Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Conclusion
Materials and methods
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(3):884-899. doi:10.7150/thno.29445 This issue Cite
Research Paper
Nanoparticle Binding to Urokinase Receptor on Cancer Cell Surface Triggers Nanoparticle Disintegration and Cargo Release
Shijie Li1,2, Cai Yuan1, Jincan Chen2, Dan Chen1, Zhuo Chen2, Wenlie Chen3, Shufeng Yan2, Ping Hu2, Jinping Xue1, Rui Li2, Ke Zheng1 ![]() , Mingdong Huang1
, Mingdong Huang1 ![]()
1. Fuzhou University, Fuzhou, Fujian 350116, China.
2. State Key Laboratory of Structure Chemistry, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou, Fujian 350002, China.
3. Fujian Academy of Integrative Medicine, Fujian University of Traditional Chinese Medicine, Fuzhou, Fujian 350122, China.
Received 2018-8-23; Accepted 2018-12-31; Published 2019-1-25
Abstract

Cancer cell expresses abundant surface receptors. These receptors are important targets for cancer treatment and imaging applications. Our goal here is to develop nanoparticles with cargo loading and tumor targeting capability.
Methods: A peptide targeting at cancer cell surface receptor (urokinase receptor, uPAR) was expressed in fusion with albumin (diameter of ~7 nm), and the fusion protein was assembled into nanoparticles with diameter of 40 nm, either in the presence or absence of cargo molecules, by a novel preparation method. An important feature of this method is that the nanoparticles were stabilized by hydrophobic interaction of the fusion protein and no covalent linking agent was used in the preparation. The stability, the cargo release, in vitro and in vivo properties of such formed nanoparticles were characterized by transmission electron microscopy, dynamic light scattering, gel shift assay, laser scanning confocal microscopy and 3D fluorescent molecular tomography.
Results: The nanoparticles were stable for more than two weeks in aqueous buffer, even in the buffer containing 10% fetal bovine serum. Interestingly, in the presence of urokinase receptor, the uPAR-targeting nanoparticle disintegrated into 7.5 nm fragments and released its cargo, but not the non-targeting nanoparticles made from albumin by the same preparation method. Such nanoparticles also showed higher uptake and cytotoxicity to the receptor-expressing cancer cells in vitro and higher tumor accumulation in xenografted tumor-bearing mice in vivo compared to the non-targeting nanoparticles.
Conclusion: Our results demonstrate a new function of cell surface receptor as a responsive trigger to disassemble nanoparticles, besides its common use to enrich targeting agents. Such nanoparticles were thus named receptor-responsive nanoparticles (RRNP).
Keywords: receptor-triggered disintegration, cargo release, urokinase receptor, amino terminal fragment of urokinase-type plasminogen activator, human serum albumin
Introduction
Nanoparticle has strong potential in biomedical applications in, for example, early detection of diseases [1] or delivery of drugs to tumor lesion site [2-8]. Nanoparticles can protect drug from degradation during distribution phase, control the accumulation and release of pharmacologically active agents at the pathological site, increase drug efficacy and reduce intensity of side effects by reducing their buildup in healthy tissues [9-11]. Over the last years, numerous theranostic nanoparticle formulations have been designed and evaluated, including liposomes, polymers, protein nanoparticles, gold or iron oxide nanoparticles [9, 12-14]. There are a number of successful examples of nanoparticles in clinical applications including Abraxane® and Doxil®. Abraxane® is albumin-bound paclitaxel nanoparticle with an approximate diameter of 130 nm and was approved by the FDA for the treatment of breast cancer, lung cancer and other solid tumors [15]. Doxil® is a polyethylene glycol coated liposome-encapsulated doxorubicin (DOX), and was approved by the FDA for treatment of AIDS-related Kaposi's sarcoma, ovarian cancer and multiple myeloma [16, 17].
A unique advantage of nanoparticle is its capability to integrate together multiple functions, e.g. tumor therapy, imaging for tumor location, and control cargo release in response to various stimuli. The stimuli include pH [18, 19], redox potential [20], or enzymatic activity [21, 22]. Here, we demonstrate that tumor surface receptor itself can be an important stimulus to trigger the disintegration of the nanoparticles besides its typical role in promoting the specific accumulation of targeting nanoparticles to tumor. Such new function of receptor targeting nanoparticles is possible because the nanoparticles are held together by non-covalent interaction, and at the same time, maintain high stability.
Cancer cell surface expresses a range of receptors different from normal cells (a complete list of receptors can be found in recent review [23]). Commonly used cancer cell surface receptors include epidermal growth factor receptor (EGFR) [24], transferrin receptor [25], folate receptor [26], integrins [27] and urokinase-type plasminogen activator (uPA) receptor (uPAR) [28-31]. In this study, we choose uPAR as the target receptor. uPAR is greatly over-expressed on the surface of invasive cancer cells, in invasive borders and in the cancer associated stromal cells. uPAR plays a critical role in cancer cell migration, adhesion, and has been recognized as a promising candidate receptor in cancer targeting therapy and/or imaging [32-34]. A peptidyl inhibitor of uPAR was used successfully in clinical trial for nuclear imaging of primary tumors and lymph node metastases in patients [35]. Previously, we developed an uPAR-targeting agent, which is a fusion protein of human serum albumin (HSA) with amino-terminal fragment (ATF) of urokinase-type plasminogen activator (uPA) [36] and binds to uPAR at very high potency (sub-nanomolar dissociation constant). ATF-HSA was demonstrated potent antitumor effect by binding to uPAR in vivo [37].
Results
Preparation and characterization of receptor-responsive nanoparticle (RRNP)
To prepare receptor-responsive nanoparticle (RRNP), urokinase receptor targeting agent (ATF-HSA, 125 μl, 200 mg/ml) was denatured by 6 M urea and 55% ethanol, in the presence of mono-substituted β-carboxy phthalocyanine zinc (ZnPc-COOH, abbreviated as CPZ). After stirred for about 10 min, and at the moment that aqueous liquid transformed into oily liquid, a large volume of deionized water (8-fold volume) was poured quickly into the mixed solution. This instantaneously reduced the denaturant to a low concentration (0.67 M) allowing for protein refolding. The protein did not refold back into native protein with this procedure, but instead, assembled into RRNP entrapping CPZ with a size of ~40 nm (Figure 1A-B). The thus-formed RRNP (ATF-HSA: CPZ@RRNP) was dialyzed to remove ethanol and urea, and was further purified through an anion exchange column to remove the unreacted ATF-HSA and CPZ. The nanoparticle held together purely by non-covalent force because no chemical crosslinking agent was used during preparation. The hydrophobic CPZ molecule was not required for the formation of nanoparticle because this procedure also worked well in the absence of CPZ.
A) Schematic illustration of the preparation of receptor-responsive nanoparticle (RRNP). Both nanoparticles ATF-HSA:CPZ@RRNP (B) and HSA:CPZ@NP (C) showed monodispersed distribution on DLS. The TEM images and photograph of the respective nanoparticles were shown as insets. The insets at lower right panels showed that the hydrophobic cargo CPZ precipitated in aqueous buffer but was solubilized inside nanoparticles (left). D) The zeta potential of both nanoparticles (10 mM Tris-HCl, pH 8.0) was about -20 mV.
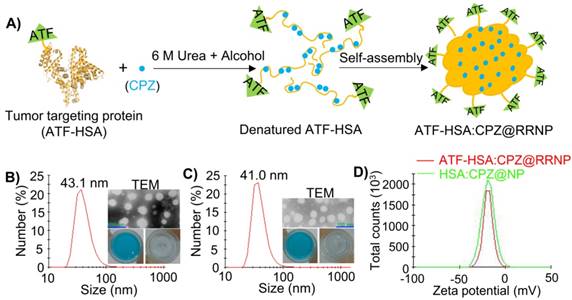
The properties of nanoparticles, such as the size and shape, affect their efficacies [38-41]. At the condition of 55% ethanol, 6 M urea and a feed ratio of 1:10, we obtained ATF-HSA:CPZ@RRNP of about 40 nm diameter (Figure 1B and Figure S1A) and the loading capacity of CPZ was 86.5 µg/mg ATF-HSA (Figure S1B), equivalent to 12.1 CPZ molecule loaded into per ATF-HSA molecule. Although the feed ratio of ATF-HSA/CPZ is 1:10, the final loading capacity of ATF-HSA/CPZ is 1:12.1, reflecting more loss of protein than CPZ during preparation.
As a control, we also used human serum albumin (HSA) as building block [42, 43] of the biodegradable nanoparticle. HSA was also formed into nanoparticle entrapping CPZ (HSA:CPZ@NP) by the current method with an optimized preparation condition of 6 M urea, 60% ethanol and a feed ratio of 1:10. This nanoparticle has a size of about 40 nm (Figure 1C and Figure S1A) and the loading capacity of CPZ/HSA was 110.37 µg/mg (Figure S1B), equivalent to 11.8 CPZ molecule loaded into per HSA molecule, demonstrating the broad applicability of the method.
The thus-formed nanoparticle (ATF-HSA:CPZ@ RRNP or HSA:CPZ@NP) appeared as clear bluish solution, an intrinsic color of CPZ, demonstrating CPZ was incorporated into nanoparticles because CPZ has no water solubility at all and was precipitate in aqueous solution (Figure 1B-C). The CPZ is a potent photosensitizer in its monomeric form, showing phototoxicity when illuminated with 680 nm, but not so in aggregate form. The CPZ inside nanoparticles had higher percentage of monomer (680 nm) than aggregate form (635 nm) based on UV-VIS absorption measurements (Figure S2A).
The size distribution and diameter of both nanoparticles in buffer solution were measured by dynamic light scattering (DLS). The results demonstrated both ATF-HSA:CPZ@RRNP and HSA:CPZ@NP had sizes of ~40 nm in diameter with a high degree of monodispersity (Figure 1B-C). The zeta potential of both nanoparticles (10 mM Tris-HCl, pH 8.0) was about -20 mV (Figure 1D), which can maintain the stability of nanoparticles. Transmission electron microscopy (TEM) images showed that both nanoparticles possessed spherical shapes with diameter of about 40 nm, which was consistent with DLS measurements (Figure 1B-C). Both nanoparticles in buffer were stable for more than two weeks (Figure S2B) without change of their sizes. Importantly, the nanoparticles were also stable even in the buffer containing 10% fetal bovine serum (Figure S2C).
