Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Methods
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(1):196-209. doi:10.7150/thno.27550 This issue Cite
Research Paper
Tumor-derived extracellular vesicles inhibit osteogenesis and exacerbate myeloma bone disease
Liming Zhang1,4*, Qian Lei1,2*, Hongxiang Wang3*, Cuiwei Xu1,5*, Teng Liu2*, Fancong Kong1,6*, Cui Yang1,7*, Guoxin Yan1, Li Sun1, Aiqi Zhao1, Wenlan Chen1, Yu Hu1, Hui Xie1, Yulin Cao1, Fenfen Fu1, Guolin Yuan8, Zhichao Chen1, An-Yuan Guo2 ![]() , Qiubai Li1
, Qiubai Li1 ![]()
1. Institute of Hematology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430022, China.
2. Department of Bioinformatics and Systems Biology, Key Laboratory of Molecular Biophysics of the Ministry of Education, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, China.
3. Department of Hematology, Wuhan Central Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China.
4. Department of Hematology, JingZhou Central Hospital, Tongji Medical College, Huazhong University of Science and Technology, Jingzhou, China.
5. Zhuhai People's Hospital of Jinan University, Zhuhai, China.
6. Department of Hematology, First Affiliated Hospital of Nanchang University, Nanchang, China.
7. Department of Hematology, Affiliated Hospital of Xinxiang Medical College, Xinxiang, China.
8. Department of Hematology, Xiangyang Central Hospital, the Affiliated Hospital of Hubei University of Arts and Science, Xiangyang 441021, China.
*These authors contributed equally to this study.
Received 2018-5-29; Accepted 2018-11-6; Published 2019-1-1
Abstract

Background: As a hallmark driver of multiple myeloma (MM), MM bone disease (MBD) is unique in that it is characterized by severely impaired osteoblast activity resulting from blocked osteogenesis in bone marrow-derived mesenchymal stem cells (BM-MSCs). The mechanisms underlying this preferential blockade are incompletely understood.
Methods: miRNA expression of MM cell-derived extracellular vesicles (MM-EVs) was detected by RNA sequencing. MM-EVs impaired osteogenesis and exacerbated MBD were in vitro and in vivo validated by histochemical staining, qPCR and micro-CT. We additionally examined the correlation between CD138+ circulating EVs (cirEVs) count and bone lesion in de novo MM patients.
Results: Here, by sequencing and bioinformatics analysis, we found that MM-EVs were enriched in various molecules negatively regulating osteogenesis. We experimentally verified that MM-EVs inhibited BM-MSC osteogenesis, induced elevated expression of miR-103a-3p inhibiting osteogenesis in BM-MSCs, and increased cell viability and interleukin-6 secretion in MM cells. In a mouse model, MM-EVs that were injected into the marrow space of the left tibia led to impaired osteogenesis and exacerbated MBD and MM progression. Furthermore, the levels of CD138+ cirEVs in the peripheral blood were positively correlated with the number of MM bone lesions in MM patients.
Conclusions: These findings suggest that MM-EVs play a pivotal role in the development of severely impaired osteoblast activity, which represents a novel biomarker for the precise diagnosis of MBD and a compelling rationale for exploring MM-EVs as a therapeutic target.
Keywords: multiple myeloma, extracellular vesicles, mesenchymal stem cells, osteogenesis, bone lesions
Introduction
Multiple myeloma (MM) is the second most frequent hematologic malignancy and is characterized by malignant plasma cells that intractably grow in the bone marrow (BM) and produce osteolytic bone lesions in more than 80% of patients with MM [1-3]. As a hallmark, myeloma bone disease (MBD) eventually leads to pathological fracturing in approximately 20% of patients at the time of diagnosis and in 85% of patients over the course of the disease [4]. Despite the use of bisphosphonates and bortezomib, the mainstay treatments for MBD, bone fractures and osteoporosis continue to persist, and the devastating skeletal complications associated with this disease are a significant cause of morbidity and mortality in MM patients [4-7].
Although osteolytic metastases from MM and other tumors induce bone resorption by osteoclasts (OC), MBD is unique in that osteoblast (OB) activity is severely impaired [8]. Because of this defect and failed self-repair functions, MBD may never heal, even in times of disease quiescence. The impairment results primarily from blocked osteogenesis in BM-derived mesenchymal stem cells (BM-MSCs) [4, 9, 10], and the undifferentiated BM-MSCs provide a much richer repertoire of cytokines that contribute to the intractable growth of MM cells [8, 11, 12]. MM cells develop via forward-feedback mechanisms with local BM-MSCs, and the cycle of interactions between MM cells and BM-MSCs contributes to the incurability of MBD and myeloma [1, 10, 12, 13].
Although both soluble factors and cell-to-cell contact between MM cells and BM-MSCs or OB progenitors have been shown to be involved in the suppression of OB differentiation in MM, the mechanisms underlying these interactions remain elusive [4, 6, 7]. Recently, a growing body of evidence has shown that extracellular vesicles (EVs) are a previously unappreciated mechanism for intercellular communication, and cancer cells can release EVs that contain a variety of molecules, such as proteins, RNAs and microRNAs (miRNAs), to influence the microenvironment [14-20]. In this study, we studied bioactive content in EVs derived from MM cells (MM-EVs) and examined the effects of MM-EVs on osteogenesis in BM-MSCs in vitro and on MBD and disease progression in vivo. We additionally identified the correlation between peripheral blood CD138+ circulating EV (cirEVs) counts and bone lesions in de novo MM patients (D-MM).
Results
MM-EVs are selectively enriched in a variety of molecules related to the regulation of MBD
MM-EVs obtained from human MM cell lines (HMCLs) were isolated and validated according to previously described methods [21, 22]. These EVs were shown to be spherical (Figure 1A and Figure S1A) with a size distribution ranging from 200 nm to 1000 nm (Figure 1B). MM-EVs were CD138-, Annexin-V- and PKH-26-positive (Figure 1C and Figure S1B-C), and validated by their expression of EV markers (Alix, TSG101, CD63, MFGE8 or CD9) (Figure 1D). Flow cytometry (FCM) analysis confirmed that calcein+CD138+ cirEVs were present both in the peripheral blood and bone marrow of MM patients (Figure 1E-F and Figure S1D), and the cirEVs were also shown to be spherical (Figure 1G).
Characteristics of the MM-EVs. (A) Transmission electron microscopy image revealing EVs (arrows) as 100-1000 nm vesicles shed from RPMI8226 cells (R-EVs). Scale bar, 500 nm (× 25600). (B) TRPS analysis of the size distribution of R-EVs and U266-EVs. (C) R-EVs were positive for Annexin-V and CD138. (D) Western blot of Alix, TSG101, CD63, MFGE8, CD9 and β-actin contained in RPMI8226, U266, R-EVs and U266-EVs (U-EVs). (E) Representative FACS analysis of PB cirEVs obtained from a de novo MM (D-MM) patient. (F) Comparison of PB and BM CD138+ cirEVs in D-MM patients (n = 61). n.s. Signifies P > 0.05. (G) Representative transmission electron micrograph of PB cirEVs obtained from a D-MM patient. Scale bar, 500 nm. (H) Distribution of expression levels for mRNAs, lncRNAs and miRNAs with RPKM (RPM) > 1 in R-EVs. The scale shown for piRNA is the read count. (I) Comparative expression levels of miRNAs in B cell-EVs, R-EVs and K562-EVs. miRNAs marked by “#” are associated with both osteogenesis and proliferation. (J) ELISA analysis of DKK1, IL-7 and sFRP2 in MM cells and MM-EVs.
