Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results and Discussion
Conclusion
Methods
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(3):837-852. doi:10.7150/thno.30228 This issue Cite
Research Paper
Anti-EGF Receptor Aptamer-Guided Co-Delivery of Anti-Cancer siRNAs and Quantum Dots for Theranostics of Triple-Negative Breast Cancer
Min Woo Kim1,2, Hwa Yeon Jeong1, Seong Jae Kang1, In Ho Jeong1, Moon Jung Choi1, Young Myoung You1, Chan Su Im1, In Ho Song4, Tae Sup Lee4, Jin Suk Lee5, Aeju Lee2,3, Yong Serk Park1 ![]()
1. Department of Biomedical Laboratory Science, Yonsei University, Wonju, Republic of Korea
2. International Research Organization for Advance Science and Technology (IROAST), Kumamoto University, Kumamoto, Japan
3. Magnesium Research Center, Kumamoto University, Kumamoto, Japan
4. Division of RI-Convergence Research, Korea Institute of Radiological and Medical Science, Seoul, Republic of Korea
5. Department of Anatomy, Yonsei University Wonju Collage of Medicine, Wonju, Republic of Korea
Received 2018-9-27; Accepted 2018-11-28; Published 2019-1-25
Abstract

Many aptamers have been evaluated for their ability as drug delivery vehicles to target ligands, and a variety of small interfering RNAs (siRNAs) have been tested for their anti-cancer properties. However, since these two types of molecules have similar physicochemical properties, it has so far been difficult to formulate siRNA-encapsulating carriers guided by aptamers. Here, we propose aptamer-coupled lipid nanocarriers encapsulating quantum dots (QDs) and siRNAs for theragnosis of triple-negative breast cancer (TNBC).
Methods: Hydrophobic QDs were effectively incorporated into lipid bilayers, and then therapeutic siRNAs were complexed with QD-lipid nanocarriers (QLs). Finally, anti-EGFR aptamer-lipid conjugates were inserted into the QLs for TNBC targeting (aptamo-QLs). TNBC-targeting aptamo-QLs were directly compared to anti-EGFR antibody-coupled immuno-QLs. The in vitro delivery of therapeutic siRNAs and QDs to target cells was assessed by flow cytometry and confocal microscopy. The in vivo targeting of siRNAs to tumors and their therapeutic efficacy were evaluated in mice carrying MDA-MB-231 tumors.
Results: Both types of EGFR-targeting QLs showed enhanced delivery to target cancer cells, resulting in more effective gene silencing and enhanced tumor imaging compared to non-targeting control QLs. Moreover, combinatorial therapy with Bcl-2 and PKC-ι siRNAs loaded into the anti-EGFR QLs was remarkably effective in inhibiting tumor growth and metastasis.
Conclusion: In general, the aptamo-QLs showed competitive in vivo delivery and therapeutic efficacy compared to immuno-QLs under the same experimental conditions. Our results show that the anti-EGFR aptamer-guided lipid carriers may be a potential theranostic delivery vehicle for RNA interference and fluorescence imaging of TNBCs.
Keywords: nanotheranostics, anti-EGFR aptamer, lipid nanocarriers, quantum dots, gene therapy
Introduction
Nucleic acid aptamers are short single-stranded oligonucleotides with high affinity and specificity to a broad range of target molecules; therefore, they are referred to as “chemical antibodies” [1]. Aptamers are artificially selected ligands that can be easily synthesized and modified [2]. Owing to certain advantages such as rapid and inexpensive synthesis, low immunogenicity, and efficient delivery to a variety of cells, aptamers have been suggested for targeted diagnostic and therapeutic applications in cancer treatment [3]. Compared to conventional antibodies, aptamers are artificially synthesized and screened by the in vitro selection method of systemic evolution of ligands by exponential enrichment (SELEX), so they can be easily modified [4]. Thus, modification of aptamers with several other types of oligonucleotides such as siRNAs, miRNAs, and anti-miRNAs is a promising method for gene delivery [5-7]. Among these, siRNAs have been the most extensively tested for aptamer-guided gene therapeutics [8].
siRNA molecules function in RNA interference (RNAi) and enable target gene expression to be knocked down by a series of reactions involving the RNA-induced silencing complex (RISC) and endoribonuclease Dicer [9]. Although the history of siRNA therapeutics is short, it has garnered remarkable attention from the pharmaceutical industry [10]. RNAi technology has several challenges, including toxicity, delivery efficiency, and stability [11]. Therefore, to allay these concerns, therapeutic siRNA molecules must be precisely delivered to the intended target tissues.
Aptamer and siRNA conjugates have exhibited specific binding to targeted cells, superior internalization into tumor tissues, as well as RNAi-mediated target gene silencing for cancer therapy [12]. Despite these advantages, the nucleic acid complexes are easily degraded by nucleases present in biological fluids [13]. Even if therapeutic genes arrive at their destination, it is difficult to deliver them to the cytoplasm, the site of gene expression, because they are easily degraded by endosomal enzymes [14]. Therefore, an appropriate siRNA delivery vehicle that protects siRNAs from biological obstacles is an essential constituent for RNAi-mediated therapy.
Various types of cationic lipid nanocarriers have been widely utilized as siRNA delivery systems since they have advantages with respect to formulation, immunogenicity, and safety [15]. The most remarkable advantages of cationic lipid nanocarriers are their efficient cellular uptake, which is comparable to viral vector systems, and endosomal escape capability. Moreover, the multi-layer structure of lipids is able to accommodate a large number of siRNA molecules and protect them when exposed to an in vivo environment [16]. However, their off-targeting remains a major hurdle to be overcome for clinical applications.
Aptamer-guided targeting of cationic lipid nanocarriers containing siRNAs is a potentially feasible approach to reduce the off-targeting of the carriers. However, aptamers and siRNAs share similar physicochemical characteristics; therefore, an accurate method of preparation is required to form a stable and effective formulation, with the aptamer exposed to the outside and the siRNA complexed inside. Recently, we reported aptamer-coupled cationic nanoparticles carrying siRNAs and quantum dots (QDs) for theranostic applications [17]. Here, we present evidence supporting the potential clinical application of this system for tumor-targeted theranostics.
Bcl-2 is a well-known apoptosis-inhibiting protein and is down-regulated in all major types of cell death [18]. On the other hand, PKC-ι is involved in cancer development [19] and cancer metastasis [20, 21]. Therefore, it is conceivable that EGF receptor (EGFR)-targeted delivery of Bcl-2 and PKC-ι siRNAs may inhibit the proliferation and metastasis of triple-negative breast cancers over-expressing EGFR. Tumor-targeting cationic nanocarriers were carefully prepared in 2 steps: complete complexation of 2 anti-cancer siRNA therapeutics (Bcl-2 and PKC-ι) with cationic lipids and simultaneous QD incorporation, and then conditioned insertion of anti-EGF receptor aptamer-lipid conjugates. The aptamer-guided theranostic efficiency was compared to an antibody-guided system in cultured cells and an animal model.
Results and Discussion
This study aimed to provide a practical example of a theranostic application of anti-EGFR aptamer-coupled cationic lipid nanocarriers containing anti-cancer siRNAs and QDs (aptamo-QLs). The anti-EGFR aptamo-QLs were compared to anti-EGFR antibody-coupled cationic nanocarriers (immuno-QLs) and non-targeting cationic nanocarriers (QLs) in terms of EGFR-directed targeting efficiency and anti-cancer therapeutic efficacy. Scheme 1 illustrates the tumor-targeted theranostics of the aptamer-coupled cationic nanocarriers containing QDs and therapeutic siRNAs.
Schematic of the theranostic strategy for RNAi gene therapy and fluorescence tumor imaging. Anti-EGFR aptamer-conjugated lipid nanocarriers encapsulating QDs and anti-cancer siRNAs were prepared and intravenously administered. The administered lipid nanocarriers presumably extravasated through the leaky tumor vasculature and then targeted primary tumors via specific recognition of EGF receptors overexpressed on MDA-MB-231 tumors. The delivered QDs and therapeutic siRNAs provide fluorescence tumor images and inhibitory effects on tumor growth.
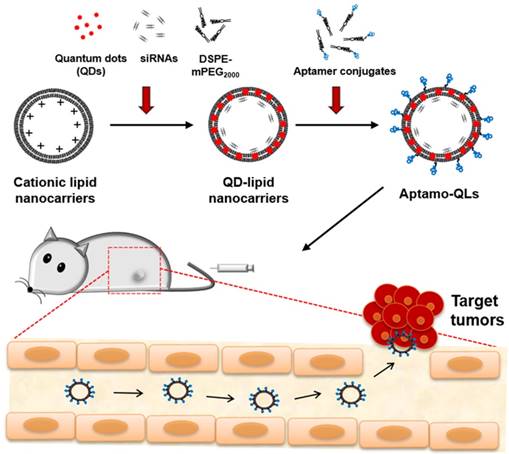
Preparation and characterization of aptamo-QLs and immuno-QLs
Preparation of aptamo-QLs was performed in 3 sequential steps: preparation of polyethylene glycol (PEG)ylated cationic liposomes containing QDs, siRNA complexation with the cationic liposomes, and a second PEGylation with insertion of aptamer-lipid conjugates to form aptamo-QLs. Addition of accurate amounts of polyethylene glycol (PEG)-lipid conjugates was required to solubilize hydrophobic QDs in the lipid bilayers. Sometimes PEGylation can be harmful for nanocarrier formulation unless it is carefully controlled in terms of amount and process [22]. The lipid nanocarriers containing 4 mol% DSPE-mPEG2000 exhibited the highest QD incorporation rate (94.9% ± 4.8%) and formed vesicles 175 ± 5 nm in diameter (Table S1), an appropriate size for in vivo delivery [23]. Moreover, this formulation still possessed a certain amount of cationic charge (8.0 ± 3.4 mV, Table S1) for interaction with anionic siRNA molecules. Formulations with a lower or higher content of DSPE-mPEG2000 exhibited inefficient QD capture or less siRNA complexation, respectively. Accommodation of QDs and siRNA molecules by the 2 separate steps was beneficial for maintaining vesicle size, high QD incorporation, and effective siRNA complexation (over 80% siRNA loading efficiency).