The current preparation method uses stringent conditions with high concentrations of ethanol and denaturant urea, will this procedure affect the targeting capability of ATF-HSA? We measured the binding of monomer ATF-HSA:CPZ from ATF-HSA: CPZ@RRNP to recombinant soluble uPAR protein by native gel shift experiment (native PAGE), and observed that uPAR indeed shifted the band of ATF-HSA:CPZ (Figure S2D), illustrating ATF-HSA: CPZ from ATF-HSA:CPZ@RRNP possessed receptor binding bioactivity comparable to native ATF-HSA.
Receptor-triggered disintegration of ATF-HSA:CPZ@RRNP
We mixed the receptor targeting RRNP with recombinant uPAR (at a protein molar ratio of 1:1) and measured the size by DLS. Interestingly, we observed that the size of RRNP decreased to ~10 nm (Figure 2A), a size comparable to ATF-HSA:uPAR complex, clearly demonstrating receptor-mediated disintegration of the RRNP. As controls, we mixed this RRNP with other proteins, including soluble protein BSA or a non-uPAR membrane surface receptor HAI-1, and observed no change of the sizes of the nanoparticles (Figure 2A). On the other hand, we mixed non-targeting nanoparticle (HSA:CPZ@NP) with uPAR, BSA, or HAI-1 and observed no change of sizes (Figure 2B). We also monitored the fluorescence change of the above samples, and found the fluorescence intensity of ATF-HSA:CPZ@RRNP increased by ~3.7 fold in the presence of receptor uPAR (at a protein molar ratio of 1:1), demonstrating reduced fluorescence self-quenching of CPZ after disintegration (Figure 2C). However, the fluorescence in other control groups didn't change basically (Figure 2C-D), demonstrating nanoparticles remained intact. To further verification, TEM experiment (Figure 2E) was done and indeed showed ATF-HSA:CPZ@RRNP disintegrated into small fragment after incubation with recombinant uPAR. In the native gel shift assay, we incubated ATF-HSA:CPZ@RRNP with recombinant uPAR at a protein molar ratio of 1:1 in the solution (50 mM Tris-HCl, pH 8.0, 50 mM NaCl) for 2.5 h. Then the mixture was loaded on to a native PAGE gel (8% native-PAGE). The result (Figure S2E) showed that the RRNP in the presence of the receptor uPAR gave a band (lane 1) at migration similar to ATF-HSA:uPAR complex (lane 3). The RRNP itself did not go into the gel (not shown), presumably due to its large size (~40 nm). Next, we measured if CPZ remained bound to the ATF-HSA after the disintegration of RRNP. We mixed the ATF-HSA:CPZ@RRNP with recombinant uPAR at 1:1 molar ratio for 2.5 h, and the mixture was purified through a Ni-NTA column which has affinity toward the hexahis-tag on the ATF-HSA protein. After the ATF-HSA was eluted out by eluent buffer (containing 500 mM imidazole), its ratio of CPZ/protein was measured, and the results showed the CPZ content in the eluted ATF-HSA protein was about the same as in the starting RRNP, demonstrating CPZ was remained bound to ATF-HSA after RRNP disintegration.
A) ATF-HSA:CPZ@RRNP (black curve, size ~40 nm) was disintegrated and reduced to ~10 nm in the presence of recombinant uPAR receptor at a protein molecular ratio of 1:1 (red curve) but no disintegration observed in the presence of non-uPAR membrane surface receptor HAI-1 (green curve) or BSA (blue curve). B) HSA:CPZ@NP (~40 nm, black curve) is stable in the presence of uPAR (red curve), HAI-1 (green curve) or BSA (blue curve). C) Fluorescence of ATF-HSA:CPZ@RRNP (black curve) increased in the presence of recombinant uPAR receptor at a protein molecular ratio of 1:1 (red curve), but remain unchanged in the presence of non-uPAR membrane surface receptor HAI-1 (green curve) or BSA (blue curve). D) Fluorescence of HSA:CPZ@NP (black curve) did not changed in the presence of uPAR (red curve), HAI-1 (green curve) or BSA (blue curve). E) The TEM images showed ATF-HSA:CPZ@RRNP disintegrated into small pieces after incubation with recombinant uPAR protein for 2.5 h. Left TEM image is for ATF-HSA:CPZ@RRNP whereas the right TEM is the nanoparticle containing 1:1 uPAR and was incubated for 2.5 h. F) Schematic illustration of receptor-triggered disintegration and cargo release of ATF-HSA:CPZ@RRNP.
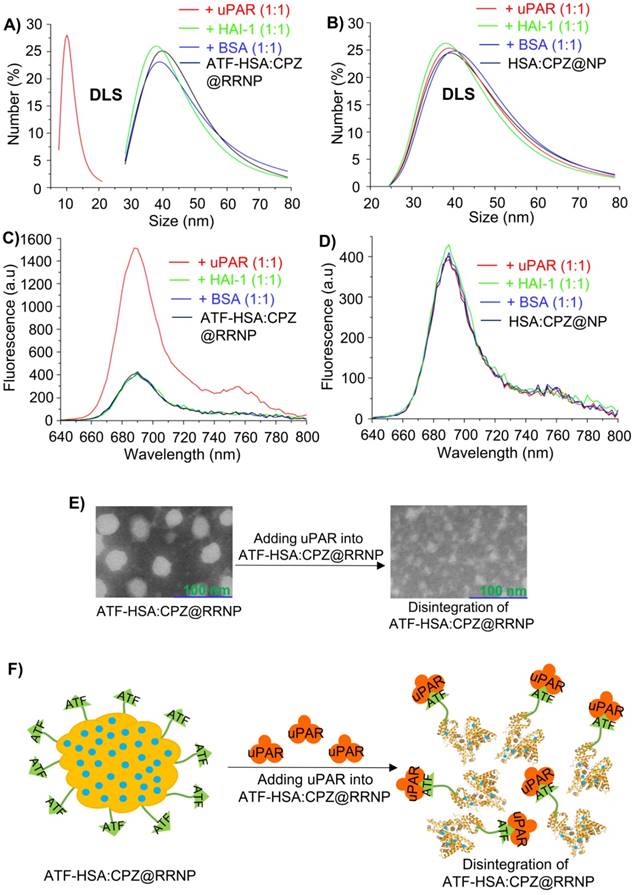
Furthermore, the amount of ATF-HSA:CPZ released from ATF-HSA:CPZ@RRNP was measured to be 86%, which was calculated as the amount of monomeric ATF-HSA:CPZ from disintegrated nanoparticles divided by the amount of feeding nanoparticles. How the receptor triggers the disintegration of ATF-HSA:CPZ@RRNP was not known mechanistically. We think this unique property of RRNP disintegration is due to its preparation method, which did not disrupt the disulfide bonds in the albumin. In addition, no crosslinking reagents, e.g., glutaraldehyde, was used to conjugate the albumin together covalently in the preparation, allowing the nanoparticle to fall apart. The RRNP is most likely held together by non-covalent hydrophobic van der Waals force contributed by the interior of albumin and CPZ, which is highly hydrophobic. Some ATF-HSA has its ATF moiety exposed outside, but some ATF are buried inside the core of RRNP. The exposed ATF most likely maintains its receptor binding conformation due to the presence of multiple disulfide bonds in its structure [36]. Thus, one potential mechanism of receptor-induced RRNP disintegration is that uPAR binds to the surface exposed ATF moiety of RRNP, causing the conformational change of the RRNP, resulting in more exposure of additional ATF moiety, finally leading to the disintegration of the RRNP (Figure 2F).
Cells expressing uPAR triggers disintegration of RRNP
We measured the uPAR expression levels on cells using a commercial ELISA kit. For H1299 cells, the uPAR level on H1299 was detected to be 5832 uPAR molecules per H1299 cell, which is similar to the value reported in the literature (7100 uPAR per H1299 cell) [44]. For HELF cells, no uPAR was detected.
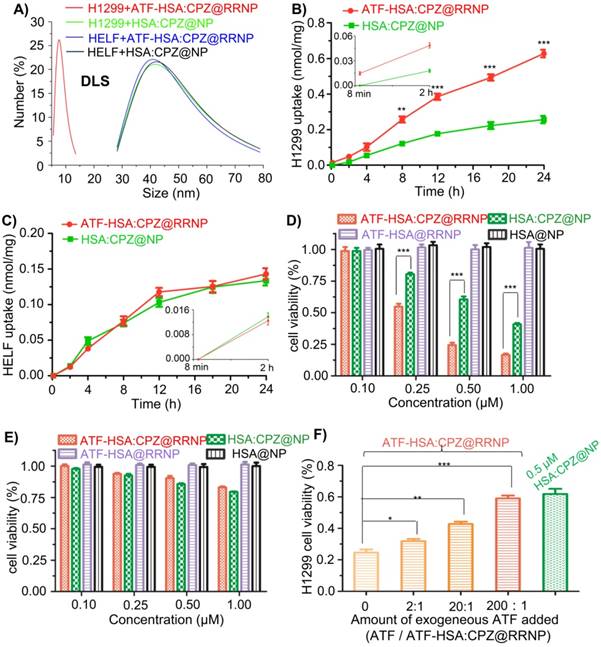
Furthermore, we incubated ATF-HSA:CPZ@RRNP or HSA:CPZ@NP with uPAR-expressing H1299 cells or no uPAR expressing HELF cells (3.0×104/ml culture medium) for 2.5 h. The supernatant of cell culture medium was collected and filtered by a 0.22 μm Millipore filter membrane prior to size measurement by DLS. The DLS result demonstrated the size of the collected culture in the nanoparticle group (uPAR-expressing H1299 cells incubated with ATF-HSA:CPZ@RRNP, Figure 3A, red curve) was about 7.5 nm, which was the size of ATF-HSA:CPZ. As controls, the sizes of other three groups (H1299 cells incubated with HSA:CPZ@NP, HELF cells (without uPAR) incubated with ATF-HSA:CPZ@RRNP, or HELF incubated with HSA:CPZ@NP, shown in Figure 3A in green, blue or black curve, respectively) were still about 40 nm, which was close to the size of nanoparticles. In another control, the size of culture medium only was found to about 6.5 nm, presumably from albumin of the serum (data was not shown). Therefore, RRNP indeed disintegrates after interacting with the uPAR on cancer cell surface.
