Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Conclusions
Materials and Methods
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(6):1580-1598. doi:10.7150/thno.30302 This issue Cite
Research Paper
Nano-Structural Effects on Gene Transfection: Large, Botryoid-Shaped Nanoparticles Enhance DNA Delivery via Macropinocytosis and Effective Dissociation
Wenyuan Zhang1,2, Xuejia Kang1,3, Bo Yuan4, Huiyuan Wang1, Tao Zhang1, Mingjie Shi1, Zening Zheng1,3, Yuanheng Zhang1, Chengyuan Peng1, Xiaoming Fan1, Huaiyu Yang1,5, Youqing Shen6, Yongzhuo Huang1,2 ![]()
1. State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China
2. University of Chinese Academy of Sciences, Beijing, 100049, China
3. Institute of Tropical Medicine, Guangzhou University of Chinese Medicine, Guangzhou 510405, China
4. Affiliated Hospital of Shandong University of Traditional Chinese Medicine, Jinan 250011, China
5. East China Normal University School of Life Sciences, Shanghai 200241, China
6. College of Chemical and Biological Engineering, Zhejiang University, Hangzhou 310027, China
Received 2018-9-30; Accepted 2019-1-3; Published 2019-2-28
Abstract

Effective delivery is the primary barrier against the clinical translation of gene therapy. Yet there remains too much unknown in the gene delivery mechanisms, even for the most investigated polymeric carrier (i.e., PEI). As a consequence, the conflicting results have been often seen in the literature due to the large variability in the experimental conditions and operations. Therefore, some key parameters should be identified and thus strictly controlled in the formulation process.
Methods: The effect of the formulation processing parameters (e.g., concentration or mixture volume) and the resulting nanostructure properties on gene transfection have been rarely investigated. Two types of the PEI/DNA nanoparticles (NPs) were prepared in the same manner with the same dose but at different concentrations. The microstructure of the NPs and the transfection mechanisms were investigated through various microscopic methods. The therapeutic efficacy of the NPs was demonstrated in the cervical subcutaneous xenograft and peritoneal metastasis mouse models.
Results: The high-concentration process (i.e., small reaction-volume) for mixture resulted in the large-sized PEI/DNA NPs that had a higher efficiency of gene transfection, compared to the small counterpart that was prepared at a low concentration. The microstructural experiments showed that the prepared small NPs were firmly condensed, whereas the large NPs were bulky and botryoid-shaped. The large NPs entered the tumor cells via the macropinocytosis pathway, and then efficiently dissociated in the cytoplasm and released DNA, thus promoting the intranuclear delivery. The enhanced in vivo therapeutic efficacy of the large NPs was demonstrated, indicating the promise for local-regional administration.
Conclusion: This work provides better understanding of the effect of formulation process on nano-structural properties and gene transfection, laying a theoretical basis for rational design of the experimental process.
Keywords: gene delivery, PEI, micropinocytosis, TRAIL, local-regional gene therapy, cervical cancer
Introduction
The common purpose of gene delivery is to deliver the therapeutic nucleic acids from the application site to the pathological tissues, following by intracellular transport, and targeting the specific cytoplasm components or the nucleus [1]. PEI has become one of the most commonly used non-viral gene vectors due to its high transfection efficiency in mammalian cells and ready availability. Despite the extensive application of the PEI, the conflicting results of PEI-mediated transfection have been often seen in literature. It could be accounted for the large variability in the experimental conditions and operations in transfection experiments [2-4]. It is known that the transfection efficiency of the PEI/DNA NPs is affected by the N/P ratio of PEI and DNA, serum content of culture medium, etc. Indeed, such changes resulted in the difference of particle size and surface charge, and consequently the transfection efficacy.
The formation of the PEI/DNA nanocomplex is first driven by the electrostatic interaction and finally by entropy, when mixing the positive charges of the PEI and the negative charges of the DNA together [5, 6]. Therefore, such formation is highly dynamic and variable, profoundly affected by the parameters of the process. There remains much room to explore because the mechanisms of PEI-mediated transfection have not been well understood yet [1, 2, 7]. Usually, much attention has been placed in how the changing ratio of the two reagents of the PEI/DNA affected the transfection efficiency. For example, a low N/P ratio of PEI/DNA tends to form the large-sized complexes, while a high N/P ratio was the opposite [8]. Moreover, the use of the different molecular weight of PEI was also a major factor for controlling particle size; for example, low-molecular-weight PEI generally exhibited weaker condensation ability to DNA than the high-molecular-weight PEI, thus resulting in larger particle size [8].
However, the operational changes of the process may affect the physicochemical properties and the transfection results as well. Unfortunately, it is little known about the influence of the operational changes on the resulting PEI/DNA NPs. For example, it is often seen in literature the variable transfection results even with the same dose of PEI/DNA and the cell lines. Usually, the PEI/DNA ratio or dose is considered as a critical parameter, but the operation concentration and liquid volume are largely ignored and missed in the reports. From an aspect of pharmaceutical engineering, the setting of the processing parameters is also an essential consideration for formulation. Yet, the important effect of the process setting on transfection has not been realized.
In this study, we demonstrated the significant effect of the operation concentration or mixture volume on the formation of the nanocomplex of PEI/ DNA, as well as their size and morphology. Moreover, it was also demonstrated that the large-sized NPs possessed enhanced gene transfection efficiency and the promising application in local-regional therapies.
Results
The mixture volume affected transfection efficiency of PEI/DNA
At a fixed dose, a small-volume mixture process was related to using a high concentration of PEI and DNA, and a large-volume process was related to low concentration (Scheme 1). It was revealed that the mixture volume of PEI and DNA is a crucial factor of the formulation process for controlling the transfection of the PEI/DNA NPs. A small-volume process (i.e., a high concentration of PEI and DNA, termed PEI/DNAhigh) resulted in better efficacy. The cellular uptake study of the NPs was conducted in the HeLa cells using flow cytometry and confocal microscopy. For both the NPs at the same dose, the positive rate was greater than 80% (i.e., PEI/DNAlow 82% and PEI/ DNAhigh 91%) (Figure 1A-C). However, the total fluorescence intensity of the PEI/DNAhigh group showed a 3.7-fold increase, compared to the PEI/DNAlow (Figure 1D). The fluorescence intensity results suggested that there was a greater amount of the PEI/ DNAhigh NPs in cellular uptake.
The scheme of self-assembly process of the two groups of PEI/DNA NPs.
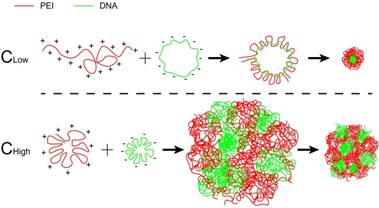
The cellular uptake results of the two groups of NPs. (A) Confocal images. The NPs are shown in green. Scale bar, 20 μm. (B-D) FACS analysis of cellular uptake, and the quantitative analysis of the cellular uptake rate and total fluorescence intensity.
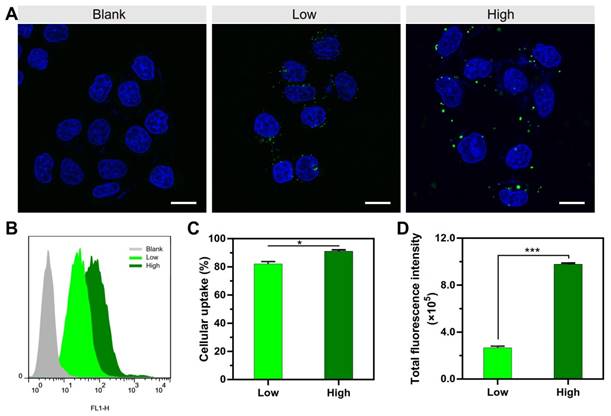
The gene transfection efficiency of these two NPs was investigated. In Figure 2A-D, peGFP-N1 was used as the reporter gene; consistent with the cellular uptake results, the transfection positive rate of the PEI/DNAhigh NPs was significantly higher, displaying 4.9-fold higher than that of the PEI/DNAlow NPs (Figure 2C). Accordingly, the total fluorescence intensity of the PEI/DNAhigh NPs, reflecting the level of GFP expression, was 11.9-fold higher than that of the PEI/DNAlow NPs (Figure 2D).
The results of transfection experiments of the two groups of PEI/DNA NPs. (A, F) Fluorescence images of GFP and RFP expression. GFP is shown in green, and RFP is shown in red. Scale bar, 100 μm. (B-D, G-H) FACS analysis of GFP and RFP expression in HeLa cells, and the quantitative analysis of positive transfection rate and total fluorescence intensity of HeLa cells. (E) Luciferase expression.
Other reporter genes (e.g., the red fluorescence protein-encoding pRFP and luciferase-encoding pGL4.13) were also applied for the transfection studies, and similar results were found (Figure 2E-H), both displaying an appropriate 10-fold increase in the PEI/DNAhigh group.
We further tested the intracellular uptake and transfection efficiency of these two groups of PEI/DNA NPs in other types of cells. Dendritic cells (DCs) are the most potent antigen-presenting cells, and the major target for cancer DNA vaccines. The results in DC2.4 cells and bone marrow-derived dendritic cells (BMDCs) also showed that the PEI/DNAhigh NPs had the enhanced cellular uptake and gene transfection compared to the PEI/DNAlow NPs (Figure S1). Moreover, it was also found that the PEI/DNAhigh NPs had the enhanced gene transfection compared to the PEI/DNAlow NPs in the MCF-7 cells, H/T cells and A549T cells (Figure S2).
