Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(3):676-690. doi:10.7150/thno.30224 This issue Cite
Research Paper
TBC1D8 Amplification Drives Tumorigenesis through Metabolism Reprogramming in Ovarian Cancer
Min Chen1*, Xiu-Jie Sheng3*, Yuan-Yi Qin1*, Song Zhu1, Qing-Xia Wu1,3, Liqing Jia1, Nan Meng1, Yu-Tian He1, Guang-Rong Yan1,2 ![]()
1. Biomedicine Research Center, the Third Affiliated Hospital of Guangzhou Medical University, Guangzhou, 510150, China
2. Key Laboratory of Protein Modification and Degradation, Guangzhou Medical University, Guangzhou, 511436, China
3. Department of Obstetrics and Gynecology, the Third Affiliated Hospital of Guangzhou Medical University, Guangzhou, 510150, China
*Equal contribution
Received 2018-9-26; Accepted 2018-12-22; Published 2019-1-24
Abstract

Cancer cells undergo metabolic reprogramming to support their energy demand and biomass synthesis. However, the mechanisms driving cancer metabolism reprogramming are not well understood.
Methods: The differential proteins and interacted proteins were identified by proteomics. Western blot, qRT-PCR and IHC staining were used to analyze TBC1D8 levels. In vivo tumorigenesis and metastasis were performed by xenograft tumor model. Cross-Linking assays were designed to analyze PKM2 polymerization. Lactate production, glucose uptake and PK activity were determined.
Results: We established two aggressive ovarian cancer (OVCA) cell models with increased aerobic glycolysis. TBC1D8, a member of the TBC domain protein family, was significantly up-regulated in the more aggressive OVCA cells. TBC1D8 is amplified and up-regulated in OVCA tissues. OVCA patients with high TBC1D8 levels have poorer prognoses. TBC1D8 promotes OVCA tumorigenesis and aerobic glycolysis in a GAP activity-independent manner in vitro and in vivo. TBC1D8 bound to PKM2, not PKM1, via its Rab-GAP TBC domain. Mechanistically, TBC1D8 binds to PKM2 and hinders PKM2 tetramerization to decreases pyruvate kinase activity and promote aerobic glycolysis, and to promote the nuclear translocation of PKM2, which induces the expression of genes which are involved in glucose metabolism and cell cycle.
Conclusions: TBC1D8 drives OVCA tumorigenesis and metabolic reprogramming, and TBC1D8 serves as an independent prognosis factor for OVCA patients.
Keywords: TBC1D, PKM2, aerobic glycolysis, ovarian cancer
Introduction
Metabolic reprogramming is one of the hallmarks of cancer cells and leads to aerobic glycolysis, also called the Warburg effect, which is characterized by increased glucose uptake and lactate production even in the presence of sufficient oxygen [1-3]. Metabolic reprogramming provides a selective advantage to cancer growth and progression [4, 5]. However, the driving mechanism of cancer cell metabolic reprogramming is still unclear.
In this metabolic pathway, pyruvate kinase (PK) is the rate-limiting enzyme that catalyzes the conversion of phosphoenolpyruvate and ADP to pyruvate and ATP during glycolysis [6, 7]. Pyruvate can be converted to lactate by aerobic glycolysis or to acetyl-CoA by mitochondrial oxidative phosphorylation in cancer cells, and the direction of this conversion is determined by the enzymatic activity of the M2 isoform of pyruvate kinase (PKM2) [1, 7]. PKM2 is crucial for aerobic glycolysis and provides a proliferative advantage during tumorigenesis. The M type PK gene (PKM) consists of 12 exons, of which exon 9 and 10 are alternatively spliced to give rise to the PKM1 and PKM2 isoforms, respectively [8]. Exon 9 and 10 each encode a 56 amino acid variable segment that confers distinctive properties to the regulation and enzymatic activity of PKM1 and PKM2 [9].
PKM2 exists in either a low-activity dimeric or high-activity tetrameric form, whereas PKM1 constantly exists in a high-activity tetrameric form. The low-activity dimeric form of PKM2 is predominantly present in cancer cells, whereas the high-activity tetrameric form is present in normal proliferating cells [1]. Various factors have been reported to regulate the PKM2 activity such as fructose 1,6-biophophate (FBP) [7], Y105 phosphorylation [10], K433 acetylation [11], and C358 oxidation [12] modification and the binding of phosphotyrosine- containing proteins and peptides to PKM2 [13]. Our study also demonstrated that the splicing of PKM mRNA into the PKM1 or PKM2 isoform regulates PK activity [8]. However, the mechanisms controlling the PKM2 switch from the tetrameric to dimeric form are not well understood in cancer cells.
Here, we established two more aggressive ovarian cancer (OVCA) cell models with increased aerobic glycolysis and found that TBC1D8 was most significantly up-regulated in more aggressive cancer cells by SILAC proteomics technology. TBC1D8 is a member of the Tre2/Bub2/Cdc16 (TBC) domain protein family, which is characterized by the presence of highly conserved TBC domains, and members of this family act as negative regulators of Rab proteins to facilitate Rab inactivation [14]. This family has 44 predicted proteins. Most TBC domain proteins have GTPase-activating protein (GAP) activity [15, 16]. Recent studies have revealed that TBC domain proteins mainly participate in intracellular trafficking, organelle biogenesis, transport and cytokinesis [14, 17]. Some TBC domain proteins have been shown to be involved in diseases, such as tumorigenesis, atopic dermatitis, viral and bacterial infection susceptibility, and diabetes [14]. Currently, only TBC1D7 [18], TBC1D16 [19], PRC17 [20] and USP6/TRE17 [21] have been shown to regulate tumorigenesis. The functional roles of TBC1D8 in cancers have not been reported.
In this study, we describe the role of TBC1D8 as a negative regulator of PKM2 tetramerization to regulate metabolic reprogramming in a GAP activity-independent manner. TBC1D8 is amplificated and up-regulated in OVCA and is significantly associated with a poor prognosis in OVCA patients. TBC1D8 promotes OVCA tumorigenesis in vitro and in vivo in a GAP activity-independent manner. The Rab-GAP TBC domain of TBC1D8 interacts with PKM2, not PKM1, hinders PKM2 tetramerization, and inhibits PK activity to promote tumorigenesis and aerobic glycolysis, but not affects the acetylation and phosphorylation modification of PKM2. Furthermore, TBC1D8 also stimulates depolymerized PKM2 translocation into the nucleus and induces the expression of genes associated with the cell cycle and cancer metabolism. Collectively, TBC1D8 promotes OVCA tumorigenesis and metabolic reprogramming by hindering PKM2 tetramerization.
Methods
Cell culture and tissue samples
OVCAR-3 and SK-OV-3 OVCA cell lines and HEK293T cell line were from American Type Culture Collection and cultured under standard conditions, and their identity is routinely monitored by short tandem repeat (STR) profiling. The OVCA cell sub-lines OVCAR-3high and SK-OV-3high, were triply screened from OVCAR-3 and SK-OV-3 cell lines, respectively. Cells were monitored regularly for mycoplasma contamination using PCR mycoplasma detection assays.
Normal ovarian tissues and OVCA tissues were collected from OVCA patients at the Third Affiliated Hospital of Guangzhou Medicine University. These cases were selected based on a clear pathological diagnosis, and the patients had not received preoperative anticancer treatment. Tissue microarray chips containing 160 OVCA tissue samples (including 141 OVCA tissues, 5 borderline ovarian tumor or adjacent non-tumor tissues and 14 distant metastases) and the associated clinicopathological information were purchased from Shanghai OUTDO Biotech Co., Ltd. (Shanghai). The collection of tissue specimens was approved by the Internal Review and Ethics Boards at the Third Affiliated Hospital of Guangzhou Medicine University. Informed consent was obtained from each patient.
Establishment of aggressive OVCA cell sublines
OVCAR-3 and SK-OV-3 cells in medium supplemented with 0.1% FBS were added in the upper transwell chambers coated with Matrigel. Invasive cells on the undersurface were suspended and cultured in medium supplement 10% FBS to a certain amount. And these cultured cells were then screened as described above. After three rounds-screening, OVCAR-3high (OV-3high) and SK-OV-3high (SK-3high) subline cells were established from the OVCAR-3 (OV-3) and SK-OV-3 (SK-3) cell lines, respectively.
SILAC labeling and quantitative proteomics analysis
OVCA-3 and OVCA-3high cells were labeled with “light” (12C6)-and “heavy”(13C6)-lysine, respectively, as previously described [8]. The peptide mixtures were analyzed using nano-LC-MS/MS (AB SCIEX TripleTOF 5600, USA), as previously described [8]. Proteins were identified using the Mascot (v2.3.02) program against the Uniprot human protein database (released Dec. 2014) with the default settings. Proteins with protein scores ≥40 and unique peptide scores ≥2 were selected. The protein ratios of heavy vs light SILAC were analyzed by Protein Pilot Software v4.5 (AB SCIEX, USA) with the default settings.