A) uPAR on H1299 cell triggers the disintegration of ATF-HSA:CPZ@RRNP (red curve), however, the sizes of other three groups, H1299 incubated with HSA:CPZ@NP (green curve), HELF incubated with ATF-HSA:CPZ@RRNP (blue curve) or HELF incubated with HSA:CPZ@NP (black curve) did not change basically. B) ATF-HSA:CPZ@RRNP shows higher cellular uptakes in H1299 cell compared with HSA:CPZ@NP. The time points (8 min and 2 h) of cells uptake were enlarged and shown as insets. C) Both nanoparticles have similar cellular uptakes in HELF cells. The time points (8 min and 2 h) of cells uptake were enlarged and shown as insets. D) ATF-HSA:CPZ@RRNP shows enhanced phototoxicity on H1299 cells compared with HSA:CPZ@NP at the same drug concentration. E) Both ATF-HSA:CPZ@RRNP and HSA:CPZ@NP display no phototoxicity on HELF cells compared to H1299 cells. F) The pre-incubation of free ATF (200-fold) with H1299 cells followed by the addition of ATF-HSA:CPZ@RRNP reduced the enhanced phototoxicity of ATF-HSA:CPZ@RRNP (left column) to the level of HSA:CPZ@NP (right column). Values represent the mean of three separate experiments; bars represent standard error of the mean (SEM). The unpaired, 2-tailed Student t test was used to analyze data; * p < 0.05, ** p < 0.01, *** p < 0.001.
Enhanced cellular uptake and cytotoxicity effects of receptor targeting RRNP in vitro
Next, we measured the uptake of nanoparticles by non-small cell lung carcinoma cells (H1299) with high expression of uPAR by incubating nanoparticles (0.5 µM CPZ) with the cells for different time periods. The results (Figure 3B) demonstrated uptake rate of the receptor-targeting RRNP was faster than the non-targeting nanoparticle. At 24 h, the uptake of receptor-targeting RRNP was more than 2.5 fold higher. As a control, the uptake of the nanoparticles on human embryo lung fibroblasts (HELF) cells expressing no uPAR was lower compared to H1299 and both nanoparticles had similar amounts of cell uptake on HELF (Figure 3C).
CPZ is a fluorescent probe (λex = 610 nm, λem = 690 nm) in its monomeric form, and can also behave as a photosensitizer and generate cytotoxicity when excited with near-infrared light at 680 nm [45, 46]. We found that the phototoxicity of receptor targeting RRNP to H1299 tumor cells was higher than the non-targeting nanoparticles, and reached 2.1-fold at concentrations of 0.5 µM (Figure 3D). For HELF cells expressing no uPAR, both nanoparticles had similar cellular toxicity (Figure 3E). No cytotoxicity to either H1299 or HELF in the absence of light was observed regardless which nanoparticles were used (Figure S3A-B).
In order to further confirm the receptor-mediated cytotoxicity of ATF-HSA:CPZ@RRNP, we carried out a competition assay (Figure 3F). We saturated the cell surface receptor uPAR by addition of exogenous ligand ATF at different concentrations (the protein molar ratio of ATF/ATF-HSA:CPZ@RRNP was about 2:1, 20:1, 200:1), and measured the cytotoxicity of ATF-HSA:CPZ@RRNP at 0.5 µM. We found that receptor-targeting RRNP gradually lost its cytotoxicity as the exogenous ATF concentration increased, and at a 200-fold molar excess of ATF, its toxicity dropped to a level comparable to the nanoparticle lacking targeting moiety.
We also used confocal fluorescence imaging to track the localization of the nanoparticles and their dynamics. Within 8 min of incubations of the nanoparticles (0.5 µM) with H1299 cells, we saw the accumulation of the receptor-targeting RRNP on the cells surface, but not inside the cells (Figure S4A). The non-targeting nanoparticles did not absorb onto the cell at this time point (Figure S4B). In addition, we also studied the locolization of both nanoparticles on H1299 cells. After 12 h incubation, both nanoparticles entered cells and co-localized with both lysosome (Figure S5B) and mitochondria (Figure S5C). Importantly, the nanoparticles did not localize in cell nucleus (Figure S5A), which is advantageous because the CPZ has large aromatic ring in its structure and can potentially intercalate with DNA and be carcinogenic.
Enhanced anti-tumor efficacy of ATF-HSA:CPZ@RRNP in vivo
To evaluate the in vivo localization of nanoparticles on tumor, we established a tumor-bearing mice model by injecting mouse hepatocellular carcinoma cells (H22) with high expression of uPAR on the back of mice. When the volume of tumor reached ~60 mm3, nanoparticles (ATF-HSA:CPZ@RRNP or HSA:CPZ@NP at 0.05 mg CPZ/kg of mice body weight) or saline was injected via tail veins. Then the mice were imaged at various time points using 3D fluorescent molecular tomography (FMT) instrument (Figure 4A) based on CPZ fluorescence signal, which allows probe quantitation (Figure 4B), slices of X/Y/Z axial profile across the center of H22 tumor (Figure 4C), three-dimensional imaging (Figure S6 and Movie 1-2). The results showed the receptor-specific RRNP gradually accumulated on tumor more than the control (HSA:CPZ@NP). At the 48 h, the amount of receptor-specific RRNP was 2.7-fold more than the nanoparticle without targeting moiety.
A) Diagram of fluorescence molecular tomography imaging instrument. B) Kinetics of cargo accumulation in the tumor sites of mice. The data were averaged from 5 mice in each group. C) Representative three-dimensional (X/Y/Z axial) profile of H22 tumor in Kunming mice post intravenous injection of nanoparticles. Slices of X/Y/Z axial profile across the center of H22 tumor in Kunming mice taken at different time points (1, 2, 4, 8, 12, 24, 48, 72, 96 h) post intravenous injection of ATF-HSA:CPZ@RRNP and HSA:CPZ@NP. D) ATF-HSA:CPZ@RRNP leads a significant reduced tumor growth rate compared with HSA:CPZ@NP-treated group and the saline-treated group. The data were averaged from 10 mice in each group. E) After 7-day photodynamic therapy, all mice were sacrificed and tumor were removed and weighed. The tumor weights of ATF-HSA:CPZ@RRNP group were significant smaller than HSA:CPZ@NP group and saline group. The data were averaged from 8 mice in each group. All bars represent standard error of the mean (SEM). The unpaired, 2-tailed Student t test was used to analyze data; * p < 0.05, ** p < 0.01, *** p < 0.001.
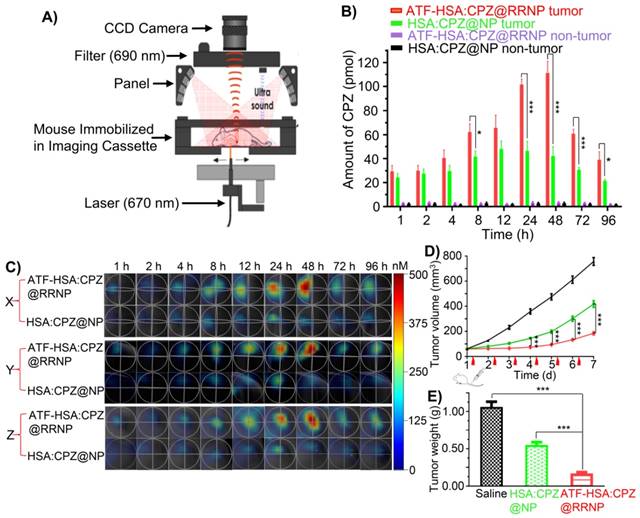
To evaluate the anti-tumor effects of nanoparticles, mice were injected with nanoparticles (the same dose as used for imaging) or saline via tail vein when the tumor volume of mice approached ~60 mm3. Subsequently, tumor sites of mice were illuminated using a 680 nm light source daily for 3 min to a dosage of 50 J/cm2. The volume of tumor (Figure 4D) and the body weight (Figure S7) of mice were monitored daily. The results showed that tumor growth of receptor-targeting RRNP group was basically stopped in the first four days. On the seven day of treatment, the tumor volume of this group was 2.4-fold and 4.4-fold lower compared to the albumin nanoparticle group and the saline group, respectively. The tumor was cautiously exfoliated and weighted at the 7th day. The results (Figure 4E) showed the tumor weight of saline group was 1.9-fold and 6.5-fold more than non-targeting nanoparticle group and the receptor-targeting RRNP group, respectively.
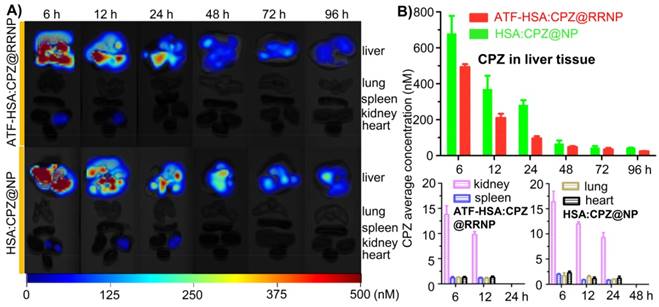
We also measure the biodistribution of nanoparticles in the tumor-bearing mice (Figure 5). Both 3D images (Figure 5A) and quantitative analysis (Figure 5B) showed ATF-HSA:CPZ@RRNP had lower accumulation on liver tissue than the NPs without uPAR targeting capability (HSA:CPZ@NP). The liver was the organ with the cargo concentration much higher than other body parts, which was commonly observed in photosensitizers [37, 47, 48]. Importantly, the cargo concentration in liver decreased quickly. Other organs (kidney, spleen, lung, and heart) have significantly lower cargo concentration (in the range of 17 nM), suggesting strong safety prospect of these nanoparticles for future application. Interestingly, the cargo concentrations in other blood-rich organ like kidney were much lower than the cargo embedded in albumin [37].
