Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results
Discussion
Conclusion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(1):290-310. doi:10.7150/thno.28671 This issue Cite
Research Paper
Direct in vivo application of induced pluripotent stem cells is feasible and can be safe
Meng Xiang1*, Meng Lu1*, Jing Quan1*, Ming Xu1*, Dan Meng1, Anfeng Cui1, Ning Li1, Yingying Liu1, Peng Lu1, Xueling Kang1, Xiaokai Wang1, Ning Sun1, Meng Zhao1, Qiujuan Liang1, Lili Le1, Xinhong Wang1, Jianyi Zhang2, Sifeng Chen1 ![]()
1. Department of Physiology & Pathophysiology, School of Basic Medical Sciences, Fudan University, Shanghai, P.R. China
2. Department of Biomedical Engineering, The University of Alabama at Birmingham, Birmingham, USA
*These authors contributed equally.
Received 2018-7-22; Accepted 2018-11-28; Published 2019-1-1
Abstract

Increasing evidence suggests the consensus that direct in vivo application of induced pluripotent stem cells (iPSCs) is infeasible may not be true.
Methods: Teratoma formation and fate were examined in 53 normal and disease conditions involving brain, lung, liver, kidney, islet, skin, hind limb, and arteries.
Results: Using classic teratoma generation assays, which require iPSCs to be congregated and confined, all mouse, human, and individualized autologous monkey iPSCs tested formed teratoma, while iPSC-derived cells did not. Intravenously or topically-disseminated iPSCs did not form teratomas with doses up to 2.5×108 iPSCs/kg and observation times up to 18 months, regardless of host tissue type; autologous, syngeneic, or immune-deficient host animals; presence or absence of disease; disease type; iPSC induction method; commercial or self-induced iPSCs; mouse, human, or monkey iPSCs; frequency of delivery; and sex. Matrigel-confined, but not PBS-suspended, syngeneic iPSCs delivered into the peritoneal cavity or renal capsule formed teratomas. Intravenously administered iPSCs were therapeutic with a dose as low as 5×106/kg and some iPSCs differentiated into somatic cells in injured organs. Disseminated iPSCs trafficked into injured tissue and survived significantly longer in injured than uninjured organs. In disease-free animals, no intravenously administered cell differentiated into an unwanted long-lasting cell or survived as a quiescent stem cell. In coculture, the stem cell medium and dominant cell-type status were critical for iPSCs to form cell masses.
Conclusion: Teratoma can be easily and completely avoided by disseminating the cells. Direct in vivo iPSC application is feasible and can be safe.
Keywords: induced pluripotent stem cells, teratoma, cellular microenvironment, monkey
Introduction
Induced pluripotent stem cells (iPSCs) have great therapeutic potential for multiple major diseases and have several advantages over other cell types [1-3]. For example, owing to their unlimited proliferation potential, iPSCs can be massively produced in tissue culture dishes and bioreactors [4]. iPSCs can be differentiated into various types of adult human cells [2, 5]. Furthermore, iPSCs can be generated from a patient's somatic cells [5] and may thus avoid the problem of immune rejection upon in vivo application. However, regenerative therapies using iPSCs encounter several major obstacles with regard to efficiency, safety, and efficacy [3, 6, 7]. These obstacles must be overcome before iPSCs can be actually applied in clinical practice.
Teratoma is benign tumor containing different types of cells spontaneously differentiated from the three embryonic germ layers [8]. Teratoma generation assays require iPSCs to be congregated and confined [9, 10]. Based on teratoma formation resulting from local injection of iPSCs in immunodeficient animals under tightly controlled artificial conditions, the direct in vivo application of iPSCs, including physician-favored intravenous and topical administration, is excluded by most researchers.
Currently, iPSC-differentiated somatic cells are favorable for iPSC therapy. However, somatic cells, other than immune, inflammatory and cancer cells, cannot migrate across the vascular wall and thus cannot be administered intravascularly. For example, iPSC-differentiated myocardial cells, endothelial cells (ECs), and smooth muscle cells in our three-dimensional (3D) printed myocardial patch did not dislocate after implantation [11, 12]. For cells to be fully functional, a proper orientation and context are required. For example, the structures and contexts on the six sides of a hepatocyte are all different. Somatic cells cannot adjust to a microenvironment as easily as stem cells can. Thus, the valuable in vivo application of iPSCs is not replaceable.
Increasing evidence indicates that teratoma formation from iPSCs can be avoided. First, stem cells can be active or quiescent for a long period. Numerous bioengineered tissues consisting of billions of iPSC-derived cells have been implanted in vivo. Because it is impossible to test whether each single iPSC has differentiated, the bioengineered tissue may contain quiescent undifferentiated or partially differentiated iPSCs due to epigenetic memory as well as inefficient purification. A recent study showed that teratoma generated from pluripotent stem cells contained quiescent stem cells [13]. iPSC-derived neural progenitor cells promoted functional and structural recovery of spinal cord injury with no tumor formation but undifferentiated cells still existed 5 [14] or 16 [15] weeks later. Thus, ensuring that solitary iPSCs do not generate teratoma is essential before iPSC-derived cells can be confidently used in vivo. Fortunately, no teratoma generated from iPSC-engineered tissues has been reported. Second, mesenchymal stem cells naturally tend to differentiate into adipocytes and osteoblasts [16]. In vivo application of mesenchymal stem cells has been widely examined in humans. No unwanted differentiation, for example into osteoblasts, in targeted organs such as heart, brain, liver, and lungs has been reported. Finally, embryonic cells in the blastula eventually develop into the human body in the absence of totipotent stem cells and teratoma. All these phenomena indicate that there must be a mechanism in the body to prevent totipotent stem cells from generating teratoma.
We hypothesized that when iPSCs dominate the local microenvironment, given their pluripotent nature, they can grow and differentiate to form a tumor containing unwanted differentiated cells. In contrast, disseminated iPSCs are controlled by their local microenvironment so that their differentiation and proliferation properties are shaped by the needs of the local lesion, which would also prevent subsequent teratoma formation. Intravenously or topically administered iPSCs spread widely and evenly across large lesions. The disseminated cells fulfill the requirement of being dominantly influenced by their local microenvironment. Intravenous and topical administrations are crucially important for cell therapies not only because of their convenience, but also because stem cell differentiation controlled by the local microenvironment at the site of injury may best meet the cellular and structural needs of disease repair and recovery.
The maintenance of iPSC pluripotency requires a strict microenvironment. The maintenance of human and mouse embryonic stem cells (ESCs) or iPSCs requires defined stem cell culture medium in addition to feeder cells or strictly formulated extracellular matrix. Without a specific microenvironment, iPSCs usually die or differentiate into downstream cells of different lineages [17]. Maldifferentiation occurred in experimental mice that received >1×107 mesenchymal stem cells (MSCs) per kilogram bodyweight [18]. However, maldifferentiation was not reported in patients receiving MSCs intravenously, and occurred only occasionally in patients receiving locally injected MSCs [19]. This difference could be explained by the fact that the dose of intravenously administered MSCs in patients is significantly lower than the dose used in mice. In addition, lineage-specific cells can be differentiated from ESCs and iPSCs by manipulating the culture conditions, which highlights the importance of the soluble microenvironment in iPSC pluripotency [4, 20, 21]. Although actively growing iPSCs can be detected, it is not feasible to confirm there is not a single quiescent iPSC present in the differentiated cells. Lineage-specific cells derived from iPSCs are likely contaminated with a small number of undifferentiated cells [13]. To date, it is not reported that the local injection of ES- or iPS-derived lineage-specific cells form teratomas [20, 22]. Finally, a minimum of 1×105 ES cells in myocardium and 1×104 cells in skeletal muscle were required for teratoma development in immunodeficient mice [23], suggesting that the number of cells congregated in a spot is a critical factor for teratoma formation by pluripotent cells. Similarly, at least 100 human ESCs per subcutaneous injection were required to form a teratoma in the presence of 1×106 mitotically inactivated feeder cells and Matrigel to promote teratoma formation [24]. Thus, iPSCs administered through physician-favored intravascularly or topically disseminated approaches may not form teratoma. Under the worst scenario, where iPSCs do occasionally form teratoma, there are strategies to limit the adverse impact thereof. First, teratoma generated from iPSCs is self-limited and is a benign tumor. Its damage to the body is limited. Second, it is possible to embed an inducible suicide gene in the cells. If teratoma does appear, one can trigger the expression of the embedded suicide gene and eliminate the teratoma cells [25].
In this study, the in vivo teratoma formation potential of iPSCs was examined in as many conditions as possible. We expected that if we could provide evidence of the safety of in vivo application of iPSCs, this would be a landmark in stem cell therapy.
Methods
Animals and human blood samples
The animal protocols were approved by the Animal Care Committee of the Fudan University Shanghai Medical College in accordance with the Guide for the Care and Use of Laboratory Animals (National Research Council of United States). All procedures involving animals and human blood samples were performed in accordance with institutional guidelines and permission of the Ethics Committee at the Fudan University Shanghai Medical College. Male and female C57BL/6 mice were purchased from Shanghai Laboratory Animal Co. Ltd. (Shanghai, China). SCID-NOD mice were purchased from Vital River Laboratory Animal Technology Co. Ltd. (Beijing, China). The mice were maintained on standard rodent chow and water ad libitum and were used for the experiments at 6-8 weeks of age. Four adult male Macaques from unrelated families were purchased from the Dongwu Experimental Monkey Farm (Ningbo, China) and housed in an animal laboratory at Fudan University affiliated Shanghai Public Health Clinical Center (Shanghai, China).
Lentivirus production and mouse iPSC induction
Subconfluent 293FT cells were transfected with FUW-OSKM lentiviral plasmids encoding doxycycline-inducible (with Tet-on transactivator) mouse Oct4, Sox2, Klf4, and c-Myc (Addgene plasmids 20328; Addgene, Cambridge, MA) along with the packaging plasmids PsPAX2 and PMD2.G (Addgene plasmids 12259 and 12260) using Lipofectamine 2000 (Invitrogen, Carlsbad, CA). The virus-containing supernatants were harvested 48 h after transfection and were used for iPSC induction according to the protocol described by Takahashi et al [26] in the presence of doxycycline. Briefly, MEFs were isolated from embryonic day 13.5 embryos and cultured in Dulbecco's modified Eagle's medium (DMEM; Thermo Fisher, Waltham, MA) containing 10% fetal bovine serum (FBS; Thermo Fisher) as described previously [26]. MEFs (5×105) were incubated in DMEM containing 10% FBS in 100-mm dishes overnight and then the medium was replaced with an equal volume of lentivirus medium supplemented with 10 µg/mL polybrene (Sigma-Aldrich, St. Louis, MO). After 24 h of lentivirus transduction, the cells were returned to fresh DMEM containing 10% FBS for an additional 48 h before being changed to ESC medium (DMEM supplemented with 2 mM L-glutamine, 0.1 mM nonessential amino acids, 1 mM sodium pyruvate, 0.1 mM β-mercaptoethanol, 50 U/mL penicillin, 50 µg/mL streptomycin, 2 μg/mL doxycycline, and 0.1 µg/mL leukemia inhibitory factor). Cells were cultured with daily changes of ESC medium. Approximately 0.15% of the cells formed clones. Cell clones were picked 12-14 days after lentivirus infection and were expanded. Twelve clones were expanded, and two clones prepared by different researchers were used for the experiment as described in Table 1. The cells were identified as iPSCs using the classic criteria of colony morphology, the expression of alkaline phosphatase and pluripotency genes, and embryoid body and teratoma formation [26].
