Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results
Discussion
Conclusions
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(3):633-645. doi:10.7150/thno.31485 This issue Cite
Research Paper
A Synthetic Receptor as a Specific Antidote for Paraquat Poisoning
Xiangjun Zhang1, Xiaoqiu Xu2, Shengke Li1, Lanlan Li2, Jianxiang Zhang2 ![]() , Ruibing Wang1
, Ruibing Wang1 ![]()
1. State Key Laboratory of Quality Research in Chinese Medicine, and Institute of Chinese Medical Sciences, University of Macau, Taipa, Macau, China.
2. Department of Pharmaceutics, College of Pharmacy, Third Military Medical University, Chongqing 400038, China.
Received 2018-11-13; Accepted 2018-12-18; Published 2019-1-21
Abstract

Accidental or suicidal ingestion of the world's most widely used herbicide, paraquat (PQ), may result in rapid multi-organ failure with a 60% fatality rate due to the absence of an effective detoxification solution. Effective, specific antidotes to PQ poisoning have been highly desired.
Methods: The binding constant of PQ and a synthetic receptor, cucurbit[7]uril (CB[7]), was first determined in various pH environments. The antidotal effects of CB[7] on PQ toxicity were firstly evaluated with in-vitro cell lines. With in-vivo mice models, the pharmacokinetics and the biodistribution of PQ in major organs were determined to evaluate the influence of CB[7] on the oral bioavailability of PQ. Major organs' injuries and overall survival rates of the mice were systemically examined to evaluate the therapeutic efficacy of CB[7] on PQ poisoning.
Results: We demonstrate that CB[7] may complex PQ strongly under various conditions and significantly reduce its toxicity in vitro and in vivo. Oral administration of PQ in the presence of CB[7] in a mouse model significantly decreased PQ levels in the plasma and major organs and alleviated major organs' injuries, when compared to those of mice administered with PQ alone. Further studies indicated that oral administration of CB[7] within 2 h post PQ ingestion significantly increased the survival rates and extended the survival time of the mice, in contrast to the ineffective treatment by activated charcoal, which is commonly recommended for PQ decontamination.
Conclusion: CB[7] may be used as a specific oral antidote for PQ poisoning by strongly binding with PQ and inhibiting its absorption in the gastrointestinal tracts.
Keywords: paraquat toxicity, cucurbit[7]uril, antidote, detoxification
Introduction
There are estimated 109,700 deaths from pesticide self-poisoning worldwide each year, and paraquat (PQ) is one of the leading pesticides causing death in many developing countries [1, 2]. Suicidal or accidental ingestion of PQ would result in rapid multi-organ failure with a mortality rate exceeding 60% [3, 4]. These poor outcomes are mainly attributed to the severe toxicity of PQ and the lack of effective detoxification therapies. Although several countries have banned or restricted its use, PQ is still being registered, legally distributed and used in more than 90 developed and developing countries [5-7], meaning that there are still large populations facing the problem of potential PQ poisoning. Without effective, specific antidotes available, only moderately effective, nonspecific, life-supporting measures are taken to detoxify PQ poisoning in clinics. These measures often include oral decontaminants such as activated charcoal (AC), for early PQ decontamination, followed by anti-oxidant therapy, immunosuppression, hemoperfusion, hemodialysis, resuscitation and supportive care [5, 8-15]. Unfortunately, no clinical benefits from these treatments have been demonstrated and there has been no specific antidotal therapy to date for PQ poisoning [13, 16-20]. Upon oral ingestion, PQ is usually rapidly absorbed into the small intestine and distributed to major organs, including the liver, lungs, kidneys and muscles [4, 21]. Therefore, preventing its intestinal absorption in the first place after oral ingestion of PQ might be the key to a successful treatment. An effective and specific oral antidote is indeed urgently needed to treat PQ poisoning.
The essential attributes of an effective oral PQ antidote should include: (i) good oral biocompatibility to allow administration at high doses; (ii) high binding affinity with PQ to allow specific and efficient scavenging of PQ in the gastrointestinal (GI) environment; (iii) ability to reduce PQ absorption upon complexation with PQ in the GI tract. Cucurbit[n]urils (CB[n]s, n = 5-8, and 10) are a series of macrocyclic, methylene-bridged glycoluril oligomers with shapes resembling that of a pumpkin and have received increasing interest as a growing family of host molecules in supramolecular chemistry [22-24]. As synthetic receptors, CB[n]s can bind with a variety of guest molecules such as aromatic and aliphatic compounds [25, 26]. Our previous studies demonstrated that the toxicity of certain guest molecules might be dramatically reduced upon their complexation with CB[7] strongly [27, 28]. However, the underlying mechanism is not clear and none of these or other examples have demonstrated that CB[7] may reverse toxicity of a guest molecule in a mammalian model. Of note, it has been previously reported that PQ and CB[7] form a stable 1:1 complex with a relatively high binding affinity [29, 30]. Moreover, the excellent oral biocompatibility of CB[7] has been recently demonstrated in mice [31-33]. Therefore, CB[7] likely fulfills all of these requirements as an effective PQ antidote.
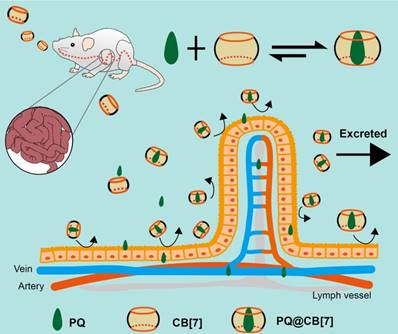
Herein, we hypothesized that CB[7] might be used as a safe oral antidote for PQ ingestion by sequestering PQ in the GI tract, thereby reducing the intestinal absorption of PQ and detoxifying PQ poisoning (Figure 1). As a proof of concept, we successfully demonstrated that complexation of PQ by CB[7] decreased its toxicity in vitro and in vivo and, more importantly, oral administration of CB[7] significantly reduced the intestinal absorption of PQ, decreased PQ levels in the plasma and major organs, and alleviated major organs' damage, resulting in highly increased survival rates and survival time of the mice.
The proposed function of CB[7] for detoxifying PQ ingestion. CB[7] is orally administered after PQ ingestion. In the stomach or intestine, CB[7] undergoes complexation with PQ, preventing intestinal injury and reducing the absorption and tissue distribution of PQ. Most of PQ will be excreted in the form of PQ@CB[7] complex.
Methods
Animals and cell lines
All animal studies were approved by the Ethical Committee for Animal Experimentation of the Third Military Medical University, and were conducted according to the Animal Management Rules of the Ministry of Health of the People's Republic of China (No. 55, 2001) and the guidelines for the Care and Use of Laboratory Animals of the Third Military Medical University. Male Balb/c mice were obtained from the Animal Center of the Third Military Medical University (Chongqing, China). Human lung carcinoma A549 cells, normal human hepatic LO2 cells and human intestinal epithelial Caco-2 cells were originally obtained from Cell Bank at the Chinese Academy of Sciences (Shanghai, China). A549 cells and LO2 cells were cultured in a 5% CO2 humidified environment at 37 °C in complete Dulbecco's modified eagle medium (DMEM, Gibco, NYC, USA) supplemented with 10% fetal bovine serum (FBS, Gibco, NYC, USA) and 1% penicillin-streptomycin (Sigma-Aldrich, St. Louis, MO, USA). In addition, Caco-2 cell line(passage numbers: 30-40) was grown in the same environment with the addition of 1% MEM non-essential amino acids (Gibco, NYC, USA) to the culture medium.
Synthesis of CB[7]
CB[7] (C42H42N28O14, molecular weight:1162.96 g/mol) was synthesized as described previously [22, 34, 35].
Modeling studies of CB[7] and PQ binding
Three-dimensional structures of CB[7] and PQ were drawn with ChemBioOffice Ultra 14.0 software. AutoDock Tools was used to generate the pdb (protein data bank) files. The binding conformations between CB[7] and PQ were simulated with AutoDock Vina [36]. A grid map of dimensions 40 Å × 40 Å × 40 Å with a grid space of 0.375 Å was set. The center of the search space was set to -3.066 Å, -0.011 Å and -0.086 Å (x, y, z). One hundred GA (genetic algorithm) runs was set, and all other parameters were the default values by AutoDock Vina. Conformational searching was performed by the Lamarckian genetic algorithm (LGA). The structure of the complex with lowest energy was re-optimized with ChemBioOffice Ultra 14.0 software.