A) Representative organs fluorescence images of H22 tumor-bearing mice taken at different time points (6, 12, 24, 48, 72, 96 h) post intravenous injection of ATF-HSA:CPZ@RRNP (top) and HSA:CPZ@NP (bottom). B) 3D quantitative analysis showed ATF-HSA:CPZ@RRNP group showed lower drug accumulation in liver tissue than HSA:CPZ@NP group. In addition, the cargo concentration in liver decreased quickly. Other organs (kidney, spleen, lung, and heart) have significantly lower cargo concentration (in the range of 17 nM), suggesting strong safety for future application. The data were averaged with the bars representing standard error of the mean (SEM).
Discussion
Unique features of receptor-responsive nanoparticle (RRNP)
The detailed mechanism of receptor-induced nanoparticle disintegration and endocytosis of cargo is not completely understood. The unique feature of RRNP may be related to the very tight binding between the receptor and the targeting agent (ATF-HSA), which has a dissociation constant at the sub-nanomolar range. The denaturing and refolding protocol used in the preparation of RRNP most likely did not affect the activity of ATF, because ATF is a compact protein, contains multiple disulfide bonds based on our previous structural study [36], and is resistant to such denaturing without reducing agent. Thus, it can be imagined intuitively that this tight binding disturbed the nanoparticle assembly, by e.g., promoting the exposure of ATF-HSA onto nanoparticle surface, and to an extent that leads to the final breakup of nanoparticles. What happen next is well-defined based on previous studies [32, 49, 50]: the ligand-occupied uPAR undergoes endocytosis in the presence of other cofactors, taking the cargo into the lysosome. The ligand or other cofactors are degraded, dumping out the cargo, while the receptor survives and is recycled back to cell surface. The HSA has seven specific binding sites for various cargo molecule, as defined by a number of groups [51] including us [52], and can facilitate the cargo entrance into cells.
Our current method of nanoparticle preparation has unique advantages compared to previous reported methods for protein-based nanoparticles. The previous protocols can be classified into five categories: desolvation [53, 54], emulsification [55-57], thermal gelation [58, 59], nab-technology [15], and hydrophobic-cargo-induced nanoparticle formation [47, 60]. In desolvation method, the hydration layer around proteins was removed by dehydrating agents to promote protein aggregation following by stabilized with cross-linking reagents like glutaraldehyde [61, 62]. With emulsification method, protein nanoparticles were formed by homogenizing the oil phase (e.g. cotton seed oil) containing the protein droplets at high speed and then were stabilized by chemical cross-linking agent or thermal treatment [55, 63, 64]. Thermal gelation method is a sequential process that involves heat-induced unfolding and formation of protein nanoparticle stabilized by van der Waals interaction and/or disulfide bond shuffling [58, 59, 65]. In nab-technology, drug (paclitaxel) is mixed with HSA in an aqueous solvent and passed under high pressure through a jet to form drug HSA nanoparticles named Abraxane (nab-paclitaxel), which was approved by FDA in 2005 for the treatment of metastatic breast cancer. The existing disulfide bonds of HSA may be disrupted and sulfhydryl group of HSA may be oxidized, leading to formation of new crosslinking disulfide bonds. In addition, a self-assembly strategy was also developed to construct protein nanoparticle via increasing the hydrophobicity of protein, e. g. addition of a lipophilic drug or diminishment of primary amine groups on protein surface. In the fifth method, small organic hydrophobic molecules were used to bring together protein molecules. Jeong et al. was the first to report the albumin-based nanoparticles by covalent conjugation of hydrophobic cargo (chlorine e6) to albumin at a stoichiometric ratio of 1:10 led to the formation of albumin:ce6 nanoparticles with a size of 88 nm [47]. Followed up on this work, Zhuang Liu group succeeded in generating nanoparticles of albumin promoted by lipophilic compound, such as PTX [60], without invoking covalent interaction. Covalent crosslinking mediated by protein disulfide bond reduction and re-oxidation was also reported to promote the formation of such albumin-drug nanoparticles [66, 67].
Our RRNP preparation bears some resemblance to last method mentioned above (hydrophobic-cargo-induced nanoparticle formation) where the aggregation of albumin into nanoparticle was promoted by hydrophobic cargo molecule. However, in our RRNP method, a hallmark feature is no hydrophobic cargo is needed to form nanoparticle - the albumin itself can be formed into nanoparticle, avoiding the use of cross-linking agents and dependence on hydrophobic cargos. The reason for such preparation is to denature the albumin and to expose its hydrophobic core completely, which will intertwine together upon removal of the denaturant, leading to the formation of nanoparticle. The main driving force for the formation of nanoparticle is most likely the non-covalent interaction among proteins with the hydrophobic interaction having prominent importance. Another observation of RRNP is its high stability, even in 10% FBS, which is not too surprised, considering the stability of hydrophobic-cargo-induced nanoparticle.
Stimuli-responsive nanoparticles for cancer therapy
A number of stimuli-responsive nano-systems were previously reported. The stimuli include: temperature [68, 69], pH [18, 19], redox potential [20], magnetism [70], light [71, 72] or enzyme [21, 22]. Tumor tissue has pH value with 0.5-1 units lower than the surrounding normal tissue due to metabolic glycolysis and lactic acid production. Greater pH differences can be found at the subcellular level such as lysosomes (pH 4.5-5), endosomes (pH 5.5-6) [73]. pH-responsive nanoparticles were designed to explore the such pH difference for controlling the release of encapsulated drugs with maximum therapeutic impact and minimum side-effects. For example, Pu et al. reported pH responsive nanoparticles consisted of poly(L-glutamic acid) dendrimers with polyhedral oligomeric silsesquioxane (POSS) as core. pH-responsive hydrazone bonds were used to graft cytotoxic drug doxorubicin and targeting moiety biotin to the surface of the dendrimers. Such nanoparticles is stable at neutral pH (< 10% drug release), but at pH 5, released 90% of its cargo. In vivo studies in xenograft breast cancer models showed enhanced tumor inhibition efficiency and reduced systemic toxicity of the nanoparticles compared with free drug [74].
Tumor tissue has dysregulated activities of enzymes, such as phospholipases, oxidoreductases, proteases, which are promising biological triggers for enzyme-responsive nanoparticle in the field of theranostics [75]. A nanoparticle responsive to proteases (matrix metalloproteinases, MMP-2 and MMP-9) was reported by Olson et al [22, 76]. In this strategy, the nanoparticle composes of a dendrimer as core covalently attached to the activatable cell-penetrating peptide (ACPP). The ACPP consists of a polyarginine (polycationic) -based cell-penetrating peptide (CPP) sequence, a MMP-sensitive peptide linker (XPLGLAG) and an inhibitory domain made up of negatively charged residues (polyglutamate, 9-mer), forming a hairpin structure. The polyanionic inhibitory domain masks the adhesiveness of the polycationic CPP until proteases (MMP-2 and MMP-9) presented in tumor cut the linker, releasing the polycationic CPP and associated payload to adhere to and be taken up into cells. The as-prepared ACPP decorated nanoparticles exhibited more than 10-fold cellular association compared to unconjugated ACPP and showed high in vivo contrast ratio of images corresponding well to the MMP distribution in tumor.
Tumor surface receptor, as a natural stimuli or trigger, can recruit our receptor-responsive nanoparticle, followed by disintegrating and releasing cargo. Our RRNP strategy is likely to be applicable to other type of targeting proteins (ligands) and the corresponding receptors. The key of this strategy is to form stable nanoparticles not invoking covalent cross-linkers, thus, the nanoparticle will fall apart upon the tight interaction from the receptor. Although exact mechanism of RRNP disintegration is not known, one possibility is that receptors bind to the targeting proteins renatured partially, leading to the conformation change of targeting proteins and more other protein targeting domains renatured partly are exposed outside from the inside of RRNP and bind to receptors, triggering the disintegration of RRNP. In addition, our RRNP method forms nanoparticle, independent of the existence of cargo, which is different from drug-induced nanoparticle assembly strategy [47, 60]. Therefore, our method is very likely applicable to other cargos and targeting proteins to prepare RRNP besides the one reported here. Such possibility will await future test.
Conclusion
To summarize, we report a novel type of receptor-responsive nanoparticle (RRNP) which not only targets to tumor receptor, but also disintegrates and releases cargos, leading to significantly improved antitumor efficiency. The receptor-triggered disintegration and cargo release was clearly demonstrated in vitro. In vivo studies also showed ATF-HSA:CPZ@ RRNP had a higher drug concentration at tumor site and better curative effect than HSA:CPZ@NP.
Materials and methods
Materials
Mono-substituted β-carboxy phthalocyanine zinc (ZnPc-COOH, abbreviated as CPZ) was synthesized according to previous procedure reported [45]. Pichia pastoris yeast strain X-33 (Invitrogen, USA) with plasmid pPICZaA encoding HSA or ATF-HSA was constructed, as previously described [37]. Recombinant soluble uPAR (extracellular domain of uPAR) and ATF were prepared as previously reported [77, 78]. Ni-chelating Sepharose Fast Flow resin and diethylaminoethyl (DEAE) anion exchange resin were purchased from GE Healthcare (Uppsala, Sweden). All chemicals and reagents were of analytical grade and were purchased either from Sigma-Aldrich (St Louis, MO, USA) or from Sinopharm Chemical Reagent Co. Ltd. (Shanghai, China) unless otherwise stated. Non-small cell lung carcinoma cell line H1299 and human embryo lung fibroblasts (HELF) were purchased from American Type Culture Collection (ATCC, Rockville, MD, USA). HELF cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and antibiotics. H1299 cells were grown in RPMI-1640 medium supplemented with 10% FBS and antibiotics. All cells were kept at 37 °C in a humidified incubator with 5% CO2 atmosphere. The viability of cells was determined by the dye Trypan blue. Cells were maintained in logarithmic phase with viability > 95%. The mouse hepatoma-22 cell line (H22) was purchased from the Shanghai Institute of Cell Biology (Chinese Academy of Sciences, Shanghai, China).