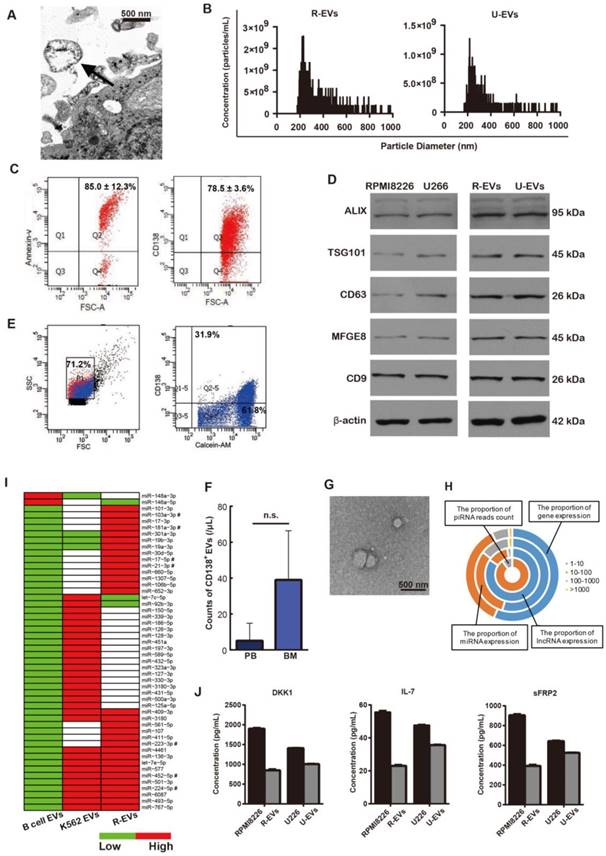
We next sequenced RNA molecules isolated from RPMI8226 cell-derived EVs (R-EVs) and K562 cell-derived EVs. We identified the expression of 7903 mRNAs in these EVs, 569 of which were highly expressed (RPKM > 100). In addition, 287 micro RNAs (miRNAs) and 676 long non-coding RNAs (lncRNAs) were expressed, and some PIWI-interacting RNAs (piRNAs) were supported by dozens of sequenced reads (Figure 1H). Because miRNAs are selectively excreted into EVs and act as core regulators [23], we further analyzed the miRNAs contained in the R-EVs and compared them to B cell-derived EVs from EVmiRNA (a database established by us) [24] and EVs derived from K562 leukemia cells (K562-EVs), which were described in our previous studies [25, 26]. There were 287 miRNAs expressed in the R-EVs and 176 miRNAs expressed in the K562-EVs. Then, we combined the miRNA expression profiles of R-EVs, K562-EVs and B cell-EVs, and identified specially highly expressed miRNAs (SHEMs) of these three kinds of EVs. We found that some miRNAs that regulate cell proliferation were highly expressed in both R-EVs and K562-EVs, such as miR-452-5p and miR-224-5p (Figure 1I). Interestingly, some miRNAs involved in OB or OC differentiation were only highly expressed in R-EVs, such as miR-103a-3p, miR-181a-3p and miR-21-3p (Figure 1I). These results suggest that R-EVs contain miRNAs that might play roles not only in cell proliferation as K562-EVs but also in MBD. We next found that the expression levels of genes in the osteogenesis-related WNT and BMP signaling pathways were very low, which were potentially regulated by 22 osteogenesis-related miRNAs (Figure S1E-F). Moreover, the proteins DKK1, IL-7 and sFRP2, which are key regulators of MBD [27], were assayed and shown to be selectively expressed by MM cells and MM-EVs (Figure 1J and Figure S1G). These results indicate that MM-EVs potentially play a supporting role in the progression of MBD and MM.
MM-EVs inhibit osteogenesis of BM-MSCs in vitro
Based on impaired BM-MSC osteogenic differentiation in MBD and the molecules found in R-EVs, we next sought to determine whether MM-EVs affect BM-MSC osteogenesis. Normal BM-MSCs were either purchased or obtained from healthy donors (n = 20; Figure S2A-B). BM-MSCs at passages 3-5 were treated with 80% osteogenic differentiation medium and 20% unfiltered MM cell medium or conditioned cell medium (EVs were removed by centrifugation and filtration). After 14 d, ALP staining showed that the application of either conditioned medium or unfiltered medium resulted in a significantly lower frequency of BM-MSC colony-forming unit fibroblasts (CFU-F) than the control group. Statistical significance for these relationships was reached for RPMI 8226 and U266 but not for KM3 medium. Thus, after day 21, Alizarin Red staining was additionally conducted, and we observed that the unfiltered medium had a more significant inhibitory effect than the conditioned medium on colony-forming unit osteoblasts (CFU-OB) in BM-MSCs (Figure 2A). These results suggest that the filtered EVs play a role in the osteo-inhibitory effect of the medium on BM-MSCs.
MM-EVs inhibit osteogenesis in BM-MSCs. (A) ALP and Alizarin Red staining images (left, × 40) and quantification (right) of BM-MSCs treated with unfiltered MM cell medium or conditioned medium after 14 and 21 d. Conditioned medium was obtained by centrifuging MM cell medium at 16,000 ×g for 60 min and then filtering using 0.22 μm filters. (B) Confocal microscopy revealing fusion of R-EVs (PKH26, red) and BM-MSCs (DAPI, blue). Arrows: R-EVs near the cell nuclei. Imaging was performed in the x-y plane by confocal microscopy (Nikon, A1Si) with a pinhole radius of 52.9 µm (× 200). (C) FACS analyses of CD138+ BM-MSCs cultured with MM-EVs for 24 h. Negative control: BM-MSCs alone and isotype control. (D-E) ALP staining (D) and Alizarin Red staining (E) images (left, × 40) and quantification (right) of BM-MSCs treated with 50 ng/mL MM-EVs and K562-EVs for 14 and 21 d. (F) Real-time PCR analysis of OB marker genes (RUNX2, ALP, osteocalcin and collagen I) in BM-MSCs treated with MM-EVs or K562-EVs (50 ng/mL) for 3 or 6 d. The results were normalized to β-actin. (G) ALP activity and secreted osteocalcin (OC) protein in the BM-MSCs treated with MM-EVs (50 ng/mL) during OB differentiation. All data are shown as the mean ± SD from at least three experiments. Experiments were confirmed in eight different donors (A, D-G) or three different donors (B-C). *** P < 0.001, ** P < 0.01, * P < 0.05, via one-way ANOVA.
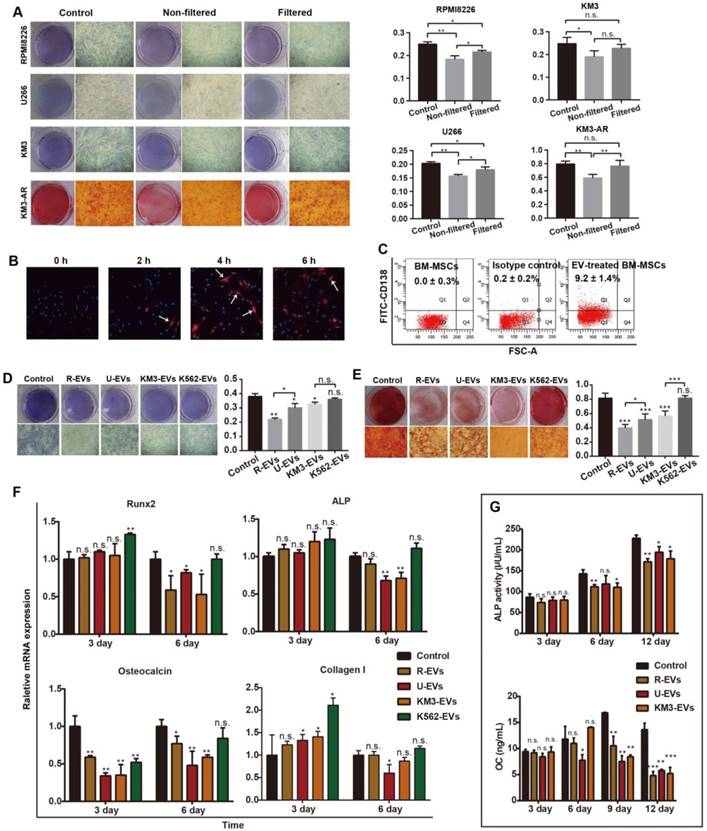
We next directly treated BM-MSCs with MM-EVs derived from RPMI8226 (R-EVs), U266 (U-EVs) or KM3 cells (K-EVs). After 6 h, a single dose of R-EVs (50 ng/mL) increased red PKH-26 staining in close proximity to the blue, DAPI-stained nuclei of BM-MSCs when viewed under a laser confocal microscope (Figure 2B). After 24 h, fluorescence-activated cell sorting (FACS) analysis confirmed the expression of CD138, a marker expressed by MM cells and MM-EVs but not BM-MSCs (Figure 2C), whereas BM-MSC activity and apoptosis were not affected (Figure S2C-D). However, Alizarin Red staining indicated that MM-EVs inhibited osteogenic differentiation of BM-MSCs in a dose-dependent manner (Figure S2E). Furthermore, there were significantly fewer BM-MSC CFU-F and CFU-OB on days 14 and 21 in the cells treated with MM-EVs than in the controls. R-EVs exerted the greatest inhibitory effect, and K562-EVs failed to induce an obvious effect (Figure 2D-E), which was consistent with the miRNA patterns we previously observed (Figure 1I).