Determination of CB[7] and PQ binding affinities
Isothermal titration calorimetry (ITC, Malvern MicroCal PEAQ, Malvern, Worcestershire, UK) was utilized to determine the binding constant and thermodynamic parameters for CB[7] and PQ (J&K Scientific, Beijing, China). Briefly, 0.2 mL of aqueous CB[7] solution (0.2 mM) was placed into the sample cell. In addition, a 2-mM aqueous PQ solution was added in a series of 19 injections (2 μL each) as the heat evolved was recorded at 25.0 °C at time intervals of 150 s for each titration. The obtained data were analyzed and fitted by the built-in software. Thermodynamic parameters analysis was conducted with the “one set of binding sites” mathematic model. Similar methods were utilized for the determination of CB[7] and PQ binding affinities in a hydrochloric acid-saline solution (pH 1.2) containing 84 mM HCl and 34 mM NaCl, and in pH 3.0, pH 4.5, pH 6.8 and pH 7.4 phosphate-buffered saline (PBS) solutions containing 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, and 1.8 mM KH2PO4 with the pH adjusted by 1 M HCl.
Cell viability assays
After culturing 1×104 cells in 96-well plates for 12 h, A549 or LO2 cells were then incubated with various concentrations of PQ (0.1, 0.2 and 0.5 mM) in the presence or absence of CB[7] (0.1, 0.2 and 0.5 mM). After 24 h, a cell counting kit-8 (Beyotime, Nantong, Jiangsu, China) was utilized to determine the cell viability. The same method was applied to determine the cell viability when cells were incubated with 0.5 mM PQ in the presence of various concentrations of CB[7] (0, 0.2, 0.5, 1.0, and 2.0 mM).
Determination of the intracellular levels of PQ
3×105 Caco-2 cells in 1.0 mL of growth medium were seeded in 12-well plates for 12 h. Cells were then incubated with PQ (0.5 mM) or PQ@CB[7] (PQ and CB[7], 0.5 and 2 mM, respectively) for 0.5, 1, 2, 4 and 8 h (n = 3). After being washed with PBS, cells were lysed with 300 μL RIPA Lysis Buffer with 1% protease inhibitor (Beyotime, Nantong, Jiangsu, China). Lysate was centrifuged at 10000 × g for 10 min, protein contents in the supernatant were determined by BCA assays (Beyotime, Nantong, Jiangsu, China) according to the manufacturer's protocol. A HPLC (high performance liquid chromatography) method was used to determine PQ concentration. 200 μL of supernatant was mixed with 50 μL of 6% perchloric acid in methanol (V/V) for 3 min and kept at -20 °C for 10 min. After centrifugation at 10000 × g for 15 min, HPLC coupled with a UV-Vis detector (HPLC-UV, Agilent 1200, Santa Clara, CA, USA) was employed to determine the PQ concentration in the supernatant. The mobile phase consisted of 10% acetonitrile (V/V) and 90% of a buffer solution containing 0.1 M orthophosphoric acid and 3.0 mM of sodium 1-octanesulphonic acid, which was adjusted to pH 3.0 with the addition of diethylamine. The stationary phase was ZORBAX Eclipse XDB-C18 column (4.6×250 mm, 5 μm), and the flow-rate was at 0.8 mL/min and the UV absorbance was detected at 258 nm. The intracellular levels of PQ were presented as the ratio of PQ/protein.
PQ poisoning mouse model
Male Balb/c mice (8-10 weeks old) with body weights of 20-25 g were used. This experimental protocol was approved by the guidelines for the Care and Use of Laboratory Animals of the Third Military Medical University. After they had been fasted for 8 h, the mice were orally administered with PQ solutions at doses ranging from 100, 150, 200, 250, 300 to 350 mg/kg. The body weights as well as the number of living mice were recorded each day. The behavior of the mice was also observed daily for any signs of illness.
Determination of the plasma levels and tissue accumulation of PQ
Sample collection
A total of 84 mice were used for the determination of PQ concentration in plasma and to detect its accumulation in different tissues. Briefly, 8-10-week-old male Balb/c mice with weights of 20-25 g were orally administered with PQ or PQ@CB[7] (molar ratio 1:2, CB[7]= 2.71 g/kg) solution at a PQ dose of 300 mg/kg. After 10 min, 30 min, 1 h, 2 h, 4 h, 8 h and 12 h, blood samples were collected into sodium heparin spray-coated tubes and plasma samples were separated and stored at -80 °C. After 4, 8 and 12 h, major organs, including the heart, liver, lungs, kidneys, spleen and intestines were collected, washed with ice-cold PBS and stored at -80 °C.
Sample pretreatment
Plasma pretreatment was conducted as described in previous literature [37]. A 5 µL of 150 μg/mL diethyl paraquat (EQ, internal standard, aladdin®, Shanghai, China) aqueous solution was added to 200 µL plasma simples, and thoroughly mixed with 45 µL of 6% perchloric acid (v/v) in methanol. Samples were then left standing for 10 min at -20 °C and centrifuged at 8000 ×g for 10 min. Supernatants were then centrifuged for once again under the same conditions for HPLC analysis.
Tissue pretreatments were performed with a previously published method, albeit with modifications[38]. Major organs, including the heart, liver, lungs, spleen and kidneys, were weighed and placed into tubes. Samples were cut into small pieces and homogenized in 1 or 2 mL (for liver tissues) pH 7.4 PBS solutions containing 0.1% Triton-X 100. Samples were then centrifuged at 8000 ×g for 10 min. Supernatants were subsequently centrifuged for one more time under the same conditions and received, using the same procedure for plasma pretreatment for HPLC analysis.
Intestines were cut into three sections, including the duodenum, jejunum and ileum, with a length of 5 cm for each part. Samples were weighed and homogenized in 1 mL of pH 7.4 PBS containing 0.1% Triton-X 100. Samples were subsequently centrifuged twice at 8000 ×g for 10 min and the supernatants were treated via the same procedure for plasma pretreatment for HPLC analysis.
Standard samples were prepared by adding PQ aqueous solution to the blank plasma or supernatants of homogenized tissues to make a series of standard samples with the PQ concentration of 0, 0.5, 1, 2, 5, 10, 20, 50 μg/mL. Standard samples were then received with the same procedure for plasma pretreatment for HPLC analysis.
HPLC analysis
The HPLC characterization was performed via a previously described method with minor changes [37]. Briefly, a HPLC system equipped with a UV-Vis detector (SHIMADZU i-Series, Shimadzu, Kyoto, Japan) was used. The mobile phase was the same as what was used in the determination of intracellular PQ content. Samples were separated on a ZORBAX Eclipse XDB-C18 column (4.6 × 250 mm, 5 μm) and a C-18 pre-column (4.6×10 mm, 5 μm) at 37 °C. PQ and an internal standard were eluted at a flow-rate of 0.8 mL/min and detected at 258 nm.
Calculation of AUC (area under the curve)
The area under the plasma concentration-time curve from time zero to time t (AUC(0-t)) was calculated with the DAS 3.2.3 pharmacokinetic program.
