Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(6):1651-1665. doi:10.7150/thno.29703 This issue Cite
Research Paper
A Novel Peptide Interfering with proBDNF-Sortilin Interaction Alleviates Chronic Inflammatory Pain
Idy H.T. Ho1*, Xiaodong Liu1*, Yidan Zou1*, Taian Liu1, Wei Hu1, Hung Chan1, Yuanyuan Tian1, Yuchen Zhang1, Qing Li1, Shanglong Kou1, Chee Sam Chan1, Tony Gin1, Christopher H.K. Cheng2, Sunny H. Wong3,4,5, Jun Yu3,4,5, Lin Zhang1,4,6, William K.K. Wu1,4,5,6 ![]() , Matthew T.V. Chan1,6
, Matthew T.V. Chan1,6 ![]()
1. Department of Anaesthesia and Intensive Care, The Chinese University of Hong Kong, Hong Kong Special Administrative Region
2. School of Biomedical Sciences, The Chinese University of Hong Kong, Hong Kong Special Administrative Region
3. Department of Medicine and Therapeutics, The Chinese University of Hong Kong, Hong Kong Special Administrative Region
4. State Key Laboratory of Digestive Diseases, The Chinese University of Hong Kong, Hong Kong Special Administrative Region
5. LKS Institute of Health Sciences, The Chinese University of Hong Kong, Hong Kong Special Administrative Region
6. CUHK Shenzhen Research Institute, Shenzhen, China.
*These authors contributed equally: I Ho, X Liu, Y Zou
Received 2018-9-4; Accepted 2019-1-27; Published 2019-2-28
Abstract

Rationale: Brain-derived neurotrophic factor (BDNF) is a key mediator in the development of chronic pain. Sortilin is known to interact with proBDNF and regulate its activity-dependent secretion in cortical neurons. In a rat model of inflammatory pain with intraplantar injection of complete Freund's adjuvant (CFA), we examined the functional role of proBDNF-sortilin interaction in dorsal root ganglia (DRG).
Methods: Expression and co-localization of BDNF and sortilin were determined by immunofluorescence. ProBDNF-sortilin interaction interface was mapped using co-immunoprecipitation and bimolecular fluorescence complementation assay. The analgesic effect of intrathecal injection of a synthetic peptide interfering with proBDNF-sortilin interaction was measured in the CFA model.
Results: BDNF and sortilin were co-localized and their expression was significantly increased in ipsilateral L4/5 DRG upon hind paw CFA injection. In vivo adeno-associated virus-mediated knockdown of sortilin-1 in L5 DRG alleviated pain-like responses. Mapping by serial deletions in the BDNF prodomain indicated that amino acid residues 71-100 supported the proBDNF-sortilin interaction. A synthetic peptide identical to amino acid residues 89-98 of proBDNF, as compared with scrambled peptide, was found to interfere with proBDNF-sortilin interaction, inhibit activity-dependent release of BDNF in vitro and reduce CFA-induced mechanical allodynia and heat hyperalgesia in vivo. The synthetic peptide also interfered with capsaicin-induced phosphorylation of extracellular signal-regulated kinases in ipsilateral spinal cord of CFA-injected rats.
Conclusions: Sortilin-mediated secretion of BDNF from DRG neurons contributes to CFA-induced inflammatory pain. Interfering with proBDNF-sortilin interaction reduced activity-dependent release of BDNF and might serve as a therapeutic approach for chronic inflammatory pain.
Keywords: analgesics, peptide drug, neurotransmitter, cell-penetrating peptide, protein transduction domain, Tat
Introduction
Chronic pain is one of the most frequently diagnosed conditions that affects at least 20% of the general population [1]. It is the result of complex interactions that persist after initial nociception, involving both neural pathways and psychosocial factors. Usual analgesics used for acute pain are only partially effective when given for chronic pain, and are often accompanied by severe side effects, such as drug dependence [2].
In order to treat chronic pain, it is necessary to consider the neuronal plastic changes that have occurred to perpetuate pain. In this respect, research into pain mechanisms and novel approaches targeting the nociceptive pathways in the spinal and supraspinal levels have been proposed to develop safer and more tolerable analgesics [3, 4] However, there have been many disappointments in the history of translational pain research [5]. Nevertheless, neurotrophin signalling in pain remains a promising area [6].
The role of brain-derived neurotrophic factor (BDNF) in nociceptive transmission has been investigated extensively. BDNF was found to regulate the development of chronic pain via central sensitization. Numerous preclinical studies showed that BDNF has a pronociceptive effect in spinal dorsal horn by acting as a pain mediator at the synapse between the primary afferents and second-order neurons. This involves binding of BDNF to its high-affinity receptor, namely tropomyosin receptor kinase B (TrkB). The subsequent intracellular signalling cascade enhances the activity of the N-methyl-D-aspartate receptor (NMDAR) by elevating intracellular calcium through phosphorylation of NR2A and NR2B subunits for synaptic efficacy potentiation [7]. In this respect, BDNF expression in small-diameter dorsal root ganglia (DRG) and their terminals in the superficial lamina of spinal dorsal horn were dramatically elevated in animal models of inflammatory pain [8, 9]. Previous studies also showed that intrathecal administration of exogenous BDNF into normal animals resulted in a potent pronociceptive effect, while inflammation-associated pain hypersensitivity could be alleviated by anti-BDNF treatments in rodents [10, 11]. The hyperalgesia induced by intrathecal administration of BDNF could also be abrogated by the TrkB receptor antagonist cyclotraxin B; or partially by pregabalin, ketamine, morphine, tapentadol or agoelatine [12]. Furthermore, our recent human study on genetic variations of BDNF showed that two single-nucleotide polymorphisms (SNPs) in BDNF were associated with chronic postsurgical pain. In two independent cohorts of 1,358 patients, those carrying allele G in BDNF (rs6265) were at higher risk [13]. These findings supported the hypothesis that the BDNF-TrkB pathway in the spinal dorsal horn was crucial and sufficient for potentiation of nociceptive responses, thus implying BDNF could be a potential target for therapeutic treatment of chronic pain.
Sortilin, encoded by SORT1, is a type 1 membrane protein that belongs to the Vps10p protein family, and has been shown to modulate proneurotrophins (proNTs) processing. Sortilin is widely expressed in neuronal and non-neuronal cells, and may function as a receptor, a co-receptor or a sorting partner to trigger protein sorting into degradation or secretory pathways [14]. The involvement of sortilin and its proNTs processing in pain perception has been proposed [15]. Sortilin-deficient mice modified the neurotensinergic system and resulted in a lower sensitivity to thermal and chemical pain [16]. These findings highlighted the potential role of sortilin for pain relief.
Endogenous BDNF from primary sensory neurons was released in an activity-dependent manner [17]. However, the exact regulatory mechanism remains to be elucidated. From human genetic and animal studies, a SNP (rs6265) in BDNF leading to a valine (Val) to methionine (Met) substitution at codon 66 (Val66Met) in the 5' prodomain of BDNF (BDNFMet), was found to reduce the activity-dependent secretion of BDNF in hippocampal and cortical neurons, resulting in diminished memory performance [13, 18-20]. Thus, the BDNF prodomain, which is essential for BDNF trafficking, is required for its activity- dependent secretion. Previous in vitro studies suggested that sortilin regulated the release of BDNF by binding to its prodomain [21]. Alteration of this interaction led to BDNF mis-sorting from the activity-dependent to the constitutive pathway but did not affect its endogenous expression [21]. As activity-dependent secretion of BDNF is crucial to chronic pain development and maintenance, we hypothesized that blocking of sortilin-mediated BDNF secretion from the initial afferents at the spinal dorsal horn could prevent and treat chronic pain with minimal side effects.
Materials and Methods
Reagents and antibodies
All peptides were commercially synthesized with a purity of >95% (GenScript, Piscataway, NJ, USA). The peptides were dissolved in normal saline to make a 2 mM stock solution. The stock solution was diluted with culture medium or normal saline before use. Chemicals were purchased from Sigma (St Louis, MO, USA) unless stated otherwise.