We then tested for alterations in osteogenesis-related markers in BM-MSCs (Figure 2F). After 3 d, the expression of osteocalcin was significantly inhibited by all three types of MM-EVs (P < 0.001 vs. control). After 6 d, the expression levels of the four genes (RUNX2, ALP, osteocalcin and collagen I) were all significantly decreased by one or all types of MM-EVs. However, the K562-EVs enhanced the expression of RUNX-2 and ALP. Further analysis of the levels of secreted osteocalcin and ALP activity in the BM-MSCs treated with MM-EVs consistently showed that from day 6 onward, both were significantly decreased and that the strongest effects were observed for the R-EVs on day 12 (Figure 2G). Taken together, the results confirm that MM-EVs inhibit osteogenesis in BM-MSCs.
MM-EVs induce elevated expression of miR-103a-3p, inhibiting osteogenesis in BM-MSCs
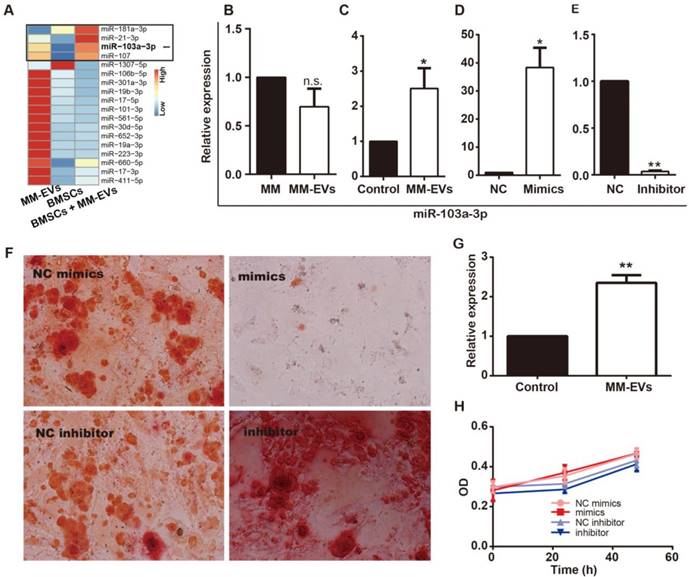
Because MM-EVs contain miRNAs that play an important role in regulating osteogenesis and cell proliferation, we examined whether MM-EVs induced miRNA expression changes in BM-MSCs by miRNA sequencing (Figure 3A). Among the most highly changed miRNAs in the BM-MSCs were miR-181a-3p, miR-21-3p, miR-103a-3p and miR-107, and miR-103a-3p is critical for osteogenesis [28, 29]. Then, through real-time PCR, the expression levels of miR-103a-3p in RPMI8226, R-EVs, BM-MSCs and BM-MSCs treated with R-EVs were examined, and R-EVs were verified to induce significantly higher expression of miR-103a-3p in BM-MSCs (Figure 3B-C). Next, miR-103a-3p expression in BM-MSCs was proved to be up-regulated after transfection with miR-103a-3p mimics, while it was down-regulated after transfection with miR-103a-3p inhibitor (Figure 3D-E). Upon their functional testing, miR-103a-3p mimics exhibited an inhibitory role on osteogenic differentiation in BM-MSCs, while inhibition of miR-103a-3p facilitated osteogenic differentiation in BM-MSCs (Figure 3F). These results suggest that miR-103a-3p may be involved in the osteo-inhibitory effects in BM-MSCs.
MM-EVs induce elevated expression of miR-103a-3p, inhibiting osteogenesis in BM-MSCs. (A) Expression heatmap of the 18 MM-EVs' specifically highly expressed miRNA (Figure 1I) in MM-EVs, BM-MSCs and BM-MSCs treated with MM-EVs. (B) Real-time PCR showing the expression of miR-103a-3p in MM cells (RPMI8226) and MM-EVs (R-EVs). (C) The expression of miR-103a-3p in BM-MSCs treated with MM-EVs for 24 h. (D) BM-MSCs were transfected with miR-103a-3p mimics and the miR-103a-3p level was increased by 38.3-fold in the mimics group compared with the NC group. (E) BM-MSCs were transfected with miR-103a-3p inhibitor and the miR-103a-3p level was significantly decreased in BM-MSCs. (F) Alizarin Red staining of BM-MSCs transfected with miR-103a-3p mimics or inhibitor after 21 d (× 200). (G) The expression of miR-103a-3p in MM cells treated with MM-EVs for 24 h. (H) MM cells were analyzed with CCK-8 assay after being transfected with miR-103a-3p mimics or inhibitor for 24 and 48 h. All data are shown as the mean ± SD from at least four independent experiments; ** P < 0.01, * P < 0.05 by two-tailed Student's t-test.
MM-EVs increase viability, interleukin-6, and expression of miR-103a-3p in MM cells
Based on previous findings [22] and the proliferation-related miRNAs in MM-EVs, we next investigated the effects of MM-EVs on their parental MM cells. We found PKH26-stained MM-EVs (Red) in close proximity to DAPI-stained nuclei (Blue) of MM cells (Figure S3A) after 6 h of treatment, and MM-EVs significantly enhanced the viability of their parental cells within 72 h in a dose-dependent manner (Figure S3B). Additionally, the expression of IL-6 was markedly higher in the cells treated with MM-EVs (Figure S3C). Next, we investigated the effects of miR-103a-3p on proliferation of MM cells and found that the level of miR-103-3p was significantly increased in RPMI8226 cells treated with R-EVs for 24 h (Figure 3G). Nonetheless, CCK-8 assay indicated the expression level of miR-103a-3p had no effect on MM cell proliferation (Figure 3H), suggesting that not miR-103a-3p but other factors, such as miR-181a-3p and miR-107, were involved.
MM-EVs impair osteogenesis and exacerbate MBD in a xenograft mouse model
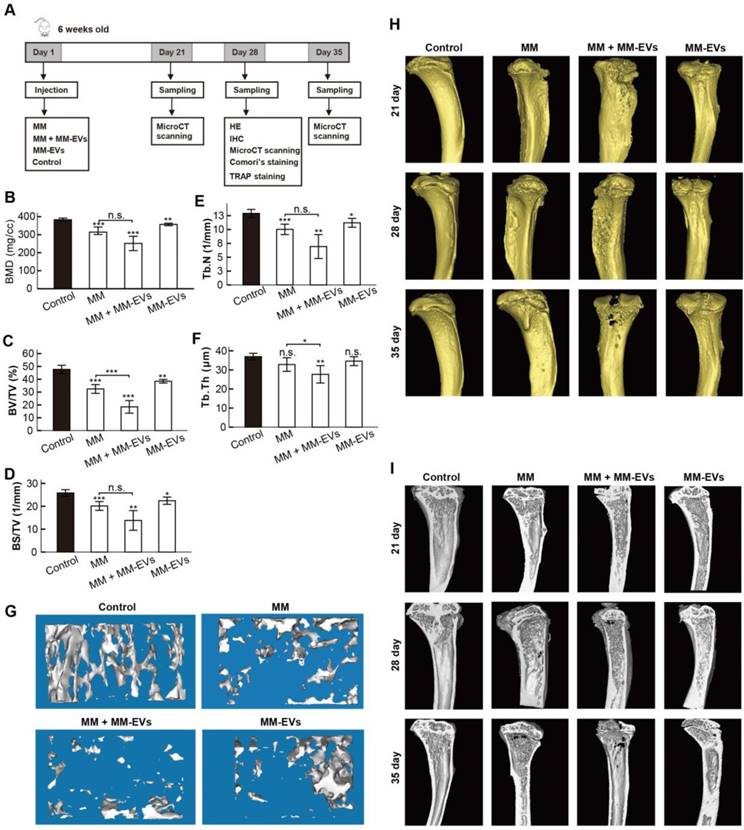
Our previous results prompted us to perform in vivo studies. Because R-EVs exerted stronger effects than other MM-EVs on BM-MSC osteogenesis and parental cell proliferation, we established a mouse model in which the tibia was injected directly with RPMI 8226 cells and/or R-EVs. RPMI 8226 cells (MM, n = 22), R-EVs (MM-EVs, n = 17), RPMI 8226 cells plus R-EVs (MM+MM-EVs, n = 22) and RPMI 1640 medium (control, n = 9) were injected, bone samples were taken (Figure 4A), and survival rate was analyzed. A micro-CT study demonstrated that BMD, BV/TV, BS/TV and Tb.N of diaphysis of the left tibia were significantly lower in the MM-EVs group and MM group than in the control group, and both BV/TV and Tb.Th were significantly lower in the MM+MM-EVs group than in the MM group. (Figures 4B-F). Representative 3D figures and section maps of the separated trabecular bones are shown in Figure 4G-I. In addition, H&E staining (Figure 5A) and anti-CD138 staining (Figure 5B) indicated more severe bone destruction and MM cell infiltration, and IHC for osteocalcin expression (Figure 5C) and tartrate-resistant acidic phosphatase staining (TRAP staining) (Figure 5D) indicated markedly fewer OBs and more OCs in the MM group and the MM+MM-EVs group than in the control group and MM-EVs group (Figure 5E). These results suggest that a single injection of R-EVs decreases the osteogenic effect in the tibia of model mice, which is consistent with our in vitro findings. These R-EVs can also enhanced MM cell activity, exacerbated MM cell-induced decreased osteogenic effect and substantially increased MM-induced osteoclastic cell differentiation in the tibia of the models.