The branched PEI25k was also used to investigate. Similarly, the b-PEI/DNAhigh NPs had a larger particle size compared to the b-PEI/DNAlow NPs (542 nm vs. 75 nm) (Figure S3A). It was also observed that the b-PEI/DNAhigh NPs improved the gene transfection compared to the b-PEI/DNAlow NPs (Figure S3B-D). Moreover, the b-PEI/DNAhigh NPs had the enhanced gene transfection compared to the b-PEI/DNAlow NPs in MCF-7, H/T and A549T cells (Figure S4).
The mixture volume affected the morphology of the PEI/DNA NPs
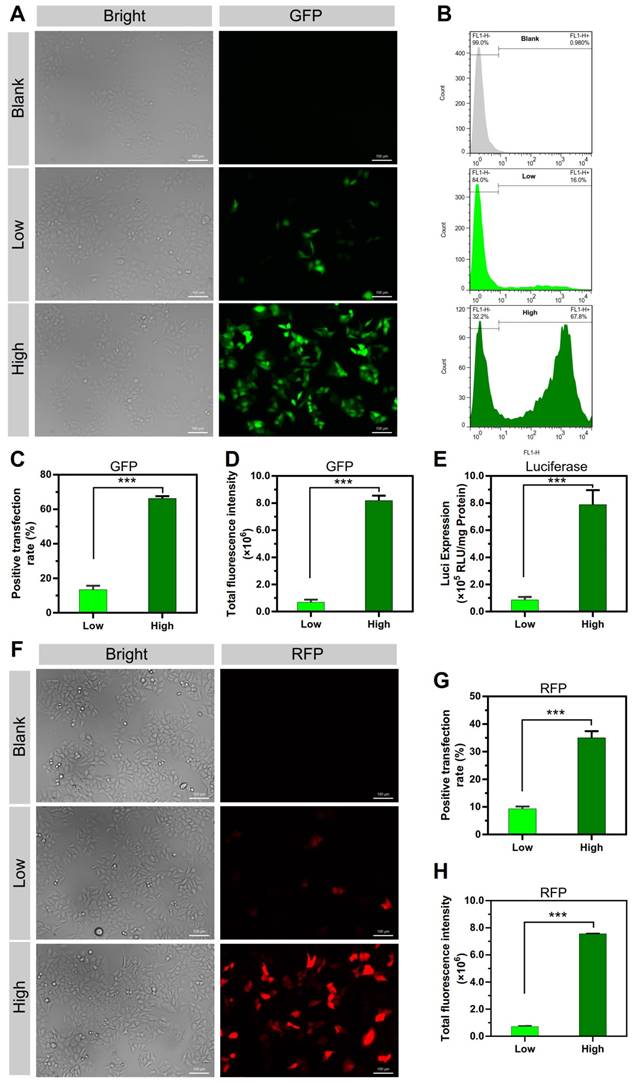
The findings above were interesting, which inspired us to further investigate the mechanisms of how the different volume process affected the transfection. The morphology of these two NPs was examined. It was revealed that a small-volume process (i.e., with high mixture concentration) resulted in the large particles (denoted as PEI/DNAhigh NPs), compared the large-volume process that led to the small particles (i.e., denoted as PEI/DNAlow NPs), displaying 581 nm vs 89 nm. Both of the PEI/DNAhigh and PEI/DNAlow NPs remained stable in water, HEPES buffer, or the culture medium with FBS (Figure S5A-C). There was no difference between the two NPs in the zeta potential (Figure S5D-F).
The microstructure of the NPs was investigated by atomic force microscopy (AFM). The 2D and 3D AFM images showed that PEI efficiently condensed DNA and form the particle-shaped nanocomplex for both PEI/DNAhigh and PEI/DNAlow (Figure 3A). The section analysis (vertical distance for 2D AFM images) and the particle peak height for 3D images were analyzed by NanoScope Analysis software. Both of the section analysis and the average value of particle peak height confirmed the larger size of the PEI/DNAhigh NPs compared to the PEI/DNAlow NPs (Figure 3B-C).
AFM measurements results. (A) The 2D and 3D AFM images of the NPs. Scale bar, 1 μm. (B) Section analysis of the NPs in the 2D AFM images. (C) The particle peak height of the two groups of NPs.
The internal microstructure of the NPs was observed using the stimulated emission depletion (STED) microscopy technique by dual-labeling (DNA, green; PEI, red). In the PEI/DNAlow NPs, DNA was almost co-localized with PEI, demonstrating the compact condensation, while the PEI/DNAhigh NPs displayed a less compact pattern and a botryoid shape (Figure 4). It suggested that the formulation process via a small-volume mixture that was at a high concentration condition resulted in the weak interaction between DNA and PEI, and the loose structure and large size.
The STED images of the PEI/DNAlow NPs (A) and the PEI/DNAhigh NPs (B). PEI is shown in red, DNA is shown in green.
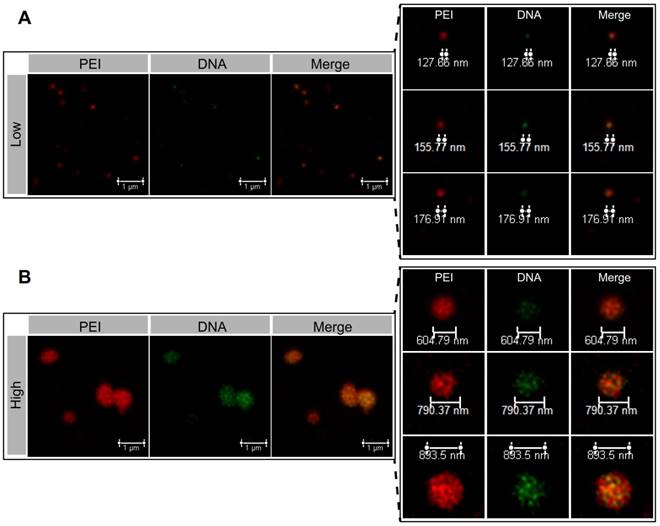
Ionic polymers contain various amount of charges (e.g., -NH3+ in PEI, and -PO4- in DNA), depending on the ionizing degree in aqueous solution. Our results demonstrated that PEI at low concentration possessed high zeta potential and large hydrodynamic size (Figure S6), compared to that at high concentration, indicating the enhanced ionization and extension of PEI polymer chains at low concentration. As a consequence, the polymers (PEI and DNA) at low concentration yielded a stronger interaction due to the adequate exposure the charged segments at each chain. Our conclusion was in accordance with a previous report that the high degree of ionization for the cationic polymer (poly[2- (dimethylamino)ethyl methacrylate] or DMAEMA) at a favorable pH resulted in greater binding affinities with DNA and the compact complexation structure [9].
The structural difference affected the cellular uptake pathway
The cellular uptake pathway was investigated using the total internal reflection fluorescence microscopy (TIRFM) for detecting the single cell tracking at a 100-nm thickness. As shown in Figure 5, Supplementary Video 1, and Figure S7, there was an interesting phenomenon that the PEI/DNAhigh NPs interacted with the pseudopod-like extension of the cell and was then engulfed by the cells. The cellular uptake of the PEI/DNAhigh NPs was analyzed by single-particle tracking (SPT) technique. SPT was a technique using the tracking algorithm to address the principal challenges such as particle motion heterogeneity, high particle density, temporary particle disappearance, and particle splitting. The algorithm first links the particles between consecutive frames, and then links the resulting track segments into complete trajectories [10].
TIRFM analysis. (A)The cellular uptake of the PEI/DNAhigh NPs was divided into three parts. (B) The trajectory analysis of the PEI/DNAhigh NPs, and the amplified trajectory of the bulky NPs. (C) The cellular uptake of the bulky NPs tabbed with the trajectory timely; (D) the normalized lifetime histogram of trajectories; (E) mean square displacement (MSD) analysis of the bulky NP trajectories in and out of the cell in Figure 5D; (F) the trajectory of the bulky NPs is shown as x coordinate, y coordinate and amplitude over time. Scale bar, 20 μm.