According to measurements made by dynamic light scattering, CLs encapsulating QDs and siRNAs exhibited 224.2 ± 6.6 nm in diameter with 51.8 ± 1.3 mV surface charge. After the first PEGylation with 4 mol% DSPE-mPEG2000 followed by siRNA complexation, QLs were 160.4 ± 7.9 nm in diameter with 0.4 ± 1.9 mV surface charge. The second PEGylation was performed with an additional 4 mol% PEG lipids containing anti-EGFR aptamer-conjugated DSPE-PEG2000 (0.2 mol%, Figures S1-S2). The final aptamo-QLs showed a -2.7 ± 0.6 mV surface charge and a vesicle size of 171.7 ± 4.0 nm. Anti-EGFR immuno-QLs that were prepared by the same procedure exhibited a -1.9 ± 0.7 mV surface charge and vesicle diameter of 175.5 ± 9.0 nm. The QD encapsulation significantly condensed the particles, but increased PDIs from 0.12 ± 0.03 to 0.37 ± 0.05. Meanwhile, the ligand coupling slightly increased the particle size without significant PDI changes, but further reduced their surface charges. The finalized QL formulations exhibited relatively broader dispersity, but still keep a certain level of homogeneity (Figure S3). These QL formulations did not show any inherent cytotoxicity up to 0.15 mM lipid and 1 μM siRNA (Figure S4).
Structural stability of aptamo-QLs and immuno-QLs
Protection of siRNAs in the QL formulations from ribonuclease digestion was verified by agarose gel electrophoresis (Figure 1A). The integrity of the siRNAs in the lipid nanocarriers was estimated by lysis of the lipid nanocarriers followed by digestion with RNase A. All the formulations showed complete complexation of siRNAs with cationic liposomes at a 4:1 N/P ratio (Figure 1A). However, the cationic lipid nanocarriers (CLs) and QLs showed only 17.1% ± 8.2% and 30.2% ± 4.5% intact siRNA remaining (Figure 1B). This implies that siRNAs are not effectively protected by the cationic liposomes at the 4:1 N/P ratio. Under the same conditions, the aptamo-QLs and immuno-QLs protected the siRNA molecules more effectively, exhibiting 81.0% ± 3.0% and 65.1% ± 11.2% intact siRNA, respectively (Figure 1B). An additional 4 mol% of PEG lipids containing anti-EGFR aptamer or anti-EGFR antibody provided QLs with further structural stability.
siRNA protection and serum stability of aptamo-QLs and immuno-QLs. (A) Lipid nanocarriers were incubated with RNase A and siRNAs were then recovered after treatment with Triton® X-100. The recovered siRNAs were electrophoresed on agarose gel. Lane 1, free siRNA; lane 2, RNase A-treated free siRNA; lane 3, lipid nanocarriers; lane 4, RNase A-treated lipid nanocarriers; and lane 5, RNase A/Triton X®-100-treated lipid nanocarriers. The bands in red squares indicate the remaining intact siRNAs. (B) Residual ratios of intact siRNA were estimated by comparison with the amount of siRNA added. Each error bar represents the mean ± S.D. for 3 separate experiments. ***p < 0.001 vs. CLs. (C) Lipid nanocarriers were incubated in the presence of 50% FBS at 37°C and their changes in size were examined by dynamic light scattering. Each error bar represents the mean ± S.D. for 3 separate experiments. (D) Lipid nanocarriers were incubated in the presence of 50% FBS for 5 days, washed, and electrophoresed on SDS-PAGE. Lane 1, markers; lane 2, FBS; lane 3, QLs; lane 4, aptamo-QLs; lane 5, immuno-QLs; lane 6, CLs.
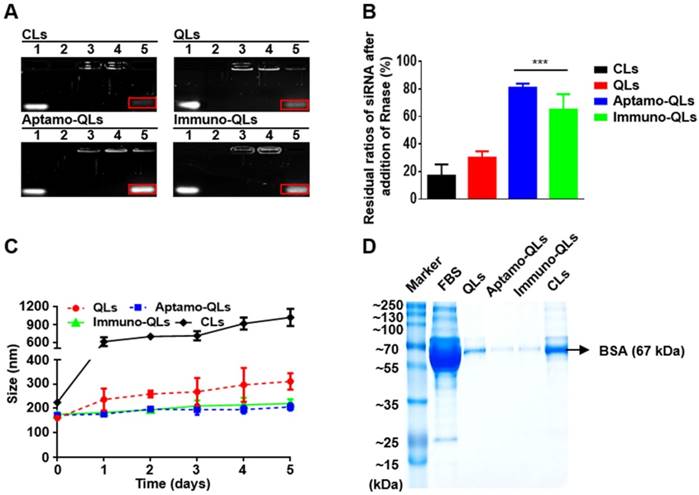
To evaluate the physiological stability of aptamo-QLs and immuno- QLs, changes in vesicle size and serum protein binding were analyzed in the presence of 50% fetal bovine serum (FBS). According to the analysis of particle size distribution and polydispersity index (PDI) (Figure 1C and Figure S3), the major portions of aptamo-QLs and immuno-QLs showed little change at least for 2 days, from 171.7 ± 4.0 nm (PDI, 0.33 ± 0.04) to 196.9 ± 2.9 nm (PDI, 0.49 ± 0.02) and from 175.5 ± 9.0 (PDI, 0.35 ± 0.03) to 193.9 ± 6.9 nm (PDI, 0.51 ± 0.05), respectively. Since then the particles began to be degraded or aggregated. Under the same conditions, a large amount of the QLs were degraded on day 1, showing their serious fragility, in addition to a certain increase in vesicle size, from 160.4 ± 8.0 nm to 310.4 ± 33.7 nm over 5 days. The CLs showed a marked increase at day 1, from 224.2 ± 6.6 nm to 610.8 ± 73.8 nm, and continuous aggregation. Serum proteins can non-specifically bind to the cationic surface of CLs and destabilize vesicles, resulting in membranous fusion. To determine the extent of serum protein binding, the prepared lipid nanocarriers were incubated with 50% FBS and then washed with saline by centrifugation. A large amount of serum proteins remained bound to the CLs even after washing (Figure 1D). Meanwhile, the almost-neutralized PEGylated QLs exhibited markedly reduced binding of serum proteins, and the aptamo-QLs and the immuno-QLs showed even further reduced amounts of serum protein binding. The most prevalent protein bound to the lipid nanocarriers appeared to be albumin (67 kDa).
In general, siRNA molecules are readily degraded in the bloodstream, and cationic lipid nanocarriers at a controlled size do not show efficient passive targeting in a biological environment because of undesirable opsonization of their surface with serum proteins, leading to their elimination by the mononuclear phagocyte system (MPS) [24]. Thus, the cationic moieties of lipid nanocarriers complexed with siRNA should be effectively covered to prevent their exposure during blood circulation. Transmission electron microscopy (TEM) of the lipid nanocarriers indicated stable QD deposition between the leaflets of lipid bilayers and siRNA molecules localized between the bilayers (Figure S5). The multi-lamellar structure of the PEGylated lipid nanocarriers provides an appropriate environment to protect against QD oxidation and siRNA degradation. These results imply that the QDs and siRNAs in the aptamo-QLs and the immune-QLs were protected from physical and chemical degradation in the biological environment.
Target cell-specific siRNA and QD delivery
EGFR-expressing MDA-MB-231 and EGFR-negative MDA-MB-453 breast cancer cell lines (Figure S6) were treated with the prepared nanocarriers containing FITC-labeled siRNA and EGFR-specific cell binding was analyzed by flow cytometry (Figure 2A). The naked QLs with a weak positive surface charge were able to efficiently bind to MDA-MB-231 (245.6 MFI; mean fluorescence intensity) as well as MDA-MB-453 (100.7 MFI). The QL binding to both cell lines was not diminished by pretreatment with the competitive inhibitor cetuximab (228.9 and 117.0 MFI, respectively). Meanwhile, the aptamo-QLs showed high binding to the target MDA-MB-231 cells (274.5 MFI), but not to non-target cell MDA-MB-453 cells (19.6 MFI). This target-specific binding affinity was equivalent to the immuno-QLs, which showed 282.1 and 21.2 MFI to MDA-MB-231 and MDA-MB-453 cells, respectively. Binding to the target cells for both nanocarriers was significantly diminished by pretreatment with competitive inhibitors, anti-EGFR aptamer, or cetuximab (38.9 and 93.9 MFI, respectively). These results imply that the strong binding to the target cell was mediated by EGF receptors on the surface of tumor cells.