RT-PCR and qRT-PCR
Total RNA was extracted from cells and tissues using the TRIzol total RNA isolation reagent (Invitrogen, USA). cDNA was prepared using a PrimeScriptTM RT reagent kit with gDNA Eraser (TaKaRa, Japan), and quantitative RT-PCR was performed with a SYBR Premix Ex Taq™ II kit (TaKaRa, Japan). The data were normalized to GAPDH expression. The primers used in this study are listed in the Supplementary Table S5.
Western blotting
In brief, cells were suspended in lysis buffer (50 mM Tris-HCl pH 8.0, 1% SDS, 1 mM EDTA, 5 mM DTT, 10 mM PMSF, 1 mM NaF, 1 mM Na3VO4, and protease inhibitor cocktail). The cellular lysates were collected and their concentration was determined by BCA assay. Cellular lysates were separated using 10% SDS-PAGE, and the indicated proteins were detected using anti-TBC1D8 (cat# Sc-376637, Santa Cruz), Flag (cat# M185-3L, MBL), HA (cat# 561, MBL), PKM2 (cat# 15822-1-AP, Proteintech), PKM1 (cat# 7067, Cell Signaling Technology), β-Actin (cat# BS6007W, Bioworld), acetylated-lysine (cat# 9814, Cell Signaling Technology), and phosphor-tyrosine (cat# 9811, Cell Signaling Technology) antibodies.
RNA interference
SiRNA against TBC1D8 or PKM2 and the corresponding scrambled siRNA (GenePharma, Suzhou, China) were transfected into cells with RNAiMAX (Invitrogen, USA) for 48 h (unless otherwise stated). The cells were subsequently harvested for further analyses. The siRNA sequences used in this study were provided in supplementary Table S5.
Migration and invasion assays
In vitro migration and invasion assays were performed using transwell chambers. Briefly, cells were transfected with the specified concentrations of the indicated siRNA or plasmids for 24 h. These cells were suspended and 1×105 cells in 100 μL serum-free medium each group were added in the upper transwell chambers for migration assay (8.0 μM pore size, BD) or the upper transwell chambers coated with Matrigel for invasion assay, and medium supplemented with 10% FBS was added to the bottom chamber. Cells on the undersurface were stained with 5% crystal violet. Images were captured from each membrane and migrated or invasive cells were counted. The migrated or invasive cells in treatment group were compared to those in negative control treatment group.
Cellular growth assay
OVCA cells were transfected with the indicated siRNAs or plasmids for 12 h. These cells were suspended and 1×104 cells were plated in 96-well culture plates and cultured each group. The cell number was counted at 24, 48, 72, 96, and 120 h. These experiments were repeated three times.
Colony formation assays
OVCA cells were transfected with the indicated siRNAs or plasmids for 24 h. 500 OVCA cells were then plated in 6-well culture plates and cultured for two weeks. These cells were then fixed with methanol and stained with crystal violet solution. The number of colonies was counted under the microscope (n=3).
Construction of cell lines with stable TBC1D8 silencing
OVCAR-3 cells were infected with the lentivirus pLV5-GFP-Luc for three days. The Luc-labeled OVCAR-3 cells were filtered using colony screening, and OVCAR-3-Luc cell line stably expressing Luc was established. The lentivirus pLV3-TBC1D8 shRNA that expresses TBC1D8 shRNA was purchased from GenePharma (Shanghai, China). The Luc-labeled OVCAR-3-Luc cells were further transduced with lentivirus pLV3-TBC1D8 shRNA for 5 days, after which the OVCAR-3-Luc-shTBC1D8-transduced cells were selected by 2 μg/ml puromycin for two weeks. TBC1D8 silencing was validated by western blotting.
Plasmid construction
To generate Flag or HA fusion protein constructs (TBC1D8-Flag or HA-TBC1D8) with the TBC1D8 ORF sequence was cloned into the pcDNA3.1(+) vector (Invitrogen). To generate eGFP fusion protein constructs (TBC1D8-GFP) and mutants (MUT1, 2, 3, 4) with the TBC1D8 ORF and the sequences containing different TBC1D8 domains was cloned into a pEGFP-N1 vector (Clontech). The Flag-tagged PKM2 (Flag-PKM2) plasmid and its mutants Flag-PKM2 K433R and Flag-PKM2 K433Q were kindly provided by Prof. Qun-Ying Lei from Key Laboratory of Molecular Medicine, Ministry of Education, and Department of Biochemistry and Molecular Biology, Fudan University Shanghai Medical College.
Construct of synonymous mutant
The anti-PKM2 siRNA-targeted sequences GCCATCTACCACTTGCAA in Flag-PKM2 and Flag-PKM2 K433R and Flag-PKM2 K433Q mutant plasmids were synonymously mutated to GCGATTTATCATCTTCAG. And the synonymous mutant Flag-sPKM2, Flag-sPKM2 K433R and Flag-sPKM2 K433Q plasmids were generated.
In vivo xenograft tumor model
Female NOD-SCID and BALB/c nude mice (3-4 weeks old) were purchased from Charles River Laboratories in China (Beijing). An in vivo tumor growth assay was performed as previously described [8, 22]. Briefly, 2×106 OVCAR-3-Luc-NC and OVCAR-3-Luc-shTBC1D8 cells were subcutaneously injected into the dorsal left and right flanks, respectively, of each BALB/c nude mouse (n=10). After four weeks, the mice were euthanized, and the tumors were dissected and weighed. In addition, an in vivo metastatic assay was performed as previously described [23, 24]. Briefly, 2×106 OVCAR-3-Luc-NC and OVCAR-3-Luc-shTBC1D8 cells were injected into the peritoneal cavity of NOD-SCID mice (n=5 per group). Three months after the injection, each mouse was injected through the tail vein with 0.15 mg/g body weight D-luciferin potassium salt for 5 min. The luminescence in the mice bearing tumors was detected using an IVIS 200 imaging system (Xenogen, USA). These mice were sacrificed and the ascites weight, tumor nodule number and body weight were assessed. The mice used in these experiments were bred and maintained under defined conditions at the Animal Experiment Center of the College of Medicine (SPF grade), Jinan University. The animal experiments were approved by the Laboratory Animal Ethics Committee of The Third Affiliated Hospital of Guangzhou Medicine University and conformed to the legal mandates and national guidelines for the care and maintenance of laboratory animals. The NOD-SCID and BALB/c nude mice were randomly divided into groups prior to the injection with the indicated OVCA cells.
Immunohistochemistry (IHC) assay
IHC assays were performed as previously described using anti-TBC1D8 antibodies [8, 23]. All IHC samples were assessed by two independent pathologists blinded to both the sample origins and the subject outcomes. Each specimen was scored according to the extent of cell staining (≤10% positive cells, 0; 11-50% positive cells, 2; 51-80% positive cells, 3; >80% positive cells, 4) and the staining intensity (no staining, 0; slight staining, 1; moderate staining, 2; strong staining, 3) [8, 23]. Scores for the extent of staining cells and the staining intensity were added. Score of 0-3 was indicative of low TBC1D8 level (low) and scores of 4-7 were indicative of high TBC1D8 level (high) in OVCA tissues.
Co-Immunoprecipitation (co-IP) assays
Co-IP or IP was conducted using anti-Flag or TBC1D8 antibodies, and the immune complexes were captured on protein A/G agarose beads (Santa Cruz, USA). The complexes were then separated, and the gels were stained with silver or were detected by Western blotting.
The co-IP gel bands and their corresponding negative gel bands were excised and digested using in-gel trypsin. The extracted peptide mixtures were analyzed using nano-LC-MS/MS (AB SCIEX TripleTOF 5600, USA), as previously described [8, 25]. Proteins were identified using the Mascot (v2.3.02) program against the Uniprot human protein database (released Dec. 2014) with the default settings. Proteins with protein scores ≥40 and unique peptide scores ≥2 were selected.
Cross-Linking assays
Cells were lysed with sodium phosphate buffer (pH 7.3) containing 0.5% Triton X-100 and protease inhibitor for 30 min at 4°C. Whole cell lysates were centrifuged at 20000 rpm for 30 min at 4°C, and the supernatants were collected. The supernatants were then treated with 0.01% glutaraldehyde for 5 min at 37°C. The reactions were terminated by using 50 mM Tris-Cl (pH 8.0). These samples were separated by 10% SDS-PAGE and analyzed by Western blotting with anti-PKM2 or anti-Flag antibodies.
In vivo PK activity assay
Cells were washed twice with ice-cold PBS and lysed with PK assay buffer. The extracts were obtained, and the PK activities in the extracts were measured with a PK activity colorimetric assay kit according to the manufacturer's instructions (K709-100, BioVision, USA). The experiments were repeated three times.
Measurement of lactate production
The lactate production was measured using a Lactate Colorimetric Assay kit II (K627-100, BioVision, USA). The background was corrected by subtracting the OD value derived from the fresh phenol red-free medium. A standard curve of nmol/well vs. OD450nm value was plotted according to the lactate standard measurements. The sample OD450nm values were applied to the standard curve to calculate the lactate concentrations of the test samples (n=3).
Glucose uptake assay
OVCA cells transfected with the indicated siRNAs or plasmids were cultured in DMEM for 36 h and then incubated with DMEM without L-glucose and phenol red for 8 h. The amount of glucose in these media was measured with a Glucose Colorimetric Assay kit according to the manufacturer's instructions (K606-100, BioVision, USA). Fresh DMEM was used for the negative control. The experiments were repeated three times.