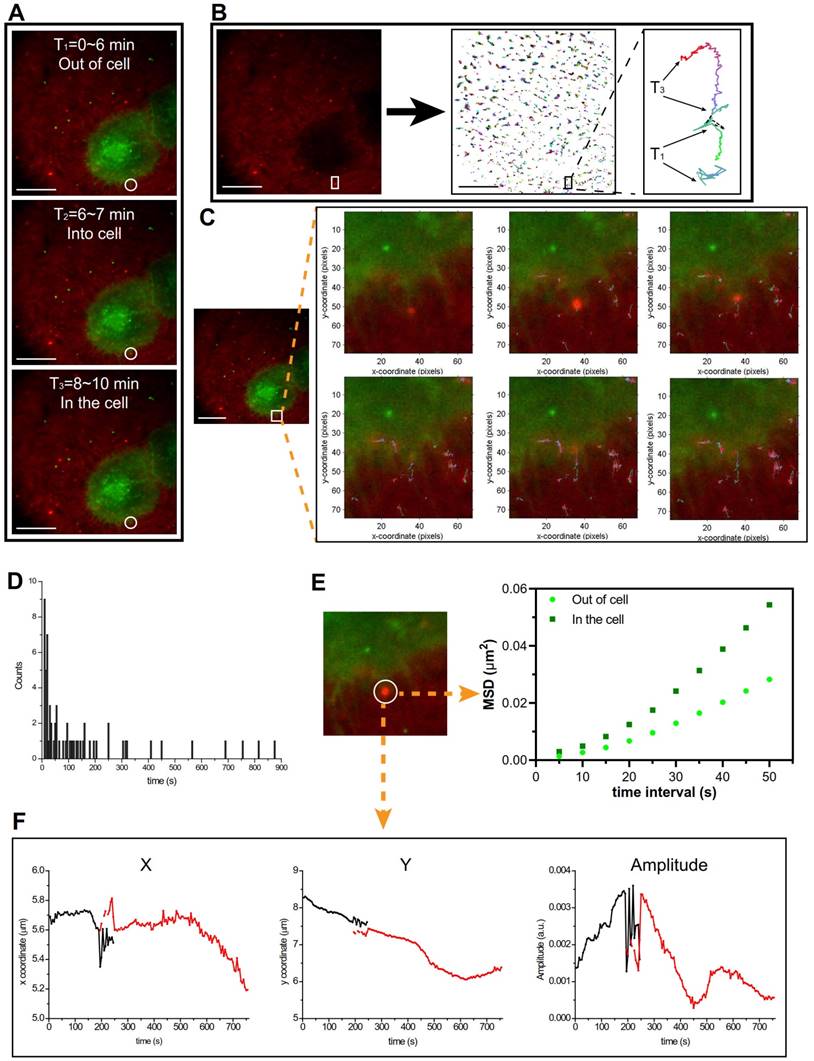
In Figure 5A, the cellular uptake of the PEI/DNAhigh NPs (red) was clearly tracked the process from outside of the cells to enter the cells; and the trajectory of the bulky NPs was shown in amplification (Figure 5B). In Figure 5C (Supplementary Video 2), the cellular uptake of the PEI/DNAhigh NPs was real-time tabbed with the trajectories. The normalized lifetime histogram of trajectories was shown in Figure 5D. The mean square displacement of the PEI/DNAhigh NPs, representing the diffusion coefficient of particles, was analyzed during its movement outside and inside the cell (Figure 5E). It indicated two different movements of the bulky NPs when it was in or out of the cell. The trajectory of the PEI/DNAhigh NPs in Figure 5C was shown as x coordinate, y coordinate and amplitude over time (Figure 5F). And no particle splitting events were found, suggesting there was no disassociation during the cellular uptake. These results indicated that the pseudopod-mediated mechanism could be involved in the cellular uptake of the large PEI/DNAhigh NPs. It has been reported that the pseudopod interaction was a pathway for the cellular uptake of a bacteria [11].
Endocytosis pathway study
To investigate the endocytosis pathway, the cells were pre-treated with various endocytosis inhibitors (chlorpromazine, an inhibitor of clathrin-mediated endocytosis [12]; nystatin, an inhibitor of caveolae- mediated endocytosis [13]; or cytochalasin D, an inhibitor of macropinocytosis) [14], and then conducted the gene transfection experiments. peGFP-N1 was used as the reporter gene. The relative positive rate of transfection and protein expression level in the cells were quantitatively analyzed by FACS.
Chlorpromazine treatment dramatically decreased the relative positive rate of transfection and protein expression level in the PEI/DNAlow NPs group, but no major impact on the PEI/DNAhigh NPs group (Figure 6A, B). It indicated that clathrin-mediated endocytosis was a primary mechanism for the PEI/ DNAlow NPs. By contrast, cytochalasin D treatment led to the remarkable decrease in the relative positive rate of transfection and protein expression level in the PEI/DNAhigh NPs group but yielded minor effect on the PEI/DNAlow NPs group (Figure 6C, D). It revealed macropinocytosis was a main pathway for the PEI/DNAhigh NPs. Nystatin (< 20 µM) yielded very minor inhibition of transfection in both the NPs, suggesting that caveolae-mediated endocytosis was not essential for both NPs (Figure 6E, F).
Effects of gene transfection by endocytosis inhibitors. (A, C, E) The transfection relative positive level. (B, D, F) The protein expression relative positive level.
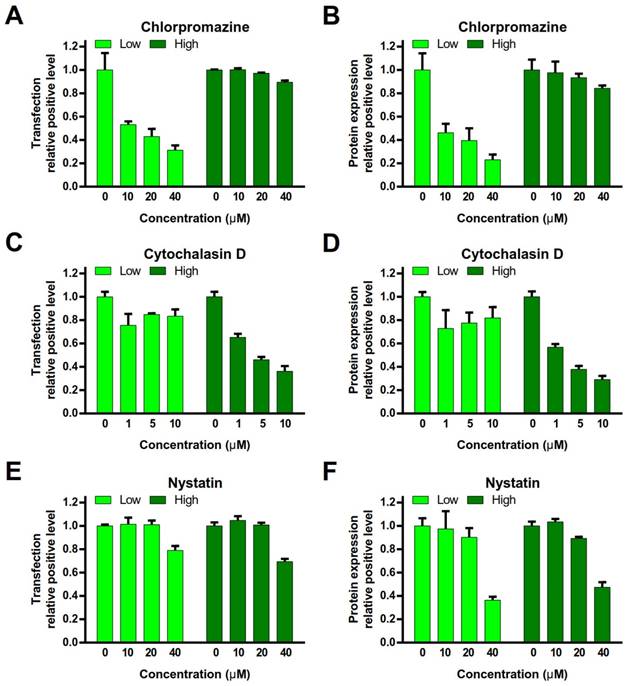
The clathrin-mediated endocytosis pathway for NPs was size-dependent, showing an observed upper limit for internalization of about 200 nm [15]; therefore, the NPs with a size under 200 nm was favorable for the clathrin pathway. However, the clathrin-coated vesicles finally enter the endo/ lysosomes, susceptible to degradation, which was disadvantageous to gene transfection [16, 17]. The macropinosomes may not mature to late endosomes or fuse with lysosomes [18]. Due to the physical characteristics of leakage of macropinosomes, the NPs can easily escape into the cytoplasm, which is helpful for facilitating gene transfection [19, 20]. Moreover, the macropinocytosis can take up the large cargo, which contains a high amount of DNA, and therefore, it is thought that the macropinocytic pathway provides a benefit of increasing uptake [21]. The analysis above provided a possible explanation that the large NPs of PEI/DNAhigh performed higher uptake amount of DNA and more efficient transfection than the PEI/DNAlow NPs.
Subcellular distribution of the two groups of PEI/DNA NPs
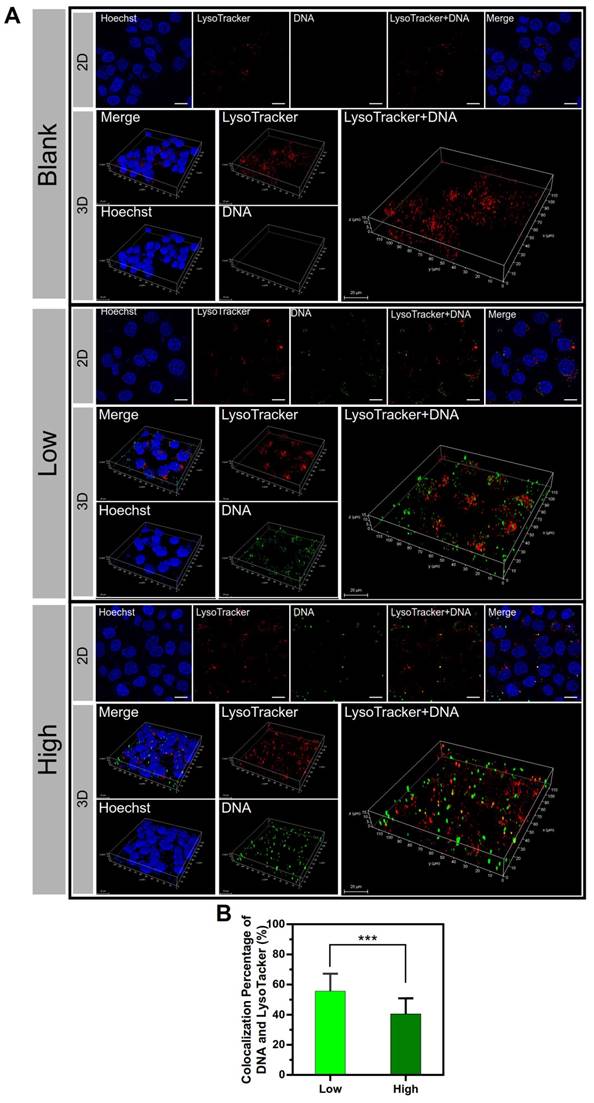
The subcellular distribution of the two groups of PEI/DNA NPs was examined. The lysosome trap is a primary barrier to effective gene delivery [22]. DNA is not only difficult to escape from lysosomes but also readily enzymatically degraded in lysosomes. After a 4-h incubation with the NPs that were labeled with YOYO-1 (green), the cells were observed by confocal laser scanning microscopy (CLSM) followed by LysoTracker Red DND-99 (red) staining. The co- localization of lysosomes and NPs was shown in 2D and 3D confocal images (Figure 7A), and the co-localization percentage was 55.6% for the PEI/ DNAlow NPs and 40.5% for the PEI/DNAhigh NPs, quantitatively analyzed by ImageJ (Figure 7B).
