merck Millipore, LOT: R6EA45140, Ireland) with PBS buffer. Subsequently, the maleimide-terminated gVLPs were reacted with 30 μL of Cys-CPP solution (0.3 mg/mL) at 25°C for 2 h in the dark, and then purified again using the same procedure as above to obtain CPP-gVLPs. To confirm the successful modification of CPP on gVLPs, the CPP-gVLPs were mixed with 2-mercaptoethanol (2-BME; Sigma-Aldrich, St. Louis, MO, USA) and incubated at 95°C for disulfide-bond breaking and protein denaturing. The denatured samples were analyzed by SDS-PAGE (15%) electrophoresis and then stained by Coomassie Brilliant Blue R-250 Dye (Sigma-Aldrich, St. Louis, MO, USA).
Preparation of epirubicin (EPI)-loaded gVLPs
The CPP-gVLPs with 2 µM were mixed with various concentrations of EPI (0.2-10 µg/µL) in PBS buffer solution (pH = 7.4) for 1 h to obtain EPI@CPP-gVLPs. To cover the EPI@CPP-gVLPs surface with PEG8000, the EPI@CPP-gVLPs were precipitated by addition of 20% PEG8000 solution and then resuspended in PBS. The EPI@CPP-gVLPs were concentrated using a 100,000 MWCO filter column (Amicon Ultra-15 Centrifugal Filter Units, Merck Millipore, Ireland). Then we measured the amount of unentrapped EPI in supernatant to determine the EPI loading efficiency by absorbance of EPI at 480 nm using NanoDrop (ThermoFisher Scientific, USA) with the EPI standard curve.
Transmission electron microscopy (TEM)
The diameter and morphology were analyzed by TEM (CM-200 TWIN instrument, Philips Co., Netherlands) with 75 keV accelerating voltage. VLP-based samples were prepared by pipetting 5 µL onto Formvar-coated copper mesh grids (200 mesh, Ted Pella, Redding, CA, USA) for 5 min, followed by exposure to 8 µL of a solution of uranyl acetate (15 mg/mL in DI-H2O) for 2 min as a negative stain. Excess stain was then removed, and the grids were left to dry in air overnight.
EPI release study
For the EPI release study, 2.4 mL of EPI@CPP-gVLPs (containing 2.4 mg of EPI and 6.2 mg of CPP-gVLPs) were dialyzed with a 10,000 MWCO dialyzing bag (SnakeSkin® ThermoFisher Scientific) in 8 mL of PBS buffer (pH = 6.0 or 7.4) or Fetal bovine serum (FBS) for a period of incubation at 37℃ (0.5, 1, 1.5, 2, 3, 4, 5, 12, 24, 48, 72, 96, 120, 144, and 168 h). The released EPI in dialysate was determined by fluorescence signal exhibited at 592 nm under excitation at 480 nm using a SpectraMax M2 microtiter plate reader (Molecular Device, USA).
In vitro studies
GBM U87-MG cells were seeded into a 12-well plate (approximately 106 cells/well) and incubated for 24 h in Dulbecco's Modified Eagle's Medium (DMEM) with 2.2 mg/mL of sodium carbonate, 10% FBS, 50 µg/mL of penicillin, and 50 µg/mL of streptomycin at 37°C for cellular uptake efficiency analysis. All cells were grown and maintained at 37°C in 5% CO2. The gVLPs or CPP-gVLPs were added to the U87-MG cells to a final concentration of 500 nM (1.3 mg/mL). Treated cells were incubated for 0, 1, 3, or 5 h; cells were detached, pelleted, and resuspended in PBS after being washed three times with PBS. The flow cytometry data calculated the number and intensity of cells with GFP signals. There were 10,000 cells collected in each dataset. The experiments were performed using NovocyteTM flow cytometry (ACEA, USA) and the data were analyzed using the NovoExpress® software package. Furthermore, we investigated the cellular EPI delivery efficiency by gVLPs or CPP-gVLPs; the U87-MG cells were incubated with EPI@gVLPs or EPI@CPP-gVLPs to a final concentration of 1.8 mg/mL containing 0.5 mg/mL of EPI for 24 h and then stained with Hoechst 33342 (Sigma-Aldrich, St. Louis, MO, USA) for 10 min after washing with PBS. Images were acquired on a Nikon® fluorescence microscope (Eclipse Ti-S Inverted Microscope System, Nikon®, Japan).
The cultured U87-MG cells (5,000 cells/well) were treated with EPI@gVLPs or EPI@CPP-gVLPs with final concentrations of 0.05, 0.075, 0.1, 0.2, 0.4, 0.6, 0.8, 1, and 1.5 µg/mL EPI followed by incubation for 24 h. The culture medium was removed, and the cells were incubated in 120 µL of XTT solution for 2 h. After that, 100 µL of XTT solution from each well was transferred to another 96-well counting plate. The survival of U87-MG cells at different time points was evaluated by OD at 490 nm using a SpectraMax M2 microtiter plate reader.
Biodistribution of CPP-gVLPs through tail-vein injection and CED
All commercially available reagents were used without further purification. The 1,4,7,10-Tetraazacyclododecane-1,4,7,10-tetraacetic acid mono-N-hydroxysuccinimide ester (DOTA-NHS-ester) was purchased from Macrocyclics, Inc. (Plano, TX). The 68GaCl3 solution was produced by eluting 0.05 N HCl through a 68Ge/68Ga generator (itG, Germany). Water and all buffers were passed through a Chelex 100 resin (50-100mesh) column (1 × 15 cm) before radiolabeling to minimize the potential metal contamination. Purification of the crude product was performed with a MiniTrap G-25 column purchased from GE Healthcare.
The eluted 68GaCl3 (166-185 MBq in 0.05 N HCl) was added to 0.1 N sodium acetate (pH = 5.5) buffer, in which the purified DOTA-conjugated CPP-gVLPs (20-30 µL/14 µM) was added and the reaction mixture was maintained at 95℃ for 5 min. The 68Ga-DOTA-labeled CPP-gVLPs were subsequently purified by gel filtration column (MiniTrap G-25), and the fraction containing the radioactive peak was collected and passed through a 0.22-µm syringe filter for in vivo imaging and biodistribution studies. The radiochemical purity of the final products was confirmed by radio-TLC. Due to the high labeling yield (>95%), the 68Ga-DOTA-labeled CPP-gVLPs could be used directly without further purification.