Expression of the receptor-targeting recombinant fusion protein: ATF-HSA
The transformed Pichia pastoris strain X-33 integrated with ATF-HSA expression vector was cultured in YPD medium (1% yeast extract, 2% peptone, 2% dextrose) containing 100 µg/ml Zeocin® at 28 °C for 2 days before it was transferred into BMGY medium (1% yeast extract, 2% peptone, 100 mM potassium phosphate, pH 6.0, 1% v/v glycerol). The X-33 strain was further cultured in BMGY medium at 28 °C for about 24 h to an OD600 of 4-5. After being transferred into BMMY medium (1% yeast extract, 2% peptone, 100 mM potassium phosphate, pH 6.0, 1% v/v methanol), the cells were induced every 24 h with methanol (at a final concentration of 1%) over the following 4 days to express the protein ATF-HSA. HSA was expressed in the same way as ATF-HSA.
Purification of ATF-HSA in Pichia pastoris strain X-33
After 4 days' induction with 1% methanol, the BMMY medium was harvested by centrifugation at 9,000 g for 20 min. After the supernatant was adjusted to pH 7.4 using 1 M Tris-HCl (pH 8.5) and centrifuged one more time. The supernatant was collected and applied to a Ni2+-chelating column that was pre-equilibrated with 20 mM Tris-HCl containing 500 mM NaCl, pH 7.4. The column was then washed by 20 mM Tris-HCl, pH 7.4, 500 mM NaCl containing 5 mM imidazole, followed by the elution of the target protein (ATF-HSA) using the above buffer containing 500 mM imidazole. The fraction containing ATF-HSA from the column was dialyzed overnight against 20 mM Tris-HCl, pH 8.0, 50 mM NaCl, at 4 °C by a dialysis membrane with molecular weight cutoff of 8-10 KDa. The molecular weight of the target protein was confirmed by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) in a Bio-Rad Biologic system (Bio-Rad Laboratories Inc. Hercules, CA, USA), and the concentration of the target protein was measured by a GE Nanovue® spectrophotometer (Cambridge, UK). HSA was purified in the same way as ATF-HSA.
Preparation of receptor-responsive nanoparticle (RRNP) entrapping CPZ molecule (ATF-HSA:CPZ@RRNP)
To prepare ATF-HSA:CPZ@RRNP, urea (180 mg) was dissolved in mixed solution including water (75 μl) and ethanol (300 μl). Then ATF-HSA (125 μl, 200 mg/ml) was added dropwise to the mixed solution. After stirring for 1.5 min, CPZ (1.8 mg) which had been dissolved in dimethylsulfoxide (DMSO) (20 μl) was added dropwise to the solution under stirring. After stirred for about 10 min at room temperature, and at the moment that aqueous liquid transformed into oily liquid, deionized water (4 ml) was added quickly to the solution under stirring. Half of an hour later, the mixed solution was dialyzed overnight against dialysate (20 mM Tris-HCl, 50 mM NaCl, pH 8.0) in the dark at 4 °C by a dialysis membrane with molecular weight cutoff of 8-10 KDa. Then the mixing solution dialyzed was applied to a DEAE column (column volume of ~10 ml) pre-equilibrated with equilibrium solution (50 mM Tris-HCl, 50 mM NaCl, pH 8.0). ATF-HSA monomer and free CPZ were adsorbed on DEAE column. The final product, ATF-HSA:CPZ@RRNP, flowed straightly through DEAE column. ATF-HSA:CPZ@RRNP was collected and stored with protein molar ratio of 1:100 (ε-poly-lysine/RRNP) at 4 °C in a dark place. As a control, HSA:CPZ@NP was prepared by similar procedure.
Control of key parameters in the preparation process of nanoparticles
To investigate the effect of ethanol concentration on the characteristics of protein nanoparticles, we added various concentrations (40%, 50%, 55%, 60%, 70%) of ethanol containing 6 M urea with a fixed feed molar ratio (ATF-HSA:CPZ = 1:10). In addition, we also added different concentrations (40%, 50%, 55%, 60%, 70%) of ethanol containing 6 M urea with a fixed feed molar ratio (HSA:CPZ = 1:10). Furthermore, to study the effect of the feeding molar ratio (ATF-HSA:CPZ) on the CPZ loading per ATF-HSA on final nanoparticle, ATF-HSA:CPZ@RRNP was prepared at various ATF-HSA:CPZ feed molar ratios (1:3, 1:5, 1:10, 1:15) in the condition of 6 M urea and 55% ethanol. Meanwhile, HSA:CPZ@NP was also prepared at different HSA:CPZ feed molar ratios (1:3, 1:5, 1:10, 1:15) in the condition of 6 M urea and 60% ethanol.
Characterization of nanoparticles
The size distribution and diameter of the ATF-HSA:CPZ@RRNP and HSA:CPZ@NP were measured by dynamic light scattering (DLS) instrument (Nano ZS ZEN 3600, Malvern Instruments, Malvern, UK) at room temperature. Meanwhile, the zeta potential of both nanoparticles (10 mM Tris-HCl, pH 8.0) was also detected by DLS. All samples were filtered by a 0.22 μm Millipore filter membrane prior to DLS measurement. The morphology of the nanoparticles was observed by a transmission electron microscope (TEM) (Hitachi H-7650) operated at an acceleration voltage of 80 KV. The samples (protein concentration: 0.5 mg/ml) were placed on a 300-mesh copper grid coated with carbon. After drying, the samples loaded on copper grid were stained using phosphotungstic acid solution (pH 7.2). Ultraviolet-visible absorption spectra of nanoparticles were measured from 500 to 800 nm using microplate reader (Synergy4, Biotek Instruments, Winooski, VT, USA). The protein concentration of nanoparticles was determined by a BCA Protein Assay Kit (Solarbio Science & Technology Co. Ltd. Beijing, China). The fluorescence intensity of nanoparticles was measured by a microplate reader (Synergy4, BioTek Instruments, Winooski, USA) with λex = 610 nm and λem = 690 nm.
Targeting specificity of ATF-HSA:CPZ@RRNP in vitro
3 ml of ATF-HSA:CPZ@RRNP was added to 6 ml of urea solution (9 M) under stirring for 12 h. After the mixed solution was applied to the Ni-NTA column pre-equilibrated with the buffer (20 mM Tris-HCl, 6 M urea, pH 8.0), 5 column volumes of the eluent buffer (20 mM Tris-HCl, 500 mM NaCl, pH 8.0, 500 mM imidazole) was added to Ni-NTA column, the monomer target protein ATF-HSA:CPZ was eluted and collected. After dialyzing in buffer solution (20 mM Tris-HCl, 50 mM NaCl, pH 8.0), we mixed recombinant soluble uPAR with the monomer ATF-HSA:CPZ or expressed ATF-HSA from Pichia Pastoris at approximately a protein molar ratio of 1:1 for 2 h before doing gel shift experiment (native PAGE), only expressed ATF-HSA from Pichia Pastoris as a control.
Receptor-responsive disintegration and cargo release of ATF-HSA:CPZ@RRNP in vitro
We incubated ATF-HSA:CPZ@RRNP or HSA:CPZ@NP with recombinant uPAR, non-uPAR membrane surface receptor HAI-1 (a control receptor targeting at matriptase) or BSA at approximately a protein molar ratio of 1:1 in the solution (50 mM Tris-HCl, pH 8.0, 50 mM NaCl) for 2.5 h under stirring at room temperature. Then the size distribution and diameter of mixing solutions were measured by DLS. Next, the fluorescence spectrums (640 nm~800 nm) of all samples were measured on microplate reader (Synergy4, Biotek Instruments, Winooski, VT, USA) with λex = 610 nm. To further observe the disintegration capacity of ATF-HSA:CPZ@RRNP, the morphology of the ATF-HSA:CPZ@RRNP incubated with recombinant uPAR for 2.5 h was observed by a transmission electron microscope (TEM) operated at an acceleration voltage of 80 KV. In addition, gel shift assay (8% native PAGE) was carried out on the mixture (incubating ATF-HSA:CPZ@RRNP with recombinant uPAR at approximately a protein molar ratio of 1:1 for 2.5 h). Recombinant ATF-HSA expressed from Pichia Pastoris was used as a negative control and the complex of ATF-HSA:uPAR (1:1) was used as a positive control.
To measure the drug release profile, we incubated ATF-HSA:CPZ@RRNP or HSA:CPZ@NP with recombinant uPAR, BSA or recombinant HAI-1 at approximately a protein molar ratio of 1:1 in the solution (50 mM Tris-HCl, pH 8.0, 50 mM NaCl) for 2.5 h with stirring at room temperature. All samples were applied to a DEAE column (column volume of ~6 ml) pre-equilibrated with equilibrium solution (50 mM Tris-HCl, 50 mM NaCl, pH 8.0). Intact nanoparticles flowed straightly through DEAE column and monomeric ATF-HSA:CPZ was absorbed on DEAE column. After adding eluent buffer (50 mM Tris-HCl, 350 mM NaCl, pH 8.0), the monomeric ATF-HSA:CPZ from the disintegrated nanoparticles was eluted and collected. The cargo release rate of nanoparticles was defined by the amount of monomeric ATF-HSA:CPZ from disintegrated nanoparticles divided by the amount of incubated nanoparticles.
