Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results
Discussion
Conclusions
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(22):6307-6321. doi:10.7150/thno.29746 This issue Cite
Research Paper
Anti-tumor macrophages activated by ferumoxytol combined or surface-functionalized with the TLR3 agonist poly (I : C) promote melanoma regression
Jiaojiao zhao1, Zhengkui Zhang2, Yaxian Xue1, Guoqun Wang3, Yuan Cheng4, Yuchen Pan1, Shuli Zhao5 ![]() , Yayi Hou1,6
, Yayi Hou1,6 ![]()
1. The State Key Laboratory of Pharmaceutical Biotechnology, Division of Immunology, Medical School, Nanjing University, Nanjing 210093, PR China
2. MOE Key Laboratory of High Performance Polymer Materials and Technology, Department of Polymer Science & Engineering, College of Chemistry & Chemical Engineering, and Jiangsu Key Laboratory for Nanotechnology, Nanjing University , Nanjing, 210093, PR China
3. Department of Oncology, First Affiliated Hospital, Nanjing Medical University, Nanjing 211166, PR China
4. Department of Biomedical Engineering, College of Engineering and Applied Sciences, Nanjing National Laboratory of Microstructures, Nanjing University, Nanjing, Jiangsu 210093, PR China
5. General Clinical Research Center, Nanjing First Hospital, Nanjing Medical University, Nanjing 210006, PR China
6. Jiangsu Key Laboratory of Molecular Medicine, Nanjing University, Nanjing 210093, PR China
Received 2018-9-6; Accepted 2018-11-3; Published 2018-11-29
Abstract

Macrophages orchestrate inflammation and control the promotion or inhibition of tumors and metastasis. Ferumoxytol (FMT), a clinically approved iron oxide nanoparticle, possesses anti-tumor therapeutic potential by inducing pro-inflammatory macrophage polarization. Toll-like receptor 3 (TLR3) activation also potently enhances the anti-tumor response of immune cells. Herein, the anti-tumor potential of macrophages harnessed by FMT combined with the TLR3 agonist, poly (I:C) (PIC), and FP-NPs (nanoparticles composed of amino-modified FMT (FMT-NH2) surface functionalized with PIC) was explored.
Methods: Proliferation of B16F10 cells co-cultured with macrophages was measured using immunofluorescence or flow cytometry (FCM). Phagocytosis was analyzed using FCM and fluorescence imaging. FP-NPs were prepared through electrostatic interactions and their properties were characterized using dynamic light scattering, transmission electron microscopy, and gel retardation assay. Anti-tumor and anti-metastasis effects were evaluated in B16F10 tumor-bearing mice, and tumor-infiltrating immunocytes were detected by immunofluorescence staining and FCM.
Results: FMT, PIC, or the combination of both hardly impaired B16F10 cell viability. However, FMT combined with PIC synergistically inhibited their proliferation by shifting macrophages to a tumoricidal phenotype with upregulated TNF-α and iNOS, increased NO secretion and augmented phagocytosis induced by NOX2-derived ROS in vitro. Combined treatment with FMT/PIC and FMT-NH2/PIC respectively resulted in primary melanoma regression and alleviated pulmonary metastasis with elevated pro-inflammatory macrophage infiltration and upregulation of pro-inflammatory genes in vivo. In comparison, FP-NPs with properties of internalization by macrophages and accumulation in the lung produced a more pronounced anti-metastatic effect accompanied with decreased myeloid-derived suppressor cells, and tumor-associated macrophages shifted to M1 phenotype. In vitro mechanistic studies revealed that FP-NPs nanoparticles barely affected B16F10 cell viability, but specifically retarded their growth by steering macrophages to M1 phenotype through NF-κB signaling.
Conclusion: FMT synergized with the TLR3 agonist PIC either in combination or as a nano-composition to induce macrophage activation for primary and metastatic melanoma regression, and the nano-composition of FP-NPs exhibited a more superior anti-metastatic efficacy.
Keywords: macrophage, ferumoxytol, poly (I : C), FP-NPs, melanoma
Introduction
Melanoma is known as "the cancer that rises with the sun" because of its high degree of malignancy, recurrence, and particularly aggressive metastasis [1]. Despite enormous progress in targeted therapies and immune checkpoint inhibitors have demonstrated some curative effects [2, 3], the limitations of these treatment modalities are also obvious. BRAFV600E/K mutations are present in around 40-50% melanomas; however, most patients receiving BRAF inhibitor treatment relapse within several months [4, 5]. The total response rate of ipilimumab, the best-studied monoclonal antibody targeting cytotoxic T-lymphocyte-associated antigen (CTLA)-4, is less than 20%, and it may also cause autoimmune toxicity [6, 7]. Though individualized tumor vaccine therapy improves the therapeutic outcome and prevents recurrence [8, 9], disadvantages of long periods of preparation and high costs limit its application. There are still a substantial number of patients with primary or acquired resistance that cannot benefit from current treatments [10]. Therefore, it is imperative to develop more efficacious and affordable approaches to eradicate primary and metastatic melanoma.
Macrophages are one of the most widely studied pleiotropic cells in the immune system, and tumor-associated macrophages (TAMs) are macrophages that infiltrate tumor stroma [11, 12]. TAMs are detrimental with melanoma-promoting functions by supporting tumor cell growth, stimulating angiogenesis, and thus assisting melanoma invasion and metastasis [13-15]. However, as innate immune cells, macrophages can directly kill tumors by shaping the acute inflammatory response for anti-proliferative and cytotoxic activities with secretion of pro-inflammatory cytokines (e.g., tumor necrosis factor α (TNF-α)) and tumoricidal species (e.g., nitric oxide (NO), reactive oxygen species (ROS)), which are critical to suppress tumorigenicity and abrogate metastasis, as well as maintain tissue homeostasis via phagocytosis by removing malignant cells [16, 17]. Increasing evidence indicates that activating macrophages to pro-inflammatory phenotype or reprogramming TAMs to M1 is promising for cancer therapy [18-20]. Thus, targeted TAMs could be an effective therapy for primary and metastatic melanoma suppression [21].
Toll-like receptors (TLRs) are pattern recognition receptors that recognize conserved molecular motifs in bacteria and viruses to initiate innate immunity [22]. TLR3 is a Toll-like family receptor that mediates antiviral responses by recognizing double-stranded RNA (dsRNA) [23]. The TLR3 agonist, polyinosinic-polycytidylic acid (poly (I:C) (PIC)), an artificial dsRNA analog, has been widely exploited for cancer treatment because of its significant potential to boost the immune system [24, 25]. Abundant studies have demonstrated that TLR3 potently enhances anti-tumor responses by mediating anti-tumor natural killer (NK) cell and tumor-specific cytotoxic T lymphocyte (CTL) activities through maturation of dendritic cells (DCs) [26-28]. However, few studies have focused on the effects of TLR3 on macrophage activation. In addition, according to the latest research, TLR3 agonists alone are not sufficient to induce macrophage-mediated tumor cell growth inhibition except when combined with IFN-γ, which can result in optimal activation of anti-tumor M1 macrophages [29]. In particular, the susceptibility of PIC to nuclease degradation leads to insufficient immune-boosting, which has hindered its clinical application [30, 31]. Therefore, it is extremely desirable to combine PIC with other macrophage activators or to develop an efficient delivery system to address these issues.
The emergence of nanotechnology has provided unprecedented opportunities to exploit nanoparticles (NPs) as a platform for delivery of vaccines and genes [32]. For example, immune nanotheranostics, as an important class of cancer nanotheranostics for immune system activation, are extremely promising [33]. Various NPs (e.g., zinc oxide nanoparticles, calcium phosphate nanoparticles, synthetic nanoparticles) have been intensively explored for PIC delivery [34-36]. However, different from all these vectors for the sole purpose of delivery, ferumoxytol (FMT), an iron oxide nano-carrier composed of an iron oxide core and dextran shell, also integrates the crucial essence of both diagnostic and therapeutic functions. FMT, with low cytotoxicity, has been approved by the FDA for clinical treatment of iron deficiency [37]. The particular characterization of FMT internalized by macrophages has been exploited for magnetic resonance imaging (MRI) [38]. Besides, FMT can facilitate tumor treatment through magnetic hyperthermia [39, 40]. More importantly, FMT is recently reported to possess an appealing feature of anti-tumor therapeutic potential by inducing pro-inflammatory macrophage polarization [41]. Due to the inherent capacity of preferential uptake by macrophages, NPs conjugated or engineered with ancillary molecules such as functional RNA or TLR agonists could further amplify the immune-modulating properties of macrophages for anti-tumor treatment [42, 43]. As a promising delivery system candidate as well as incorporating features for macrophage activation, FMT may protect PIC from degradation and facilitate its immune-stimulatory properties for anti-tumor immunotherapy. Therefore, the untapped potential of macrophages activated by the combination of FMT and PIC or nano-particles composited by FMT surface functionalized with PIC provides an impetus to discover whether it is an efficient strategy against melanoma.
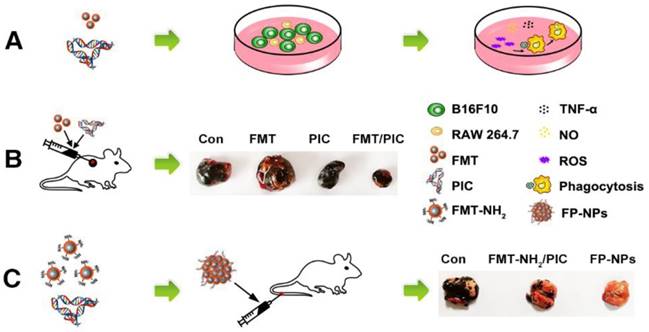
In the present study, we identified that FMT combined or surface functionalized with the TLR3 agonist PIC (FP-NPs) could induce macrophages to be tumoricidal for both subcutaneous and pulmonary metastasis in melanoma regression (Scheme 1). Notably, FP-NPs displayed greater therapeutic benefit with diminished or even disappeared metastatic nodules in mice than amino-modified FMT combined with PIC (FMT-NH2/PIC).