In vivo positron emission tomography/ computed tomography (PET/CT) imaging experiments were conducted to monitor the biodistribution of prepared 68Ga-DOTA-labeled CPP-gVLPs across several time points, and athymic male nude mice bearing orthotopic U87-MGLu tumor xenografts were randomly divided into intratumoral injection (n = 5) and tail-vein injection (n=6) groups. In the intratumoral injection group (using the ECD delivery strategy), approximately 0.74-1.29 MBq of 68Ga-DOTA-labeled CPP-gVLPs were administered, and sequential static PET/CT scans were acquired at 60 min and 120 min post-injection. These time points were selected because of the limited short physical half-life of 68Ga (68 min). In addition, the intravenous injection of 68Ga-DOTA-labeled CPP-gVLPs (approximately 6.7-7.4 MBq; 60 min and 120 min postinjection) scans were conducted in tumor-bearing mice for comparison studies. PET/CT studies were performed on an Inveon micro-PET rodent model scanner (Siemens Medical Solutions USA, Inc.). Anesthesia was induced with 3.5% isoflurane and maintained at 2% in O2, and the mice were placed near the center of the field of view, where the highest resolution and sensitivity values were obtained. Micro-CT imaging was then performed for all animals for anatomic registration after the micro-PET imaging. The reconstructed image matrix size was 128 × 128 × 159 with 0.14 × 0.14 × 0.14 mm3 with the two-dimensional ordered-subset expectation maximum iterative method. All imaging was performed with scatter correction, random correction, and attenuation correction (AC). AC was performed by transmission scanning with a 68Ge/68Ga rotating line source. The regions of interest (ROIs) were determined according to 50% of the maximum minus minimum tumor activity in the coronal plane using image analysis software (PMOD, version 3.2; PMOD Technologies, Zurich, Switzerland). The average radioactivity concentration within the selected organs region was obtained from mean pixel values within the multiple ROI volume. Assuming a tissue density of 1 g/mL, the ROIs were converted to microcuries per gram and then divided by the total administered activity to obtain the imaging ROI-derived percentage administered activity per gram of tissue (% ID/g). To validate the complete distribution profile of the VLPs, U87-MGLu tumor-bearing mice were sacrificed immediately after the PET/CT image scans at 2 h postinjection. Selected major tissues and organs were excised and weighed, and the radioactivity was counted with an automatic γ-counter (Perkin-Elmer). The biodistribution data are presented as the percentage of the injected dose per gram (%ID/g; mean ± SD).
Animal procedures
For the animal experiments, luciferase expression plasmid transfected U87-MGLu cell-implanted pathogen-free male NU/NU mice (5-7 weeks old, 20-25 g, from BioLASCO, Taiwan) were employed in this study. U87-MGLu cells were cultured at 37°C with 5% CO2 in MEM with 10% fetal bovine serum and 1% penicillin/streptomycin (Invitrogen). Animals were anesthetized with 2% isoflurane gas and immobilized on a stereotactic frame to implant the U87-MGLu cells. A sagittal incision in the skin overlying the calvarium was created. The U87-MGLu cell implantation was performed by creating a hole in the exposed cranium 1.5 mm anterior and 2 mm lateral to the bregma using a 27G needle. A total volume of 5 µL of U87-MGLu cell suspension (1×105 cell/µL) was injected at a depth of 3 mm from the brain surface over a 5-min period. The needle was withdrawn over 2 min. MRI was performed to monitor the brain tumor growth for 10 days after tumor cells implantation.
CED procedure
The details of the CED procedure are as described in our previous study. Briefly, infusion cannulas were fabricated with silica tubing (Polymicro Technologies, Phoenix, AZ) fused to a 0.1 mL syringe (Plastic One, Roanoke, VA) with a 0.5 mm stepped-tip needle that protruded from the silica guide base. CPP-gVLPs or EPI@CPP-gVLPs were loaded into the syringes and attached to a microinfusion pump (Bioanalytical Systems, Lafayette, IN). The syringe with a silica cannula was mounted onto a stereotactic holder and then lowered through a puncture hole made in the skull to the implanted tumor. The sample solution was infused at a rate of 1 µL/min until a volume of 5 µL had been delivered, and the cannula was removed 2 min later.
Histopathological studies
Histopathological studies were performed on 10-µm sections of paraformaldehyde-fixed, paraffin-embedded mice brains. Slides were soaked in hydrochloric acid-potassium ferrocyanide solution for 30 min at room temperature. Brain tissue-damaging situations after different treatments (saline, free EPI, CPP-gVLPs, and EPI@CPP-gVLPs) were evaluated using hematoxylin and eosin staining, CD68 staining (macrophages), and ly6G staining (neutrophils). The distribution of EPI (red) and gVLPs (green) was evaluated through fluorescence microscopy imaging after staining nuclei with DAPI.
Antitumor efficiency of EPI@CPP-gVLPs infused by CED
To evaluate intracerebral tumor growth, we used an IVIS Spectrum system (XENOGEN IVIS 100) to monitor the tumor-bearing mice. Before imaging, D-luciferin (3 mg/mouse) was injected intraperitoneally; 8 min after injection, luminescent signals from the tumor were obtained by IVIS to determine the tumor progression. We conducted imaging twice a week to assess the efficacy and dose-response to CPP-gVLPs and EPI@CPP-gVLPs infused by CED in animals with brain tumor xenografts. We anaesthetized mice with vaporized isoflurane and divided them into five experimental groups (10 mice in each group). The mice in Group 1 received a CED infusion of saline after transplantation of U87-MGLu cells, and served as the control. The mice in Group 2 received a CED infusion of 3.6 mg/kg CPP-gVLPs on day 5 after tumor inoculation. The mice in Group 3 received two CED infusions of 3.6 mg/kg CPP-gVLPs on days 5 and 12 after tumor inoculation. The mice in Group 4 received CED infusion of 5.0 mg/kg EPI@CPP-gVLPs (containing 1.4 mg of EPI/kg) on day 5 after tumor inoculation. The mice in the final group received two CED infusions of 5.0 mg/kg of EPI@CPP-gVLPs (containing 1.4 mg of EPI/kg) on days 5 and 12 after tumor inoculation. The survival time was calculated from the day of U87-MGLu cell inoculation (0 day) to the day of death. Kaplan-Meier survival curves were plotted for each group. The body weights of mice were monitored at determined time intervals.