Micro-CT analysis of tibiae collected. (A) A schematic diagram illustrating the experimental design. (B-F) Micro-CT bone quantitative analysis of BMD (B), BV/TV (C), BS/TV (D), Tb.N (E), and Tb.Th (F) in tibiae collected. (G) Representative 3D reconstructive images of separated trabecular bones. (H-I) Representative 3D reconstructive images (H) and section maps (I) of the collected tibiae. All data are shown as the mean ± SD. *** P< 0.001, ** P < 0.01, * P < 0.05 by one-way ANOVA followed by Sidak's multiple comparison test.
Decreased osteogenesis and exacerbated MBD in mice with MM-EVs. H&E (A), anti-CD138 (B), IHC (C), and TRAP (D) staining of sections of tibiae collected from the indicated groups of mice. Scale bars for (A), 200 μm (left) and 50 μm (right). Scale bars for (B-D), 20 μm. Osteocalcin is stained yellow in (C). (E) OB and OC numbers (left) and ratios (right). (F) Survival of the MM+MM-EVs group (n = 13) and the MM group (n = 13) compared by the log rank test. HE: H&E staining; IHC: immunohistochemistry; TRAP: tartrate resistant acid phosphatase. All data are shown as the mean ± SD. *** P< 0.001, ** P < 0.01, * P < 0.05 by one-way ANOVA followed by Sidak's multiple comparison test.
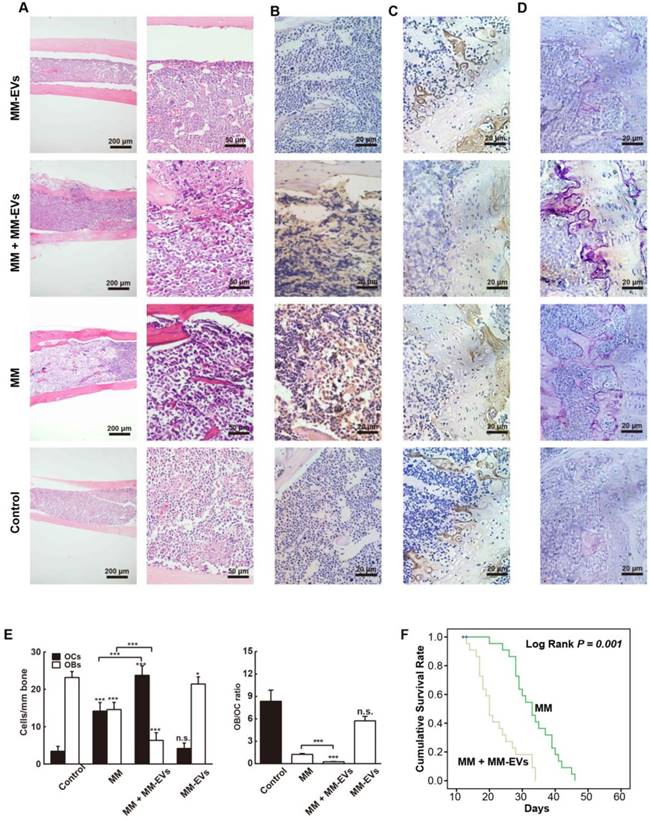
We further compared the survival rates between the MM group (n = 13) and the MM+MM-EVs group (n = 13); the log rank test showed that the latter had significantly lower survival rates than the former (log rank: P = 0.001) (Figure 5F). These findings give additional support to the aggravated role of R-EVs in disease progression of the models.
Peripheral blood CD138+ cirEV counts indicate bone lesions in D-MM patients
The clinical role of circulating tumor-derived EVs has been previously demonstrated [19, 30-33]. We next isolated cirEVs from EDTA-anticoagulated peripheral plasma obtained from D-MM patients to evaluate the associations of the level of these cirEVs with BMD and other clinical MM features. A cohort of 61 D-MM and 29 healthy donors (Table 1) was involved. We found that the CD138+ cirEV counts distinguished the D-MM patients from the normal donors (P = 0.000) with an ROC value of 0.734 (Figure 6A-B), which was in agreement with the result of a recent report [34]. Furthermore, we additionally found that the CD138+CD38+ cirEV counts were significantly higher in the D-MM patients (n = 40, Table S1) than in the healthy donors (Figures 6C-D, ROC value = 0.906).
Characteristics of the de novo patients and healthy donors used to analyze CD138+ cirEVs.
| Number or value | |
|---|---|
| De novo patients (n) | 61 |
| Sex (n) | |
| Male | 40 |
| Female | 21 |
| Age, years, median (range) | 59 (40-82) |
| Durie-Salmon stage (n = 52) | |
| 1 | 3 |
| 2 | 11 |
| 3 | 38 |
| International staging system (n = 58) | |
| 1 | 6 |
| 2 | 25 |
| 3 | 27 |
| Type of monoclonal Ig (n = 54) | |
| IgG | 27 |
| IgA | 16 |
| IgD | 3 |
| LC | 8 |
| Bone lesions (n = 56) | |
| None | 19 |
| ≤ 3 | 15 |
| > 3 | 22 |
| C-reactive protein (n = 49) | |
| 1 | 27 |
| 2 | 22 |
| β2-microglobulin, median (range), mg/L, n = 57 | 4.5 (1.2-20) |
| LDH, median (range), mM, n = 48 | 153 (56-463) |
| Calcium, median (range), mM, n = 56 | 2.21 (1.37-4.3) |
| Albumin, median (range), mg/L, n = 59 | 31.1 (17.2-44.8) |
| BMPC, median (range), %, n = 26 | 25 (2.5-91) |
| Relapsed patients (n) | 5 |
| Sex (n) | |
| Male | 3 |
| Female | 2 |
| Median age, years (range) | 60 (56-70) |
| Healthy donors (n) | 29 |
| Sex (n) | |
| Male | 18 |
| Female | 11 |
| Median age, years (range) | 52 (45-76) |
Peripheral CD138+ cirEV counts indicate bone lesions in D-MM patients. CD138+ cirEV counts in D-MM patients (n = 61) and healthy donors (n = 29) (A) and corresponding ROC curve (n = 56) (B) CD138+CD38+ cirEV counts in D-MM patients (n = 40) and healthy donors (n = 29) (C) and corresponding ROC curve (D). (E) The correlation between CD138+ cirEV count and bone lesion number in D-MM patients (n = 56). (F) The levels of PB CD138+ cirEVs among patients with 0 (n = 19), ≤ 3 (n = 15) or > 3 bone lesions (n = 22). CD138+ cirEV counts in D-MM patients with ≤ 3 lesions (n = 34) and those with > 3 lesions (n = 22) (G) and corresponding ROC curve (H). (I) Comparative analysis of CD138+CD38+ cirEV counts in D-MM patients with ≤ 3 lesions (n = 32) and those with > 3 lesions (n = 8). (J) Proposed MM-EV-mediated cycles between BM-MSCs and MM cells.