Schematic illustration of FMT combined or surface functionalized with PIC for melanoma regression. (A) FMT/PIC synergistically inhibited B16F10 cell proliferation in co-culture with macrophages. (B) FMT/PIC prominently reduced tumor burden in melanoma-bearing mice. (C) System delivery of FP-NPs alleviated lung metastases more significantly compared to FMT-NH2/PIC.
Methods
Cell culture and reagents
B16F10 melanoma cells and RAW 264.7 macrophages purchased from Shanghai Cell Bank, Chinese Academy of Sciences were cultured in complete Dulbecco's modified Eagle's medium (DMEM) (Gibco, Carlsbad, CA) with 10% fetal bovine serum (FBS) (Gibco) and 1% v/v penicillin and streptomycin (Gibco) at 37 °C in a 5% CO2 incubator.
Cell viability
Cell viability was measured using a Cell Counting Kit-8 (CCK-8) assay. Briefly, tumor cells were seeded at a density of 5 × 103 per well in 96-well plates in quintuplicate (for measurement of co-cultured cell viability, B16F10 and RAW 264.7 cells were seeded at a ratio of 2:1 at a density of 3 × 103 and 1.5 × 103 per well in 96-well plates, respectively) and incubated with ferumoxytol (FMT), poly (I:C) (PIC) (InvivoGen, San Diego, USA), and a combination of both for 48 h. The supernatants were then discarded and fresh medium containing 10 μL of WST-8 reagent (Dojindo, Kumamoto, Japan) was added. After incubation for another 4 h at 37 °C, absorbance was determined with a multi-well spectrophotometer (BioTek, VT, USA) at 450 nm.
Generation of Bone Marrow-Derived Macrophages (BMDMs)
The femurs and tibias of C57BL/6 mice (female, 6-8 weeks) were aseptically dissected and flushed with a 27 G syringe to obtain bone marrow cells. Next, the bone marrow cells were cultured in complete RPMI-1640 medium (10% FBS, 1% v/v penicillin and streptomycin) (Gibco) and incubated with 20 ng/mL recombinant mouse macrophage colony-stimulating factor (M-CSF) (R&D Systems, Minneapolis, MN) at 37 °C in a 5% CO2 incubator for 6 days. Non-adherent cells were discarded and adherent cells were collected for further experiments.
Cell proliferation analysis
For cell proliferation analysis, tumor cells were pre-stained with the cell proliferation dye CFSE (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions at a concentration of 10 μM. After the last wash with PBS, cells were co-cultured with RAW 264.7 cells at a ratio of 2:1 (for measuring B16F10 proliferation upon co-culture with BMDMs, the ratio was adjusted to 1:2) and respectively incubated with 100 μg/mL FMT, 10 μg/mL PIC, or 100 μg/mL +10 μg/mL FMT/PIC for 48 h. Cells were then harvested, washed twice with PBS, and analyzed by flow cytometry (FCM). The proliferation index (PI) of cells was analyzed using Mod Fit LT 3.0 software, and the cell growth inhibition rate was calculated by the following equation: cell growth inhibition rate = (1-PI (groups with different treatment)/PI (control group without any treatment) ) × 100%.
Phagocytosis assay
To measure the phagocytic ability of macrophages, B16F10 and RAW 264.7 cells were respectively fluorescence-labeled with CFSE and the Cell Proliferation Dye eFluor 670 (Invitrogen). After the last wash with PBS, cells were co-cultured as above mentioned. Cells were then harvested, washed twice with PBS, and detected by FCM. The relative phagocytosis index was analyzed using FlowJo software and was represented by double positive cells for CFSE and Dye 670. In an effort to assess the phagocytosis visually, GFP-B16F10 co-cultured with RAW 264.7 at a ratio of 2:1 were incubated with FMT/PIC for 48 h, and the phagocytosis was detected using immunofluorescence microscopy.
Isolation of total RNA and quantitative RT-PCR (qRT-PCR)
Total RNA was extracted from different treated cells or tissues with TRIzol (Invitrogen). Then, the collected mRNA was reverse-transcribed to cDNA using HiScript II 1st Strand cDNA Synthesis Kit (Vazyme Biotech Co., Ltd, Nanjing, China) according to the manufacturer's instructions. SYBR Green PCR Kit (Bio-Rad, Hercules, CA) was next used to measure the target genes on a StepOne-Plus Real-Time PCR System (Applied Biosystems, Foster City, CA). Finally, the relative expression ratio of target genes was calculated by the 2-ΔΔCt method. The sequences of the qRT-PCR primers are provided in supplemental Table 1.
Enzyme linked immunosorbent assay (ELISA)
TNF-α in the cell culture supernatant or blood of mice subjected to different treatments was measured by ELISA (Dakewe, Nanjing, China). Briefly, culture medium supernatant or blood was collected and centrifuged. Aliquots (50 µL) of diluted samples were then measured according to the manufacturer's instructions, and the absorbance was measured at 450 nm.
Western blotting (WB) analysis
The specific procedures for WB have been described in a previous report [44]. Briefly, cells cultured with various treatments were collected, washed, and lysed with RIPA lysis buffer (Beyotime, Haimen, China). The acquired protein was quantified with a BCA protein assay kit (Thermo Fisher Scientific, Waltham, MA). Next, equal amounts of protein were subjected to SDS-PAGE and then transferred onto polyvinylidene fluoride membranes (Millipore, Bedford, MA). Immunoblotting was performed with rabbit anti-NOX2 (Abcam, Cambridge, UK), rabbit anti-P65 (Cell Signaling Technology (CST), Danvers, MA), rabbit anti-p-P65 (CST), rabbit anti-IKBα (CST), rabbit anti-p-IKBα (CST), mouse anti-β-actin (Thermo Fisher Scientific), and mouse anti-GAPDH (Thermo Fisher Scientific) primary antibodies followed by horseradish peroxidase (HRP)-conjugated anti-rabbit or anti-mouse secondary antibodies (Thermo Fisher Scientific). Finally, the targeted proteins were visualized with ECL Plus WB detection reagents (Millipore).
Detection of NO and ROS
NO was measured by Griess Test using commercial kits (Beyotime). Briefly, 50 µL of cell culture supernatant was immediately added to 50 µL of Griess reagent A. The mixture was incubated with 50 µL of Griess reagent B, and absorbance at 540 nm was measured with a microplate reader. ROS production was determined by staining cells with 10 μM of 2,7-Dichlorodihydrofluorescein diacetate (DCFH-DA) (Beyotime) for 30 min at 37 °C. Cells were then washed twice with RPMI-1640 medium and suspended in PBS for FCM analysis.
Synthesis of amino-modified ferumoxytol nanoparticles (FMT-NH2)
FMT synthesis has been described in previous reports [45, 46]. The procedures used to prepare the amino-modified FMT are as follows. FMT nanoparticles (100 mg, containing carboxyl groups on the surface) were first dissolved in a solution of 100 mL dimethyl sulfoxide containing 12 mg of dimethylaminopyridine (DMAP, 0.1 mmol) (Sigma-Aldrich, St. Louis, MO), and were then added to 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC) (J&K Scientific Ltd, Shanghai, China) (192 mg, 1 mmol), and N-hydroxysuccinimide (NHS) (J&K Scientific Ltd) (115 mg, 1 mmol). The resulting mixture was then stirred at room temperature for 2 h and 1, 3-diaminopropane (J&K Scientific Ltd) (150 mg, 2 mmol) was added to the mixture and stirred for another 24 h. Thereafter, the reaction mixture was added with excess acetone to obtain crude FMT-NH2 nanoparticles. The crude products were washed thrice with acetone and dried under reduced pressure.
Fluorescence-labeled FMT-NH2
The procedures for preparing Rhodamine B (RhB) (Sigma-Aldrich) or NIR 797 (Sigma-Aldrich) labeled FMT-NH2 were similar. Briefly, FMT-NH2 (20 mg), RhB (0.2 mg), and dimethyl sulfoxide (J&K Scientific Ltd) (5 mL) were added into a 10-mL flask. After stirring at room temperature overnight, the mixture was diluted with 20 mL deionized water and dialyzed against 2 L of deionized water in a 3.5 kDa MWCO for two days to remove the unreacted fluorescence dye and then lyophilized.
Gel retardation assay
FMT-NH2 was dissolved in RNase free diethyl pyrocarbonate-treated H2O and subtly complexed with PIC at w/w ratios of 10/1, 20/1, and 40/1, and gently mixed using a vortex mixer for 5 min at room temperature to obtain the FMT-NH2-PIC (FP-NPs) nanocomposites. FP-NPs mixed with bromophenol blue were then loaded onto a 1% w/v agarose gel containing ethidium bromide and electrophoresed for 30 min at 120 V. Finally, the gel was analyzed using an ultraviolet light transilluminator (Bio-Rad, Hercules, CA) and was imaged using the Tanon Gel Image System 2500 (Tanon, Shanghai, China).
In vitro uptake of FP-NPs by macrophages
Macrophages used for the measurement of cellular uptake were seeded on coverslips in 24-well plates at a density of 1 × 105 cells/well. Then, cells were treated with RhB labeled FP-NPs and incubated for another 6 h. After staining the nuclei with DAPI for 5 min, fluorescence images were obtained using the FV3000 laser scanning confocal microscope (LSCM) (Olympus, Japan).
Bio-distribution analysis of FMT-NH2 and FP-NPs
FMT-NH2 fluorescently labeled with NIR797 and its composite with PIC (FP-NPs) were intravenously injected into mice, and the ex vivo imaging of major organs after an 8-h incubation was performed using the IVIS Spectrum In Vivo Imaging System (PerkinElmer, USA). In addition, FP-NPs pre-labeled with RhB were intravenously injected into mice for 4 h, and the lung tissues were isolated, minced and incubated with collagenase (Sigma-Aldrich) and DNase I (Sigma-Aldrich) dissolved in RPMI-1640 medium for 30 min at 37 °C. Then, the homogenates were filtered through a 70-μm nylon mesh filter and lysed with RBC lysis buffer to obtain single cell suspension for fluorescent antibody staining. Finally, FP-NPs uptake by the lung macrophages were measured by LSCM and FCM.