EGFR-mediated delivery of QDs and siRNA by aptamo-QLs and immuno-QLs. MDA-MB-231 and MDA-MB-453 cells were treated with the lipid nanocarriers containing QDs and FITC-siRNA for 1 h, with or without 1 h competitor (anti-EGFR aptamer or antibody) pretreatment. (A) Cellular binding was verified with flow cytometry analysis of QDs. (B) MDA-MB-231 and (C) MDA-MB-453 cells were further incubated for 8 h and observed with a fluorescence confocal microscope (scale bars, 50 µm; inset scale bar, 10 µm). Cells were treated with QLs (upper), aptamo-QLs (middle), and immuno-QLs (lower). Nuclei were stained with DAPI (blue). (D) MDA-MB-231 cells were treated with the aptamo-QLs or immuno-QLs and then incubated in the presence of LysoTracker Red. The treated cells were observed 1 h and 8 h later with a fluorescence confocal microscope (scale bars, 10 µm).
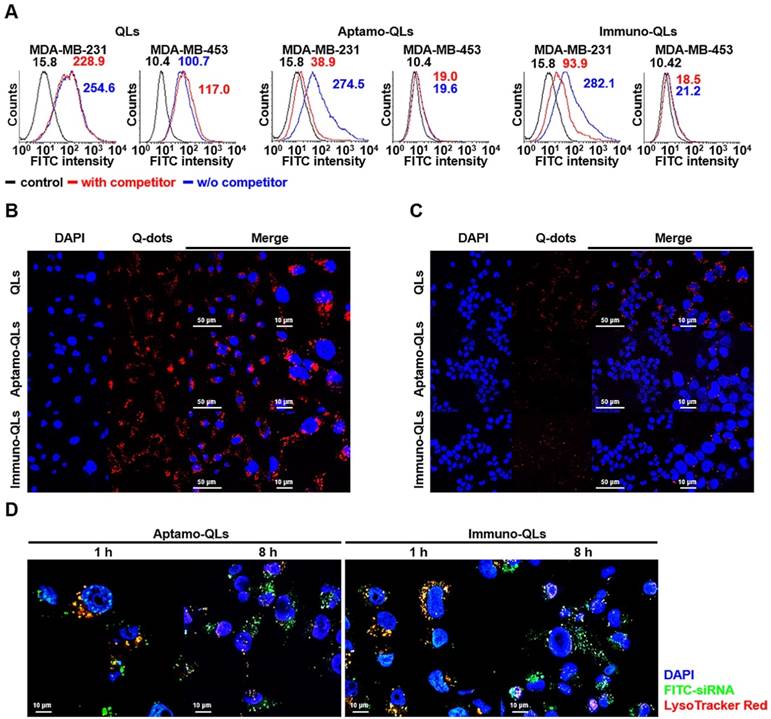
The same treated cancer cells were also examined with a fluorescence confocal microscope to assess the target-specific QD delivery (Figure 2B-C). Generally, the CLs showed substantial QD delivery to both MDA-MB-231 target cancer cells and MDA-MB-453 non-target cells. Meanwhile, the tumor-targeting aptamo-QLs and immuno-QLs were able to deliver QDs to MDA-MB-231 cells more efficiently than the nontargeting QLs. However, these systems were not efficient in QD delivery to the non-target cells. MDA-MB-453 cells treated with the aptamo-QLs or immuno-QLs showed only minor fluorescence signal localized in the marginal area of plasma membranes. These data clearly show that the non-targeting CLs and QLs were able to deliver their cargo to tumor cells, regardless of the expression level of the EGF receptor, presumably via non-specific charge interactions. However, the aptamo-QLs could specifically recognize cancer cells over-expressing EGFR and their cell binding was mediated by interactions between anti-EGFR aptamers and their target molecules on the cell surface. The EGFR-targeted binding capability of the aptamo-QLs was comparable to that of the immuno-QLs.
Many previous studies have emphasized the importance of establishing a strategy for intracellular uptake following nanoparticle arrival at targeted cells [25]. Generally, the EGF receptor-mediated intracellular entry is known to be clathrin-mediated endocytosis, also commonly referred to as receptor-mediated endocytosis. Specific ligand binding to EGF receptors on the plasma membranes triggers endocytosis into the cell. During the process of endocytosis, therapeutics must escape from endosomal vesicles, otherwise they are degraded in lysosomes [26]. In this study, siRNAs in the prepared aptamo-QLs and immuno-QLs were efficiently translocated into target tumor cells, and then escaped from the endosomes. At 1 h after treatment of MDA-MB-231 cells with the aptamo-QLs or immuno-QLs, siRNA molecules were largely localized in the endosomes (Figure 2D). However, 7 h later, the amount of siRNA in the endosomes decreased, while siRNA outside the endosomes increased and spread throughout the cytoplasm. These results show the efficient endosomal escape of siRNAs delivered by the aptamo-QLs and immuno-QLs. Presumably, the cationic lipid carriers enter the cells by EGF receptor-mediated endocytosis and the fully protonated cationic head groups of lipids in early endosomes interact with the anionic lipids in the endosomal membranes, resulting in reorganization of the lipid bilayers. Hence, siRNAs become dissociated from the lipid nanocarriers and then diffuse into the cytosol [27].
In vitro anti-cancer efficacy of Bcl-2 and PKC-ι siRNA
To evaluate the gene silencing activity of siRNAs, MDA-MB-231 cells were transfected with varied amounts of QLs of Bcl-2 or PKC-ι siRNA. After incubation for 24 h, the mRNA and protein levels of Bcl-2 and PKC-ι were determined by RT-PCR (Figure 3A) and western blotting (Figure 3B), respectively. The cells treated with the QLs encapsulating Bcl-2 or PKC-ι siRNAs showed a dose-dependent reduction in their target gene expression. These results imply that the siRNA molecules were efficiently transfected by the QLs and the transfected siRNA molecules were functionally active in terms of target mRNA interference.
Anti-cancer effects of Bcl-2 and PKC-ι siRNA transfected with aptamo- or immune-QLs. The mRNA and protein levels of Bcl-2 and PKC-ι in the transfected MDA-MB-231 cells were determined by RT-PCR (A) and western blotting (B). The viability of the transfected MDA-MB-231 cells (200 nM siRNA each) was measured by the CCK-8 assay after 48 h incubation (C). Each bar represents the mean ± S.D. from 5 separate experiments. ***p < 0.001 vs. control. (D) Invasion of MDA-MB-231 cells transfected with PKC-ι siRNA was measured by counting the invaded cells through membranes per field. *p < 0.05 vs. control. Migration of the transfected cells was also compared with that of control cells by examining the wound healing with a microscope (E) and analyzing the percentage of wound closure with Image J software (F). Each bar represents the mean ± S.D. from 3 separate experiments. ***p < 0.001 vs. 0 h.
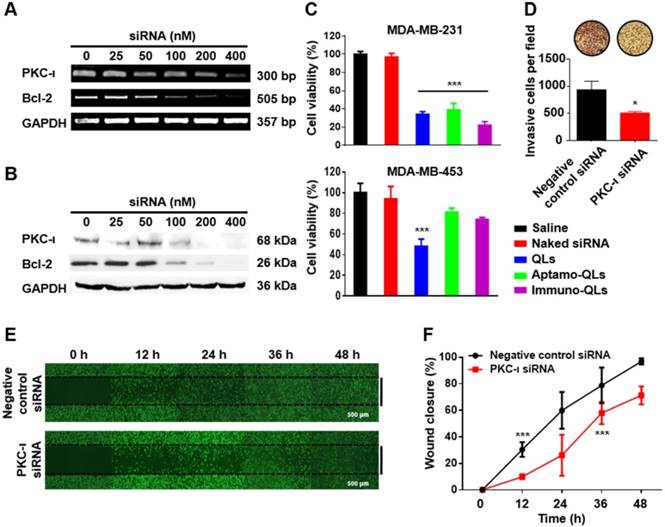
In fact, the inhibition of Bcl-2 and PKC-ι expression yielded anti-cancer activities in the target MDA-MB-231 cells, which are known to be highly invasive. To verify these anti-cancer activities, the cells were transfected with Bcl-2 and/or PKC-ι siRNAs using the aptamo- or immune-QLs and the cell viability was measured 48 h later (Figure S7). The EGFR-targeted transfection of Bcl-2 siRNA using aptamo- or immune-QLs showed the same level of cytotoxicity in the target cells as that induced by non-specific transfection by QLs. The same treatment did not elicit a strong cytotoxicity in the control MDA-MB-453 cells, while the transfection with QLs was still significantly toxic (Figure 3C). This implies that the anti-EGFR aptamo-QLs as well as immune-QLs were able to actively deliver siRNAs, and the delivered Bcl-2 siRNAs were toxic to the target cancer cells. Meanwhile, PKC-ι siRNA alone did not show strong cytotoxicity even at the highest concentration (400 nM siRNA) at which PKC-ι expression was completely abolished (Figure S7). Interestingly, PKC-ι siRNA transfection provided an additive toxicity to the cells treated with Bcl-2 siRNA.