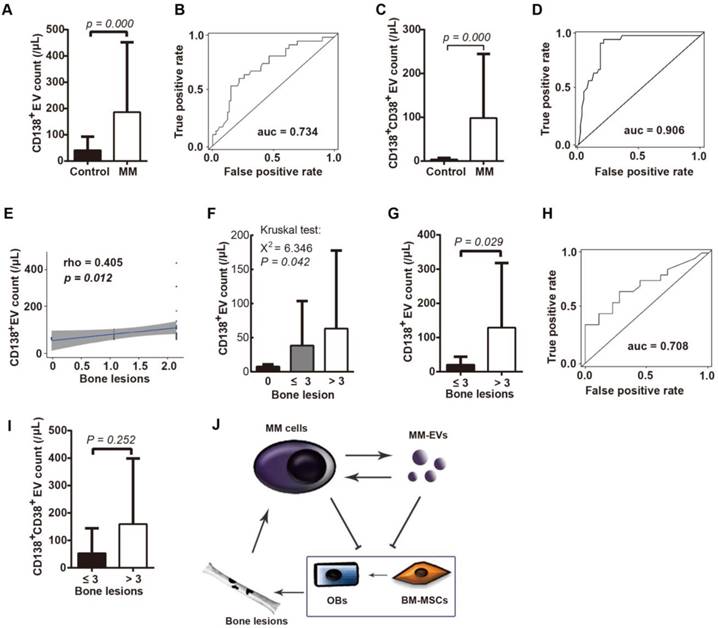
In BMD, we found that the CD138+ cirEV counts were correlated with the number of bone lesions (detected using MRI or PET-CT, n = 56) (Figure 6E) and that they were significantly different (P = 0.042) among the patients with 0 (n = 19), ≤ 3 (n = 15) and > 3 (n = 22) bone lesions (Figure 6F). Notably, the CD138+ cirEV levels were higher in patients with > 3 lesions (n = 22) than in patients with ≤ 3 lesions (n = 34, P = 0.029) (Figure 6G). The ROC value for using CD138+ cirEV counts to diagnose bone lesions was 0.708 (Figure 6H). However, no statistical difference was found between the two groups of patients (≤ 3 vs. > 3 lesions) when CD138+CD38+ cirEV counts were considered (Figure 6I). These results indicate that PB CD138+ cirEV counts are a potential diagnostic biomarker in MBD.
We also investigated the correlations between CD138+ or CD138+CD38+ cirEV counts (n = 40, Table S1) and other clinical features in D-MM, such as disease stage (Durie-Salmon system or ISS system), serum levels of lactate dehydrogenase (LDH), β2-microglobulin (β2-MG), c-reactive protein (CRP), calcium, albumin, the monoprotein subtypes of IgG, IgA, IgD and light chain, the percentage of bone marrow malignant plasma cells (BMPC%) and genetic lesions. The findings are shown in Figures S4-S6 and most of them are not significant. Moreover, based on phenotyping according to markers of MM side population (SP) or stem cells [35], we confirmed PB CD138-CD19+ and CD38+CD45- cirEVs to be present in the D-MM patients (n = 22, Table S2); correlations between the levels of each type of cirEV and the clinical issues are shown in Figures S7-S8.
Discussion
MBD is increasingly recognized as an intractable clinical problem in MM patients. Despite improved outcomes from novel drugs and regimens, patients continue to suffer from morbidity and mortality caused by MBD. Thus, there is an increasing need to understand the underlying mechanisms for more effective treatment of MBD. Consequently, understanding the interactions between tumor cells and other cells within the bone marrow microenvironment has a potential to uncover novel or more effective treatments. Though EVs derived from BM-MSCs or MM cells have been reported to play different roles in MM [21, 22, 34, 36-41], we have presented the first evidence showing that MM-EVs inhibit osteogenesis in BM-MSCs in vitro and exacerbate MBD in vivo, and PB CD138+ cirEV counts are of diagnostic significance in MBD of D-MM patients.
EVs are composed of a lipid bilayer containing transmembrane and cytosolic proteins and RNA. Generally, cells generate two subtypes of EVs according to their sub-cellular origin. Microvesicles (100-1,000 nm in diameter) are formed and released by budding from the plasma membrane, while exosomes (smaller than 150 nm) are derived from the endolysosomal pathway. In our study, the conditioned CM was prepared by filtration with a 0.22 μm filter to remove vesicles larger than 220 nm. The conditioned CM had a significantly weaker osteo-inhibitory effect on BM-MSCs than unfiltered CM, indicating the inhibitory effect is possibly caused by the larger vesicles not exosomes that are smaller than 150 nm. Of note, the conditioned CM still showed an inhibitory effect on the osteogenic differentiation of BM-MSCs, though it was markedly weaker than that of the CM of HCMLs. This could be explained by MM-exosomes, other important EVs already reported in MM bone disease [42]. In addition, though R-EVs had the most obvious inhibitory function on osteogenesis of BM-MSCs, EVs from these different and heterogeneous MM cell lines shared similar effects, reflecting the common characteristics and functions of these MM-EVs to affect osteogenesis in BM-MSCs. It is extremely important to isolate MM-EVs from PB or bone marrow of MM patients to verify these functions of MM-EVs from HMCLs; however, there are no available standards or reliable methods to successfully harvest enough MM-EVs from patients. This deserves further basic and clinical investigations.
The key roles of these miRNAs enriched in EVs that are specifically released by parental cells and transferred to target cells have also been demonstrated by many studies [43-47]. Interestingly, we observed that many osteogenesis-related miRNAs were highly expressed in MM-EVs compared to K562-EVs and B cell-EVs. miR-30 family members negatively regulated BMP-2-induced osteoblast differentiation by targeting Smad1 and Runx2 [48]. In our result, miR-30b/d/e-5p were all expressed at much higher levels (3-6-fold) in MM-MVs than in K562-MVs. miR-103 inhibits osteoblast proliferation under simulated microgravity condition through regulating Cav1.2 and functions as a mechanosensitive miRNA to inhibit bone formation through targeting Runx2 [28, 29]. miR-17-5p modulates osteoblastic differentiation and cell proliferation by targeting SMAD7 in non-traumatic osteonecrosis [49]. miR-126-5p and miR-21-5p regulate osteolysis formation or osteoclast differentiation by inhibiting different target genes [50-52]. Therefore, we further sequenced RNA of MM-EVs, BM-MSCs and BM-MSCs treated with MM-EVs. Based on the 18 SHEMs of MM-EVs, we found that miR-181a-3p, miR-21-3p, miR-103a-3p and miR-107 were upregulated in BM-MSCs after treatment with MM-EVs. Then, we focused on miR-103a-3p and found that this miRNA is one potential molecule that plays an important role in the osteo-inhibitory effect in BM-MSCs induced by MM-EVs (Figure 3). Additionally, we showed that MM-EVs express heterogeneous markers of MM cells. Thus, we propose that the inhibitory functions of MM-EVs on osteogenesis in BM-MSCs are most probably mediated through transfer of the bioactive cargoes, including selective miRNAs and/or membrane markers. It is difficult to determine the specific role played by a single factor out of the numerous molecules that are involved in the inhibitory functions of MM-EVs, which are likely to be mediated by a “cluster of molecules” that operates as a whole rather than by one or two specific molecules.
OBs are highly specific bone cells that are generated via the osteogenic differentiation of MSCs [8, 53]. Our data support the notion that MM-EVs are a potential pathway for MM-to-BM-MSC cell communication. Although the precise role of undifferentiated BM-MSCs in our model was not directly evaluated, we show that a single dose of MM-EVs can induce a dramatic increase in MM cell activity through a “vicious” cycle of MM-to-MM interactions. This cycle consequently results in the generation of many more undifferentiated BM-MSCs, severe bone lesions and MM progression. We propose that these two cycles, tightly linked through MM-EVs, substantially promote MBD and MM progression (Figure 6J). Further studies are needed to validate this hypothesis. Currently, there is an ongoing dilemma in the management of patients with smoldering MM (SMM), which is an intermediate clinically-defined stage associated with a much higher risk of progression to malignancy (~10% per year) than monoclonal gammopathy of undetermined significance (MGUS; ~1% per year) [54, 55]. Though some patients with SMM are premalignant and relatively safe, some have true malignancy in whom it is only a matter of time before end-organ damage occurs [54]. So, the use of specific biomarkers is urgently needed, and modern imagining tools such as positron emission tomography-computed tomography (PET-CT) have been used in the diagnostic criteria of the International Myeloma Working Group (IMWG). Thus, our findings support the level of PB circulating CD138+ cirEVs as a novel and precise biomarker to define MM and distinguish SMM patients with a high likelihood of true malignancy and to monitor disease progression of SMM to overt MM, which is worthy of further investigation. Meanwhile, with fewer invasions, this indicator will act as a novel “liquid biopsy” and be more easily accepted by patients.