Subcutaneously delivered iPSCs formed teratomas. s.c.: subcutaneous.
| iPSC type | Recipient animal | Route of administration | Dose | Frequency of administration | Matrigel | Disease model | Time of observation | Number of animals | Rate of teratoma formation |
|---|---|---|---|---|---|---|---|---|---|
| C57BL/6-1 | Male, C57BL/6 | s.c. | 1×106/spot | Single | Yes, 100 μL | None | 28 days | 10 | 100% |
| C57BL/6-1 | Male, C57BL/6 | s.c. | 1×106/spot | Single | No, but in 100 μL PBS | None | 28 days | 10 | 100% |
| C57BL/6-1 | Female, C57BL/6 | s.c. | 1×106/spot | Single | Yes, 100 μL | None | 28 days | 10 | 100% |
| C57BL/6-1 | Male, SCID-NOD | s.c. | 1×106/spot | Single | Yes, 100 μL | None | 28 days | 10 | 100% |
| C57BL/6-1 derivatives | Male, C57BL/6 | s.c. | 1×106/spot | Single | Yes, 100 μL | None | 180 days | 10 | 0% |
| C57BL/6-2 | Male, C57BL/6 | s.c. | 1×106/spot | Single | Yes, 100 μL | None | 28 days | 10 | 100% |
| C57BL/6-2 | Female, C57BL/6 | s.c. | 1×106/spot | Single | Yes, 100 μL | None | 28 days | 10 | 100% |
| C57BL/6-2 | Male, SCID-NOD | s.c. | 1×106/spot | Single | Yes, 100 μL | None | 28 days | 10 | 100% |
| Healthy human iPSC-1 | Male, SCID-NOD | s.c. | 1×106/spot | Single | Yes, 100 μL | None | 28 days | 10 | 80% |
| Healthy human iPSC-2 | Male, SCID-NOD | s.c. | 1×106/spot | Single | Yes, 100 μL | None | 28 days | 10 | 90% |
| iPSCs from 4 different type 2 diabetic patients | Male, SCID-NOD | s.c. | 5×106/spot | Single | Yes, 100 μL | None | 28 days | 4 | 100% |
| Autologous iPSCs from 4 different monkeys | Individualized, monkey from which cells were used for iPSC induction | s.c. | 1×105/spot | Single | No, but in 100 μL PBS | None | 180 days | 4 | 0% |
| Autologous iPSCs from 4 different monkeys | Individualized, monkey from which cells were used for iPSC induction | s.c. | 5×106/spot | Single | Yes, 100 μL | None | 28 days | 4 | 100% |
| C57BL/6-1 | Male, C57BL/6 | s.c. | 1×105/spot | Single | No, but in 100 μL PBS | None | 28 days | 10 | 0% |
| C57BL/6-1 | Male, C57BL/6 | s.c. | 1×105/spot | Single | Yes, 100 μL | None | 28 days | 3 | 30% |
Human iPSC induction
Peripheral blood nucleated cells were isolated from 2 healthy subjects and 4 diabetic patients using Ficoll-Paque PREMIUM (GE Healthcare, Pittsburgh, UK). The isolated cells were resuspended in a serum-free medium for lymphocytes (Nobimpex, Herbolzheim, Germany) and cultured with anti-CD3 monoclonal antibody (20 ng/mL, T&L Biological Technology, Beijing, China) and interleukin-2 (10 ng/mL, T&L Biological Technology, Beijing, China). At day 5, the cells were used for iPSC induction. Individualized iPSC lines were generated from 1 healthy subject and 4 diabetic patients using Sendai virus carrying human Klf4, Oct3/4, Sox2, and c-Myc. Cells from the remaining healthy subject were used for iPSC induction using a method developed by one of the co-authors [4, 29], with minor modifications. iPSC clones were amplified and fully characterized by pluripotency marker expression assay, alkaline phosphatase activity assay, karyotyping, in vitro differentiation assay, and teratoma formation assay [30]. On the basis of positive assay results, one iPSC colony from each human donor was selected for the experiments.
Generation and culture of monkey iPSCs
Ear skin samples were obtained from four adult male macaques (Macaca mulatta) using biopsy punch (FRAY, Buffalo, NY) and were used for iPSC induction separately. The samples were minced and cultured in DMEM supplemented with 10% FBS. Cells of third passage were used for iPSC induction. MEFs were isolated from embryos at embryonic day 12.5 as described. Cells of 4-6th passage were pretreated with 10 μg/mL mitomycin C at 37 °C in air containing 5% CO2 for 2.5 h. Immediately after mitomycin C treatment and washing, the MEFs were seeded on 0.1% gelatin-coated 6-well plates (2.5×106 cells per well) and served as feeder cells in macaque iPSC induction. For macaque iPSC induction, cells from macaque skin were digested with trypsin, centrifuged, and resuspended with a Sendai virus (Thermo Fisher) mixture at multiplicities of infection (MOIs) of 5, 5, and 3 for KOS, hc-Myc, and hKlf4, respectively, at 37 °C in air containing 5% CO2 for 40 min. Then, the cells were plated in Matrigel-coated 6-well plates and cultured in DMEM with 10% FBS and 1 mM valproic acid (Sigma-Aldrich, St. Louis, MO) for 3-4 days until significant morphological changes were observed. Then, the cells were trypsinized and replated onto fresh mitomycin C-treated MEFs at a 1:6 ratio. The medium was replaced with macaque iPSC medium without valproic acid. The macaque iPSC medium was KSR medium containing 85% DMEM/F12 (DF12, Thermo Fisher), 15% knockout serum replacement (KSR, Thermo Fisher), 1 mM L-glutamine (Thermo Fisher), 0.1 mM non-essential amino acids (Thermo Fisher), 0.1 mM β-mercaptoethanol (Sigma-Aldrich, St. Louis, MO) and 5 ng/mL basic fibroblast growth factor (Thermo Fisher). The medium was changed every day until the colonies grew sufficiently large to be picked up.
For macaque iPSC expansion, clone-like macaque iPSC masses were manually cut into smaller cell masses with a 10 µL pipette tip and transferred onto fresh MEFs. When the induced cells could be converted, i.e., when cell growth stabilized and colonies were sufficiently large, the induced cells were incubated with collagenase IV (1 mg/mL) (Thermo Fisher) at 37 °C for 15 min and replated onto fresh MEFs. The macaque iPSCs were passaged every 4-6 days. Alkaline phosphatase staining, qPCR and immunohistochemical staining for pluripotency markers, and teratoma generation assay were performed using cells of passage 10 for iPSC identification. Cells were used for experiments after 18 passages to minimize the heterogeneity among cells from different animals.
Embryoid body formation assay
Mouse iPSCs were cultured in ESC medium in the absence of leukemia inhibitory factor. The medium was changed daily. Embryoid bodies on day 6 are shown in Figure S1C.
Mouse iPSC purification
After iPSCs were washed with PBS 3 times, iPSCs and feeder MEFs were dissociated into single cells in medium containing 0.25% trypsin and 0.038% ethylenediaminetetraacetic acid. The cells were suspended in ESC medium and were plated onto culture dishes pretreated with 0.2% gelatin. After incubation at 37 °C for 30 min, unattached cells, mainly MEFs, were removed by aspirating the supernatant. iPSCs were more likely to adhere and, therefore, remained on the dishes. The weakly attached iPSCs were then detached and suspended in PBS. After centrifugation, the cells were resuspended in PBS for injection.
Teratoma formation from subcutaneously injected iPSCs
To confirm their pluripotency and capability to form teratomas, iPSCs (1×106 cells per site) in 100 µL Matrigel (BD Biosciences, San Jose, CA) were injected subcutaneously into xenogeneic immunodeficient, allogenic immunodeficient, and syngeneic mice of either sex as described in Table 1. Additional 10 mice were injected subcutaneously with syngeneic iPSCs suspended in PBS instead of Matrigel. Four weeks after subcutaneous iPSC injection, teratomas were confirmed using small animal positron emission tomography/computed tomography (micro-PET/CT) and palpation, and then collected. H&E staining was used to determine whether teratomas contained tissues from all three germ layers.
Immunofluorescence staining of mouse pluripotency markers
ESC marker expression in iPSCs was assessed by immunofluorescence staining with primary antibodies against Oct3/4 (Santa Cruz Biotechnology, Santa Cruz, CA) and SSEA-1 (Cellular Signaling Technology, Beverly, MA). Corresponding fluorescein-labeled secondary antibodies raised in donkeys were purchased from Jackson ImmunoResearch Laboratories, Inc. (West Grove, PA). Nuclei were counterstained with 4',6-diamidino-2-phenylindole (DAPI; Sigma-Aldrich). Images were acquired using a Leica Axiovert 200 inverted fluorescence microscope (Leica, Bannockburn, IL).
Immunofluorescent staining for human and monkey pluripotency markers
Cells were fixed in 4% paraformaldehyde at room temperature for 8 min, rinsed with PBS, and blocked with 5% donkey serum at room temperature for 60 min. For cytoplasmic protein staining, 0.3% Triton X-100 was added to permeabilize the cell membrane. The cells were incubated with primary antibodies diluted in 5% donkey serum at 4 °C overnight. The cells were washed with PBS three times, exposed to secondary antibodies at room temperature for 60 min, and washed. The nuclei were counterstained with 1 µg/mL DAPI.
Immunohistochemical staining for SSEA-4 and STEM 121
Paraffin sections of heart, lung, kidney, and liver from NOD-SCID mice 180 days after a bolus intravenous injection of 2.5×108 hiPSCs/kg body weight and kidney sections from NOD-SCID mice 3 days after receiving single intra-kidney injection of 1×106 hiPSCs/kidney were stained for SSEA-4 and STEM 121. The sections (5 μm thick) were subjected to antigen retrieval with citrate buffer at 95 °C for 8 min, and incubated with 0.3% hydrogen peroxide for 15 min, 5% normal donkey serum for 1 h, primary antibodies, and HRP-conjugated donkey anti-mouse IgG secondary antibodies. The primary antibodies were mouse monoclonal anti-human SSEA-4 antibodies (Thermo Fisher) and mouse monoclonal anti-human STEM121 (Takara Bio Inc., Shiga, Japan). Peroxidase activity was visualized using diaminobenzidine. Sections were counterstained with hematoxylin before observation. For the negative control, the primary antibody was replaced with 5% normal donkey serum. Dispersed iPSCs smeared on glass slides were stained and they served as additional positive and negative controls. Images were captured with a Leica microscope.