Cell Culture
Human embryonic kidney cells, HEK-293 cells (CRL-1573) and HEK-293T cells (CRL-3216) were purchased from American Type Culture Collection (Manassas, VA, USA). The cells were maintained in high glucose Dulbecco's modified Eagle's medium (DMEM; Gibco, Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS; Life Technologies, Calsbad, CA, USA) and 1% penicillin/streptomycin (Gibco, Waltham, MA, USA) in a humidified incubator at 37 °C and 5% CO2. The primary rat cortical neurons were extracted from the cortex of prenatal rat fetuses (E18.5), rinsed and dissociated in Trypsin-EDTA (0.25%), phenol red (Life Technologies, Carlsbad, CA, USA) for 15 min in a 37 °C water bath. The cells were washed with DMEM supplemented with 10% FBS. DNaseI (Roche, Basel, Switzerland) was then added for 5 min before washing and triturated with neuron culture medium (Neurobasal- A medium, Thermo Fisher Scientific Inc., Rockford, IL, USA), 2% B27, 1% GlutaMAX (Thermo Fisher Scientific Inc., Rockford, IL, USA) and 1% Penicillin- Streptomycin-Neomycin (PSN) antibiotic mixture (Gibco, Waltham, MA, USA). Number of neurons was counted with a hemocytometer and cells were plated into Poly-D-lysine (PDL) pre-coated 24-well plates at a concentration of 3 x 106 cells/mL. Half of the culture medium was changed every 3-4 days to feed the cells. For primary DRG neuronal cultures, DRGs were extracted from rats weighing 130 g and dissociated in dispase (100 mg/mL) and collagenase Type 1A (200 mg/mL), rinsed and triturated in DMEM/F12 medium. Sunk neurons were re-suspended and plated into PDL and laminin pre-coated 24-well plates in DRG culture medium [DMEM/F12 with 2% B27 (Thermo Fisher Scientific Inc., Rockford, IL, USA), and 1% penicillin/streptomycin (Gibco, Waltham, MA, USA)].
Plasmid construction and cell transfection
Full length (Fl) human BDNF and SORT1 complementary DNA were subcloned into pcDNA3.1 vector (Invitrogen, San Diego, CA, USA) with HA and Myc epitope tag added to the 3′ end, respectively. For plasmid construction of the BDNF prodomain variants, pcDNA-3.1-Fl-HA was used as template and the mutant constructs were generated with a 2-step PCR method. Cell transfection was performed using Lipofectamine 2000 (Life Technologies, Carlsbad, CA, USA) or Polyethylenimine (PEI) (Sigma-Aldrich, St. Louis, MO, USA) according to the instruction of the manufacturers. The cells were seeded at a concentration of 1×106 cells per well in a 6-well plates the night before transfection.
Immunofluorescence
Whole animal perfusion fixation with 4% paraformaldehyde (PFA) was performed for immunofluorescence. Tissues were harvested and post-fixed in 4% PFA overnight and cryopreserved in 30% sucrose (w/v) for at least 3 days before cryosectioning. For cell culture, the cells plated on coverslips were fixed in 4% PFA for 10 min before immunofluorescence. All samples were washed 3 times with Phosphate Buffered Saline (PBS) and blocked in Phosphate Buffered Saline-Tween (PBST, PBS and 0.3% Triton X-100) supplemented with 10% FBS for 1 h. The sections were then incubated with the primary antibodies at 4°C overnight. Following washing for 3 times with PBS, the sections were incubated with Alexa Fluor secondary antibodies (Thermo Fisher Scientific Inc., Rockford, IL, USA), rinsed and mounted with Prolong Diamond Antifade Mountant with DAPI (Invitrogen, San Diego, CA, USA). The primary antibodies used included anti-proBDNF antibody (#ANT-006-AG, Alomone Labs Ltd, Jerusalem, Israel), anti-phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) antibody (Cell signalling Technology, Danvers, MA, USA), anti-NeuN antibody (MAB377, EMD Millipore Corporation, Billerica, MA, USA) and anti-sortilin antibody (Abcam, Cambridge, UK). The images were captured using a Leica TCS SP8 Confocal Platform (Leica Microsystems, Wetzlar, Germany) with a 10× or 63× objective.
Co-immunoprecipitation
Co-immunoprecipitation (co-IP) was performed with Dynabeads Protein G for Immunoprecipitation (10004D, Thermo Fisher Scientific Inc., Rockford, IL, USA) according to the manufacturer's instruction. Proteins extracted in IP lysis buffer were allowed to bind with anti-HA antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA) at 4 °C overnight with rotation.
Bimolecular fluorescence complementation (BiFC) assay
HEK293T cells were cultivated in 24-well plates containing coverslip at 2×106 cells per well overnight before transfection. The cells were co-transfected with the pBiFC-VN155(I152L) plasmids with mutant genes encoding proBDNF and pBiFC-VC155 with sortilin (125 ng each) using PEI. After 24 h, live fluorescence images were captured.
BDNF Enzyme-Linked ImmunoSorbent Assay
BDNF-mCherry was over-expressed in the primary cultures by lentiviral transduction 24 h after plating for 72 h. The cells were pretreated with the peptides for 30 min and washed 3 times with Kreb's-Ringer's Henseleit (KRH) buffer (125 mM NaCl, 4.8 mM KCl, 2.6 mM CaCl2, 25 mM HEPES, 1.2 mM MgSO4, 5.6 mM glucose, 1 mM sodium ascorbic acid and 1.2 mM KH2PO4, pH7.4) followed by incubation in KRH or high-potassium KRH buffer (modified KRH buffer with a concentration of 75 mM NaCl and 56 mM KCl) at 37 °C for 10 min. Secreted BDNF levels in supernatants of primary cortical or DRG neuronal cultures were measured by enzyme- linked immunosorbent assay (ELISA) using the ChemiKine BDNF, Sandwich ELISA Kit (CYT306, EMD Millipore Corporation, Billerica, MA, USA) according to the manufacturer's instruction.
Animals
Sprague-Dawley rats were provided by the Laboratory Animal Services Centre, the Chinese University of Hong Kong. The animals were housed under constant 12 h light/dark cycle at 23 ± 2 °C with food and water available ad libitum. Pregnant rats and rats with intrathecal catheter implantation were housed individually in cages. Following treatments, animals were euthanized by decapitation for tissue collection, unless otherwise stated. All animal experimental procedures in this study were approved by the Animal Experimentation Ethics Committee at the Chinese University of Hong Kong.
Plantar complete Freund's adjuvant injection nociception model in rats
Male rats (220-250 g) were anesthetized under 3% isoflurane. Unilateral peripheral inflammation of the left hind paw was induced by intraplantar injection of 100 µL of emulsified complete Freund's adjuvant (CFA; Sigma-Aldrich, St. Louis, MO, USA) into the plantar surface.
DRG injection
Male rats (130 g) were used for DRG injection. Under anesthesia, left lumbar hemilaminectomy was performed to expose L5 DRG. For each DRG, 1.8 µL of rAAV-U6-BBSI-shRNA-CMV-mCherry-pA or rAAV-U6-shRNA2-Sort1-CMV-mCherry (2.55×1012 GC/mL, Brainvta, Wuhan, China) was injected at 0.2 µL/min using an infusion syringe pump and stereotaxic syringe holder with a glass micropipette.
Intrathecal catheter implantation and intrathecal injection
During 3% isoflurane anesthesia, an 18-gauge guiding needle was inserted into the intrathecal space between the L5 and L6 vertebrae. The inner end of a PE10-polyethylene catheter was gently implanted through a guiding needle until it reached the lumbar enlargement of the spinal cord. The outer end of the catheter was plugged and transfixed to the nuchal skin for later intrathecal injection. Synthetic peptides were injected through the indwelling catheter 30 min before the behavioural test. Capsaicin (30 ng suspended in 10% Tween-80 in 0.9% saline) was injected with the same method 3 min before sacrifice of the rat.
Pain-related behavioral analyses
The rats were placed individually in opaque acrylic chambers upon a metallic mesh and allowed to accustom to the test environment for 20 min before testing. The rigid tip of the electronic von Frey Anesthesiometer (2390 series, IITC Inc. Life Science, Woodland Hills, CA, USA) was applied to the mid-plantar surface of the left hind paw. A stimulus-related withdrawal of the tested paw was considered a positive withdrawal response. The maximum amount of pressure applied until a positive withdrawal response occurred was recorded as the pain withdrawal thresholds (PWT). The test was performed on each rat for 7 times at intervals of 5 min. For thermal hyperalgesia, each rat was placed individually in opaque acrylic chambers on a transparent glass plate above the light box which emitted a focused and non-noxious visible light beam of the plantar analgesia meter (Model 390G, IITC Inc. Life Science, Woodland Hills, CA, USA). The radiant heat light beam was aimed at the mid-plantar surface of the left hind paws for applying thermal stimulation. The light source was immediately switched off when a positive withdrawal response of the tested hind paw was observed. The time required for the withdrawal response was recorded as the pain withdrawal latency (PWL). The investigators were blinded to the grouping of rats. Specifically, blinding and randomization always involved two investigators for all behavioral tests. The first investigator randomized the rats of different groups into the numbered cages where they stayed until the end of the behavioral tests. The second investigator, who performed the tests, entered the room only after the rats had accustomed to the environment. There was no external skin marking, so the second investigator was blinded to the group allocation.