Determination of inflammatory cytokines and ROS (reactive oxygen species) levels
Male Balb/c mice (8-10 weeks old) with weights of 20-25 g were orally administered with PQ or PQ@CB[7] (molar ratio 1:2, CB[7] = 2.71 g/kg) solution at a PQ dose of 300 mg/kg. After 8 h, lungs and intestine samples were collected and washed with ice-cold PBS. Samples of the jejunum and ileum with a length of 5 cm for each part were cut off from the intestine. Tissues including lungs, jejunum and ileum were then placed into tubes and homogenized in 1 mL ice-cold pH 7.4 PBS. Samples were then centrifuged twice at 8000 ×g for 10 min and supernatants were saved for the determination of inflammatory cytokines and ROS levels. The levels of various cytokines including tumor necrosis factor-α (TNF-α), interferon-γ (IFN-r) and interleukin-1α (IL-1α) and myeloperoxidase (MPO, Boster Biological Technology co. ltd, Wuhan, Hubei, China) were determined by using ELISA kit (Neobioscience, Shenzhen, Guangdong, China) according to the manufacturer's protocols. Relative superoxide levels were determined using an assay kit purchased from Beyotime (Nantong, Jiangsu, China). A fluorescent hydrogen peroxide (H2O2) assay kit (Sigma-Aldrich, St. Louis, MO, USA) was used to detect the H2O2levels. After peroxidase substrate reacted with H2O2, the red fluorescent product (excitation wavelength = 540 nm and emission wavelength = 590 nm) was analyzed with a multifunctional microplate reader (TECAN Infinite M200, Tecan, Mannedorf, Switzerland).
Determination of tissue injuries
To determine the protection of CB[7] on PQ-induced tissue injuries, 8-10-week-old male Balb/c mice with weights of 20-25 g were orally administered with PQ or PQ@CB[7] (molar ratio of PQ/CB[7] = 1:2, CB[7] = 2.71 g/kg) solution at a PQ dose of 300 mg/kg. Mice were orally administered with 100 μL saline/10 g weight as the control group. After 8 h, major organs, including the heart, liver, spleen, lungs and kidneys, were collected and washed with PBS. GI tissues were also collected and washed with PBS. Sections of major organs and GI tissues were prepared and stained with hematoxylin and eosin (H&E) for histopathological analysis.
Treatment protocols
Male Balb/c mice (8-10 weeks old) with weights of 20-25 g were orally administered with PQ at a dose of 300 mg/kg. After 10 min, CB[7]/saline suspensions (1, 2 and 3 equiv. to PQ , 1.36, 2.71, and 4.07 g/kg) were gavaged. For the time-dependent treatment study, mice were gavaged with 300 mg/kg of PQ first, and CB[7]/saline suspensions (2 equiv. to PQ, 2.71 g/kg) were subsequently administered after 0.5, 1 and 2 h, respectively. The weights as well as the number of living mice were recorded each day. After 21 days, all the remaining animals were sacrificed. Major organs were collected and weighed. Sections of major organs and GI tissues were prepared and stained with H&E for histopathological analysis. For the time-dependent treatment study of PQ-poisoned mice by AC, mice were gavaged with 300 mg/kg PQ and AC (~200 meshes, specific surface area ~ 1500 m2/g, Xiya Reagent, Linyi, Shandong, China) were subsequently administered after 10 min, 0.5 h, 1 h and 2 h at a dose of 1.5 g/kg. The weights and the number of living mice were recorded each day.
Statistical analysis
Data are presented as means ± SEM (standard error of mean). Two-tailed, unpaired Student's t-tests were performed for the comparisons between two sample sets and one-way ANOVA was used for experiments consisting of more than two groups with PASW Statistics 18.0. Kaplan-Meier survival analysis was assessed via a log-rank test. Statistical significance was considered as P < 0.05.
Results
PQ-CB[7] binding affinity
In this study, we firstly simulated the binding of PQ and CB[7] (Figure 2A) utilizing AutoDock Vina[36] and the complex structure with the lowest binding energy is shown in Figure 2B. It has been reported previously that in pH 7.0 tris-buffer solution, CB[7] and PQ formed a stable 1:1 complex with a binding constant of 2.0×105 M-1 [29, 30]. However, the pH values encountered within the GI tract can vary from ~1.0 to ~7.4 for humans depending on their ages or health situations [39-41]. We, therefore, determined the binding affinities of PQ and CB[7] in pH 1.2 hydrochloric acid-saline solution, in pH 3.0, pH 4.5, pH 6.8, and pH 7.4 PBS solutions (Figure 2C, Table 1) as well as in water, by ITC. The binding constants between CB[7] and PQ in pH 1.2~7.4 buffers are all above ~105 M-1 , indicating that CB[7] likely exhibits a high binding affinity with PQ in the GI tract.
Complexation of PQ by CB[7]. (A) Structures of CB[7] (left) and PQ (right). (B) Energy-optimized inclusion complexes of CB[7] and PQ2+ by AutoDock Vina. The left panel is the side view and the right panel is the top view. (C) Representative of ITC titration of CB[7] with PQ in pH 7.4 phosphate buffered saline (PBS) at 25 °C. One binding site model was utilized to fit the data, affording an association constant Ka of 1.57 (±0.15) × 105 M-1 (ΔH: -8.81 ± 0.15 kJ•mol-1, ΔG: -29.7 kJ•mol-1, -T Δ S: -20.9 kJ•mol-1).
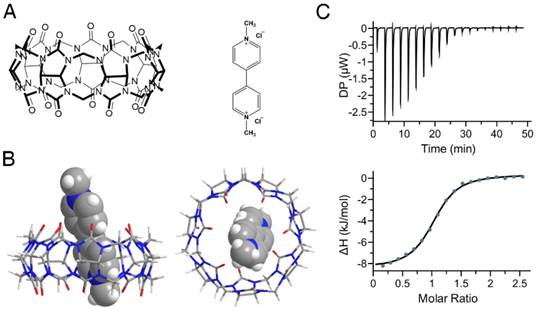
Binding constants between CB[7] and PQ in various pH environments.
| Solutions | pH 1.2 | pH 3.0 | pH 4.5 | pH 6.8 | pH 7.4 | water |
|---|---|---|---|---|---|---|
| Ka (M-1) | 6.25×105 | 1.39×105 | 1.08×105 | 1.77×105 | 1.57×105 | 2.80×106 |
In vitro relieving effects of CB[7] on PQ poisoning
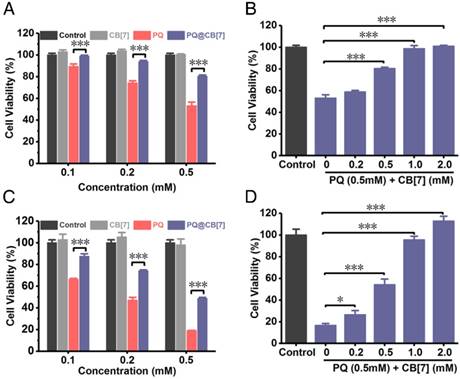
After having demonstrated that CB[7] may still effectively bind PQ in various pH environments, we firstly evaluated the relief effects of CB[7] on PQ poisoning in vitro. We analyzed the protective effect of CB[7] (molar ratio to PQ = 1:1) at different PQ concentrations on A549 cells (a cell line that has been previously used as a model for lung cells in PQ toxicity studies [42, 43]) and LO2 cells (a human hepatic cell line), as PQ would induce severe injuries on lungs and liver. As shown in Figure 3A, PQ induced A549 cellular death in 24 h in a dose-dependent manner, whereas its cytotoxicity was effectively inhibited with the addition of CB[7] (PQ@CB[7], molar ratio 1:1). In addition, the protective effects of CB[7] on cellular viability against PQ's cytotoxicity also showed a dose-dependent behavior (Figure 3B). Similar results were obtained in LO2 cells (Figure 3C-D).
Antidotal effects of CB[7] on PQ poisoning in A549 cells and LO2 cells. (A) Viability of A549 cells which were incubated with CB[7], PQ or PQ@CB[7] (molar ratio=1:1) at the various PQ concentration. Data are presented as means ± SEM, n = 5, ***P<0.001. (B) Viability of A549 cells which were incubated with PQ (0.5mM) or PQ@CB[7] at the various CB[7] concentration for 24 h. Data are presented as means ± SEM, n = 5, ***P<0.001. (C) Viability of LO2 cells which were incubated with CB[7], PQ or PQ@CB[7] (molar ratio=1:1) at the various PQ concentration. Data are presented as means ± SEM, n = 5, ***P<0.001. (D) Viability of LO2 cells which were incubated with PQ (0.5mM) or PQ@CB[7] at the various CB[7] concentration for 24 h. Data are presented as means ± SEM, n = 5, *P<0.05, ***P<0.001.