Tumor growth inhibition
All animal studies were performed using protocols approved by institutional guidelines (Nanjing University Institutional Animal Care and Use Committee) and also conformed to the Guidelines for the Care and Use of Laboratory Animals published by the National Institutes of Health. Female C57BL/6 mice (6-8 weeks) were purchased from Model Animal Research Center of Nanjing University (Nanjing, China). Next, 5 × 105 B16F10 cells suspended in 100 μL sterile PBS were transplanted subcutaneously into the right flank of mice. When tumors reached an average size of about 100 mm3, mice were randomized into four groups and intratumorally injected with PBS, FMT (10 mg/kg), PIC (1 mg/kg), and FMT/PIC (10 mg/kg+1 mg/kg) every other day for three times. The body weight of mice and two perpendicular diameters of tumors measured by digital calipers were recorded, and tumor size was calculated as per the formula: V (volume) = (L × W2)/2, where L and W represent the tumor length and width, respectively.
Isolation of tumor-associated macrophages (TAMs)
To isolate the TAMs from solid tumors, the tumor tissues were minced and incubated with the Tumor Dissociation Kit (Miltenyi Biotec, Germany) dissolved in RPMI-1640 medium for 30 min at 37 °C. Next, the cell lysate were washed with PBS, passed through a 70-μm nylon mesh to acquire single-cell suspensions, layered on Percoll gradients, and centrifuged. Then, the lymphomononuclear correspondent layer was isolated and cultured in RPMI-1640 medium for 40 min at 37 °C. Next, PBS was used to wash away the non-adherent cells to obtain the adherent cells [47].
In vivo anti-metastatic efficacy measurements
To establish a B16F10 lung metastases model, 5 × 105 B16F10 cells were suspended in 100 μL sterile PBS and intravenously injected into C57BL/6 mice. For the establishment of 4T1 lung metastases model, 5 × 105 GFP-4T1 cells were suspended in 100 μL sterile PBS and intravenously injected into female BALB/c mice (6-8 weeks). On the following day, mice were randomly grouped and intravenously treated with PBS, FP-NPs (FMT-NH2 200 μg composited with PIC 10 μg), and FMT-NH2 (200 μg) combined with PIC (10 μg), every other day for three times. It should be emphasized that in order to avoid complexation of FMT-NH2 and PIC in the group of combined therapy, PIC was concomitantly administered 5 min after the intravenous injection of FMT-NH2.
Analysis of tumor-infiltrating immunocytes
Tumors dissected from tumor-bearing mice were cut into pieces with a scissors and incubated with a cocktail of enzymes from a Tumor Dissociation Kit (Miltenyi Biotec) dissolved in RPMI-1640 medium for 30 min at 37 °C. The homogenates were then washed with PBS and passed through a 70-μm nylon mesh to acquire single-cell suspensions. Prior to staining, the cells were incubated with FcR blocking reagent (Miltenyi Biotec) for 10 min. Subsequently, the suspensions were divided into various parts for immunocyte detection. Antibodies against CD45, CD11b, F4/80, and Gr-1 were purchased from BioLegend (San Diego, USA).
Immunofluorescence staining
For immunofluorescence analysis, tumor tissue sections immunostained with mouse anti-CD86 (Novus, Littleton, USA) and goat anti-CD206 antibody (R&D) were respectively labeled with goat anti-mouse IgG (H+L) conjugated to Alexa Fluor 647 (Invitrogen) and donkey anti-goat IgG (H+L) conjugated to Alexa Fluor 488 (Invitrogen). All sections were imaged using a LSCM.
Histologic analysis of the microvasculature
For angiogenesis staining, mice were intravenously injected with 100 μL AF594-conjugated streptavidin tomato lectin (1 mg/mL in PBS; Vector Labs). After 5 min, mice were sacrificed and tumors were dissected. Images of frozen sections of tumor samples were taken on a LSCM and quantitatively analyzed using Image J.
Statistical analysis
All experiments were repeated at least three times. Statistical analysis was performed using one-way ANOVA followed by Student's t-test with GraphPad Prism. Statistical differences were defined as significant for *p < 0.05 and highly significant for **p < 0.01 and ***P < 0.001.
Results
Combined effect of FMT and PIC on macrophage activation and tumor cell growth inhibition in co-culture system
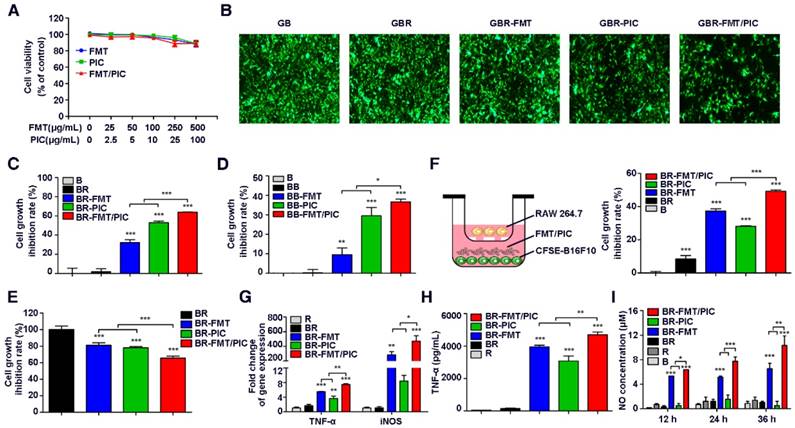
CCK-8 analysis of mouse melanoma B16F10 cell viability showed that FMT, PIC, or FMT combined with PIC (FMT/PIC) at a low dose had no effects on cell viability and only exhibited moderate cytotoxicity at the maximum dose (Figure 1A). To investigate the potential cytotoxic and cytostatic activity of macrophages toward tumor cells, RAW 264.7 and GFP-B16F10 stably expressing green fluorescent protein (GFP) were co-cultured at a ratio of 1:2 and treated with FMT and PIC solely or in combination. Tumor cell growth was then determined by fluorescence intensity. As shown in Figure 1B, introduction of RAW 264.7 cells barely influenced the tumor cell growth compared to B16F10 cells alone. Co-incubation of B16F10 and RAW 264.7 with FMT or PIC only slightly inhibited B16F10 cell growth, whereas FMT was found to synergize with PIC resulting in a more significant growth retardation of B16F10 cells. To accurately verify the inhibition rate of cell proliferation, B16F10 cells were pre-stained with the cell proliferation dye, CFSE, and the cell growth with different treatments was detected by FCM. As shown in Figure 1C, B16F10 co-cultured with RAW 264.7 yielded minimal growth inhibition (inhibition rate of 1.8%) compared to B16F10 alone. Stimulation with FMT or PIC resulted in cell growth inhibition rates of 32.2% and 53.0%, respectively, whereas FMT/PIC collaboratively generated more significant growth retardation with an inhibition rate of 63.9%. To further confirm the above results, C57BL/6-derived BMDMs, a source of primary macrophages, were co-cultured with B16F10. It should be noted that in vitro, BMDMs are non-proliferating, whereas the numbers of macrophages in specific tissues can be maintained by circulating monocytes derived from bone marrow, spleen, hematopoietic stem and progenitor cells, or local macrophages amplified in vivo [48-51]. Herein, the ratio of BMDMs and B16F10 was adjusted to 2:1. Consistently, incubation of FMT/PIC synergistically inhibited B16F10 cell proliferation in this co-culture system (Figure 1D). In addition, CCK-8 assay of the total co-culture cell viability indirectly suggested that FMT/PIC synergized to inhibit tumor cell growth (Figure 1E). To further determine whether this inhibition of proliferation was dependent on direct cell-to-cell contact, a dual-chamber Transwell system with macrophages seeded in the upper chamber and tumor cells seeded in the lower chamber was adopted. As shown in Figure 1F, unlike the direct contact co-culture, B16F10 proliferation was marginally retarded in the co-culture group (8.5%), and incubation with FMT further strengthened this effect with an inhibition rate of 37.3%. Incubation with PIC prohibited cell growth by 28.1%, and addition of FMT/PIC still exhibited the most significant inhibitory effect with an inhibition rate of 49.2%, both of which were much smaller than those in the direct contact co-culture system (53.0% and 63.9%, respectively), indicating that inhibition of the growth of B16F10 incubated with PIC and FMT/PIC was dependent on direct cell-to-cell contact to some extent. Further, macrophages co-cultured in the upper chamber were isolated and their transcript expression was examined by qRT-PCR. As shown in Figure 1G, compared with separate treatment, the presence of FMT/PIC significantly synergized to upregulate the mRNA expression of TNF-α and iNOS. In addition, incubation with FMT/PIC synergistically promoted the release of TNF-α (Figure 1H) and showed a time-dependent increase in NO concentration in the co-culture system (Figure 1I). The increase in concentration of NO was thirty-five times more than that in B16F10 cells at 12 h and continued to be ten times more than that in B16F10 cells at 24 h and 36 h, which was enough to inhibit B16F10 cell growth [29]. Taken together, FMT combined with PIC synergistically inhibited the proliferation of B16F10 cells by converting macrophages to tumoricidal phenotype with enhanced production of cytotoxic TNF-α and NO.
