Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
PGC-1 signaling network
The role of PGC-1 in myocardial...
The role of PGC-1 in...
The role of PGC-1 in heart...
Research progress regarding...
Conclusions
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(2):466-476. doi:10.7150/thno.29130 This issue Cite
Review
Blossoming 20: The Energetic Regulator's Birthday Unveils its Versatility in Cardiac Diseases
Jianjun Lv1,2*, Chao Deng3*, Shuai Jiang4*, Ting Ji1, Zhi Yang2, Zheng Wang5,6, Yang Yang1 ![]()
1. Key Laboratory of Resource Biology and Biotechnology in Western China, Ministry of Education. Faculty of Life Sciences, Northwest University, 229 Taibai North Road, Xi'an 710069, China
2. School of Basic Medicine, The Fourth Military Medical University, 169 Changle West Road, Xi'an 710032, China
3. Department of Cardiovascular Surgery, The First Affiliated Hospital of Xi'an Jiaotong University, 277 Yanta West Road, Xi'an 710061, Shaanxi, China
4. Department of Aerospace Medicine, The Fourth Military Medical University, 169 Changle West Road, Xi'an 710032, China
5. Department of Cadio-Thoracic Surgery, Wuhan General Hospital of The People's Liberation Army, 627 Wuluo Road, Wuhan 430070, China
6. Department of Cardiovascular Surgery, Xijing Hospital, The Fourth Military Medical University, 127 Changle West Road, Xi'an 710032, China
*These authors contributed equally to this work.
Received 2018-8-9; Accepted 2018-10-3; Published 2019-1-1
Abstract

The peroxisome proliferator-activated receptor γ (PPARγ) coactivator-1α (PGC-1α) was first identified in 1998 as a PGC-1 family member that regulates adaptive thermogenesis and mitochondrial function following cold exposure in brown adipose tissue. The PGC-1 family has drawn widespread attention over the past two decades as the energetic regulator. We recently summarized a review regarding PGC-1 signaling pathway and its mechanisms in cardiac metabolism. In this review, we elaborate upon the PGC-1 signaling network and highlight the recent progress of its versatile roles in cardiac diseases, including myocardial hypertrophy, peripartum and diabetic cardiomyopathy, and heart failure. The information reviewed here may be useful in future studies, which may increase the potential of this energetic regulator as a therapeutic target.
Keywords: PGC-1, energetic regulator, cardiac diseases
Introduction
In response to stimuli the heart exerts compensatory effects at first, such as altered heart rates and size of cardiomyocytes. However, the sustaining duration of the stress in turn worsens the imbalance between energy transformation and energy demand to satisfy the contractile function of cardiac muscle, which is characterized by an initial phase of compensation with subsequent progression to decompensation, thus leading to irreversible myocardial damage and energy failure [1-4]. Accumulating evidence suggests that dysfunction of mitochondrial biogenesis, maladaptive fuel shift, and reactive oxygen species (ROS) are three primary contributors to cardiac abnormalities, ultimately resulting in cardiac diseases [5]. The underlying mechanisms appear to involve the transcriptional coactivators termed peroxisome proliferator-activated receptor γ (PPARγ) coactivator-1s (PGC-1s).
PGC-1s are a family of transcriptional coactivators that consist of PGC-1α, PGC-1β, and PGC-1-related coactivator (PRC). The PGC-1 family can be regulated at both the transcriptional and post-translational levels, and they activate a variety of coactivated genes, such as those encoding estrogen-related receptors (ERRs), PPARs, and nuclear respiratory factors (NRFs), critical for proper cardiac function. Take one of the PPAR members for example; abnormality of PPARγ in the heart can result in many detrimental effects, such as lipid accumulation, mitochondrial dysfunction, ultimately causing cardiac hypertrophy, heart failure, and premature death [5]. Increasing evidence suggests that long chain fatty acid serves as one of the ligands for the activation of PGC-1 signaling pathway [6]. Moreover, the availability to uptake of fatty acids by the cardiomyocyte mediates this activation and the expression of its target genes for metabolic enzymes [7]. We previously summarized PGC-1 signaling pathway and its mechanisms in cardiac metabolism [5]. However, a comprehensive understanding of the latest literature regarding PGC-1 signaling network and its profound roles in various cardiac diseases has not been specifically depicted, and the present review aims to fill this gap.
Since we previously elaborated the classical PGC-1 signaling pathways, in the current review, we prominently focus on the interaction between PGC-1α and other signaling. Furthermore, we investigate the latest research progress on the versatile effects of the PGC-1 coactivators in various cardiac diseases. Additionally, we address whether this energetic regulator may be a useful target and a potential predictive biomarker for cardiac diseases. Collectively, the information compiled here should serve as a comprehensive repository of the existing knowledge in this area, which should aid in the design for further studies about PGC-1 in cardiac diseases.
PGC-1 signaling network
The PGC-1 program can be activated in variable forms of cardiac stress by the transduction of multiple upstream mediators, such as β-adrenergic receptor (β- AR)/3'-5'-cyclic adenosine monophosphate (cAMP) and AMP-activated protein kinase (AMPK) [5, 8-10]. In addition, PGC-1α can be induced by some external stimuli that increase energy demand and ATP production, such as cold exposure, which could well be relevant in heart. The underlying mechanism involves the promotion of uncoupling protein 1 (UCP1) by PGC-1α, further regulating adaptive thermogenesis and mitochondrial function in brown adipose tissue [11]. Upon activation, PGC-1s initiate the expression of a number of coactivated genes, such as those encoding ERRs, PPARs, and NRFs, which regulate virtually all aspects of mitochondrial energy metabolism in the heart [5]. Notably, apart from classical PGC-1 signaling pathway, there remains several important interplays with other signaling (Fig. 1).
Interaction between PGC-1 and other signaling. A. PGC-1α and Ca2+. PGC-1α can be activated by two Ca2+-dependent signaling pathways, CAMK-CREB and calcineurin-MEF2, further regulating glucose utilization and mitochondrial respiratory function as well as fatty acid utilization, respectively. The induction of PGC-1α can also promote cytosolic Ca2+ clearance, moderating cardiac Ca2+ cycling and promoting cardiac output in response to physiological stress. B. PGC-1α and KLF4. KLF4 modulates metabolic and mitochondrial pathways through the formation of a KLF4/PGC-1/ERR trimolecular complex. C. PGC-1α and NCoR1. NCoR1 regulates mitochondrial metabolism together with PGC-1α in a Yin-Yang fashion. D. PGC-1α and mTOR. mTOR drives mitochondrial function by inhibiting 4E-BPs and simultaneously orchestrating the PGC-1α program.
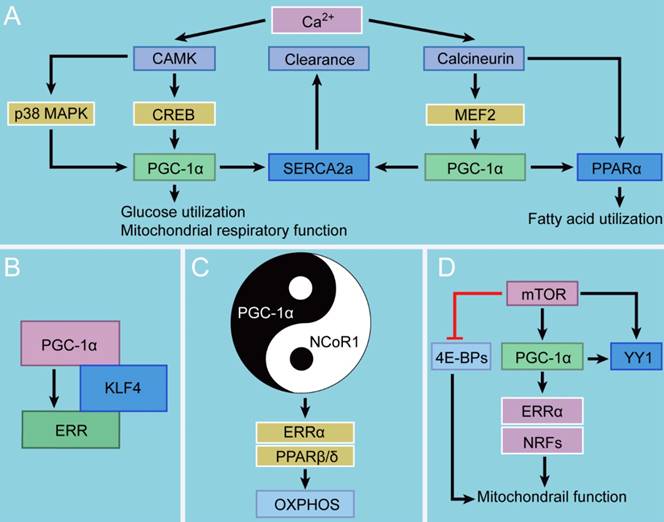
Role of PGC-1α in Ca2+ homeostasis
Cytosolic Ca2+ signals are one of the most vital regulators in energy production and cardiomyocyte contraction. They regulate the activity of the critical enzymes of the tricarboxylic acid (TCA) cycle and initiate the contractile function of cardiac myocytes in a concentration-dependent manner [12, 13]. There are a myriad of kinases and phosphatases regulated by cytosolic Ca2+ signals, which are sensitive to Ca2+ location, size, and frequency [14]. One such Ca2+ sensor, Ca2+/calmodulin-dependent enzyme (CAMK), controls cardiomyocyte growth, differentiation, and development, which are also important factors in the pathophysiology of various cardiac diseases [15, 16]. The other, calcineurin, has recently been demonstrated to have beneficial effects on myocardial hypertrophy and ventricular function [17]. CAMK and calcineurin, two central enzymes in Ca2+-dependent signaling pathways, exhibit distinct features and functionalities in metabolic programs. CAMK activates glucose utilization and mitochondrial respiratory function, whereas calcineurin directs the expression of genes that encode fatty acid utilization enzymes [18].