The combinatorial transfection aimed at 2 different target genes, Bcl-2 and PKC-ι, may be a suitable strategy to enhance the effectiveness of anti-cancer therapeutics. It has been well documented that Bcl-2, an apoptosis inhibiting protein, is highly correlated with autophagic cell death, apoptosis, and necrosis [18, 28]. In contrast, PKC-ι is involved in various cell signaling pathways regulating cell plasticity, cell-cell adhesion, cancer metastasis, and even autophagy [20, 29-31]. Therefore, it is conceivable that the simultaneous reduction of Bcl-2 and PKC-ι expression may synergistically inhibit cancer proliferation and metastasis, and promote stronger autophagic cell death. In fact, the combined siRNA treatment exhibited the lowest cytotoxicity at all tested concentrations of siRNA.
Additionally, in order to further study the role of PKC-ι expression in cancer progression, MDA-MB-231 cells were transfected with PKC-ι siRNA, and their migration was examined using transwell invasion assay and wound healing assay. The MDA-MB-231 cells transfected with PKC-ι siRNA showed less penetration through the well membranes compared to the cells transfected with control siRNA (Figure 3D). Moreover, the wound healing assay revealed that the MDA-MB-231 cells transfected with PKC-ι siRNA exhibited slower migration than the control cells, resulting in slower closure of the scratches made in cell cultures (Figure 3E). The scratched area of MDA-MB-231 cells transfected with negative control siRNA were almost completely recovered in 48 h, while those transfected with PKC-ι siRNA remained unrecovered (Figure 3F). Therefore, it can be presumed that PKC-ι downregulation may be one approach to inhibit tumor invasion and migration.
In vivo fluorescence imaging and biodistribution
The active targeting capability of aptamo-QLs was evaluated in MDA-MB-231 xenografts in mice. In vivo time lapse images of MDA-MB-231 xenografts were taken after intravenous administration of QLs, aptamo-QLs, or immuno-QLs (Figure 4A). The mice injected with aptamo-QLs or immuno-QLs showed higher levels of red QD fluorescence in tumor tissues grown on the ventral side, compared to those treated with untargeted QLs. The fluorescence intensity was the most obvious in the tumors among the organs. The fluorescence in tumors was not strong at 1 h post-injection regardless of the type of QLs administered. However, the fluorescence signals of tumors were highest at 4 h post-injection and then decreased. Images of excised tumors and frozen-sectioned tumors also showed that aptamo-QLs and immuno-QLs provided more efficient QD delivery to the tumor tissues when compared with the non-targeting QLs (Figure S8).
In vivo fluorescence imaging of MDA-MB-231 xenografts, and biodistribution of aptamo-QLs and immuno-QLs in mice. (A) In vivo fluorescence images of tumor-bearing mice intravenously treated with QLs (upper), aptamo-QLs (middle), and immuno-QLs (lower) (0.2 mg lipid/mouse, n=3) were taken with a Maestro 2 in vivo imaging system at 1, 4, 8, and 24 h post-injection. The auto-fluorescence from mice is pseudocolored white and the unmixed QD signal is pseudocolored red. Black arrows indicate tumors. (B) After fluorescence imaging of the whole mouse body, major organs including tumors were excised and their fluorescence signals were measured at varied time points. (C) Fluorescence images of excised organs from the mice treated with QLs (upper), aptamo-QLs (middle), and immuno-QLs (lower) were captured at 4 h post-injection. (D) Changes in fluorescence signal in tumor tissues was also measured as time elapsed. (E) The tumor-to-liver ratios were compared at varied time points. Each bar represents the mean ± S.D. (n=3). *p < 0.05, **p < 0.01, ***p < 0.001 vs. QLs or †p < 0.05 between groups.
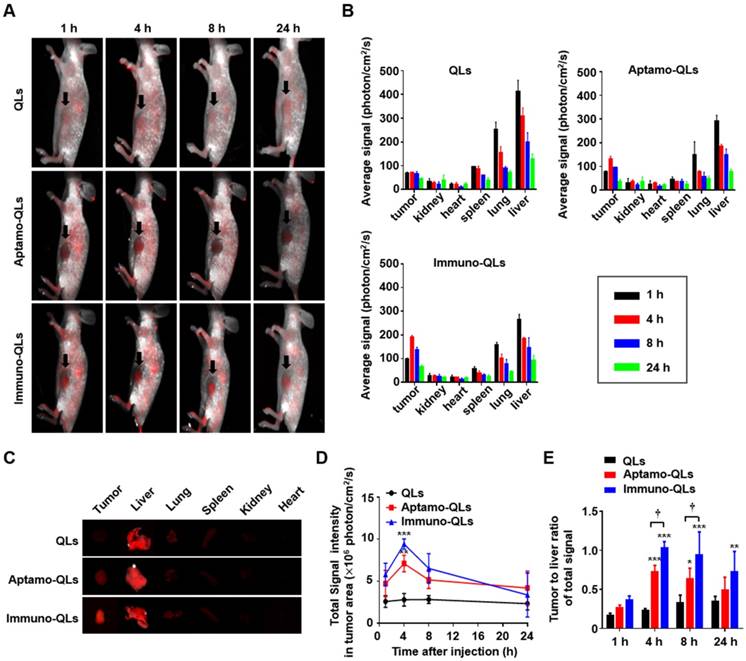
The pharmacokinetics and biodistribution of the prepared nanocarriers were assessed by QD fluorescence. According to the biodistribution analysis (Figure 4B), all nanocarriers largely accumulated in the liver and lungs at the earliest time point (1 h post-injection). As time elapsed, QD accumulation in the tumors increased, but there was still a considerable amount of QDs in the reticuloendothelial organs, particularly in the liver. The strong QD signals in the liver, lungs, and spleen slowly decreased as time elapsed regardless of the type of QD carrier. The QD fluorescence became obvious in the tumors resected from the mice treated with aptamo-QLs or immuno-QLs at 4 h post-injection (Figure 4C-D). At this time point, the aptamo-QLs exhibited a relatively weaker QD fluorescence than that of the immune-QLs. Nevertheless, the highest fluorescence signal was still observed in the liver regardless of the QL formulation. However, there were statistically significant changes in terms of the tumor-to-liver ratio among the QL formulations (Figure 4E). In general, the tumor-targeting QLs exhibited higher tumor-to-liver ratios compared to the non-targeting QLs. Between the two formulations, the anti-EGFR immuno-QLs exhibited a markedly higher tumor-to-liver ratio of fluorescence signal than that of the aptamo-QLs. However, it has to be aware that the surface fluorescence imaging has evident limitations for pharmacokinetic analysis of the QL formulations because the fluorescence surface images cannot quantitatively represent the total fluorescence of organs.
The in vitro binding (Figure 2) and siRNA delivery (Figure 3) to MDA-MB231 target cells directly supported EGFR-mediated cell binding and siRNA transfection. These in vivo tumor imaging data also indirectly suggest that the EGFR-targeting aptamers and antibodies were functionally exposed by their carriers so that they could effectively interact with EGF receptors on the surface of TNBC. Between the targeting ligands, the anti-EGFR antibody (cetuximab) appeared to be more effective in tumor targeting than the aptamer molecules. In fact, under the same experimental conditions, antibodies coupled to QLs were more efficient in delivering QDs and siRNAs to target cells. The stronger tumor-targeting activity of the anti-EGFR antibody could be attributed to its inherent higher binding affinity (Kd: 0.15-0.2 nM) [32], compared to the anti-EGFR aptamer (Kd: 0.62 nM) [33]. The difference in binding affinity may result from the different numbers of epitope binding sites. The antibody molecules retaining 2 antigen determinant sites would presumably have a stronger binding affinity to target cells than the aptamer molecules, which have one binding site. Therefore, at the same concentration of ligand on the nanoparticle surface (approximately 80 ligands per particle) aptamo-QLs may be less effective in target cell binding, compared to immune-QLs. Moreover, the anionicity of aptamers may negatively affect their outward positioning on the surface of nanocarriers consisting of cationic lipids.
In vivo anti-cancer efficacy of Bcl-2 and PKC-ι siRNA
The anti-cancer therapeutic efficacies of tumor-targeting QLs encapsulating Bcl-2 and/or PKC-ι siRNA were evaluated in MDA-MB-231 tumor-bearing mice (Figure 5A). Mice were intravenously treated 3 times at intervals of 3 days and tumor growth in each group of mice was monitored for 31 days (Figure 5B). Compared to the mice treated with saline or the non-targeting QLs, mice with tumor-targeting QLs encapsulating Bcl-2 siRNA exhibited inhibition of tumor growth during treatment for 31 days. Moreover, tumor-targeting aptamo-QLs or immuno-QLs co-encapsulating Bcl-2 and PKC-ι siRNA inhibited the tumor growth even further. In fact, the mice treated with both of the siRNAs showed a slower tumor growth compared to mice treated with Bcl-2 siRNA, regardless of QL formulations. However, in vitro transfection with PKC-ι alone had little effect on the proliferation of MDA-MB-231 cells (Figure S7). A mechanism of the additive anti-cancer effect of Bcl-2 and PKC-ι siRNA remains to be verified in detail.