To measure if CPZ remained bound to the ATF-HSA after the disintegration of RRNP, ATF-HSA:CPZ@RRNP was incubated with recombinant uPAR at approximately a protein molar ratio of 1:1 in the solution (50 mM Tris-HCl, pH 8.0, 50 mM NaCl) for 2.5 h under stirring at room temperature. The mixture was applied to the Ni-NTA column pre-equilibrated with the buffer (20 mM Tris-HCl, 50 mM NaCl, pH 8.0), and 10 column volumes of the equilibrated buffer was added to wash the column. Then the eluent buffer (20 mM Tris-HCl, 50 mM NaCl, pH 8.0, 500 mM imidazole) was added to Ni-NTA column, protein in Ni-NTA column was eluted and collected. The contents of protein and CPZ were each quantified based BCA Protein Assay Kit and microplate reader with λex = 610 nm and λem = 690 nm respectively, to obtain the ratio of CPZ/protein.
Measurement of uPAR expression level on cells by human uPAR ELISA kit
The cells (H1299 or HELF, 5x105/ml) were harvested and disrupted by ultrasonic cell disruptor. The supernatant containing uPAR was obtained by centrifugation at 8000 rpm for 5 min and detected by human uPAR ELISA kit (Shanghai JingKang Biotech, http://www.jkbio.cn) following the manufacturer's instructions. Basically, the purified human uPAR antibody was coated on microtiter plate wells, followed by the addition of the samples. The detection antibody, conjugated with horseradish peroxidase (HRP), was added, and the wells were washed five times, followed by the addition of HRP substrate (TMB). After 15 min, the reaction was terminated by the addition of a sulphuric acid solution and the color change was measured spectrophotometrically at a wavelength of 450 nm. The concentration of uPAR in the samples was then determined by comparing the OD value of the samples to the standard curve.
Measurements of nanoparticle size after incubating with cells expressing with surface uPAR
200 µl suspended H1299 or HELF cell (3x104/ml) were seeded onto 96-multiwell plates and incubated overnight at 37 °C under 5% CO2. One blank control with only culture medium was set up in each plate. The cells were incubated with ATF-HSA:CPZ@RRNP or HSA:CPZ@NP (final concentration: 1 µM) for 2.5 h, and the cell culture medium in the wells were transferred to centrifuge tubes and filtered through a 0.22 μm Millipore filter membrane. The samples were then placed on DLS instrument to measure the size of the particles in the samples.
Cellular uptake of ATF-HSA:CPZ@RRNP in vitro
200 µL of exponentially growing cells (H1299 or HELF, 1.5×105/ml culture medium) were cultured in 96-multiwell plates and were placed overnight under 5% CO2 at 37 °C. Then the cells were incubated with ATF-HSA:CPZ@RRNP (final concentration: 0.5 µM) for different time periods (8 min, 2 h, 4 h, 8 h, 12 h, 18 h, 24 h). The culture medium only containing nanoparticles (final concentration: 0.5 µM) for various incubation times were designed as controls. Subsequently, we washed the cells with sterile PBS solution (pH 7.2) three times before we lysed the cells with 100 μl cell lysis buffer (1% SDS, 0.1 M NaOH) to get a homogenous solution. The cell protein concentration was measured by the BCA Protein Assay Kit with bovine serum albumin as standard. The fluorescence of the cell lysates was determined using a microplate reader (Synergy4, Biotek Instruments, Winooski, VT, USA) by excitation and emission at 610 nm and 690 nm, respectively. HSA:CPZ@NP was designed in the same way in parallel as a comparison. Results were showed as nmol CPZ per mg cellular protein by four replicates in each time point. Each experiment was repeated three times.
Phototoxicity and dark toxicity of ATF-HSA:CPZ@RRNP in vitro
200 µl suspended H1299 or HELF cell (3x104/ml) were seeded onto 96-multiwell plates and incubated overnight at 37 °C under 5% CO2. One blank controller in the plate was placed with only culture medium. The cells were incubated with ATF-HSA:CPZ@RRNP at different final concentrations (0.1, 0.25, 0.5, 1 µM) for 24 h. We washed the cells with sterile PBS (pH 7.2) twice before fresh medium was added. Then we gave an illumination for 1 min at a light dosage of 1.5 J/cm2 by a 680 nm LED light source (100 mW, Sundynamic Inc, Qingdao, Shandong, China) and put the plates into the incubator. After 4 h incubation, the cell viability was detected with a MTT colorimetric assay using the BioTek microplate reader at 544 nm. Dark toxicity was operated in parallel except illumination procedure. HSA:CPZ@NP was designed in the same way. Four replicates were made at each concentration and each experiment was repeated three times.
Phototoxicity of ATF-HSA:CPZ@RRNP on H1299 cells competed by free ATF
H1299 cells (at a density of 3×104/ml) at a volume of 200 μl per well were seeded in 96-multiwell plates and cultured overnight at 37 °C under 5% CO2. Before incubation with ATF-HSA:CPZ@RRNP (final concentration: 0.5 μM) about 24 h, we added different concentrations of free ATF (the protein molar ratio of free ATF/ATF-HSA:CPZ@RRNP was about 2:1, 20:1, 200:1) with H1299. As a control, HSA:CPZ@NP (final concentration: 0.5 μM) was also incubated with H1299 cells about 24 h. In addition, one blank controller was filled with 200 μl culture medium only. We washed H1299 cells twice with sterile PBS (pH 7.2) before fresh medium was added. Then we illuminated the plates for 1 min at a light dosage of 1.5 J/cm2 with the 680 nm LED light source and subsequently put it into incubator. After 4 h incubation, we measured the cell viability using the MTT colorimetric assay on a microplate reader (Synergy4, Biotek Instruments, Winooski, VT, USA) at 544 nm. Four replications were used at each free ATF dosage and the experiment was repeated three times.
Cellular localization of nanoparticles in H1299 cell
1 ml suspended H1299 cells (1x104/ml) were seeded in confocal chamber slides (NEST) for 24 h at 37 °C under 5% CO2. Then the cells were incubated with ATF-HSA:CPZ@RRNP or HSA:CPZ@NP (final concentration: 0.5 µM) for 8 min. After washing with PBS (pH 7.2) three times, we observed these cell specimens by the Olympus FluoViewTMFV1000 laser scanning confocal microscope (Olympus Corp, Tokyo, Japan). In addition, the suspended H1299 cells (1x104/ml) were seeded in confocal chamber slides (NEST) for 24 h. Then the cells were incubated with 1 µM ATF-HSA:CPZ@RRNP or HSA:CPZ@NP for 12 h and washed with PBS (pH 7.2). Subsequently, the cells were incubated with a DNA fluorescent dye, DAPI (4', 6-diamidino-2-phenylindole) for 5 min, Lyso-Tracker for 60 min or Mito-Tracker for 30 min at room temperature, respectively (the ratio and incubation time were followed under the manufacturer's instruction). After washing with PBS (pH 7.2), we observed these cell specimens by the laser scanning confocal microscope. The fluorescence of cell specimens in the confocal chamber slides was excited by a laser light (λ = 633 nm for CPZ, 405 nm for DAPI, 577 nm for Lyso-Tracker and 490 nm for Mito-Tracker), while the emitted fluorescence was filtered with barrier filters (650/100 nm, 450/30 nm, 510/30 nm, 590/30 nm band pass, respectively). All parameters including the laser line intensity, photometric gain, settings of photo-multiplier tube and filter attenuation were kept constant throughout the entire experiment. All images were analyzed by Olympus Fluoview v2.1 software.
Establishment of Hepatoma-22 (H22) tumor-bearing Kunming mice model
Male Kunming mice (four weeks old, 18-22 g, purchased from Shanghai SLAC Laboratory Animal Co. Ltd., Shanghai, China) were fed and handled in accordance with the recommendations of the institutional animal care and use committee (IACUC) during the whole course of the experiment. The animals were allowed free access to water and food throughout all experimental processes. The mouse hepatocellular carcinoma cell line H22 was passaged weekly through Kunming mice with a body weight of 18-22 g in the form of ascites from the mouse abdominal cavity. To establish the tumor-bearing model, ascites containing H22 cells were harvested from the peritoneal cavity of a passaged mice about 6 days after inoculation. Subsequently, 200 μl of the ascites diluted with sterilized saline containing H22 cells (1.0×107/ml) were subcutaneously injected on the back. The tumors reached a volume about 60 mm3 in 4-5 days after the inoculation. For imaging and therapy purpose, the hair of the tumor-bearing mice was removed by shaving and chemical depilating cream (Shibi, Shanghai, China).
Fluorescent molecular tomography (FMT) imaging of ATF-HSA:CPZ@RRNP on H22 tumor-bearing mice
H22 tumor-bearing Kunming mice were established as above and randomly divided into three groups (5 mice in each group) with equivalent average starting tumor volume (~60 mm3) and body weight (~22 g). Here, we used HSA:CPZ@NP as a positive control and saline as a negative control. Mice with tumor were injected intravenously via the caudal vein with 200 μl nanoparticle (ATF-HSA:CPZ@RRNP or HSA:CPZ@NP, 0.050 mg CPZ/kg of mouse body weight) or saline. After the mice were anaesthetized with isoflurane, the tumor targeting property of the nanoparticles within the mice was detected at various time points (1 h, 2 h, 4 h, 8 h, 12 h, 24 h, 48 h, 72 h, 96 h) by using a fluorescent molecular tomography (FMT) 2500™ LX instrument (PerkinElmer, Waltham, USA). A 680 nm laser diode was used to excite the CPZ molecules in the mice body and the regions of interest (ROIs) were scanned with 50-60 source locations and default scan parameters (3 mm spacing between adjacent source locations). Long wavelength fluorescence emission (690-740 nm) was detected. To quantify the CPZ content, 1 μM ATF-HSA:CPZ@RRNP in saline was used as a standard to calibrate the instrument. The recorded images were reconstructed by the software TrueQuant v3.0 (PerkinElmer, Waltham, USA) to get the quantitative information in terms of the total amount (in picomoles) of fluorochrome by drawing ROIs around the tumor sites, non-tumor sites after the subtraction of auto-fluorescence from the tumor-bearing mice treated with saline. In order to prevent photosensitizer degradation, all mice were maintained in the dark environment after the injection of nanoparticles during the whole experiment period except the process of FMT measurements.