The results of co-localization of the lysosomes and NPs. (A) The 2D and 3D confocal images. DNA is shown in green, lysosomes stained with LysoTracker Red DND-99 are shown in red, and the cell nuclei are shown in blue. (B) The co-localization percentage of the lysosomes and NPs. Scale bar, 20 μm.
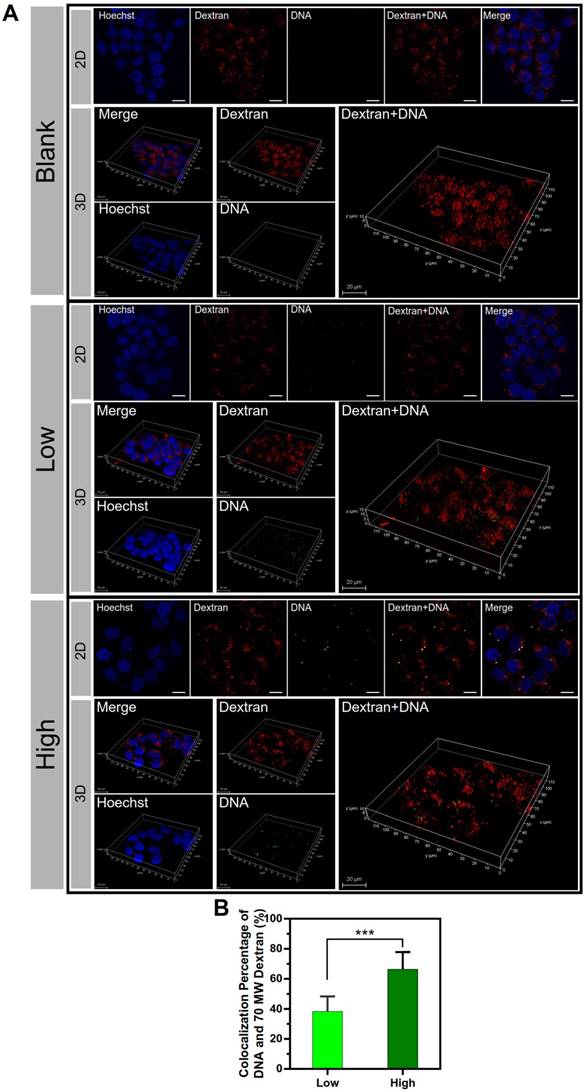
Furthermore, the co-localization of the macropinosomes and the NPs was examined by using the macropinosome probe dextran rhodamine B and 2D and 3D confocal imaging (Figure 8A). The significant co-localization indicated by yellow color was observed in the PEI/DNAhigh NP group. The co-localization percentage was 66.4% for the PEI/DNAhigh NP group, compared to 38.3% for PEI/DNAlow NP group (Figure 8B). The results further demonstrated that the macropinocytosis was the major mechanism for the enhanced transfection efficiency of the large PEI/ DNAhigh NPs.
The results of co-localization of the macropinosomes and NPs. (A) The 2D and 3D confocal images. DNA is shown in green, macropinosomes labeled with Dextran Rhodamine B are shown in red, and the cell nuclei are shown in blue. (B) The co-localization percentage of the macropinosomes and NPs. Scale bar, 20 μm.
Observation of the disassociation of DNA and PEI in cells
As the PEI/DNAhigh NPs displayed a less compact pattern and a botryoid shape, it was thus hypothesized that such structure could facilitate the disassociation of DNA from the complex inside the cells and the subsequent gene transfection.
The disassociation process was observed by fluorescence resonance energy transfer (FRET) technique. DNA and PEI were labeled with Cy3 (green) and Cy5 (red) dye, respectively, which were a pair of donor and acceptor used for FRET. The PEICy5/DNACy3 NPs with different mass ratios were measured using a fluorospectrometer. The results showed that the complexation of PEICy5/DNACy3 resulted in the significantly decreased emission fluorescence intensity of Cy3 and the increased fluorescence intensity of Cy5 (Figure 9A).
Observation of the disassociation of DNA and PEI by FRET. (A) The FRET effect was present in the PEICy5/DNACy3 in varying mass ratios. (B) The 3D confocal images of the disassociation of the two groups of NPs in the cell at 4, 8, and 12 h. PEI was shown in red, DNA was shown in green, the cell nuclei were shown in blue, and the co-localization of PEI and DNA was shown in yellow. Scale bar, 20 μm. (C) Quantitative analysis of the FRET efficiency.
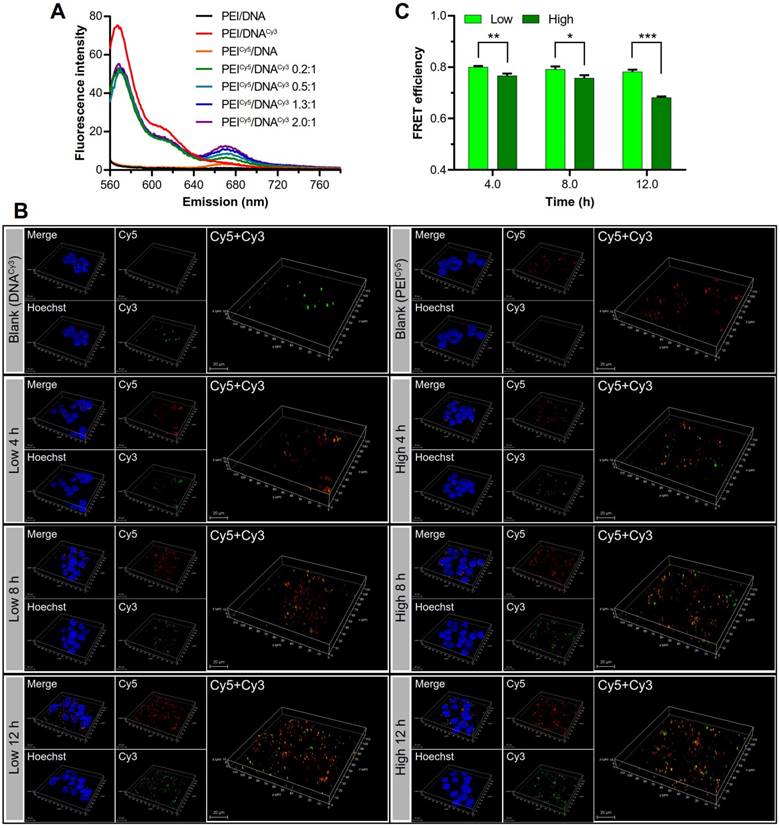
Then the disassociation of the two groups of NPs in the cell was observed by CLSM at 4, 8, and 12 h. Over the time, along with the gradual dissociation of DNACy3 (green) from PEICy5 (red), and the FRET effect weakened. The 3D confocal images (Figure 9B) showed the fluorescence recovery of DNACy3, which was more evident in the PEI/DNAhigh NPs group than in the PEI/DNAlow NPs group, suggesting the higher intracellular disassociation efficiency for the PEI/DNAhigh NPs.
As shown in Figure 9C, the FRET of the two NPs was gradually decreased from 4 to 12 h, indicating that DNA gradually dissociated from PEI in the cells. The FRET (quenched) efficiency of the PEI/DNAhigh NPs was lower than the PEI/DNAlow NPs over the time. For instance, at the 12-h mark, the quenched rate was 68% for the PEI/DNAhigh NPs compared to 78% for the PEI/DNAlow NPs, demonstrating the higher cytoplasmic disassociation of the PEI/DNAhigh NPs.
Nuclear uptake of the PEI/DNA NPs
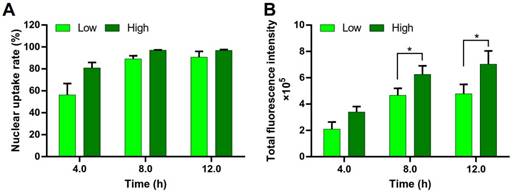
DNA must be delivered into the nucleus to access to the transcriptional machinery. The nuclear uptake of DNA is a key step for transfection. The DNA nuclear uptake percentage of the PEI/DNAhigh NPs was higher than that of the PEI/DNAlow NPs, analyzed by FACS (Figure 10A). Importantly, the DNA nucleic uptake amount, represented by the total fluorescence intensity, was also significantly higher in the PEI/DNAhigh NPs group (Figure 10B). Therefore, the increased intranuclear delivery of DNA was a mechanism underlying its enhanced transfection efficiency of the PEI/DNAhigh NPs group.
Nuclear uptake of DNA in the two groups of NPs. (A) Quantitative analysis of the nuclear uptake rate. (B) The total fluorescence intensity of the nuclei.
The in vitro antitumor activity of the PEI/DNA NPs
The pTRAIL DNA was used as a therapeutic gene in this study. After transfection, the Western blot analysis showed that the expression of TRAIL protein was higher in the large PEI/pTRAILhigh NPs group (Figure 11A). Accordingly, the PEI/pTRAILhigh NPs also showed higher antitumor efficacy in the HeLa cells, compared to the PEI/pTRAILlow NPs (Figure 11B).
(A) Western blot analysis results of TRAIL expression level in HeLa cells after gene transfection of the two groups of PEI/pTRAIL NPs. (B) The results of anti-tumor activity of the two groups of PEI/pTRAIL NPs in vitro.