Blood biochemical analysis and immunoassay
One group of mice (n = 3) were injected with 5 µL of CPP-gVLPs (18.1 mg/mL) per mouse via CED; another group of uninjected mice served as the control; 1-mL blood samples were collected by retro-orbital venous plexus puncture at 0 min (before injection) and 7 days after injection. Clotted blood samples were centrifuged at 7,000 rpm for 20 min to obtain serum for blood-biochemistry analysis and immunoassay.
Statistical analysis
The data were expressed as mean ± SD on the basis of at least three independent experiments. Statistical analysis was performed using a Student's t-test. Differences were considered statistically significant if p < 0.05.
Results and Discussion
Producing and characterization of EPI@CPP-gVLPs
First, we characterized the physical properties of the EPI@CPP-gVLPs system complex and studied its cell uptake, physical release, and therapeutic efficacy in vitro. We used fluorescence microscopy and positron emission tomography/computed tomography (PET/CT) to examine the distribution of EPI@CPP-gVLPs in the brain and evaluate its in vivo biodistribution in tumor-bearing mice. Furthermore, we examined the biological toxicity and immunogenicity of EPI@CPP-gVLPs and its in vivo therapeutic response with CED using an in vivo imaging system (IVIS).
EPI is a highly potent anticancer drug but its use is severely limited by its poor aqueous solubility and stability as well as high cardiotoxicity [28]. Herein, we developed gVLP-based EPI formulations to overcome these limitations. The gVLPs were generated through transforming Escherichia coli (E. coli) with pCDFDuet-1-QβCP-GFP plasmid, which can coexpress GFP and Qβ coat protein (QβCP) and then self-assemble in E. coli after protein induction; the primers used for pCDFDuet-1-QβCP- GFP construction are shown in Table S1. The prepared gVLPs exhibited green color and green fluorescence (GFP packaging), which can be easily observed by naked eyes to extract the final products after ultracentrifuge sucrose gradient purification for preventing serious yield loss as well as imaging tracking in cells or tissues compared with synthetic drug vehicles; no extra purchasing of a tracing agent is required, nor is a labelling process (Figure 2A(a)). Subsequently, the purified gVLPs were modified with CPP on the surface to form CPP-gVLPs (Figure S1) to facilitate the transduction of CPP-gVLPs across the plasma membrane into the cytoplasm for the controlled release of EPI in cells. EPI is known to have a high affinity for binding to DNA and RNA, thus the obtained CPP-gVLPs contained nonfunctioning RNAs from E. coli, which are able to pull more EPI inside CPP-gVLPs through the pores of the QβCP shell by electrostatic attraction to form EPI@CPP-gVLPs. In addition, the entrapped EPI might also intercalate into double-stranded regions of RNAs inside CPP-gVLPs through noncovalent intercalation and form a physical complex to prevent serious leakage [29]. The amount of EPI loaded in CPP-gVLPs increased with increasing amounts of EPI, reaching a maximum concentration of 0.39 mg EPI/mg CPP-gVLPs (39 wt%) above 0.9 mg of added EPI (Figure S2), which is much higher than in a doxorubicin (DOX)-loaded micelles system (the highest DOX loading in micelles is ∼8 wt%) [30]. When EPI was loaded in the CPP-gVLPs, its fluorescence was quenched as a result of intercalations within the RNAs, but fluorescence recovery was observed when the CPP-gVLPs and RNAs were denatured using SDS and urea, respectively, indicating the successful intercalation of EPI within the RNAs inside CPP-gVLPs (Figure 2B and Figure S3). In this study, the prepared EPI@gVLPs were then purified by PEG8000 precipitation, no free EPI signal remained on the top of column after ultracentrifuge sucrose gradient purification (Figure 2A(b)) when the EPI@gVLPs' surface was covered with few PEG8000, indicating that PEG8000 can be gatekeeper to prevent serious EPI leakage compared with free EPI (Figure 2A(c)) and EPI@gVLPs without PEG8000 covering (Figure S4). Quantification of EPI release by dialysis confirmed a slow and extended release with approximately 57.3% and 50.2% of EPI released from the EPI@CPP-gVLPs after 132 h of incubation at pH 6.0 and in FBS respectively, which were both lower than that of incubation at pH 7.4 (approximately 75.2%). This is most likely because the folding structure of RNAs tends to be more stable in lower pH environments and is protected by CPP-gVLPs from cleavage by nuclease (Figure S5). This provides therapeutic benefits for CED infusion of EPI@CPP-gVLPs with slow release in an acidic tumor environment, which would not release an excess of EPI and damage normal brain tissues.
(A) White light photographs of ultracentrifuge tubes with sucrose density gradient ultracentrifugation to concentrate and partially purify (a) CPP-gVLPs (green color), (b) EPI@CPP-gVLPs (red color), and (c) free EPI (orange color). (B) Fluorescence intensities of EPI+SDS+Urea, EPI@gVLPs, and EPI@gVLPs+SDS+Urea under excitation at 480 nm; the SDS (10%) and Urea (8 M) were added for protein and RNAs were denatured to simulate EPI release from CPP-gVLPs in U87-MG cells (inset: fluorescence images for each sample). (C) Transmission electron microscope images of gVLPs (top) and EPI@CPP-gVLPs (bottom) (scale bar = 20 nm). (D) Serum stability of EPI@CPP-gVLPs after incubation in mouse blood serum for 3 days; values are expressed as means ± SD (n = 3). (E) Characterization of purified gVLPs (lane 1), CPP-gVLPs (lane 2), and EPI@CPP-gVLPs (lane 3) by 15% SDS-PAGE. The molecular weight of QβCP monomer is 14.4 kDa (blue arrow) and QβCP dimer is 28.8 kDa (green arrow). Red dots: QβCP conjugated with single CPP. Yellow dots: QβCP conjugated with two CPPs.