Alkaline phosphatase activity assay
For alkaline phosphatase staining to assay pluripotency, we used an alkaline phosphatase detection kit (Sigma-Aldrich, St. Louis, MO), according to the manufacturer's instructions.
qRT-PCR for mouse pluripotency markers
mRNA levels of Oct4, Nanog, Sox2, Klf4, Myc, Rex1, and Fbx15 were determined by qRT-PCR. In brief, total RNA from our C57BL/6 iPSCs, ESCs (TACK, Manassas, VA), purchased mouse iPSCs (miPSC; SiDanSai Ltd., Shanghai, China), and MEFs was isolated using TRIzol Reagent® (Life Technologies, Carlsbad, CA), following the manufacturer's instructions. RNA concentrations and purities were determined using a Nanodrop (Implen GmbH, Munich, Germany) at wavelengths of 260/280 nm. The total RNA was reverse-transcribed using a SuperScript Preamplification kit (Life Technologies). qPCRs were carried out using 1× SYBR Green PCR Master Mix (Applied Biosystems) and the following thermal cycles: initial denaturation 95 ºC for 5 min, followed by 40 cycles of 20 s at 95 ºC, 45 s at 57 ºC, and 30 s at 72 ºC.
qRT-PCR for human and monkey pluripotency markers
Total RNA was extracted from cultured iPSCs using TRIzol Reagent (Thermo Fisher). cDNA was synthesized from 1 µg of total RNA with SuperScript III (Thermo Fisher), according to the manufacturer's instructions. The cDNA was diluted to 10 ng/μL with DNase-free water. qPCRs were carried out in a Bio-Rad iQ Real-Time PCR Detection System (Bio-Rad, Hercules, CA) using the IQ SYBR Green SuperMix. mRNA levels of target genes were normalized to that of GAPDH as the internal control and were expressed relative to the quantity of the control group.
iPSCs were cultured in complete EC growth medium (ScienCell Research Laboratories, Carlsbad, CA, USA) in the absence of feeder cells and then subcultured once on day 4. On day 6, the cells were mixed with Matrigel and injected subcutaneously (1×106 cells/site) to observe their capability to form teratomas.
Mouse iPSC maintenance
Mouse iPSCs were grown on mitomycin C-treated MEF feeders in standard ESC medium containing 10% KSR. All culture reagents were obtained from Invitrogen unless otherwise stated.
Effects of culture media and iPSC/EC ratio on the formation of iPSC masses
To determine the effect of the iPSC/EC ratio on the formation of iPSC masses, red fluorophore-labeled iPSCs were mixed with primary cultured C57BL/6 aortic ECs isolated, as we described previously [31], at a ratio of 1:9 or 9:1 for a total of 2×105 cells/well in a 6-well plate. The cells were cultured in ESC medium for 48 h. To determine the effects of different media, the same experiment was conducted using complete EC growth medium instead of ESC medium. At 48 h, cells with red fluorescence were imaged and counted using a fluorescence microscope (Leica).
Teratoma examination
Teratoma generation was examined regularly by palpation and micro-PET/CT scans. All mice were subject to full-body necropsy and histological examination of major tissues. Organs were excised, fixed in 4% paraformaldehyde, and subjected to histological examination by H&E staining to determine if teratomas were present.
Small animal PET/CT imaging
18F-Fluorodeoxyglucose ([18F]FDG) was obtained from the Department of Nuclear Medicine at Fudan University (Shanghai, China). PET and CT images were obtained with a small animal PET/CT (Inveon PET/CT; Siemens Healthcare Global, Erlangen, Germany). Mice received tail vein injections of [18F]FDG (200 µCi = 7.4 MBq) off the scanner bed. One hour after injection, the mice were placed in the mouse imaging chamber for CT acquisition for 10 min. The animals were maintained under 2% isoflurane anesthesia from the time after injection until the end of the scanning period. The CT scans were performed at two overlapping bed positions by using a detector-source rotating 360 degrees around the animal and projections were acquired every 2° (180° angles). CT images were reconstructed using a cone beam algorithm (bilinear interpolation, Shepp-Logan filter) with a 206 µm pixel size. Following CT acquisition, each mouse received a PET scan. The images were reconstructed using a 3D OSEM/MAP algorithm with spatial resolution of 1.4 mm. Following the reconstruction, the CT images were spatially aligned to the PET images using a fixed, predetermined transformation matrix. The CT data were also used for attenuation correction of PET images. Image analysis was performed using Inveon Research Workstation (Siemens Medical Solutions, Knoxville, TN, USA).
In vivo mouse iPSC quantification
The Tet-on gene was carried into iPSCs by lentiviruses used for iPSC induction. Because the Tet-on gene is a non-expressed gene in iPSCs, but not in recipient cells, it can be used as a marker for iPSC identification and quantification. The following Tet-on-specific PCR primers were designed and confirmed using BLASTN searches (U.S. National Center for Biotechnology Information): sense 5′-AGCACAACTACGCCGCACCC-3′; antisense 5′-ATGCACCAGAGTTTCGAAGC-3′. PCR was conducted using genomic DNA isolated from cells or tissues as a template. The PCR product of 18S rDNA (18S rRNA gene) was used as an internal control. To calculate the number of Tet-on gene-containing cells in tissues, standard curves depicting the relationship of Tet-on gene and cell number were generated by plotting PCR product abundance versus cell number. The cell numbers used to isolate Tet-on DNA followed by PCR measurement to generate the curve were 10, 30, 60, 600, 6000, and 60000.
Paraquat intoxication
For teratoma observation, paraquat (Sigma-Aldrich, St Louis, MO) was injected intraperitoneally (15 mg/kg body weight). One hour after paraquat injection, 5×106 iPSCs/kg (n = 30) or 2.5×108 cells/kg (n = 20) were injected via the tail vein and mice were monitored for 180 days.
To observe the homing and survival of injected syngeneic iPSCs in the injured lungs, the lung tissue was harvested for Tet-on gene measurement and fluorescence observation of the PKH26-labeled iPSCs at 1, 3, 28, 90 and 180 days (n = 3 for each time point) after intraperitoneal paraquat injection and intravenous iPSC injection (5×106 iPSCs/kg). Animals receiving PBS instead of iPSCs served as an untreated control.
Measurement of pulmonary function
To determine if intravenous administration of iPSCs at a dose of 5×106 iPSCs/kg was effective, additional syngeneic mice receiving PBS, paraquat, and paraquat followed by iPSC administration after 1 h were sacrificed at 24 h and 48 h after treatment with 5 mice per group per time point. Mice (male C57BL/6 mice, 8-week-old, 25-27 g) were anesthetized with pentobarbital sodium (70-90 mg/kg). A 20-gauge tracheal cannula was surgically inserted 2.5 mm into the trachea and was secured with silk suture. The tracheal cannula was connected to a Y-shaped tube in order to connect the mouse to a lung function analytical system (Anires2005 system, BioLab Tech Co. Ltd, Peking, China) and a ventilator (RES3020, BioLab Tech Co. Ltd). After the surgery, the mouse was placed in a supine position with its entire body inside the plethysmographic chamber (550 mL volume) to analyze pulmonary function. Natural air was provided to the animal through the ventilator with a rate of 110 breaths/min. The lung function analytical system was calibrated for each measurement with three normal mice under a peak airway positive pressure of 12 cm H2O. By detecting the ventilation-associated pressure change inside the chamber, the Anires2005 system automatically calculated and displayed pulmonary function parameters such as dynamic pulmonary compliance, inspiratory resistance, and expiratory resistance.
LPS-induced lung injury
Tracheotomies were performed on mice and 14-gauge catheters were inserted into their tracheas. LPS (Escherichia coli O55:B5; Sigma-Aldrich, 50 µg in 50 μL saline) was administered into the lungs via the catheter. Two hours after LPS administration, iPSCs (2.5×108 cells/kg body weight) were injected via the tail vein.
Fluorescent labeling and observation of iPSCs
To track the cells in vivo, mouse iPSCs were labeled with the red fluorescent dye PKH26 before being injected into the tail veins of healthy, LPS-induced lung-injured or paraquat-injured mice. The fluorescence signal can stay on the cells for approximately 1 month; DAPI was used to counter-stain nuclei. Labeled cells on culture ware or in frozen sections were observed using an inverted Leica microscope.
Co-localization of iPSC labeling and type 2 pneumocytes
To show if any iPSCs that homed to injured tissue differentiated into somatic parenchymal cells of the organ, lungs of syngeneic mice injected with 5×106 iPSCs/kg after paraquat injection were harvested on day 3. The lung tissue was fixed with 3.7% formaldehyde plus 1% methanol for 24 h, serially dehydrated in 10%, 20% and 30% sucrose, embedded in optimal cutting temperature compound (OCT) and frozen. Frozen sections (5 μm-thick) of the lungs were incubated in 3% H2O2 for 10 min at room temperature to block endogenous peroxidase activity, microwaved for 6 min in target retrieval solution (DM828, Agilent Technologies, Santa Clara, California) and allowed to cool at room temperature for 1 h. The sections were blocked with 5% normal donkey serum (NC9624464, Jackson ImmunoResearch Labs, Waltham, MA) and incubated with goat anti-mouse SP-C (M-20, pulmonary surfactant-associated protein C) polyclonal antibodies (sc-7706, Santa Cruz Biotechnology, CA) overnight at 4 °C followed by incubation with HRP-labeled donkey anti-goat IgG (1:1000, 705-035-003, Jackson ImmunoResearch Labs) at room temperature for 2 h. Negative controls were processed by omitting the primary antibody. Diaminobenzidine (DAB, P0203, Beyotime Biotechnology, Shanghai, China) was used for visualization of the sections. After dehydration and mounting, images were captured by an inverted fluorescence microscope (Leica SCN400, Leica Microsystems, Wetzlar, Germany) and the colocation cell count for the positive cells and PKH26-miPSCs was assessed using ImageJ (version 1.49a, National Institutes of Health, Bethesda, Maryland).
Atherosclerosis
Thirty 6-week-old homozygous apoE-deficient (apoE-/-) mice on a C57BL/6 genetic background were purchased from the Department of Laboratory Animal Science at the Peking University Health Science Center (Beijing, China). After one week of acclimation, the mice were fed a high-fat diet (Shanghai Laboratory Animal Center, Chinese Academy of Science, Shanghai, China) containing 42% kcal from fat, 43% kcal from carbohydrates, 15% kcal from protein, and 0.2% cholesterol for 7 weeks. The mice were injected via the tail vein with 5×106 iPSCs/kg body weight in 0.2 mL PBS one time (n = 10) or once every 3 days for 60 days (n = 30). The mice were sacrificed 180 days after the first iPSC injection, and the presence of teratomas was examined.
Acute kidney ischemia-reperfusion injury
Male C57BL/6J mice were anesthetized by pentobarbital sodium (Sigma-Aldrich, St. Louis, MO) intraperitoneal (i.p.) injection. The left kidney was exposed by left flank incision and left renal ischemia was induced by clamping the renal artery with a non-traumatic microvessel clamp (size B-1 V; S&T, Neuhausen, Switzerland) for 50 min. After the clamp was released and reperfusion of blood flow was confirmed visually, the left flank incision was closed using 3-0 silk sutures. All mice received 30 µL saline/g body weight subcutaneously post-surgery to replenish fluid loss. One hour post-operation, iPSCs (5×106 cells/kg body weight, n = 30) in 0.2 mL PBS were administered slowly via the tail vein. The mice were observed for teratoma formation for 180 days.
Full-thickness excisional wounding
Excisional wounds were made as previously described [32]. Briefly, mice were anesthetized with isoflurane prior to the wound-creating surgery. Mouse dorsal skins were prepared by removing hair with depilatory cream. Full-thickness excision wounds were created by excising the skin on the mid-back using a 4-mm biopunch (FRAY, Buffalo, NY). After the skin edges retracted (day 0), iPSCs were delivered topically or intravenously as described in Table 1. The mice were sacrificed 180 days later.