Immunofluorescence staining
Cells were fixed with 4% paraformaldehyde for 20 min and permeabilized with 0.1% Triton X-100 for 7 min. These cells were incubated with anti-PKM2 antibody and subsequently incubated with the corresponding Alexa Fluor 488-conjugated secondary IgG antibodies. Nuclei were stained with DAPI. The cells were examined with a confocal laser-scanning microscope (Nikon, Japan).
Subcellular fractionation
Cytosolic and nuclear extract fractionation was performed by using a Nuclear and Cytoplasmic Protein Extraction kit (P0027, Beyotime, China) according to the manufacturer's instructions.
Statistics
Statistical analyses were performed using Prism 5 software or SPSS program. Two-tailed unpaired Student's t-tests or Mann Whitney U tests were used for comparisons between two groups. Two-way ANOVA was used for the growth curve comparison. Survival curves were analyzed using the Kaplan-Meier method and compared using the log-rank test. The treatment groups were compared to the control, except where stated otherwise. The data are presented as the means ± SEM, except where stated otherwise. Differences with *p<0.05, **p<0.01 or ***p<0.001 were considered statistically significant.
Results
Establishment of more aggressive OVCA cell models with higher aerobic glycolysis rates
To investigate the possible driving mechanism of metabolism reprogramming in cancer cells, we established two aggressive OVCA cell subline models OVCAR-3high (OV-3high) and SK-OV-3high (SK-3high) from the OVCAR-3 (OV-3) and SK-OV-3 (SK-3) cell lines by triple Matrigel-coated transwell screening, respectively (Figure 1A). We further confirmed that colony formation, migration and invasion, and cell growth were higher in the OVCAR-3high and SK-OV-3high cells than in the parental OVCAR-3 and SK-OV-3 cells, respectively (Figure 1B-1D). Collectively, OVCAR-3high and SK-OV-3high cells exhibited a more aggressive phenotype than their corresponding parent cells.
Two more aggressive OVCA cell models were established and more aggressive OVCA cells had a higher aerobic glycolysis rate. (A) The OVCA cell models, OVCAR-3high and SK-OV-3high, were triple screened from the OVCAR-3 and SK-OV-3 cell lines, respectively, using Matrigel-coated Transwell chambers. (B-D) Colony formation (B), the migration and invasion (C) and cell growth (D) of the indicated cells were determined. (E-G) The glucose uptake (E), lactate production (F), and PK activity (G) of the indicated cells were detected (n=3).
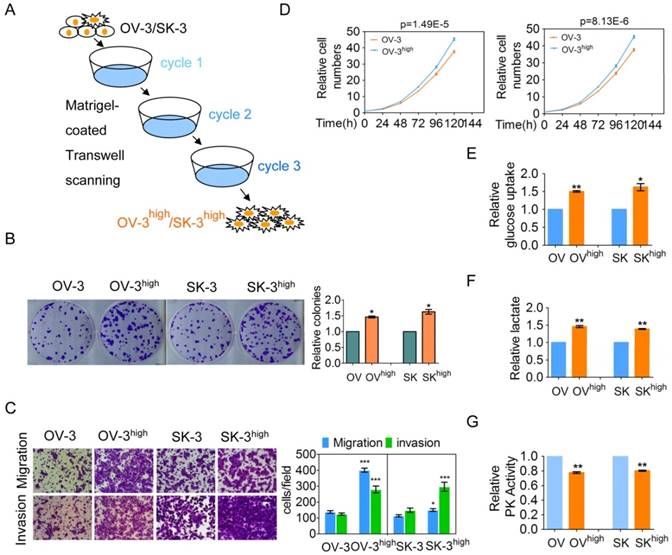
One of the hallmarks of cancer cells is their altered metabolism, referred to as aerobic glycolysis or the Warburg effect, which provides a selective advantage to tumors [1]. We demonstrated that more glucose uptake and lactate production was observed in the OVCAR-3high and SK-OV-3high cells than in the OVCAR-3 and SK-OV-3 cells, respectively (Figure 1E and 1F). Pyruvate kinase (PK) is the final rate-limiting enzyme in aerobic glycolysis, and low PK activity promotes the Warburg effect. PK activity was significantly decreased in the OVCAR-3high and SK-OV-3high cells compared with the OVCAR-3 and SK-OV-3 cells, respectively (Figure 1G). Taken together, our results indicate that more aggressive OVCA cells have a greater Warburg effect.
TBC1D8 expression is increased in more aggressive OVCA cells
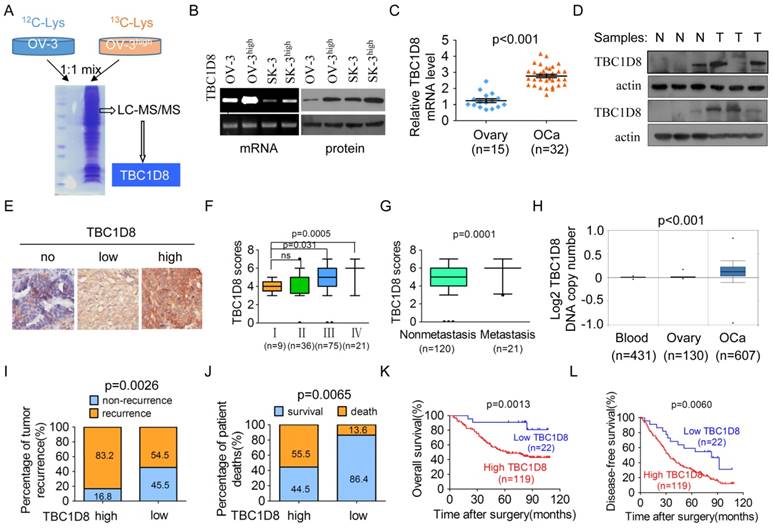
To identify potential mechanisms by which the more aggressive OVCA cells have a greater Warburg effect, we examined the differences in the protein expression profiles between OVCAR-3high and OVCAR-3 cells by a stable isotope labeling with amino acids in cell culture (SILAC) quantitative proteomics method (Figure 2A) [26]. We analyzed the fold-change differences in protein expression between the two cell lines and found that a 5-fold cut-off included 54 proteins with altered expression levels (including 24 upregulations and 30 downregulations) (Supplementary Table S1). Interestingly, among the top 10 proteins that were up-regulated in OVCAR-3high cells, we found that the roles of TBC1D8 in tumorigenesis have not reported, whereas the other 9 proteins HOOK3, MID1, CUL1, CLIP2, FBXL12, LPCAT2, USP22, KRAS and C2CD5 have been reported to be involved in regulating various cancer hallmarks [27-33]. Therefore, we selected TBC1D8 for further detailed investigation. We further confirmed that TBC1D8 protein and mRNA levels were markedly up-regulated in the more aggressive OVCAR-3high and SK-OV-3high cells compared with the OVCAR-3 and SK-OV-3 cells, respectively (Figure 2B). Therefore, a potential driver for OVCA tumorigenesis and metabolic reprogramming was identified.
TBC1D8 is amplified and up-regulated in the more aggressive OVCA cells and OVCA tissues, and high TBC1D8 level is significantly associated with a poor prognosis for OVCA patients. (A) SILAC quantitative proteomics analysis identified that TBC1D8 expression is markedly up-regulated in the more aggressive OVCA cells OVCAR-3high compared with the OVCAR-3 cells. (B) The TBC1D8 mRNA and protein levels of the more aggressive OVCA cells and their parental cells were determined. (C, D) The TBC1D8 mRNA (C) and protein (D) levels between OVCA tissues and normal ovarian tissues were determined. (E) The TBC1D8 protein levels in 141 OVCA tissues were detected by IHC assay. Representative IHC images of TBC1D8 level in OVCA tissues. (F) Differences in TBC1D8 score in OVCA with clinical stage I, II, III and IV are presented as a box plot. (G) Differences in TBC1D8 score in OVCA samples between metastasis and nonmetastasis are presented as a box plot. (H) TBC1D8 gene copy number was analyzed in OVCA tissues, whole blood and normal ovarian tissues in the TCGA ovarian dataset. (I, J) Associations between TBC1D8 levels and the percentage of tumor recurrence (I) and patient death (J) were analyzed in 141 OVCA samples. (K, L) Kaplan-Meier plots for the overall patient survival rate (K) and the tumor-free survival rate (L) of patients with OVCA in TBC1D8 low and TBC1D8 high groups. Score of 0-3 was indicative of low TBC1D8 level (low) and scores of 4-7 were indicative of high TBC1D8 level (high) in OVCA tissues.
TBC1D8 is amplified and up-regulated in OVCA
To investigate the role of TBC1D8 in OVCA tumorigenesis, we analyzed TBC1D8 mRNA and protein levels in fresh-frozen OVCA tissue samples (T) and normal ovarian tissue samples (N). TBC1D8 mRNA and protein levels were frequently up-regulated in OVCA tissues compared with normal ovarian tissues (Figure 2C and 2D).