Western blotting analysis
The freshly separated tissue was homogenized and lysed in radio-immunoprecipitation assay (RIPA) lysis and extraction buffer (50 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA, 1% (v/v) Nonidet P-40 (NP-40, pH 7.6)) containing 1× proteinase and phosphatase inhibitors cocktail (Roche, Basel, Switzerland) for 30 min at 4°C with rotation. Lysate was centrifuged and supernatant was collected for total protein concentration determination using Pierce BCA protein assay kit (Thermo Fisher Scientific Inc., Rockford, IL, USA). The protein samples were separated with a 10% SDS-polyacrylamide gel and transferred onto PVDF membrane (EMD Millipore Corporation, Billerica, MA, USA). The membrane was then blocked in 5% (w/v) skim milk powder in TBST (Tris-buffered saline, 0.1% Tween 20) and immunobloted with primary antibodies. The antibodies used were anti-BDNF, anti-HA and anti-Myc antibodies (Santa Cruz Biotechnology, Santa Cruz, CA, USA), purified mouse anti-NTR3 antibody (BD Biosciences, Franklin Lakes, NJ, USA), anti-p44/42MAPK and anti- phospho-p44/ 42 MAPK (Erk1/2) (Thr202/Tyr204) antibodies (Cell signalling Technology, Danvers, MA, USA).
Statistical Analyses
Details of group means, standard error of the mean (SEM), statistical test and results of statistical analyses for each experiment were given in the Results section. Results were expressed as means ± SEM. Statistical significance between groups were examined with unpaired t-test, while multiple comparisons involving more than 2 groups were assessed by analysis of variance (ANOVA) using GraphPad Prism 5 (Graphpad Software, La Jolla, CA, USA). P<0.05 was considered as statistically significant. All surgical procedures, as well as quantitative histological and behavioral analyses were conducted in a blinded fashion.
Results
Co-expression of BDNF and sortilin in the primary afferents and their induction in rat model of CFA-induced inflammatory pain
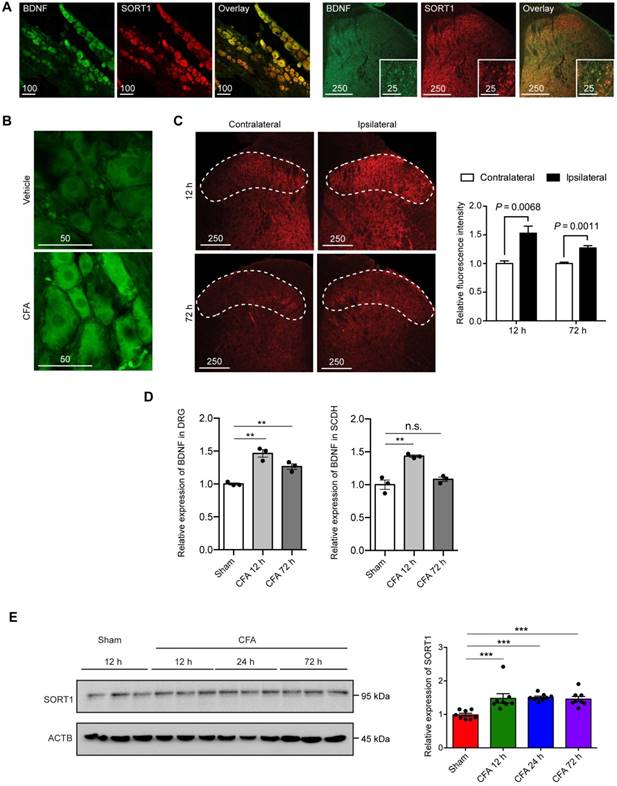
Immunofluorescence staining was used to determine the expression of BDNF and sortilin in primary afferents. BDNF and sortilin were co-expressed in cortical and DRG neuronal cultures in rats and mice, as well as in untreated adult rat DRG tissues and the central terminals in lumbar spinal dorsal horn (Figure 1A). We hypothesized that sortilin serves to regulate inflammation-induced pain hypersensitivity. To test this hypothesis, we first evaluated expression of sortilin in CFA-induced chronic inflammatory pain model. We previously reported significantly increased thermal hyperalgesia after intraplantar injection of CFA [22]. Classical signs, including edema and erythema of the left hind paws, with CFA injection were observed within 1 h and throughout the entire experiment (Supplementary Figure S1). In addition, BDNF was markedly upregulated in ipsilateral DRG tissues and superficial lamina of lumbar dorsal spinal horn (Figure 1B to 1D, DRG ELISA: P=0.0007; dorsal horn of lumbar spinal cord ELISA: P=0.0012, one-way ANOVA, n = 3). Total protein from ipsilateral dorsal spinal horn (L4 and L5) harvested for Western blot analysis of sortilin in the CFA-induced chronic inflammatory pain model showed that sortilin was significantly upregulated at least 1.5-fold compared to control animals (Figure 1E, P=0.0004, one-way ANOVA, n = 8). The upregulation lasted for at least 72 h after CFA injection.
Co-expression of BDNF and sortilin in the primary afferents and their induction in rat model of CFA-induced inflammatory pain. (A) Fluorescence microscopy of BDNF (green) and Sortilin (SORT1) (red). Left, L4-L5 DRGs. Scale bar represents 100 µm. Right, lumbar dorsal horn. Overview and magnification are shown. Scale bars represent 250 µm (overview) and 25 µm (insert). Tissues were extracted from untreated animals. (B) Fluorescence microscopy of BDNF (green) at 12 h after intraplantar injection of vehicle or CFA. (C) Left, fluorescence microscopy of BDNF (red) in lumbar dorsal horn at 12 and 72 h. Dotted line indicates superficial lamina. Right, quantification of fluorescence intensity of superficial layers of lumbar dorsal horn. Values are mean ± SEM; Student's t-test. n = 4. (D) Left, BDNF expression in L4-L5 DRGs measured with ELISA. Right, BDNF expression in dorsal horn of lumbar spinal cord measured with ELISA. Values are mean ± SEM; P=0.0007 for DRG, P=0.0012 for SCDH, one-way ANOVA. n = 3. **P<0.01; n.s., non-significant. DRG, dorsal root ganglion; SCDH, spinal cord dorsal horn. (E) Left, representative immunoblots of ipsilateral L4-L5 DRG tissues of CFA-induced inflammatory pain model obtained with antibodies against sortilin or β-actin. Each lane corresponds to the protein sample extracted from one rat. Right, histogram shows relative sortilin expression normalized to β-actin. Values are mean ± SEM; P=0.004, one-way ANOVA. n = 8. ***P<0.001; Ctrl, Control.
Sortilin in primary afferents is required for development of pain hypersensitivity
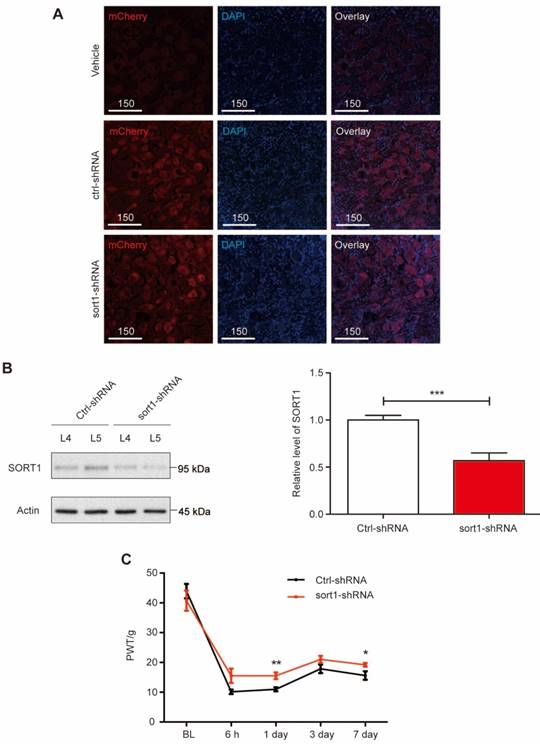
Next, we performed in vivo knockdown of sortilin in L5 DRG to evaluate whether changes in sortilin expression were required for pain hypersensitivity. AAV6-mediated shRNA with mCherry expression was delivered into L5 DRG tissues (Figure 2A). DRG tissues were collected to examine the sortilin knockdown efficacy. At 4 weeks after L5 DRG injection of AAV6/shRNA, compared with scrambled shRNA-treated rats, the sort1-shRNA construct led to >50% reduction of sortilin expression at the protein level (P<0.001) after intraplantar CFA injection (Figure 2B and Supplementary Figure S2). Mechanical allodynia was evaluated in the CFA model for 1 week. The elevated PWT confirmed that sortilin knockdown in DRG neurons alleviated inflammation-induced chronic pain (Figure 2C). Taken together, our data showed that sortilin contributed to the development of pain hypersensitivity.