These two critical enzymes play distinct but overlapping roles in PGC-1α activation: CAMK activates the PGC-1α promotor via binding of the transcription factor cAMP response element-binding protein (CREB), whereas calcineurin does by binding to another transcription factor myocyte enhancer factor 2 (MEF2) [18]. Moreover, CAMK induces p38 mitogen activated protein kinase (MAPK) signaling, additively or synergistically activating PGC-1α gene transcription [19-21]. Additionally, calcineurin can directly activate the PPARα gene promoter, a PGC-1α-coactivated partner that regulates fatty acid utilization and facilitates cardiac energy producing capacity [22]. This could also be one mechanism by which CAMK and calcineurin activate distinct metabolic programs. In other words, this selectivity may occur because PGC-1α acts in concert with other transcription factors instead of stimulating gene transcription alone.
The short-term effects of CAMK and calcineurin signaling promote mitochondrial energy production and cardiac growth during physiological and developmental forms of cardiac hypertrophic growth. However, chronic expression of constitutively active forms of CAMK or calcineurin in cardiac myocytes leads to myocardial hypertrophy, heart failure, and even death [22]. Notably, under physiological condition in cardiac myocytes, PGC-1α stimulates the expression of sarcoplasmic reticulum (SR) Ca2+-ATPase type 2a (SERCA2a), an important protein that mediates Ca2+ reuptake into SR and governs the normal intracellular Ca2+ handling process [23-25]. The molecular mechanism of the PGC-1α-dependent upregulation of SERCA2a may involve the coactivation of two transcription factors, MEF-2 and PPAR/ retinoid X receptor (RXR), the binding sites of which present in conserved regulatory regions in the promotor of the human SERCA2a gene [23, 26]. PGC-1α confers a special effect on the process of cytosolic Ca2+ clearance, which follows the induction of SERCA2a by PGC-1α (90%) but not the activity of Na+/Ca2+ exchange (10%) [23]. Moreover, PGC-1α boosts energy production to accommodate for the demand for the high expression of SERCA2a [27]. Their study strongly indicates that PGC-1α improves the capacity of ventricular systole and diastole by stabilizing Ca2+ handling and accelerating Ca2+ clearance. Together, these data reveal a protective role of PGC-1α in moderating cardiac Ca2+ cycling and promoting cardiac output in response to physiological stress, shedding new light on the role of the Ca2+-PGC-1α handing pathway in the treatment of cardiac diseases.
PGC-1α and Kruppel-like factor 4 (KLF4)
KLFs are a subfamily of the zinc-finger class of transcription factors that play important roles in the regulation of cellular metabolism and muscle function [28]. Among them, KLF4 is critical for the heart's adaptation to stress [29]. Recent studies have shown that KLF4 governs mitochondrial biogenesis, metabolic function, dynamics, and autophagic clearance. The related mechanism may involve the formation of a KLF4/PGC-1/ERR trimolecular complex which KLF4 binds to, cooperates with, and is requisite for optimal function of the PGC-1/ERR transcriptional regulatory module on metabolic and mitochondrial targets [30]. Furthermore, the cooperativity between KLFs and nuclear receptors also depends on upstream regulation of PPARα [31]. However, there are several aspects of KLF4 in cardiac myocytes and mitochondrial biogenesis and function that remain incompletely understood and therefore require further investingation. For instance, whether the KLF4/PGC-1/ERR pathway is operative in these processes to coordinate the entire mitochondrial life cycle is an essential unsolved question.
PGC-1α and NR corepressor 1 (NCoR1)
NCoR1, a transcriptional corepressor, can antagonize the effects of PGC-1α. NCoR1 was originally identified as a mediator of ligand-independent transcriptional repression of thyroid hormone and retinoic acid receptor [32]. It is a ubiquitously expressed corepressor that lacks intrinsic histone deacetylases (HDAC) activity [33]. NCoR1 regulates mitochondrial metabolism together with PGC-1α in a Yin-Yang fashion [34]. Specifically, NCoR1 expression is suppressed when PGC-1α is induced, and the knockout of its expression results in a phenotype similar to that of PGC-1α overexpression in terms of the regulation of mitochondrial oxidative metabolism [35]. In short, there is a high overlap between the effects of NCoR1 deletion and PGC-1α overexpression on oxidative metabolism. More importantly, ERRα and PPARβ/δ have been identified as common targets of NCoR1 and PGC-1α with opposing effects on the transcriptional activity of these PGC-1α-coactivated nuclear receptors [36]. In fact, the inhibitory effect of NCoR1 on oxidative phosphorylation (OXPHOS) gene expression specifically antagonizes PGC-1α-mediated coactivation of ERRα [37]. However, these mechanistic investigations have only been done in skeletal muscle and adipocytes [38], and the detailed mechanisms remain poorly understood. Notably, neither PGC-1α nor NCoR1 interacts directly with DNA; instead, they rely on docking transcription factors and coactivator complexes to regulate mitochondrial function.
PGC-1α and mammalian target of rapamycin (mTOR)
mTOR is an important kinase in the energy and nutrient pathways that regulates cardiac development, growth, and function [39]. Moreover, mTOR is also necessary for the maintenance of cardiac metabolism and mitochondrial oxidative function [40]. Accumulating evidence shows that mTOR controls mitochondrial activity and biogenesis largely through inhibition of the eukaryotic translation initiation factor 4E (eIF4E)-binding proteins (4E-BPs) [41]. Cardiac-specific knockout of mTOR results in heart failure and the mice exhibit uncontrolled apoptosis, excess autophagy, and altered mitochondrial structure [42]. mTOR can control ketogenesis in response to fasting in the mice models. Inhibition of mTOR contributes to a fasting-resistant increase in liver size, and to a remarkable defect in ketone body production on fasting. This is required for the fasting-induced activation of PPARα by controlling the subcellular localization of NCoR1 [43]. Furthermore, the mTOR inhibitor rapamycin decreases the expression of PGC-1α and its coactivated partners ERRα and NRFs, leading to a decrease in mitochondrial gene expression and oxygen consumption [44]. Treatment with rapamycin in humans having organ transplants and in rodents is linked to elevated levels of blood triacylglycerols and cholesterol, which might contribute to insulin resistance [45] The rapamycin-dependent downregulation of those genes could involve the regulation of the transcription factor yin yang 1 (YY1), a common downstream target of mTOR and PGC-1α. Thus, it is conceivable that on top of inhibiting 4E-BPs, mTOR drives mitochondrial function by simultaneously orchestrating the PGC-1α program that modulates expression of mitochondria-related genes. This could also open new possibilities for potential pharmacological interventions to increase mitochondrial activity in cardiometabolic diseases.