An extensive tissue microarray analysis of 141 OVCA tissue samples was performed using an IHC assay (Figure 2E). A higher TBC1D8 level was observed in OVCA tissues with more clinical stage III and IV compared to stage I and II (Figure 2F). And TBC1D8 levels were also significantly up-regulated in primary OVCA tissues with distant metastasis compared to in those without distant metastasis (Figure 2G). The IHC data deposited in the publicly database (The Human Protein Atlas) also demonstrated that TBC1D8 protein levels were significantly up-regulated in the OVCA tissues compared with the normal ovarian tissues (p<0.0001) (Supplementary Figure S1A and S1B). Furthermore, the publicly accessible datasets of OVCA patients deposited in the TCGA database were analyzed for differences in TBC1D8 DNA copy numbers between OVCA and normal tissues. The TBC1D8 DNA copy numbers were significantly increased in OVCA tissues compared with whole blood and normal ovarian tissues (p=5.61 E-30) (Figure 2H), indicating that the TBC1D8 gene had been amplified, resulting in the up-regulation of TBC1D8 mRNA and protein levels in OVCA tissues.
Increased TBC1D8 expression is associated with a poorer prognosis
The correlations between TBC1D8 levels and OVCA patient prognosis were further investigated in the 141 OVCA samples. Increased TBC1D8 levels in OVCA tissues were positively associated with tumor recurrence, histological stage, pT status and pN status of OVCA (Supplementary Table S2). Higher patient death and tumor recurrence rates were observed in OVCA patients with high TBC1D8 level compared with the patients classified as low TBC1D8 level (Figure 2I and 2J). Kaplan-Meier survival analyses revealed the TBC1D8 levels correlated significantly with disease-free survival rate (p=0.0060, log-rank test, Figure 2L) as well as overall patient survival rate (p=0.0013, log-rank test, Figure 2K). The mean overall survival time for OVCA patients with high TBC1D8 was 57.3 months, whereas that for OVCA patients with low TBC1D8 was 80.8 months. Compared with patients with lower TBC1D8 levels, OVCA patients with higher TBC1D8 levels were at an increased risk for OVCA-related death from 1,435 OVCA patients data analysis in a publicly available OVCA protein database (Supplementary Figure S1C, p=0.009, log-rank test). These data indicate that patients with high TBC1D8 levels were at increased risk of cancer-related death compared with patients with low TBC1D8 levels. Further multivariate Cox regression analysis indicated that high TBC1D8 level is an independent prognostic factor for poor survival of patients with OVCA (HR=2.11, 95% CI=1.61-2.78, p=0.000, Supplementary Table S3). Collectively, our results indicate high TBC1D8 level is significantly correlated with a poor prognosis for OVCA patients, and TBC1D8 serves as an independent prognosis factor for OVCA patients.
TBC1D8 promotes the OVCA aggressive phenotypes
TBC1D8 belongs to a member of the TBC domain family. The functional roles of TBC1D8 in tumorigenesis have not been reported. To investigate the functional roles of TBC1D8 in cancer, TBC1D8 expression was silenced and over-expressed. Silencing of TBC1D8 significantly reduced OVCA cell growth, migration and invasion, colony formation in vitro (Figure 3A-3D). However, TBC1D8 overexpression in OVCAR-3 and SK-OV-3 cells, where TBC1D8 was lowly expressed, promoted tumor growth, colony formation, migration and invasion (Supplementary Figure S2).
Silencing TBC1D8 expression inhibited OVCA cell growth, colony formation, migration and invasion in vitro and tumorigenesis and progression in vivo. (A) TBC1D8 expression in OV-3high and SK-3high cells was silenced by two anti-TBC1D8 siRNAs. (B) OV-3high and SK-3high cells were transfected with two anti-TBC1D8 siRNAs for the indicated times, and the number of cells was measured (n=3). (C) OV-3high and SK-3high cells were transfected with two anti-TBC1D8 siRNAs for the indicated times, and migration and invasion abilities were determined using Transwell assays (left panel). The migrated and invasive cells were counted (right panel) (n=3). (D) OV-3high and SK-3high cells were transfected with two anti-TBC1D8 siRNAs for the indicated times, and their colony-forming abilities were measured after two weeks (left panel). The colony number was counted (right panel) (n=3). (E) The in vivo growth of OVCA cells with stably-silenced TBC1D8 expression was examined. Mouse xenograft tumors are shown in the upper panel and the xenograft tumor weights were measured (n=10) (low panel). (F-J) Luc-labeled OVCA cells (2×106 cells/mouse) were injected into the peritoneal cavity of NOD-SCID mice; luciferase activity was visualized at three months post transplantation (F), fluorescence levels in Figure 3F (G), ascites weight (H), nodule number (I) and nodule weight (J) were analyzed (n=5).
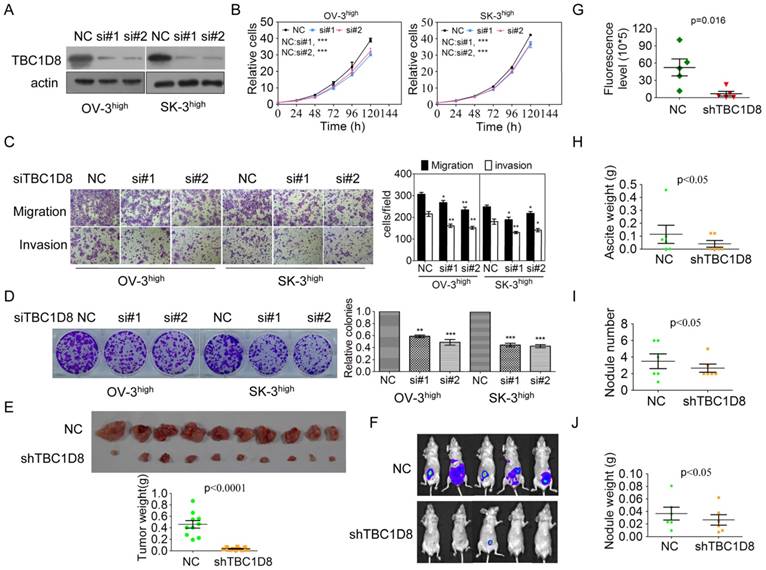
BALB/c nude mice were subcutaneously injected with TBC1D8 knockdown cells and control cells on the left and right flanks, respectively. As shown in Figure 3E and Supplementary Figure S3A and S3B, the in vivo growth was clearly impaired in the OVCA xenografts composed of TBC1D8-stably silenced cells compared with those composed of control cells. Furthermore, TBC1D8-knockdown cells and control cells were injected into the peritoneal cavity of BALB/c nude mice. OVCA tumor growth within the peritoneal cavity and ascites development were monitored. After three months, a dramatic decrease in the luminescence of the mice bearing tumors derived from the TBC1D8-knockdown OVCA cells was observed compared with the luminescence of those with control OVCA cells (Figure 3F and 3G). These mice were sacrificed and inspected for ascites and tumor nodules. TBC1D8 knockdown impaired OVCA cell metastases to the intestinal wall sites (Supplementary Figure S3C). Moreover, TBC1D8 knockdown decreased the ascites weights (Figure 3H), nodule number (Figure 3I), and tumor nodule weights (Figure 3J). Collectively, our results indicate that TBC1D8 promotes the aggressive OVCA cell phenotypes in vitro and in vivo.
TBC1D8 promotes the OVCA aggressive phenotypes in a GAP activity-independent manner
TBC1D8 is a member of the TBC domain protein family, most of which have GTPase-activating protein (GAP) activity [15, 16]. Sequence comparison analyses demonstrated that the TBC domain of TBC1D8 contains conserved R, Y, and Q residues, which form the “R” and “Q” fingers that are critical for GTP hydrolysis in other TBC proteins [34] (Supplementary Figure S4A). Therefore, the R, Y, and Q residues in TBC domain in TBC1D8 were mutated to the A residue (TBC1D8 RYQ/AAA) (Supplementary Figure S4B). Both wild type TBC1D8 and TBC1D8 RYQ/AAA mutant exhibited the same stimulatory effects on OVAC cell growth, colony formation, migration and invasion (Supplementary Figure S4C-S4E), suggesting that the mutation of the GTPase-activating sites in TBC domain in TBC1D8 did not change the stimulatory roles of TBC1D8 on tumorigenesis. Collectively, TBC1D8 promotes the OVCA aggressive phenotypes in a GAP activity-independent manner.
TBC1D8 interacts with PKM2 via Rab-GAP TBC domain
To further investigate the molecular mechanisms by which TBC1D8 acts in tumorigenesis, the proteins that interact with the TBC1D8 protein were identified (Figure 4A). A total of 29 proteins that interacted with TBC1D8 were identified (Supplementary Table S4). Among them, we found PKM2 to be particularly interesting because PKM2 is a critical determinant of the aerobic glycolysis metabolic phenotype and confers a selective proliferative advantage to tumor cells in vivo [1, 7]. We further confirmed that PKM2, not PKM1, was present in the anti-TBC1D8 co-IP complexes (Figure 4B). We also confirmed that TBC1D8 was present in the Flag-PKM2 complexes but not in the Flag complexes (Figure 4C). Collectively, our data indicate that TBC1D8 interacted with PKM2, not PKM1.