L5 DRG-specific knockdown of sortilin alleviated inflammatory-induced chronic pain. (A) Fluorescence microscopy of mCherry (red) and DAPI (blue) in L5 DRG tissues injected with vehicle, control-shRNA (ctrl-shRNA) or sortilin-targeted shRNA (sort1-shRNA). Scale bars represent 150 µm. (B) Left, representative immunoblots of L4 or L5 DRG tissues at 4 weeks after L5 DRG injection obtained with antibodies against sortilin or β-actin. Samples were collected on day 7 after CFA injection. Right, histogram shows relative sortilin expression in L5 DRGs normalized to β-actin. ***P<0.001 versus control (ctrl), Student's t-test. Values are means ± SEM. n = 7. (C) Effect of sortilin knockdown at L5 DRG on CFA-induced reduction of paw withdrawal threshold (PWT) to Von-Frey stimuli in the ipsilateral paw. P=0.0208, two-way ANOVA with repeated measures; *P<0.05; **P<0.01. Values are means ± SEM. n = 6. BL, Baseline.
Mapping of proBDNF-sortilin interaction interface
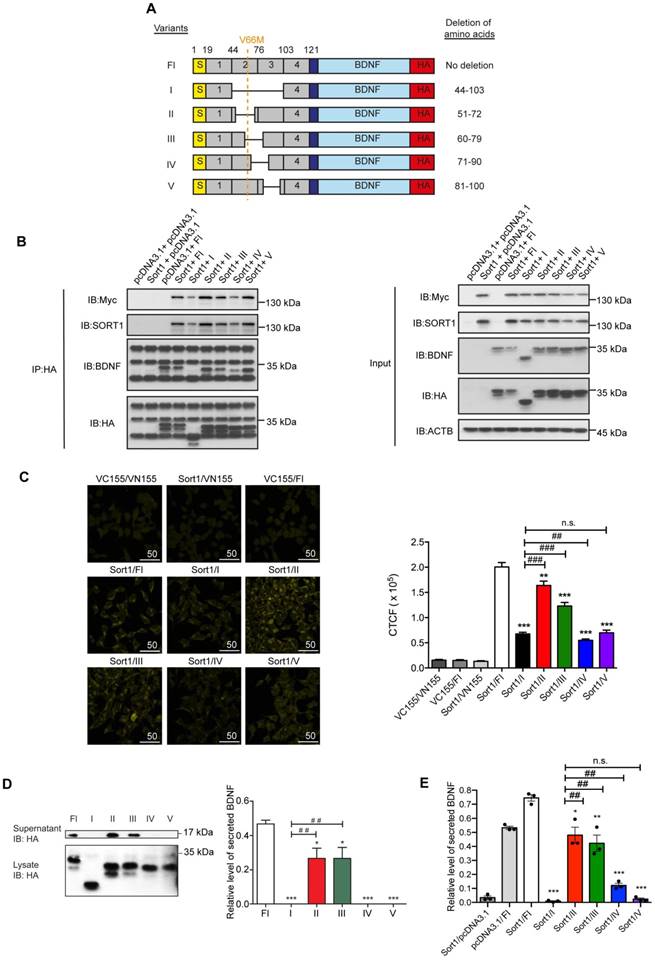
For generating a peptide blocking the regulatory secretion of BDNF, precise mapping of the proBDNF-sortilin binding interface is a prerequisite. Chen et al. reported that the deletion of Box 2 and Box 3 (amino acid residues 44-102) in the BDNF prodomain attenuated its interaction with sortilin [21]. In light of this, we fine-mapped the interacting interface exactly to 20 amino acid residues for the design of a novel peptide. A series of deletions in the BDNF prodomain were designed to further divide Box 2 and Box 3 into 4 regions according to amino acid sequence, namely variants II (deletion of amino acid residues 51-72), III (deletion of amino acid residues 60-79), IV (deletion of amino acid residues 71-90) and V (deletion of amino acid residues 81-100) (Figure 3A). Mutant proBDNF constructs with HA epitope tag and sortilin construct with Myc epitope tag were co-expressed in HEK293 cells. Co-IP was performed to analyse the association of each mutant with sortilin. A strong association was observed with sortilin in the full-length proBDNF group, while the association with variant I (removal of amino acid residues 44-103) was very weak. These findings were consistent with the current understanding that Box 2 and Box 3 in the BDNF prodomain are crucial for mediating proBDNF-sortilin interaction. The interaction with sortilin was most significantly disrupted by deleting amino acid residues as in construct variant IV (removal of amino acid residues 71-90) but not by variants II (removal of amino acid residues 51-72), III (removal of amino acid residues 60-79) and V (removal of amino acid residues 81-100) (Figure 3B). These results suggested that amino acid residues 81-90 supported proBDNF-sortilin interaction. In vitro binding assays were performed to confirm the core sequence for the molecular interaction in live cells under physiological conditions. Our BiFC assay showed a markedly reduced fluorescence intensity of Venus fluorophore when variants IV and V were expressed with sortilin, respectively (Figure 3C). This finding confirmed that the interaction interface was located at amino acid residues 71-100 in the BDNF prodomain. Overexpression assay was then performed to evaluate the effect of the variants on BDNF secretion. Secreted BDNF in the supernatant was detected only in the Fl group but not in the variant I group, which acted as a negative control. Secreted BDNF was only detected in supernatants of cells expressing variants II and III. In contrast, no secreted BDNF was detected from cells expressing variants IV and V (Figure 3D and 3E). Taken together, these results confirmed our co-IP and BiFC studies and provided evidence at a functional level to substantiate the precise crucial region of proBDNF-sortilin interaction was located within amino acid residues 71-100.
Mapping of proBDNF-sortilin interaction interface. (A) Schematic diagram illustrating the design of human BDNF prodomain variants. Six variants with a series of amino acids deletions were used in this study. V66M, Val66Met; S, Signal peptide (amino acid 1-18); BDNF, mature BDNF (amino acid 129-247); HA, C-terminal HA epitope. (B) Immunoblots of co-immunoprecipitation (Left) or inputs (Right) of sortilin and proBDNF variants obtained with antibody against HA or Myc epitope. IP, Immunoprecipitation; IB, Immunoblot. (C) Left, live fluorescence micrograph of BiFC mapping assay (Yellow) using HEK293T cells with VC155-sortilin (Sort1) and VN155(I152L)-BDNF prodomain variants. Scale bar represents 50 µm. Right, quantification of fluorescence intensity of BiFC assay. P<0.0001, one-way ANOVA. **P<0.01 versus Fl group; ***P<0.0001 versus Fl group; ##P<0.001 versus variant I; ###P<0.0001 versus variant I group; n.s., non-significant. Values are means ± SEM. n = 100 individual cells from 4 images. (D) Left, representative immunoblots of supernatant and cell lysates of HEK293 cells overexpressing sortilin and BDNF prodomain variants obtained with antibodies against HA. Right, densitometry analyses of secreted BDNF in supernatants, normalized to input in lysates. P<0.001, one-way ANOVA. *P<0.05, versus Fl group; ***P<0.001, versus Fl group; ##P<0.05, versus I group. Values are means ± SEM. n = 4. IB, Immunoblot. (E) BDNF ELISA quantification of supernatant collected from HEK293 cells overexpressing sortilin and BDNF prodomain variants. P<0.0001, one-way ANOVA. *P<0.05; **P<0.01; ***P<0.001, versus Sort1/Fl group; ##P<0.05, versus Sort1/variant I group. Values are means ± SEM. Each data point represents the average of 3 independent experiments.
Construction of a novel peptide targeting proBDNF-sortilin interaction
To develop a practical reagent with potential to translate into clinical practice that interfere with the binding of proBDNF to sortilin, we designed a series of overlapping 10-amino-acid peptides targeting amino acid residues 79-98 of proBDNF (Figure 4A). Our mapping studies indicated that region III (amino acid 60-79) had no effect on the proBDNF-sortilin interaction. Therefore, we intentionally neglected the sequences (amino acids 71-79) in region IV that shared with region III, leaving amino acids 79-98, covering regions IV and V, as our target. These peptides were acquired from commercially available custom peptide synthesis services (GeneScript, Piscataway, NJ, USA). Further co-IP studies were performed to screen the most effective peptide among the three 10-amino- acid-long candidates covering region V in HEK293 cells. Among the three candidates with amino acid sequences overlapping the core interaction surface, proBDNF-sortilin interaction was significantly decreased by the peptide bdnf89-98 in the co-IP assay with anti-HA antibody and reverse co-IP assay with anti-Myc antibody (Figure 4B). This suggested that bdnf89-98 had the most significant effect on interfering proBDNF-sortilin interaction.