Evaluation of PQ poisoning in vivo.
To determine the toxicity of PQ with in vivo models, mice were orally administered with PQ at the doses of 100, 150, 200, 250, 300 and 350 mg/kg. The body weight of the mice decreased rapidly (Figure S1A) and fatality took place when the mice were orally administered with PQ at the dose of 150 mg/kg or above (Figure S1B). When the given dose was increased to 250, 300 or 350 mg/kg, the mice lost weight much more rapidly, and all the animals in these groups died within 3 to 4 days post administration (Figure S1B). Unexpectedly, the body weight of the surviving mice in low-dose groups (e.g. 100-200 mg/kg dosage) began to increase after day 5 and the weight of the surviving animals at 21 days after PQ ingestion exhibited negligible differences with those of the control group, indicating that the mice exhibited some level of self-healing ability if they survived from PQ poisoning. Based on these results, we chose 300 mg/kg, a supralethal dose, as the administered dosage of PQ ingestion to evaluate the oral antidotal efficacy of CB[7].
Decreased intestinal accumulation of PQ and intestine injury
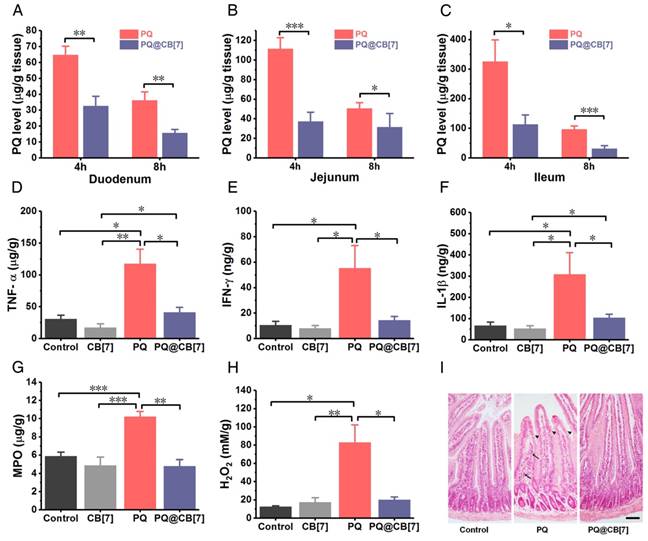
Once taken orally, PQ is known to induce extensive corrosive injuries to the intestine, which, in turn, increases further absorption of PQ [5]. To test whether CB[7] may inhibit PQ accumulation in the intestine and reduce its injury, we firstly used Caco-2 cells as a cellular model of the intestine to determine the influence of CB[7] on the cellular uptake of PQ. Cells were incubated with PQ or PQ@CB[7] for 0.5, 1, 2, 4 and 8 h, and the intracellular content of PQ was significantly lower for the PQ@CB[7] group compared with the PQ group (Figure S2), implying that the cellular uptake of PQ was inhibited with the addition of CB[7]. To determine whether CB[7] could decrease the intestinal accumulation of PQ and the intestinal injury induced by PQ ingestion, mice were orally administered with PQ or co-administered with PQ and CB[7] suspension (PQ@CB[7], at a molar ratio of PQ/CB[7] = 1:2, CB[7] = 2.71 g/kg) at the PQ dose of 300 mg/kg, a supralethal dose. Intestinal bleeding was observed at 8 h post administration for mice that received PQ and the symptoms were completely alleviated with the co-administration of CB[7]. Subsequently, the accumulation of PQ in the duodenum, jejunum and ileum was approximately 60, 110 and 325 μg/g, respectively, at 4 h post PQ ingestion, whereas the values for the PQ@CB[7]-treated group were approximately 30, 40 and 100 μg/g, respectively (Figure 4A-C), suggesting that PQ accumulation decreased significantly in various intestinal tissues when CB[7] was present. A similar trend was also observed at 8 h post-ingestion. It should be noted that CB[7] does not influence the quantitation of PQ via HPLC characterization (Figure S3) and the amount of free PQ or bound PQ could not be distinguished from one another in the tissues for the PQ@CB[7]-treated group. Furthermore, the concentrations of the typical inflammatory cytokines, including TNF-α, IFN-γand IL-1β, were also reduced in the jejunum in the mice treated with PQ in the presence of CB[7] (Figure 4D-F). Additionally, PQ induced high levels of MPO as well as ROS, including H2O2 and other superoxide substances in the jejunum (Figure 4G-H and Figure S4). However, when PQ was ingested in the presence of CB[7], these values were dramatically lower. Similar results were also observed in the ileum (Figure S5A-B). Moreover, epithelial erosion and inflammatory cells infiltration in the lamina propria were shown in the H&E-stained sections of the duodenum for the group of mice that ingested PQ, whereas in both the control group of mice and that which had ingested PQ@CB[7], no discernible injuries were observed (Figure 4I). More significantly, while PQ was readily detected in the intestinal tissues of the mice that ingested PQ, no increase of the pro-inflammatory cytokines and ROS levels was observed in the intestinal tissues of the mice that ingested PQ@CB[7], which was comparable with the control group with no detectable injuries observed. This might be attributed to significantly reduced intestinal absorption of PQ upon its encapsulation by CB[7], and the bound form may not induce ROS generation, inflammation and erosion in the intestine. Taken together, all of these results suggested that CB[7] may significantly reduce intestinal injuries by binding with PQ and inhibiting its accumulation in the intestine.
CB[7] reduces the accumulation of PQ in intestine and protects the intestine from injury. (A-C) The accumulation of PQ in the duodenum, jejunum and ileum 4 and 8 h after PQ or PQ@CB[7] (molar ratio of PQ/CB[7] = 1:2, CB[7] = 2.71 g/kg) ingestion at the PQ dose of 300 mg/kg. Data are presented as means ± SEM, n = 6, and *P < 0.05, **P < 0.01. (D-H) Typical pro-inflammatory cytokines (D-F) and ROS related levels (G and H) in jejunum determined at 8 h after mice had ingested saline (control group), CB[7] (2.71 g/kg), PQ or PQ@CB[7] (molar ratio of PQ/CB[7] = 1:2, CB[7] = 2.71 g/kg) at the PQ dose of 300 mg/kg. Data are presented as means ± SEM, n = 6, and *P < 0.05, **P < 0.01, ***P < 0.001. (I) H&E-stained sections of duodenum collected from the mice sacrificed at 8 h after oral administration with PQ or PQ@CB[7] (molar ratio of PQ/CB[7] = 1:2, CB[7] = 2.71 g/kg) at the PQ dose of 300 mg/kg. The arrows show inflammatory cells infiltration and the triangular arrow showed regions exhibiting epithelial erosion. Scale bar = 200 μm.
Reduced absorption of PQ and PQ poisoning on major organs in a mouse model
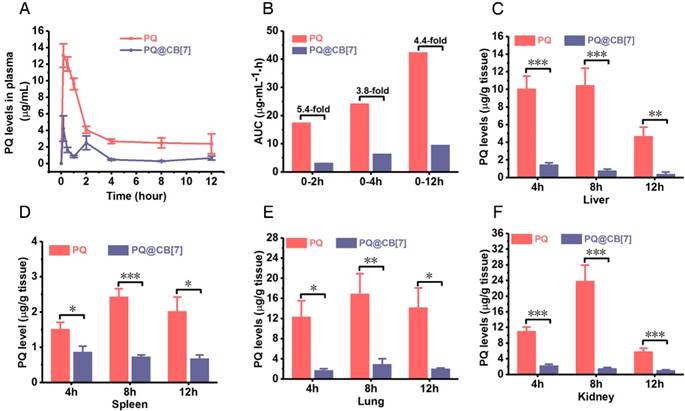
In addition to the intestine, we further investigated the influence of CB[7] on the pharmacokinetics, absorption and tissue distribution of PQ as well as the toxicity of PQ on major organs among the mice that had been orally administered PQ in the absence and presence of CB[7]. Mice were orally administered with PQ solution and PQ@CB[7] suspension (molar ratio PQ/CB[7] = 1:2, CB[7] = 2.71 g/kg), respectively, at a PQ dosage of 300 mg/kg, and Figure 5A shows the pharmacokinetic profiles of PQ and PQ@CB[7]. In the mice that were orally administered with PQ, the concentration of PQ in plasma reached the maximum at the first pre-determined time point (10 min) and kept at relatively high levels within 2 h after PQ ingestion, indicating that PQ was rapidly absorbed into the systemic circulation. When fed with PQ in the presence of CB[7], the plasma levels of PQ in the mice were significantly reduced in comparison to those of the free PQ group at 10 min, 0.5, 1, 2, 4, 8 and 12 h post ingestion. Accordingly, the area under the pharmacokinetic curve AUC(0-2h), AUC(0-4h), AUC(0-12h) and AUC(0-∞) of the PQ@CB[7] group was 3.3, 6.5, 9.6 and 10.2 μg•mL-1•h, respectively , which was ~ 5.4-fold, ~3.8-fold, ~ 4.4-fold and 19.4-fold lower than those of the free PQ treated group (17.5, 24.3, 42.5 and 197.9 μg•mL-1•h, respectively, as shown in Figure 5B). The accumulation of PQ in major organs, including the heart, liver, spleen, lungs and kidneys, at 4, 8 and 12 h post ingestion was also significantly lower for the PQ@CB[7] group, when compared with the free-PQ-treated group (Figure 5C-F and Figure S6). The decreased absorption of PQ may be attributed to the complexation of CB[7] that may exhibit a very little absorption via oral administration in rats, similar to CB[8] [44].