Interaction of TBC1D8 with PKM2, not PKM1, hinders PKM2 tetramerization. (A) Proteins that interacted with TBC1D8 were identified by combining co-IP and mass spectrometry methods. (B) The TBC1D8 complexes were co-IPed using an anti-TBC1D8 antibody, and PKM2 in the complexes was detected. (C) A Flag-PKM2 vector was transfected into HEK293T cells, the Flag-PKM2 complexes were co-IPed, and TBC1D8 in the complexes was detected. (D) Diagram of TBC1D8 wild type and mutation constructs with the different domains. (E) The indicated TBC1D8-GFP mutants were transfected into HEK293T cells; these mutants were detected. (F) The indicated TBC1D8-GFP mutants were transfected into HEK293T cells, and TBC1D8-GFP mutant complexes were co-IPed; PKM2 was then detected. (G) The indicated TBC1D8 vectors were transfected into HEK293T cells, and TBC1D8-GFP mutant complexes were co-IPed; PKM2 was then detected. (H) Cell lysates were treated by cross-linking, PKM2 was detected. (I) PKM2 tetramers, dimers and monomers were detected as described in (H) in cells with TBC1D8 expression stably silenced (left panel) and in cells transfected with anti-TBC1D8 siRNAs (right panel). (J) PKM2 tetramers, dimers and monomers were detected in mouse xenograft tumors in Figure 2E. (K) The indicated TBC1D8-GFP mutants together with Flag-PKM2 were co-transfected into HEK293T cells, PKM2 tetramers, dimers and monomers were detected. (L) The indicated vectors were transfected into HEK293T cells, PKM2 tetramers, dimers and monomers were detected.
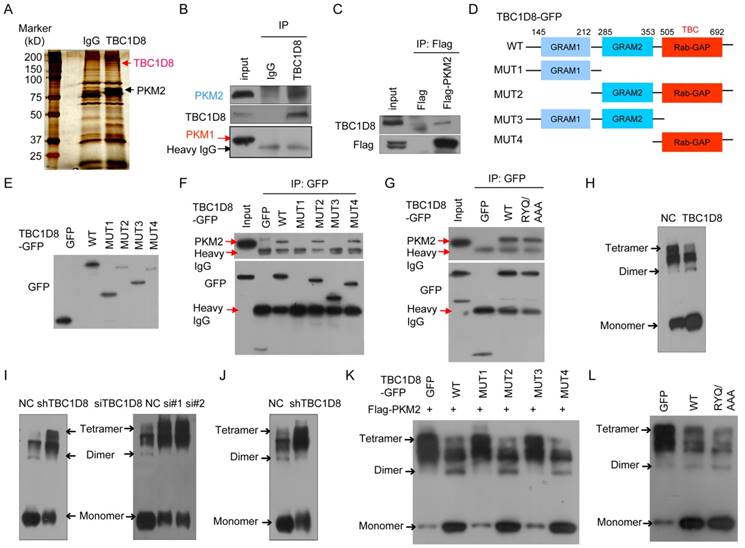
TBC1D8 consists of two GRAM (from Glucosyltransferases, Rab-like GTPase activators and Myotubularins) domain 1 and 2 and a Rab-GAP TBC domain. To determine which domains interact with PKM2, we generated truncated TBC1D8 constructs with a C-terminal GFP tag (Figure 4D). When these constructs were co-expressed with Flag-PKM2 in cells, only the constructs containing the TBC1D8 C-terminal Rab-GAP TBC domain, but no other GRAM1 and GRAM2 domain, could interact with PKM2, indicating that the C-terminal Rab-GAP TBC domain is essential for PKM2 binding (Figure 4E and 4F).
The Rab-GAP TBC domain of TBC1D8 contains conserved R, Y, and Q residues, which are critical for GTP hydrolysis in other TBC proteins. We further investigate whether the R, Y, and Q residues in the domain is critical for PKM2 binding. PKM2 bound both the wide type TBC1D8 and the TBC1D8 RYQ/AAA mutant (Figure 4G), indicating that the R, Y, and Q residues in the Rab-GAP TBC domain is not essential for PKM2 binding, consistent with our above results in which the R, Y, and Q residues in the Rab-GAP TBC domain is not essential for TBC1D8 functions in tumorigenesis.
TBC1D8 hinders tetrameric PKM2 formation via the Rab-GAP TBC domain
Next, we wondered if the interaction between TBC1D8 and PKM2 affects the PKM2 protein level. Neither silencing TBC1D8 expression nor overexpressing TBC1D8 changed the PKM2 protein level (Supplementary Figure S5). In cells, PKM2 exists as either a high PK activity tetramer or a low PK activity dimmer. The ratio of dimer to tetramer is positively correlated with cell proliferation, survival, transformation, migration and invasion [1, 6, 7]. To determine whether TBC1D8 can change the dimer to tetramer ratio, TBC1D8 was overexpressed or silenced in OVCA cells, followed by cross-linking experiments with glutaraldehyde as previously described methods [35]. Tetrameric PKM2 (240 kD) was notably decreased and was replaced by dimeric (120 kD) and monomeric (60 kD) forms when TBC1D8 was overexpressed (Figure 4H). However, silencing TBC1D8 expression increased the tetrameric form of PKM2, with a concomitant decrease in dimeric and monomeric PKM2 levels (right panel in Figure 4I). Similar results were also observed in OVCAR cells with TBC1D8 expression stably silenced (left panel in Figure 4I). Furthermore, we also found that dimeric and monomeric PKM2 levels were markedly reduced and were replaced by the tetrameric PKM2 form in the mouse xenograft tumors composed of TBC1D8-stably silenced cells compared with those with control cells (Figure 4J).
Furthermore, we found that only TBC1D8 containing the C-terminal Rab-GAP TBC domain, but no other GRAM1 and GRAM2 domain, decreased tetrameric PKM2 and increased dimeric/monomeric PKM2 (Figure 4K). Mutation of the R, Y, and Q residues in the Rab-GAP TBC domain in TBC1D8 did not change the conversion of PKM2 from tetramer to dimmer/monomer (Figure 4L). Together, TBC1D8 hinders PKM2 tetrameric assembly via the Rab-GAP TBC domain.
TBC1D8 hinders PKM2 tetrameric assembly by blocking the transition from dimeric to tetrameric PKM2, but not by dissociating the tetramer to dimers
We wanted to determine how TBC1D8 reduces the tetrameric PKM2 form and increases the dimeric and monomeric form. Two mechanisms may be possible: TBC1D8 may hinder the assembly of the dimeric/monomeric PKM2 form into the tetrameric form or dissociate tetrameric PKM2 to form dimeric/monomeric PKM2. Previous studies revealed that acetylation at K433 and phosphorylation at Tyr105 on PKM2 inhibits the tetrameric form and causes the accumulation of dimers and monomers [11]. The influence of TBC1D8 on PKM2 acetylation and tyrosine-phosphorylation levels were further investigated. We found that the acetylation and tyrosine-phosphorylation levels of PKM2 did not change when TBC1D8 was ectopically expressed (Supplementary Figure S6A and S6B), suggesting that TBC1D8 dissociates tetrameric PKM2 to form dimers not by acetylating or tyrosine-phosphorylating PKM2.
Furthermore, we used PKM2 acetylation mutants K433R (nonacetylatable arginine) and K433Q (acetyl-mimic glutamine) as PKM2 tetramer and dimer/monomer models, respectively [11], to investigate the binding of TBC1D8 to the dimeric or tetrameric PKM2 forms. The PKM2 K433Q mutant exhibited more dimeric and monomeric PKM2 forms, while the PKM2 K433R mutant exhibited more tetrameric PKM2 forms (Supplementary Figure S7A and S7G). We demonstrated that wild-type PKM2 and the K433Q mutant, but not PKM2 K433R mutant, interacted with TBC1D8 (Supplementary Figure S7B and S7C).
To preclude the influences of endogenous wild-type PKM2 on the roles of the exogenous PKM2 K433Q and K433R mutants, synonymously mutated sPKM2, sPKM2 K433Q mutant, and sPKM2 K433R mutant were constructed, in which the PKM2 sequences targeted by anti-PKM2 siRNA were synonymously mutated so that exogenous sPKM2, sPKM2 K433Q and K433R mutant expression was not silenced by anti-PKM2 siRNA, but the endogenous PKM2 expression was silenced (Supplementary Figure S7D). The sPKM2 K433Q mutant exhibited more dimeric and monomeric PKM2 forms, while the sPKM2 K433R mutant exhibited more tetrameric PKM2 forms (Supplementary Figure S7H). In the absence of the influences of endogenous wild-type PKM2, we also showed that wild-type sPKM2 and the sPKM2 K433Q mutant, but not the sPKM2 K433R mutant, interacted with TBC1D8 (Supplementary Figure S7E and S7F). Taken together, our results indicate that TBC1D8 interacted with dimeric/ monomeric PKM2 but not tetrameric PKM2, suggesting that TBC1D8 hinders the transition of the dimer/monomer form to a tetramer by binding dimeric/monomeric PKM2.