Construction of a novel peptide targeting proBDNF-sortilin Interaction. (A) Schematic diagram illustrating the design of a series of blocking peptides targeting proBDNF-sortilin interaction interface. Scr-bdnf84-83 (scr), scrambled peptide with permutation of peptide sequence at amino acid 84-93. (B) Immunoblots of co-immunoprecipitation (upper) or reverse co-immunoprecipitation (lower) of sortilin and proBDNF variants in cell lysates overexpressing full-length proBDNF and sortilin followed by peptide incubation obtained with antibodies against HA or Myc. IP, Immunoprecipitation; IB, Immunoblot. (C) Schematic diagram of the design of the Tat-tagged interfering peptide. Scr-bdnf89-98-Tat, Tat-tagged scrambled peptide with permutation of peptide sequence at amino acid 89-98. (D) Live fluorescence micrographs of 5FAM (green) or bright field in rat DRG neuronal cultures before (0 min) and incubated with 5 µM 5FAM-tagged bdnf89-98-Tat (bdnf-Tat) for 2 h. Scale bar represents 150 µm. (E) Fluorescence micrograph of 5FAM (green), sortilin (red) or DAPI (blue) in rat DRG neuronal cultures with peptides pre-treatment for 30 min. Scale bars represent 25 µm. (F) Left, fluorescence micrograph of BiFC assay (yellow) of HEK293T cells overexpressing full-length BDNF and sortilin with vehicle, 10 µM scr-Tat or bdnf-Tat pre-treatment for 30 min. Scale bars represent 25 µm. Right, Determination of the corrected total cell fluorescence (CTCF) of the BiFC signal. P<0.0001, one-way ANOVA. ***P<0.0001, scr-Tat treatment versus bdnf-Tat treatment. Mean values ± SEM. n = 100 individual cells from 5 images. (G) Effect of 30 min peptide pre-treatment on activity-dependent secretion of BDNF in rat DRG neuronal cultures with high-potassium stimulus using ELISA. P<0.05, one-way ANOVA. *P<0.05, vehicle versus scr-Tat treatment or scr-Tat treatment versus bdnf-Tat treatment. Values are means ± SEM. Each data point represents the average of 3 independent experiments.
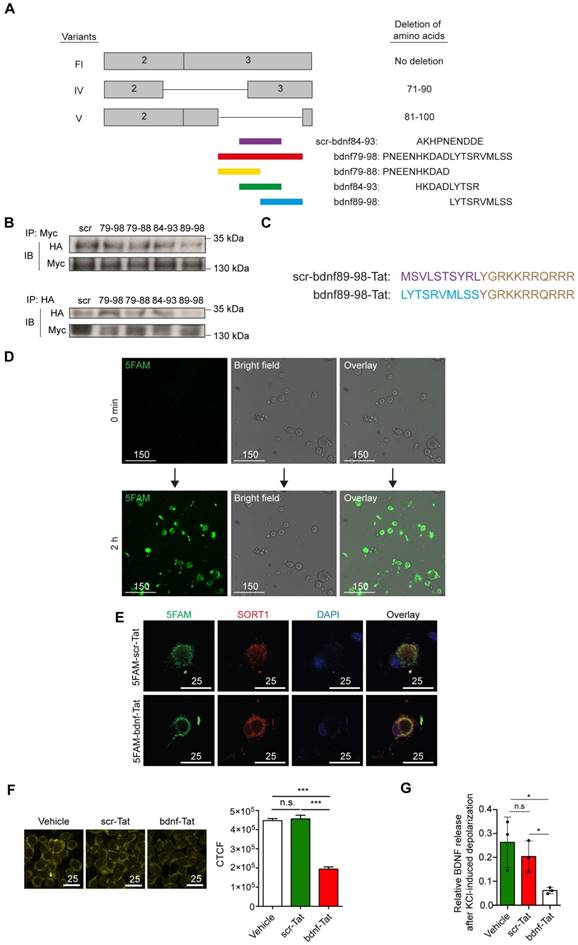
Since bdnf89-98 was predicted to be membrane impermeant, we fused the peptide to the protein transduction domain generated from the human immunodeficiency virus (HIV) transactivator of transcription (Tat, Sequence: GRKKRRQRRR) at the C-terminals [23]. Conjugation to protein transduction domain represents an approach for intracellular access [24]. Scrambled peptide was redesigned according to the amino acid sequence at position 89-98 with the same combination but different sequence (Figure 4C). The peptides, preceded by a 5-carboxyfluorescein (5FAM) in their N-terminals, were synthesized to determine if the Tat-tagged peptides could be delivered into the cells. A significant 5FAM signal could be observed in the live or fixed HEK293 cells and rat cortical neuronal cultures at 30 min and 2 h, with the peak at 30 min (Supplementary Figure S3A, S3B and S3D). 5FAM signal was also observed in the live or fixed DRG neuronal culture incubated with 5FAM-scr-bdnf89-98-Tat (5FAM-scr-Tat) at 30 min and 2 h, demonstrating that Tat-tagged peptides were able to penetrate small-sized DRG neurons (Figure 4D and Supplementary Figure S3C).
We then examined the ability of the synthetic peptide to compete with endogenous BDNF for the binding with sortilin. Fluorescence images showed positive green fluorescence signal for 5FAM, which was co-localized with endogenous sortilin in primary hippocampal cultures and primary culture of DRG neurons incubated with either 1 µM 5FAM-scr-Tat or 5FAM-bdnf89-98-Tat for 30 min (Figure 4E and Supplementary Figure S3E). BiFC assays were performed to demonstrate the interference of proBDNF-sortilin interaction by the peptide under physiological condition. Incubation with bdnf-Tat, instead of scr-Tat, reduced proBDNF-sortilin interaction under physiological condition in HEK293T cells transfected with VN155-BDNF-Fl and VC155(I152L)- SORT1 (Figure 4F). The functional effect of the peptide on attenuating the activity-dependent release of BDNF was validated with an in vitro assay using high-potassium KRH as a depolarization stimulus for BDNF release. The cells with lentiviral-mediated BDNF overexpression were pretreated with either 10 µM scr-Tat or bdnf89-98-Tat for 30 min before high-potassium KRH-induced depolarization. ELISA results showed a significant reduction in secreted BDNF in cortical neuronal cultures with conditioned medium (Supplementary Figure S4A to S4C) and DRG neuronal cultures (Figure 4G) treated with bdnf-Tat compared with scr-Tat. The decreases in BDNF secretion were consistent in both cortical neuronal culture and DRG neuronal cultures. These data demonstrated that the activity-dependent secretion of BDNF from neurons could be attenuated by addition of bdnf-Tat.