The role of PGC-1 in myocardial hypertrophy
PGC-1α has been confirmed to be a divergent regulator in physiological and pathological forms of myocardial hypertrophy. Physiological myocardial hypertrophy under conditions of increased energy demand, such as during postnatal development and endurance exercise training, is associated with increased PGC-1α expression, which results in enhanced mitochondrial biogenesis, oxygen consumption, ATP production, and cardiac contraction [1, 2, 46-48]. Conversely, during pathological myocardial hypertrophy [3], as in hypertension, aortic stenosis, and post-infarction heart failure, PGC-1α expression is repressed, and many energetic abnormalities occur in cardiac myocytes, such as decreases in fatty acid β-oxidation (FAO) and the mitochondrial oxidative capacity. This is further supported by the downregulation of PGC-1α and its downstream targets in response to pressure overload [2, 49, 50]. Notably, the suppression of PGC-1α signaling pathway occurs during the very early stage of pathologically hypertrophic response, indicating that it is a primary event rather than an indirect consequence of pathological hypertrophy. Interestingly, Hu and colleagues [51] have reported that the PGC-1α mRNA level is increased following pressure overload-induced myocardial hypertrophy in rodents, in contrast with reports indicating that myocardial hypertrophy causes decreased PGC-1α mRNA expression. This discrepancy between studies may result from differences in the severity of left ventricular dysfunction and the duration of pressure overload (Fig. 2).
The roles of PGC-1 in myocardial hypertrophy and heart failure. There is an initial increase in the expression and activity of PGC-1α signaling as an adaptive reaction, contributing to a fuel shift to generate more ATP and eventually exerting a cardioprotective effect. However, continuous metabolic stress impairs caridiac function, accompanied with decreased PGC-1α levels. Moreover, suppression of PGC-1α inhibits MnSOD generation, overwhelming the ROS detoxifying system yet again and ultimately resulting in progression to heart failure.
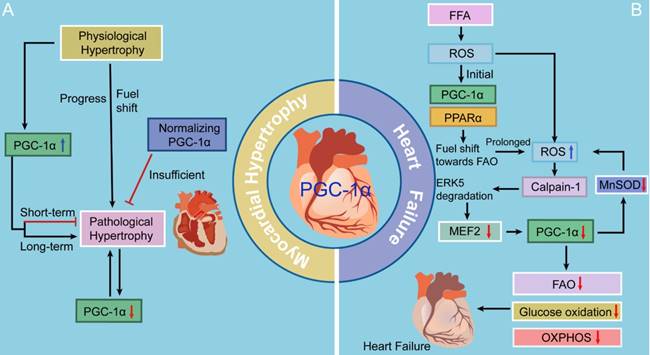
Increasing evidence indicates that deactivation of the PGC-1-PPAR axis is involved in the downregulation of FAO genes and contribution to a fuel shift from FAO toward increased glucose utilization in the pathologically hypertrophied heart [3]. Moreover, chronic progressive deactivation of the PGC-1-ERR signaling pathway reduces ATP production and may ultimately exacerbate myocardial hypertrophy, resulting in heart failure [52]. Furthermore, suppression of the upregulation of PGC-1α signaling pathway can markedly decrease the levels of mitochondrial transcription factor A (TFAM) and mitochondrial DNA (mtDNA), thereby attenuating the increases in mitochondrial biogenesis in the setting of left ventricular hypertrophy induced by pressure overload [53]. Therefore, inhibition of the PGC-1 signaling pathway results in decompensated pressure overload-induced myocardial hypertrophy.
Recent studies have demonstrated the beneficial effects of preventing the development of myocardial hypertrophy by promoting the PGC-1 signaling pathway. Planavila and colleagues [54] have found that silent information regulator 1 (SIRT1), a novel cardioprotective and longevity factor, can protect the heart from hypertrophy, metabolic dysregulation, and inflammation. SIRT1 is induced by an increase in the nicotinamide adenine dinucleotide (NAD+) level in the presence of reduced cellular energy stores. SIRT1 activation deacetylates and activates PGC-1α, subsequently acting in association with PPARα. Co-immunoprecipitation studies have revealed that the SIRT1-PGC-1α-PPARα complex binds to and deacetylates the p65 subunit of nuclear factor-κB (NF-κB) in neonatal cardiac myocytes, which promotes the transcription of FAO genes, inhibits NF-κB pro-inflammatory pathways, and thereby prevents hypertrophy [55]. Furthermore, Luan and colleagues [56] have studied an experimental model of isoproterenol (Iso)-induced myocardial hypertrophy and have reported that administration of astragalus polysaccharide (APS), a Chinese medicine extracted from Astragalus membranaceus, regulates energy biosynthesis and further attenuates myocardial hypertrophy [37]. This cardioprotective role may involve the upregulation of PGC-1α expression by tumor necrosis factor-α (TNF-α) inhibition, however, the precise regulatory mechanisms need to be further clarified.
On the other hand, several lines of gain-of-function studies indicate that PGC-1α may be bad for cardiac responses to stressors and that the complex effects may vary significantly between neonatal and adult hearts. Russell and colleagues [57] have shown that cardiac-specific overexpression of PGC-1α markedly induces mitochondrial proliferation in neonatal mice. However, in long-term studies, chronic induction of PGC-1α expression in adult cardiac myocytes drives a modest increase in mitochondrial number, aberrant mitochondrial structures, and the development of a reversible dilated cardiomyopathy, which can be fully reversed when PGC-1α expression is knocked out. There are three possible mechanisms for the pathologic phenotypes associated with PGC-1α overexpression: 1) certain components of the mitochondrial biogenesis program may be inactive in adult cardiomyocytes, which contributes to a state of energy depletion; 2) increased mitochondrial flux could augment ROS production, further causing cardiomyocyte injury; and 3) a metabolic program could alter expression of structural and sarcomeric proteins in the heart. Considering the reversible nature of the cardiomyopathy, the current scientific view favors the first proposed mechanism. Altogether, these results strongly suggest that upregulation of PGC-1α has a diverse effect on the heart in developmental stage-dependent manner, which is beneficial during the neonatal period.
Notably, a recent study has revealed that following transverse aortic constriction (TAC)-induced myocardial hypertrophy, sustaining a physiological level of PGC-1α expression does not prevent mitochondrial and cardiac contractile dysfunction, but it can preserve myocardial vascularity [58]. This result suggests that maintaining PGC-1α expression in the physiological range in models of pathological myocardial hypertrophy could not be sufficient to increase mitochondrial biogenesis or counteract the impaired contractile or mitochondrial function in the failing heart. Taken together, facilitating PGC-1α signaling in a short term may play a cardioprotective role against pathological myocardial hypertrophy.
As for the other two family members, PGC-1β expression is repressed in rodent models of pressure overload-induced myocardial hypertrophy, whereas PRC expression is reportedly unchanged [59, 60]. PGC-1β deficiency accelerates the transition to heart failure in pressure overload hypertrophy [59]. Moreover, both in vivo and in vitro studies have indicated that melatonin confers a protective role against the pathological cardiac hypertrophy induced by TAC via activation of PGC-1β [61]. Therefore, the therapeutic potency of melatonin as a pharmacological primer for the PGC-1β pathway to prevent cardiac hypertrophy deserves intensive research in the future.