TBC1D8 inhibits PK activity and promotes aerobic glycolysis in a GAP activity-independent manner
Tetrameric PKM2 has a significantly higher affinity for the substrate PEP and is highly active, while dimeric PKM2 is almost inactive. If TBC1D8 truly hinders PKM2 tetramer formation, PK activity should be decreased in the presence of TBC1D8. We showed that TBC1D8 overexpression significantly decreased PK activity (Figure 5A), whereas silencing TBC1D8 expression increased PK activity (Figure 5B). Similar results were also observed when OVCAR cells with TBC1D8 expression stably knocked down were compared with the control cells (Figure 5C). Furthermore, PK activity was significantly increased in the mouse xenograft tumors composed of TBC1D8-stably silenced cells compared with those composed of control cells (Figure 5D). As same as wild-type TBC1D8, TBC1D8 RYQ/AAA mutant inhibited PK activity, suggesting that the elimination of Rab-GAP activity of TBC1D8 did not influence the inhibitory effects of TBC1D8 on PK activity (Supplementary Figure S8A). Taken together, our data indicate that TBC1D8 decreased PK activity in a GAP activity-independent manner.
TBC1D8 expression inhibits OVCA cell PK activity and increases cell glucose uptake and lactate production. (A) OV-3 cells were transfected with the indicated amount of TBC1D8 vector, and the PK activity was evaluated (n=3). (B) OV-3high cells were transfected with two anti-TBC1D8 siRNAs, and the PK activity was determined (n=3). (C) PK activity was determined in OV-3high cells with TBC1D8 expression stably silenced (n=3). (D) PK activity was determined in mouse xenograft tumors composed of OV-3high cells with TBC1D8 expression stably silenced, as described in Figure 3E (n=3). (E, G) OV-3 cells were transfected with the indicated amount of TBC1D8 vector, and glucose uptake (E) and lactate production (G) were determined (n=3). (F, H) OV-3high cells were transfected with two anti-TBC1D8 siRNAs, and glucose uptake (F) and lactate production (H) were determined (n=3).
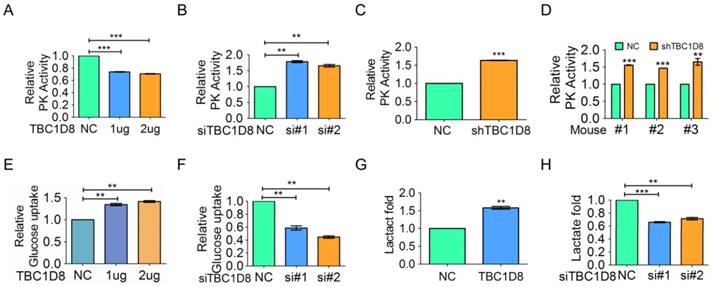
Low PKM2 activity promotes pyruvate conversion to lactate and leads to the Warburg effect, whereas high PKM2 activity promotes pyruvate conversion to acetyl-CoA and leads to oxidative phosphorylation (OXPHOS). The Warburg effect/ aerobic glycolysis is characterized by increased glucose uptake and lactate production. We showed that TBC1D8 overexpression significantly increased glucose uptake and lactate production in OVCA cells (Figure 5E and 5G), whereas silencing TBC1D8 expression down-regulated glucose uptake and lactate production (Figure 5F and 5H). Furthermore, as same as wild-type TBC1D8, TBC1D8 RYQ/AAA mutant increased glucose uptake and lactate production, indicating that the Rab-GAP activity of TBC1D8 did not participate into aerobic glycolysis (Supplementary Figure S8B and S8C). In summary, TBC1D8 inhibits PK activity and drives aerobic glycolysis in a GAP activity-independent manner.
TBC1D8 stimulates the nuclear translocation of PKM2
PKM2 normally functions in the cytoplasm as a glycolytic enzyme, but recent studies have also discovered a nonmetabolic function for PKM2 in the nucleus as a protein kinase and transcriptional co-activator to regulate gene transcription. Previous studies have shown that dimeric/monomeric PKM2 could translocate to nucleus [6, 7, 36]. We further wondered whether TBC1D8-induced PKM2 tetramerization disruption is linked to PKM2 nuclear translocation. When TBC1D8 was overexpressed in OVCAR cells, the nuclear PKM2 signal was increased (Figure 6A), whereas silencing TBC1D8 expression markedly decreased the nuclear PKM2 signal (Figure 6B). Cell fractionation analysis also showed that PKM2 was enriched in the nucleus when TBC1D8 was overexpressed (Figure 6C), whereas PKM2 was rarely detected in the nucleus when TBC1D8 expression was silenced (Figure 6D). In summary, TBC1D8 promotes PKM2 nuclear translocation.
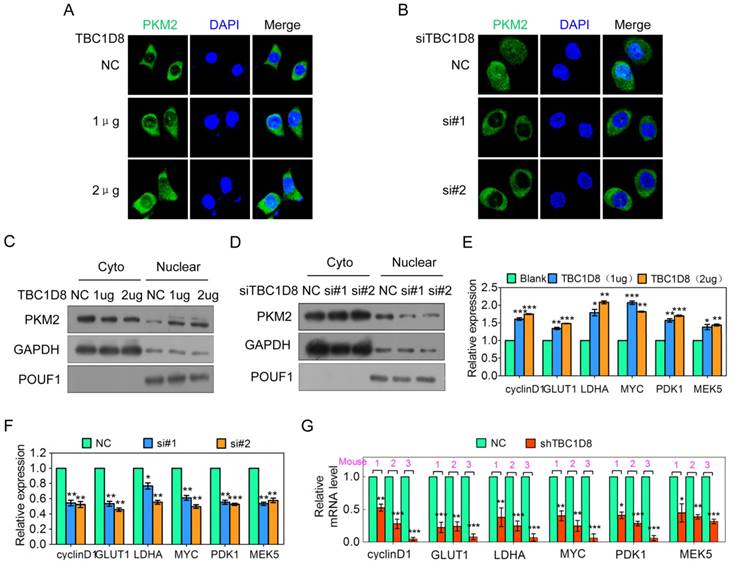
TBC1D8 promotes PKM2 nuclear translocation, and nuclear PKM2 subsequently exerts nonmetabolic functions. (A, B) OV-3 and OV-3high cells were transfected with the indicated amount of TBC1D8 vector (A) and anti-TBC1D8 siRNAs (B), respectively, and the subcellular localization of PKM2 was detected using immunostaining with an anti-PKM2 (green) antibody. The nucleus was stained with DAPI (red). (C, D) OV-3 and OV-3high cells were transfected with the indicated amount of TBC1D8 vector (C) and anti-TBC1D8 siRNAs (D), respectively; the cell cytoplasm and nucleus were fractionated; and PKM2 was then analyzed by Western blotting. (E, F) OV-3 and OV-3high cells were transfected with the indicated amount of TBC1D8 vector (E) and anti-TBC1D8 siRNAs (F), respectively, and the mRNA levels of the indicated genes were determined by qRT-PCR (n=3). (G) The mRNA levels of the indicated genes were determined by qRT-PCR in mouse xenograft tumors composed of OV-3high cells with TBC1D8 expression stably silenced (n=3).
TBC1D8 promotes nuclear PKM2-dependent gene expression
PKM2 translocation into the nucleus promotes both HIF1 transactivation, leading to GLUT1, PDK1 and LDHA expression, β-catenin transactivation, leading to cyclin D and c-Myc expression [6], and STAT3 transactivation, leading to MEK5 transcription [6, 37], which participates in tumorigenesis, tumor growth, the cell cycle and aerobic glycolysis. TBC1D8 overexpression up-regulated cyclin D, GLUT, LDHA, c-myc, PDK1 and MEK5 expression in a dose-dependent manner (Figure 6E), whereas silencing TBC1D8 expression down-regulated the expression level of these genes (Figure 6F). The expression level of these genes in the mouse xenograft tumors was further investigated. Cyclin D, GLUT, LDHA, c-myc, PDK1 and MEK5 expression levels were significantly down-regulated in the mouse xenograft tumors composed of TBC1D8-stably silenced cells compared with those composed of control cells (Figure 6G). Collectively, TBC1D8 promotes the expression of genes associated with tumorigenesis, the cell cycle and aerobic glycolysis.
TBC1D8 promotes tumorigenesis and metabolic reprogramming through PKM2
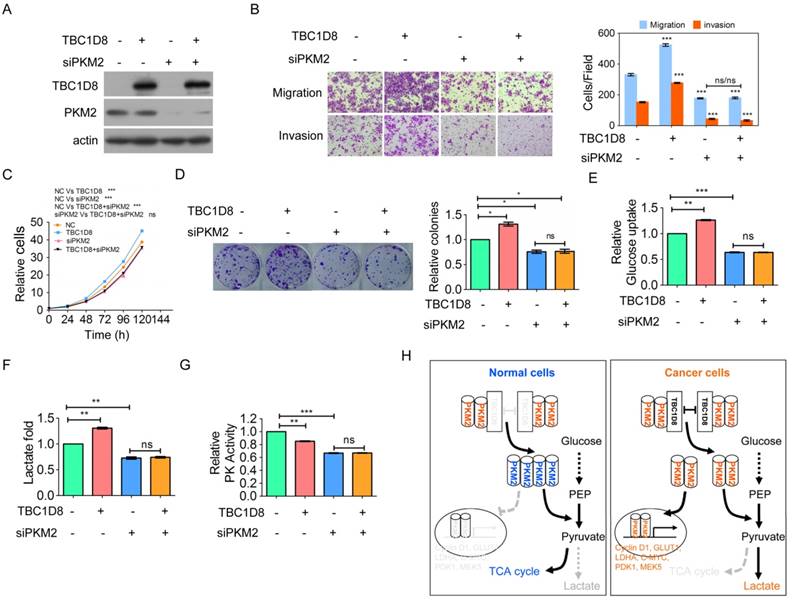
Finally, we furthermore investigate whether TBC1D8 promotes tumorigenesis and aerobic glycolysis through PKM2. The enhancement of OVCA migration and invasion, cell growth, and colony formation induced by TBC1D8 over-expression was completely blocked by PKM2 silencing (Figure 7A-7D). In addition, the inhibitory effects of TBC1D8 over-expression on PK activity and the stimulatory effects of TBC1D8 over-expression on lactate production and glucose production were completely antagonized when PKM2 expression was silenced in OVCA cells (Figure 7E-7G). These results indicated that PKM2 is critical for TBC1D8 functions in tumorigenesis and metabolic reprogramming. Taken together, TBC1D8 promotes OVCA cell tumorigenesis and metabolic reprogramming through PKM2.