Diabetes mellitus
A diabetes mellitus mouse model was established by daily i.p. injection of 80 mg/kg streptozotocin in 50 mM sodium citrate (pH 4.5) for three consecutive days. Fasting blood glucose was measured daily from the tail vein using an Optimum Xceed Blood Glucose Meter (Abbott Laboratories, Abbott Park, IL). Mice with fasting blood glucose levels higher than 16 mM on day 4 were considered diabetic and were used for analysis of teratoma generation in response to tail vein injection of iPSCs and topical application to skin wounds or insulitic pancreases as indicated in Table 1.
Topical application of iPSCs on pancreases
After median incisions were made to the upper abdomens, 1×106 iPSCs were suspended in 200 μL 2% low melting point agarose (Biowest, Madrid, Spain) at 37 ºC and smeared onto the surface of insulitic pancreases. After confirming the agarose gelatinized by room temperature, the incision was closed. The mice were examined for 180 days.
Intrarenal capsule iPSC implantation
Under an operating microscope, a small tube with one end connected to a 1 mL insulin syringe was inserted into the renal capsule on the lateral surface of the lower pole of the left kidney. One million iPSCs suspended in 100 µL PBS or Matrigel were injected beneath the renal capsule. The breach in the capsule was cauterized, the kidney was repositioned, and the incision was closed in layers as we described previously [33].
Wire injury in femoral arteries
Endoluminal injury to the common femoral artery was achieved by passing a 0.015-inch-diameter angioplasty guidewire three times (Cook Medical LLC., Bloomington, IN, USA) as described by [34]. Single intravenous injections of 5×106 iPSCs/kg were performed 1 h after the incisions were closed.
Hind limb ischemia-reperfusion injury
General anesthesia was achieved by intraperitoneal injection of pentobarbital sodium at 40 mg/kg body weight. Under a surgical microscope, groin incisions were made. The left femoral artery was clamped at the level of the inguinal ligament for 5 h. Then, the clamp was removed and the incisions were closed. Single injections of 5×106 iPSCs/kg were administered 1 h after reperfusion.
Carbon tetrachloride-induced liver injury
A single dose of 2.0 mL/kg of body weight (2:5 v/v in mineral oil) was administered by intraperitoneal injection. Four hours after carbon tetrachloride injection, 5×106 iPSCs/kg were injected via the tail vein. The mice were observed for 180 days.
Left-brain middle-artery ischemia-reperfusion injury
Under an operating microscope, small incisions were made in the left external carotid arteries. Nylon filaments (diameter, 0.36 mm) were inserted into the left internal carotid arteries through the openings and were advanced further to occlude the left middle cerebral arteries for 45 min. Then, the nylon filaments were removed to restore blood flow of the left internal carotid arteries as we described previously [35]. One hour after restoration of blood flow, 5×106 iPSCs/kg were injected via the tail vein and the mice were observed for 180 days.
Skin burn wounding
Cylindrical stainless-steel balance weights (5 g) were preheated to 150 °C in an oven and placed onto the moistened and depilated dorsal skin for 10 s to produce a round skin wound 0.9 cm in diameter. Single injections of 2.5×108 cells/kg or 5×106 cells/kg were delivered intravenously and the mice were monitored for 180 days.
Intraperitoneal administration
After median incisions were made to the lower abdomens, 5×106 cells in 200 µL PBS or Matrigel were delivered to the lower abdomen behind the bladder. To ensure no teratomas were generated, the mice were observed for 28 or 180 days for iPSCs suspended in Matrigel or PBS, respectively.
Materials
The key reagents, resources, virus stains, oligonucleotides and recombinant DNA, etc. are list at Table S1.
Statistical analyses
All data are presented as the mean ± standard error of the mean. Means were compared by analysis of variance and the Student-Newman-Keuls post-hoc test. Statistical significance was defined as P < 0.05.
Results
Induction of mouse, monkey, and human iPSCs
To ensure the immune compatibility between administered iPSCs and host animals, we established a series of in house-induced iPSC lines. Two new mouse iPSC lines were induced by transducing C57BL/6 mouse embryonic fibroblasts (MEFs) with lentiviruses carrying Tet-On inducible fusion genes of Klf4, Oct3/4, Sox2, and c-Myc according to methods described by [26]. We used the four classic Yamanaka genes to ensure that the iPSCs used for evaluating teratoma formation capability are highly representative and are not induced by side-effect-reduced induction assays [27, 28]. C57BL/6 mouse iPSC lines generated by two colleagues were tested to increase the diversity of the iPSCs used for the study.
Two vector systems were successfully used to develop human iPSCs. First, human iPSC lines were generated with lentiviruses carrying human Klf4, Oct3/4, Sox2, and c-Myc from peripheral blood nucleated cells from a healthy subject according to previous reports [29]. Second, individualized iPSC lines were generated from a healthy subject and four diabetic patients from peripheral blood nucleated cells using Sendai virus carrying human Klf4, Oct3/4, Sox2, and c-Myc.
Individualized monkey iPSCs were induced from skin fibroblasts of four monkeys using Sendai virus carrying human Klf4, Oct3/4, Sox2, and c-Myc.
All iPSC lines were identified according to commonly accepted criteria. They expressed characteristic pluripotency markers, exhibited strong alkaline phosphatase activity, and formed embryoid bodies in vitro and teratomas in vivo under regular teratoma generation conditions (Table 1, Figure 1, and Figures S1-S3).
Conditional teratoma formation. (A) Subcutaneous teratomas generated in xenogeneic immunodeficient mice, allogenic immunodeficient mice, and syngeneic mice of either sex. sub-cu: subcutaneous injection. (B) Single intravenous administration of a high dose of induced pluripotent stem (iPS) cells did not generate teratomas in xenogeneic immunodeficient mice, allogenic immunodeficient mice, syngeneic mice of either sex, or in lung-injured mice. IV: intravenous injection. (C) Histological examination showed that the heart, lung, kidney, and liver of mice receiving intravenously injected iPSCs had no teratomas. (D) Representative histology of skin wound lesions that had topically or subcutaneously received syngeneic iPSCs. (E) Syngeneic iPSCs suspended in Matrigel generated teratomas (red arrows) when administered intraperitoneally or under the renal capsule (left panels). Replacing Matrigel with PBS prevented teratoma formation (right panels). (F) Representative gross images of the major organs of mice that had intravenously or intrapleurally received syngeneic iPSCs for 18 months (lower left and right panels). The organs harvested from mice 28 days after intra-kidney injection of syngeneic iPSCs served as positive control (upper left panel).
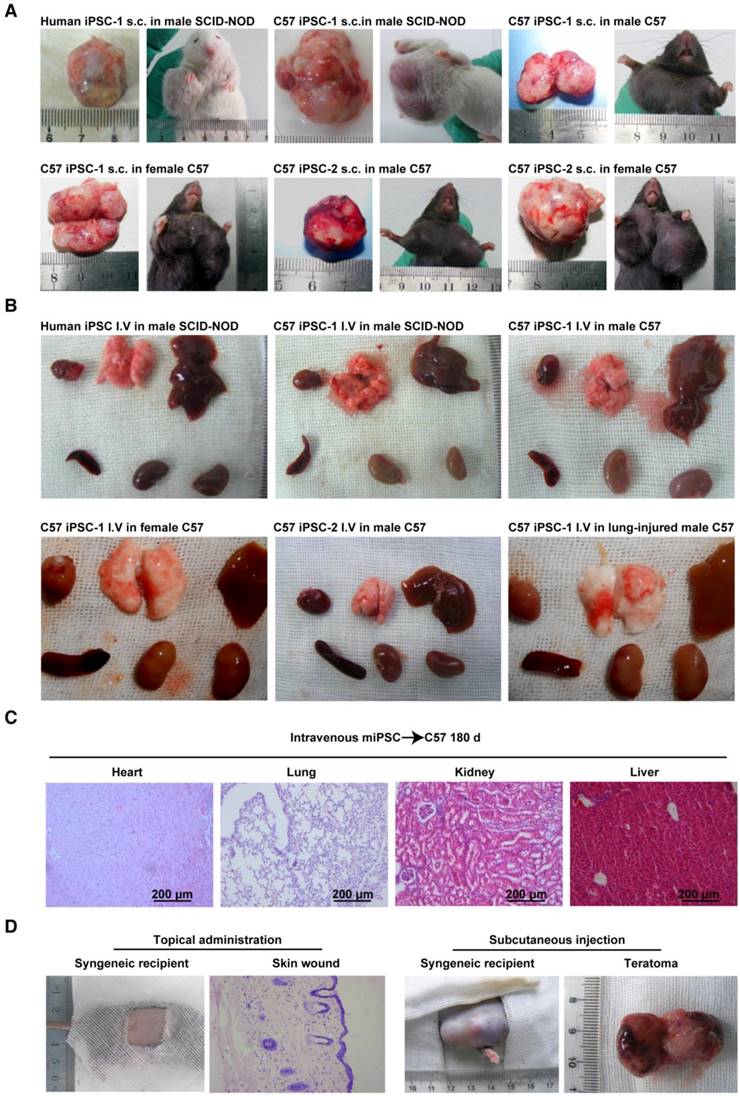
Extracellular matrix-embedded congregated iPSCs form teratomas in immune-deficient mice
Using a classic teratoma generation assay, the teratoma generation capability of all the in house-induced iPSCs and commercially available mouse iPSCs was confirmed (Figure 1A and Figure S2A-C). A classical subcutaneous injection assay using 1×106 cells mixed in 100 μL Matrigel per injection was performed. One hundred percent teratoma formation rates were observed in 28 days in severe combined immunodeficiency-non-obese diabetic (SCID-NOD) mice that received either the C57BL/6 iPSCs or the commercial mouse iPSCs (Table 1).
All in house iPSCs, including those generated from two healthy subjects, four diabetic patients, and four monkeys, developed teratoma under the same condition at a dose of 5×106 cells mixed in 100 μL Matrigel per injection (Figure S3 and Table 1).
Congregated mouse iPSCs form teratoma in syngeneic mice
To assess the immune compatibility between administered C57BL/6 iPSCs and host C57BL/6 mice, iPSCs from the two in house-developed mouse cell lines were injected into male and female C57BL/6 mice subcutaneously, with 1×106 cells/injection in Matrigel (n = 10/group). All 40 mice formed teratomas in 28 days, suggesting that the cells were tolerant to syngeneic mice, regardless of sex. All mice (n = 10) subcutaneously injected with 1×106 iPSCs/injection suspended in PBS instead of Matrigel also formed teratomas (Table 1). However, when the dose of iPSCs was decreased by 90% (1×105 cells per injection spot) and when iPSCs were suspended in PBS instead of Matrigel, they did not generate teratomas (Table 1).
Autologous monkey iPSCs injected subcutaneously form teratomas in a dose-dependent manner
With a bolus dose of 1×105 iPSCs per spot injected subcutaneously, none of the iPSCs induced from the four monkeys formed teratoma in 180 days in the monkey the individualized autologous iPSC line was generated from, either with or without embedding in Matrigel. When the dose was increased to 5×106 iPSCs per spot, all autologous monkey iPSCs generated teratoma in 28 days when they were mixed with Matrigel. Similar results were achieved when the cells were subcutaneously injected in immunodeficient mice (Table 1 and Figure S3).