The role of PGC-1 in cardiomyopathy
PGC-1 in peripartum cardiomyopathy (PPCM)
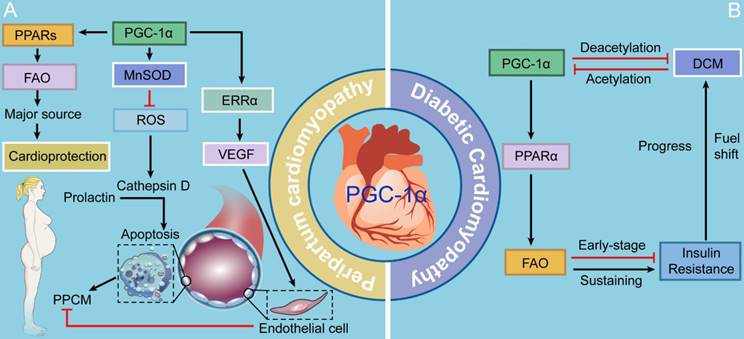
PPCM is a potentially life-threatening pregnancy-associated disease that typically occurs in the peripartum period. Accumulating evidence strongly proposes that PPCM is a vascular disease triggered by the disturbed hormonal milieu of the peripartum, which is characterized by left ventricular dysfunction and heart failure [62]. ROS is one of the chief culprits of the vasculotoxicity in the course of PPCM. Increased ROS production contributes to the secretion of cathepsin D, subsequently cleaving prolactin, a hormone specific to late pregnancy, into a 16-kDa fragment that facilitates apoptosis in endothelial cells [62]. Patten and colleagues [63] have found that PGC-1α is an energetic inhibitor of vasculotoxicity, as evidenced by profound PPCM caused by cardiac-specific PGC-1α deletion. PGC-1α drives the expression of a ROS scavenger, manganese-dependent superoxide dismutase (MnSOD) (also known as SOD2), thus inhibiting superoxides generated by beating cardiomyocytes with robust mitochondrial activity [5, 62, 64, 65]. Additionally, PGC-1α triggers the expression of the vascular endothelial growth factor (VEGF), the most widely studied angiogenic factor, via coactivation of ERRα [66, 67]. Furthermore, PGC-1α potently activates its coactivated partners, PPARs, which play significant roles in the regulation of FAO in the heart [5]. Since there is an increasing fuel shift towards high reliance on FAO in the gestational heart, aberrant FAO is believed to contribute to PPCM [5, 68]. Thus, PCG-1α provides cardioprotection against PPCM through three mechanisms: 1) activating anti-oxidative enzyme MnSOD that further inhibits apoptosis of vascular endothelial cells; 2) triggering the provascular VEGF-mediated signaling; and 3) meeting the need of a fuel shift towards FAO driven by PPARs (Fig. 3).
The roles of PGC-1 in PPCM and DCM. PCG-1α provides cardioprotection against PPCM through three mechanisms: 1) activating anti-oxidative enzyme MnSOD that further inhibits apoptosis of vascular endothelial cells; 2) triggering the provascular VEGF-mediated signaling; and 3) meeting the need of a fuel shift towards FAO driven by PPARs. In the condition of DCM, short- and long-term increases in PGC-1α pathway inhibit and promote insulin resistance, respectively.
PGC-1 in diabetic cardiomyopathy (DCM)
The heart is considered as a "metabolic omnivore", making use of various fuel to produce ATP. There is a significant shift in fuel utilization from glucose and lactate metabolism toward high dependence on FAO in postnatal period [69]. Accumulating evidence shows that PGC-1 primarily interacts with PPARα, further mediating the shift of substrate utilization [5]. DCM develops in the setting of chronically high FAO, impaired glucose uptake and oxidation, and myocardial insulin resistance. PGC-1s and PPARs are critical for the increases in FAO and lipid uptake in the diabetic heart, as evidenced by metabolic dysregulation of the activities of both PGC-1α and PPARα [70]. Notably, the activities and expression of PGC-1α and PPARα are increased during the early stage of insulin resistance in the heart, which is probably an adaptive response of the heart to the increase in fatty acid transport [71]. However, as the disease progresses, sustaining activation of the PGC-1α-PPARα signaling pathway excessively promotes the expression of genes encoding enzymes involved in FAO, thereby setting the stage for the development of lipotoxicity and driving DCM[72, 73]. Insulin resistance and DCM in turn diminish PGC-1α protein levels in the later stage [74]. Several lines of evidence show that insulin resistance gives a boost to PGC-1α acetylation, which represses PGC-1α transcriptional activity [5, 74]. Conversely, promotion of PGC-1α deacetylation can play a protective role against insulin resistance-induced cardiac contractile dysfunction [74]. Although the fuel shift may initially play an adaptive role, its effects of fatty acid utilization and the loss of substrate utilization flexibility may be maladaptive and ultimately contribute to the etiology of the disease. Therefore, additional studies are necessary to predict the effects of regulating cardiac metabolism via mediating PGC-1s and PPARs on the prevention and treatment of DCM (Fig. 3).
The role of PGC-1 in heart failure
Cardiac dysfunction is caused by various diseases, such as hypertension, ischemic heart disease, and myocarditis, which describes a functional deficit of systolic heart failure [37]. The development and progression of cardiac dysfunction and heart failure are multifactorial and can be influenced by genetic mutations. Mutations in nuclear-encoded genes involved in the oxidative respiratory chain may lead to cardiac dysfunction. In addition, targeted mutations that affect myocardial processes, including FAO, OXPHOS, mtDNA proofreading, and protection from mitochondrial ROS, can also lead to profound cardiac dysfunction [75]. Mounting evidence indicates that both the level and transfer capacity of ATP are markedly reduced in failing hearts [76]. Moreover, mitochondrial dysfunction has been demonstrated in human cardiomyopathy and animal heart failure [77, 78]. These alterations may largely result from suppressed replication of the mitochondrial genome and the decreased expression of numerous nuclear genes encoding mitochondrial proteins [79, 80]. Thus, the coordinated downregulation of mitochondrial pathways may occur in failing hearts.
Accumulating studies have suggested that the PGC-1α level is significantly decreased in various models of heart failure [81, 82]. Additionally, activation of PGC-1β gene has been shown to also be reduced in response to chronic pressure overload, but is also dependent on, and responsive to, the chemical form of exogenous and dietary long chain fatty acids [6]. Thus, can the decreased activity or expression of PGC-1 coactivators explain the above observations? Although it remains unclear whether the extent of changes in PGC-1 activity and expression is related to the development of heart failure, several studies have shown that PGC-1 knockout mice subjected to TAC exhibit cardiac dysfunction, which finally progresses to heart failure, suggesting that this might be the case [60]. Moreover, inhibition of PGC-1 expression by Cdk9 leads to a reduction in mitochondrial gene expression and eventually to heart failure [83]. Similarly, suppressing the PGC-1-PPAR and PGC-1-ERR signaling pathways exacerbates heart failure [84]. Therefore, these data strongly indicate that an association exists between heart failure and the decreased activity and expression of PGC-1 coactivators.
Several latest studies may fill the knowledge gap of the molecular mechanisms whereby PGC-1 levels fall in heart failure by presenting a previously unappreciated theory in the free fatty acid (FFA)-stressed heart (Fig. 2). There is an initial increase in the activity of PGC-1α-PPARα signaling under ROS as an adaptive reaction, contributing to a fuel shift towards FAO to generate more ATP [5]. Accumulating offensive lipids further intensify ROS production through a mechanism called “ROS-induced ROS release” [85]. If metabolic stress continues, this vicious cycle impairs mitochondria, accompanied with decreased PGC-1α levels, and thereafter, cardiac myocytes perish and heart failure ensues [86]. Moreover, suppression of PGC-1α inhibits MnSOD generation, overwhelming the ROS detoxifying system yet again and ultimately resulting in progression to heart failure [87]. Liu and colleagues [86] have focused on the extracellular signal-regulated protein kinase 5 (ERK5) and claimed that ERK5 is an intermediate signal that connects FFA-stimulated ROS signaling to the regulation of PGC-1α. When faced with a stress challenge, ERK5/MEF2 cascade is a prerequisite for upregulating PGC-1α in the heart. However, on account of unrelenting FFA stress, the breakdown of ERK5 results in PGC-1α downregulation, which confers detrimental effects on mitochondrial function. Therefore, these data strongly indicate that ERK5's positive regulation of PGC-1α plays a protective role in cardiac mitochondrial function and explain how prolonged FFA-caused ERK5 disruption leads to inevitable progression to heart failure.