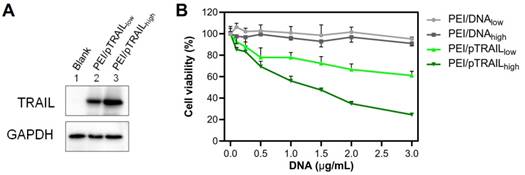
Besides, the materials-associated cytotoxicity of the PEI/DNA NPs was investigated by using an empty plasmid DNA. Both groups of NPs did not show apparent cytotoxicity (cell viability > 90%) at a concentration up to 3 µg/mL, and there was no significant difference between the two groups (Figure 11B).
The in vivo therapeutic effects of the PEI/DNA NPs in a cervical tumor xenograft model
The therapeutic efficacy of the two groups of NPs was investigated in a HeLa cervical tumor xenograft mouse model. Our previous demonstrated that pTRAIL was a potent therapeutic gene in various cancers [23-25]. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) is the key protein inducing apoptosis through action on the death receptors DR4 and DR5 [26].
Both kinds of NPs inhibited tumor growth and prolonged survival time. The PEI/DNAhigh NPs showed a higher inhibition rate of tumor growth of 74.4%, in sharp contrast to 44.8% for the PEI/DNAlow NPs (Figure 12A-D). The animals receiving PEI/ DNAhigh NPs all survived during the treatment.
(A) Tumor volume changes in the treatment regimen (n=8). (B) The presentation of tumor tissues after treatment. (C, D) The tumor weight at the endpoint and the tumor inhibition rate. (E) The survival rate in the treatment regimen. (F) Western blot analysis results of TRAIL expression and DR5 level in tumor tissues. (G) Images of immunohistochemical staining for the examination of TRAIL protein expression. (H) Images of TUNEL-stained tumor sections for evaluating apoptosis. Scale bar, 20 μm.
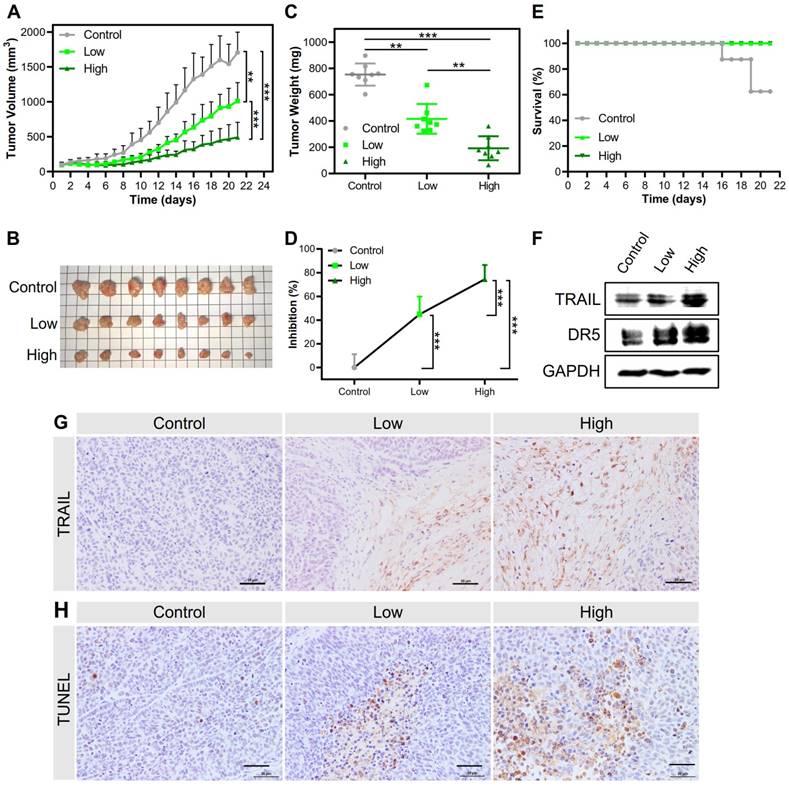
Accordingly, the TRAIL expression in the tumors dissected from the mice treated with the PEI/DNAhigh NPs was higher than that observed in the PEI/DNAlow NPs group, as determined by both Western blotting and immunohistochemistry (Figure 12F-G). Furthermore, the TUNEL assay in the tumors also exhibited significant apoptosis induced by the PEI/DNAhigh NPs (Figure 12H). These results were in good agreement with the results of the tumor inhibition rate.
Interestingly, DR5 upregulation was found in the PEI/DNAhigh NPs (Figure 12F). It was reported that virus transfection can stimulate the upregulation of DR5 expression [27]. It was thus suspected that the PEI-mediated transfection could have a similar effect on DR5 upregulation. Also, the generation of reactive oxygen species (ROS) in the apoptosis process could contribute to the up-regulation of DR5 [28]. We also found the PEI/DNAhigh NPs induced the higher intracellular ROS level than the counterpart (Figure S8).
The side toxicity of the NPs was preliminarily evaluated. During the treatment course, there were no obvious changes in body weight in both NPs groups (Figure S9A). The organ coefficient (i.e., a ratio of the weight of an organ to the body weight) of the mice at the experimental endpoint was not significantly different compared to the control group, except for a slight increase in the liver coefficient in both groups (Figure S9B). Histological examination showed no significant pathological changes (Figure S9C). Although PEI is known for its cytotoxicity, our method that involved a low dose for local application was not found with unwanted toxicities, rendering it potential for translation. Overall, the results indicated that the PEI/DNAhigh NPs had better in vivo therapeutic effects than the PEI/DNAlow NPs due to the enhanced gene transfection efficiency.
The in vivo therapeutic effects of the PEI/DNA NPs in a peritoneal HeLa tumor model
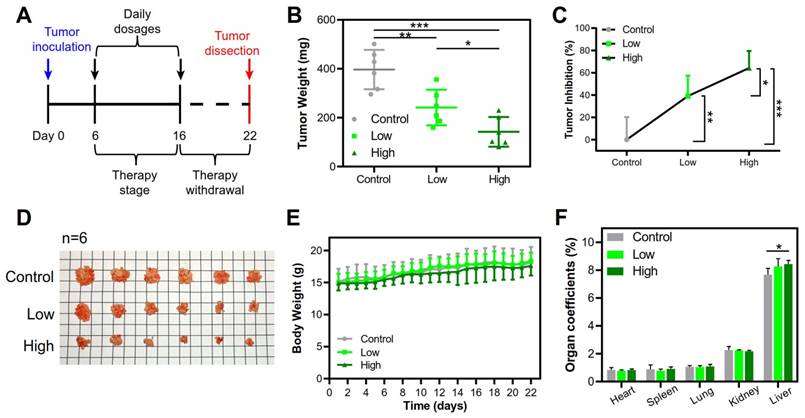
The clinical use of local-regional administration in cancer treatment has also been gradually increased [29]. PEI-based nanomedicines have been used as local-regional gene therapy for patients with bladder, ovarian and pancreatic cancer in clinical trials [30]. The therapeutic potential of the large PEI/pTRAIL NPs was further demonstrated by using a peritoneal HeLa tumor model, a mimic of a late stage of cervical metastasis of peritoneum [31]. Intraperitoneal therapy has been used in peritoneal metastasis for the optimized local drug delivery and improved clinical outcome [32]. Our results revealed that the intraperitoneal administration of both the PEI/pTRAIL NPs inhibited the tumor growth. The PEI/pTRAILhigh NPs showed a high inhibition rate of tumor growth of 64.2%, in contrast to 39.1% for the PEI/pTRAILlow NPs (Figure 13A-D), without obvious side toxicity signs (Figure 13E-F).
Therapeutic efficacy in the peritoneal HeLa tumor model treated with the PEI/pTRAIL NPs. (A) Treatment regime. (B) The average tumor weights. (C) The tumor inhibition rate. (D) The dissected tumor nodules of mice (n=6). (E) The body weight changes over the regimen. (F) The organ coefficients.
Discussion
At the cellular level, non-viral vectors consistently are hindered by numerous extra- and intracellular obstacles, such as cellular uptake, endosomal escape, nuclear entry, transcription and protein expression [22, 33]. This presented work revealed that the formulation process parameters (e.g., the mixture volume or concentration) yielded a significant impact on gene transfection of PEI/DNA NPs. Our work showed the small operation volume (i.e., high reaction concentration) resulted in larger particle size due to the low-degree ionization of the polymers and the consequent less compact interaction.
Though the conventional perception was that the small NPs are superior to the large ones in systemic delivery because the large ones are prone to be cleared away from the bloodstream [34], it is still difficult to reach a conclusion in gene transfection. Size is also a key factor controlling the intracellular fate [35]. For example, the small NPs could have higher cellular uptake efficiency than the large NPs, but the latter could exhibit higher endosome escape capacity and thus yield enhanced gene transfection [36]. However, the mechanisms for enhanced transfection of the large NPs have not been fully elucidated yet. Multiple factors could be involved in the gene transfection, such as physical and biological issues. For example, it was reported that gravitational sedimentation of NPs was an important factor that benefited the large NPs for the in vitro transfection [2].
PEI and DNA are polyelectrolytes, and their interaction can be treated as polyelectrolyte complex. The chain conformations in linear polymers with a uniformly distributed charge reflect a balance among the intramolecular repulsion of charged segments [37]. Therefore, the high ionization results in a flexible linear conformation, while the polymers at low ionization tend to form a curl conformation [38, 39], because of insufficient intramolecular repulsion. Low concentration facilitates the ionization of the charged polymers [40], and the highly ionic and extended polymer chains can readily bind with other polymers with the counterion with strong interaction. Conversely, the polymers with low ionization and curled confirmation tend to form less compact polyelectrolyte complex with the counterion polymers.