CB[7] reduces the absorption and tissue distribution of PQ in a mouse model. (A) The PQ levels in plasma from mice that had ingested PQ or PQ@CB[7] (molar ratio of PQ/CB[7] = 1:2, CB[7] = 2.71 g/kg) at the PQ dose of 300 mg/kg for various times post-administration. Data are presented as means ± SEM, n = 6. (B) AUC results calculated with the data from (A). (C-F) PQ accumulation in the liver, spleen, lungs and kidney from mice that had ingested PQ or PQ@CB[7] (molar ratio of PQ/CB[7]=1:2) at the PQ dose of 300 mg/kg for various times. Data are presented as means ± SEM, n = 6, and *P < 0.05, **P < 0.01, ***P < 0.001.
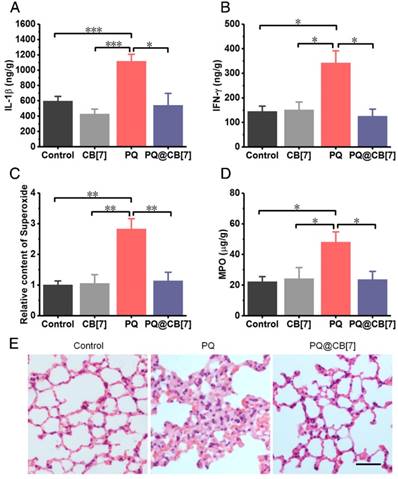
Additionally, we evaluated lung injuries in the mice that were administered with PQ in the absence and presence of CB[7], respectively, as PQ has been well known to preferentially accumulate in this organ[5]. As expected, PQ induced high levels of typical pro-inflammatory cytokines, including IL-1β and IFN-γ, as well as MPO and ROS, including H2O2 and superoxide substances, in the lungs at 8 h post PQ administration at a dosage of 300 mg/kg, while the levels of these species were significantly lower for the PQ@CB[7] (molar ratio PQ/CB[7] =1:2, CB[7] = 2.71 g/kg) treated group (Figure 6A-D and Figure S7). Moreover, H&E-stained sections of the liver, heart, spleen, lungs and kidneys showed obvious damage caused by PQ poisoning at 8 h post ingestion, in contrast to the very minor injuries that were observed for the PQ@CB[7]-treated group (Figure 6E and Figure S8).
CB[7] alleviated the PQ poisoning to lungs in mouse model. (A-D) Typical pro-inflammatory cytokines and ROS-related levels in lungs from mice sacrificed 8 h after ingesting PQ or PQ@CB[7] (molar ratio of PQ/CB[7] = 1:2, CB[7] = 2.71 g/kg) at the PQ dose of 300 mg/kg. Data are presented as means ± SEM, n = 6, and *P < 0.05, **P < 0.01, ***P < 0.001. (E) H&E-stained histopathological sections of lungs from mice sacrificed 8 h after they had ingested saline (control group), PQ or PQ@CB[7] (molar ratio of PQ/CB[7] = 1:2) at the PQ dose of 300 mg/kg. Scale bar = 100 μm.
Survival rate in a supralethal PQ poisoning mouse model
In emergency circumstances, patients who ingest PQ may receive medical managements in minutes or a few hours after PQ ingestion. We subsequently evaluated the most optimal dose and time that CB[7] may be used as an antidote to reverse PQ toxicity. All of the mice orally administered with CB[7] suspension (molar ratio 1:1 to PQ, CB[7] = 1.36 g/kg) at only 10 min after PQ ingestion at a PQ dose of 300 mg/kg died in 10 days (Figure 7A-B ), in contrast to less than 3-day survival of the group of mice without CB[7] treatment post PQ ingestion. Very significantly, a 40% survival rate was obtained when the mice were orally administered CB[7] suspension (molar ratio = 2:1 to PQ, CB[7] = 2.71g/kg) at 10 min after PQ ingestion (at the dose of 300 mg/kg) (Figure 7A-B ). However, the oral administration of CB[7] (molar ratio 3:1 to PQ, CB[7] = 4.07 g/kg) at 10 min post PQ ingestion did not increase the overall survival rate, although it did moderately prolong the survival time in comparison with the PQ-alone-treated group (Figure 7A-B ). This was likely attributed to the physical injury of the stomach caused by over-feeding of the mice during the administration of 3:1 CB[7]-PQ dose, as the mice were orally administered with a volume dose of 20 mL/kg, which significantly exceeded the maximum volume (10 mL/kg) recommended for mice[45]. A lower volume was not possible to administer due to the high-concentration-induced thickness of the sample. Collectively, these results suggested that the dosage of CB[7] at a molar ratio of 2:1 to PQ might be most suitable to detoxify PQ and to improve the survival rate of mice poisoned by PQ. Further studies showed that the survival rate and time were both increased when the mice were orally administered with CB[7] (molar ratio = 2:1 to PQ) at 30 min, 1 h and 2 h after PQ ingestion (Figure 7C). Moreover, H&E sections of the intestinal tissues and major organs (Figure S9-10) of survived mice on Day 21 post poisoning showed no detectable injures, implying that the survived PQ-poisoning mice underwent healing after treating with CB[7].
CB[7] increased the survival rate in a supralethal PQ poisoning mouse model. (A) Scheme depicting the administration methods. (B) Kaplan-Meier survival curves of mice orally administered with CB[7] (molar ratio of PQ/CB[7] = 1:1, 1:2 or 1:3, CB[7] = 1.36, 2.71 or 4.07 g/kg) at 10 min after the mice had ingested PQ at a dose of 300 mg/kg. n = 8, *P < 0.05, ***P < 0.001, comparing CB[7] groups to the PQ group. (C) Kaplan-Meier survival curves of mice orally administered with CB[7] (molar ratio to PQ = 2:1, CB[7] = 2.71 g/kg) at 30 min, 1 h and 2 h after the mice had ingested PQ at a dose of 300 mg/kg. n = 8, *P < 0.05, **P < 0.01, ***P < 0.001, comparing CB[7] groups to the PQ group. (D) Kaplan-Meier survival curves of mice orally administered with AC (1.5 g/kg) at 10 min, 30 min, 1 h and 2 h after the mice had ingested PQ at a dose of 300 mg/kg. n = 8.