Photodynamic efficacy of ATF-HSA:CPZ@RRNP on H22 tumor-bearing mice
H22 tumor bearing mice (established as above) were randomly divided into four groups (10 mice in each group) with equivalent average starting tumor volume (~60 mm3) and body weight (~22 g) for photodynamic therapy (PDT). Two treated groups were injected 200 μl of ATF-HSA:CPZ@RRNP or HSA:CPZ@NP separately (both at a dosage of 0.050 mg CPZ/kg of mice body weight) via the caudal vein followed by continuous daily light illumination with a 680 nm light source (1 W, Luma Care Medical Group, Newport Beach, USA) for 3 min to get a light dosage of 50 J/cm2. Another control group was injected 200 μl of saline with daily light illumination as a control. The hair of tumor regions was shaved before light treatment in order to reduce the interference from hair at the tumor surface. The subcutaneous tumor volume was measured daily with a caliper and calculated using the ellipsoid volume formula π/6 x (length x width x height). Body weight was also recorded daily with a precise balance during 7 days experiment period. Mice were kept away from light during the experiment period in order to prevent CPZ degradation. After the completion of the photodynamic therapy, mice were sacrificed and the tumor was carefully dissected out and weighted.
Biodistribution and clearance of nanoparticles in H22 tumor-bearing mice
The H22 tumor-bearing Kunming mice were established and divided into a total of twelve groups with equivalent average starting tumor volume (~60 mm3) and body weight (~22 g) randomly. Tumor-bearing mice were injected intravenously via the caudal vein with 200 μl nanoparticle (ATF-HSA:CPZ@RRNP or HSA:CPZ@NP, 0.050 mg CPZ/kg of mouse body weight). At 6, 12, 24, 48, 72, 96 h post the injection, mice were sacrificed and their primary organs (liver, lung, spleen, kidneys, heart) were dissected out and washed by saline. After dried using a paper towel, these organs were put on sampling cassette and imaged by the FMT instrument using the same acquisition settings as for in vivo imaging.
Statistical analysis
All data represent group means and standard errors of the mean (SEM). The experimental data in vitro and in vivo were analyzed using the unpaired, 2-tailed Student t test. Differences at the 95% confidence level (p < 0.05) were considered to be statistically significant.
Abbreviations
ACPP: activatable cell-penetrating peptide; ATF: amino-terminal fragment of urokinase; Ce6: chlorine e6; CPP: cell-penetrating peptide; CPZ: mono-substituted β-carboxy phthalocyanine zinc; DAPI: 4', 6-diamidino-2-phenylindole; DEAE: diethylaminoethyl; DLS: dynamic light scattering; DMEM: dulbecco's modified eagle's medium; DMSO: dimethylsulfoxide; EGFR: epidermal growth factor receptor; FBS: fetal bovine serum; FMT: fluorescent molecular tomography; HELF: human embryo lung fibroblasts; HRP: horseradish peroxidase; HSA: human serum albumin; H1299: non-small cell lung carcinoma cell line H1299; H22: mouse hepatoma-22 cell line; IACUC: institutional animal care and use committee; MMP: matrix metalloproteinases; PDT: photodynamic therapy; POSS: polyhedral oligomeric silsesquioxane; ROIs: regions of interest; RRNP: receptor-responsive nanoparticle; SDS-PAGE: sodium dodecyl sulphate-polyacrylamide gel electrophoresis; TEM: transmission electron microscope; uPA: urokinase-type plasminogen activator; uPAR: urokinase-type plasminogen activator receptor.
Supplementary Material
Supplementary figures.
Acknowledgements
Our research work is financially supported by grants from National Key R&D Program of China (2017YFE0103200), Natural Science Foundation of China (31370737, 31400637, 31570745, 31670739, U1405229), and Natural Science Foundation of Fujian Province (2018J01897 and 2018J01729).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Swierczewska M, Liu G, Lee S, Chen X. High-sensitivity nanosensors for biomarker detection. Chem Soc Rev. 2012;41:2641-55
2. Ferrari M. Cancer nanotechnology: opportunities and challenges. Nat Rev Cancer. 2005;5:161-71
3. Wagner V, Dullaart A, Bock AK, Zweck A. The emerging nanomedicine landscape. Nat Biotechnol. 2006;24:1211-7
4. Davis ME, Zuckerman JE, Choi CH, Seligson D, Tolcher A, Alabi CA. et al. Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature. 2010;464:1067-70
5. Davis ME, Chen ZG, Shin DM. Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat Rev Drug Discov. 2008;7:771-82
6. He Q, Guo S, Qian Z, Chen X. Development of individualized anti-metastasis strategies by engineering nanomedicines. Chem Soc Rev. 2015;44:6258-86
7. Kamaly N, Yameen B, Wu J, Farokhzad OC. Degradable controlled-release polymers and polymeric nanoparticles: mechanisms of controlling drug release. Chem Rev. 2016;116:2602-63
8. Shi J, Kantoff PW, Wooster R, Farokhzad OC. Cancer nanomedicine: progress, challenges and opportunities. Nat Rev Cancer. 2017;17:20-37
9. Kiessling F, Mertens ME, Grimm J, Lammers T. Nanoparticles for imaging: top or flop? Radiology. 2014;273:10-28
10. Duncan R, Gaspar R. Nanomedicine(s) under the microscope. Mol Pharm. 2011;8:2101-41
11. Rizzo LY, Theek B, Storm G, Kiessling F, Lammers T. Recent progress in nanomedicine: therapeutic, diagnostic and theranostic applications. Curr Opin Biotechnol. 2013;24:1159-66
12. Kunjachan S, Jayapaul J, Mertens ME, Storm G, Kiessling F, Lammers T. Theranostic systems and strategies for monitoring nanomedicine-mediated drug targeting. Curr Pharm Biotechnol. 2012;13:609-22
13. Kircher MF, De La Zerda A, Jokerst JV, Zavaleta CL, Kempen PJ, Mittra E. et al. A brain tumor molecular imaging strategy using a new triple-modality MRI-photoacoustic-Raman nanoparticle. Nat Med. 2012;18:829-34
14. Kaittanis C, Shaffer TM, Ogirala A, Santra S, Perez JM, Chiosis G. et al. Environment-responsive nanophores for therapy and treatment monitoring via molecular MRI quenching. Nat Commun. 2014;5:3384
15. Miele E, Spinelli GP, Miele E, Tomao F, Tomao S. Albumin-bound formulation of paclitaxel (Abraxane ABI-007) in the treatment of breast cancer. Int J Nanomedicine. 2009;4:99-105
16. Barenholz Y. Doxil(R)-the first FDA-approved nano-drug: lessons learned. J Control Release. 2012;160:117-34
17. Rose PG. Pegylated liposomal doxorubicin: optimizing the dosing schedule in ovarian cancer. Oncologist. 2005;10:205-14
18. Chen H, Moore T, Qi B, Colvin DC, Jelen EK, Hitchcock DA. et al. Monitoring pH-triggered drug release from radioluminescent nanocapsules with X-ray excited optical luminescence. ACS Nano. 2013;7:1178-87
19. Shchukin DG, Sukhorukov GB, Mohwald H. Smart inorganic/organic nanocomposite hollow microcapsules. Angew Chem Int Ed Engl. 2003;42:4472-5
20. Li YL, Zhu L, Liu Z, Cheng R, Meng F, Cui JH. et al. Reversibly stabilized multifunctional dextran nanoparticles efficiently deliver doxorubicin into the nuclei of cancer cells. Angew Chem Int Ed Engl. 2009;48:9914-8
21. Wang C, Chen Q, Wang Z, Zhang X. An enzyme-responsive polymeric superamphiphile. Angew Chem Int Ed Engl. 2010;49:8612-5
22. Olson ES, Jiang T, Aguilera TA, Nguyen QT, Ellies LG, Scadeng M. et al. Activatable cell penetrating peptides linked to nanoparticles as dual probes for in vivo fluorescence and MR imaging of proteases. Proc Natl Acad Sci U S A. 2010;107:4311-6
23. Santos R, Ursu O, Gaulton A, Bento AP, Donadi RS, Bologa CG. et al. A comprehensive map of molecular drug targets. Nat Rev Drug Discov. 2017;16:19-34
24. Perona R. Cell signalling: growth factors and tyrosine kinase receptors. Clin Transl Oncol. 2006;8:77-82
25. Sutherland R, Delia D, Schneider C, Newman R, Kemshead J, Greaves M. Ubiquitous cell-surface glycoprotein on tumor cells is proliferation-associated receptor for transferrin. Proc Natl Acad Sci U S A. 1981;78:4515-9
26. Hilgenbrink AR, Low PS. Folate receptor-mediated drug targeting: from therapeutics to diagnostics. J Pharm Sci. 2005;94:2135-46
27. Hallahan D, Geng L, Qu SM, Scarfone C, Giorgio T, Donnelly E. et al. Integrin-mediated targeting of drug delivery to irradiated tumor blood vessels. Cancer Cell. 2003;3:63-74
28. Li D, Liu SL, Shan H, Conti P, Li ZB. Urokinase plasminogen activator receptor (uPAR) targeted nuclear imaging and radionuclide therapy. Theranostics. 2013;3:507-15
29. O'Halloran TV, Ahn R, Hankins P, Swindell E, Mazar AP. The many spaces of uPAR: delivery of theranostic agents and nanobins to multiple tumor compartments through a single target. Theranostics. 2013;3:496-506
30. Yang LL, Sajja HK, Cao ZH, Qian WP, Bender L, Marcus AI. et al. uPAR-targeted optical imaging contrasts as theranostic agents for tumor margin detection. Theranostics. 2014;4:106-18