TBC1D8 promotes OVCA tumorigenesis and aerobic glycolysis through PKM2. (A-G) TBC1D8 plasmid together with anti-PKM2 siRNA were co-transfected into OVCAR-3 cells, the indicated proteins (A), cell growth (B), migration and invasion (C), colony formation (D), glucose uptake (E), lactate production (F) and PK activity (G) were determined. (H) A proposed model illustrating that TBC1D8 promotes aerobic glycolysis and tumorigenesis by hindering PKM2 tetramerization (n=3).
Discussion
TBC domain-containing proteins traditionally act as GAPs to inactivate Rabs, which are essential for the precise coordination of budding, transport, cytokinesis, membrane trafficking and vesicle fusion. Here, a novel functional role of the TBC domain-containing protein TBC1D8 is elucidated. In addition to functioning as a GAP, TBC1D8 is an important regulator of PKM2 polymerization that controls cellular metabolism and tumorigenesis in a GAP activity-independent manner in OVCA. The Rab-GAP TBC domain of TBC1D8 interacts with dimeric/ monomeric PKM2, not PKM1, to impede PKM2 tetramerization and inhibit PK activity, leading to aerobic glycolysis and promoting the nuclear translocation of PKM2 to regulate the expression of genes associated with aerobic glycolysis and the cell cycle, indicating that TBC1D8 drives OVCA tumorigenesis and metabolic reprogramming (Figure 7H).
Metabolic reprogramming is currently recognized as one of the hallmarks of cancer. However, the driving mechanism of cancer cell metabolism reprogramming is not still clear. In this study, we found that TBC1D8 gene is amplified in OVCA tissues, resulting in the up-regulation of TBC1D8 mRNA and protein levels, and TBC1D8 impedes the tetramerization of PKM2 to suppress its PK activity and promote aerobic glycolysis and tumorigenesis in OVCA. Whether TBC1D8 is amplified and drives tumorigenesis through similar mechanism in other cancers will be worthwhile to be investigated in next step. Amplification of oncogenes and loss of tumor suppressers are frequent in cancer cells and tissues due to genetic abnormality and chromosomal instability, and are drivers for tumorigenesis. A novel driving model of cancer cell metabolism reprogramming is here elucidated due to oncogene amplification.
PKM2, but not the spliced variant PKM1, has a low PK activity that favors the Warburg effect and provides selective advantages for the rapid proliferation and survival of cancer cells. PKM1, with high PK activity, promotes the mitochondrial oxidative phosphorylation pathway and inhibits aerobic glycolytic phenotypes and tumor growth. The integrative capability of PKM2 relies on a specific alternative splicing segment encoded by exon 10, which is responsible for the tetrameric assembly and allosteric regulation that enables PKM2 to induce the Warburg effect [8, 38]. Previous studies have demonstrated that PK activity of PKM2, not PKM1, is modulated by the allosteric activators FBP, serine and SAICAR; tyrosine-peptide binding and posttranslational modifications such as acetylation, phosphorylation and oxidation [1, 6, 7, 10, 12, 13]. In this context, the interaction of TBC1D8 with PKM2, not PKM1, at this region, to disrupt the formation of an allosterically controllable tetramer, represents a novel model of PK tetramer and activity and metabolic reprogramming, in which TBC1D8 interacts with dimeric/monomeric PKM2 but not PKM1, hindering PKM2 tetrameric assembly, decreasing PK activity, and inducing the nuclear translocation of PKM2, to promote the Warburg effect and tumorigenesis. Unlike the other PKM2 interactors HPV16-E7, which inhibits PKM2 tetramerization by acetylating PKM2 at K433 [11], and CD44, which inhibits PKM2 tetramerization by phosphorylating PKM2 at Tyr105 [39], TBC1D8 does not change the acetylation or phosphorylation levels of PKM2. We found that TBC1D8 binds to dimeric PKM2, not tetrameric PKM2, suggesting that the binding of TBC1D8 to the allosteric regulatory region in PKM2 cause a steric hindrance of FBP binding, leading to an decreased PKM2 dimer-to-tetramer conversion. Therefore, a different way of regulating PKM2 activity than previously reported is here elucidated.
Previous study revealed that ERK1/2 phosphorylates PKM2 at Ser37, leading to PIN1-dependent cis-trans isomerization, and PKM2 tetramer-to-dimer/monomer conversion [40]. PKM2 monomer formation exposes the sterically buried nuclear localization signal (NLS) of PKM2, makes the NLS accessible for binding to importin α to induce PKM2 nuclear translocation [40, 41]. In this study, we found that TBC1D8 inhibits the conversion of PKM2 from tetramer to dimmer/monomer. Subsequently, the NLS of monomeric PKM2 can be easily recognized by importin α, thereby inducing PKM2 nuclear translocation.
Among the 44 TBC-containing proteins, only a few, including TBC1D7 [18], TBC1D14 [17], TBC1D16 [19], PRC17 [20] and USP6/TRE17 [21], were reported to be associated with cancer progression; the functional roles of the majority of the other members are still unclear. In this study, we found that TBC1D8 amplification resulted into increased TBC1D8 expression in OVCA, and drove OVCA tumorigenesis and metabolic reprogramming in a GAP activity-independent manner. We also demonstrated that TBC domain proteins, such as TBC1D8, participate in regulating glucose metabolism and induce the nuclear translocation of some proteins, in addition to controlling different trafficking routes and organelle biogenesis and autophagy [15, 17, 19, 34]. And the regulation of other TBC proteins on trafficking routes and organelle biogenesis and autophagy was dependent on the Rab-GAP activity of these TBC proteins. However, we found that TBC1D8 drives tumorigenesis and metabolic reprogramming in a Rab-GAP activity-independent manner. Our results enrich the understanding of the functional roles of TBC domain proteins. TBC1D8 may have potential to serve as an independent prognostic factor for OVCA and anti-OVCA drug target.
In summary, our findings provide a driving fashion of metabolism reprogramming in OVCA and delineate the functional roles of a TBC domain protein TBC1D8 in tumorigenesis and cancer metabolism reprogramming in Rab-GAP TBC activity-independent manner. TBC1D8 promotes OVCA tumorigenesis and aerobic glycolysis by binding to PKM2 to hinder PKM2 tetramerization, inhibit PK activity, and promote PKM2 nuclear translocation to perform nonmetabolic functions. OVCA patients with TBC1D8high exhibit more aggressive clinicopathological features and poorer prognoses. TBC1D8 amplification is critical oncogenic event for OVCA tumorigenesis and metabolic reprogramming.
Abbreviations
OVCA: ovarian cancer; PK: pyruvate kinase; PKM: M type PK; PKM2: M2 isoform of pyruvate kinase; TBC: Tre2/Bub2/Cdc16; GAP: GTPase-activating protein; NLS: nuclear localization signal.
Supplementary Material
Supplementary table S1.
Supplementary figures and tables S2-3.
Supplementary table S4.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81772998, 81672393), the R&D Plan of Guangzhou (201704020115), the Yangcheng Scholars program from the Ministry of Education of Guangzhou (1201561583), Innovative Research Team of Ministry of Education of Guangzhou (1201610015), the R&D Plan of Guangdong (2017A020215096), the National Funds of Developing Local Colleges and Universities (B16056001).