The interaction of ionic components can be described using the Coulomb's law:
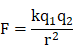
where k is Coulomb's constant (k ≈ 9×109 N m2 C-2), q1 and q2 are the signed magnitudes of the charges, and the scalar r is the distance between the charges. Generally, the increased charges and the reduced distance between the ionic components result in the strong interaction force, forming the compact complex. This can be accounted for the formation of small and compact NP in the low concentration group (PEI/DNAlow) but large NP in the high concentration group.
An interesting finding from our studies was that the cellular uptake percentages (represented by the positive rate) of both NPs were close, but the uptake amount (represented by the total fluorescence intensity) was significantly higher in the PEI/DNAhigh NPs than the PEI/DNAlow NPs. It suggested that the macropinocytosis pathway was more highly efficient to engulf the NPs compared to the clathrin-mediated uptake. Macropinocytosis is associated with the generation of the large endocytic vesicles by driving by the actins and plasma membrane evaginates, thus enabling to engulf a large number of extracellular substances.
Our results indicated that macropinocytosis facilitated a greater amount of NPs into a cell than other endocytosis pathways. Moreover, the large PEI/DNAhigh NPs had higher cytoplasmic dissociation efficiency and led to enhanced intranuclear delivery.
PEI-based vectors have poor systemic delivery efficiency in the blood circulation, which results in low overall delivery efficiency in vivo and hinders the systemic therapy in clinical application [41]. Recently, gene therapy via local-regional administration has obtained great success, representing the first directly administered gene therapy product (Luxturna) approved in the U.S. for hereditary retinal dystrophies via subretinal injection. Therefore, the local-regional administration provides a promising strategy for bypassing the inherent shortcomings of PEI.
Conclusions
It was first revealed that the formation of PEI/DNA NPs was significantly affected by the reaction concentration. The PEI and DNA that complexed at a high concentration tended to form a large and less compact NPs, compared to that at a low concentration. As a result, the large PEI/DNAhigh NPs performed the enhanced gene transfection via the mechanisms of macropinocytosis-mediated cellular uptake, effective dissociation and DNA release, and the enhanced intranuclear delivery. By contrast, the PEI and DNA that complexed at a low concentration tended to form a small and compact NPs, which preferred the clathrin-mediated endocytosis pathway and confronted the endosome barrier against effective cytoplasmic delivery. These findings shed light on the importance of formulation process optimization (e.g., reaction concentration) that has long since been ignored, and thus provide a better understanding of the formulation process effect on nano-structural properties and gene transfection, laying a theoretical basis for rational design of the experimental process.
Materials and Methods
Materials
Linear PEI25K was purchased from Polysciences (USA). Chlorpromazine, nystatin, and cytochalasin D were purchased from Sigma-Aldrich (USA). Nuc-Cyto-Mem Preparation Kit was purchased from Applygen Technologies (Beijing, China). The plasmids of peGFP-N1 encoding enhanced green fluorescent protein, pRFP encoding red fluorescent protein, and pGL4.13 encoding luciferase were propagated in a DH5-strain of E. coli and purified by using a QIAGEN Plasmid Maxi Kit (Germany). YOYO-1, LysoTracker Red DND-99, DIO, dextran rhodamine B (MW 70 kDa) were purchased from Invitrogen (USA). The 5-TAMRA-SE was purchased from MedChem Express (USA). Fetal bovine serum (FBS) and RPMI 1640 medium were purchased from Thermo Fisher Scientific (USA). Hoechst 33342 Staining Solution for Live Cells, DAPI staining solution, RIPA lysis buffer, 0.25% trypsin-EDTA, BCA microplate protein assay kit, and ROS detection kit were obtained from Beyotime Institute of Biotechnology (Haimen, China). Luciferase Assay System Kit was purchased from Promega (USA). Anti-DR5, anti- TRAIL, and anti-GAPDH were purchased from Cell Signaling Technology (USA). All of the other reagents were of analytical grade and purchased from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). Branch PEI25K was purchased from Sigma-Aldrich (USA).
Cell culture and animals
Human cervical cancer HeLa cells, MCF-7 cells, H/T cells, and A549T cells were obtained from the Cell Bank of Chinese Academy of Sciences (Shanghai, China). DC2.4 cells, a murine dendritic cell line [42], were kindly provided by Prof. Ying Hu (Wenzhou Medical University, Zhejiang, China). The cells were cultured in Gibco® RPMI 1640 medium containing 10% FBS, 100 μg/mL streptomycin, and 100 U/mL penicillin in a humidified atmosphere at 37°C and 5% CO2. The female BALB/c nude mice (3-4 weeks old) were purchased from SLAC Animal Inc. (Shanghai, China) and housed in specific pathogen-free conditions. The in vivo experimental procedures were approved by the Animal Care and Use Committee of Shanghai Institute of Materia Medica.
Preparation of two types of the PEI/DNA NPs
Two types of the PEI/DNA NPs were prepared using the same procedure and the same amount of PEI and DNA in pure water but at different concentrations. The high-concentration group (PEI/ DNAhigh) was prepared using 0.2 mg/mL of plasmid DNA and PEI (equal to a total 20 μL), while the low-concentration group (PEI/DNAlow) using 0.02 mg/mL of DNA and PEI (equal to a total 200 μL). In specific, the PEI aqueous solution was added to the equal volume of DNA aqueous solution (1.3:1, w/w), and after gentle mixing, the mixture was incubated at 37°C for 30 min. The NPs were freshly prepared before use.
Particle size and zeta potential analysis
The particle size and zeta potential of the PEI/DNA NPs were measured in water or serum-containing medium using the ZetaSizer Nano ZS90 Size Analyzer (Malvern Panalytical, Brighton, UK), and the stability of the NPs was evaluated by detecting the particle size change at varying time points. The final concentration was diluted to equally 3 μg/mL DNA with the final volume 1 mL for particle size and zeta potential analysis of each formulation.
Transfection experiments
HeLa cells were seeded in the 12-well plates at a density of 1 × 105 cells per well. After 20-h culture, the cells were treated with the same amount (1 μg DNA per well) of the PEI/DNA NPs dispersed in the fresh RPMI 1640 medium without FBS for 4 h at 37 °C, and then cultured with the fresh completed medium for 24 h to allow gene expression. The plasmid DNA reporter (peGFP-N1, pRFP, or pGL4.13) was used in the transfection studies. The transfection efficiency of peGFP-N1 or pRFP was measured using a flow cytometer (BD Pharmingen, San Jose, CA, USA), and data analysis was performed using FlowJo software. For the transfection efficiency assay of pGL4.13, the cells were harvested and treated using 200 μL of the RIPA lysis buffer. After centrifugation at 12,000 g for 20 min, the luciferase activity of the supernatants was detected using the Luciferase Assay Kit, and luminescence was measured with the EnSpire Multimode Plate Reader (PerkinElmer, Waltham, MA, USA). Luminescence was normalized to the protein concentration of each sample, which was measured using the BCA Microplate Protein Assay Kit. Luciferase activity was designated as RLU/mg protein. Moreover, DC2.4 cells, BMDCs, MCF-7 cells, H/T cells and A549T cells were also performed the transfection experiment as the HeLa cells.
Cellular uptake of the two groups of PEI/DNA NPs
The plasmid DNA was labeled with YOYO-1 dye. The HeLa cells were seeded into the 12-well plates at a density of 1 × 105 cells per well. After 20-h culture, they were treated with the same amount of the PEI/DNA NPs in 1 mL fresh RPMI 1640 medium without FBS for 4 h at 37 °C. The cells were thoroughly washed with PBS and fixed in 4% paraformaldehyde. After staining with DAPI, the cells were observed by CLSM (TCS-SP8 STED; Leica Microsystems, Mannheim, Germany). For quantitative measurement, the cells were thoroughly washed and collected, and then analyzed with a flow cytometer using the FL1 channel. Moreover, the cellular uptake experiments were also performed in DC2.4 cells and BMDCs.
Observation of cellular uptake
The PEI/DNA NPs were labeled with 5-TAMRA-SE dye. The HeLa cells were seeded into the glass plates. After 20-h culture, the cell membrane was labeled with DIO dye and then treated with the same amount of the PEI/DNA NPs in 1 mL fresh RPMI 1640 medium without FBS. The living cells were immediately observed by TIRFM (Olympus, Japan). The fluorescence images were captured every 5 s, and the nanoparticles were analyzed by single-particle tracking (SPT) technique programmed by MATLAB software.
Influence of polyelectrolyte concentrations on the structure of PEI chains via zeta potential and hydrodynamic diameter analysis
Zeta potential and hydrodynamic diameter of the NPs were measured to investigate the structure of PEI chains at various polyelectrolyte concentrations. In brief, the PEI aqueous solution was prepared at different concentrations (0.02, 0.026, 0.05, 0.1, 0.2, 0.26, 0.5, and 1.0 mg/mL). The zeta potential and hydrodynamic diameter of the PEI molecules were measured using the ZetaSizer Nano ZS90 Size Analyzer (Malvern Panalytical Ltd, UK). In addition, the zeta potential and hydrodynamic diameter of the PEI/DNAlow NPs (CPEI = 0.026 mg/mL) and the PEI/DNAhigh NPs (CPEI = 0.26 mg/mL), were also measured.