Combined effect of FMT and PIC on macrophage activation and tumor cell growth inhibition. (A) B16F10 cells were incubated with FMT, PIC, or FMT/PIC for 48 h and cell viability was analyzed by CCK-8 assay. (B) GFP-B16F10 (GB) cells co-cultured with RAW 264.7 (R) at a ratio of 2:1 were incubated with FMT, PIC, or FMT/PIC for 48 h and the fluorescence intensity of GFP was captured. (C) B16F10 (B) cells pre-labeled with CFSE and co-cultured with RAW 264.7 (R) at a ratio of 2:1 were incubated with FMT, PIC, or FMT/PIC for 48 h, and cell proliferation was then analyzed by FCM. (D) B16F10 (B) cells pre-labeled with CFSE, and co-cultured with BMDMs (B) at a ratio of 1:2 were incubated with FMT, PIC, or FMT/PIC for 48 h, and cell proliferation was then analyzed by FCM. (E) B16F10 (B) cells co-cultured with RAW 264.7 (R) at a ratio of 2:1 were incubated with FMT, PIC, or FMT/PIC for 48 h and total cell viability was analyzed using CCK-8 assay. (F) Left: Schematic diagram of the co-culture. Right: B16F10 (B) cells pre-labeled with CFSE co-cultured with RAW 264.7 (R) at a ratio of 2:1 were incubated with FMT, PIC, or FMT/PIC in a dual-chamber Transwell system for 48 h, and cell proliferation of B16F10 was then analyzed by FCM. (G) RAW 264.7 (R) cells co-cultured with B16F10 (B) at a ratio of 2:1 were incubated with FMT, PIC, or FMT/PIC in a dual-chamber Transwell system for 12 h and then the gene expression of TNF-α and iNOS in RAW 264.7 was analyzed by qRT-PCR. (H) RAW 264.7 (R) cells co-cultured with B16F10 (B) at a ratio of 2:1 were incubated with FMT, PIC, or FMT/PIC in a dual-chamber Transwell system for 24 h and TNF-α secretion was then detected using ELISA. (I) B16F10 (B) cells co-cultured without or with RAW 264.7 (R) at a ratio of 2:1 were incubated with FMT, PIC, or FMT/PIC and the NO in the medium was analyzed using Griess test at the indicated time. All representative data are from three independent experiments. Error bars, SD. *P < 0.05, **P < 0.01, ***P < 0.001. BMDMs: bone marrow-derived macrophages; FCM: flow cytometry; FMT: ferumoxytol; NO: nitric oxide; PIC: poly I:C, polyinosinic-polycytidylic acid; TNF-α: tumor necrosis factor α.
Impact of FMT/PIC on ROS production and phagocytosis
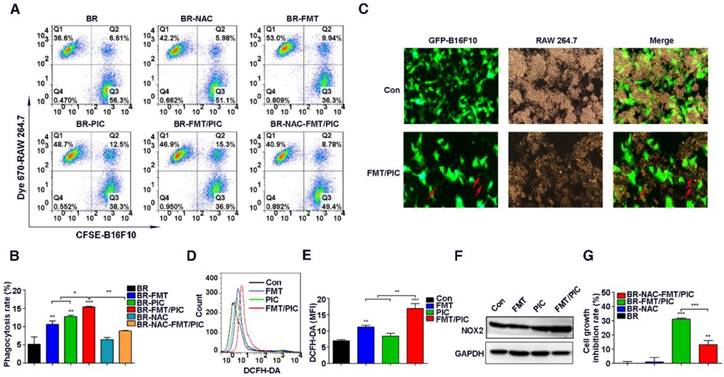
It has been well documented that as professional phagocytes, macrophages play a crucial role in clearing tumor cells [52]. Given that the prohibited growth of B16F10 incubated with PIC and FMT/PIC was partially mediated by direct cell contact, it was of great interest to observe whether their addition could enhance the capacity of macrophages to engulf tumor cells. Next, CFSE pre-labeled B16F10 cells were co-cultured with Dye 670 pre-stained RAW 264.7 cells at a ratio of 2:1 and incubated with FMT, PIC, or FMT/PIC. Phagocytosis was then analyzed by FCM. As expected, PIC treatment apparently increased the percentage of phagocytic cells compared to the control or FMT groups, and there was a statistically elevated percentage of phagocytosis in the group administered with FMT/PIC (Figure 2A-B). In an effort to assess the phagocytosis visually, GFP-B16F10 were co-cultured with RAW 264.7 and the results are presented in Figure 2C where FMT/PIC significantly increased the phagocytosis of GFP-B16F10 cells by macrophages with a reduced cell number of GFP-B16F10 compared to the control group. Reactive oxygen species (ROS) are an important product of macrophage activation, which play critical roles in modulating phagocytosis [53]. Our experiments showed that treatment with FMT or PIC alone only slightly promoted the production of ROS, whereas their combination dramatically enhanced the ROS production level in RAW 264.7 (Figure 2D-E). In addition, ROS-induced phagocytic activity was distinctly blunted with the addition of ROS scavenger N-acetyl-cysteine (NAC) (Figure 2A-B), demonstrating that ROS indeed played a critical role in the phagocytosis. However, it seemed contradictory that a higher expression level of ROS with an attenuated phagocytosis was observed in the group treated with FMT rather than with PIC. It is well known that endogenous ROS production in cells is mainly associated with mitochondria and the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX) family [54]. Of the latter, NOX2 NADPH oxidase in particular has been assigned with pivotal roles in regulating inflammation and phagocytosis by generating ROS [53, 55]. Further experiments showed that combined treatment synergistically promoted NOX2 expression, while neither FMT nor PIC altered the expression (Figure 2F). Therefore, NOX2-derived ROS was predicted to boost phagocytosis. An abnormal level of ROS is known to induce tumor cell apoptosis and damage tumor cell proliferation [56, 57]. Consistently, suppression of cell growth was partly recovered by injection with NAC in the co-stimulation group (Figure 2G), which indicated that ROS was as well involved in macrophage activation-mediated tumor cell growth inhibition. Collectively, FMT/PIC synergistically promoted ROS generation, which was involved in modulating the phagocytosis of RAW 264.7, and played roles in arresting tumor cell growth. In light of the above results, it was anticipated that macrophage-mediated inhibition of tumor cell growth might not be restricted to the B16F10 cell line. Next, the 4T1 breast cancer cells were co-cultured with RAW 264.7 similar to the above conditions. As shown in Figure S1A, FMT, PIC, or FMT/PIC had no obvious impact on cell viability of tumor cells. However, in the co-culture system, FMT/PIC synergistically inhibited the proliferation of 4T1 cells (Figure S1B).
Impact of FMT/PIC on ROS production and phagocytosis. (A) CFSE pre-labeled B16F10 were co-cultured with Dye 670 pre-stained RAW 264.7 cells and incubated with FMT, PIC, or FMT/PIC; phagocytosis was analyzed by FCM. (B) Quantified phagocytosis (double positive of CFSE and Dye 670) in (A). (C) GFP-B16F10 were co-cultured with RAW 264.7 cells and incubated of FMT/PIC; phagocytosis was analyzed by fluorescence microscopy. Red arrows indicate GFP-B16F10 tumor cells phagocytized and digested by macrophages. (D) RAW 264.7 cells were incubated with FMT, PIC, or FMT/PIC for 12 h and ROS production was detected by FCM. (E) Quantitative data of (D). (F) RAW 264.7 was incubated with FMT, PIC, or FMT/PIC for 24 h and the protein expression of NOX2 was detected by WB. (G) The co-culture system was pre-treated with a ROS scavenger, NAC, and quantitative cell proliferation was determined by FCM. All representative data are from three independent experiments. Error bars, SD. *P < 0.05, **P < 0.01, ***P < 0.001. DCFH-DA: dichloro-dihydro-fluorescein diacetate; FCM: flow cytometry; FMT: ferumoxytol; MFI: mean fluorescence intensity; NAC: n-acetyl-cysteine; PIC: poly I:C, polyinosinic-polycytidylic acid; ROS: reactive oxygen species; WB: western blotting.
Superior therapeutic effect of FMT/PIC in vivo
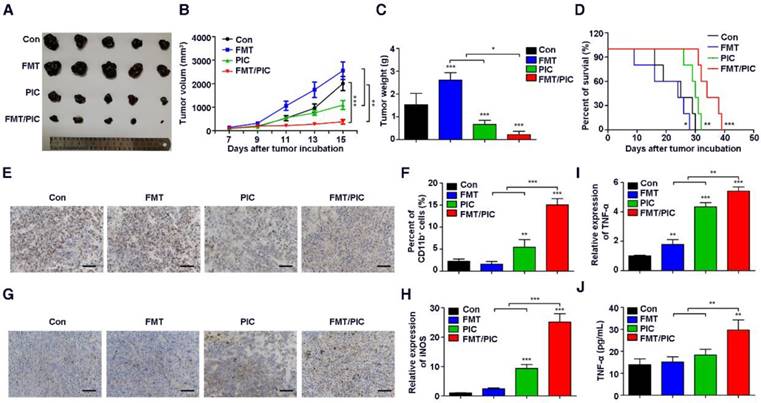
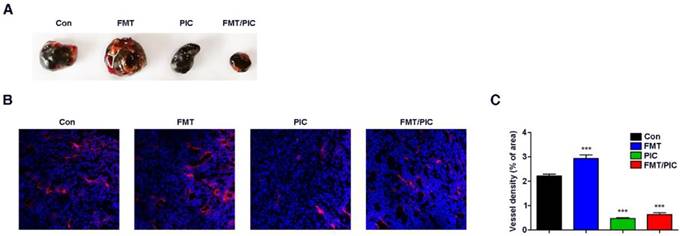
Inspired by the above results, we tested the hypothesis that FMT/PIC would induce an anti-tumor immune response capable of executing tumor regression in vivo. In our study, B16F10 tumor-bearing mice were randomized and intratumorally administered with either FMT or PIC solely or collectively. As depicted in Figure 3A-D, treatment with PIC exhibited remarkable therapeutic benefits with reduced tumor burden, and co-administration of PIC with FMT more prominently led to tumor regression with decreased tumor weight and extended survival of tumor-bearing mice. In sharp contrast to the in vitro results, sole treatment of FMT substantially accelerated melanoma progression with aggravated tumor burden and reduced survival of animals. Immunohistochemistry of Ki67 in tumor tissues (Figure 3E) also showed that FMT exposure promoted tumor cell proliferation, whereas PIC exhibited a therapeutic potency with reduced cell proliferation, and FMT/PIC most notably retarded tumor cell growth. FCM results indicated that treatment with PIC increased the CD11b+ immune cell infiltration compared to that in the control, and this trend was particularly prominent in the combined treatment group (Figure 3F). Immunohistochemistry results further revealed that macrophage infiltration in tumor tissues was significantly increased after FMT/PIC treatment (Figure 3G). In addition, the results of qRT-PCR demonstrated that local administration of FMT/PIC substantially elevated the cytotoxic gene expression of iNOS and TNF-α in TAMs (Figure 3H-I). ELISA also confirmed an increased TNF-α concentration in the serum of mice treated with FMT/PIC (Figure 3J). It is well known that the spread and growth of tumor cells is dependent on the formation of new blood vessels and thus on angiogenesis [58]. Figure 4 illustrated a markedly increased vascular disorganization both surrounding and within the tumor tissues in the group with FMT treatment. Diametrically, a diminished blood vessel density was observed both in the groups of PIC and FMT/PIC treatment. Therefore, these results together suggested that local application of FMT/PIC resulted in vigorous melanoma regression via an immune-activated tumor microenvironment.