Recently, Krishnan and colleagues [34] reported that the levels of CD138+ cirEVs were significantly higher in MM patients and were correlated with tumor burden. Here, we demonstrated the diagnostic potential of not only CD138+ but also CD138+CD38+ cirEV counts in MM. Importantly, we confirmed the diagnostic significance of CD138+ but not CD138+CD38+ cirEV counts in MBD. Notably, based on our data, these EVs are a driver, rather than a passenger, biomarker. Our findings also suggest that cirEVs may reflect the inherent heterogeneity of subgroups of MM cells, including MM stem cells, and provide researchers with a new window for observing and analyzing these cells.
Collectively, our findings reveal that MM-EVs play a pivotal role in the functional communications between MM cells and BM-MSCs, which results in the development of severely impaired OB activity. These results indicate that MM-EVs represent a previously undescribed factor as a driver biomarker for the precise diagnosis of MBD and a compelling rationale for exploring MM-EVs as a therapeutic target. We also provide researchers with novel insights regarding the importance of EVs in investigations of osteoporosis and bone disease caused by other tumors that originate from the breast or prostate.
Methods
Detailed methods are available in Supplementary Material.
Cells
RPMI8226 and U266 cell lines were purchased from American Type Culture Collection (ATCC; Manassas, VA, USA), and KM3 and K562 cell lines were purchased from the China Center for Type Culture Collection (Wuhan, China). Low-passage stocks were used and cultured fewer than 3 months in RPMI 1640 containing 10% fetal bovine serum at 37 °C in 5% CO2. Primary BM-MSCs were purchased from Cyagen Biosciences Inc. (Guangzhou, China) or obtained from healthy adult donors. The BM-MSCs were cultured and validated as we previously described [56]. Primary peripheral blood mononuclear cells (PBMCs) were obtained from the donors.
Patients and healthy donors
Ethical approval for this study was obtained from the Review Board of Tongji Medical College, Huazhong University of Science and Technology (No 2012S119). Patients who were diagnosed and staged with de novo or relapsed MM [57] were included in the analyses of cirEVs, and those de novo patients who had received any anti-MM drugs were excluded. Bone lesions were detected using MRI or PET-CT, and genetic lesions were detected using FISH (fluorescence in situ hybridization). Healthy adult volunteers who had no considerable disease history according to the Medical Examination Center of Union Hospital, Tongji Medical College were involved. Both patients and donors provided informed written consent in accordance with the Declaration of Helsinki.
Isolation and validation of EVs
EVs derived from the cell lines were isolated and validated based on previously reported methods [21, 25, 26], and cirEVs were isolated as reported [15]. Briefly, For MM-EVs isolation, myeloma cells were cultured in RPMI-1640 with 10% FBS (we also removed FBS-derived EVs by centrifugation at 16,000 ×g for 60 min). After 24 h of culture, cells and debris were removed by centrifugation at 200 ×g for 5 min and 1,500 ×g for 30 min. The supernatant was centrifuged at 16,000 ×g for 60 min at 4 °C to obtain EVs. After being washed in PBS, EVs were lysed in ice-cold RIPA buffer (Beyotime, China) for 30 min at 4 °C and quantified by Bradford protein assay kit (Beyotime, China). 1 μg EV protein corresponded to ~1.5×107 EVs.
RNA extraction, sequencing and analysis
To investigate RNA content in EVs, EVs were incubated (37 °C, 15 min) with 2 mg/mL protease-free RNaseA (Sigma-Aldrich) followed by addition of 10× concentrated RNase inhibitor (Ambion). Total RNA was extracted from the EVs using TRIzol reagent (Invitrogen Life Technologies, CA, USA). The cDNA and small sequencing libraries were prepared according to the Illumina sequencing protocol. RNA-Seq and small RNA-Seq were performed and the data were analyzed as we previously reported [25, 26]. The FASTQ files of the RNA-Seq and small RNA-Seq data of the R-EVs and K562-EVs were submitted to NCBI Sequence Read Archive (SRA) under accession numbers SRP092289 and SRP057826, respectively. Specifically highly expressed miRNAs (SHEM) were identified by SEGtools with default parameters.
Conditioned HMCL medium preparation
Cell medium was collected from HMCLs (5×105) cultured for 24 h. The cultures were centrifuged at 3,000 ×g for 10 min. The acquired cell medium was then centrifuged at 16,000 ×g for 60 min and filtered through a 0.22 μm filter to remove all vesicles larger than 0.22 μm. The medium was stored at 4 °C until use.
Flow cytometry analysis of BM-MSCs
BM-MSCs were harvested from plates using trypsin digestion and then washed with PBS. Phycoerythrin (PE)-labeled anti-CD73, CD29, CD34, CD44, CD45, CD90, CD105, HLA-DR, FITC-CD138 and IgG isotype control were incubated with BM-MSCs for 20 min. After washing three times, BM-MSCs were detected by flow cytometry.
In vitro osteogenic differentiation of BM-MSCs
BM-MSCs were cultured in osteogenic differentiation medium (Cyagen Biosciences Inc., Guangzhou, China), osteogenic gene expression was detected using real-time PCR (Bio-Rad, Hercules, CA, USA), the level of secreted osteocalcin was detected using ELISA (Abcam, Cambridge, UK), ALP activity was examined using ELISA (Boster, Wuhan, China), and ALP staining (Baso Cell Science & Technology Co., Ltd., Zhuhai, China) and Alizarin Red staining (Cyagen Biosciences Inc., Guangzhou, China) were respectively used to quantify the number of colony-forming units with a fibroblastic phenotype (CFU-F) and CFU-OBs.
Cell apoptosis and viability analyses
Apoptotic BM-MSCs were detected using FCM with an Annexin-V FITC kit (Bender MedSystems, Vienna, Austria) according to the manufacturer's instructions. Cell activity was analyzed using MTT (Beyotime Biotechnology, China) or CCK-8 assay (Dojindo, Japan).
Confocal microscopy
To visualize and characterize EVs confocal microscopy (NikonA1Si, Nikon, Tokyo, Japan) were used as we previously described [21].
Transmission electron microscopy
The isolated PB cirEVs were fixed in 200 μL of 3% glutaraldehyde for 10 min. A drop (20 μL) of cirEVs was transferred to a formvar-carbon-coated grid. After rinsing, the grids were stained with 1% uranyl acetate solution for 5 min. Then, the grids were washed twice and examined with transmission electron microscopy (Hitachi HT7700, Tokyo, Japan) at 80 kV.
Tunable resistive pulse sensing
The size distribution and concentration of MM-EVs were measured with TRPS (qNano, Izon Science Ltd, Christchurch, New Zealand) using a NP400 (number A42746) nanopore at a 47 mm stretch. The concentration of MM-EVs was standardized using calibration with CPC400 carboxylated polystyrene beads (size 350 nm, concentration 7.5×1011 particles/mL).
Cell transfection
Cells were plated and cultured in complete medium at 37 °C for 24 h. Next, cells were transfected with 5 μL 20 μM miR-103a-3p-5p mimics and 10 μL 20 μM miR-103a-3p inhibitor using riboFECTTM CP Reagent (RiboBio, Guangzhou, China). Transfection efficiency was detected after 48 h.
Western blot analysis
The extracted proteins were quantified using a BCA protein assay kit (Beyotime, Suzhou, China), resolved on SDS-PAGE gels, and then transferred onto PVDF membranes (Millipore). Primary antibodies and secondary antibodies are shown in Supplementary Material. Membranes were visualized using an enhanced chemiluminescence kit (Beyotime).