Inhibition of tumor growth and TUNEL staining of tumor sections from mice treated with aptamo-QLs and immuno-QLs. (A) Schematic of cancer treatment protocol and analysis over time. (B) Tumor-xenograft mice (n=5) were intravenously administered saline, QLs encapsulating Bcl-2 siRNA, aptamo-QLs encapsulating Bcl-2 siRNA, immuno-QLs encapsulating Bcl-2 siRNA, aptamo-QLs encapsulating Bcl-2 and PKC-ι siRNA, and immuno-QLs encapsulating Bcl-2 and PKC-ι (10 mg siRNA/kg), every 3 days. Tumor growth was measured every 3 days. *p < 0.05, **p < 0.01 and ***p < 0.001 between experimental groups. (C) All mice were sacrificed at 31 days after injection, tumor weights were measured, and (D) the gross appearance of tumors was also examined. **p < 0.01 and ***p < 0.001 vs. saline-treated. (E) TUNEL-positive nuclei in representative tumor sections were counted. Scatter dot plots with mean ± S.D. (n=6) represent the percent TUNEL-positive nuclei to total nuclei counts. ***p < 0.001 vs. saline-treated.
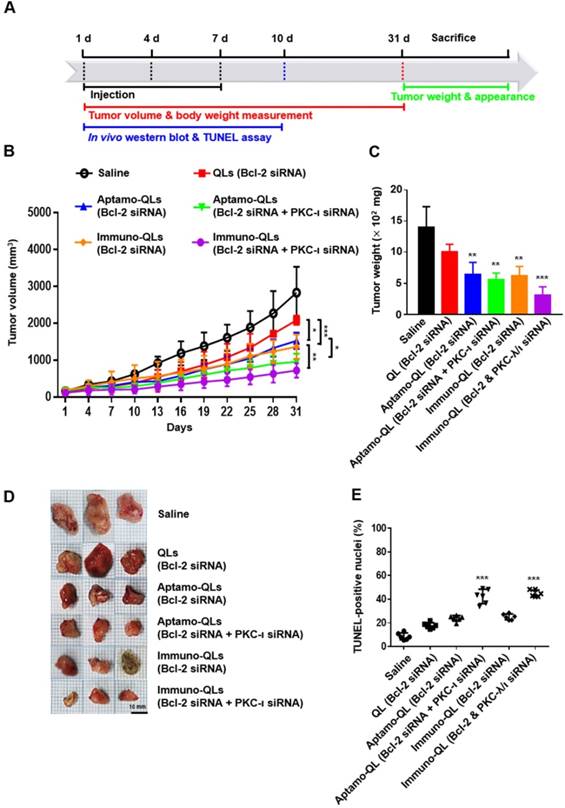
Between the tumor-targeting carriers, the anti-EGFR immune-QLs appeared to be more effective in terms of tumor growth inhibition, exhibiting a slower tumor growth (Figure 5B) and smaller tumors at day 31 (Figure 5C-D). On average, the tumors taken from the mice treated with the immuno-QLs encapsulating Bcl-2 and PKC-ι siRNAs were the smallest, even compared to those from mice treated with aptamo-QLs. These in vivo data also suggest that combinatorial transfection of the 2 siRNAs showed smaller tumors compared to transfection of Bcl-2 alone. And, under the same conditions, the tumor-targeting QLs were more efficient siRNA carriers compared to the non-targeting QLs.
To verify the reduction in target gene expression, in vivo protein levels of Bcl-2 and PKC-ι in tumors was analyzed by western blotting (Figure S9). Expression of Bcl-2 and PKC-ι proteins were markedly reduced by 3 repeated administrations of aptamo-QLs and immuno-QLs encapsulating the respective siRNAs. This suggests that the anti-cancer therapeutic siRNAs were efficiently transfected to the target tumors by the EGFR-targeting QLs, and the effective siRNA transfection presumably resulted in growth inhibition of MDA-MB-231 xenografts in mice.
According to TUNEL assay performed on the 10th day after the first treatment (Figure 5E and Figure S10), tumor growth inhibition was presumably due to apoptosis of the transfected tumor cells. TUNEL-positive cells in the tumor tissues of mice treated with tumor-targeting QLs encapsulating Bcl-2 and PKC-ι siRNAs were higher (aptamo-QLs; 42.37% ± 6.07%, immuno-QLs; 44.89% ± 2.87%) than those of all other groups. Mice transfected with only Bcl-2 siRNA using aptamo-QLs, immuno-QLs, or non-targeting QLs also showed higher numbers of TUNEL-positive cells in their tumors (23.94% ± 2.38%, 25.10% ± 2.52%, and 19.33% ± 2.46%, respectively), compared to saline-treated mice (8.63% ± 2.93%). The reduced tumor growth could be due to increased apoptosis induced by simultaneous reduction of Bcl-2 and PKC-ι expressions.
According to the in vitro transfection data (Figure 3 and Figure S7), transfection with PKC-ι siRNA almost completely blocked expression of the gene but did not affect the proliferation of MDA-MB-231 cells on its own. However, reduction of both Bcl-2 and PKC-ι exhibited an additive effect on tumor growth in vitro and in vivo. In addition, blocking of PKC-ι expression markedly affected pulmonary metastasis of MDA-MB-231 cells in mice and growth of metastasized tumors (Figures S11-S12). The mechanism underlying the additive anti-cancer effect of Bcl-2 and PKC-ι siRNA remains to be verified in detail. However, the results of animal studies also suggest that the combinatorial knockdown of Bcl-2 and PKC-ι genes could be a feasible strategy for cancer treatment.
In vivo toxicity of aptamo-QLs and immuno-QLs
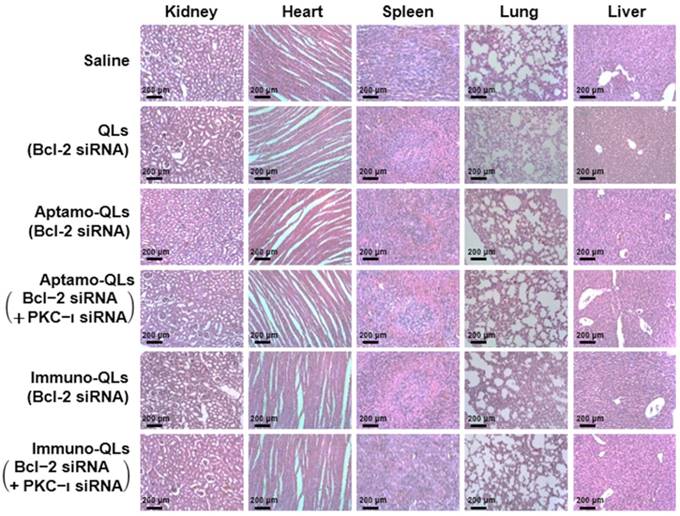
To examine the toxicity of the QL formulations, body weights of treated mice were measured during the 31 days of the experiment (Figure S13). The body weights of QL-treated mice were initially slightly increased and then became steady as time elapsed, similar to those of saline-treated control mice. There was no significant body weight loss resulting from administration of the QL formulations. Moreover, the major organs, including kidneys, heart, spleen, lungs, and liver, resected from the treated mice on day 31 did not exhibit any substantial histological abnormalities such as inflammation and premature cell death (Figure 6).
Histopathological examination of internal organs of mice treated with aptamo-QLs and immuno-QLs. Major organs including kidneys, heart, spleen, lungs, and liver were resected on day 31 after treatment. Paraffin-embedded sections of the organs stained with H&E were observed with a light microscope (scale bar, 200 µm).
Even though the EGFR-targeted QL nanocarriers exhibited no substantial inherent toxicity in vitro and normal histology in vivo, it is too early to state the QL formulations are a safe siRNA delivery vehicle for human use. There has been also concern expressed regarding the safety of cadmium QDs utilized in this study, but we did not observe any toxicity related to the QDs. A number of reports have claimed that cadmium QDs are biocompatible and safely secreted from the body [34]. Nevertheless, it remains uncertain if lipid nanocarriers containing QDs have long-term toxicity. QDs that are safer and more biocompatible than heavy metal-based QDs would be a more feasible option for application in molecular imaging. In recent years, a variety of QD modification methods have been suggested and different types of QDs have been developed for nanomedicines [35, 36]. For example, carbon-based QDs emitting in the second near infrared II region (λ1,000-1,700) may offer the advantages of safety, deeper penetration, and low auto-fluorescence [37]. Further advances in QD technologies will eliminate issues regarding QD toxicity.
Conclusion
This study demonstrates an example of combinatorial anti-cancer treatment using Bcl-2 siRNA interfering with tumor cell proliferation and PKC-ι inhibiting tumor cell migration. We formulated anti-EGFR aptamo-QLs simultaneously containing two different types of siRNA therapeutics for tumor-targeted delivery. These vehicles were able to efficiently deliver targeting anti-cancer therapeutic siRNAs as well as fluorescent QDs to tumor tissues. Aptamo-QLs were comparable with cetuximab-coupled immune-QLs in terms of cargo delivery efficiency and inhibitory effect on cancer progression. The delivered siRNAs effectively reduced target gene expression, resulting in inhibition of tumor growth. Moreover, simultaneously delivered QDs provided fluorescence signals in internal organs and tumors. These fluorescence signals revealed valuable information regarding the biodistribution of the lipid nanocarriers. The anti-EGFR aptamo-QLs and immuno-QLs could be utilized as theranostic delivery systems capable of treatment and fluorescence imaging of cancers, especially TNBCs.