Analysis of the effects of gene transfection by endocytosis inhibitors
The plasmid peGFP-N1 was used as a reporter gene. The HeLa cells were seeded in the 12-well plates at a density of 1 × 105 cells per well. After 20-h culture, the cells were treated with various endocytosis inhibitors for 1 h (chlorpromazine at 10 μM, 20 μM, or 40 μM; nystatin at 10 μM, 20 μM, or 40 μM and cytochalasin D at 1 μM, 5 μM, or 10 μM). The cells were then treated with the same amount of PEI/DNAlow or PEI/DNAhigh NPs in the fresh RPMI 1640 medium without FBS for 4 h at 37 °C. After 4-h treatment of the NPs, the cells were cultured with the fresh complete RPMI 1640 medium for 24 h to allow gene expression. The fluorescence images were observed by fluorescence microscope (Zeiss, Germany). Meanwhile, the cells were harvested and analyzed with a flow cytometer.
Observation of intracellular distribution of the NPs
The plasmid DNA was labeled with the YOYO-1 dye. The HeLa cells were seeded into the glass plates. After 20-h culture, the cell nuclei were stained with Hoechst 33342. The cells were then treated with the NPs for 4 h at 37 °C, and at the mark of 2 h during the cellular uptake experiment, a macropinosome dye (dextran rhodamine B, 70 kDa) was added to the culture medium with another 2-h incubation with the cells. The cells were stained with LysoTracker Red DND-99 for 30 min. The cells were thoroughly washed with PBS, fixed in 4% paraformaldehyde, and observed by CLSM. The co-localization percentage was analyzed by ImageJ software.
Atomic force microscopy (AFM) observation
The PEI/DNA NPs were added to the surface of freshly cleaved mica for 20 min. The mica surface was then gently rinsed with sterile water for 10 times, followed by AFM observation (Bruker Scientific Instruments, Bremen, Germany) in the peakforce mode using ScanAsyst-Fluid+ probes with standard silicon tips (Bruker). The PEI solution was used as the control group. AFM images were analyzed using NanoScope Analysis software. The average value of particle peak height was a result of counting 589 particles in the PEI/DNAhigh group and 2379 particles in the PEI/DNAlow group.
Stimulated emission depletion (STED) microscopy
The plasmid DNA was labeled with YOYO-1 dye, and PEI was labeled with 5-TAMRA-SE dye. The PEI/DNA NPs were added to the surface of freshly cleaved mica and incubated for 20 min. The mica surface was gently rinsed with distilled water 10 times and dried at room temperature. The samples were observed by CLSM operating in the STED mode.
Fluorescence resonance energy transfer (FRET) effect
PEI was chemically labeled with Cy5 NHS ester, and the plasmid DNA was chemically labeled with Cy3 dye. PEICy5 and DNACy3 were mixed at the same volume but at different mass ratios (0.2:1, 0.5:1, 1.3:1, 2:1). The mixtures were incubated at 37 °C for 30 min. The observation of FRET effect was performed by measuring the emission fluorescence intensity of Cy3 at 547 nm using a fluorospectrometer (F4600; Hitachi, Japan). The HeLa cells were seeded into the glass slices at a density of 1 × 105 cells per well. After 20-h culture, the cell nuclei were stained with Hoechst 33342 Staining Solution for Live Cells for 10 min. The cells were then treated with the same amount of the PEICy5/DNACy3 NPs in the fresh RPMI 1640 medium without FBS for 4 h at 37 °C. To investigate the disassociation of PEI and DNA, the cells were thoroughly washed with PBS and fixed in 4% paraformaldehyde at the time points of 4, 8, and 12 h, followed by CLSM observation. For quantitative analysis of the FRET efficiency, the cells were analyzed by flow cytometry. The PEI/DNA and PEI/DNACy3 NPs were set as the unlabeled and donor-labeled controls.
The FRET efficiency of the two groups of NPs in the cell was quantitatively analyzed by flow cytometry [43]. The FRET efficiency (E) was calculated according to the following equation:
,
where I0 was the fluorescence intensity of the unlabeled sample, ID was the fluorescence intensity of the donor-labeled sample, and IDA was that of the double-labeled sample.
Nuclear transport of the two groups of PEI/DNA NPs
The plasmid DNA was chemically labeled with Cy3 dye. The HeLa cells were seeded into the 12-well plates at a density of 1 × 105 cells per well. After 20-h culture, they were treated with the same amount of PEI/DNA NPs in the fresh RPMI 1640 medium without FBS for 4 h at 37 °C, and then cultured with the fresh complete RPMI 1640 medium. The cells were thoroughly washed with PBS and harvested at 4, 8, and 12 h time points. The cell nuclei were isolated using the Nuc-Cyto-Mem Preparation Kit and analyzed using flow cytometry.
In vitro transfection and cytotoxicity assay of PEI/pTRAIL NPs
As a therapeutic gene, pTRAIL was used to investigate the in vitro treatment efficacy. The HeLa cells were seeded in the 12-well plates at a density of 1 × 105 cells per well. After 20-h culture, the cells were treated with the same amount (1 μg pTRAIL per well) of the PEI/pTRAIL NPs dispersed in the fresh RPMI 1640 medium for 4 h at 37 °C, and then cultured with the fresh completed medium for 48 h to allow gene expression. After transfection, the cells were harvested for Western blot analysis to detect the TRAIL expression.
For the in vitro cytotoxicity assay, the HeLa cells were seeded in the 96-well plates. For the control groups, the PEI/pDNA NPs were prepared by using empty plasmid. The cells were treated with the NPs dispersed in the fresh RPMI 1640 medium without FBS for 4 h at 37 °C, and then cultured with the fresh completed medium for 48 h. Cytotoxicity was then measured by a standard MTT assay. Briefly, MTT solution (20 μL, 5 mg/mL) was added to each well, after which the cells were incubated for another 4 h. After removal of the medium, 200 μL of DMSO was added to each well, and the absorbance was measured at 490 nm using a microplate reader (Thermo Fisher Scientific, Waltham, MA, USA). The cell viability (%) was calculated according to the following equation:
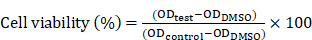
Measurement of intracellular ROS
After 24-h transfection of HeLa cells with the PEI/pTRAIL NPs as the procedure described above, the cells were harvested and stained using a ROS detection kit according to the manufacturer's protocol. The samples were analyzed by flow cytometry.
In vivo antitumor experiments with local administration
The subcutaneous tumor xenograft model was developed by s.c. injection of the HeLa cells (4 × 106 cells/mouse). When the tumor volume reached about 100 nm3, the mice were daily treated with PBS, PEI/pTRAILlow, and PEI/pTRAILhigh NPs via intratumoral administration at a dose of 50 µg/kg of pTRAIL per mouse (eight mice per group), around the 1/10 dose of the commonly used dose of i.v. injection [23, 25]. After injection, the pinhole was sealed by medical glue to accelerate the healing. The body weight and tumor size (measured with a caliper) were recorded. The estimated tumor volume was calculated as follows:
,
where a is the shortest diameter and b is the longest diameter.
At the experimental endpoint, the mice were sacrificed and the major organs and tumors were harvested to measure the organ coefficient and tumor weight. The organ coefficient was calculated by comparing each organ weight between a treatment group and the PBS control group, and the tumor inhibition rate was calculated by comparing the tumor weight between a treatment group and the PBS control group. The organs were fixed in 4% paraformaldehyde for histological examination by hematoxylin and eosin staining. Immunohistochemical staining of the tumor slices was performed to observe TRAIL expression, and Western blot analysis was conducted to detect the TRAIL and DR5 expression. The TUNEL assay was performed to detect apoptosis.
In vivo gene therapy experiments with regional administration
Abdominally disseminated cervical tumor model was established by i.p. inoculation of the HeLa cells (4 × 106 cells/mouse) into the nude mice. Six days after inoculation, the mice were treated with water, PEI/pTRAILlow, and PEI/pTRAILhigh NPs via i.p. injection at a daily dose of 0.25 mg/kg of pTRAIL per mouse (six mice per group). After eleven times treatment, the mice received no further additional treatment for another six days. The body weight was detected every day. At the experimental endpoint, the mice were sacrificed and the major organs and tumors in the peritoneal cavity were carefully harvested to measure the organ coefficient and tumor weight.
Statistical analysis
All quantitative data are presented as the mean ± standard deviation (SD), all of which were repeated at least three times. Statistical significance was analyzed with the Student's t-test. P values less than 0.05 were considered statistically significant.
Acknowledgements
We are thankful for the support from 973 Program, China (2014CB931900) and NFSC, China (81673382, 81402883, 81422048, and 81521005), the Strategic Priority Research Program of CAS (XDA12050307), National Special Project for Significant New Drugs Development (2018ZX09711002-010-002), CAS Scientific Research and Equipment Project (YZ201437), SANOFI-SIBS Scholarship Program, Youth Innovation Promotion Association of CAS, and the Fudan-SIMM Joint Research Fund (FU-SIMM20174009). We also thank the technical assistance from the Molecular Imaging and Electron Microscopy Core Facilities, SIMM.
Supplementary Material
Supplementary figures and tables.