Peptide targeting proBDNF-sortilin interaction attenuates chronic pain
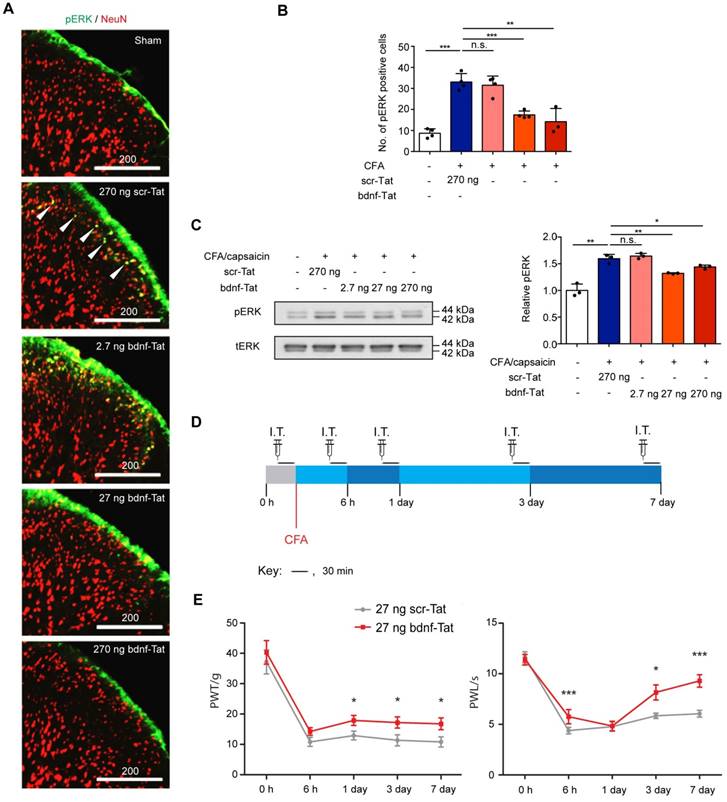
We then validated if the blocking peptide could functionally attenuate pain development with in vivo experiments. 5FAM signals were observed in rat DRG tissues after intrathecal injection, suggesting that the Tat-tagged peptide could be efficiently transported to DRG neurons (Supplementary Figure S5). Since activation of ERK1/2 pathway is a hallmark of BDNF-mediated TrkB activation, we studied the effect of the peptide on ERK1/2 phosphorylation. Rats with intraplantar CFA injection-induced chronic inflammatory pain were pretreated with different dosages of the peptide and challenged with capsaicin for 3 min. The ratio of phospho-ERK1/2-to-total ERK1/2 was measured by immunofluorescence staining. Our results showed that after challenging with capsaicin in CFA-injected rats pretreated with either 270 ng scr-Tat, the number of phosphor-ERK1/2-positive neurons at the superficial laminae were significantly increased when compared to the sham group. In contrast, pre-treatment with bdnf-Tat led to a dose- dependent decrease in the number of phosphor- ERK1/2-positive neurons (Figure 5A and B). The immunofluorescence staining results were further confirmed with Western blot analysis (Figure 5C). The effect of the peptide on the development of chronic inflammatory pain was then evaluated using thermal and mechanical hypersensitivity evoked by intraplantar injection of CFA. Each rat received vehicle or peptide intrathecally 30 min before the behavioural tests at each time point for a total of 7 days (Figure 5D). We chose to administer 27 ng of peptides for behavioural studies, because it was effective in reducing pain-related molecular markers. There were no significant changes of PWL and PWT between the groups of animals receiving vehicle or scr-Tat peptide throughout the 7-day behavioural test (n = 8 rats from 3 independent experiments, P=0.948 for PWT, P=0.566 for PWL, two-way ANOVA with repeated measures) (Supplementary Figure S6A and S6B), confirming that scr-Tat peptide was an appropriate control. Differences in thermal hyperalgesia and mechanical allodynia were tested in CFA-injected rats receiving 27 ng of scr-Tat or bdnf89-98-Tat peptides. The rats receiving bdnf89-98-Tat were less sensitive to noxious mechanical stimuli, with a higher withdrawal threshold compared to the rats receiving scr-Tat. The CFA model-induced decrease in PWT was significantly attenuated on days 1, 3 and 7 in rats pretreated with bdnf89-98-Tat but not in those with scr-Tat (Figure 5E). Similarly, The CFA-induced decrease in PWL was significantly attenuated by bdnf89-98-Tat (Figure 5F), showing that the rats receiving bdnf89-98-Tat were less sensitive to noxious heat than the rats receiving scr-Tat. In order to further develop the peptide for practical use in humans where intrathecal injection is not feasible, delivery of the peptide into DRG neurons by intravenous injection was tested with 5FAM-bdnf89-98-Tat in rats. Compared to rats receiving tail vein injection of vehicle, significant green fluorescence signals were detected in a subset of neurons in DRG tissues of the rat receiving 5FAM-bdnf89-98-Tat (Supplementary Figure S6C).
Pain responses in groups of rats pre-treated with scr-tat (270 ng) or bdnf-Tat (2.7 ng, 27 ng and 270 ng). (A), Fluorescence micrograph of pERK1/2 (green) or NeuN (red) in superficial lumbar dorsal horn of rat CFA model of chronic inflammatory pain receiving intrathecal injection of scr-Tat or bdnf-Tat. Arrows indicate pERK1/2 positive neurons. Scale bar represents 200 µm. (B), Quantification of fluorescence intensity of pERK1/2 positive cells in (A). 3D images for cryo-sections of each lumbar spinal cord were obtained by confocal microscopy. Maximal projections of the 3D images were processed for cell counting (only green cells were counted). P<0.0001, one-way ANOVA. Groups with rats pre-treated with scr-Tat (270 ng) and treated with CFA and capsaicin were compared with sham animals, while groups with rats pre-treated with bdnf-Tat were compared with rats pre-treated with 270 ng scr-Tat. **P<0.01, ***P<0.001, n.s. non-significant. Values are means ± SEM. n = at least 3 rats, at least 20 slices of lumbar spinal cords were counted. (C) Left, immunoblots of ipsilateral lumbar dorsal horn obtained with antibodies against p-pERK1/2 (pERK) or total pERK1/2 (tERK). Right, histogram shows relative p-pERK1/2 expression normalized to total pERK1/2. P<0.0001, one-way ANOVA. Values are means ± SEM. Immunoblots (in triplicates) with samples obtained from 5 rats in each group. *P<0.05, **P<0.01, ***P<0.001, n.s. non-significant (D) Timeline for behavioural testings. I.T., intrathecal injection of the interfering peptides; CFA, intraplantar Complete Freund's Adjuvant injection. (E) Effect of intrathecal injection of the peptides on mechanical allodynia of the rat CFA model. Results are presented as pain withdrawal threshold (PWT) in grams. P=0.036, two-way ANOVA with repeated measures. Values are means ± SEM. *P<0.05, n = 11 rats in scr-Tat group, n = 12 rats in bdnf-Tat group. (F), Effect of intrathecal injection of the peptides on thermal hyperalgesia of the rat CFA model. Results are presented as pain withdrawal latency (PWT) in seconds. P=0.001, two-way ANOVA with repeated measures. *P<0.05. ***P<0.001. Values are means ± SEM. n = 11 rats in scr-Tat group, n = 12 rats in bdnf-Tat group.
Discussion
BDNF is an important molecule in the development of chronic inflammatory pain and postsurgical pain. Our findings showed that sortilin contributed to the modulation of pain hypersensitivity through its interaction with BDNF. We also found that the exact binding interface for the proBDNF-sortilin interaction was located on amino acid 71-100. The bdnf-Tat covering amino acid residues 89-98 competed with endogenous BDNF, bound to endogenous sortilin and attenuated activity-dependent release of BDNF in hyperpolarized neurons. Our data showed that targeting proBDNF-sortilin interaction improved pain-related behavior in the CFA-induced chronic inflammatory pain model. Given that the prodomain of BDNF is conserved across human and rat, and that intravenous injection of synthetic peptide penetrates readily into DRG tissues, our bdnf-Tat peptide has potential to serve as a novel treatment for chronic inflammatory pain clinically.
In the pain pathway, BDNF is released from primary afferents after pain stimulation. BDNF is then secreted to the first synaptic process and bound to TrkB for pain signalling. Consistent with previous findings, our data showed that BDNF expression was enhanced in DRG tissues and the respective central terminals in dorsal spinal horn. We also found that BDNF co-localized with regulated secretory granules and presynaptic vesicles (Supplementary Figure S7). These data showed that the release of BDNF from DRG into the first synaptic process was in a regulated secretory manner, contributing to chronic pain development.
The interaction interface between BDNF and sortilin-Box 2 and -Box 3 (amino acids 44-102) on the BDNF prodomain, has been located to the luminal domain of sortilin [21]. In our study, we provided further details of the core region of the interaction, amino acid residues 71-100. Interestingly, the BDNF SNP Val66Met, that reduces BDNF secretion, was located out of the core sequence. This is due to the fact that there are multiple levels of regulation for BDNF sorting. For instance, the sorting motif (E146 and D234) located at the mature domain of BDNF interacts with sortilin-independent carboxypeptidase E and contributes to BDNF sorting. Concerning proBDNF- sortilin interaction, both Val66Met and the core sequence mapped in our study contributed to the BDNF trafficking to the appropriate pathways, with the core sequence exerting a more significant binding ability to sortilin based on our mapping results.
The interaction between sortilin and proNTs in the development of pain has also been suggested [15]. In our study, the findings that DRG-specific sortilin knockdown attenuated pain hypersensitivity was consistent with a prior sortilin knockout study [16]. In addition to thermal hyperalgesia, we further showed that sortilin-deficiency in DRG resulted in a decrease in mechanical allodynia. Therefore, strategies and compounds targeting proNTs-sortilin interaction may be useful in managing chronic pain. Recently, a sortilin-specific cell-based assay has been developed to identify compounds that interfere with the interaction between sortilin and proNTs [25]. The use of the proBDNF-sortilin interfering peptide in our study provided in vivo data demonstrating the feasibility of interfering proNTs-sortilin interaction. Our peptide attenuated the rapid effect of mature BDNF in spinal synapses, possibly by reducing the release of mature BDNF from excited primary sensory neurons.
Peptide-based approaches represent a novel class of biotherapeutics, and they have been used in various disorders and diseases, such as Cushing's disease, multiple myeloma, anaemia, short bowel syndrome, respiratory distress syndrome and constipation-predominant irritable bowel syndrome. Peptide-based drugs have been introduced into the clinical pipeline and have been approved for therapy by the United States Food and Drug Administration [26]. The use of cell-penetrating peptides for nociceptive modulation have been demonstrated previously. Pain response was decreased after administration of cell-penetrating peptide/nuclei acids complementary to galanin receptor type 1 mRNA [27]. Intrathecal application of Tat-fused isozyme specific protein kinase C inhibitors also modified formalin-induced pain perception [28]. Furthermore, antagonism of BDNF by the delivery of a neutralizing anti-BDNF antibody has been shown to reduce pain-related behaviour [29-31]. In our study, bdnf-89-98 was effective in interfering with proBDNF- sortilin interaction. It was conjugated to a HIV Tat- derived sequence for efficient cellular delivery by endocytosis. The conjugation could occur either at the N- or C-terminal. Attachment of the Tat peptide to the N- or C-terminal may produce differences in cellular uptake [32]. We conjugated the Tat peptide to both the N- and C-terminal of the peptides and demonstrated positive penetrations for both combinations. It should be noted that whether N-terminal Tat-conjugated peptide could be a better alternative was unclear. Herein, we proved that at least the C-terminal Tat- conjugated peptide interacted with sortilin and functionally blocked activity- dependent release of BDNF.