Subcutaneously injected iPSC-derived cells do not form teratoma
To examine whether cells differentiated from iPSCs are still able to form teratoma, C57BL/6 iPSCs were cultured in complete EC culture medium for 6 days, without feeder cells. About 80% of the cells died, and the remaining cells lost iPSC morphology and no longer expressed pluripotent markers (data not shown). Cells (1×106/injection) were then mixed with Matrigel and injected into C57BL/6 mice subcutaneously. No teratomas formed over 180 days (n = 10) (Table 1). Hematoxylin and eosin (H&E) staining of a Matrigel plug harvested at day 28 is shown in Figure S1 and showed the cells had lost iPSC morphology. These results suggested that when iPSCs start losing their pluripotency capacity, they are no longer able to form teratoma, even when locally injected into a syngeneic environment.
Intravenously injected mouse iPSCs do not form teratoma in healthy syngeneic mice and immunodeficient mice
In future clinical applications of iPSCs or iPSC derivatives, intravenous administration will be one of the most acceptable routes. However, there are no reports on whether intravenously injected iPSCs can form teratoma. To assess teratoma formation by intravenously administered iPSCs, the two C57BL/6 iPSC lines in PBS were injected into healthy C57BL/6 mice and SCID-NOD mice via the tail vein, using a single dose of 2.5×108 cells/kg body weight. This dose equates to ~5×106 cells/mouse, or 6,000 100-mm dishes of cells for a 60 kg individual. In contrast to the teratoma formation by subcutaneously injected iPSCs, no teratomas in any part of the body were found over 18 months post injection, regardless of mouse sex (n = 10/group) (Table 2). Representative gross morphological images of major organs are shown in Figure 1B.
Intravenously and topically delivered iPSCs did not formed teratomas. IV: intravenous.
| iPSC type | Recipient animal | Route of administration | Dose | Frequency of administration | Matrigel | Disease model | Time of observation | Number of animals | Rate of teratoma formation |
|---|---|---|---|---|---|---|---|---|---|
| C57BL/6-1 | Male, C57BL/6 | IV | 2.5×108/kg | Single | No, but in 200 μL PBS | None | 180 days | 20 | 0% |
| C57BL/6-1 | Male, C57BL/6 | IV | 2.5×108/kg | Single | No, but in 200 μL PBS | None | 18 months | 20 | 0% |
| C57BL/6-1 | Female, C57BL/6 | IV | 2.5×108/kg | Single | No, but in 200 μL PBS | None | 180 days | 20 | 0% |
| C57BL/6-2 | Male, C57BL/6 | IV | 2.5×108/kg | Single | No, but in 200 μL PBS | None | 180 days | 20 | 0% |
| Healthy human iPSC-1 | SCID-NOD | IV | 2.5×108/kg | Single | No, but in 200 μL PBS | None | 180 days | 20 | 0% |
| Healthy human iPSC-1 | SCID-NOD | IV | 2.5×108/kg | Single | No, but in 200 μL PBS | None | 18 months | 10 | 0% |
| C57BL/6-2 | Male, C57BL/6 | IV | 2.5×108/kg | Single | No, but in 200 μL PBS | Lung injury caused by intratracheally administered LPS | 180 days | 20 | 0% |
| C57BL/6-2 | Male, C57BL/6 | IV | 5×106/kg | Single | No, but in 200 μL PBS | Paraquat intoxication (lung) | 180 days | 30 | 0% |
| C57BL/6-1 | Male, C57BL/6 | IV | 5×106/kg | Single | No, but in 200 μL PBS | Carbon tetrachloride-induced liver injury | 180 days | 30 | 0% |
| C57BL/6-1 | Male, C57BL/6 | IV | 5×106/kg | Single | No, but in 200 μL PBS | Kidney ischemia-reperfusion injury | 180 days | 30 | 0% |
| C57BL/6-1 | Male, C57BL/6 | IV | 5×106/kg | Single | No, but in 200 μL PBS | Stroke, brain middle artery ischemia-reperfusion injury | 180 days | 30 | 0% |
| C57BL/6-1 | Male, C57BL/6 | IV | 5×106/kg | Single | No, but in 200 μL PBS | Femoral artery wire injury | 180 days | 30 | 0% |
| C57BL/6-1 | Male, C57BL/6 | IV | 5×106/kg | Single | No, but in 200 μL PBS | Hind limb ischemia-reperfusion injury | 180 days | 30 | 0% |
| C57BL/6-2 | Male, C57BL/6 | IV | 5×106/kg | Single | No, but in 200 μL PBS | Streptozotocin-induced insulitis and diabetes | 180 days | 10 | 0% |
| C57BL/6-2 | Male, C57BL/6 | IV | 5×106/kg | 3 times (once daily for 3 days) | No, but in 200 μL PBS | Streptozotocin-induced insulitis and diabetes | 180 days | 30 | 0% |
| C57BL/6-1 | Male, C57BL/6 | IV | 5×106/kg | Single | No, but in 200 μL PBS | Atherosclerosis | 180 days | 10 | 0% |
| C57BL/6-1 | Male, C57BL/6 | IV | 5×106/kg | 20 times (once every 3 days for 60 days) | No, but in 200 μL PBS | Atherosclerosis | 180 days | 30 | 0% |
| C57BL/6-1 | Male, C57BL/6 | IV | 5×106/kg | Single | No, but in 200 μL PBS | Burned skin wound | 180 days | 30 | 0% |
| C57BL/6-1 | Male, C57BL/6 | IV | 2.5×108/kg | Single | No, but in 200 μL PBS | Burned skin wound | 180 days | 10 | 0% |
| C57BL/6-1 | Male, C57BL/6 | IV | 5×106/kg | Single | No, but in 200 μL PBS | Excisional skin wound | 180 days | 30 | 0% |
| C57BL/6-1 | Male, C57BL/6 | Topically smeared on wound surface | 1×105 | Single | No, but in 15 μL PBS | Excisional skin wound | 180 days | 30 | 0% |
| C57BL/6-1 | Male, C57BL/6 | Topically smeared on wound surface | 1×105 | Single | No, but in 15μL PBS | Excisional skin wound in streptozotocin-induced insulitis and diabetic mice | 180 days | 30 | 0% |
| C57BL/6-1 | Male, C57BL/6 | Topically smeared on pancreas | 1×106 | Single | No, but in 200 μL low melting point agarose | Streptozotocin-induced insulitis and diabetes | 180 days | 30 | 0% |
| Autologous iPSCs from 4 different monkeys | Individualized, monkey from which cells were used for iPSC induction | Topically smeared on wound surface | 1×106 | Single | No, but in 15 μL PBS | Excisional skin wound | 180 days | 4 | 0% |
| iPSCs from 4 different type 2 diabetic patients | Male, SCID-NOD | IV | 2.5×108/kg | Single | No, but in 200 μL PBS | None | 180 days | 4 | 0% |
| Autologous iPSCs from 4 different monkeys | Male, SCID-NOD | IV | 2.5×108/kg | Single | No, but in 200 μL PBS | None | 180 days | 4 | 0% |
Intravenously injected human and monkey iPSCs do not form teratomas in immunodeficient mice
Commercial, widely accepted human iPSCs did not form teratomas when injected via the tail vein using a single dose of 2.5×108 cells/kg body weight in SCID-NOD mice. Consistently, none of the iPSCs prepared in house from three healthy and four type-2 diabetic individuals as well as from four monkeys generated teratomas under the same conditions (Table 2).
Intravenously injected syngeneic iPSCs do not form teratomas in injured organs
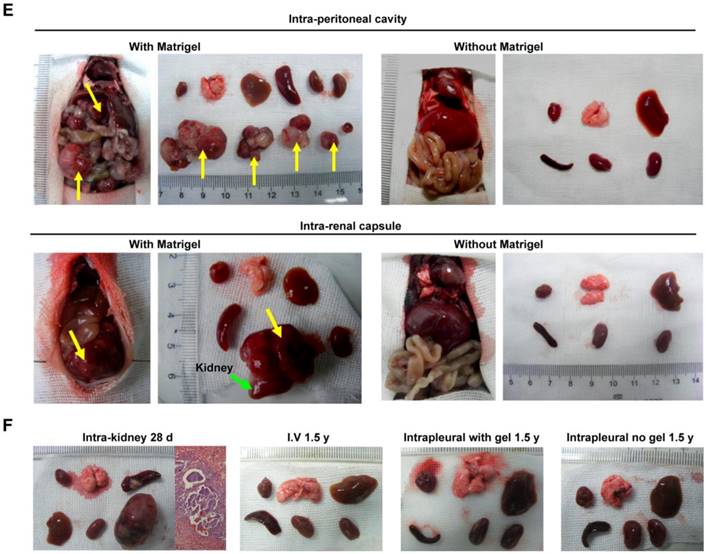
It is important to examine in vivo teratoma formation in disease models because iPSCs tend to migrate to the site of inflammation and injury. For example, 8 h after intravenous iPSC administration (2.5×108 cells/kg body weight), significantly more iPSCs were detected in mouse lungs injured by intratracheally delivered lipopolysaccharide (LPS) than in lungs of healthy mice (Figure 2). Because disease lesions in other organs are usually smaller than those in lungs, a single injection of the more clinically relevant dose of 5×106 cells/kg body weight, equivalent to ~1×105 per mouse, or 120 100-mm dishes of cells for a 60 kg individual, was applied. No teratoma formation was observed within 180 days in mouse models involving major tissues and organs, including paraquat intoxication with severe lung injury, carbon tetrachloride-induced liver injury, ischemia-reperfusion kidney injury, stroke, hindlimb ischemia-reperfusion injury, streptozotocin-induced diabetes, burned and excisional skin wounds, and atherosclerosis (Table 2). In addition, a single dose of 50 times higher (2.5×108 C57BL/6 iPSCs/kg) was injected intravenously to syngeneic mice with burned skin wounds and lung injury induced by intratracheally delivered LPS. No teratoma was detected after 180 days in these mice (Figure 1B-C and Table 2). Furthermore, immunocompetent mice that received syngeneic iPSCs or immunodeficient mice that received human iPSCs generated from 4 separate people with a dose of 2.5×108 iPSCs/kg body weight did not generate teratoma (Table 2). Tumorigenesis was examined by three ways: autopsy, micro PET-CT and HE staining. As teratoma is benign tumor, its border with other tissue is clear and it is large enough to be visible by naked eyes and PET-CT. Autopsy (Figure 1) and PET-CT (Figure S4A) were performed to examine the presence of teratoma throughout the animal. To observe a designated area, high-resolution focal images were captured. Representative PET-CT images of brain and spinal cord are shown in Figure S4B.
Distribution of injected cells in major organs. (A) Fluorescently labeled iPSCs in the hearts, livers, lungs, and kidneys of healthy (left panels) and lung-injured (right panels) mice 8 h after receiving bolus injections of 2.5×108 cells/kg body weight via tail vein. (B) PCR analysis of Tet-On and 18S (18S rRNA gene) DNA levels in the organs. The Tet-On gene was carried into iPSCs by lentiviruses used for the induction of iPSCs from mouse fibroblasts.
Because multiple iPSC administrations may be required in many disease therapies, consecutive intravenous injections of 5×106 cells/kg for 3 days and multiple injections of 5×106 cells/kg every 3 days for 60 days were applied to mouse models of streptozotocin-induced insulitis and atherosclerosis, respectively. Not a single teratoma was detected within 180 days in any part of these mice (Table 2). These data confirmed that intravenous injection of iPSCs, even with 20 consecutive injections or a significantly larger dose than that used for subcutaneous injection, did not lead to teratoma formation in vivo (Table 2).