Most studies have reported that there is a decrease in the expression of PGC-1s and their downstream targets in the pathophysiology of human heart diseases, however some have not. For instance, recent studies have suggested that ERRα is downregulated, whereas PGC-1α expression is increased in failing human heart [81]. The discrepancy in findings might be attributed to the complex post-translational regulation of PGC-1α activity, which may not correspond with expression levels. Furthermore, Karamanlidis and colleagues [80] have collected left ventricular tissue from end-stage heart failure patients and have found that the PGC-1α protein level was remarkably increased in the failing hearts. Among the downstream targets of PGC-1α, the expression of ERRα is markedly decreased, whereas that of PPARα is unchanged, in the failing hearts. D-loop formation in mtDNA is normal, but D-loop extension occurs, which significantly contributes to decreased mtDNA replication and severely impaired mitochondrial biogenesis and function, in human heart failure. In addition to the above reason, other strong possibilities to consider in understanding the differences are the severity of heart failure and lack of age-matched control group used in previous studies. It is also noteworthy that in many studies on human myocardium of patients with systolic heart failure, the activity of the respiratory chain and the number and structure of cardiac mitochondria is normal and not impaired [88, 89]. Altogether, the role of PGC-1s in human heart diseases is still controversial and this crucial question requires further investigation.
Research progress regarding PGC-1 in cardiac diseases
Of the many recent studies that target PGC-1 signaling pathways, some promising findings suggest potential directions for future research and might be useful for therapeutic intervention in cardiac diseases. Neuropilin-1 (NRP-1), a multi-domain receptor with functional roles in cardiovascular, nervous, and immune system, has recently been demonstrated to specifically control PGC-1α and PPARγ expression in cardiomyocytes through crosstalk with the Notch1 and Smad2 signaling pathways [90]. NRP-1 deletion in cardiomyocytes and vascular smooth muscle cells is accompanied by developed cardiomyopathy and deteriorated ischemia-induced heart failure. Profuse expression of PGC-1α is caused by NRP-1 deletion in these cells, eventually leading to dysregulation of cardiac mitochondrial accumulation and induction of cardiac hypertrophy-related markers. In addition to a negative regulator of PGC-1α, NRP-1 serves as a coreceptor for transforming growth factor-β (TGF-β) and activates the TGF-β-Smad2 pathway in fibroblasts and cardiomyocytes, which further downregulates PPARγ expression and its downstream effectors [90, 91]. While PPARγ ligands counteract TGF-β-mediated fibrosis, whether the regulation of TGF-β-Smad2 axis by PGC-1 impairs extracellular matrix composition and determines cardiac fibrosis and dysfunction remains unclear. In brief, negatively regulating excessive expression PGC-1α and PPARγ is significant in maintaining normal cardiovascular health by balancing mitochondrial homeostasis.
Li and colleagues [92] have identified miR-199 as a negative upstream regulator of PGC-1α. The specific disruption of miR-199 in the heart induces a mild increase in PGC-1α levels, consistent with a tempered upregulation of its downstream metabolic genes, contributing to physiological myocardial hypertrophy in response to physiological stimuli, such as exercise. In addition, PGC-1α deacetylation may participate into the enhancement of PGC-1α activity and further exert this cardioprotection [93]. Increasing evidence suggests that the positive effect of exercise training on cardiac diseases involve the induction of PGC-1α expression. The underlying mechanisms may involve 1) the induction of endothelial nitric oxide synthase (eNOS) that regulates mitochondrial biogenesis via NO-PGC-1α-NRF1-TFAM signaling pathway [94]; 2) the suppression of forkhead box O1 (FoxO1) activity by increased PGC-1α expression [95-98]; and 3) improved myocardial SERCA2a performance that normalizes Ca2+ handling and promotes cardiomyocyte contractility [23, 99]. Furthermore, the loss of PGC-1α in non-coronary artery disease (CAD) arterioles generates a CAD phenotype characterized by a switch from NO- to mitochondrial H2O2-mediated dilation, whereas the phenotype returns to NO-mediated dilation existing in healthy human vessels after PGC-1α restoration probably via caveolin-1 [100]. Altogether, these results suggest that moderately increased PGC-1α expression tend to be beneficial for the maintenance of cardiac homeostasis. The future focus will probably be on determining whether a proper increase in PGC-1α expression can improve cardiac function under physiological or pathological conditions.
PGC-1α has similar modular structures and partly overlapping functions. Variants of these proteins, such as PGC-1α-a (also named PGC-1α1), b, and c, NT-PGC-1α-a, b, and c, and PGC-1α2, 3, and 4, can form via alternative splicing [101]. Among these variants, PGC-1α1 has been demonstrated to regulate metabolic pathways in the heart. In addition to its well-known targets, PGC-1α1 can form coactivator complexes with variable binding partners and is likely to exert various cellular functions [94]. Therefore, some of the adverse effects of PGC-1α may be attributed to expression of diverse gene isoforms in the heart. Currently, it remains unclear if cardiac diseases involve specific PGC-1α isoforms, but they may shed light on the interaction between metabolic and contractile remodeling in cardiac diseases.
Conclusions
The PGC-1 family in cardiac metabolism and diseases is a rather large topic that has been studied for exactly two decades, with hundreds of papers published. However, how the PGC-1 signaling pathway interacts with other signaling has not been specifically proffered. Moreover, there is not enough discussion to reconcile the published differences in cardiac diseases. We recently summarized a review regarding PGC-1 signaling pathway and its mechanisms in cardiac metabolism. In this review, we elaborate upon the PGC-1 signaling network and its versatile roles in cardiac diseases, including myocardial hypertrophy, cardiomyopathy, and heart failure.
To be specific, we 1) dissected two Ca2+-dependent signaling pathways that play distinct but overlapping roles in PGC-1α activation (Fig. 1); 2) described short- and long-term effects of Ca2+ signals, cytosolic Ca2+ clearance under physiological condition, and involved Ca2+-PGC-1α handing pathway; 3) probed the KLF4/PGC-1/ERR transcriptional regulatory module, how NCoR1 and PGC-1α interact with each other in a Yin-Yang fashion, and how mTOR drives mitochondrial function cooperating with PGC-1α; 4) introduced PGC-1 changes in the courses of physiological and pathological myocardial hypertrophy and that PGC-1 loss-of-function results in decompensated myocardial hypertrophy (Fig. 2); 5) proposed that PGC-1 gain-of-function can exert either protective or detrimental effect on the heart in a developmental stage-dependent manner and that there may be three possible mechanisms for the pathologic hypertrophy associated with PGC-1α overexpression; 6) showed that maintaining PGC-1α expression in the physiological range might not be sufficient to counteract pathological myocardial hypertrophy; 7) suggested that PCG-1α provides cardioprotection against PPCM through three mechanisms (Fig. 3); 8) illustrated how PGC-1α may participate into the progression of insulin resistance and DCM; 9) discussed existing major controversial results in heart failure; and 10) highlighted the research progress regarding this energetic regulator in various cardiac diseases.