Superior therapeutic effect of FMT/PIC in vivo. C57BL/6 mice were subcutaneously inoculated with 5 × 105 B16F10 cells and upon reaching an average volume of 100 mm3, they were intratumorally treated with PBS, FMT (10 mg/kg), PIC (1 mg/kg), and FMT/PIC (10 mg/kg + 1 mg/kg) (5 per group) every other day for three times. (A) Photograph of dissected tumor tissues from mice with the indicated treatment. (B) Tumor growth curves of B16F10 tumors with the indicated treatment. (C) B16F10 tumor weight of mice with the indicated treatment. (D) Survival curves of B16F10 tumor-bearing mice with the indicated treatment. (E) Histopathologic analyses of tumor cells stained with Ki67 in tumor tissues with the indicated treatment, Scale bar: 50 μm. (F) FCM analysis of CD11b+ cell populations in tumor tissues with the indicated treatment. (G) Histopathologic analyses of macrophages stained with F4/80 in tumor tissues with the indicated treatment. (H, I) Relative gene expression of iNOS and TNF-α in TAMs quantified by qRT-PCR. (J) ELISA analysis of TNF-α secretion in the blood of B16F10 tumor-bearing mice with the indicated treatment. All representative data are from three independent experiments. Error bars, SD. *P < 0.05, **P < 0.01, ***P < 0.001. ELISA: enzyme linked immunosorbent assay; FCM: flow cytometry; FMT: ferumoxytol; PIC: poly I:C, polyinosinic-polycytidylic acid; TAMs: tumor-associated macrophages. TNF-α: tumor necrosis factor α.
Effect of FMT/PIC combined therapy on tumor blood vessels. C57BL/6 mice were subcutaneously inoculated with 5 × 105 B16F10 cells and when the tumors reached an average volume of 100 mm3, they were intratumorally treated with PBS, FMT (10 mg/kg), PIC (1 mg/kg), and FMT/PIC (10 mg/kg + 1 mg/kg) (5 per group) every other day for three times. (A) Photograph of dissected tumor tissue covered with vascular tissues from B16F10 tumor-bearing mice with the indicated treatment. (B) LSCM images of blood vessels in tumor sections of mice with the indicated treatment. DAPI, blue. AF594-coupled streptavidin marked lectin, red. (C) Corresponding quantitative percentage of blood vessel area in the total stained area displayed in (B). All representative data are from three independent experiments. Error bars, SD. *P < 0.05, **P < 0.01, ***P < 0.001. DAPI: 4',6-diamidino-2-phenylindole; FCM: flow cytometry; FMT: ferumoxytol; LSCM: laser scanning confocal microscopy; PIC: poly I:C, polyinosinic-polycytidylic acid.
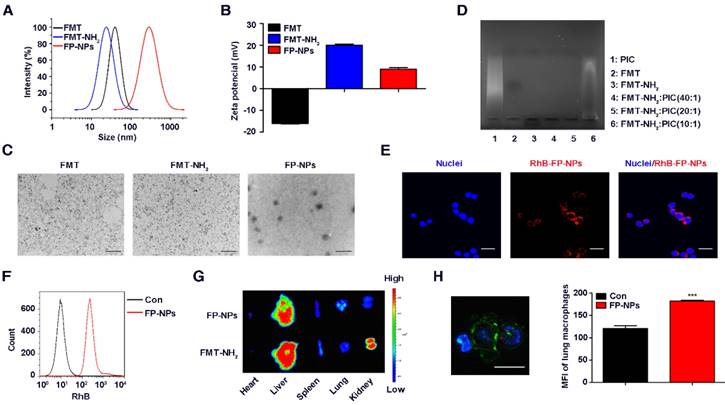
Synthesis and characterization of FP-NPs
For systemic delivery of PIC, an engineered nanocomposite of FMT and PIC was constructed. Firstly, EDC-activated propylene diamine was modified to conjugate with the carboxyl group of dextran to obtain a positively charged amino group. Dynamic light scattering (DLS) showed that the zeta potential of FMT (-16 mV) successfully grafted with the amino group (FMT-NH2) was +20 mV, and its hydration particle size was decreased slightly from 70 nm to 60 nm (Figure 5A-B). TEM images revealed a homogeneous dispersion of spherical FMT-NH2 nanoparticles with an average diameter of 7 nm, approximately the same as that of FMT (Figure 5C). Next, FMT-NH2 NPs were combined with PIC at various w/w ratios via electrostatic adsorption to form FMT-NH2-PIC nano-complexes (FP-NPs), and PIC binding ability was tested using an agarose gel retardation assay. As depicted in Figure 5D, FMT-NH2 fully averted PIC migration at the mass ratio of 20/1 and 40/1, which proved a complete complex formation between FMT and PIC. Meanwhile, FMT and FMT-NH2 in gel electrophoresis showed opposite migration directions, which indirectly verified a successful graft of amino groups in the FMT-NH2. Further, the decreased zeta potential from +20 mV to +9 mV for FMT-NH2 grafted with PIC at a mass ratio of 20/1 also illustrated the efficient recombination of FMT and PIC (Figure 5B). Images from TEM further revealed that the engineered FP-NPs were uniform particles with diameter of approximately 250 nm (Figure 5C), and the results of DLS indicated that its size was about 290 nm (Figure 5A). To monitor the efficiency of FP-NPs internalized by macrophages, FMT-NH2 was fluorescently labeled with RhB, and the cellular uptake of FP-NPs was visualized by LSCM and quantified by FCM. LSCM analysis showed that FP-NPs were efficiently internalized into RAW 264.7 cells (Figure 5E), and FCM also verified this phenomenon (Figure 5F). In addition, ex vivo imaging of dissected tissues showed that despite both being abundantly accumulated in the liver, FP-NPs had significantly higher lung accumulation than FMT-NH2, which was more localized in the kidney and spleen (Figure 5G). The results of LSCM and FCM further showed that the FP-NPs were taken up by the lung macrophages (Figure 5H). Collectively, these data suggested the successful preparation of FP-NPs with properties of efficient uptake by macrophages and accumulation in the lungs of mice.
Synthesis and characterization of FP-NPs. (A, B) The size distribution and zeta potential profile of FMT, FMT-NH2, and FP-NPs was determined by DLS. (C) The images of FMT, FMT-NH2 nanoparticles, and FP-NPs nanocomposites were obtained by TEM. Scale bar: FMT, FMT-NH2: 100 nm, FP-NPs: 1 μm. (D) FMT-NH2 complexed with PIC at w/w ratios of 10/1, 20/1, and 40/1 was tested by the agarose gel retardation assay. (E) RAW 264.7 cells were incubated with RhB labeled FP-NPs for 6 h and the uptake of FP-NPs was detected by LSCM, scale bar: 20 μm. (F) RAW 264.7 cells were incubated with RhB labeled FP-NPs for 6 h and the uptake of FP-NPs was measured by FCM. (G) FP-NPs fluorescently labeled with NIR797 were intravenously injected into mice pre-incubated with B16F10 cells for 8 h and the ex vivo imaging of major organs was carried out on the IVIS Spectrum In Vivo Imaging System. (H) FP-NPs fluorescently labeled with RhB were intravenously injected into mice pre-incubated with B16F10 cells for 4 h and the uptake of FP-NPs by lung macrophages was analyzed by LSCM (left) and FCM (right). Blue: DAPI, Green: F4/80, Red: FP-NPs. Scale bar: 10 μm. All representative data are from three independent experiments. Error bars, SD. *P < 0.05, **P < 0.01, ***P < 0.001. DAPI: 4',6-diamidino-2-phenylindole; DLS: dynamic light scattering; FCM: flow cytometry; FMT: ferumoxytol; FP-NPs: FMT-NH2-poly I:C; LSCM: laser scanning confocal microscope; MFI: mean fluorescence intensity; PIC: poly I:C, polyinosinic-polycytidylic acid; RhB: rhodamine B; TEM: transmission electron microscopy.
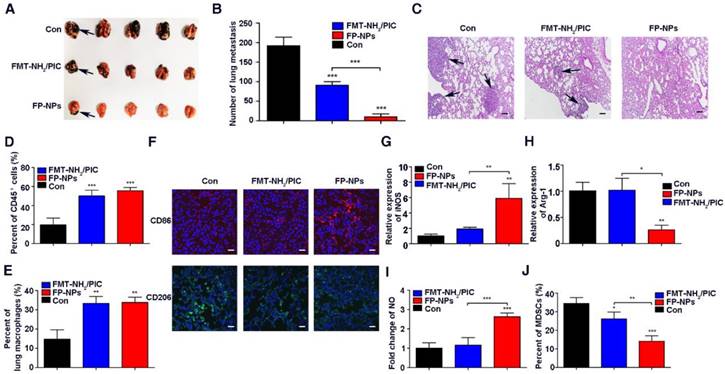
Therapeutic benefits of FP-NPs for lung metastasis
Encouraged by the synergistic therapeutic effect of FMT and PIC on subcutaneous tumors, the therapeutic potential of FP-NPs for metastatic melanoma was evaluated. FMT-NH2 combined with PIC (FMT-NH2/PIC) was used for comparison. First, C57BL/6 mice with lung metastasis were prepared by intravenous injection with B16F10 melanoma cells and then, the randomized mice were treated with PBS, FP-NPs or FMT-NH2/PIC. As shown in Figure 6A-B, administration of FMT-NH2/PIC and FP-NPs both exhibited therapeutic benefits with reduced pulmonary metastasis compared to the control. Nevertheless, the number of lung colonies was drastically reduced or even absent in FP-NPs-treated mice. Hematoxylin and eosin staining (H&E) of lung sections (Figure 6C) also revealed that FP-NPs were superior compared to FMT-NH2/PIC with better anti-metastasis activity and reduced metastatic tumor deposits in the lung. In further pursuit of the potential mechanism by which FMT-NH2/PIC and FP-NPs efficaciously mediated pulmonary metastasis regression, immune cells in the lung were examined. FCM analysis revealed that both FMT-NH2/PIC and FP-NPs exerted excellent immunostimulatory effects, leading to significant accumulation of both CD45+ immune cells and macrophages in the lung and that there were no differences between the two treatments (Figure 6D-E). Strikingly, further immunofluorescence analysis by LSCM indicated that systemic administration of FP-NPs significantly increased the number of macrophages highly expressing CD86 with loss of the M2 marker, CD206 (Figure 6F). Treatment with FP-NPs dramatically increased the M1 marker gene, iNOS, decreased the M2 marker Arg-1, and notably upregulated the secretion of NO (Figure 6G-I). Myeloid-derived suppressor cells (MDSCs) are thought to impair antitumor immunity [59, 60]. In addition, systemic delivery of FP-NPs also resulted in remarkably reduced numbers of MDSCs in blood compared to the combined treatment of FMT-NH2/PIC (Figure 6J).