Methods
Materials
1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (DSPE-mPEG2000), cholesterol, and 1,2-distearoyl-sn-glycero-3- phosphoethanolamine-N-[maleimide(polyethylene glycol)-2000] (DSPE-PEG2000-maleimide) were purchased from Avanti Polar Lipid, Inc. (Alabaster, AL, USA). O,O′-dimyristyl-N-lysyl glutamate (DMKE) cationic lipid was provided by KOMA Biotech (Seoul, Republic of Korea). CdSe/ZnS High Quality Organic NSQDs (6 nm in diameter, λemit = 620 nm) were purchased from Nanosquare Inc. (Seoul, Republic of Korea). Anti-EGFR antibodies (Cetuximab, Erbitux®) were purchased from Merck KgaA (Darmstadt, Germany). Anti-EGFR DNA aptamer with a sulfhydryl group at the 3'-end was purchased from Aptamer Science Inc. (Pohang, Republic of Korea). AccuTarget™ fluorescein-labeled scrambled siRNA (SN-1023), AccuTarget™ scrambled siRNA (SN-1003), and predesigned Bcl-2 siRNA (No.1011940) were purchased from BioNeer Inc. (Daejeon, Republic of Korea). PKC-ι siRNA (SC-36257) was purchased from Santa Cruz Biotechnology (Dallas, TX, USA).
Cell lines and cell culture
Human breast adenocarcinoma MDA-MB-231 (HTB-26™) and human breast metastatic carcinoma MDA-MB-453 (HTB-131™) cells were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). MDA-MB-231 cells expressing firefly luciferase, MDA-MB-231-Luc, were kindly provided by Dr. Tae-Sup Lee (Korea Institute of Radiological and Medical Sciences, Seoul, Republic of Korea). MDA-MB-231 and MDA-MB-453 cells were maintained in Leibovitz's L-15 Medium (Gibco, Carlsbad, CA, USA) supplemented with 10% FBS (Gibco), 100 IU/mL penicillin (Gibco) and 100 μg/mL streptomycin (Gibco) in a humidified atmosphere and CO2-free at 37 °C. MDA-MB-231-Luc cells were maintained in Dulbecco's modified Eagle's medium (DMEM, Gibco) with 10% FBS, 100 IU/mL penicillin and 100 μg/mL streptomycin in a humidified atmosphere of 95% air and 5% CO2 at 37 °C.
Primers and antibodies
The mRNA levels of GAPDH, Bcl-2, and PKC-ι were determined by PCR amplification by using GAPDH primers (forward; 5'-CGGGAAGCTTGTCATCAATGG-3', reverse; 5'-GGCAGTGATGGCATGGACTG-3'), Bcl-2 primers (forward; 5'-CATTTCCACGTCAACAATCATTTCCACGTCAACAATTG-3', reverse; 5'-AGCACAGGATTGGATATTCCAT-3'), and PKC-ι (sc-36257-PR, Santa Cruz Biotechnology). The protein levels of GAPDH, Bcl-2, PKC-ι, and EGFR were determined by western blotting using GAPDH (CB1001, Millipore, Darmstadt, Germany), Bcl-2 (sc-7382, Santa Cruz Biotechnology), PKC-ι (sc-17837, Santa Cruz Biotechnology), and EGFR (2232S, Cell Signaling Technology, Danvers, MA, USA) antibodies.
Conjugation of aptamer or antibody
Equal volumes of thiolated aptamers (100 μM) and dithiothreitol (DTT, 10 mM) were mixed in TE buffer (pH 7.5) and then incubated at room temperature for 1 h. The reaction solution was passed through a PD-10 column (GE Healthcare, Chicago, IL, USA) to remove the remnant DTT. Subsequently, the reactive aptamers were added to DSPE-mPEG2000- maleimide in a chloroform and methanol mixture (2:1, v/v) at a molar ratio of 1:1 and then incubated for 16 h at room temperature with continuous stirring. The mixture was centrifuged in a 100 kDa Amicon tube (Merck Millipore, Darmstadt, Germany) for 10 min at 4,000 ×g. The aptamer conjugation yield and its purity were determined by electrophoresis on a 1.5% agarose gel.
Cetuximab (2 mg) was thiolated for 1 h at room temperature by reacting with 0.01 mg Traut's Reagent (Thermo Scientific, Rockford, IL, USA) in HEPES-EDTA buffer (25 mM HEPES, 140 mM NaCl, 2 mM EDTA, pH 8.0). Unreacted Traut's Reagent was removed by passing the reaction through a PD-10 column. The thiolated antibodies were immediately added to a mixture of DSPE-PEG2000-maleimide/ DSPE-mPEG2000 (1:4 molar ratio) at a molar ratio of 0.2:1 (cetuximab:lipid) and incubated overnight at 4 °C with continuous stirring. The conjugate of cetuximab and lipid was purified using chromatography on Sepharose CL-4B columns (GE Healthcare) in HEPES buffer (pH 7.4). The conjugated antibody was verified in 6% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gels.
Preparation of lipid nanocarriers encapsulating QDs and siRNAs
Lipid nanocarriers for QDs and siRNAs were prepared according to a previously reported procedure [17]. DMKE, cholesterol, DSPE-mPEG2000, and QDs (5:1, weight ratio of lipids to QDs) were dissolved in a mixture of chloroform and methanol (2:1, v/v) (Table S2). The organic solvent was evaporated under a stream of N2 gas and completely removed by vacuum-drying for 1 h. The dried residuals containing 1 mg of lipids and 200 μg of QDs were hydrated in 1 mL of saline and then vigorously mixed. The hydrated solution was sonicated 3 times for 20 min each at 10 min intervals. The QD concentration was estimated by measuring the absorbance at 310 nm wavelength with an Infinite 200 Pro NanoQuant (TECAN Group Ltd., Männedorf, Switzerland). Then, siRNAs were added to the lipid nanocarriers QDs (4:1 N/P ratio) and incubated with continuous mixing at 1,000 rpm (IKA vortex genius 3, Sigma-Aldrich, St. Louis, MO, USA) for 30 min at room temperature to ensure stable complexation of siRNAs and cationic lipid nanocarriers encapsulating QDs. The cationic lipid nanocarriers encapsulating QDs and siRNAs were referred to as QLs. Finally, additional DSPE-mPEG2000 (3.8 mol%) and the DSPE-mPEG2000-aptamer (0.2 mol%) (or DSPE- mPEG2000-antibody) were added to the prepared QLs followed by incubation for 4 h at 37°C [38]. The anti-EGFR aptamer-conjugated lipid nanocarriers encapsulating QDs and siRNAs were referred to as aptamo-QLs and their antibody-coupled version was referred to as immuno-QLs (Table 1).
Vesicular size and ζ-potential of QLs (Mean ± SD)
| Particle size (nm) | ζ -potential (mV) | |
|---|---|---|
| a) CLs b) QLs c) Aptamo-QLs d) Immuno-QLs | 224.2 ± 6.6 | 51.8 ± 1.3 |
| 160.4 ± 7.9 | 0.4 ± 1.9 | |
| 171.7 ± 4.0 | -2.7 ± 0.6 | |
| 175.5 ± 9.0 | -1.9 ± 0.7 |
a) Commercial cationic lipid nanocarriers, EzWay™ transfection
b) Cationic lipid nanocarriers encapsulating QDs and siRNA
c) Anti-EGFR aptamer conjugated QLs d) Anti-EGFR antibody conjugated QLs
Characterization of QLs, aptamo-QLs, and immuno-QLs
During preparation of the lipid nanocarriers, the sizes and ζ-potentials of DMKE cationic lipid nanocarriers (CLs), QLs, aptamo-QLs, and immuno- QLs were measured using dynamic light scattering (DLS) with a Zetasizer Nano-ZS90 (Malvern Instruments Ltd., Malvern, UK). All measurements were repeated 5 times.
Analysis of RNase protection and serum-stability
To evaluate siRNA protection from RNase digestion, 4 μL of ribonuclease A (RNase A, 20 μg/mL) was added to the lipid nanocarriers (25 pmol siRNA) and incubated for 1 h. The reaction was stopped by adding 10 μL stopping solution (0.5 M EDTA) and then further incubated for 1 h at room temperature. The reaction mixtures were incubated in the presence of 0.1% Triton® X-100 detergent for 2 h to release siRNA from the lipid nanocarriers and then electrophoresed as described above.
At the same time, CLs, QLs, aptamo-QLs, and immuno-QLs were added to FBS (1:1, v/v) and then incubated for varied time periods (24, 48, 72, 96, and 120 h) at 37°C. At the scheduled time points, changes in vesicle size were measured with a Zetasizer Nano-ZS90. At the last time point, the mixture of FBS and lipid nanocarriers was centrifuged at 10,000 × g for 10 min and washed with saline 3 times. The washed precipitates were collected and electrophoresed on a 10% SDS-PAGE gel at 120 V for 1.5 h. Profiles of serum proteins bound to lipid nanocarriers were observed by Coomassie blue staining of the gel.