Topically applied iPSCs do not form teratoma in syngeneic mice and autologous monkeys
We originally hypothesized that 3D congregation of iPSCs is required for these cells to dominate the local environment and to form teratomas, while topically spread iPSCs would not form teratomas because they lack the capacity to control the microenvironment in three dimensions. To test this hypothesis, we used surface wound models and spread iPSCs on syngeneic animals. As expected, no teratoma in the skin lesion or any other part of the body was observed 180 days after spreading 1×105 iPSCs in 15 μL PBS on the surface of excisional skin wounds in normal and diabetic syngeneic mice, nor from 1×106 iPSCs in 200 μL 2% low melting point agarose on the pancreatic surface of syngeneic mice that received streptozotocin 24 h before iPSC administration (Table 2). Representative histological images of skin wounds are shown in Figure 1D. Similar results were obtained when autologous iPSCs were topically spread onto monkey skin wound lesions (Table 2). These results further confirmed that teratoma formation requires a relatively strict condition and 3D congregation.
Matrigel is critical for iPSCs to form teratoma in body cavities
iPSCs applied to organ surfaces did not form teratomas if they were suspended in PBS. Syngeneic iPSCs administered under the kidney capsule (1×106 cells in 100 μL PBS), intrapleural cavity (5×106 cells in 200 μL PBS), or intraperitoneal cavity (5×106 cells in 200 μL PBS) did not form teratomas in 180 days and 18 months. In contrast, syngeneic iPSCs suspended in Matrigel instead of PBS formed teratomas in all mice that had received cells under the kidney capsule, and in 60% of mice that had received the cells intraperitoneally (Table 3 and Figure 1E) in 28 days. However, teratoma was not detected in the pleural cavity over 180 days and 18 months, even though the cells were injected with Matrigel (Figure 1F). These results are in accordance with previous reports and indicated that Matrigel, a unique scaffold of extra cellular matrix that embeds and confines cells in the scaffold, is critical for teratoma formation.
Matrigel promoted teratoma formation of iPSCs delivered into the body cavity.
| iPSC type | Recipient animal | Route of administration | Dose | Frequency of administration | Matrigel | Disease model | Time of observation | Number of animals | Rate of teratoma formation |
|---|---|---|---|---|---|---|---|---|---|
| C57BL/6-1 | Male, C57BL/6 | Topically smeared on wound surface | 1×105 | Single | No, but in 15 μL PBS | Excisional skin wound | 180 days | 30 | 0% |
| C57BL/6-1 | Male, C57BL/6 | Topically smeared on wound surface | 1×105 | Single | No, but in 15 μL PBS | Excisional skin wound in streptozotocin-induced insulitis and diabetic mice | 180 days | 30 | 0% |
| C57BL/6-1 | Male, C57BL/6 | Topically smeared on Pancreas | 1×106 | Single | No, but in 200 μL low melting point agarose | Streptozotocin-induced insulitis and diabetes | 180 days | 30 | 0% |
| C57BL/6-2 | Male, C57BL/6 | Beneath kidney capsule | 1×106 | Single | No, but in 100 μL PBS | None | 180 days | 30 | 0% |
| C57BL/6-2 | Male, C57BL/6 | Beneath kidney capsule | 1×106 | Single | Yes, 100 μL | None | 28 days | 10 | 100% |
| C57BL/6-2 | Male, C57BL/6 | Intraperitoneally | 5×106 | Single | No, but in 200 μL PBS | None | 180 days | 20 | 0% |
| C57BL/6-2 | Male, C57BL/6 | Intraperitoneally | 5×106 | Single | No, but in 200 μL PBS | None | 18 months | 10 | 0% |
| C57BL/6-1 | Male, C57BL/6 | Intraperitoneally | 5×106 | Single | Yes, 200 μL | None | 28 days | 10 | 60% |
| C57BL/6-2 | Male, C57BL/6 | Intrapleurally | 5×106 | Single | No, but in 200 μL PBS | None | 180 days | 20 | 0% |
| C57BL/6-2 | Male, C57BL/6 | Intrapleurally | 5×106 | Single | No, but in 200 μL PBS | None | 18 months | 10 | 0% |
| C57BL/6-2 | Male, C57BL/6 | Intrapleurally | 5×106 | Single | Yes, 200 μL | None | 180 days | 10 | 0% |
iPSCs do not generate teratoma outside the injected area
When sacrificed, all animals that had received iPSCs were examined to determine the presence of teratoma. No teratoma was detected outside the injected area as examined by autopsy. The results in mice were confirmed by small animal positron emission tomography/computed tomography (PET/CT) (Figure S4).
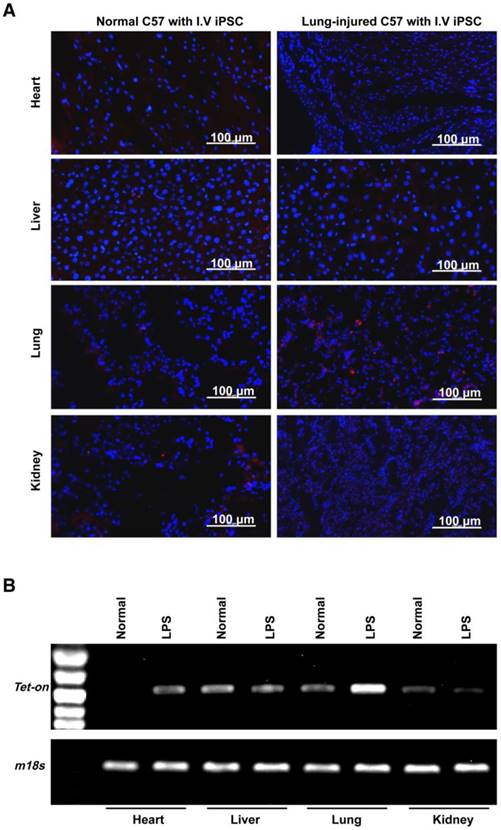
Homing, survival, and therapeutic function of intravenously administered syngeneic iPSCs in paraquat-injured lungs
Using paraquat-induced lung injury as an example, trafficking of intravenously administered syngeneic iPSCs in the injured/diseased tissue was demonstrated. The injected cells were observed in the injured lungs on days 1, 3, and 28 after administration (Figure 3A-D and Figure S5) but were not detectable at 90 days (Figure 3B-C and Figure S5) after administration. The number of the cells in the injured lungs peaked on day 3 after lung injury and iPSC administration. Conversely, iPSCs in organs outside the lungs were not observed on days 3 and 28 after administration (Figure 3D-E). Co-localization of iPSCs and type 2 pneumocytes revealed a small portion of the iPSCs that trafficked into injured tissue differentiated into type 2 pneumocytes, a type of pulmonary parenchymal cell (Figure 3F).
Homing, survival, differentiation and therapeutic effect of intravenously injected iPSC in paraquat-injured lungs. (A) Representative fluorescence images of lungs harvested 1, 3, 7, or 28 days after paraquat injection along with PBS or iPSC administration. iPSCs were labeled with the red fluorescent dye PKH26 before injection. The slides were counterstained with DAPI. (B) The relationship between iPSC number and abundance of PCR product from the Tet-on gene, which was embedded in the mouse iPSCs (left half), and abundance of PCR product from the Tet-on gene isolated from paraquat-injured lungs of syngeneic iPSC-treated mice 1, 3, 7, 28, or 90 days after treatment (right half). The image was captured with the best exposure for showing the relationship between iPSC number and abundance of PCR product. (C) Statistical results of iPSC number in paraquat-induced lungs based on abundance of PCR product from the Tet-on gene. n =3 per time point. Data are represented as the mean ± SEM. (D) Abundance of PCR product from the Tet-on gene isolated from heart, liver, spleen, lung and kidney of paraquat-injected mice receiving syngeneic iPSCs 3 and 28 days before sample harvesting. (E) Detectable rate of Tet-on gene in major organs shown in paraquat-injected mice receiving syngeneic iPSCs. (F) Co-localization of injected cells and type 2 pneumocytes. Red: injected cells. Green: type 2 pneumocytes. Yellow: red and green fluorescence signals co-localized. (G) Intravenously injected syngeneic iPSCs improved pulmonary function of paraquat-injured lungs as shown by inspiratory and expiratory resistances and dynamic pulmonary compliance.
To observe if the intravenously administered iPSCs were therapeutic, syngeneic mouse iPSCs with a bolus dose of 5×106/kg body weight (~1×105 per mouse) were injected 4 h after intraperitoneal injection of paraquat. Paraquat decreased dynamic pulmonary compliance, total lung resistance, and expiratory lung resistance significantly at 1 and 3 days after iPSC treatment (Figure 3G).
Intravenously administered iPSCs did not differentiate into unwanted long-lasting cells or survive as quiescent stem cells
STEM 121 is a cytoplasmic protein expressed in a variety of tissues. STEM 121 staining detects the migration, engraftment, and differentiation of human cells transplanted into rodents. To separate host cells from differentiated iPSC cells, immunohistochemistry of STEM 121 was performed in multiple tissues of immune-deficient SCID-NOD mice 180 days after receiving human iPSCs via tail vein injection of a bolus dose of 2.5×108/kg. The results showed no positive staining for STEM 121 in tissues including heart, lung, spleen, kidney, and liver (Figure 4A). In contrast, numerous cells in the kidneys that received intra-kidney injection of 1×106 iPSCs/kidney 3 days before harvesting were positive for STEM 121 (Figure 4A). The negative results for STEM 121 in tissues harvested 180 days after iPSC injection indicated that the injected iPSCs did not differentiate into long-lasting stromal cells such as neural and myocardial cells.
Intravenously administered iPSCs did not differentiate into unwanted long-lasting cells or survive as quiescent stem cells. (A) Heart, lung, kidney, and liver of immunodeficient mice were free of human STEM 121 180 days after intravenous iPSC injection (the first four panels on the upper left). In the meantime, many cells in kidneys that received an intra-kidney injection 3 days before harvesting were positive for STEM 121 (upper right corner panel). (B) Pluripotent marker SSEA-4 was absent in all tissues harvested 180 days after tail vein iPSC injection and kidneys harvested 3 days after receiving intra-kidney injections.
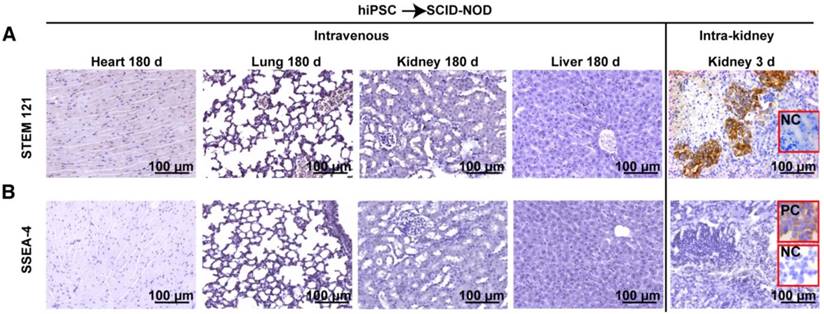
To determine if iPSCs survive without differentiating and remain quiescent in the tissue, immunohistochemistry for SSEA-4 (a pluripotent marker for embryonic cells or iPSCs) was performed. The result showed that both the tissues harvested 180 days after tail vein injection of iPSC and kidneys harvested 3 days after receiving intra-kidney injection were negative for SSEA-4 (Figure 4B).