Despite its great strides over recent two decades, many unresolved questions remain. For instance, 1) to what extent is the activity and expression of PGC-1 coactivators altered in human cardiac diseases? 2) How could the PGC-1 signaling be controlled in a manner that exerts powerful and highly regulatory effects without causing concurrent adverse side effects? 3) Do delivery strategies such as the use of an intermittent dosing regimen represent breakthroughs for therapeutically enhancing the activities of PGC-1s without causing apparent toxicity? 4) Does reactiveation of PGC-1s restore mitochondrial biogenesis and improve cardiac function? Such questions must be answered before the pharmacological modulation of this energetic regulator can be considered as a promising and valuable strategy for tackling cardiac diseases. Undoubtedly, with elucidation of the cellular and molecular mechanisms underlying cardiac diseases, we will gain a better understanding of the essential roles of this energetic regulator in human cardiac diseases and further elucidate the significant clinical implications.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81871607, 81700236, 81600306, 81500263), China Postdoctoral Science Foundation (2016T90973 and 2015M572681), and Natural Science Foundation of Shaanxi Province (S2018-JC-YB-1960).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Lehman JJ, Barger PM, Kovacs A, Saffitz JE, Medeiros DM, Kelly DP. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J Clin Invest. 2000;106:847-56
2. Finck BN, Kelly DP. Peroxisome proliferator-activated receptor gamma coactivator-1 (PGC-1) regulatory cascade in cardiac physiology and disease. Circulation. 2007;115:2540-8
3. Abel ED, Doenst T. Mitochondrial adaptations to physiological vs. pathological cardiac hypertrophy. Cardiovasc Res. 2011;90:234-42
4. Li T, Jiang S, Yang Y. Letter by Li et al Regarding Article, "Particulate Matter Exposure and Stress Hormone Levels: A Randomized, Double-Blind, Crossover Trial of Air Purification". Circulation. 2018;137:1207-8
5. Di W, Lv J, Jiang S, Lu C, Yang Z, Ma Z. et al. PGC-1: The Energetic Regulator in Cardiac Metabolism. Curr Issues Mol Biol. 2018;28:29-46
6. Lahey R, Wang X, Carley AN, Lewandowski ED. Dietary fat supply to failing hearts determines dynamic lipid signaling for nuclear receptor activation and oxidation of stored triglyceride. Circulation. 2014;130:1790-9
7. Haemmerle G, Moustafa T, Woelkart G, Buttner S, Schmidt A, van de Weijer T. et al. ATGL-mediated fat catabolism regulates cardiac mitochondrial function via PPAR-alpha and PGC-1. Nat Med. 2011;17:1076-85
8. Jiang S, Li T, Yang Z, Yi W, Di S, Sun Y. et al. AMPK orchestrates an elaborate cascade protecting tissue from fibrosis and aging. Ageing Res Rev. 2017;38:18-27
9. Gu C, Li T, Jiang S, Yang Z, Lv J, Yi W. et al. AMP-activated protein kinase sparks the fire of cardioprotection against myocardial ischemia and cardiac ageing. Ageing Res Rev. 2018;47:168-75
10. Jiang S, Li T, Ji T, Yi W, Yang Z, Wang S. et al. AMPK: Potential Therapeutic Target for Ischemic Stroke. Theranostics. 2018;8:4535-51
11. Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92:829-39
12. Maack C, O'Rourke B. Excitation-contraction coupling and mitochondrial energetics. Basic Res Cardiol. 2007;102:369-92
13. Val-Blasco A, Piedras MJ, Ruiz-Hurtado G, Suarez N, Prieto P, Gonzalez-Ramos S. et al. Role of NOD1 in Heart Failure Progression via Regulation of Ca(2+) Handling. J Am Coll Cardiol. 2017;69:423-33
14. Louch WE, Koivumaki JT, Tavi P. Calcium signalling in developing cardiomyocytes: implications for model systems and disease. J Physiol. 2015;593:1047-63
15. Frey N, McKinsey TA, Olson EN. Decoding calcium signals involved in cardiac growth and function. Nat Med. 2000;6:1221-7
16. Shahin R, Shaheen O, El-Dahiyat F, Habash M, Saffour S. Research advances in kinase enzymes and inhibitors for cardiovascular disease treatment. Future Sci OA. 2017;3:FSO204
17. Padron-Barthe L, Villalba-Orero M, Gomez-Salinero JM, Acin-Perez R, Cogliati S, Lopez-Olaneta M. et al. Activation of Serine One-Carbon Metabolism by Calcineurin Abeta1 Reduces Myocardial Hypertrophy and Improves Ventricular Function. J Am Coll Cardiol. 2018;71:654-67
18. Handschin C, Rhee J, Lin J, Tarr PT, Spiegelman BM. An autoregulatory loop controls peroxisome proliferator-activated receptor gamma coactivator 1alpha expression in muscle. Proc Natl Acad Sci U S A. 2003;100:7111-6
19. Scharf M, Neef S, Freund R, Geers-Knorr C, Franz-Wachtel M, Brandis A. et al. Mitogen-activated protein kinase-activated protein kinases 2 and 3 regulate SERCA2a expression and fiber type composition to modulate skeletal muscle and cardiomyocyte function. Mol Cell Biol. 2013;33:2586-602
20. Wang S, Dougherty EJ, Danner RL. PPARgamma signaling and emerging opportunities for improved therapeutics. Pharmacol Res. 2016;111:76-85
21. Nakamura TY, Nakao S, Wakabayashi S. Neuronal Ca(2+) sensor-1 contributes to stress tolerance in cardiomyocytes via activation of mitochondrial detoxification pathways. J Mol Cell Cardiol. 2016;99:23-34
22. Schaeffer PJ, Wende AR, Magee CJ, Neilson JR, Leone TC, Chen F. et al. Calcineurin and calcium/calmodulin-dependent protein kinase activate distinct metabolic gene regulatory programs in cardiac muscle. J Biol Chem. 2004;279:39593-603
23. Chen M, Wang Y, Qu A. PGC-1 alpha accelerates cytosolic Ca2+ clearance without disturbing Ca2+ homeostasis in cardiac myocytes. Biochem Biophys Res Commun. 2010;396:894-900
24. Zhang Y, Jiao L, Sun LH, Li Y, Gao Y, Xu C. et al. LncRNA ZFAS1 as a SERCA2a Inhibitor to Cause Intracellular Ca(2+) Overload and Contractile Dysfunction in a Mouse Model of Myocardial Infarction. Circ Res. 2018
25. Ma Z, Yang Y, Di S, Feng X, Liu D, Jiang S. et al. Pterostilbene exerts anticancer activity on non-small-cell lung cancer via activating endoplasmic reticulum stress. Sci Rep. 2017;7:8091
26. Zarain-Herzberg A, Alvarez-Fernandez G. Sarco(endo)plasmic reticulum Ca2+-ATPase-2 gene: structure and transcriptional regulation of the human gene. ScientificWorldJournal. 2002;2:1469-83
27. Jiang S, Yang Z, Di S, Hu W, Ma Z, Chen F. et al. Novel role of forkhead box O 4 transcription factor in cancer: Bringing out the good or the bad. Semin Cancer Biol. 2018;50:1-12
28. McConnell BB, Yang VW. Mammalian Kruppel-like factors in health and diseases. Physiol Rev. 2010;90:1337-81
29. Yoshida T, Yamashita M, Horimai C, Hayashi M. Kruppel-like factor 4 protein regulates isoproterenol-induced cardiac hypertrophy by modulating myocardin expression and activity. J Biol Chem. 2014;289:26107-18
30. Liang Z, Wu G, Fan C, Xu J, Jiang S, Yan X. et al. The emerging role of signal transducer and activator of transcription 3 in cerebral ischemic and hemorrhagic stroke. Prog Neurobiol. 2016;137:1-16