31. LeBeau AM, Sevillano N, King ML, Duriseti S, Murphy ST, Craik CS. et al. Imaging the urokinase plasminongen activator receptor in preclinical breast cancer models of acquired drug resistance. Theranostics. 2014;4:267-79
32. Smith HW, Marshall CJ. Regulation of cell signalling by uPAR. Nat Rev Mol Cell Biol. 2010;11:23-36
33. Blasi F, Carmeliet P. uPAR: a versatile signalling orchestrator. Nat Rev Mol Cell Biol. 2002;3:932-43
34. Christensen A, Juhl K, Persson M, Charabi BW, Mortensen J, Kiss K. et al. uPAR-targeted optical near-infrared (NIR) fluorescence imaging and PET for image-guided surgery in head and neck cancer: proof-of-concept in orthotopic xenograft model. Oncotarget. 2017;8:15407-19
35. Persson M, Skovgaard D, Brandt-Larsen M, Christensen C, Madsen J, Nielsen CH. et al. First-in-human uPAR PET: imaging of cancer aggressiveness. Theranostics. 2015;5:1303-16
36. Huai Q, Mazar AP, Kuo A, Parry GC, Shaw DE, Callahan J. et al. Structure of human urokinase plasminogen activator in complex with its receptor. Science. 2006;311:656-9
37. Li R, Zheng K, Hu P, Chen Z, Zhou S, Chen J. et al. A novel tumor targeting drug carrier for optical imaging and therapy. Theranostics. 2014;4:642-59
38. Gratton SE, Ropp PA, Pohlhaus PD, Luft JC, Madden VJ, Napier ME. et al. The effect of particle design on cellular internalization pathways. Proc Natl Acad Sci U S A. 2008;105:11613-8
39. Sykes EA, Chen J, Zheng G, Chan WC. Investigating the impact of nanoparticle size on active and passive tumor targeting efficiency. ACS Nano. 2014;8:5696-706
40. Chauhan VP, Stylianopoulos T, Martin JD, Popovic Z, Chen O, Kamoun WS. et al. Normalization of tumour blood vessels improves the delivery of nanomedicines in a size-dependent manner. Nat Nanotechnol. 2012;7:383-8
41. Geng Y, Dalhaimer P, Cai S, Tsai R, Tewari M, Minko T. et al. Shape effects of filaments versus spherical particles in flow and drug delivery. Nat Nanotechnol. 2007;2:249-55
42. Kratz F. Albumin as a drug carrier: design of prodrugs, drug conjugates and nanoparticles. J Control Release. 2008;132:171-83
43. Kratz F, Elsadek B. Clinical impact of serum proteins on drug delivery. J Control Release. 2012;161:429-45
44. Abi-Habib RJ, Singh R, Liu SH, Bugge TH, Leppla SH, Frankel AE. A urokinase-activated recombinant anthrax toxin is selectively cytotoxic to many human tumor cell types. Mol Cancer Ther. 2006;5:2556-62
45. Chen JC, Chen NS, Huang JF, Wang JD, Huang MD. Derivatizable phthalocyanine with single carboxyl group: synthesis and purification. Inorg Chem Commun. 2006;9:313-5
46. Van Lier JE, Spikes JD. The chemistry, photophysics and photosensitizing properties of phthalocyanines. Ciba Found Symp. 1989;146:17-26
47. Jeong H, Huh M, Lee SJ, Koo H, Kwon IC, Jeong SY. et al. Photosensitizer-conjugated human serum albumin nanoparticles for effective photodynamic therapy. Theranostics. 2011;1:230-9
48. Zhou X, Zheng K, Li R, Chen Z, Yuan C, Hu P. et al. A drug carrier targeting murine uPAR for photodynamic therapy and tumor imaging. Acta Biomater. 2015;23:116-26
49. Czekay RP, Kuemmel TA, Orlando RA, Farquhar MG. Direct binding of occupied urokinase receptor (uPAR) to LDL receptor-related protein is required for endocytosis of uPAR and regulation of cell surface urokinase activity. Mol Biol Cell. 2001;12:1467-79
50. Nykjaer A, Conese M, Christensen EI, Olson D, Cremona O, Gliemann J. et al. Recycling of the urokinase receptor upon internalization of the uPA: serpin complexes. EMBO J. 1997;16:2610-20
51. Ghuman J, Zunszain PA, Petitpas I, Bhattacharya AA, Otagiri M, Curry S. Structural basis of the drug-binding specificity of human serum albumin. J Mol Biol. 2005;353:38-52
52. Zhu L, Yang F, Chen L, Meehan EJ, Huang M. A new drug binding subsite on human serum albumin and drug-drug interaction studied by X-ray crystallography. J Struct Biol. 2008;162:40-9
53. Langer K, Balthasar S, Vogel V, Dinauer N, Von Briesen H, Schubert D. Optimization of the preparation process for human serum albumin (HSA) nanoparticles. Int J Pharm. 2003;257:169-80
54. Weber C, Coester C, Kreuter J, Langer K. Desolvation process and surface characteristics of protein nanoparticles. Int J Pharm. 2000;194:91-102
55. Patil GV. Biopolymer albumin for diagnosis and in drug delivery. Drug Develop Res. 2003;58:219-47
56. Crisante F, Francolini I, Bellusci M, Martinelli A, D'Ilario L, Piozzi A. Antibiotic delivery polyurethanes containing albumin and polyallylamine nanoparticles. Eur J Pharm Sci. 2009;36:555-64
57. Yang L, Cui F, Cun D, Tao A, Shi K, Lin W. Preparation, characterization and biodistribution of the lactone form of 10-hydroxycamptothecin (HCPT)-loaded bovine serum albumin (BSA) nanoparticles. Int J Pharm. 2007;340:163-72
58. Yu S, Yao P, Jiang M, Zhang G. Nanogels prepared by self-assembly of oppositely charged globular proteins. Biopolymers. 2006;83:148-58
59. Qi JN, Yao P, He F, Yu CL, Huang CO. Nanoparticles with dextran/chitosan shell and BSA/chitosan core-doxorubicin loading and delivery. Int J Pharm. 2010;393:176-84
60. Chen Q, Wang X, Wang C, Feng L, Li Y, Liu Z. Drug-induced self-assembly of modified albumins as nano-theranostics for tumor-targeted combination therapy. ACS Nano. 2015;9:5223-33
61. Jenita JL, Chocalingam V, Wilson B. Albumin nanoparticles coated with polysorbate 80 as a novel drug carrier for the delivery of antiretroviral drug-efavirenz. Int J Pharm Investig. 2014;4:142-8
62. Merodio M, Arnedo A, Renedo MJ, Irache JM. Ganciclovir-loaded albumin nanoparticles: characterization and in vitro release properties. Eur J Pharm Sci. 2001;12:251-9
63. Sundar S, Kundu J, Kundu SC. Biopolymeric nanoparticles. Sci Technol Adv Mater. 2010;11:014104
64. Jahanshahi M, Babaei Z. Protein nanoparticle: a unique system as drug delivery vehicles. Afr J Biotechnol. 2008;7:4926-34
65. Dissanayake M, Kelly AL, Vasiljevic T. Gelling properties of microparticulated whey proteins. J Agric Food Chem. 2010;58:6825-32
66. Sheng Z, Hu D, Zheng M, Zhao P, Liu H, Gao D. et al. Smart human serum albumin-indocyanine green nanoparticles generated by programmed assembly for dual-modal imaging-guided cancer synergistic phototherapy. ACS Nano. 2014;8:12310-22
67. Lin T, Zhao P, Jiang Y, Tang Y, Jin H, Pan Z. et al. Blood-brain-barrier-penetrating albumin nanoparticles for biomimetic drug delivery via albumin-binding protein pathways for antiglioma therapy. ACS Nano. 2016;10:9999-10012
68. Jeong B, Bae YH, Kim SW. Drug release from biodegradable injectable thermosensitive hydrogel of PEG-PLGA-PEG triblock copolymers. J Control Release. 2000;63:155-63
69. Sun Y, Wang Q, Chen J, Liu L, Ding L, Shen M. et al. Temperature-sensitive gold nanoparticle-coated pluronic-PLL nanoparticles for drug delivery and chemo-photothermal therapy. Theranostics. 2017;7:4424-44
70. Thomas CR, Ferris DP, Lee JH, Choi E, Cho MH, Kim ES. et al. Noninvasive remote-controlled release of drug molecules in vitro using magnetic actuation of mechanized nanoparticles. J Am Chem Soc. 2010;132:10623-5
71. Skirtach AG, Munoz Javier A, Kreft O, Kohler K, Piera Alberola A, Mohwald H. et al. Laser-induced release of encapsulated materials inside living cells. Angew Chem Int Ed Engl. 2006;45:4612-7
72. You J, Zhang GD, Li C. Exceptionally high payload of doxorubicin in hollow gold nanospheres for near-infrared light-triggered drug release. ACS Nano. 2010;4:1033-41
73. Karimi M, Eslami M, Sahandi-Zangabad P, Mirab F, Farajisafiloo N, Shafaei Z. et al. pH-sensitive stimulus-responsive nanocarriers for targeted delivery of therapeutic agents. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2016;8:696-716
74. Pu YJ, Chang S, Yuan H, Wang G, He B, Gu ZW. The anti-tumor efficiency of poly(L-glutamic acid) dendrimers with polyhedral oligomeric silsesquioxane cores. Biomaterials. 2013;34:3658-66
75. Hu QY, Katti PS, Gu Z. Enzyme-responsive nanomaterials for controlled drug delivery. Nanoscale. 2014;6:12273-86
76. Jiang T, Olson ES, Nguyen QT, Roy M, Jennings PA, Tsien RY. Tumor imaging by means of proteolytic activation of cell-penetrating peptides. Proc Natl Acad Sci U S A. 2004;101:17867-72
77. Yuan C, Huai Q, Bian CB, Huang MD. The expression, purification and crystallization of monomeric soluble human urokinase receptor. Sheng Wu Hua Xue Yu Sheng Wu Wu Li Jin Zhan. 2006;33:277-81
78. Zhao G, Yuan C, Bian C, Hou X, Shi X, Ye X. et al. Protein expression and preliminary crystallographic analysis of amino-terminal fragment of urokinase-type plasminogen activator. Protein Expr Purif. 2006;49:71-7
Author contact
![]() Corresponding authors: HMD_labedu.cn or zhengke_qustcom
Corresponding authors: HMD_labedu.cn or zhengke_qustcom