Therapeutic benefits of FP-NPs for lung metastasis. C57BL/6 mice bearing lung metastasis were prepared by intravenous injection with 5 × 105 B16F10 melanoma cells overnight and were intravenously treated with PBS, FP-NPs (FMT-NH2 200 μg composited with PIC 10 μg), and FMT-NH2 (200 μg) combined with PIC (10 μg) every other day for three times. (A) Photograph of dissected lung tissues from tumor-bearing mice after the indicated treatment. The blank arrows indicated the metastatic nodules. (B) Corresponding quantitative metastatic nodules displayed in (A). (C) Histopathologic analysis of H&E-stained tissue sections of the lung in tumor-bearing mice with the indicated treatment. The blank arrows indicated the metastatic nodules. (D, E) FCM analysis of leukocyte and macrophage populations within lung tissues of tumor-bearing mice with the indicated treatment. (F) Representative immunofluorescence staining for CD86 (red) and CD206 (green) in lung metastatic mice with the indicated treatment, Scale bar: 30 μm. (G-I) Relative gene expression of iNOS and Arg-1, and NO concentration in lung tissues quantified by qRT-PCR and Griess test, respectively. (J) FCM analysis of MDSC populations in the blood of tumor bearing mice with the indicated treatment. All representative data are from three independent experiments. Error bars, SD. *P < 0.05, **P < 0.01, ***P < 0.001. FCM: flow cytometry; FMT: ferumoxytol; FP-NPs: FMT-NH2-poly I:C; iNOS: inducible nitric oxide synthase; MDSCs: myeloid-derived suppressor cells; NO: nitric oxide. PIC: poly I:C, polyinosinic-polycytidylic acid.
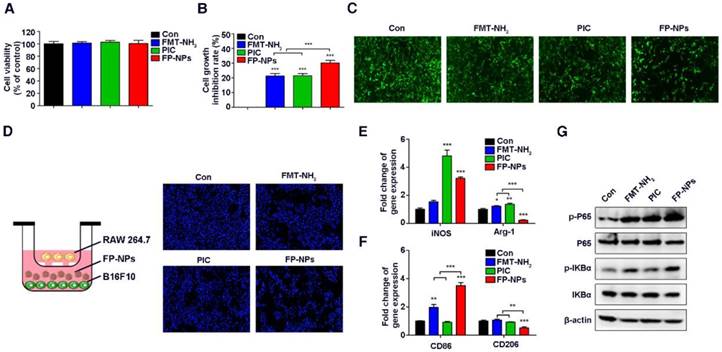
To further test whether FP-NPs are better for anti-metastasis treatment than FMT-NH2/PIC, mice with 4T1 breast cancer lung metastasis were grouped and treated as mentioned above. As data showed, although combined therapy with FMT-NH2/PIC effectively alleviated the pulmonary metastasis, the therapeutic benefit of FP-NPs was more significant and displayed even less lung tumor nodules and 4T1 cell numbers in the lungs (Figure S2A-D) as well as declined MDSC numbers in blood (Figure S2E). Nanoparticle-based remedies are always accompanied by major concerns about biological safety [61]. In our study, no obvious toxicity was observed in 4T1 breast lung metastatic mice exposed to FP-NPs, and the treated mice maintained the increased body weight (Figure S2F), indicating that the dosage of FP-NPs used in our study was fairly appropriate.
To further investigate the mechanism of FP-NPs inhibition in lung metastases, the effect of FMT-NH2, PIC, and FP-NPs on tumor cells and macrophages was examined in vitro. It is worth mentioning here that although the FMT-NH2/PIC combined treatment group was set up in vivo, due to the sequential injection of FMT-NH2 and PIC, they were less likely to act on tumor cells and macrophages together. Therefore, we set up the above groups to study the mechanism in vitro. Similar to the work performed above, we found that FMT-NH2, PIC, or FP-NPs alone barely affected the viability of B16F10 cells (Figure 7A); however, the treatment inhibited their proliferation when co-cultured with macrophages, wherein the performance of FP-NPs was more distinct than that of either FMT-NH2 or PIC (Figure 7B-C). Fluorescence images of B16F10 nuclei co-cultured with RAW 264.7 in the lower dual-chamber Transwell system directly confirmed these findings (Figure 7D). In addition, qRT-PCR results showed that compared with FMT-NH2 and PIC, FP-NPs efficaciously promoted the expression of M1 related genes such as iNOS and CD86 and significantly downregulated the expression of M2 marker genes, Arg-1 and CD206 (Figure 7E-F). It is well known that NF-κB activation positively mediates the tumoricidal M1-like property of macrophages [62]. Results of WB analysis showed that FP-NPs significantly promoted the phosphorylation of IκBα and P65 compared to the monotherapies with FMT-NH2 or PIC (Figure 7G), indicating that FP-NPs promoted macrophage polarization to M1 for tumor cell growth inhibition through the NF-κB signaling pathway.
Effect of FP-NPs on tumor cell proliferation and macrophage polarization. (A) B16F10 cells were incubated with FMT-NH2, PIC, or FP-NPs for 48 h and the cell viability was analyzed by CCK-8 assay. (B) B16F10 cells pre-labeled with CFSE co-cultured with RAW 264.7 at a ratio of 2:1 were incubated with FMT-NH2, PIC, or FP-NPs for 48 h and cell proliferation was then analyzed by FCM. (C) GFP-B16F10 cells co-cultured with RAW 264.7 at a ratio of 2:1 were incubated with FMT-NH2, PIC, or FP-NPs for 48 h and the fluorescence intensity of GFP was captured. (D) Left: Schematic diagram of the co-culture. Right: B16F10 cells co-cultured with RAW 264.7 at a ratio of 2:1 were incubated with FMT-NH2, PIC, or FP-NPs in a dual-chamber Transwell system for 48 h and the tumor cells in the lower chamber were stained with DAPI and analyzed by fluorescence microscopy. (E, F) RAW 264.7 were incubated with FMT-NH2, PIC, or FP-NPs for 12 h and the expression of macrophage M1 (iNOS, CD86) and M2 (Arg-1, CD206) related genes was analyzed by qRT-PCR. (G) RAW 264.7 cells were incubated with FMT-NH2, PIC, or FP-NPs for 15 min and the expression of target proteins was analyzed by WB. All representative data are from three independent experiments. Error bars, SD. *P < 0.05, **P < 0.01, ***P < 0.001. FCM: flow cytometry; FMT: ferumoxytol; FP-NPs: FMT-NH2-poly I:C; iNOS: inducible nitric oxide synthase; PIC: poly I:C, polyinosinic-polycytidylic acid. WB: western blotting.
Discussion
An increasing number of researches have shown that iron oxide NPs, commonly used carriers for anticancer agent delivery, are gaining momentum as potent carriers of genes or adjuvants for cancer immunotherapy [63-65]. However, it has been overlooked that iron oxide NPs themselves can be used as an immunopotentiator to boost immunity and suppress tumor growth [41]. Currently, considerable attention has been focused on the impact of combination immunotherapy with the potential to achieve additive or synergistic effects for tumor treatment [16, 66]. All these clues seem to predict that employing combined treatment with FMT and the TLR3 agonist, PIC, one of the agents with high potential to boost cancer immunotherapy as ranked by the National Cancer Institute [67], or systemic delivery of PIC based on FMT would be more efficient for macrophage activation and tumor regression compared to either treatment alone. Melanoma is a systemic, malignant tumor with high incidence of metastasis, and the extraordinarily high cost of treatment with an extremely low cure rate brings great pain to patients [10]. In our present study, an innovative strategy for melanoma treatment based on the fairly universal, economical, and favorably secure FMT was explored. FMT, when used in combination with the TLR3 agonist PIC barely affected the viability of B16F10 melanoma cells, but synergistically attenuated cell proliferation in a co-culture system by converting macrophages to a tumoricidal phenotype with enhanced tumor-cytotoxic activities and phagocytosis. Further in vivo studies showed that FMT or FMT-NH2 combined with PIC, respectively, reduced the subcutaneous melanoma burden and alleviated the lung metastases of melanoma. Notably, in comparison, when the FMT was surface functionalized with PIC to form a nanostructure for systemic delivery, the novel nano-composition of FP-NPs demonstrated a more distinguished anti-metastasis effect. This was consistent with literature reports that targeted delivery of TLR agonist by nano-based systems could provide enhanced antitumor immunity compared to the individual treatment [68].