Intravenously injected iPSCs did not form stem cell thromboses
As demonstrated by autopsy and histological examination, none of the animals in Table 2 had stem cell thrombosis (Figure 1, Figure 2A, Figure 3A, Figure 4 and Figure S3F).
Stem cell medium and dominant cell type are critical for iPSCs to form a cell mass
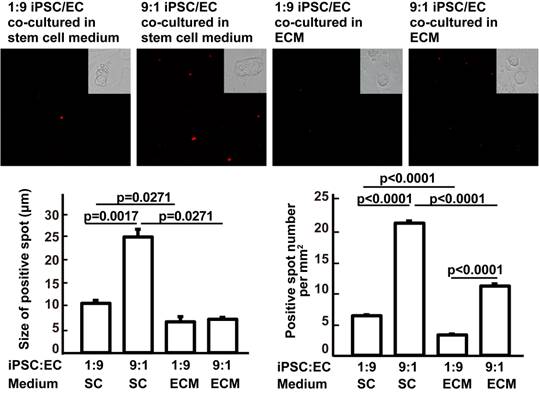
In vitro mechanistic data are not sufficient to support our hypothesis that it is possible to avoid teratoma formation during in vivo iPSC therapy. Therefore, we assessed the effects of the culture medium and of cocultured cells on iPSC mass formation to demonstrate the effect of the soluble microenvironment and adjacent cells. Compared with ESC medium, complete EC growth medium significantly decreased the ability of iPSCs to form a stem cell mass (three or more iPSCs clustered together). Compared with 9:1 iPSC:EC coculture, 1:9 iPSC/EC coculture significantly inhibited the ability of iPSCs to form a stem cell mass in ESC medium. At an iPSC:EC ratio of 1:9 in complete EC growth medium, no cell mass was found (Figure 5). These data further supported the idea that iPSC congregation is critical for teratoma formation, while teratoma formation can be avoided by intravenous injection, which does not favor iPSC aggregation.
Effects of culture media and iPSC/EC ratio on the formation of iPSC masses. Upper panels: red fluorophore-labeled iPSCs and their derivatives visualized under a fluorescence microscope. Insets are bright-field images of the cells. Lower left: size of the iPSC masses. Lower right: number of red fluorescent spots per mm2. Data are represented as mean ± SEM.
Discussion
Currently, the most acceptable strategy for in vivo iPSC application is to differentiate iPSCs into downstream specific cell types of interest before injection. However, this strategy has limitations, including limited cell functionality and mechanical injury. Cell functionality may be hampered by improper structural, orientational, and dimensional integration with the host structure and cells. For example, there are more than 10 different types of cells in the lungs, kidneys, liver, and digestive systems; therefore, integrating the injected cells with these native cells is challenging. Furthermore, like mature parenchymal cells, differentiated cells have lost trans-endothelial migration capacity, and therefore are ineffective when administered intravascularly. Tissue patches engineered from iPSC-derived cells provide a good alternative for treating large tissue defects. We previously produced functionally engineered human cardiac patches using human iPSC-derived cardiac cells and decellularized natural heart extracellular matrix as scaffolds. The patches exhibited normal contractile and electrical physiology in vitro. When patched on the infarcted area, they improved heart function in rats with acute myocardial infarction [12]. However, implantation of a tissue patch is actually tissue transplantation instead of tissue repair, which is needed for most diseases. In addition, because the migratory distance of non-inflammatory and immune cells is limited, the local administration of iPSC-derived cells requires multiple injections. This is feasible in small animals, such as mice. However, significant mechanical injury caused by the needle itself as well as surgical exposure of major organs may occur in patients. Therefore, finding alternative strategies is necessary. A promising finding in our in vivo therapeutic study using tissue patches and intra-parenchymal injection of iPSC-derived cells, along with the results obtained by other researchers, was that no teratoma was observed [36]. However, to clear the safety concern of in vivo application of ES- or iPSC-differentiated cells, fundamental study of teratoma formation upon in vivo application of iPSCs or iPSC-differentiated cells is needed.
To ensure immune compatibility between injected iPSCs and recipient mice in evaluation of teratoma formation, two different syngeneic iPSC lines were established for C57BL/6 recipient mice by two different researchers and from mice from different companies. Considering the fact that numerous clones of mouse antibodies against green fluorescent protein (GFP) and luciferase are commercially available, reporter genes such as GFP and luciferase were not used to label the injected cells because they are foreign genes for mice and may induce immune response. Further, it was critical to confirm that the iPSC lines we used in the study had full capacity to generate teratomas in conventional assays. Our results showed that when injected subcutaneously, teratoma generation efficiency of all human, mouse, and monkey iPSCs was 80-100% in the presence of Matrigel in immune-deficient mice. The capacity of syngeneic mouse iPSCs to form teratoma was also confirmed by injecting them into the kidney capsule and peritoneal cavity in the presence of Matrigel. At the highest dose tested, the syngeneic mouse iPSCs formed teratoma after subcutaneous or intra-kidney injection in the absence of Matrigel. Furthermore, individualized autologous monkey iPSCs formed teratoma at the sites of subcutaneous injections. These results indicated that all our iPSCs had full teratoma-forming capacity when evaluated by conventional assays. Therefore, the data on intravenously and topically administered iPSCs are reliable.
To test the relationship between teratoma formation and the number of cells injected, syngeneic mouse iPSCs or autologous monkey iPSCs were mixed with Matrigel and injected subcutaneously in different numbers in adjacent spots. At a dose of 1×105 cells/spot, neither syngeneic mouse iPSCs nor autologous monkey iPSCs formed teratoma. At a dose of 5×106 cells/spot for syngeneic mouse iPSCs or for autologous monkey iPSCs, teratoma was formed.
Teratoma generation by intravenously injected iPSCs needs to be examined in healthy and diseased mice. In healthy mice, mouse iPSCs did not generate teratoma in syngeneic mice of both sexes even with a dose unlikely to be achieved in humans. None of the intravenously injected human, monkey, or mouse iPSCs formed teratomas in immunodeficient mice. Trafficking to the site of inflammation is one of the most important features of iPSCs. Because the in vivo cellular microenvironment differs among tissues, organs, and diseases, we examined teratoma formation in multiple disease conditions involving the brain, lungs, liver, kidneys, islets, skin, hindlimb (muscle), and arteries. Teratomas were not identified in a single animal intravenously injected with syngeneic mouse iPSCs. For intravenously delivered cells to function properly, the cells should adhere to and migrate across the vascular wall to arrive at the diseased tissue. Lesion size differs from disease to disease, and thus, some diseases may require a larger number of iPSCs to achieve optimal therapeutic effect. Because cell adhesion speed and capability are higher than cell trans-endothelial migration speed and capability, multiple small-dose injections at appropriate intervals may be necessary to achieve iPSC adhesion and extravasation balance to prevent overdosed iPSCs from clogging in the microcirculation of the disease lesion.
In disease-/wound-free animals, targeted homing of iPSCs was not expected to occur. The injected cells would instead distribute unselectively across the body. Thus, the animals can tolerate more cells. In our study, intravenously disseminated iPSCs did not form teratomas with doses up to 2.5×108 iPSCs/kg in mice, a dose unlikely to be achieved in humans. For injured organs with large vascular beds, such as the lungs, large doses of iPSCs may also be tolerable. In this study, we found intravenously injected iPSCs did not generate teratomas in mice with lung injuries caused by intratracheally administered LPS, even with a dose as high as 2.5×108 iPSCs/kg body weight. However, to achieve a therapeutic effect, a dose as low as 5×106 iPSCs/kg is sufficient in many cases. If necessary, multiple administrations with appropriate intervals can be applied. This dose is safe because a mouse with a body weight of 20 g needs only 1×105 iPSCs. When iPSCs were suspended in PBS without Matrigel, the mice receiving a subcutaneous injection of 1×105 iPSCs/spot were free of teratomas. Furthermore, no teratomas were found in mice 180 days after receiving iPSCs (5×106 iPSCs/kg) for 3 consecutive days.
Many diseases are chronic and progressive. Long-term repeated iPSC administration might be required to repair continuously injured tissues. We demonstrated that 20 intravenous administrations (once every 3 days for 60 days) of syngeneic iPSCs did not generate teratomas in atherosclerotic mice, suggesting that direct intravenous administration of iPSCs is feasible.
Immune staining for human STEM 121 in multiple tissues of immunodeficient mice 180 days after receiving human iPSCs via tail vein injection was performed. STEM 121 is a cytoplasmic protein expressed in a variety of tissues. STEM 121 staining detects the migration, engraftment, and differentiation of human cells transplanted into rodents [37]. The results showed no positive staining for STEM 121 in tissues including heart, lung, spleen, kidney, and liver. The negative results indicated that the injected iPSCs did not differentiate into undesirable long-lasting stromal cells such as neural and myocardial cells. Unwanted somatic cells that are replaced by the body periodically, such as type 2 pneumocytes, are not as worrisome because they will disappear after their natural life span. Our result of tracking embedded Tet-on genes confirmed that type 2 pneumocytes that differentiated from injected syngeneic iPSCs were observed on day 28, but not on day 90 after injection. In contrast, many cells in the kidney that received intra-kidney iPSC injections 3 days before harvesting were positive for STEM 121.
In the perspective of long term safety, it is important to ensure the injected iPSCs do not survive as quiescent stem cells. SSEA-4 is a pluripotent marker for embryonic cells or iPSCs. Immune staining showed all tissue including tissues harvested 180 days after intravenous iPSC injection and kidneys harvested 3 days after receiving intra-kidney PSC injection were negative for SSEA-4. The result showed that the injected iPSCs lost their pluripotent capability in vivo within 3 days.
It is important to determine if the intravenously injected iPSCs that selectively trafficked into and survived in injured tissue differentiated into somatic cells and had therapeutic effects. Using a focal lung injury model induced by intratracheal instillation of LPS, we found significantly more iPSCs in lungs than in other organs. This difference was not seen in injury-/disease-free mice. A lung injury model caused by systemic inflammation was achieved by intraperitoneal injection of paraquat. Paraquat-activated neutrophils were trapped in lungs and they caused significant lung injury. Intravenously administered syngeneic iPSCs homed to the lungs, and some of the cells differentiated into type 2 pneumocytes surviving for at least 28 days and significantly improving lung functions. In contrast, the injected cells were not detectable in other organs 3 days after injection.
Skin wounds, including burn, diabetic, traumatic, and surgical wounds, are one of the most common medical conditions. They are often difficult to heal because of loss of large numbers of epidermal cells, angiogenesis impairment, and infection [38]. Although stem cell-derived epidermal cells or ECs may be effective, iPSCs may better fulfill the multiple cellular and paracrine needs for wound repair. Moreover, skin wounds are exposed and easily accessible for wound surface smearing and repetitive deliveries of cells. More importantly, unwanted tissue in skin wound, if any, can easily be detected early and removed. Together with the fact that teratoma is benign and not metastatic, difficult-to-heal skin wounds may be the condition of choice for testing the therapeutic values of iPSCs in clinical trials in patients. As expected, unlike subcutaneous injection, topically smeared mouse iPSCs tested in syngeneic mice or autologous monkey iPSCs tested in monkeys did not form teratoma in skin wounds.