Supplementary Video.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ogris M, Steinlein P, Kursa M, Mechtler K, Kircheis R, Wagner E. The size of DNA/transferrin-PEI complexes is an important factor for gene expression in cultured cells. Gene Ther. 1998;5:1425-33
2. Pezzoli D, Giupponi E, Mantovani D, Candiani G. Size matters for in vitro gene delivery: investigating the relationships among complexation protocol, transfection medium, size and sedimentation. Sci Rep. 2017;7:44134
3. Pezzoli D, Olimpieri F, Malloggi C, Bertini S, Volonterio A, Candiani G. Chitosan-Graft-Branched Polyethylenimine Copolymers: Influence of Degree of Grafting on Transfection Behavior. PloS One. 2012;7:e34711
4. Malloggi C, Pezzoli D, Magagnin L, De Nardo L, Mantovani D, Tallarita E. et al. Comparative evaluation and optimization of off-the-shelf cationic polymers for gene delivery purposes. Polym Chem. 2015;6:6325-39
5. Lucotti A, Tommasini M, Pezzoli D, Candiani G. Molecular interactions of DNA with transfectants: a study based on infrared spectroscopy and quantum chemistry as aids to fluorescence spectroscopy and dynamic light scattering analyses. RSC Adv. 2014;4:49620-7
6. Pack DW, Hoffman AS, Pun S, Stayton PS. Design and development of polymers for gene delivery. Nat Rev Drug Discov. 2005;4:581-93
7. Godbey WT, Wu KK, Mikos AG. Poly(ethylenimine) and its role in gene delivery. J Control Release. 1999;60:149-60
8. Kunath K, von Harpe A, Fischer D, Peterson H, Bickel U, Voigt K. et al. Low-molecular-weight polyethylenimine as a non-viral vector for DNA delivery: comparison of physicochemical properties, transfection efficiency and in vivo distribution with high-molecular-weight polyethylenimine. J Control Release. 2003;89:113-25
9. Rungsardthong U, Ehtezazi T, Bailey L, Armes SP, Garnett MC, Stolnik S. Effect of polymer ionization on the interaction with DNA in nonviral gene delivery systems. Biomacromolecules. 2003;4:683-90
10. Jaqaman K, Loerke D, Mettlen M, Kuwata H, Grinstein S, Schmid SL. et al. Robust single-particle tracking in live-cell time-lapse sequences. Nat Methods. 2008;5:695-702
11. Clemens DL, Lee BY, Horwitz MA. Francisella tularensis enters macrophages via a novel process involving pseudopod loops. Infect Immun. 2005;73:5892-902
12. Rejman J, Bragonzi A, Conese M. Role of clathrin- and caveolae-mediated endocytosis in gene transfer mediated by lipo- and polyplexes. Mol Ther. 2005;12:468-74
13. Iwabuchi K, Handa K, Hakomori S. Separation of "glycosphingolipid signaling domain" from caveolin-containing membrane fraction in mouse melanoma B16 cells and its role in cell adhesion coupled with signaling. J Biol Chem. 1998;273:33766-73
14. Kruth HS, Jones NL, Huang W, Zhao B, Ishii I, Chang J. et al. Macropinocytosis is the endocytic pathway that mediates macrophage foam cell formation with native low density lipoprotein. J Biol Chem. 2005;280:2352-60
15. Rejman J, Oberle V, Zuhorn IS, Hoekstra D. Size-dependent internalization of particles via the pathways of clathrin- and caveolae-mediated endocytosis. Biochem J. 2004;377:159-69
16. McMahon HT, Boucrot E. Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nat Rev Mol Cell Biol. 2011;12:517-33
17. El-Sayed A, Harashima H. Endocytosis of gene delivery vectors: from clathrin-dependent to lipid raft-mediated endocytosis. Mol Ther. 2013;21:1118-30
18. Hamasaki M, Araki N, Hatae T. Association of early endosomal autoantigen 1 with macropinocytosis in EGF-stimulated A431 cells. Anat Rec A Discov Mol Cell Evol Biol. 2004;277:298-306
19. Love KT, Mahon KP, Levins CG, Whitehead KA, Querbes W, Dorkin JR. et al. Lipid-like materials for low-dose, in vivo gene silencing. Proc Natl Acad Sci USA. 2010;107:1864-9
20. Amyere M, Mettlen M, Van DS, Platek A. Origin, originality, functions, subversions and molecular singalling of macropinocytosis. Int J Med Microbiol. 2002;291:487-94
21. Khalil IA, Kogure K, Akita H, Harashima H. Uptake pathways and subsequent intracellular trafficking in nonviral gene delivery. Pharmacol Rev. 2006;58:32-45
22. Mintzer MA, Simanek EE. Nonviral Vectors for Gene Delivery. Chem Rev. 2009;109:259-302
23. Pan Z, Kang X, Zeng Y, Zhang W, Peng H, Wang J. et al. A mannosylated PEI-CPP hybrid for TRAIL gene targeting delivery for colorectal cancer therapy. Polym Chem. 2017;8:5275-85
24. Tan J, Wang HY, Xu F, Chen YZ, Zhang M, Peng HG. et al. Poly-gamma-glutamic acid-based GGT-targeting and surface camouflage strategy for improving cervical cancer gene therapy. J Mater Chem B. 2017;5:1315-27
25. Xu F, Zhong H, Chang Y, Li D, Jin H, Zhang M. et al. Targeting death receptors for drug-resistant cancer therapy: Codelivery of pTRAIL and monensin using dual-targeting and stimuli-responsive self-assembling nanocomposites. Biomaterials. 2018;158:56-73
26. Wang SL, El-Deiry WS. TRAIL and apoptosis induction by TNF-family death receptors. Oncogene. 2003;22:8628-33
27. Jang JY, Kim SJ, Cho EK, Jeong SW, Park EJ, Lee WC. et al. TRAIL enhances apoptosis of human hepatocellular carcinoma cells sensitized by hepatitis C virus infection: therapeutic implications. PLoS One. 2014;9:e98171
28. Park EJ, Min KJ, Choi KS, Kubatka P, Kruzliak P, Kim DE. et al. Chloroquine enhances TRAIL-mediated apoptosis through up-regulation of DR5 by stabilization of mRNA and protein in cancer cells. Sci Rep. 2016;6:22921
29. Budker VG, Monahan SD, Subbotin VM. Loco-regional cancer drug therapy: present approaches and rapidly reversible hydrophobization (RRH) of therapeutic agents as the future direction. Drug Discov Today. 2014;19:1855-70
30. Chen J, Guo Z, Tian H, Chen X. Production and clinical development of nanoparticles for gene delivery. Mol Ther Methods Clin Dev. 2016;3:16023
31. Qiu N, Liu X, Zhong Y, Zhou Z, Piao Y, Miao L. et al. Esterase-Activated Charge-Reversal Polymer for Fibroblast-Exempt Cancer Gene Therapy. Adv Mater. 2016;28:10613-22
32. Odendahl K, Solass W, Demtroder C, Giger-Pabst U, Zieren J, Tempfer C. et al. Quality of life of patients with end-stage peritoneal metastasis treated with Pressurized IntraPeritoneal Aerosol Chemotherapy (PIPAC). Eur J Surg Oncol. 2015;41:1379-85
33. Yin H, Kanasty RL, Eltoukhy AA, Vegas AJ, Dorkin JR, Anderson DG. Non-viral vectors for gene-based therapy. Nat Rev Genet. 2014;15:541-55
34. Zhou Z, Liu X, Zhu D, Wang Y, Zhang Z, Zhou X. et al. Nonviral cancer gene therapy: Delivery cascade and vector nanoproperty integration. Adv Drug Deliv Rev. 2017;115:115-54
35. Behzadi S, Serpooshan V, Tao W, Hamaly MA, Alkawareek MY, Dreaden EC. et al. Cellular uptake of nanoparticles: journey inside the cell. Chem Soc Rev. 2017;46:4218-44
36. Zhu D, Yan H, Zhou Z, Tang J, Liu X, Hartmann R. et al. Detailed investigation on how the protein corona modulates the physicochemical properties and gene delivery of polyethylenimine (PEI) polyplexes. Biomater Sci. 2018;6:1800-17
37. Chremos A, Douglas JF. Influence of higher valent ions on flexible polyelectrolyte stiffness and counter-ion distribution. J Chem Phys. 2016;144:164904
38. Dobrynin AV, Rubinstein M. Theory of polyelectrolytes in solutions and at surfaces. Prog Polym Sci. 2005;30:1049-118
39. Stevens MJ, Kremer K. Form-Factor of Salt-Free Linear Polyelectrolytes. Macromolecules. 1993;26:4717-9
40. Tsuchida E. Formation of Polyelectrolyte Complexes and Their Structures. JMS-Pure Appl Chem. 1994 A31: 1-15
41. Zhou ZX, Liu XR, Zhu DC, Wang Y, Zhang Z, Zhou XF. et al. Nonviral cancer gene therapy: Delivery cascade and vector nanoproperty integration. Adv Drug Deliver Rev. 2017;115:115-54
42. Shen ZH, Reznikoff G, Dranoff G, Rock KL. Cloned dendritic cells can present exogenous antigens on both MHC class I and class II molecules. J Immunol. 1997;158:2723-30
43. Nagy P, Vereb G, Damjanovich S, Matyus L, Szollosi J. Measuring FRET in flow cytometry and microscopy. Curr Protoc Cytom. 2006 Chapter 12: Unit12 8
Author contact
![]() Corresponding author: Yongzhuo Huang, Ph.D., Professor of Pharmaceutics, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, 501 Haike Rd, Shanghai 201203, China. Tel/Fax: +86-21-2023-1981; Email: yzhuangac.cn
Corresponding author: Yongzhuo Huang, Ph.D., Professor of Pharmaceutics, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, 501 Haike Rd, Shanghai 201203, China. Tel/Fax: +86-21-2023-1981; Email: yzhuangac.cn