There are problems associated with peptide delivery mediated by Tat domain. Fixation of cells leading to soluble protein re-localization and may overestimate Tat-fused cargo [33, 34]. The delivery of Tat-fused cargo occurs to a variable extent, depending on the cargo size, cell type, delivery conditions and in vivo application. Although Tat cell-penetrating peptides are capable of passing through the blood- brain barrier [33], their affinity and efficiency for specific tissues, cells and cellular compartments depend on the design [35]. High concentrations of HIV-Tat stimulate oxidative stress and may lead to neuronal cell death. In contrast, few toxic effects have been reported for Tat-protein transduction domain. Therefore, the best design to deliver our BDNF interfering peptide specifically into the DRG neurons, and avoiding other parts of the nervous system to prevent undesirable effects, remains an empirical process.
A wide variety of cellular assays were used in our study. Since our mapping assays required the transfection of mutated BDNF prodomain constructs, HEK293 cells were used because of the higher efficiency for transfection compared with neuronal cell lines or primary cultures. We demonstrated that the interfering peptide was able to penetrate into the neuronal (cortical neuronal cultures and DRG neuronal cultures) and non-neuronal cells (HEK293 cells). Our ultimate goal was to develop a peptide to targeting primary sensory neurons for pain relief. Thus, both primary cortical and DRG neuronal cultures were used for functional viral transfection followed by ELISA to examine the interfering effect of the peptide. In order to demonstrate the cell penetration of our Tat-tagged peptide into neuronal cells, we used live imaging to avoid membrane passage artefacts (Figure 4D and Supplementary Figure S3A).
Nerve growth factor (NGF) and BDNF are highly homologous. We have considered the interference of the designed peptide with NGF. The BLAST results of human BDNF and NGF prodomain showed that the sequences in our peptide are not conserved (Supplementary Figure S8). Also, the regions may serve different functions for BDNF and NGF. In this regard, the prodomain of NGF is required for protein folding [36]. Deletion of Box 3 in NGF prodomain leads to a decrease in NGF expression while deletion in BDNF prodomain does not affect its expression [21]. This suggested that our focused region within the prodomain of NFG may not affect sortilin binding as in BDNF. In addition, endogenous NGF prodomain has not been found in neural tissue or primary neurons. It remains possible that the peptide may interfere with NGF signaling. The exact effect of the peptide on NGF signaling will require further investigations.
The novel peptide bdnf89-98 was shown to produce antinociceptive effect in a CFA-induced chronic inflammatory pain model. This is a standard chronic inflammatory pain model for research and resembles closely to the time course of chronic postsurgical pain development. We showed that the administration of bdnf-Tat, but not scrambled peptide, alleviated BDNF release from DRG through the MAP kinase ERK1/2 pathway. Although we do not know whether it affects the activity-dependent secretion of BDNF from cells other than DRG neurons, the overall effect of the peptide appears to alleviate chronic pain development.
The use of this novel peptide is a promising approach in humans for relief of chronic pain. As a pharmacological treatment, peptide therapeutics are growing in clinical significance. Synthetic peptides mimic naturally occurring biological molecules and therefore act with higher efficacy, specificity and selectivity towards their physiological targets [37]. Synthetic peptide fragments are amino acids in nature, and should be degraded into their components without toxic consequences. The risk of accumulation of the peptide or its degraded product can be minimized. The developmental cost of a peptide drug is also low. The wide variety of peptide structural modifications, such as linkage to lipids or polyethylene glycol groups, allow it to be optimized for disease treatment. The novel peptide should gain access to the DRG for selective action but not the central nervous system to avoid any undesirable effect. Previous studies have shown that sodium fluorescein injected intravenously gained access to the DRG but not into the spinal cord nor the cerebral cortex, possibly due to the minimal blood-nerve barrier for peripheral nerves [38, 39]. Our peptide could be administered systemically for penetration into DRG. Efforts have been made to develop oral peptides to facilitate drug administration. Calcitonin is an example of an orally available peptide in phase 3 clinical trial for treating hypercalcemia and osteoporosis [40]. There is thus clinical potential for our peptide to be administered orally for chronic pain relief.
Persistent interference of the proBDNF-sortilin interaction might have side effects on other functions. Other than chronic pain development, BDNF is important for survival, growth and differentiation of neurons. It is also important for learning, long-term memory, emotion and higher thinking. Studies using knockin mice or human carriers with BDNFMet/Met produced impairment in hippocampal dependent memory [18], anxiety-like behaviors [41, 42] and affective disorders, such as depression [43]. Nevertheless, as noted above, there are differences in penentration across different tissues [39], and the peptide would not cause the effects similar to the phenotype of BDNFMet/Met if it was restricted to the peripheral nervous system. All the rats treated with the synthetic peptide acted normally in the same way as those receiving saline (vehicle). There was no observable side effect in our experiments. Nevertheless, it has been reported that binding of proBDNF with p75NTR and sortilin elicited post-synaptic neuronal apoptosis [44]. Although there was no observable cell death in our in vitro nor in vivo assays, we cannot exclude the possible neuronal death on our results.
There are limitations of our study. We did not evaluate the analgesic effect of our peptide at the brainstem or brain levels, and may have missed important targets at these sites. In addition, we did not investigate the binding affinity of our peptide to endogenous sortilin. Finally, we acknowledge the overall analgesic effect of our peptide, albeit significant, is not overwhelming. Specifically, analgesia in the rat inflammatory pain model was incomplete. This may be due to the fact that the BDNF cascade is only one of the many pro-nociceptive mechanisms. In this respect, analgesic efficacy of antagonists targeting other pain mediators at the primary afferents, such as prostanoids, tumour necrosis factor-alpha, interleukin-1β and NGF, have been demonstrated clinically. It is plausible that a combination of drugs would be required for managing chronic pain.
In summary, we showed that a proBDNF-sortilin interaction in the primary afferents contributed to pain hypersensitivity, and a peptide that interferes with this interaction can reduce the hypersensitivity. Owing to the lack of effective analgesics for the treatment of chronic pain, our study has important implication on the pathogenesis and management of chronic inflammatory pain. The proBDNF/sortilin interaction is a possible therapeutic target for the treatment of chronic pain.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
Supported by project grant No. 464212 from the General Research Fund, Research Grant Council (Hong Kong Special Administrative Region, China) and project grant No. LTP001/14 and academic enhancement grant No. AEG13/001 from the Anesthesia and Pain Medicine Foundation, Australian and New Zealand College of Anaesthetists (Melbourne, Victoria, Australia). Idy Ho is supported by the Lee Hysan Postdoctoral Fellowship in Clinical Neurosciences.