Enzyme-linked immunosorbent assay
MM cells and MM-EVs were obtained and subsequently disrupted by sonication. The removed particulate supernatants were obtained by centrifugation at 1,500 ×g for 10 min. The supernatants of MM cells and MM-EVs-treated MM cells were filtered through 0.22 μm filters (Millipore) to remove contaminating EVs and cell debris. Then, supernatants were collected and centrifuged at 1,500 ×g for 10 min. The levels of DKK1, IL-7, sFRP2 and secreted IL-6 were detected by commercial ELISA kits (Bio-Swamp, Shanghai, China) according to the manufacturer's instructions. Results were obtained by measuring absorbance at 450 nm.
Animal studies
The xenograft mouse models were established based on a published paper [58]. On days 21, 35 and day 28, the tibiae were collected from the mice for ex vivo micro-computed tomography (micro-CT) imaging and bone histological and cytochemical analyses. BALB/c-nude mice were purchased from HuaFukang Bioscience Company (Beijing, China) and animal studies were performed in accordance with protocols approved by the animal ethics committee of Tongji Medical College, Huazhong University of Science and Technology.
CirEVs detection using FCM
Calcein-AM (Life Technologies Co.) and monoclonal anti-human PE-CD138, APC-CD38, PE/Cy5-CD45 and APC/Cy7-CD19 antibodies (BD Biosciences, San Jose, CA, USA) were applied to label intact cirEVs that were counted using FCM (Figure S9) [21, 59].
Statistical analysis
Unless otherwise noted, all experiments were conducted in at least triplicate. GraphPad Prism version 6.0 (GraphPad, La Jolla, CA, USA) and R version 3.2.2 were used for all calculations and figures. Two-tailed Student's t-test was performed to compare two groups, and analysis of variance (ANOVA) was used for analyses that involved more than two groups. Sidak's multiple comparison test was used when multiple comparisons were performed. The Wilcoxon rank test was used for comparisons of the cirEV counts between two groups, and Kruskal-Wallis test was used for analyses that involved more than two groups. Lifespan curves were analyzed with Kaplan-Meier survival curves and p-values were obtained using log-rank (Mantel-Cox) test. Differences with P < 0.05 were considered statistically significant and denoted as *** P < 0.001, ** P < 0.01, * P < 0.05. Data are presented as mean ± SD.
Data availability
The FASTQ files of the RNA-Seq and small RNA-Seq data of R-EVs and K562-EVs were submitted to NCBI Sequence Read Archive (SRA) under accession numbers SRP092289 and SRP057826, respectively.
Abbreviations
BM: bone marrow; BMPC: bone marrow malignant plasma cells; CFU-F: colony-forming unit fibroblast; CFU-OB: colony-forming unit osteoblasts; cirEVs: circulating EVs; CRP: c-reactive protein; D-MM: de novo MM; EVs: extracellular vesicles; FACS: fluorescence-activated cell sorting; HMCLs: human MM cell lines; IHC: immunohistochemistry; LDH: lactate dehydrogenase; MBD: MM bone disease; MM: multiple myeloma; MSCs: mesenchymal stem cells; OB: Osteoblasts; OC: osteoclasts; TRAP: tartrate-resistant acidic phosphatase staining; β2-MG: β2-microglobulin.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
We thank professor Yong Song Gho (Pohang University of Science and Technology) for his conceptual advice and valuable suggestions in this study. We thank Professor Jian Hou (Second Military Medical University, China) for his kind gift of the KM3 cell line.
Author contributions
QL, AYG, and ZC conceived and designed the research; LZ, CY, CX, GY, QL, and YH performed the in vitro experiments; FK, HW, AZ, WC, YC, LC, HX, WC, YC, and GY conducted the human clinical study and data analyses; LZ, CY, CX, FK, and TL analyzed the data; and QL and AYG wrote the paper.
Funding
This work was supported by grants from the National Nature Science Foundation of China (NSFC; 81071943, 81272624 to Q Li, 31801154 to Q Lei, 31771458 and 31471247 to A-Y. Guo, and 81170497 and 81470330 to Z Chen), and China Postdoctoral Science Foundation funded project (2017M622455 to Q Lei). Funding for open access charge: NSFC.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Röllig C, Knop S, Bornhäuser M. Multiple myeloma. Lancet. 2015;385:2197-208
2. Johnson SK, Stewart JP, Bam R, Qu P, Barlogie B, van Rhee F. et al. CYR61/CCN1 overexpression in the myeloma microenvironment is associated with superior survival and reduced bone disease. Blood. 2014;124:2051-60
3. Roodman GD. Mechanisms of bone metastasis. N Engl J Med. 2004;350:1655-64
4. Roodman GD. Osteoblast function in myeloma. Bone. 2011;48:135-40
5. Terpos E, Berenson J, Cook RJ, Lipton A, Coleman RE. Prognostic variables for survival and skeletal complications in patients with multiple myeloma osteolytic bone disease. Leukemia. 2010;24:1043-9
6. Raje N, Roodman GD. Advances in the biology and treatment of bone disease in multiple myeloma. Clin Cancer Res. 2011;17:1278-86
7. Toscani D, Bolzoni M, Accardi F, Aversa F, Giuliani N. The osteoblastic niche in the context of multiple myeloma. Ann N Y Acad Sci. 2015;1335:45-62
8. Reagan MR, Liaw L, Rosen CJ, Ghobrial IM. Dynamic interplay between bone and multiple myeloma: emerging roles of the osteoblast. Bone. 2015;75:161-9
9. Giuliani N, Mangoni M, Rizzoli V. Osteogenic differentiation of mesenchymal stem cells in multiple myeloma: identification of potential therapeutic targets. Exp Hematol. 2009;37:879-86
10. Chen Z, Orlowski RZ, Wang M, Kwak L, McCarty N. Osteoblastic niche supports the growth of quiescent multiple myeloma cells. Blood. 2014;123:2204-8
11. Giuliani N, Rizzoli V. Myeloma cells and bone marrow osteoblast interactions: role in the development of osteolytic lesions in multiple myeloma. Leuk Lymphoma. 2007;48:2323-9
12. McGranahan N, Swanton C. Biological and therapeutic impact of intratumor heterogeneity in cancer evolution. Cancer Cell. 2015;27:15-26
13. Mahindra A, Laubach J, Raje N, Munshi N, Richardson PG, Anderson K. Latest advances and current challenges in the treatment of multiple myeloma. Nat Rev Clin Oncol. 2012;9:135-43
14. Antonyak MA, Li B, Boroughs LK, Johnson JL, Druso JE, Bryant KL. et al. Cancer cell-derived microvesicles induce transformation by transferring tissue transglutaminase and fibronectin to recipient cells. Proc Natl Acad Sci U S A. 2011;108:4852-7
15. Ghosh AK, Secreto CR, Knox TR, Ding W, Mukhopadhyay D, Kay NE. Circulating microvesicles in B-cell chronic lymphocytic leukemia can stimulate marrow stromal cells: implications for disease progression. Blood. 2010;115:1755
16. Minciacchi VR, Freeman MR, Di Vizio D. Extracellular vesicles in cancer: exosomes, microvesicles and the emerging role of large oncosomes. Semin Cell Dev Biol. 2015;40:41-51
17. Kim DJ, Linnstaedt S, Palma J, Park JC, Ntrivalas E, Kwak-Kim JY. et al. Plasma components affect accuracy of circulating cancer-related microRNA quantitation. J Mol Diagn. 2012;14:71-80
18. Tkach M, Théry C. Communication by extracellular vesicles: where we are and where we need to go. Cell. 2016;164:1226-32
19. D'Souza-Schorey C, Clancy JW. Tumor-derived microvesicles: shedding light on novel microenvironment modulators and prospective cancer biomarkers. Genes Dev. 2012;26:1287-99