31. Liao X, Zhang R, Lu Y, Prosdocimo DA, Sangwung P, Zhang L. et al. Kruppel-like factor 4 is critical for transcriptional control of cardiac mitochondrial homeostasis. J Clin Invest. 2015;125:3461-76
32. Yang Z, Jiang S, Cheng Y, Li T, Hu W, Ma Z. et al. FOXC1 in cancer development and therapy: deciphering its emerging and divergent roles. Ther Adv Med Oncol. 2017;9:797-816
33. Horlein AJ, Naar AM, Heinzel T, Torchia J, Gloss B, Kurokawa R. et al. Ligand-independent repression by the thyroid hormone receptor mediated by a nuclear receptor co-repressor. Nature. 1995;377:397-404
34. Fan W, Evans R. PPARs and ERRs: molecular mediators of mitochondrial metabolism. Curr Opin Cell Biol. 2015;33:49-54
35. Yamamoto H, Williams EG, Mouchiroud L, Canto C, Fan W, Downes M. et al. NCoR1 is a conserved physiological modulator of muscle mass and oxidative function. Cell. 2011;147:827-39
36. Perez-Schindler J, Summermatter S, Salatino S, Zorzato F, Beer M, Balwierz PJ. et al. The corepressor NCoR1 antagonizes PGC-1alpha and estrogen-related receptor alpha in the regulation of skeletal muscle function and oxidative metabolism. Mol Cell Biol. 2012;32:4913-24
37. Yu LM, Di WC, Dong X, Li Z, Zhang Y, Xue XD. et al. Melatonin protects diabetic heart against ischemia-reperfusion injury, role of membrane receptor-dependent cGMP-PKG activation. Biochim Biophys Acta Mol Basis Dis. 2018;1864:563-78
38. Li P, Fan W, Xu J, Lu M, Yamamoto H, Auwerx J. et al. Adipocyte NCoR knockout decreases PPARgamma phosphorylation and enhances PPARgamma activity and insulin sensitivity. Cell. 2011;147:815-26
39. Jhanwar-Uniyal M, Amin AG, Cooper JB, Das K, Schmidt MH, Murali R. Discrete signaling mechanisms of mTORC1 and mTORC2: Connected yet apart in cellular and molecular aspects. Adv Biol Regul. 2017;64:39-48
40. Sciarretta S, Forte M, Frati G, Sadoshima J. New Insights Into the Role of mTOR Signaling in the Cardiovascular System. Circ Res. 2018;122:489-505
41. Morita M, Gravel SP, Chenard V, Sikstrom K, Zheng L, Alain T. et al. mTORC1 controls mitochondrial activity and biogenesis through 4E-BP-dependent translational regulation. Cell Metab. 2013;18:698-711
42. Zhang D, Contu R, Latronico MV, Zhang J, Rizzi R, Catalucci D. et al. MTORC1 regulates cardiac function and myocyte survival through 4E-BP1 inhibition in mice. J Clin Invest. 2010;120:2805-16
43. Sengupta S, Peterson TR, Laplante M, Oh S, Sabatini DM. mTORC1 controls fasting-induced ketogenesis and its modulation by ageing. Nature. 2010;468:1100-4
44. Cunningham JT, Rodgers JT, Arlow DH, Vazquez F, Mootha VK, Puigserver P. mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature. 2007;450:736-40
45. Li T, Jiang S, Yang Y. Database Selection and Heterogeneity-More Details, More Credibility. JAMA Oncol. 2018;4:1295
46. Taegtmeyer H, Sen S, Vela D. Return to the fetal gene program: a suggested metabolic link to gene expression in the heart. Ann N Y Acad Sci. 2010;1188:191-8
47. Tao L, Bei Y, Lin S, Zhang H, Zhou Y, Jiang J. et al. Exercise Training Protects Against Acute Myocardial Infarction via Improving Myocardial Energy Metabolism and Mitochondrial Biogenesis. Cell Physiol Biochem. 2015;37:162-75
48. Riehle C, Abel ED. PGC-1 proteins and heart failure. Trends Cardiovasc Med. 2012;22:98-105
49. Oka S, Zhai P, Yamamoto T, Ikeda Y, Byun J, Hsu CP. et al. Peroxisome Proliferator Activated Receptor-alpha Association With Silent Information Regulator 1 Suppresses Cardiac Fatty Acid Metabolism in the Failing Heart. Circ Heart Fail. 2015;8:1123-32
50. Gomez-Arroyo J, Mizuno S, Szczepanek K, Van Tassell B, Natarajan R, dos Remedios CG. et al. Metabolic gene remodeling and mitochondrial dysfunction in failing right ventricular hypertrophy secondary to pulmonary arterial hypertension. Circ Heart Fail. 2013;6:136-44
51. Hu X, Xu X, Huang Y, Fassett J, Flagg TP, Zhang Y. et al. Disruption of sarcolemmal ATP-sensitive potassium channel activity impairs the cardiac response to systolic overload. Circ Res. 2008;103:1009-17
52. Huss JM, Imahashi K, Dufour CR, Weinheimer CJ, Courtois M, Kovacs A. et al. The nuclear receptor ERRalpha is required for the bioenergetic and functional adaptation to cardiac pressure overload. Cell Metab. 2007;6:25-37
53. Tornatore TF, Dalla Costa AP, Clemente CF, Judice C, Rocco SA, Calegari VC. et al. A role for focal adhesion kinase in cardiac mitochondrial biogenesis induced by mechanical stress. Am J Physiol Heart Circ Physiol. 2011;300:H902-12
54. Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F. et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell. 2006;127:1109-22
55. Planavila A, Iglesias R, Giralt M, Villarroya F. Sirt1 acts in association with PPARalpha to protect the heart from hypertrophy, metabolic dysregulation, and inflammation. Cardiovasc Res. 2011;90:276-84
56. Luan A, Tang F, Yang Y, Lu M, Wang H, Zhang Y. Astragalus polysaccharide attenuates isoproterenol-induced cardiac hypertrophy by regulating TNF-alpha/PGC-1alpha signaling mediated energy biosynthesis. Environ Toxicol Pharmacol. 2015;39:1081-90
57. Russell LK, Mansfield CM, Lehman JJ, Kovacs A, Courtois M, Saffitz JE. et al. Cardiac-specific induction of the transcriptional coactivator peroxisome proliferator-activated receptor gamma coactivator-1alpha promotes mitochondrial biogenesis and reversible cardiomyopathy in a developmental stage-dependent manner. Circ Res. 2004;94:525-33
58. Pereira RO, Wende AR, Crum A, Hunter D, Olsen CD, Rawlings T. et al. Maintaining PGC-1alpha expression following pressure overload-induced cardiac hypertrophy preserves angiogenesis but not contractile or mitochondrial function. FASEB J. 2014;28:3691-702
59. Riehle C, Wende AR, Zaha VG, Pires KM, Wayment B, Olsen C. et al. PGC-1beta deficiency accelerates the transition to heart failure in pressure overload hypertrophy. Circ Res. 2011;109:783-93
60. Arany Z, Novikov M, Chin S, Ma Y, Rosenzweig A, Spiegelman BM. Transverse aortic constriction leads to accelerated heart failure in mice lacking PPAR-gamma coactivator 1alpha. Proc Natl Acad Sci U S A. 2006;103:10086-91
61. Zhai M, Liu Z, Zhang B, Jing L, Li B, Li K. et al. Melatonin protects against the pathological cardiac hypertrophy induced by transverse aortic constriction through activating PGC-1beta: In vivo and in vitro studies. J Pineal Res. 2017:63
62. Arany Z, Elkayam U. Peripartum Cardiomyopathy. Circulation. 2016;133:1397-409
63. Patten IS, Rana S, Shahul S, Rowe GC, Jang C, Liu L. et al. Cardiac angiogenic imbalance leads to peripartum cardiomyopathy. Nature. 2012;485:333-8
64. Garcia-Quintans N, Prieto I, Sanchez-Ramos C, Luque A, Arza E, Olmos Y. et al. Regulation of endothelial dynamics by PGC-1alpha relies on ROS control of VEGF-A signaling. Free Radic Biol Med. 2016;93:41-51
65. Lv J, Jiang S, Yang Z, Hu W, Wang Z, Li T. et al. PGC-1alpha sparks the fire of neuroprotection against neurodegenerative disorders. Ageing Res Rev. 2018;44:8-21