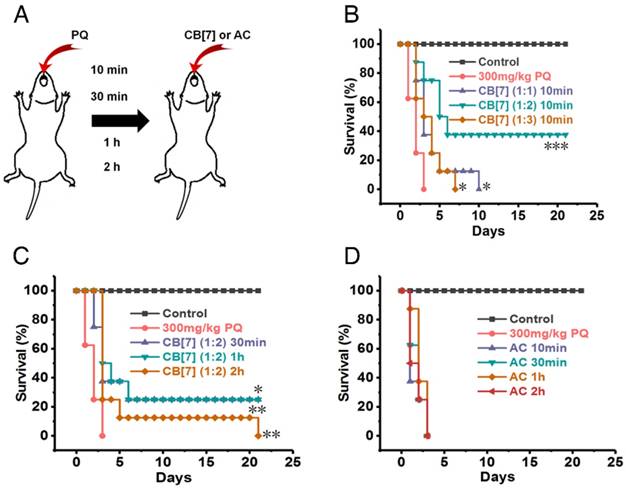
In the current clinical practice, patients suffering from PQ poisoning are usually orally administered with AC as a “gold-standard” (commonly recommended in clinics) absorbent for PQ decontamination, and 50 to 100 g of AC is commonly prescribed for adults who have taken lethal doses of many drugs or poisons[13, 46]. To compare the antidotal effects of CB[7] with AC on PQ poisoning, the antidotal effects of AC were also evaluated against supralethal doses of ingested PQ in this study. The mice receiving AC at 10 min, 30 min, 1 h and 2 h after PQ ingestion all died within 3 days post treatment (Figure 7D), suggesting that AC had poor antidotal effects towards PQ poisoning. Taken together, these results strongly suggested that CB[7] may be developed as an effective and safe oral antidote for the treatment of PQ poisoning, which is likely replacing AC in the future.
Discussion
Currently, PQ is widely used as an economic, extremely effective herbicide, being registered and legally used in over 90 countries. However, accidental or suicidal ingestion of PQ would result in a high mortality rate, exceeding 60%, making PQ the leading pesticide causing fatality in many countries [2, 4]. The current clinical management of PQ poisoning often relies on gastric decontamination aiming to prevent its absorption before receiving other treatments, and AC is commonly recommended in clinical practice for the decontamination of PQ [13]. However, in the present studies AC exhibited much less promising antidotal effects in mouse models with supralethal dose of PQ poisoning, in line with several studies reported previously [17, 20, 47].
Researchers have been actively searching and developing new approaches to treat PQ poisoning in order to save people's lives. PQ-specific antibodies have been designed and developed to reverse the toxicity of PQ post ingestion, but failed to prevent PQ from accumulating in tissues [48-50]. Pillar[6]arene was reported to bind with PQ and detoxify PQ poisoning at cellular levels without any preclinical in vivo study [51]. Similarly, p-Sulfonatocalix[n]arenes have also been demonstrated to bind with PQ and improve the survival rate of PQ-poisoned mice [52, 53]. However, the detailed antidotal mechanisms at the molecular and pharmacokinetic levels were not investigated in that study. In addition, researchers also revealed the antidotal effects of lysine acetylsalicylate, sodium salicylate and quinone oxidoreductase 2 inhibitor on PQ-poisoned rats [54-56]. However, PQ was administered intraperitoneally in these studies, which is not a typical ingestion route for PQ poisoning, as oral administration is the main route of ingestion in nearly all incidents of PQ poisoning.
What makes PQ ingestion so toxic? It is now well appreciated that upon entry into cells, PQ undergoes redox cycling that induces a series of biochemical events, including the generation of free radicals and oxidative stress, oxidation of NADPH, mitochondrial toxicity, oxidation of cellular thiol groups and oxidation damage to the lipids, protein and DNA, and eventually PQ would induce apoptosis and cell death [5, 13]. Upon ingestion in vivo, PQ is rapidly absorbed primarily from the intestines. The accumulation of PQ in the alveolar epithelium would induce an acute alveolitis followed by pulmonary oedema, lung fibrosis and lung failure [57, 58]. PQ accumulation in the kidneys would also induce large vacuolation in proximal convoluted tubules and necrosis [59]. Death would take place within several hours or a few days, by multi-organ failure, including pulmonary oedema, cardiac, renal and hepatic failure, once a large amount of PQ is ingested [13]. With CB[7] as a potential oral antidote, much less PQ would be absorbed in the intestine, and the extent of poisoning and damage in all major organs would be alleviated accordingly, as demonstrated by our results presented here in mice.
In the present studies, we demonstrated that CB[7] inhibited the cellular uptake of PQ, and decreased the PQ-induced ROS generation and apoptosis, resulting in improved cell viability. The relatively high binding affinity of PQ with CB[7] in buffers of pH values varying from 1.2 to 7.4 promoted rapid PQ binding and sequestering in the GI tract by CB[7], which subsequently decreased the accumulation of PQ in the intestine and reduced intestinal injuries accordingly. The pharmacokinetics studies revealed that PQ levels in the blood and its bio-distribution in major organs were all lowered by CB[7] complexation, and PQ-induced multi-organ injuries were alleviated accordingly. Finally, the survival rate of PQ-poisoned mice was significantly increased when they were treated with CB[7] as an antidote, in contrast to the ineffective treatment observed with AC decontamination.
In spite of the extremely promising results obtained with CB[7] as an oral antidote against PQ poisoning on a mouse model, there are still some limitations inherent to our studies. We demonstrated that CB[7] could bind with PQ in the GI tract effectively and subsequently reduced its absorption and tissue distribution. However, CB[7] did not reverse the overall mortality rate of the mice when given at 2 h post-PQ ingestion. Thus, the administration of CB[7] at time points beyond 2 h post-PQ ingestion would unlikely offer any benefits on overall survival rate. This is likely attributed to the fact that PQ was rapidly absorbed into the blood circulation within 2 h post-PQ ingestion in mice (Figure 5A). Therefore, to maximize the life-saving possibility in the emergency treatment of PQ ingestion, CB[7] should be made available in ambulances, village clinics and peripheral hospitals in rural towns and cities where people have easy access to PQ. PQ-poisoned patients would be quickly treated orally with CB[7] as a specific PQ antidote in the field, by staff in ambulances, in rural clinics or the emergency room of hospitals to inhibit the intestinal absorption of PQ as early as possible, before they are further treated with other commonly recommended procedures such as hemoperfusion and hemofiltration. In a recent Chinese case series, the median time for the hospital presentation and treatment of the patients was 7 h with an interquartile range of 5 to 10 h [60]. In fact, it may have taken several hours for the patients to be transferred to the study hospital (a tertiary hospital in a provincial capital) from rural clinics or hospitals where the patients were first checked and likely decontaminated by AC, similar to a reported case study in Sri Lanka, in which 90% of PQ poisoned patients were transferred to the study hospitals from the peripheral hospitals where they were checked and cared for initially with AC [61]. Therefore, the use of CB[7] as an oral antidote in rural clinics and hospitals as well as those field ambulances in agricultural regions, as the first treatment option, would increase the likelihood of patients' survival. Regarding the administration dose of CB[7] in humans, for the sake of discussing potential clinical translation, the LD50 of PQ in humans is approximately 35 mg/kg [62]. Therefore, the dose of CB[7] used as an antidote would be approximately 316 mg/kg (molar ratio 2:1 to PQ), which is equivalent to 19 g for a 60-kg adult in principle. In one of our recent studies, a single oral dose of 5 g/kg CB[7] exhibited excellent safety profile in mice, as shown in body weight changes, organ indices, hematological parameters, histological sections analysis of major organs as well as liver and kidney functions after 21 days of the administration [33]. Also, another study published in Chinese demonstrated a good safety profile of CB[7] in mice based on body weight changes at a single oral dose of 15 g/kg [63]. Therefore, a 60-kg adult may likely tolerate approximately 73 g CB[7] via the oral route, calculated with body surface area differences considered between mice and humans [64].
In addition to the investigation of CB[7] as a potential oral antidote, we also conducted preliminary experiments to evaluate whether i.v. injection of CB[7] may detoxify PQ-induced toxicity in mouse models. Immediately after PQ ingestion, the mice were i.v. injected with 150 mg/kg CB[7] (the maximum tolerable dose of CB[7] in mice via i.v. injection [33]), and all the mice died within 4 days. Therefore, i.v. administration of CB[7] at this dose level might not have significant therapeutic effects against PQ poisoning. This may be attributed to the high concentrations of ions and a variety of proteins in the plasma, which may competitively bind with CB[7], resulting in significantly less complexation capacity towards PQ.