Numerous studies have verified that cationic liposomes and other carriers of PIC can effectively activate DCs and augment tumor-specific CTL responses [69, 70]. However, unlike existing studies, the FMT used here acted as a carrier for systemic PIC delivery and synergized with PIC for macrophage activation, facilitating synergistic tumor suppression. In addition, different from previous reports focusing on the co-delivery of tumor antigens and DCs for T cell activation, the FP-NPs used here could directly inhibit tumor cell growth by steering macrophages to the M1 phenotype. Although it has been reported that TAM-targeted transport of PIC with a ZnPP-grafted nanovector is a potential strategy to repolarize TAMs to the M1 phenotype for substantial tumor suppression, this strategy may only be limited to solid tumors [71]. In our study, the combined treatment of FMT and PIC could synergistically reduce the subcutaneous melanoma burden as well as eliminate metastatic colonies, and the engineered nano-composition of FP-NPs showed a better performance for anti-metastasis. Interestingly, the effect of FP-NPs on tumor metastasis was not limited to aggressive melanoma, as they could also effectively inhibit 4T1 breast cancer metastasis, indicating that they are a versatile reagent with a broad spectrum of anti-tumor effects. Tumor associated antigens are known to be capable of eliciting effective antitumor T-cell responses. Co-delivery of vaccine formulations including antigens and immunopotentiators like PIC using biomaterials such as injectable polypeptide hydrogel or cationic polypeptide micelles can activate DCs and stimulate CTL responses against cancer [72]. However, although tumor cell lysates are effective for activating immunity, they are insufficient to overcome the tumor as they are different from neo-antigens that are unique, mutated antigens with a higher likelihood of recognition by host immune cells. Human melanoma cell lines with a little amount of PIC delivered to the cytosol reportedly become highly sensitive to apoptosis [73]. Therefore, it is predicted that FP-NPs can efficiently kill human melanoma cells because they can easily be transfected into cells and that the antigens released from killed cells would further stimulate antigen presenting cells (APCs, such as macrophages and DCs) to augment the immunotherapy. More intriguingly, the preparation of FP-NPs is simple and easily achievable through the electrostatic interaction between FMT-NH2 and PIC. As the TLR3 agonist, Poly-ICLC, another form of PIC, has already been widely used in pre-clinical practice and FMT used here is a clinically approved nanoparticle with a little or no effect on cell viability, it is predicted that this will provide a further framework for future clinical translation. Thus, the novel strategy for tumor treatment not only enriches the therapeutic nature of FMT but also provides a meaningful reference for advanced melanoma in the clinical setting.
Conclusions
To our knowledge, this is the first report that FMT could be used to synergize with the TLR3 agonist either in combination or in the form of a composite to induce macrophage activation for primary as well as metastatic melanoma regression. Our results indicate that the transport of TLR3 immunologic adjuvants with FMT-grafted nanovectors can be a potential approach for effective immune nanotheranostics.
Abbreviations
BMDMs: bone marrow-derived macrophages; CTLA-4: cytotoxic T-lymphocyte-associated antigen-4; CTL: cytotoxic T lymphocyte; DAPI: 4',6-diamidino-2-phenylindole; DCFH-DA: dichloro-dihydro-fluorescein diacetate; DCs: dendritic cells; dsRNA: double-stranded RNA; DLS: dynamic light scattering; ELISA: enzyme linked immunosorbent assay; FCM: flow cytometry; FMT: ferumoxytol; FP-NPs: FMT-NH2-poly I:C; iNOS: inducible nitric oxide synthase; LSCM: laser scanning confocal microscope; MDSCs: myeloid-derived suppressor cells; NAC: N-acetyl-cysteine; NO: nitric oxide; NPs: nanoparticles; Poly I:C (PIC): polyinosinic-polycytidylic acid; RhB: rhodamine B; ROS: reactive oxygen species; TAMs: tumor-associated macrophages; TEM: transmission electron microscopy; TLR3: toll-like receptor 3; TNF-α: tumor necrosis factor α; WB: western blotting.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This work was supported by National Key Research and Development Program of China (2017YFA0104303) and grants from the National Natural Science Foundation of China (No. 81872114). We are grateful to Prof. Ning Gu from Southeast University, Nanjing, PR China for providing us with the FMT. Jiaojiao Zhao and Zhengkui Zhang contributed equally to this work. All authors participated in the paper preparation and approved the final paper submission.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Holmes D. The cancer that rises with the sun. Nature. 2014;515:S110-1
2. Jang S, Atkins MB. Treatment of BRAF-mutant melanoma: the role of vemurafenib and other therapies. Clin Pharmacol Ther. 2014;95:24-31
3. Franklin C, Livingstone E, Roesch A, Schilling B, Schadendorf D. Immunotherapy in melanoma: Recent advances and future directions. Eur J Surg Oncol. 2017;43:604-11
4. Mandala M, Voit C. Targeting BRAF in melanoma: biological and clinical challenges. Crit Rev Oncol Hematol. 2013;87:239-55
5. Kudchadkar R, Paraiso KH, Smalley KS. Targeting mutant BRAF in melanoma: current status and future development of combination therapy strategies. Cancer J. 2012;18:124-31
6. Shih K, Arkenau HT, Infante JR. Clinical impact of checkpoint inhibitors as novel cancer therapies. Drugs. 2014;74:1993-2013
7. Fecher LA, Agarwala SS, Hodi FS, Weber JS. Ipilimumab and its toxicities: a multidisciplinary approach. Oncologist. 2013;18:733-43
8. Sahin U, Derhovanessian E, Miller M, Kloke BP, Simon P, Lower M. et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature. 2017;547:222-6
9. Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ. et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. 2017;547:217-21
10. Coricovac D, Dehelean C, Moaca EA. Cutaneous Melanoma-A Long Road from Experimental Models to Clinical Outcome: A Review. Int J Mol Sci. 2018:19
11. Shapouri-Moghaddam A, Mohammadian S, Vazini H, Taghadosi M, Esmaeili SA, Mardani F. et al. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol. 2018;233:6425-40
12. Franklin RA, Liao W, Sarkar A, Kim MV, Bivona MR, Liu K. et al. The cellular and molecular origin of tumor-associated macrophages. Science. 2014;344:921-5
13. Wang H, Yang L, Wang D, Zhang Q, Zhang L. Pro-tumor activities of macrophages in the progression of melanoma. Hum Vaccin Immunother. 2017;13:1556-62
14. Kale S, Raja R, Thorat D, Soundararajan G, Patil TV, Kundu GC. Osteopontin signaling upregulates cyclooxygenase-2 expression in tumor-associated macrophages leading to enhanced angiogenesis and melanoma growth via alpha9beta1 integrin. Oncogene. 2014;33:2295-306
15. Izraely S, Ben-Menachem S, Sagi-Assif O, Telerman A, Zubrilov I, Ashkenazi O. et al. The Metastatic Microenvironment: Melanoma-Microglia Cross-Talk Promotes the Malignant Phenotype of Melanoma Cells. Int J Cancer. 2018 [Epub ahead of print]
16. Perry CJ, Munoz-Rojas AR. Myeloid-targeted immunotherapies act in synergy to induce inflammation and antitumor immunity. J Exp Med. 2018;215:877-93
17. Mantovani A, Allavena P. The interaction of anticancer therapies with tumor-associated macrophages. J Exp Med. 2015;212:435-45
18. Komohara Y, Fujiwara Y, Ohnishi K, Takeya M. Tumor-associated macrophages: Potential therapeutic targets for anti-cancer therapy. Adv Drug Deliv Rev. 2016;99:180-5
19. Wu Q, Allouch A, Paoletti A, Leteur C, Mirjolet C, Martins I. et al. NOX2-dependent ATM kinase activation dictates pro-inflammatory macrophage phenotype and improves effectiveness to radiation therapy. Cell Death Differ. 2017;24:1632-44
20. Sengupta M, Pal R, Nath A, Chakraborty B, Singh LM, Das B. et al. Anticancer efficacy of noble metal nanoparticles relies on reprogramming tumor-associated macrophages through redox pathways and pro-inflammatory cytokine cascades. Cell Mol Immunol. 2018 [Epub ahead of print]
21. Cassetta L, Kitamura T. Macrophage targeting: opening new possibilities for cancer immunotherapy. Immunology. 2018;155:285-293
22. Kollmann TR, Levy O, Montgomery RR, Goriely S. Innate immune function by Toll-like receptors: distinct responses in newborns and the elderly. Immunity. 2012;37:771-83
23. Tatematsu M, Seya T, Matsumoto M. Beyond dsRNA: Toll-like receptor 3 signalling in RNA-induced immune responses. Biochem J. 2014;458:195-201
24. De Waele J, Marcq E, Van Audenaerde JR, Van Loenhout J, Deben C, Zwaenepoel K. et al. Poly(I:C) primes primary human glioblastoma cells for an immune response invigorated by PD-L1 blockade. Oncoimmunology. 2018;7:e1407899
25. Kyi C, Roudko V, Sabado R, Saenger YM, Loging W, Mandeli J. et al. Therapeutic Immune Modulation Against Solid Cancers with Intratumoral Poly-ICLC: A Pilot Trial. Clin Cancer Res. 2018;24:4937-4948
26. Matsumoto M, Takeda Y, Tatematsu M, Seya T. Toll-Like Receptor 3 Signal in Dendritic Cells Benefits Cancer Immunotherapy. Front Immunol. 2017;8:1897
27. Takeda Y, Azuma M, Matsumoto M, Seya T. Tumoricidal efficacy coincides with CD11c up-regulation in antigen-specific CD8(+) T cells during vaccine immunotherapy. J Exp Clin Cancer Res. 2016;35:143