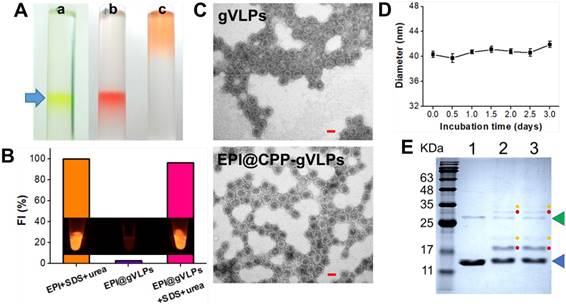
The packaging of EPI in the capsid and surface modification with CPP left the morphology of the VLPs unchanged when compared with the gVLPs analyzed by transmission electron microscopy (TEM) with a narrow diameter range (29.8 ± 1.6 nm for gVLPs and 36.9 ± 2.8 nm) (Figure 2C). Through dynamic light scattering (DLS), the diameter of EPI@gVLPs was slightly increased to 33.2 ± 0.2 nm from 32.1 ± 0.6 nm (gVLPs), and was further increased to 40.3 ± 0.5 nm after CPP conjugation on the surface (Table S2), which also exhibited excellent suspension stability in mouse blood serum without obvious aggregation observed across 3 days (Figure 2D). The diameter measured by DLS was slightly bigger than that measured by TEM, most likely because the TEM gives the diameter of nanoparticles in dried form while DLS tells the hydrodynamic diameter in the solvated state where there will be solvent molecules associated with the nanoparticles [31]. Furthermore, successful CPP conjugation was confirmed by SDS-PAGE; new bands appeared at 17.1 kDa (single CPP conjugated on one QβCP monomer) and 19.8 kDa (two CPPs conjugated on one QβCP monomer) (Figure 2E, lanes 2 and 3) after CPP modification compared with nonmodified gVLPs (Figure 2E, lane 1), thereby proving the successful immobilization of CPP on a QβCP monomer, reaching a maximum density of 140 ± 14 CPPs per gVLPs calculated by BCA Protein Assay kit. In addition, the results also indicated that the conjugated CPPs would not detach from the surface of gVLPs during the EPI encapsulation process (Figure 2E, lane 3).
In vitro cell uptake and cytotoxicity studies
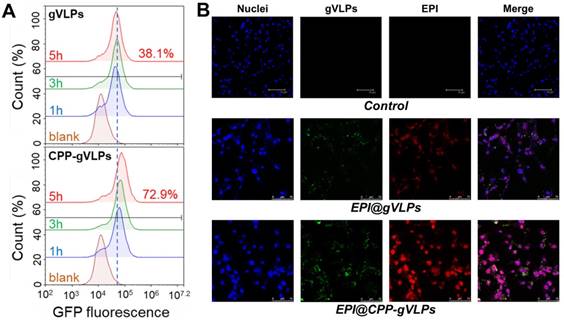
To demonstrate the cell-uptake effectiveness of this approach, U87-MG cells were exposed to gVLPs and CPP-gVLPs; the efficiency was close to 72.9% (as measured by counting GFP+ cells) for CPP-gVLPs, which was almost twice as high as that of gVLPs (38.1%) after 5 h of incubation with U87-MG cells (Figure 3A). Furthermore, as shown in Figure 3B, more EPI was delivered in U87-MG cells when the cells were exposed to EPI@CPP-gVLPs for 24 h. Conversely, only small amounts of GFP (green fluorescence) and EPI (red fluorescence) were detectable in the cells when they were exposed to EPI@gVLPs in the absence of CPP modification. These results demonstrated that more gVLPs can cross the plasma membrane into the cytoplasm after CPP modification by receptor-mediated endocytosis to deliver more EPI into U87-MG cells. Many groups have proposed that cell membrane heparan sulfate proteoglycans are negatively charged and present on the surface of many cell types, which act as receptors for extracellular CPP uptake [32]. Next, we investigated the cytotoxicity toward U87-MG cells induced by CPP-gVLPs, EPI@gVLPS, and EPI@CPP-gVLPs (Figure S6). No significant inhibition of cell proliferation was observed in the CPP-gVLPs-treated group for 24 h, indicating that CPP-gVLPs do not have an off-targeting effect to induce toxicity toward U87-MG cells. Conversely, treatment with EPI@gVLPs and EPI@CPP-gVLPs exhibited significant cell cytotoxicity toward U87-MG cells in a dose-dependent manner. A 50% concentration of inhibition of growth (IC50) for the EPI@CPP-gVLPs-treated group was only 0.07 µg/mL of EPI after 24 h of incubation, which was much lower than that when treated by EPI@gVLPs (IC50 = 1.5 µg/mL of EPI). Furthermore, this result confirmed that more EPI can be delivered into U87-MG cells by gVLPs in the presence of CPP modification for more efficient cancer cell inhibition.
(A) Flow cytometry analysis was performed to quantify the cellular uptake efficiency of gVLPs or CPP-gVLPs in U87-MG cells. The data are shown as histograms of the GFP-channel (auto fluorescence of the gVLPs) with the analysis of the Y mean fluorescence and the percentage of positive cells. (B) The cellular uptake of EPI@gVLPs (top) and EPI@CPP-gVLPs (bottom) in U87-MG cells using fluorescence microscopy analysis. U87-MG cells were incubated with 1.8 mg/mL EPI@gVLPs or EPI@CPP-gVLPs containing 0.5 mg/mL EPI for a period of 24 h in 0.2 mL of culture medium. Scale bar = 75 µm. Green color indicates gVLPs and red color indicates EPI; nuclei were stained blue with Hochest 33342.