Conclusions
In conclusion, the current studies suggested that CB[7] may act as a potential oral antidote against PQ poisoning, being superior to AC in PQ decontamination. In addition to reducing the cellular uptake of PQ, CB[7] may also undergo complexation with PQ intracellularly, inhibit the redox cycling and protect cells against PQ-induced poisoning. Our systemic studies in mice suggested that CB[7] may be used as an oral antidote for PQ poisoning, which could be readily administered in the field, remote clinics, or in emergency rooms without the need for sophisticated equipment or skills, providing a potential life-saving approach to human poisoning by PQ, particularly in rural or remote areas, or in developing nations.
Abbreviations
AC: activated charcoal; AUC: area under the curve; CB[7]: cucurbit[7]uril; CB[n]s: cucurbit[n]urils; DMEM: dulbecco's modified eagle medium; EQ: diethyl paraquat; FBS: fetal bovine serum; GA: genetic algorithm; GI: gastrointestinal; H2O2: hydrogen peroxide; HPLC: high performance liquid chromatography; H&E: hematoxylin and eosin; IFN-γ: interferon-γ; IL-1α: interleukin-1α; ITC: isothermal titration calorimetry; LGA: lamarckian genetic algorithm; MPO: myeloperoxidase; PBS: phosphate-buffered saline; PQ: paraquat; pdb: protein data bank; PQ@CB[7]: PQ and CB[7]; ROS: reactive oxygen species; SEM: standard error of mean; TNF-α : tumor necrosis factor-α.
Acknowledgements
This work was financially supported by the Macau Science and Technology Development Fund (Grant No.: FDCT 030/2015/A1) and the Research Committee at the University of Macau (MYRG2016-00008-ICMS-QRCM, MYRG2017-00010-ICMS, and MYRG2019-00059-ICMS). We are grateful to Jing Jing for her assistance with AutoDock Vina molecular modeling.
Supplementary Material
Supplementary figures.
Competing interests
X.Z., S.L. and R.W. have filed a patent relating to the content of this work. All other authors have no competing interest to declare.
References
1. Mew EJ, Padmanathan P, Konradsen F, Eddleston M, Chang S-S, Phillips MR. et al. The global burden of fatal self-poisoning with pesticides 2006-15: systematic review. J Affect Disord. 2017;219:93-104
2. Dawson AH, Eddleston M, Senarathna L, Mohamed F, Gawarammana I, Bowe SJ. et al. Acute human lethal toxicity of agricultural pesticides: a prospective cohort study. PLoS Med. 2010;7:e1000357
3. Wilks MF, Fernando R, Ariyananda P, Eddleston M, Berry DJ, Tomenson JA. et al. Improvement in survival after paraquat ingestion following introduction of a new formulation in Sri Lanka. PLoS Med. 2008;5:e49
4. Wunnapuk K, Mohammed F, Gawarammana I, Liu X, Verbeeck RK, Buckley NA. et al. Prediction of paraquat exposure and toxicity in clinically ill poisoned patients: a model based approach. Br J Clin Pharmacol. 2014;78:855-66
5. Dinis-Oliveira RJ, Duarte JA, Sanchez-Navarro A, Remiao F, Bastos ML, Carvalho F. Paraquat poisonings: mechanisms of lung toxicity, clinical features, and treatment. Crit Rev Toxicol. 2008;38:13-71
6. Kervegant M, Merigot L, Glaizal M, Schmitt C, Tichadou L, de Haro L. Paraquat poisonings in france during the european ban: Experience of the poison control center in marseille. J Med Toxicol. 2013;9:144-7
7. Bang YJ, Kim J, Lee WJ. Paraquat use among farmers in Korea after the ban. Arch Environ Occup Health. 2016:1-5
8. Okonek S, Hofmann A, Henningsen B. Efficacy of gut lavage, hemodialysis, and hemoperfusion in the therapy of paraquat or diquat intoxication. Arch Toxicol. 1976;36:43-51
9. Levy G. Gastrointestinal clearance of drugs with activated charcoal. N Engl J Med. 1982;307:676-8
10. Idid S, Lee C. Effects of Fuller's Earth and activated charcoal on oral absorption of paraquat in rabbits. Clin Exp Pharmacol Physiol. 1996;23:679-81
11. Suntres ZE. Role of antioxidants in paraquat toxicity. Toxicology. 2002;180:65-77
12. Eddleston M, Wilks M, Buckley N. Prospects for treatment of paraquat-induced lung fibrosis with immunosuppressive drugs and the need for better prediction of outcome: a systematic review. QJM. 2003;96:809-24
13. Gawarammana IB, Buckley NA. Medical management of paraquat ingestion. Br J Clin Pharmacol. 2011;72:745-57
14. Wang HR, Pan J, Shang AD, Lu YQ. Time-dependent haemoperfusion after acute paraquat poisoning. Sci Rep. 2017;7:2239
15. Zhao X, Xiao Y, Zhu J, Xu Z, Liu L, Zhang J. Prognostic comparison of goal-oriented hemoperfusion and routine hemoperfusion combined with continuous venovenous hemofiltration for paraquat poisoning. J Int Med Res. 2018;46:1091-102
16. Koo JR, Kim JC, Yoon JW, Kim GH, Jeon RW, Kim HJ. et al. Failure of continuous venovenous hemofiltration to prevent death in paraquat poisoning. Am J Kidney Dis. 2002;39:55-9
17. Eddleston M, Juszczak E, Buckley NA, Senarathna L, Mohamed F, Dissanayake W. et al. Multiple-dose activated charcoal in acute self-poisoning: a randomised controlled trial. The Lancet. 2008;371:579-87
18. Juurlink DN. Activated charcoal for acute overdose: a reappraisal. Br J Clin Pharmacol. 2016;81:482-7
19. Gawarammana I, Buckley NA, Mohamed F, Naser K, Jeganathan K, Ariyananada PL. et al. High-dose immunosuppression to prevent death after paraquat self-poisoning - a randomised controlled trial. Clin toxicol (phila). 2017:1-7
20. Ardagh M, Flood D, Tait C. Limiting the use of gastrointestinal decontamination does not worsen the outcome from deliberate self-poisoning. N Z Med J. 2001;114:423-5
21. Houze P, Baud F, Mouy R, Bismuth C, Bourdon R, Scherrmann J. Toxicokinetics of paraquat in humans. Hum Exp Toxicol. 1990;9:5-12
22. Kim J, Jung I-S, Kim S-Y, Lee E, Kang J-K, Sakamoto S. et al. New cucurbituril homologues: syntheses, isolation, characterization, and X-ray crystal structures of cucurbit [n] uril (n= 5, 7, and 8). J Am Chem Soc. 2000;122:540-1
23. Lagona J, Mukhopadhyay P, Chakrabarti S, Isaacs L. The cucurbit[n]uril family. Angew Chem Int Ed Engl. 2005;44:4844-70
24. Ma X, Zhao Y. Biomedical Applications of Supramolecular Systems Based on Host-Guest Interactions. Chem Rev. 2015;115:7794-839
25. Assaf KI, Nau WM. Cucurbiturils: from synthesis to high-affinity binding and catalysis. Chem Soc Rev. 2015;44:394-418
26. Barrow SJ, Kasera S, Rowland MJ, del Barrio J, Scherman OA. Cucurbituril-Based Molecular Recognition. Chem Rev. 2015;115:12320-406
27. Li S, Chan JY-W, Li Y, Bardelang D, Zheng J, Yew WW. et al. Complexation of clofazimine by macrocyclic cucurbit [7] uril reduced its cardiotoxicity without affecting the antimycobacterial efficacy. Org Biomol Chem. 2016;14:7563-9
28. Huang Q, Kuok KI, Zhang X, Yue X, Lee SM, Zhang J, Wang R. Inhibition of drug-induced seizure development in both zebrafish and mouse models by a synthetic nanoreceptor. Nanoscale. 2018;10:10333-10336