Author Contributions
GRY conceived the project, designed the experiments and wrote the paper; MC, YYQ, QXW, LJ, NM and YTH conducted the in vitro and in vivo experiments; XJS and QXW collected patient samples; SZ performed IHC analysis; MC and GRY provided the statistical analysis.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Israelsen WJ, Vander Heiden MG. Pyruvate kinase: Function, regulation and role in cancer. Semin Cell Dev Biol. 2015;43:43-51
2. Bu P, Chen KY, Xiang K, Johnson C, Crown SB, Rakhilin N. et al. Aldolase B-Mediated Fructose Metabolism Drives Metabolic Reprogramming of Colon Cancer Liver Metastasis. Cell Metab. 2018;27:1249-62 e4
3. Li J, He Y, Tan Z, Lu J, Li L, Song X. et al. Wild-type IDH2 promotes the Warburg effect and tumor growth through HIF1alpha in lung cancer. Theranostics. 2018;8:4050-61
4. Redis RS, Vela LE, Lu W, Ferreira de Oliveira J, Ivan C, Rodriguez-Aguayo C. et al. Allele-Specific Reprogramming of Cancer Metabolism by the Long Non-coding RNA CCAT2. Mol Cell. 2016;61:520-34
5. Mishra P, Tang W, Putluri V, Dorsey TH, Jin F, Wang F. et al. ADHFE1 is a breast cancer oncogene and induces metabolic reprogramming. J Clin Invest. 2018;128:323-40
6. Tamada M, Suematsu M, Saya H. Pyruvate kinase M2: multiple faces for conferring benefits on cancer cells. Clin Cancer Res. 2012;18:5554-61
7. Yang W, Lu Z. Pyruvate kinase M2 at a glance. J Cell Sci. 2015;128:1655-60
8. Huang JZ, Chen M, Chen, Gao XC, Zhu S, Huang H. et al. A Peptide Encoded by a Putative lncRNA HOXB-AS3 Suppresses Colon Cancer Growth. Mol Cell. 2017;68:171-84 e6
9. Clower CV, Chatterjee D, Wang Z, Cantley LC, Vander Heiden MG, Krainer AR. The alternative splicing repressors hnRNP A1/A2 and PTB influence pyruvate kinase isoform expression and cell metabolism. Proc Natl Acad Sci U S A. 2010;107:1894-9
10. Hitosugi T, Kang S, Vander Heiden MG, Chung TW, Elf S, Lythgoe K. et al. Tyrosine phosphorylation inhibits PKM2 to promote the Warburg effect and tumor growth. Sci Signal. 2009;2:ra73
11. Lv L, Xu YP, Zhao D, Li FL, Wang W, Sasaki N. et al. Mitogenic and oncogenic stimulation of K433 acetylation promotes PKM2 protein kinase activity and nuclear localization. Mol Cell. 2013;52:340-52
12. Anastasiou D, Poulogiannis G, Asara JM, Boxer MB, Jiang JK, Shen M. et al. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science. 2011;334:1278-83
13. Christofk HR, Vander Heiden MG, Wu N, Asara JM, Cantley LC. Pyruvate kinase M2 is a phosphotyrosine-binding protein. Nature. 2008;452:181-6
14. Frasa MA, Koessmeier KT, Ahmadian MR, Braga VM. Illuminating the functional and structural repertoire of human TBC/RABGAPs. Nat Rev Mol Cell Biol. 2012;13:67-73
15. Gabernet-Castello C, O'Reilly AJ, Dacks JB, Field MC. Evolution of Tre-2/Bub2/Cdc16 (TBC) Rab GTPase-activating proteins. Mol Biol Cell. 2013;24:1574-83
16. Pan X, Eathiraj S, Munson M, Lambright DG. TBC-domain GAPs for Rab GTPases accelerate GTP hydrolysis by a dual-finger mechanism. Nature. 2006;442:303-6
17. Lamb CA, Nuhlen S, Judith D, Frith D, Snijders AP, Behrends C. et al. TBC1D14 regulates autophagy via the TRAPP complex and ATG9 traffic. Embo J. 2016;35:281-301
18. Sato N, Koinuma J, Ito T, Tsuchiya E, Kondo S, Nakamura Y. et al. Activation of an oncogenic TBC1D7 (TBC1 domain family, member 7) protein in pulmonary carcinogenesis. Genes Chromosomes Cancer. 2010;49:353-67
19. Vizoso M, Ferreira HJ, Lopez-Serra P, Carmona FJ, Martinez-Cardus A, Girotti MR. et al. Epigenetic activation of a cryptic TBC1D16 transcript enhances melanoma progression by targeting EGFR. Nat Med. 2015;21:741-50
20. Pei L, Peng Y, Yang Y, Ling XB, Van Eyndhoven WG, Nguyen KC. et al. PRC17, a novel oncogene encoding a Rab GTPase-activating protein, is amplified in prostate cancer. Cancer Res. 2002;62:5420-4
21. Quick L, Young R, Henrich IC, Wang X, Asmann YW, Oliveira AM. et al. Jak1-STAT3 Signals Are Essential Effectors of the USP6/TRE17 Oncogene in Tumorigenesis. Cancer Res. 2016;76:5337-47
22. Xiao CL, Zhu S, He M, Chen, Zhang Q, Chen Y. et al. N6-Methyladenine DNA Modification in the Human Genome. Mol Cell. 2018;71:306-18 e7
23. Xu SH, Huang JZ, Xu ML, Yu G, Yin XF, Chen D. et al. ACK1 promotes gastric cancer epithelial-mesenchymal transition and metastasis through AKT-POU2F1-ECD signalling. J Pathol. 2015;236:175-85
24. Huang JZ, Chen M, Zeng M, Xu SH, Zou FY, Chen D. et al. Down-regulation of TRPS1 stimulates epithelial-mesenchymal transition and metastasis through repression of FOXA1. J Pathol. 2016;239:186-96
25. Xu SH, Zhu S, Wang Y, Huang JZ, Chen M, Wu QX. et al. ECD promotes gastric cancer metastasis by blocking E3 ligase ZFP91-mediated hnRNP F ubiquitination and degradation. Cell Death Dis. 2018;9:479
26. Yan GR, Tan Z, Wang Y, Xu ML, Yu G, Li Y. et al. Quantitative proteomics characterization on the antitumor effects of isodeoxyelephantopin against nasopharyngeal carcinoma. Proteomics. 2013;13:3222-32
27. Selmansberger M, Feuchtinger A, Zurnadzhy L, Michna A, Kaiser JC, Abend M. et al. CLIP2 as radiation biomarker in papillary thyroid carcinoma. Oncogene. 2015;34:3917-25
28. Tan R, Nakajima S, Wang Q, Sun H, Xue J, Wu J. et al. Nek7 Protects Telomeres from Oxidative DNA Damage by Phosphorylation and Stabilization of TRF1. Mol Cell. 2017;65:818-31 e5
29. Banck MS, Kanwar R, Kulkarni AA, Boora GK, Metge F, Kipp BR. et al. The genomic landscape of small intestine neuroendocrine tumors. J Clin Invest. 2013;123:2502-8
30. Mai TT, Lito P. A treatment strategy for KRAS-driven tumors. Nat Med. 2018;24:902-4
31. Williams KA, Lee M, Hu Y, Andreas J, Patel SJ, Zhang S. et al. A systems genetics approach identifies CXCL14, ITGAX, and LPCAT2 as novel aggressive prostate cancer susceptibility genes. PLoS Genet. 2014;10:e1004809
32. Kohler A, Demir U, Kickstein E, Krauss S, Aigner J, Aranda-Orgilles B. et al. A hormone-dependent feedback-loop controls androgen receptor levels by limiting MID1, a novel translation enhancer and promoter of oncogenic signaling. Mol Cancer. 2014;13:146
33. Atanassov BS, Mohan RD, Lan X, Kuang X, Lu Y, Lin K. et al. ATXN7L3 and ENY2 Coordinate Activity of Multiple H2B Deubiquitinases Important for Cellular Proliferation and Tumor Growth. Mol Cell. 2016;62:558-71
34. Gallo LI, Liao Y, Ruiz WG, Clayton DR, Li M, Liu YJ. et al. TBC1D9B functions as a GTPase-activating protein for Rab11a in polarized MDCK cells. Mol Biol Cell. 2014;25:3779-97
35. Wang HJ, Hsieh YJ, Cheng WC, Lin CP, Lin YS, Yang SF. et al. JMJD5 regulates PKM2 nuclear translocation and reprograms HIF-1alpha-mediated glucose metabolism. Proc Natl Acad Sci U S A. 2014;111:279-84
36. Yang W, Xia Y, Hawke D, Li X, Liang J, Xing D. et al. PKM2 phosphorylates histone H3 and promotes gene transcription and tumorigenesis. Cell. 2012;150:685-96
37. Gao X, Wang H, Yang JJ, Liu X, Liu ZR. Pyruvate kinase M2 regulates gene transcription by acting as a protein kinase. Mol Cell. 2012;45:598-609
38. David CJ, Chen M, Assanah M, Canoll P, Manley JL. HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature. 2010;463:364-8
39. Tamada M, Nagano O, Tateyama S, Ohmura M, Yae T, Ishimoto T. et al. Modulation of glucose metabolism by CD44 contributes to antioxidant status and drug resistance in cancer cells. Cancer Res. 2012;72:1438-48
40. Yang W, Zheng Y, Xia Y, Ji H, Chen X, Guo F. et al. ERK1/2-dependent phosphorylation and nuclear translocation of PKM2 promotes the Warburg effect. Nat Cell Biol. 2012;14:1295-304
41. Yang W, Lu Z. Nuclear PKM2 regulates the Warburg effect. Cell Cycle. 2013;12:3154-8
Author contact
![]() Corresponding author: Guang-Rong Yan, E-mail: jxygr007com, Phone: 86-020-81298832.
Corresponding author: Guang-Rong Yan, E-mail: jxygr007com, Phone: 86-020-81298832.