In vivo bio-distribution analysis of CPP-gVLPs
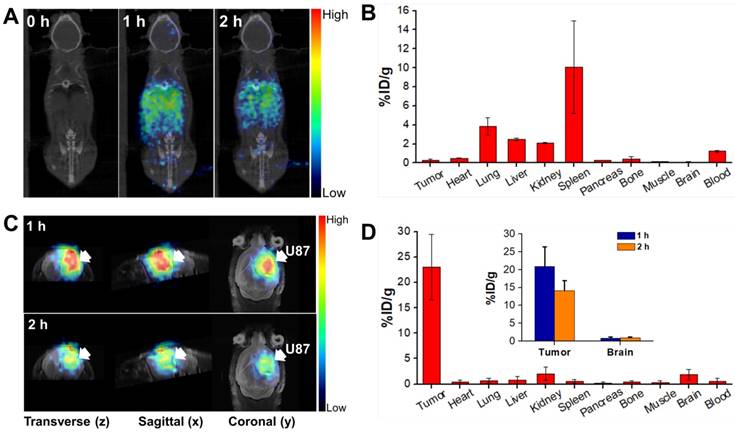
To understand the biodistribution pattern of CPP-gVLPs in mice, the CPP-gVLPs were labelled with 68Ga-DOTA, the results showed the successful labelling in sailing by radio-TLC analysis (Figure S7A). We then performed in vivo PET/CT imaging of 68Ga-DOTA-labeled CPP-gVLPs, either with intratumoral (CED) or tail-vein injection and compared the resulting images, and the sequential static PET/CT scans were acquired at 60 min and 120 min post-injection. As expected, a high level of radioactivity was quickly accumulated in the liver (1.12 ± 0.02% ID/g; 1.18 ± 0.01% ID/g), kidney (0.75 ± 0.06% ID/g; 0.69 ± 0.04% ID/g), and heart (0.64 ± 0.21% ID/g; 0.55 ± 0.25% ID/g) at 1 and 2 h after 68Ga-DOTA-labeled CPP-gVLPs administration through the tail vein (Figure 4A and B). However, because of the BBB, the radioactive uptakes in the brain and xenograft luciferase-stable U87-MG (U87-MGLu) brain tumors of the tail-vein injection group were difficult to ascertain (all were below 0.14% ID/g) based on the images. In the intratumoral injection group by CED, PET/CT imaging confirmed that once the labeled 68Ga-DOTA-labeled CPP-gVLPs were injected into the xenograft brain tumor, apparent radioactivity accumulation was observed within tumor lesions at both 1 h and 2 h after tracer injection, as indicated by arrows (Figure 4C). Quantitative analysis of PET images indicated a particle uptake of 20.89 ± 5.46% ID/g at 1 h and 14.10 ± 2.84% ID/g at 2 h, respectively. The majority of particles were retained inside the tumor and slowly diffused to surrounding normal brain tissue (0.79 ± 0.31% ID/g at 1 h and 0.96 ± 0.15% ID/g at 2 h) indicating that CED might be an effective therapeutic delivery strategy for brain tumor treatment (Figure 4D). In addition, the concentrations of 68Ga-DOTA-labeled CPP-gVLPs accumulation in other organs were all below 2% ID/g by CED, indicating that EPI@CPP-gVLPs administration by CED would not induce serious systemic toxicity compared with tail-vein injection. The organ-based biodistribution results from both groups were comparable with the PET/CT imaging analysis. The high agreement of quantitative parameters between the PET/CT imaging and biodistribution results from direct tissue sampling suggested that the %ID/g values derived from static PET/CT imaging can be effectively employed to monitor particle distribution profiling. Furthermore, the results indicated that the 68Ga-DOTA-labeled CPP-gVLPs can facilitate direct monitoring, not only of the drug distribution, but also of semiquantitative measurements of the drug in brain tumor and normal brain tissues. We cannot exclude the possibility of dissociation and biodegradation of the complex, especially in the late phase. Thus, the radio-TLC analysis of blood and urine samples found that the radioactivity mostly came from both intact 68Ga-DOTA- labeled CPP-gVLPs and its metabolites, about 5.45% of unknown metabolites was identified in the late phase radio-TLC profile. (Figure S7B). Further fragment identification studies may be required to identify and characterize these metabolites. In addition, this is the first study to determine the biodistribution of a VLPs-based nanotherapeutic agent through tail-vein and CED administration.
In vivo PET/CT imaging in the orthotopic brain tumor model after injection of 68Ga-DOTA-labeled CPP-gVLPs: (A) Decay-corrected whole-body planar coronal PET/CT images of the U87-MGLu tumor-bearing animal model at 1 h and 2 h postinjection of 6-8 MBq of 68Ga-DOTA-labeled CPP-gVLPs through tail veins. (B) The distribution of major organ uptake assessed directly from tissue sampling was expressed as a percentage of the injected dose/gram of tissue (% ID/g) through tail veins. (C) Representative micro-PET-MR images of U87-MGLu brain tumor mice intratumorally injected (by CED) with 68Ga-DOTA-labeled CPP-gVLPs at 1 h postinjection (top row) and 2 h postinjection (bottom row); the tumor is indicated by an arrow. (D) The distribution of major organ uptake assessed directly from tissue sampling was expressed as a percentage of the injected dose/gram of tissue (% ID/g) through CED (inset: the distribution of tumor and normal brain uptake at 1 and 2 h post-injection).
Toxicity and drug distribution in brain by CED
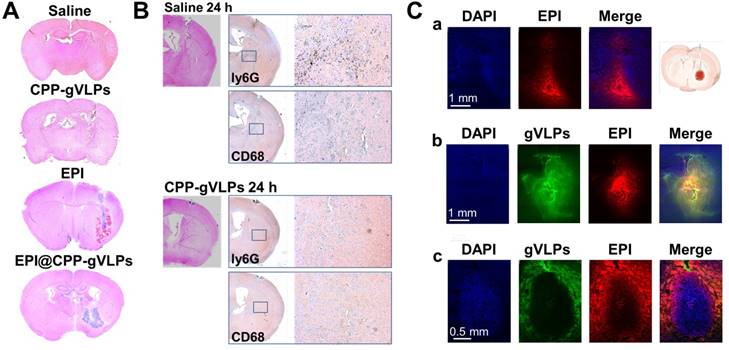
To evaluate the toxicity of EPI@CPP-gVLPs, mice received 5-µL CED infusions of saline, free EPI, CPP-gVLPs, or EPI@CPP-gVLPs (containing 90.7 µg of gVLPs and 35.6 µg of EPI) into their left hemispheres. Histopathologic examination revealed no obvious differences in the brain tissue sections of the saline- and CPP-gVLPs-treated groups of mice, the minor damage is induced by the mechanical puncture procedure for CED process, no specific character of CPP-gVLPs induced toxicity was observed compared with saline group. However, serious tissue damage and bleeding were observed in the CED infusion group with free EPI. Nevertheless, the damage was reduced significantly, and only slight irritation was observed in brain tissue when the mice received CED infusion of EPI@CPP-gVLPs, most likely because the slow release of loaded EPI from CPP-gVLPs prevented directly contact of concentrated EPI with tissues (Figure 5A). These results indicated an absence of neurological toxicity during CED infusion of EPI@CPP-gVLPs over a 3-day observation period. We further utilized the immunohistochemical staining to check the infiltrated macrophages (CD68) and neutrophils (ly6G) for evaluation of the potential immune responses against CPP-gVLPs (Figure 5B). Our data demonstrated only few immune cells were detected in the brain tissue, reveals no acute inflammatory responses were induced by CPP-gVLPs administration. We further visualized the distribution of free EPI or EPI@CPP-gVLPs delivered by CED to explore their feasibility as a nanocarrier platform for localized delivery in the brain of U87-MGLu tumor-bearing mice. The EPI@CPP-gVLPs system could provide broader distribution of EPI as compared with free EPI 3 h after administration by CED. As shown in Figure 5C (a) and (b), the red fluorescence in normal mice brain hemisphere clearly demonstrated the distribution of EPI is broader than free EPI administration. Moreover, the distribution of EPI@CPP-gVLPs influenced by the dense cellular composition of brain tumor, and the loaded EPI was released from CPP-gVLPs to penetrate into the tumor tissue from the junction of tumor tissue and normal brain tissue for complete tumor eradication (Figure 5C (c)). The toxicity and distribution profiles of EPI@CPP-gVLPs indicated stable encapsulation of EPI in CPP-gVLPs with minimal release in the concentrated protein environment of normal brain tissue. These results clearly show that CPP-gVLPs can be locally delivered by CED and easily monitored without extra tracer labeling, as well as that tissue half-life of EPI can be extended with reduced toxicity toward healthy brain tissue after incorporation in CPP-gVLPs. These findings have significant implications for the therapeutic efficacy of the nanotherapeutic agent based on CPP-gVLPs delivered by CED for brain tumors.