Analysis of cell binding and cellular uptake
QLs, aptamo-QLs, and immuno-QLs encapsulating 50 pmol FITC-siRNA were prepared as described above. The prepared lipid nanocarriers were added to EGFR-positive MDA-MB-231 cells and EGFR-negative MDA-MB-453 cells (5×105 cells in 200 μL per tube). The lipid nanocarriers were also added to the cancer cells pretreated with 100 nM anti-EGFR aptamer or cetuximab for 1 h at 4 °C. The treated cells were washed twice in cold PBS (pH 7.4) containing 0.1% bovine serum albumin (BSA). The lipid nanocarrier binding to the cancer cells was analyzed using FACS Calibur flow cytometer (BD Biosciences, San Jose, CA, USA) and CellQuest software (BD Biosciences).
MDA-MB-231 and MDA-MB-453 cells (5×105 cells/well) were seeded on cover-slips in 6-well plates and cultured for 24 h. The prepared QLs, aptamo- QLs, and immuno-QLs (3 μg of QDs) were added to the cells and then incubated for 4 h at 37 °C in serum-free medium. After incubation, the cells were washed twice with cold PBS (pH 7.4) and fixed with 2% paraformaldehyde. The cells were stained with one drop of 4′,6-diamidino-2-phenylindole (DAPI) solution (Vector lab, Burlingame, CA, USA) for 30 min in the dark and mounted on slides. The slides were observed using a confocal laser scanning microscope (LSM 510; Zeiss, Heidenheim, Germany).
In a separate experiment, the cancer cells were treated with aptamo-QLs or immuno-QLs encapsulating FITC-siRNA (100 pmol) and then incubated at 37°C for varied times. The dye for tracking acidic endosomes, Lysotracker Red DND-99 (100 nM; Invitrogen, Carlsbad, CA, USA), was added to each well and then incubated for 1 h. The treated cells were observed using a confocal laser scanning microscope as described above.
Measurement of cell toxicity
Cell growth inhibition by transfection with anti-cancer therapeutic siRNAs treated with QLs, aptamo-QLs, and immuno-QLs was measured using the Cell Counting Kit-8 (CCK-8, Dojindo Laboratories, Kumamoto, Japan). MDA-MB-231 and MDA-MB-453 cells were seeded into 96-well plates (1×104 cells/well). The cells were treated with lipid nanocarriers of Bcl-2 and/or PKC-ι siRNAs in serum-free medium for 4 h and then further incubated for 48 h. After incubation, 10 μL of CCK-8 solution was added to the cells, which were incubated for a further 2 h. The absorbance at 450 nm was measured with the Infinite 200 Pro NanoQuant.
In vitro transfection of Bcl-2 and PKC-ι siRNAs
MDA-MB-231 cells seeded in 6-well plates (5×105 cells/well) were transfected with QLs encapsulating Bcl-2 or PKC-ι siRNA in serum-free medium for 4 h and then further incubated in medium with 10% FBS for 24 h. Total RNA was isolated from MDA-MB-231 cells using Trizol reagent (Invitrogen) according to the manufacturer's instructions. cDNA was then synthesized by reverse transcription with 2 μg of the isolated RNA, 1 μL 2.5 mM dNTPs (Cosmo Genetech, Seoul, Republic of Korea), 0.25 μg random primer (Invitrogen), and 200 U Moloney murine leukemia virus reverse transcriptase (M-MLV RT, Invitrogen) for 50 min at 37 °C and then 10 min at 70 °C. Subsequent reverse transcription polymerase chain reaction (RT-PCR) using HiPi PCR PreMix (ELPIS Biotech, Daejeon, Republic of Korea) was performed in a thermocycler according to the manufacturer's recommendations. PCR products were electrophoresed on 2% agarose gels prepared in TBE buffer, stained for 10 min with ethidium bromide (EtBr), and de-stained for 20 min in tap water. Gel images were captured using a Gel Doc EQ system (Bio-Rad Laboratories, Hercules, CA, USA).
Western blot analyses were performed to verify reduction of target gene expression by siRNA transfection. The cells were transfected as described above and then lysed in RIPA buffer (Sigma-Aldrich) with Halt™ protease inhibitor (Thermo Scientific). The lysate was centrifuged at 19,000 ×g for 5 min at 4 °C. The total concentration of cellular proteins was determined using a protein standard curve. The protein samples were electrophoresed on 10% PAGE gel (120 V, 1.5 h), and then transferred to nitrocellulose blotting membranes (Pall Corp., Port Washington, NY, USA) for 1.5 h at 350 mA. Protein blots were detected by SuperSignal® West Pico (Thermo Scientific) or Dogen EZ-Western Lumi Femto (Daeil Lab Service Co., Ltd, Seoul, Republic of Korea) using Fusion Solo Chemidoc (Vilber Lourmat).
In vitro wound healing and transwell invasion assay
MDA-MB-231 cells (5×105 cells) were treated with QLs encapsulating PKC-ι siRNA (200 nM) in serum-free medium for 4 h and then further incubated in medium with 10% FBS for 24 h. The transfected cells were harvested, seeded in SPL Scar™ Block (SPL, Pocheon, Republic of Korea) (5×104 cells/block), and cultured in serum-free medium for 12 h. Then, a straight scratch (500 μm in width) was made in each well by removing a block stuck on the plate using tweezers. The cells were washed with PBS three times and further incubated at 37 °C in medium with 0.1% FBS. Healing of scratched wounds was observed under a light microscope (CKX41, Olympus, Tokyo, Japan) using image analysis software (IMT i-Solution Inc., Quebec, Canada). The wound healing images were captured at the same site 0, 12, 24, 36, and 48 h later. The experiment was repeated three times.
The MDA-MB-231 cells transfected with PKC-ι siRNA as described above were seeded in the chambers of SPL Insert™ Hanging transwells (8 μm, SPL Life Science) (5×104 cells/chamber). The chambers were inserted in 24-well plates and incubated in serum-free medium for 12 h. Medium containing 0.1% FBS was added to the upper chambers while the lower well was filled with medium containing 20% FBS. Then, the transwell system was incubated for 48 h at 37 °C in an atmosphere with 5% CO2. After incubation, the remaining cells in the upper side of the chamber membranes were removed with a swab. The membrane was trimmed and stained with 0.1% crystal violet and observed under a light microscope (Olympus) using image analysis software (IMT i-Solution Inc.). Three fields were randomly selected from each membrane and the number of invaded cells was counted at a magnification of ×200.
In vivo fluorescence imaging of tumor xenografts
All animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of Yonsei University at Wonju (YWCI-201605-002-01) and performed in accordance with their guidelines and regulations. To prepare tumor xenografts, 6-week-old female BALB/c nude mice (Orient Bio, Seongnam, Republic of Korea) were subcutaneously inoculated with 200 μL MDA-MB-231 cells (1 × 107) in medium mixed with Matrigel (BD Biosciences) (1:1 volume ratio) at the 4th mammary fat pad. Tumor volumes were measured with calipers and calculated using the equation: tumor volume = length × width2 / 2. When the tumors grew to ~ 200 mm3, the mice were injected with the prepared QLs, aptamo-QLs, and immuno-QLs via the tail vein (10 mg lipid/kg). The treated mice were placed in a prone position on the bed and kept under anesthesia with 2% isoflurane. Fluorescence images of tumors were acquired using the Maestro 2 in vivo imaging system (Caliper Life Sciences, Hopkinton, MA, USA) (λex: 550 nm, λemit: 620 nm) at 1, 4, 8, and 24 h post-injection. After in vivo fluorescence imaging, the mice were immediately sacrificed by CO2 asphyxiation, and major organs including tumor tissues were resected. The fluorescence signal from each organ was measured using the Maestro 2 in vivo imaging system.
In vivo tumor growth inhibition
When volumes of MDA-MB-231 tumor xenografts reached ~200 mm3, tumor-bearing mice were randomly divided into 6 groups (n=5). The mice were intravenously administered saline, QLs encapsulating Bcl-2, aptamo-QLs encapsulating Bcl-2 siRNA, immuno-QLs encapsulating Bcl-2 siRNA, aptamo-QLs encapsulating Bcl-2 and PKC-ι siRNA, and immuno-QLs encapsulating Bcl-2 and PKC-ι siRNA (2:1 molar ratio Bcl-2 siRNA to PKC-ι siRNA) 3 times with 2-day intervals (10 mg/kg of lipid nanocarriers, equivalent to 1.67 nmol of siRNA). For 31 days, tumor volumes and body weights of the mice were measured once every 3 days. All mice were sacrificed at day 31 after injection, and the tumor weight and dimensions were measured.
Histological study
Mice treated with various QLs were sacrificed at 31 days post-injection and their major organs including tumors were resected. The organs were formalin-fixed, embedded in paraffin, and sectioned at 8 μm thickness. The tissue sections were stained with hematoxylin and eosin (H&E) and tissue slides were photographed by optical microscopy (Leica, Wetzlar, Germany) and rendered using Leica software.
Statistical analysis
Data are represented as mean ± standard deviation (S.D.). Statistical analysis was performed with one-way or two-way ANOVA using Prism 6 (GraphPad Software, Inc., La Jolla, CA, USA). *p < 0.05, **p < 0.01 and ***p < 0.001 vs. control or between experimental groups. †p < 0.05 between experimental groups.