28. Han HD, Byeon Y, Kang TH, Jung ID, Lee JW, Shin BC. et al. Toll-like receptor 3-induced immune response by poly (d,l-lactide-co-glycolide) nanoparticles for dendritic cell-based cancer immunotherapy. Int J Nanomedicine. 2016;11:5729-42
29. Muller E, Christopoulos PF, Halder S, Lunde A, Beraki K, Speth M. et al. Toll-Like Receptor Ligands and Interferon-gamma Synergize for Induction of Antitumor M1 Macrophages. Front Immunol. 2017;8:1383
30. Hafner AM, Corthesy B, Merkle HP. Particulate formulations for the delivery of poly(I:C) as vaccine adjuvant. Adv Drug Deliv Rev. 2013;65:1386-99
31. Ammi R, De Waele J, Willemen Y, Van Brussel I, Schrijvers DM, Lion E. et al. Poly(I:C) as cancer vaccine adjuvant: knocking on the door of medical breakthroughs. Pharmacol Ther. 2015;146:120-31
32. Chen H, Zhang W, Zhu G, Xie J, Chen X. Rethinking cancer nanotheranostics. Nat Rev Mater. 2017:2
33. Irvine DJ. Materializing the future of vaccines and immunotherapy. Nat Rev Mater. 2016;1:15008
34. Ramani M. Zinc Oxide Nanoparticle-Poly I:C RNA Complexes: Implication as Therapeutics against Experimental Melanoma. Mol Pharm. 2017;14:614-25
35. Sokolova V, Shi Z, Huang S, Du Y, Kopp M, Frede A. et al. Delivery of the TLR ligand poly (I:C) to liver cells in vitro and in vivo by calcium phosphate nanoparticles leads to a pronounced immunostimulation. Acta Biomater. 2017;64:401-10
36. Kim SY, Noh YW, Kang TH, Kim JE, Kim S, Um SH. et al. Synthetic vaccine nanoparticles target to lymph node triggering enhanced innate and adaptive antitumor immunity. Biomaterials. 2017;130:56-66
37. Lu M, Cohen MH, Rieves D, Pazdur R. FDA report: Ferumoxytol for intravenous iron therapy in adult patients with chronic kidney disease. Am J Hematol. 2010;85:315-9
38. Aghighi M, Theruvath AJ, Pareek A. Magnetic Resonance Imaging of Tumor Associated Macrophages: Clinical Translation. Clin Cancer Res. 2018;24:4110-4118
39. Shi J, Yu X, Wang L, Liu Y, Gao J, Zhang J. et al. PEGylated fullerene/iron oxide nanocomposites for photodynamic therapy, targeted drug delivery and MR imaging. Biomaterials. 2013;34:9666-77
40. Niculaes D, Lak A, Anyfantis GC, Marras S, Laslett O, Avugadda SK. et al. Asymmetric Assembling of Iron Oxide Nanocubes for Improving Magnetic Hyperthermia Performance. ACS Nano. 2017;11:12121-33
41. Zanganeh S, Hutter G, Spitler R, Lenkov O, Mahmoudi M, Shaw A. et al. Iron oxide nanoparticles inhibit tumour growth by inducing pro-inflammatory macrophage polarization in tumour tissues. Nat Nanotechnol. 2016;11:986-94
42. Huang YJ, Hung KC, Hung HS, Hsu SH. Modulation of Macrophage Phenotype by Biodegradable Polyurethane Nanoparticles: Possible Relation between Macrophage Polarization and Immune Response of Nanoparticles. ACS Appl Mater Interfaces. 2018;10:19436-48
43. Kim H, Niu L, Larson P, Kucaba TA, Murphy KA, James BR. et al. Polymeric nanoparticles encapsulating novel TLR7/8 agonists as immunostimulatory adjuvants for enhanced cancer immunotherapy. Biomaterials. 2018;164:38-53
44. Zhao J, Pan Y, Li X, Zhang X, Xue Y, Wang T. et al. Dihydroartemisinin and Curcumin Synergistically Induce Apoptosis in SKOV3 Cells Via Upregulation of MiR-124 Targeting Midkine. Cell Physiol Biochem. 2017;43:589-601
45. Chen B, Sun J, Fan F, Zhang X, Qin Z, Wang P. et al. Ferumoxytol of ultrahigh magnetization produced by hydrocooling and magnetically internal heating co-precipitation. Nanoscale. 2018;10:7369-76
46. Chen B, Li Y, Zhang X, Liu F, Liu Y, ji M. et al. An efficient synthesis of ferumoxytol induced by alternating-current magnetic field. Mater Lett. 2016;170:93-6
47. Wanderley CW, Colon DF, Luiz JPM, Oliveira FF, Viacava PR, Leite CA. et al. Paclitaxel Reduces Tumor Growth by Reprogramming Tumor-Associated Macrophages to an M1 Profile in a TLR4-Dependent Manner. Cancer res. 2018;78:5891-900
48. Shi C, Jia T, Mendez-Ferrer S, Hohl TM, Serbina NV, Lipuma L. et al. Bone marrow mesenchymal stem and progenitor cells induce monocyte emigration in response to circulating toll-like receptor ligands. Immunity. 2011;34:590-601
49. Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez-Retamozo V, Panizzi P. et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science. 2009;325:612-6
50. Cortez-Retamozo V, Etzrodt M, Newton A, Rauch PJ, Chudnovskiy A, Berger C. et al. Origins of tumor-associated macrophages and neutrophils. Proc Natl Acad Sci U S A. 2012;109:2491-6
51. Jenkins SJ, Ruckerl D, Cook PC, Jones LH, Finkelman FD, van Rooijen N. et al. Local macrophage proliferation, rather than recruitment from the blood, is a signature of TH2 inflammation. Science. 2011;332:1284-8
52. Feng M, Chen JY, Weissman-Tsukamoto R, Volkmer JP, Ho PY, McKenna KM. et al. Macrophages eat cancer cells using their own calreticulin as a guide: roles of TLR and Btk. Proc Natl Acad Sci U S A. 2015;112:2145-50
53. Singel KL, Segal BH. NOX2-dependent regulation of inflammation. Clin Sci. 2016;130:479-90
54. Kovac S, Angelova PR, Holmstrom KM, Zhang Y, Dinkova-Kostova AT, Abramov AY. Nrf2 regulates ROS production by mitochondria and NADPH oxidase. Biochim Biophys Acta. 2015;1850:794-801
55. Martinez J, Malireddi RK, Lu Q, Cunha LD, Pelletier S, Gingras S. et al. Molecular characterization of LC3-associated phagocytosis reveals distinct roles for Rubicon, NOX2 and autophagy proteins. Nat Cell Biol. 2015;17:893-906
56. Raj L, Ide T, Gurkar AU, Foley M, Schenone M, Li X. et al. Selective killing of cancer cells by a small molecule targeting the stress response to ROS. Nature. 2011;475:231-4
57. Sun Y, Li J, Zhang Y, Tu Y, Huang C, Tao J. et al. The Polysaccharide Extracted from Umbilicaria esculenta Inhibits Proliferation of Melanoma Cells through ROS-Activated Mitochondrial Apoptosis Pathway. Biol Pharm Bull. 2018;41:57-64
58. Bergers G, Benjamin LE. Tumorigenesis and the angiogenic switch. Nat rev Cancer. 2003;3:401-10
59. Ostrand-Rosenberg S, Fenselau C. Myeloid-Derived Suppressor Cells: Immune-Suppressive Cells That Impair Antitumor Immunity and Are Sculpted by Their Environment. J Immunol. 2018;200:422-31
60. Veglia F, Perego M. Myeloid-derived suppressor cells coming of age. Nat Immunol. 2018;19:108-19
61. Fenton OS, Olafson KN, Pillai PS, Mitchell MJ, Langer R. Advances in Biomaterials for Drug Delivery. Adv Mater. 2018 e1705328
62. Biswas SK, Lewis CE. NF-κB as a central regulator of macrophage function in tumors. J Leukoc Biol. 2010;88:877-84
63. Lee JY, Son HY, Park JC, Park J, Nam YS. Paclitaxel-induced formation of 3D nanocrystal superlattices within injectable protein-based hybrid nanoparticles. Chem Commun. 2018;54:11586-11589
64. Cho HY, Han J, Pasquale NJ, Rabie H, Kim JH, Choi JW. Overcoming Chemoresistance in Cancer via Combined MicroRNA Therapeutics with Anticancer Drugs Using Multifunctional Magnetic Core-Shell Nanoparticles. ACS Appl Mater Interfaces. 2018;10:26954-26963
65. Zhao Y, Zhao X, Cheng Y, Guo X, Yuan W. Iron Oxide Nanoparticles-Based Vaccine Delivery for Cancer Treatment. Mol Pharm. 2018;15:1791-9
66. Wiehagen KR, Girgis NM, Yamada DH, Smith AA, Chan SR, Grewal IS. et al. Combination of CD40 Agonism and CSF-1R Blockade Reconditions Tumor-Associated Macrophages and Drives Potent Antitumor Immunity. Cancer Immunol Res. 2017;5:1109-21
67. Cheever MA. Twelve immunotherapy drugs that could cure cancers. Immunol Rev. 2008;222:357-68
68. Mahjub R, Jatana S, Lee SE, Qin Z, Pauli G, Soleimani M. et al. Recent advances in applying nanotechnologies for cancer immunotherapy. J Control Release. 2018;288:239-63
69. Luo Z, Li P, Deng J, Gao N, Zhang Y, Pan H. et al. Cationic polypeptide micelle-based antigen delivery system: a simple and robust adjuvant to improve vaccine efficacy. J Control Release. 2013;170:259-67
70. Bocanegra Gondan AI, Ruiz-de-Angulo A, Zabaleta A, Gomez Blanco N, Cobaleda-Siles BM, Garcia-Granda MJ. et al. Effective cancer immunotherapy in mice by polyIC-imiquimod complexes and engineered magnetic nanoparticles. Biomaterials. 2018;170:95-115
71. Liu L, He H, Liang R. ROS-Inducing Micelles Sensitize Tumor-Associated Macrophages to TLR3 Stimulation for Potent Immunotherapy. Biomacromolecules. 2018;19:2146-55
72. Song H, Huang P, Niu J, Shi G, Zhang C, Kong D. et al. Injectable polypeptide hydrogel for dual-delivery of antigen and TLR3 agonist to modulate dendritic cells in vivo and enhance potent cytotoxic T-lymphocyte response against melanoma. Biomaterials. 2018;159:119-29
73. Besch R, Poeck H, Hohenauer T, Senft D, Hacker G, Berking C. et al. Proapoptotic signaling induced by RIG-I and MDA-5 results in type I interferon-independent apoptosis in human melanoma cells. J Clin Invest. 2009;119:2399-411
Author contact
![]() Corresponding author: Yayi Hou, The State Key Laboratory of Pharmaceutical Biotechnology, Division of Immunology, Medical School, Nanjing University, Nanjing 210093, China. Tel: +86-25-8968-8441; Fax: +86-25-8968-8441. E-mail: yayihouedu.cn. Or Shuli Zhao, General Clinical Research Center, Nanjing First Hospital, Nanjing Medical University, Nanjing 210006, PR China. E-mail: shulizhao79com.
Corresponding author: Yayi Hou, The State Key Laboratory of Pharmaceutical Biotechnology, Division of Immunology, Medical School, Nanjing University, Nanjing 210093, China. Tel: +86-25-8968-8441; Fax: +86-25-8968-8441. E-mail: yayihouedu.cn. Or Shuli Zhao, General Clinical Research Center, Nanjing First Hospital, Nanjing Medical University, Nanjing 210006, PR China. E-mail: shulizhao79com.