(A) Brain damage and toxicity from CED treatment with saline, CPP-gVLPs, free EPI, and EPI@CPP-gVLPs infusion, as shown in four representative sections of brains after 3 days. (B) Immunohistochemistry staining demonstrated the infiltration of neutrophils (Ly6G) and macrophages (CD68) at 24 h post saline and CPP-gVLPs administration by CED. (C) Distribution and retention of free EPI or EPI@CPP-gVLPs in the brain by CED infusion was monitored using fluorescence imaging to detect fluorescence generated from DAPI (indicated nuclei), GFP (indicated gVLPs), and EPI. The images show the distribution of free EPI or EPI@CPP-gVLPs in cross-sections of tissue surrounding the brain glioma 3 h after infusion. (a) CED of free EPI in normal mice brain, (b) CED of EPI@CPP-gVLPs in normal mice brain, (c) CED of EPI@CPP-gVLPs in brain tumor mice.
In vivo anti-tumor efficiency of EPI@CPP-gVLPs infused by CED
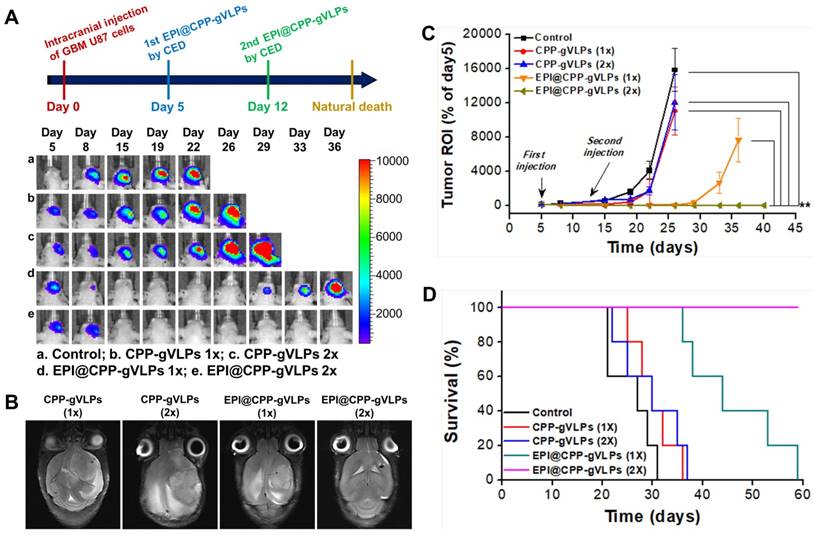
The promising toxicity and distribution outcomes instigated our exploration into whether EPI@CPP-gVLPs performed therapeutic response well to tumors in vivo. The tumor-bearing mice, through transplantation of U87-MGLu cells into their brains, were infused with 5 µL of CPP-gVLPs or EPI@CPP-gVLPs (containing 90.7 µg and 35.6 µg EPI) with an additional 5 µL administered 7 days after the initial infusion by CED. In the free EPI group, the mice died after 3 days of infusion with 5 µL of free EPI (containing 35.6 µg of EPI), most likely because the concentrated free EPI induced serious brain damage and bleeding. Figure 6A presents IVIS brain images from each animal subgroup, and the representative tumor sizes are shown as regions-of-interest (ROI) ratios. The usability of the tumor size measurements using IVIS were approved by MR imaging for each group at the end-time point, with a strong ROI signal indicating a large brain tumor (Figure 6B). The control group (15835.9 ± 2526.7% at day 26) and the mice that received one (11035.9 ± 2826.7% at day 26) or two doses (12084.1 ± 3238.5% at day 26) of CPP-gVLPs (by CED) all developed large brain tumors in the treated hemisphere, whereas the tumor-bearing mice that received one dose of EPI@CPP-gVLPs (by CED) developed significantly smaller brain tumors (7655.9 ± 2559.4.7% at day 36) compared with the control and CPP-gVLPs treated groups; however, recurrence was observed after 30 days of treatment. Overall, CED infusion of EPI@CPP-gVLPs enhanced therapeutic efficacy and reduced systemic toxicity and neurological damage of normal brain tissues. Additionally, we examined animal survival and used a Kaplan-Meier plot to present our results (Figure 6D). The tumor-bearing animals without any treatment (control) died by day 31, with a median survival time of 27 days. No obvious improvement in survival rate was observed in the mice that received CED infusions of one or two doses of CPP-gVLPs (median survival = 30 days), indicating that CPP-gVLPs possess no benefits for tumor inhibition and no neurological toxicity in normal brain tissues. The mice that received CED infusion of one dose of EPI@CPP-gVLPs at a volume of 5 µL (containing 90.7 µg CPP-gVLPs and 35.6 µg of EPI) all survived over 30 days, with a median survival of 44 days, whereas four of the ten animals (40%) in the group with CED infusion of one dose of EPI@CPP-gVLPs survived till day 53 to day 59. Notably, the tumors were completely eradicated and no recurrence was observed when the mice received CED infusion of two doses of EPI@CPP-gVLPs, which resulted in all treated mice surviving beyond day 50. Clinically, it has been reported that intratumoral DOX concentrations reaching 819 ± 482 ng/mL tissue correlate with partial or complete responses in breast cancer patients [33]. However, the present study showed that the proposed EPI@CPP-gVLPs provide an excellent drug-loading efficiency of 7.11 ± 1.37 mg/mL, resulting in the U87-MGLu tumor-implanted mice responding well to the infused EPI@CPP-gVLPs through CED.