Body cavities are not only a route to deliver therapeutic cells but also provide a perfect setting to demonstrate our hypothesis that congregation and being the dominant cell type in the microenvironment are required for teratoma formation. iPSCs suspended in PBS were free to move in the body cavity while iPSCs delivered with Matrigel were confined in the gel matrix. It was found that iPSCs suspended in Matrigel could easily form teratoma under the renal capsule [9]. Our study demonstrated that PBS-suspended syngeneic iPSCs delivered into the pleural cavity, peritoneal cavity, or renal capsule, did not form teratoma. In contrast, all the mice that had received Matrigel-suspended syngeneic iPSCs in the peritoneal cavity or renal capsule had teratoma. Unexpectedly, no teratoma was found when syngeneic iPSCs were injected into the pleural cavity in 180 days, even in the presence of Matrigel. We believe that the vigorous and continuous movement of the heart and lungs prevented the iPSCs from congregating.
Because it is practically impossible to confirm that each single iPSC has differentiated into the wanted somatic cell type before in vivo transplantation, we examined whether iPSC-derived cells formed teratoma under the conventional teratoma generation condition with Matrigel. No teratoma was formed in syngeneic mice by the iPSC-derived cells of endothelial linage, while all iPSCs injected side-by-side with the iPSCs formed teratoma subcutaneously. Consistent with the results in vivo, in vitro experiments showed that stem cell medium and being the dominant cell type are required for iPSCs to form a cell mass.
Conclusion
This study demonstrated that teratoma generation is not a natural characteristic of iPSCs in vivo. Appropriate microenvironment, congregation, and dominant cell-type status are required for teratoma formation. Our results conceptually and practically demonstrated that teratoma is avoidable when iPSCs are administered appropriately.
Abbreviations
apoE-/-: apoE-deficient; DAPI: 4',6-diamidino-2-phenylindole; DMEM: Dulbecco's modified Eagle's medium; ESCs: embryonic stem cells; FBS: fetal bovine serum; [18F]FDG: 18F-fluorodeoxyglucose; GFP: green fluorescent protein; H&E: hematoxylin and eosin; i.p.: intraperitoneal; iPSCs: induced pluripotent stem cells; LPS: lipopolysaccharide; MEFs: embryonic fibroblasts; MOIs: multiplicities of infection; MSCs: mesenchymal stem cells; PET/CT: positron emission tomography/computed tomography.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
We thank Dr. Jianyin Zhang at the Center for Biomedical Imaging, Fudan University for performing PET/CT imaging. This study was supported by Great Research Plan Program (91539120 to S. Chen), International Cooperation and Exchanges (81220108002 to S. Chen) and General Program (81470260 to M. Xiang) of the National Natural Science Foundation of China, and the National Key R&D Program of China (2016YFC1305101 to S. Chen).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Howden SE, Thomson JA, Little MH. Simultaneous reprogramming and gene editing of human fibroblasts. Nat Protoc. 2018;13:875-98
2. Chari S, Mao S. Timeline: iPSCs—The First Decade. Cell. 2016;164:580
3. Robinton DA, Daley GQ. The promise of induced pluripotent stem cells in research and therapy. Nature. 2012;481:295-305
4. Sun N, Yazawa M, Liu J, Han L, Sanchez-Freire V, Abilez OJ. et al. Patient-specific induced pluripotent stem cells as a model for familial dilated cardiomyopathy. Sci Transl Med. 2012;4:130ra47
5. Jacob A, Morley M, Hawkins F, McCauley KB, Jean JC, Heins H. et al. Differentiation of Human Pluripotent Stem Cells into Functional Lung Alveolar Epithelial Cells. Cell Stem Cell. 2017;21(e10):472-88
6. Apostolou E, Hochedlinger K. Stem cells: iPS cells under attack. Nature. 2011;474:165-6
7. Zhao T, Zhang ZN, Rong Z, Xu Y. Immunogenicity of induced pluripotent stem cells. Nature. 2011;474:212-5
8. Zhang WY, de Almeida PE, Wu JC. Teratoma formation: A tool for monitoring pluripotency in stem cell research. StemBook. Internet. Cambridge (MA): Harvard Stem Cell Institute. 2012
9. Prokhorova TA, Harkness LM, Frandsen U, Ditzel N, Schroder HD, Burns JS. et al. Teratoma formation by human embryonic stem cells is site dependent and enhanced by the presence of Matrigel. Stem Cells Dev. 2009;18:47-54
10. Miura K, Okada Y, Aoi T, Okada A, Takahashi K, Okita K. et al. Variation in the safety of induced pluripotent stem cell lines. Nat Biotechnol. 2009;27:743-5
11. Ye L, Chang YH, Xiong Q, Zhang P, Zhang L, Somasundaram P. et al. Cardiac repair in a porcine model of acute myocardial infarction with human induced pluripotent stem cell-derived cardiovascular cells. Cell Stem Cell. 2014;15:750-61
12. Wang Q, Yang H, Bai A, Jiang W, Li X, Wang X. et al. Functional engineered human cardiac patches prepared from nature's platform improve heart function after acute myocardial infarction. Biomaterials. 2016;105:52-65
13. Pei Y, Yue L, Zhang W, Xiang J, Ma Z, Han J. Murine pluripotent stem cells that escape differentiation inside teratomas maintain pluripotency. PeerJ. 2018;6:e4177
14. Oh J, Lee KI, Kim HT, You Y, Yoon DH, Song KY. et al. Human-induced pluripotent stem cells generated from intervertebral disc cells improve neurologic functions in spinal cord injury. Stem Cell Res Ther. 2015;6:125
15. Kobayashi Y, Okada Y, Itakura G, Iwai H, Nishimura S, Yasuda A. et al. Pre-evaluated safe human iPSC-derived neural stem cells promote functional recovery after spinal cord injury in common marmoset without tumorigenicity. PLoS One. 2012;7:e52787
16. Chen Q, Shou P, Zheng C, Jiang M, Cao G, Yang Q. et al. Fate decision of mesenchymal stem cells: adipocytes or osteoblasts? Cell Death Differ. 2016;23:1128-39
17. Lorenz C, Lesimple P, Bukowiecki R, Zink A, Inak G, Mlody B. et al. Human iPSC-Derived Neural Progenitors Are an Effective Drug Discovery Model for Neurological mtDNA Disorders. Cell Stem Cell. 2017;20(e9):659-74
18. Kunter U, Rong S, Boor P, Eitner F, Muller-Newen G, Djuric Z. et al. Mesenchymal stem cells prevent progressive experimental renal failure but maldifferentiate into glomerular adipocytes. J Am Soc Nephrol. 2007;18:1754-64
19. Amariglio N, Hirshberg A, Scheithauer BW, Cohen Y, Loewenthal R, Trakhtenbrot L. et al. Donor-derived brain tumor following neural stem cell transplantation in an ataxia telangiectasia patient. PLoS Med. 2009;6:e1000029
20. Lim SS, Vos T, Flaxman AD, Danaei G, Shibuya K, Adair-Rohani H. et al. A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990-2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380:2224-60
21. Levi B, Hyun JS, Montoro DT, Lo DD, Chan CK, Hu S. et al. In vivo directed differentiation of pluripotent stem cells for skeletal regeneration. Proc Natl Acad Sci U S A. 2012;109:20379-84
22. Liu Z, Tang Y, Lu S, Zhou J, Du Z, Duan C. et al. The tumourigenicity of iPS cells and their differentiated derivates. J Cell Mol Med. 2013;17:782-91
23. Lee AS, Tang C, Cao F, Xie X, van der Bogt K, Hwang A. et al. Effects of cell number on teratoma formation by human embryonic stem cells. Cell Cycle. 2009;8:2608-12
24. Gropp M, Shilo V, Vainer G, Gov M, Gil Y, Khaner H. et al. Standardization of the teratoma assay for analysis of pluripotency of human ES cells and biosafety of their differentiated progeny. PLoS One. 2012;7:e45532
25. Itakura G, Kawabata S, Ando M, Nishiyama Y, Sugai K, Ozaki M. et al. Fail-Safe System against Potential Tumorigenicity after Transplantation of iPSC Derivatives. Stem Cell Reports. 2017;8:673-84
26. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663-76
27. Cao S, Yu S, Li D, Ye J, Yang X, Li C. et al. Chromatin Accessibility Dynamics during Chemical Induction of Pluripotency. Cell Stem Cell. 2018;22(e5):529-42
28. Hou P, Li Y, Zhang X, Liu C, Guan J, Li H. et al. Pluripotent stem cells induced from mouse somatic cells by small-molecule compounds. Science. 2013;341:651-4
29. Sun N, Panetta NJ, Gupta DM, Wilson KD, Lee A, Jia F. et al. Feeder-free derivation of induced pluripotent stem cells from adult human adipose stem cells. Proc Natl Acad Sci U S A. 2009;106:15720-5
30. Nelakanti RV, Kooreman NG, Wu JC. Teratoma formation: a tool for monitoring pluripotency in stem cell research. Curr Protoc Stem Cell Biol. 2015;32(4A 8):1-17
31. Chen S, Kapturczak M, Loiler SA, Zolotukhin S, Glushakova OY, Madsen KM. et al. Efficient transduction of vascular endothelial cells with recombinant adeno-associated virus serotype 1 and 5 vectors. Hum Gene Ther. 2005;16:235-47
32. Yamasaki K, Edington HD, McClosky C, Tzeng E, Lizonova A, Kovesdi I. et al. Reversal of impaired wound repair in iNOS-deficient mice by topical adenoviral-mediated iNOS gene transfer. J Clin Invest. 1998;101:967-71
33. Curtis LM, Chen S, Chen B, Agarwal A, Klug CA, Sanders PW. Contribution of intrarenal cells to cellular repair after acute kidney injury: subcapsular implantation technique. Am J Physiol Renal Physiol. 2008;295:F310-4
34. Zhang J, Zou F, Tang J, Zhang Q, Gong Y, Wang Q. et al. Cyclooxygenase-2-derived prostaglandin E(2) promotes injury-induced vascular neointimal hyperplasia through the E-prostanoid 3 receptor. Circ Res. 2013;113:104-14
35. Le LL, Li XY, Meng D, Liang QJ, Wang XH, Li N. et al. Heme oxygenase-1 mediated memorial and revivable protective effect of ischemic preconditioning on brain injury. CNS Neurosci Ther. 2013;19:963-8
36. Zhu W, Zhao M, Mattapally S, Chen S, Zhang J. CCND2 Overexpression Enhances the Regenerative Potency of Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes: Remuscularization of Injured Ventricle. Circ Res. 2018;122:88-96
37. Sugai K, Fukuzawa R, Shofuda T, Fukusumi H, Kawabata S, Nishiyama Y. et al. Pathological classification of human iPSC-derived neural stem/progenitor cells towards safety assessment of transplantation therapy for CNS diseases. Mol Brain. 2016;9:85
38. Armstrong DG, Boulton AJM, Bus SA. Diabetic Foot Ulcers and Their Recurrence. N Engl J Med. 2017;376:2367-75
Author contact
![]() Corresponding author: Sifeng Chen, MD, Department of Physiology & Pathophysiology, School of Basic Medical Sciences, Fudan University, Shanghai 200032, P.R. China. Tel: 86-021-54237623; Fax: 86-021-54237623; chen1216edu.cn.
Corresponding author: Sifeng Chen, MD, Department of Physiology & Pathophysiology, School of Basic Medical Sciences, Fudan University, Shanghai 200032, P.R. China. Tel: 86-021-54237623; Fax: 86-021-54237623; chen1216edu.cn.