66. Arany Z, Foo SY, Ma Y, Ruas JL, Bommi-Reddy A, Girnun G. et al. HIF-independent regulation of VEGF and angiogenesis by the transcriptional coactivator PGC-1alpha. Nature. 2008;451:1008-12
67. Sawada N, Jiang A, Takizawa F, Safdar A, Manika A, Tesmenitsky Y. et al. Endothelial PGC-1alpha mediates vascular dysfunction in diabetes. Cell Metab. 2014;19:246-58
68. Liu LX, Arany Z. Maternal cardiac metabolism in pregnancy. Cardiovasc Res. 2014;101:545-53
69. Janssen CI, Kiliaan AJ. Long-chain polyunsaturated fatty acids (LCPUFA) from genesis to senescence: the influence of LCPUFA on neural development, aging, and neurodegeneration. Prog Lipid Res. 2014;53:1-17
70. Duncan JG, Finck BN. The PPARalpha-PGC-1alpha Axis Controls Cardiac Energy Metabolism in Healthy and Diseased Myocardium. PPAR Res. 2008;2008:253817
71. Duncan JG, Bharadwaj KG, Fong JL, Mitra R, Sambandam N, Courtois MR. et al. Rescue of cardiomyopathy in peroxisome proliferator-activated receptor-alpha transgenic mice by deletion of lipoprotein lipase identifies sources of cardiac lipids and peroxisome proliferator-activated receptor-alpha activators. Circulation. 2010;121:426-35
72. Burkart EM, Sambandam N, Han X, Gross RW, Courtois M, Gierasch CM. et al. Nuclear receptors PPARbeta/delta and PPARalpha direct distinct metabolic regulatory programs in the mouse heart. J Clin Invest. 2007;117:3930-9
73. Huss JM, Kelly DP. Nuclear receptor signaling and cardiac energetics. Circ Res. 2004;95:568-78
74. Hu N, Ren J, Zhang Y. Mitochondrial aldehyde dehydrogenase obliterates insulin resistance-induced cardiac dysfunction through deacetylation of PGC-1alpha. Oncotarget. 2016;7:76398-414
75. Rowe GC, Jiang A, Arany Z. PGC-1 coactivators in cardiac development and disease. Circ Res. 2010;107:825-38
76. Ingwall JS. Energy metabolism in heart failure and remodelling. Cardiovasc Res. 2009;81:412-9
77. Cimolai MC, Alvarez S, Bode C, Bugger H. Mitochondrial Mechanisms in Septic Cardiomyopathy. Int J Mol Sci. 2015;16:17763-78
78. Goldenthal MJ. Mitochondrial involvement in myocyte death and heart failure. Heart Fail Rev. 2016;21:137-55
79. Patten IS, Arany Z. PGC-1 coactivators in the cardiovascular system. Trends Endocrinol Metab. 2012;23:90-7
80. Karamanlidis G, Nascimben L, Couper GS, Shekar PS, del Monte F, Tian R. Defective DNA replication impairs mitochondrial biogenesis in human failing hearts. Circ Res. 2010;106:1541-8
81. Sihag S, Cresci S, Li AY, Sucharov CC, Lehman JJ. PGC-1alpha and ERRalpha target gene downregulation is a signature of the failing human heart. J Mol Cell Cardiol. 2009;46:201-12
82. Cao Y, Song J, Shen S, Fu H, Li X, Xu Y. et al. Trimedazidine alleviates pulmonary artery banding-induced acute right heart dysfunction and activates PRAS40 in rats. Oncotarget. 2017;8:92064-78
83. Sano M, Wang SC, Shirai M, Scaglia F, Xie M, Sakai S. et al. Activation of cardiac Cdk9 represses PGC-1 and confers a predisposition to heart failure. EMBO J. 2004;23:3559-69
84. Gurha P, Wang T, Larimore AH, Sassi Y, Abreu-Goodger C, Ramirez MO. et al. microRNA-22 promotes heart failure through coordinate suppression of PPAR/ERR-nuclear hormone receptor transcription. PLoS One. 2013;8:e75882
85. Dai DF, Rabinovitch PS, Ungvari Z. Mitochondria and cardiovascular aging. Circ Res. 2012;110:1109-24
86. Liu W, Ruiz-Velasco A, Wang S, Khan S, Zi M, Jungmann A. et al. Metabolic stress-induced cardiomyopathy is caused by mitochondrial dysfunction due to attenuated Erk5 signaling. Nat Commun. 2017;8:494
87. Wu SP, Kao CY, Wang L, Creighton CJ, Yang J, Donti TR. et al. Increased COUP-TFII expression in adult hearts induces mitochondrial dysfunction resulting in heart failure. Nat Commun. 2015;6:8245
88. Holzem KM, Vinnakota KC, Ravikumar VK, Madden EJ, Ewald GA, Dikranian K. et al. Mitochondrial structure and function are not different between nonfailing donor and end-stage failing human hearts. FASEB J. 2016;30:2698-707
89. Chidsey CA, Weinbach EC, Pool PE, Morrow AG. Biochemical studies of energy production in the failing human heart. J Clin Invest. 1966;45:40-50
90. Wang Y, Cao Y, Yamada S, Thirunavukkarasu M, Nin V, Joshi M. et al. Cardiomyopathy and Worsened Ischemic Heart Failure in SM22-alpha Cre-Mediated Neuropilin-1 Null Mice: Dysregulation of PGC1alpha and Mitochondrial Homeostasis. Arterioscler Thromb Vasc Biol. 2015;35:1401-12
91. Cao Y, Szabolcs A, Dutta SK, Yaqoob U, Jagavelu K, Wang L. et al. Neuropilin-1 mediates divergent R-Smad signaling and the myofibroblast phenotype. J Biol Chem. 2010;285:31840-8
92. Li Z, Liu L, Hou N, Song Y, An X, Zhang Y. et al. miR-199-sponge transgenic mice develop physiological cardiac hypertrophy. Cardiovasc Res. 2016;110:258-67
93. Du JK, Cong BH, Yu Q, Wang H, Wang L, Wang CN. et al. Upregulation of microRNA-22 contributes to myocardial ischemia-reperfusion injury by interfering with the mitochondrial function. Free Radic Biol Med. 2016;96:406-17
94. Tuomainen T, Tavi P. The role of cardiac energy metabolism in cardiac hypertrophy and failure. Exp Cell Res. 2017
95. Kavazis AN, Smuder AJ, Powers SK. Effects of short-term endurance exercise training on acute doxorubicin-induced FoxO transcription in cardiac and skeletal muscle. J Appl Physiol (1985). 2014;117:223-30
96. Ma Z, Xin Z, Hu W, Jiang S, Yang Z, Yan X. et al. Forkhead box O proteins: Crucial regulators of cancer EMT. Semin Cancer Biol. 2018;50:21-31
97. Xin Z, Ma Z, Hu W, Jiang S, Yang Z, Li T. et al. FOXO1/3: Potential suppressors of fibrosis. Ageing Res Rev. 2018;41:42-52
98. Jiang S, Li T, Yang Z, Hu W, Yang Y. Deciphering the roles of FOXO1 in human neoplasms. Int J Cancer. 2018
99. Kemi OJ, Ceci M, Condorelli G, Smith GL, Wisloff U. Myocardial sarcoplasmic reticulum Ca2+ ATPase function is increased by aerobic interval training. Eur J Cardiovasc Prev Rehabil. 2008;15:145-8
100. Kadlec AO, Chabowski DS, Ait-Aissa K, Hockenberry JC, Otterson MF, Durand MJ. et al. PGC-1alpha (Peroxisome Proliferator-Activated Receptor gamma Coactivator 1-alpha) Overexpression in Coronary Artery Disease Recruits NO and Hydrogen Peroxide During Flow-Mediated Dilation and Protects Against Increased Intraluminal Pressure. Hypertension. 2017;70:166-73
101. Correia JC, Ferreira DM, Ruas JL. Intercellular: local and systemic actions of skeletal muscle PGC-1s. Trends Endocrinol Metab. 2015;26:305-14
Author contact
![]() Corresponding author: Yang Yang MD., PhD., Key Laboratory of Resource Biology and Biotechnology in Western China, Ministry of Education, Faculty of Life Sciences, Northwest University, 229 Taibai North Road, Xi'an 710069, China. Telephone: +86 13379217366; Email address: yang200214yycom
Corresponding author: Yang Yang MD., PhD., Key Laboratory of Resource Biology and Biotechnology in Western China, Ministry of Education, Faculty of Life Sciences, Northwest University, 229 Taibai North Road, Xi'an 710069, China. Telephone: +86 13379217366; Email address: yang200214yycom