(A) Treatment protocols assessing nanotherapeutic agent-based therapy by CED infusion. Animals were monitored by IVIS imaging at the indicated time points to measure tumor growth; one representative mouse from each of the indicated groups is shown. (B) MR images of U87-MGLu bearing tumors after CED infusions of various nanotherapeutic agents at the end-time point to verify the tumor size measured by IVIS. (C) Tumor progression observation among groups; corresponding tumor volume ratio determined from IVIS ROI value in (B) for each time point compared with day 5. Values are expressed as means ± SD (n = 10). **indicates a significant difference (Student's t-test, **p ≤ 0.05). (D) Kaplan-Meier curve shows the survival of mice implanted with U87-MGLu cells at 5 × 105 cells per mouse. On day 5 after transplantation, animals were infused with one or two doses of CPP-gVLPs (3.6 mg of CPP-gVLPs/kg; n = 10) or EPI@CPP-gVLPs (5.0 mg of EPI@CPP-gVLPs/kg containing 1.4 mg of EPI/kg; n = 10) by CED. The animals that did not receive any treatment were the control group (n = 10). 1x: 1 dose; 2x: 2 doses.
Blood biochemical analysis
In addition, no significant body weight loss was observed in the mice treated with one or two doses of CPP-gVLPs and EPI@CPP-gVLPs (Figure S8). In addition, blood biochemical analyses showed that both liver and renal functions (Figure S9) were unaffected, and there were no signs of inflammation or antigenicity (IgG: <0.34 mg/dL in the control group and <0.34 mg/dL in the treated group by CED; IgM: 8.0 ± 3.2 mg/dL in the control group and 5.8 ± 0.9 mg/dL in the treated group by CED) after 7 days of administration with CPP-gVLPs by CED infusion twice a week (3.6 mg/kg at each administration). This indicated that CED infusion of CPP-gVLPs-based therapeutics is safe enough and a potential alternative treatment modality for human glioma patients.
Conclusions
In summary, for the first time, we demonstrated that EPI@CPP-gVLPs (a dual-imaging virus-like nanotherapeutic agent) based on in vivo one-pot fabrication of gVLPs is an ideal CED infusate, which can be formulated in aqueous buffer in a reproducible and scale-up manner. The EPI@CPP-gVLPs exhibited high stability in terms of size in a protein-rich environment and surrounded the whole tumor with a high concentration of EPI by CED delivery, which can bypass the BBB to reduce systemic toxicity. By delivering EPI@CPP-gVLPs through CED in the brain tumor twice, once per week, tumors were eradicated completely without tumor recurrence or serious brain damage until 70 days after treatment. The excellent safety and tumor eradication efficiency of the treatment observed in the animal brain tumor model suggested that CED infusion of EPI@CPP-gVLPs can serve as a platform for translating the molecular understanding of brain tumors achieved in the laboratory into effective clinical treatments.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This work was financially supported by the Ministry of Science and Technology, Chang Gung Memorial Hospital, and National Health Research Institutes, Taiwan (ROC), for the financial assistance provided (MOST106-2628-E-110-001-MY3, MOST106-2628-B-110-001-MY4, CMRPG2G0722, CMRPG3F1733, NHRI-EX108-10502NI). Furthermore, we would like to thank the Chang Gung Memorial Hospital Microscopy Core Laboratory and the Center for Advanced Molecular Imaging and Translation for their assistance with TEM, IVIS and PET/CT imaging.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Holland EC. Glioblastoma multiforme: the terminator. Proc Natl Acad Sci USA. 2000;97:6242-4
2. Ramirez YP, Weatherbee JL, Wheelhouse RT, Ross AH. Glioblastoma multiforme therapy and mechanisms of resistance. Pharmaceuticals (Basel). 2013;6:1475-506
3. Bondy ML, Scheurer ME, Malmer B, Barnholtz-Sloan JS, Davis FG, Il'yasova D. et al. Brain tumor epidemiology: consensus from the brain tumor epidemiology consortium. Cancer. 2008;113:1953-68
4. Agarwala SS, Kirkwood JM. Temozolomide, a novel alkylating agent with activity in the central nervous system, may improve the treatment of advanced metastatic melanoma. Oncologist. 2000;5:144-51
5. Sudhal T, Bharali DJ, Sell S, Darwish NHE, Davis PJ, Mousa SA. Nanoparticulate tetrac inhibits growth and vascularity of glioblastoma xenografts. Horm Cancer. 2017;8:157-65
6. Jungk C, Chatziaslanidou D, Ahmadi R, Capper D, Bermejo JL, Exner J. et al. Chemotherapy with BCNU in recurrent glioma: Analysis of clinical outcome and side effects in chemotherapy-naïve patients. BMC Cancer. 2016;16:81
7. Blanchette M, Fortin D. Blood-brain barrier disruption in the treatment of brain tumors. Methods Mol Biol. 2011;686:447-63
8. van Tellingen O, Yetkin-Arik B, de Gooijer MC, Wesseling P, Wurdinger T, de Vries HE. Overcoming the blood-brain tumor barrier for effective glioblastoma treatment. Drug Resist Updat. 2015;19:1-12
9. Bhowmik A, Khan R, Ghosh MK. Blood Brain Barrier: a challenge for effectual therapy of brain tumors. Biomed Res Int. 2015;2015:320941
10. Bobo RH, Laske DW, Akbasak A, Morrison PF, Dedrick RL, Oldfield EH. Convection-enhanced delivery of macromolecules in the brain. Proc Natl Acad Sci USA. 1994;91:2076-80