Abbreviations
aptamo-QLs: anti-EGFR aptamer-coupled QLs; ATCC: American Type Culture Collection; BSA: bovine serum albumin; CLs: cationic lipid nanocarriers; DAPI: 4′,6-diamidino-2-phenylindole; DLS: dynamic light scattering; DMEM: Dulbecco's modified Eagle's medium; DMKE: O,O′-dimyristyl-N- lysyl glutamate; DSPE-mPEG2000: 1,2-distearoyl-sn- glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000]; DSPE-PEG2000-maleimide: 1,2- distearoyl-sn-glycero-3-phosphoethanolamine-N-[maleimide(polyethylene glycol)-2000]; DTT: dithiothreitol; EtBr: ethidium bromide; FBS: fetal bovine serum; H&E: hematoxylin and eosin; immune-QLs: anti- EGFR antibody-coupled QLs; MFI: mean fluorescence intensity; M-MLV RT: moloney murine leukemia virus reverse transcriptase; MPS: mononuclear phagocyte system; PEG: polyethylene glycol; QDs: quantum dots; QLs: QD-lipid nanocarriers; RISC: RNA-induced silencing complex; RNAi: RNA interference; RNase A: ribonuclease A; RT-PCR: reverse transcription polymerase chain reaction; SELEX: systemic evolution of ligands by exponential enrichment; S.D.: standard deviation; SDS-PAGE: sodium dodecyl sulfate-polyacrylamide gel electrophoresis; siRNAs: small interfering RNAs; TEM: transmission electron microscopy; TNBC: triple negative breast cancer.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This research was supported by the National Research Foundation of Korea (NRF-2016R1D1A1B03935847).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Keefe AD, Pai S, Ellington A. Aptamers as therapeutics. Nat Rev Drug Discov. 2010;9:537-50
2. Chen K, Liu B, Yu B, Zhong W, Lu Y, Zhang J. et al. Advances in the development of aptamer drug conjugates for targeted drug delivery. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2017:9
3. Hori SI, Herrera A, Rossi JJ, Zhou J. Current advances in aptamers for cancer diagnosis and therapy. Cancers (Basel). 2018:10
4. Darmostuk M, Rimpelova S, Gbelcova H, Ruml T. Current approaches in SELEX: An update to aptamer selection technology. Biotechnol Adv. 2015;33:1141-61
5. Esposito CL, Nuzzo S, Kumar SA, Rienzo A, Lawrence CL, Pallini R. et al. A combined microRNA-based targeted therapeutic approach to eradicate glioblastoma stem-like cells. J Control Release. 2016;238:43-57
6. Wang T, Gantier MP, Xiang D, Bean AG, Bruce M, Zhou SF. et al. EpCAM aptamer-mediated survivin silencing sensitized cancer stem cells to doxorubicin in a breast cancer model. Theranostics. 2015;5:1456-72
7. Catuogno S, Rienzo A, Di Vito A, Esposito CL, de Franciscis V. Selective delivery of therapeutic single strand antimiRs by aptamer-based conjugates. J Control Release. 2015;210:147-59
8. Xiang D, Shigdar S, Qiao G, Wang T, Kouzani AZ, Zhou SF. et al. Nucleic acid aptamer-guided cancer therapeutics and diagnostics: the next generation of cancer medicine. Theranostics. 2015;5:23-42
9. Elbashir SM, Lendeckel W, Tuschl T. RNA interference is mediated by 21- and 22-nucleotide RNAs. Genes Dev. 2001;15:188-200
10. Titze-de-Almeida R, David C, Titze-de-Almeida SS. The race of 10 synthetic RNAi-based drugs to the pharmaceutical market. Pharm Res. 2017;34:1339-63
11. Barros SA, Gollob JA. Safety profile of RNAi nanomedicines. Adv Drug Deliv Rev. 2012;64:1730-7
12. Hu Y, Duan J, Zhan Q, Wang F, Lu X, Yang XD. Novel MUC1 aptamer selectively delivers cytotoxic agent to cancer cells in vitro. PLoS One. 2012;7:e31970
13. Volkov AA, Kruglova NS, Meschaninova MI, Venyaminova AG, Zenkova MA, Vlassov VV. et al. Selective protection of nuclease-sensitive sites in siRNA prolongs silencing effect. Oligonucleotides. 2009;19:191-202
14. Whitehead KA, Langer R, Anderson DG. Knocking down barriers: advances in siRNA delivery. Nat Rev Drug Discov. 2009;8:129-38
15. Dass CR, Choong PF. Selective gene delivery for cancer therapy using cationic liposomes: in vivo proof of applicability. J Control Release. 2006;113:155-63
16. Akbarzadeh A, Rezaei-Sadabady R, Davaran S, Joo SW, Zarghami N, Hanifehpour Y. et al. Liposome: classification, preparation, and applications. Nanoscale Res Lett. 2013;8:102
17. Kim MW, Jeong HY, Kang SJ, Choi MJ, You YM, Im CS. et al. Cancer-targeted nucleic acid delivery and quantum dot imaging using EGF receptor aptamer-conjugated lipid nanoparticles. Sci Rep. 2017;7:9474
18. Yip KW, Reed JC. Bcl-2 family proteins and cancer. Oncogene. 2008;27:6398-406
19. Kampfer S, Windegger M, Hochholdinger F, Schwaiger W, Pestell RG, Baier G. et al. Protein kinase C isoforms involved in the transcriptional activation of cyclin D1 by transforming Ha-Ras. J Biol Chem. 2001;276:42834-42
20. Gunaratne A, Thai BL, Di Guglielmo GM. Atypical protein kinase C phosphorylates Par6 and facilitates transforming growth factor beta-induced epithelial-to-mesenchymal transition. Mol Cell Biol. 2013;33:874-86
21. Paul A, Gunewardena S, Stecklein SR, Saha B, Parelkar N, Danley M. et al. PKClambda/iota signaling promotes triple-negative breast cancer growth and metastasis. Cell Death Differ. 2014;21:1469-81
22. Ho EA, Osooly M, Strutt D, Masin D, Yang Y, Yan H. et al. Characterization of long-circulating cationic nanoparticle formulations consisting of a two-stage PEGylation step for the delivery of siRNA in a breast cancer tumor model. J Pharm Sci. 2013;102:227-36
23. Ngoune R, Peters A, von Elverfeldt D, Winkler K, Putz G. Accumulating nanoparticles by EPR: A route of no return. J Control Release. 2016;238:58-70
24. Koning GA, Morselt HW, Kamps JA, Scherphof GL. Uptake and intracellular processing of PEG-liposomes and PEG-immunoliposomes by kupffer cells in vitro 1 *. J Liposome Res. 2001;11:195-209
25. Chou LY, Ming K, Chan WC. Strategies for the intracellular delivery of nanoparticles. Chem Soc Rev. 2011;40:233-45
26. Khalil IA, Kogure K, Akita H, Harashima H. Uptake pathways and subsequent intracellular trafficking in nonviral gene delivery. Pharmacol Rev. 2006;58:32-45
27. Zhou X, Huang L. DNA transfection mediated by cationic liposomes containing lipopolylysine: characterization and mechanism of action. Biochim Biophys Acta. 1994;1189:195-203
28. Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N. et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927-39
29. Campbell PM, Der CJ. Oncogenic Ras and its role in tumor cell invasion and metastasis. Semin Cancer Biol. 2004;14:105-14
30. Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274-93
31. Viloria-Petit AM, Wrana JL. The TGFbeta-Par6 polarity pathway: linking the Par complex to EMT and breast cancer progression. Cell Cycle. 2010;9:623-4
32. Goldstein NI, Prewett M, Zuklys K, Rockwell P, Mendelsohn J. Biological efficacy of a chimeric antibody to the epidermal growth factor receptor in a human tumor xenograft model. Clin Cancer Res. 1995;1:1311-8
33. Kim K, Lee S, Ryu S, Han D. Efficient isolation and elution of cellular proteins using aptamer-mediated protein precipitation assay. Biochem Biophys Res Commun. 2014;448:114-9
34. Wang J, Liu R, Liu B. Cadmium-containing quantum dots: current perspectives on their application as nanomedicine and toxicity concerns. Mini Rev Med Chem. 2016;16:905-16
35. Wang Q, Chao YM. Multifunctional quantum dots and liposome complexes in drug delivery. J Biomed Res. 2018;32:91-106
36. Xu G, Zeng S, Zhang B, Swihart MT, Yong KT, Prasad PN. New generation cadmium-free quantum dots for biophotonics and nanomedicine. Chem Rev. 2016;116:12234-327
37. Hong G, Dai H. In vivo fluorescence imaging in the second near-infrared window using carbon nanotubes. Methods Mol Biol. 2016;1444:167-81
38. Allen TM, Sapra P, Moase E. Use of the post-insertion method for the formation of ligand-coupled liposomes. Cell Mol Biol Lett. 2002;7:889-94
Author contact
![]() Corresponding author: Professor YS Park, Department of Biomedical Laboratory Science, Yonsei University, Wonju, Gangwon 220-710, Republic of Korea. Telephone & Fax: 82-33-760-2448 / 82-33-760-2561; E-mail: parkysac.kr
Corresponding author: Professor YS Park, Department of Biomedical Laboratory Science, Yonsei University, Wonju, Gangwon 220-710, Republic of Korea. Telephone & Fax: 82-33-760-2448 / 82-33-760-2561; E-mail: parkysac.kr