Author contributions
I.H.T.H., X.L., W.K.K.W. and M.T.V.C designed the study analyzed the data and wrote the manuscript. I.H.T.H., X.L., Y.Z. and T.L. conducted the animal experiments and pain-related behavioral testing. I.H.T.H., X.L., Y.Z., T.L. W.H., H.C., Q.L., Y.T., Y.Z., S.K. and C.S.C. prepared the cell lines and performed the cellular experiments. C.H.K.C., T.G., S.H.W. L.Z. and J.Y. helped with data analysis and critically revised the paper. All authors read and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Macfarlane GJ. The epidemiology of chronic pain. Pain. 2016;157:2158-9
2. Volkow N, Benveniste H, McLellan AT. Use and misuse of opioids in chronic pain. Annu Rev Med. 2018;69:451-65
3. Alles SRA, Smith PA. Etiology and pharmacology of neuropathic pain. Pharmacol Rev. 2018;70:315-47
4. Price TJ, Gold MS. From mechanism to cure: Renewing the goal to eliminate the disease of pain. Pain Med. 2018;19:1525-49
5. Yezierski RP, Hansson P. Inflammatory and neuropathic pain from bench to bedside: What went wrong? J Pain. 2018;19:571-88
6. Khan N, Smith MT. Neurotrophins and neuropathic pain: Role in pathobiology. Molecules. 2015;20:10657-88
7. Crozier RA, Bi C, Han YR, Plummer MR. BDNF modulation of NMDA receptors is activity dependent. J Neurophysiol. 2008;100:3264-74
8. Cho HJ, Kim JK, Zhou XF, Rush RA. Increased brain-derived neurotrophic factor immunoreactivity in rat dorsal root ganglia and spinal cord following peripheral inflammation. Brain Res. 1997;764:269-72
9. Cho HJ, Kim SY, Park MJ, Kim DS, Kim JK, Chu MY. Expression of mRNA for brain-derived neurotrophic factor in the dorsal root ganglion following peripheral inflammation. Brain Res. 1997;749:358-62
10. Merighi A, Salio C, Ghirri A, Lossi L, Ferrini F, Betelli C. et al. BDNF as a pain modulator. Prog Neurobiol. 2008;85:297-317
11. Matayoshi S, Jiang N, Katafuchi T, Koga K, Furue H, Yasaka T. et al. Actions of brain-derived neurotrophic factor on spinal nociceptive transmission during inflammation in the rat. J Physiol. 2005;569:685-95
12. M'Dahoma S, Barthelemy S, Tromilin C, Jeanson T, Viguier F, Michot B. et al. Respective pharmacological features of neuropathic-like pain evoked by intrathecal BDNF versus sciatic nerve ligation in rats. Eur Neuropsychopharmacol. 2015;25:2118-30
13. Tian Y, Liu X, Jia M, Yu H, Lichtner P, Shi Y. et al. Targeted genotyping identifies susceptibility locus in brain-derived neurotrophic factor gene for chronic postsurgical pain. Anesthesiology. 2018;128:587-97
14. Carlo AS, Nykjaer A, Willnow TE. Sorting receptor sortilin-a culprit in cardiovascular and neurological diseases. J Mol Med (Berl). 2014;92:905-11
15. Lewin GR, Nykjaer A. Pro-neurotrophins, sortilin, and nociception. Eur J Neurosci. 2014;39:363-74
16. Devader C, Moreno S, Roulot M, Deval E, Dix T, Morales CR. et al. Increased brain neurotensin and NTSR2 lead to weak nociception in NTSR3/Sortilin knockout mice. Front Neurosci. 2016;10:542
17. Balkowiec A, Katz DM. Activity-dependent release of endogenous brain-derived neurotrophic factor from primary sensory neurons detected by ELISA in situ. J Neurosci. 2000;20:7417-23
18. Egan MF, Kojima M, Callicott JH, Goldberg TE, Kolachana BS, Bertolino A. et al. The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell. 2003;112:257-69
19. Chen ZY, Patel PD, Sant G, Meng CX, Teng KK, Hempstead BL. et al. Variant brain-derived neurotrophic factor (BDNF) (Met66) alters the intracellular trafficking and activity-dependent secretion of wild-type BDNF in neurosecretory cells and cortical neurons. J Neurosci. 2004;24:4401-11
20. Hariri AR, Goldberg TE, Mattay VS, Kolachana BS, Callicott JH, Egan MF. et al. Brain-derived neurotrophic factor val66met polymorphism affects human memory-related hippocampal activity and predicts memory performance. J Neurosci. 2003;23:6690-4
21. Chen ZY, Ieraci A, Teng H, Dall H, Meng CX, Herrera DG. et al. Sortilin controls intracellular sorting of brain-derived neurotrophic factor to the regulated secretory pathway. J Neurosci. 2005;25:6156-66
22. Liu X, Tian Y, Meng Z, Chen Y, Ho IH, Choy KW. et al. Up-regulation of cathepsin G in the development of chronic postsurgical pain: An experimental and clinical genetic study. Anesthesiology. 2015;123:838-50
23. Schwarze SR, Ho A, Vocero-Akbani A, Dowdy SF. In vivo protein transduction: Delivery of a biologically active protein into the mouse. Science. 1999;285:1569-72
24. Koren E, Torchilin VP. Cell-penetrating peptides: breaking through to the other side. Trends Mol Med. 2012;18:385-93
25. Malik I, Christensen S, Stavenhagen JB, Dietz GPH. Development of a cell-based assay to assess binding of the proNGF prodomain to sortilin. Cell Mol Neurobiol. 2018;38:827-40
26. Fosgerau K, Hoffmann T. Peptide therapeutics: current status and future directions. Drug Discov Today. 2015;20:122-8
27. Pooga M, Soomets U, Hallbrink M, Valkna A, Saar K, Rezaei K. et al. Cell penetrating PNA constructs regulate galanin receptor levels and modify pain transmission in vivo. Nat Biotechnol. 1998;16:857-61
28. Sweitzer SM, Wong SM, Peters MC, Mochly-Rosen D, Yeomans DC, Kendig JJ. Protein kinase C epsilon and gamma: involvement in formalin-induced nociception in neonatal rats. J Pharmacol Exp Ther. 2004;309:616-25
29. Theodosiou M, Rush RA, Zhou XF, Hu D, Walker JS, Tracey DJ. Hyperalgesia due to nerve damage: role of nerve growth factor. Pain. 1999;81:245-55
30. Fukuoka T, Kondo E, Dai Y, Hashimoto N, Noguchi K. Brain-derived neurotrophic factor increases in the uninjured dorsal root ganglion neurons in selective spinal nerve ligation model. J Neurosci. 2001;21:4891-900
31. Li CQ, Xu JM, Liu D, Zhang JY, Dai RP. Brain derived neurotrophic factor (BDNF) contributes to the pain hypersensitivity following surgical incision in the rats. Mol Pain. 2008;4:27
32. Zhang P, Cheetham AG, Lock LL, Cui H. Cellular uptake and cytotoxicity of drug-peptide conjugates regulated by conjugation site. Bioconjug Chem. 2013;24:604-13
33. Dietz GP, Bahr M. Peptide-enhanced cellular internalization of proteins in neuroscience. Brain Res Bull. 2005;68:103-14
34. Melan MA, Sluder G. Redistribution and differential extraction of soluble proteins in permeabilized cultured cells. Implications for immunofluorescence microscopy. J Cell Sci. 1992;101( Pt 4):731-43
35. Dietz GP, Bahr M. Delivery of bioactive molecules into the cell: the Trojan horse approach. Mol Cell Neurosci. 2004;27:85-131
36. Suter U, Heymach JV Jr, Shooter EM. Two conserved domains in the NGF propeptide are necessary and sufficient for the biosynthesis of correctly processed and biologically active NGF. EMBO J. 1991;10:2395-400
37. Kaspar AA, Reichert JM. Future directions for peptide therapeutics development. Drug Discov Today. 2013;18:807-17
38. Malmgren LT, Olsson Y. Differences between the peripheral and the central nervous system in permeability to sodium fluorescein. J Comp Neurol. 1980;191:103-7
39. Abram SE, Yi J, Fuchs A, Hogan QH. Permeability of injured and intact peripheral nerves and dorsal root ganglia. Anesthesiology. 2006;105:146-53
40. Craik DJ, Fairlie DP, Liras S, Price D. The future of peptide-based drugs. Chem Biol Drug Des. 2013;81:136-47
41. Ieraci A, Madaio AI, Mallei A, Lee FS, Popoli M. Brain-derived neurotrophic factor Val66Met human polymorphism impairs the beneficial exercise- induced neurobiological changes in mice. Neuropsychopharmacology. 2016;41:3070-9
42. Chen YW, Surgent O, Rana BS, Lee F, Aoki C. Variant BDNF-Val66Met polymorphism is associated with layer-specific alterations in GABAergic innervation of pyramidal neurons, elevated anxiety and reduced vulnerability of adolescent male mice to activity-based anorexia. Cereb Cortex. 2017;27:3980-93
43. Bath KG, Chuang J, Spencer-Segal JL, Amso D, Altemus M, McEwen BS. et al. Variant brain-derived neurotrophic factor (Valine66Methionine) polymorphism contributes to developmental and estrous stage-specific expression of anxiety-like behavior in female mice. Biol Psychiatry. 2012;72:499-504
44. Teng HK, Teng KK, Lee R, Wright S, Tevar S, Almeida RD. et al. ProBDNF induces neuronal apoptosis via activation of a receptor complex of p75NTR and sortilin. J Neurosci. 2005;25:5455-63
Author contact
![]() Corresponding authors: Matthew Chan, Department of Anaesthesia and Intensive Care, The Chinese University of Hong Kong, Shatin, NT, Hong Kong. Email: mtvchanedu.hk and William Wu, Department of Anaesthesia and Intensive Care, The Chinese University of Hong Kong, Shatin, NT, Hong Kong. Email: wukakeiedu.hk
Corresponding authors: Matthew Chan, Department of Anaesthesia and Intensive Care, The Chinese University of Hong Kong, Shatin, NT, Hong Kong. Email: mtvchanedu.hk and William Wu, Department of Anaesthesia and Intensive Care, The Chinese University of Hong Kong, Shatin, NT, Hong Kong. Email: wukakeiedu.hk