11. Yun J, Rothrock RJ, Canoll P, Bruce JN. Convection-enhanced delivery for targeted delivery of antiglioma agents: the translational experience. J Drug Deliv. 2013;2013:107573
12. Yamashita Y, Krauze MT, Kawaguchi T, Noble CO, Drummond DC, Park JW. et al. Convection-enhanced delivery of a topoisomerase I inhibitor (nanoliposomal topotecan) and a topoisomerase II inhibitor (pegylated liposomal doxorubicin) in intracranial brain tumor xenografts. Neuro Oncol. 2007;9:20-8
13. van Woensel M, Wauthoz N, Rosiere R, Amighi K, Mathieu V, Lefranc F. et al. Formulations for intranasal delivery of pharmacological agents to combat brain disease: a new opportunity to tackle GBM? Cancers (Basel). 2013;5:1020-48
14. Yang W, Huo T, Barth RF, Gupta N, Weldon M, Grecula JC. et al. Convection enhanced delivery of carboplatin in combination with radiotherapy for the treatment of brain tumors. J Neurooncol. 2011;101:379-90
15. Chen PY, Yeh CK, Hsu PH, Lin CY, Huang CY, Wei KC. et al. Drug-carrying microbubbles as a theranostic tool in convection-enhanced delivery for brain tumor therapy. Oncotarget. 2017;8:42359-71
16. Miranpuri GS, Kumbier L, Hinchman A, Schomberg D, Wang A, Marshall H. et al. Gene-based therapy of Parkinson's Disease: Translation from animal model to human clinical trial employing convection enhanced delivery. Ann Neurosci. 2012;19:133-46
17. Jiang P, Mukthavavam R, Chao Y, Bharati IS, Fogal V, Pastorino S. et al. Novel anti-glioblastoma agents and therapeutic combinations identified from a collection of FDA approved drugs. J Transl Med. 2014;12:13
18. (a) Zylberberg C, Matosevic S. Pharmaceutical liposomal drug delivery: a review of new delivery systems and a look at the regulatory landscape. Drug Deliv. 2016; 23: 3319-29. (b) Bobo D, Robinson KJ, Islam J, Thurecht KJ, Corrie SR. Nanoparticle-based medicines: a review of FDA-approved materials and clinical trials to date. Pharm Res. 2016;33:2373-87
19. (a) Chen PY, Ozawa T, Drummond DC, Kalra A, Fitzgerald JB, Kirpotin DB. et al. Comparing routes of delivery for nanoliposomal irinotecan shows superior anti-tumor activity of local administration in treating intracranial glioblastoma xenografts. Neuro Oncol. 2013; 15: 189-97. (b) Dickinson PJ, LeCouteur RA, Higgins RJ, Bringas JR, Roberts B, Larson RF, et al. Canine model of convection-enhanced delivery of liposomes containing CPT-11 monitored with real-time magnetic resonance imaging: laboratory investigation. J Neurosurg. 2008;108:989-98
20. Pattni BS, Chupin VV, Torchilin VP. New developments in liposomal drug delivery. Chem Rev. 2015;115:10938-66
21. Elsabahy M, Wooley KL. Design of polymeric nanoparticles for biomedical delivery applications. Chem Soc Rev. 2012;41:2545-61
22. Corem-Salkmon E, Ram Z, Daniels D, Perlstein B, Last D, Salomon S. et al. Convection-enhanced delivery of methotrexate-loaded maghemite nanoparticles. Int J Nanomedicine. 2011;6:1595-602
23. Roldao A, Mellado MC, Castilho LR, Carrondo MJ, Alves PM. Virus-like particles in vaccine development. Expert Rev Vaccines. 2010;9:1149-76
24. Huang X, Wang X, Zhang J, Xia N, Zhao Q. Escherichia coli-derived virus-like particles in vaccine development. NPJ Vaccines. 2017;2:3
25. Rhee JK, Hovlid M, Fiedler JD, Brown SD, Manzenrieder F, Kitagishi H. et al. Colorful virus-like particles: fluorescent protein packaging by the Qβ capsid. Biomacromolecules. 2011;12:3977-81
26. Ashley CE, Carnes EC, Phillips GK, Durfee PN, Buley MD, Lino CA. et al. Cell-specific delivery of diverse cargos by bacteriophage MS2 virus-like particles. ACS Nano. 2011;5:5729-45
27. Doll TA, Raman S, Dey R, Burkhard P. Nanoscale assemblies and their biomedical applications. J R Soc Interface. 2013;10:20120740
28. Beijnen JH, van der Houwen OAGJ, Underberg WJM. Aspects of the degradation kinetics of doxorubicin in aqueous solution. Int J Pharm. 1986;32:123-31
29. (a) Alexander CM, Maye MM, Dabrowiak JC. DNA-capped nanoparticles designed for doxorubicin drug delivery. Chem Commun (Camb). 2011; 47: 3418-20. (b) Zeng Q, Wen H, Wen Q, Chen X, Wang Y, Xuan W, et al. Cucumber mosaic virus as drug delivery vehicle for doxorubicin. Biomaterials. 2013;34:4632-42
30. Dube N, Shu JY, Dong H, Seo JW, Ingham E, Kheirolomoom A. et al. Evaluation of doxorubicin-loaded 3-helix micelles as nanocarriers. Biomacromolecules. 2013;14:3697-705
31. Yang HW, Hua MY, Liu HL, Huang CY, Tsai RY, Lu YJ. et al. Self-protecting core-shell magnetic nanoparticles for targeted, traceable, long half-life delivery of BCNU to gliomas. Biomaterials. 2011;32:6523-32
32. Schmidt N, Mishra A, Lai GH, Wong GC. Arginine-rich cell-penetrating peptides. FEBS Lett. 2010;584:1806-13
33. Cummings J, McArdle CS. Studies on the in vivo disposition of adriamycin in human tumours which exhibit different responses to the drug. Br J Cancer. 1986;3:835-8
Author contact
![]() Corresponding authors: Hung-Wei Yang. Tel: +886 7 5252000 (ext. 5842); Fax: +886 7 5250151; Email: howardyangnsysu.edu.tw and Chiung-Yin Huang. Tel: +886 3 3281200 (ext. 2412); Fax: +886 3 3285818; Email: chyinhuangcom
Corresponding authors: Hung-Wei Yang. Tel: +886 7 5252000 (ext. 5842); Fax: +886 7 5250151; Email: howardyangnsysu.edu.tw and Chiung-Yin Huang. Tel: +886 3 3281200 (ext. 2412); Fax: +886 3 3285818; Email: chyinhuangcom