29. Kim HJ, Jeon WS, Ko YH, Kim K. Inclusion of methylviologen in cucurbit[7]uril. Proc Natl Acad Sci U S A. 2002;99:5007-11
30. Ling Y, Mague JT, Kaifer AE. Inclusion complexation of diquat and paraquat by the hosts cucurbit[7]uril and cucurbit[8]uril. Chemistry. 2007;13:7908-14
31. Uzunova VD, Cullinane C, Brix K, Nau WM, Day AI. Toxicity of cucurbit[7]uril and cucurbit[8]uril: an exploratory in vitro and in vivo study. Org Biomol Chem. 2010;8:2037-42
32. Oun R, Floriano RS, Isaacs L, Rowan EG, Wheate NJ. The ex vivo neurotoxic, myotoxic and cardiotoxic activity of cucurbituril-based macrocyclic drug delivery vehicles. Toxicol Res (Camb). 2014;3:447-55
33. Zhang X, Xu X, Li S, Wang L-H, Zhang J, Wang R. A systematic evaluation of the biocompatibility of cucurbit[7]uril in mice. Sci Rep. 2018;8:8819
34. Day A, Arnold AP, Blanch RJ, Snushall B. Controlling factors in the synthesis of cucurbituril and its homologues. J Org Chem. 2001;66:8094-100
35. Bardelang D, Udachin KA, Leek DM, Margeson JC, Chan G, Ratcliffe CI. et al. Cucurbit[n]urils (n= 5-8): A Comprehensive Solid State Study. Cryst Growth Des. 2011;11:5598-614
36. Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455-61
37. Paixao P, Costa P, Bugalho T, Fidalgo C, Pereira L. Simple method for determination of paraquat in plasma and serum of human patients by high-performance liquid chromatography. J Chromatogr B. 2002;775:109-13
38. Corasaniti MT, Strongoli MC, Nisticò G. Determination of paraquat in rat brain using ion-pair solid-phase extraction and reversed-phase high-performance liquid chromatography with ultraviolet detection. J Chromatogr B. 1990;527:189-95
39. Evans D, Pye G, Bramley R, Clark A, Dyson T, Hardcastle J. Measurement of gastrointestinal pH profiles in normal ambulant human subjects. Gut. 1988;29:1035-41
40. Fallingborg J, Christensen LA, Ingeman-Nielsen M, Jacobsen BA, Abildgaard K, Rasmussen HH. et al. Measurement of gastrointestinal pH and regional transit times in normal children. J Pediatr Gastroenterol Nutr. 1990;11:211-4
41. Press A, Hauptmann I, Hauptmann L, Fuchs B, Fuchs M, Ewe K. et al. Gastrointestinal pH profiles in patients with inflammatory bowel disease. Aliment Pharmacol Ther. 1998;12:673-8
42. Podder B, Kim Y-S, Zerin T, Song H-Y. Antioxidant effect of silymarin on paraquat-induced human lung adenocarcinoma A549 cell line. Food Chem Toxicol. 2012;50:3206-14
43. Zerin T, Kim Y-S, Hong S-Y, Song H-Y. Protective effect of methylprednisolone on paraquat-induced A549 cell cytotoxicity via induction of efflux transporter, P-glycoprotein expression. Toxicol Lett. 2012;208:101-7
44. Li F, Gorle AK, Ranson M, Vine KL, Kinobe R, Feterl M. et al. Probing the pharmacokinetics of cucurbit[7, 8 and 10]uril: and a dinuclear ruthenium antimicrobial complex encapsulated in cucurbit[10]uril. Org Biomol Chem. 2017;15:4172-9
45. Diehl KH, Hull R, Morton D, Pfister R, Rabemampianina Y, Smith D. et al. A good practice guide to the administration of substances and removal of blood, including routes and volumes. J Appl Toxicol. 2001;21:15-23
46. Neuvonen PJ. Clinical pharmacokinetics of oral activated charcoal in acute intoxications. Clin Pharmacokinet. 1982;7:465-89
47. Li Y, Tse ML, Gawarammana I, Buckley N, Eddleston M. Systematic review of controlled clinical trials of gastric lavage in acute organophosphorus pesticide poisoning. Clin Toxicol (Phila). 2009;47:179-92
48. Cadot R, Descotes J, Grenot C, Cuilleron C, Evreux J. Increased plasma paraquat levels in intoxicated mice following antiparaquat F (ab') 2 treatment. J Immunopharmacol. 1985;7:467-77
49. Nagao M, Takatori T, Wu B, Terazawa K, Gotouda H, Akabane H. Immunotherapy for the treatment of acute paraquat poisoning. Hum Toxicol. 1989;8:121-3
50. Chen N, Bowles MR, Pond SM. Prevention of paraquat toxicity in suspensions of alveolar type II cells by paraquat-specific antibodies. Hum Exp Toxicol. 1994;13:551-7
51. Yu G, Zhou X, Zhang Z, Han C, Mao Z, Gao C. et al. Pillar[6]arene/paraquat molecular recognition in water: high binding strength, pH-responsiveness, and application in controllable self-assembly, controlled release, and treatment of paraquat poisoning. J Am Chem Soc. 2012;134:19489-97
52. Wang K, Guo DS, Zhang HQ, Li D, Zheng XL, Liu Y. Highly effective binding of viologens by p-sulfonatocalixarenes for the treatment of viologen poisoning. J Med Chem. 2009;52:6402-12
53. Wang GF, Ren XL, Zhao M, Qiu XL, Qi AD. Paraquat detoxification with p-sulfonatocalix-[4]arene by a pharmacokinetic study. J Agric Food Chem. 2011;59:4294-9
54. Dinis-Oliveira RJ, Sousa C, Remiao F, Duarte JA, Navarro AS, Bastos ML. et al. Full survival of paraquat-exposed rats after treatment with sodium salicylate. Free Radic Biol Med. 2007;42:1017-28
55. Dinis-Oliveira RJ, Pontes H, Bastos ML, Remiao F, Duarte JA, Carvalho F. An effective antidote for paraquat poisonings: the treatment with lysine acetylsalicylate. Toxicology. 2009;255:187-93
56. Janda E, Parafati M, Aprigliano S, Carresi C, Visalli V, Sacco I. et al. The antidote effect of quinone oxidoreductase 2 inhibitor against paraquat-induced toxicity in vitro and in vivo. Br J Pharmacol. 2013;168:46-59
57. Smith L. Mechanism of paraquat toxicity in lung and its relevance to treatment. Hum Toxicol. 1987;6:31-6
58. Rockey DC, Bell PD, Hill JA. Fibrosis—a common pathway to organ injury and failure. N Engl J Med. 2015;372:1138-49
59. Fowler BA, Brooks RE. Effects of the herbicide paraquat on the ultrastructure of mouse kidney. Am J Path. 1971;63:505
60. Li Y, Wang M, Gao Y, Yang W, Xu Q, Eddleston M. et al. Abnormal pancreatic enzymes and their prognostic role after acute paraquat poisoning. Sci Rep. 2015;5:17299
61. Senarathna D. How the level of resources and hospital staff attitude in primary care hospitals in rural Sri Lanka affect poisoning patient outcome. Minor thesis): University of Newcastle, Australia. 2006
62. Saravu K, Sekhar S, Pai A, Barkur AS, Rajesh V, Earla JR. Paraquat-A deadly poison: Report of a case and review. Indian journal of critical care medicine: peer-reviewed, official publication of Indian Society of Critical Care Medicine. 2013;17:182
63. X. Fu, X. Shen, Y. Huang, Z. Tao, S. Xue and Q. Zhu, An experimental study of acute toxicity of CB[7] on mice. Journal of Guizhou University (Natural Science). 2007;24(6):650-2 (In Chinese)
64. Nair AB, Jacob S. A simple practice guide for dose conversion between animals and human. J Basic Clinical Pharm. 2016;7:27
Author contact
![]() Corresponding authors: Ruibing Wang or Jianxiang Zhang. Email: rwangmo (Ruibing Wang); jxzhangedu.cn (Jianxiang Zhang).
Corresponding authors: Ruibing Wang or Jianxiang Zhang. Email: rwangmo (Ruibing Wang); jxzhangedu.cn (Jianxiang Zhang).