20. Peinado H, Aleckovic M, Lavotshkin S, Matei I, Costa-Silva B, Moreno-Bueno G. et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat Med. 2012;18:883-91
21. Sun L, Wang HX, Zhu XJ, Wu PH, Chen WQ, Zou P. et al. Serum deprivation elevates the levels of microvesicles with different size distributions and selectively enriched proteins in human myeloma cells in vitro. Acta Pharmacol Sin. 2014;35:381-93
22. Liu Y, Zhu XJ, Zeng C, Wu PH, Wang HX, Chen ZC. et al. Microvesicles secreted from human multiple myeloma cells promote angiogenesis. Acta Pharmacol Sin. 2014;35:230-8
23. Chen X, Liang H, Zhang J, Zen K, Zhang CY. Secreted microRNAs: a new form of intercellular communication. Trends Cell Biol. 2012;22:125-32
24. Liu T, Zhang Q, Zhang J, Li C, Miao YR, Lei Q. et al. EVmiRNA: a database of miRNA profiling in extracellular vesicles. Nucleic Acids Res. 2018
25. Zhu X, You Y, Li Q, Zeng C, Fu F, Guo A. et al. BCR-ABL1-positive microvesicles transform normal hematopoietic transplants through genomic instability: implications for donor cell leukemia. Leukemia. 2014;28:1666
26. Zhang HM, Li Q, Zhu X, Liu W, Hu H, Liu T. et al. miR-146b-5p within BCR-ABL1-positive microvesicles promotes leukemic transformation of hematopoietic cells. Cancer Res. 2016;76:2901-11
27. Roodman GD. Pathogenesis of myeloma bone disease. Leukemia. 2009;23:435-41
28. Sun Z, Cao X, Hu Z, Zhang L, Wang H, Zhou H. et al. MiR-103 inhibits osteoblast proliferation mainly through suppressing Cav1.2 expression in simulated microgravity. Bone. 2015;76:121-8
29. Zuo B, Zhu J, Li J, Wang C, Zhao X, Cai G. et al. microRNA-103a functions as a mechanosensitive microRNA to inhibit bone formation through targeting Runx2. J Bone Miner Res. 2015;30:330-45
30. Anderson HC, Mulhall D, Garimella R. Role of extracellular membrane vesicles in the pathogenesis of various diseases, including cancer, renal diseases, atherosclerosis, and arthritis. Lab Invest. 2010;90:1549-57
31. Taylor DD, Gerceltaylor C. MicroRNA signatures of tumor-derived exosomes as diagnostic biomarkers of ovarian cancer. Gynecol Oncol. 2008;110:13-21
32. Umezu T, Ohyashiki K, Kuroda M, Ohyashiki JH. Leukemia cell to endothelial cell communication via exosomal miRNAs. Oncogene. 2013;32:2747-55
33. Shao H, Chung J, Balaj L, Charest A, Bigner DD, Carter BS. et al. Protein typing of circulating microvesicles allows real-time monitoring of glioblastoma therapy. Nat Med. 2012;18:1835-40
34. Krishnan SR, Luk F, Brown RD, Suen H, Kwan Y, Bebawy M. Isolation of human CD138+ microparticles from the plasma of patients with multiple myeloma. Neoplasia. 2016;18:25-32
35. Ghosh N, Matsui W. Cancer stem cells in multiple myeloma. Cancer Lett. 2009;277:1-7
36. Arendt BK, Walters DK, Wu X, Tschumper RC, Jelinek DF. Multiple myeloma cell-derived microvesicles are enriched in CD147 expression and enhance tumor cell proliferation. Oncotarget. 2014;5:5686-99
37. Raimondi L, De Luca A, Amodio N, Manno M, Raccosta S, Taverna S. et al. Involvement of multiple myeloma cell-derived exosomes in osteoclast differentiation. Oncotarget. 2015;6:13772-89
38. Roccaro AM, Sacco A, Maiso P, Azab AK, Tai YT, Reagan M. et al. BM mesenchymal stromal cell-derived exosomes facilitate multiple myeloma progression. J Clin Invest. 2013;123:1542-55
39. Boswell EN, Glavey S, Sacco A, Maiso P, Mishima Y, Aljaway Y. et al. Mirna rxpression profiling and proteomic analysis of circulating exosomes from multiple myeloma patients. Am Soc Hematology. 2013;122:3086
40. Wang J, Hendrix A, De Bruyne E, Van Valckenborgh E, Himpe E, De Beule N. et al. Bone marrow stromal cell-derived exosomes facilitate multiple myeloma cell survival through inhibition of the JNK pathway. Am Soc Hematology. 2013;122:679
41. Wang J, Hendrix A, Hernot S, Lemaire M, De Bruyne E, Van Valckenborgh E. et al. Bone marrow stromal cell-derived exosomes as communicators in drug resistance in multiple myeloma cells. Blood. 2014;124:555-66
42. Raimondi L, Luca AD, Amodio N, Manno M, Raccosta S, Taverna S. et al. Involvement of multiple myeloma cell-derived exosomes in osteoclast differentiation. Oncotarget. 2015;6:13772-89
43. Stoorvogel W. Functional transfer of microRNA by exosomes. Blood. 2012;119:646-8
44. Hergenreider E, Heydt S, Tréguer K, Boettger T, Horrevoets AJG, Zeiher AM. et al. Atheroprotective communication between endothelial cells and smooth muscle cells through miRNAs. Nature Cell Biology. 2012;14:249-5
45. Pegtel DM, Cosmopoulos K, Thorley-Lawson DA, van Eijndhoven MAJ, Hopmans ES, Lindenberg JL. et al. Functional delivery of viral miRNAs via exosomes. Proc Natl Acad Sci USA. 2010;107:6328-33
46. Zhang L, Valencia CA, Dong B, Chen M, Guan P-J, Pan L. Transfer of microRNAs by extracellular membrane microvesicles: a nascent crosstalk model in tumor pathogenesis, especially tumor cell-microenvironment interactions. J Hematol Oncol. 2015;8:14
47. Zhang L, Zhang S, Yao J, Lowery FJ, Zhang Q, Huang W-C. et al. Microenvironment-induced PTEN loss by exosomal microRNA primes brain metastasis outgrowth. Nature. 2015;527:100-4
48. Wu T, Zhou H, Hong Y, Li J, Jiang X, Huang H. miR-30 Family members negatively regulate osteoblast differentiation. J Biol Chem. 2012;287:7503-11
49. Jia J, Feng X, Xu W, Yang S, Zhang Q, Liu X. et al. MiR-17-5p modulates osteoblastic differentiation and cell proliferation by targeting SMAD7 in non-traumatic osteonecrosis. Exp Mol Med. 2014;46:e107
50. Zhou W, Yin H, Wang T, Liu T, Li Z, Yan W. et al. MiR-126-5p regulates osteolysis formation and stromal cell proliferation in giant cell tumor through inhibition of PTHrP. Bone. 2014;66:267-76
51. Wu Z, Yin H, Liu T, Yan W, Li Z, Chen J. et al. MiR-126-5p regulates osteoclast differentiation and bone resorption in giant cell tumor through inhibition of MMP-13. Biochem Biophys Res Commun. 2014;443:944-9
52. Sugatani T, Vacher J, Hruska KA. A microRNA expression signature of osteoclastogenesis. Blood. 2011;117:3648-57
53. Capulli M, Paone R, Rucci N. Osteoblast and osteocyte: games without frontiers. Arch Biochem Biophys. 2014;561:3-12
54. Rajkumar SV. Evolving diagnostic criteria for multiple myeloma. Hematology Am Soc Hematol Educ Program. 2015;2015:272-8
55. Kyle RA, Remstein ED, Therneau TM, Dispenzieri A, Kurtin PJ, Hodnefield JM. et al. Clinical course and prognosis of smoldering (asymptomatic) multiple myeloma. N Engl J Med. 2007;356:2582-90
56. Zhao ZG, Liang Y, Li K, Li WM, Li QB, Chen ZC. et al. Phenotypic and functional comparison of mesenchymal stem cells derived from the bone marrow of normal adults and patients with hematologic malignant diseases. Stem Cells Dev. 2007;16:637-48
57. Kyle RA, Rajkumar SV. Criteria for diagnosis, staging, risk stratification and response assessment of multiple myeloma. Leukemia. 2009;23:3-9
58. Labrinidis A, Diamond P, Martin S, Hay S, Liapis V, Zinonos I. et al. Apo2L/TRAIL inhibits tumor growth and bone destruction in a murine model of multiple myeloma. Clin Cancer Res. 2009;15:1998-2009
59. Jayachandran M, Miller VM, Heit JA, Owen WG. Methodology for isolation, identification and characterization of microvesicles in peripheral blood. J Immunol Methods. 2012;375:207-14
Author contact
![]() Corresponding author: Q.B.L (qiubailiedu.cn) or A.Y.G. (guoayedu.cn).
Corresponding author: Q.B.L (qiubailiedu.cn) or A.Y.G. (guoayedu.cn).