Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results
Discussion
Conclusions
Supplementary Material
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(21):6070-6087. doi:10.7150/thno.27275 This issue Cite
Research Paper
T-cell functionality testing is highly relevant to developing novel immuno-tracers monitoring T cells in the context of immunotherapies and revealed CD7 as an attractive target
Kristine E. Mayer1*, Sabine Mall1,2*, Nahid Yusufi3, Dario Gosmann1, Katja Steiger2,4, Lisa Russelli3, Henrique de Oliviera Bianchi1, Stefan Audehm1, Ricarda Wagner1, Eva Bräunlein1, Anja Stelzl1, Florian Bassermann1,2, Wilko Weichert2,4, Wolfgang Weber2,3, Markus Schwaiger2,3, Calogero D'Alessandria3, Angela M. Krackhardt1,2 ![]()
1. Clinic and Policlinic for Internal Medicine III, Klinikum rechts der Isar, Technical University of Munich, Munich, Germany
2. German Cancer Consortium (DKTK), partner-site Munich; and German Cancer Research Center (DKFZ), Heidelberg, Germany
3. Department of Nuclear Medicine, Klinikum rechts der Isar, Technical University of Munich, Munich, Germany
4. Institute of Pathology, Klinikum rechts der Isar, Technical University of Munich, Munich, Germany
*K.E.M. and S.M. contributed equally to the work
Received 2018-5-15; Accepted 2018-10-14; Published 2018-11-28
Abstract

Cancer immunotherapy has proven high efficacy in treating diverse cancer entities by immune checkpoint modulation and adoptive T-cell transfer. However, patterns of treatment response differ substantially from conventional therapies, and reliable surrogate markers are missing for early detection of responders versus non-responders. Current imaging techniques using 18F-fluorodeoxyglucose-positron-emmission-tomograpy (18F-FDG-PET) cannot discriminate, at early treatment times, between tumor progression and inflammation. Therefore, direct imaging of T cells at the tumor site represents a highly attractive tool to evaluate effective tumor rejection or evasion. Moreover, such markers may be suitable for theranostic imaging.
Methods: We mainly investigated the potential of two novel pan T-cell markers, CD2 and CD7, for T-cell tracking by immuno-PET imaging. Respective antibody- and F(ab´)2 fragment-based tracers were produced and characterized, focusing on functional in vitro and in vivo T-cell analyses to exclude any impact of T-cell targeting on cell survival and antitumor efficacy.
Results: T cells incubated with anti-CD2 and anti-CD7 F(ab´)2 showed no major modulation of functionality in vitro, and PET imaging provided a distinct and strong signal at the tumor site using the respective zirconium-89-labeled radiotracers. However, while T-cell tracking by anti-CD7 F(ab´)2 had no long-term impact on T-cell functionality in vivo, anti-CD2 F(ab´)2 caused severe T-cell depletion and failure of tumor rejection.
Conclusion: This study stresses the importance of extended functional T-cell assays for T-cell tracer development in cancer immunotherapy imaging and proposes CD7 as a highly suitable target for T-cell immuno-PET imaging.
Keywords: T-cell imaging, cancer immunotherapy, T-cell function, immuno-PET
Introduction
Immunotherapy, especially the application of immune checkpoint modulators, has become a highly effective treatment strategy for diverse tumor entities [1,2]. In addition, other immunotherapeutic approaches such as adoptive transfer of T cells transgenic for defined chimeric antigen receptors (CAR) or T-cell receptors (TCR) have proven efficacy in diverse malignancies [3-7]. The US Food and Drug Administration recently approved the use of CD19 CAR T cells for the treatment of advanced B-cell-derived malignancies. However, pharmacodynamics and pharmacokinetics of immune modulators or cellular immunotherapies differ substantially from conventional chemotherapy or molecular therapy. Animal models provide only limited information because of the highly species-specific and individual specificities of the tumor immune microenvironment. Thus, until now, many questions remain unresolved, including the understanding and prediction of response versus primary and adoptive immune resistance, as well as the occurrence of adverse effects at early time points [1,8].
In fact, current computer tomography (CT) imaging techniques and the response evaluation criteria in solid tumors (RECIST) [9] have shown major limitations. In particular, the early distinction between progression and pseudo-progression, as well as a reliable evaluation of response heterogeneity is difficult and requires novel evaluation criteria better adapted to these immunotherapies [10,11]. Similarly, novel evaluation criteria have been established for 18FDG-PET to address these issues [12-15]. Although 18FDG-PET stands out for its high sensitivity [15,16], it has the intrinsic limit that radiolabeled glucose is incorporated by not only metabolically active tumor cells but also metabolically active immune cells. Thus, innovative imaging techniques that can be used as predictive and early surrogate markers covering the whole spectrum of immunotherapeutic response patterns are urgently needed.
In addition, safe tracking of immune cells represents another essential demand when introducing a novel imaging tracer to monitor immunomodulatory therapies. Thus far, imaging methods focusing on tumor detection, such as PSMA imaging for prostate cancer [17], have not raised high safety issues because radioactive labeling can also be used therapeutically with respective tracers and patients may benefit from induction of target cell death.
Currently, several strategies are being tested for their potential as functional immune-imaging approaches in immunotherapies. For genetically modified cells, reporter genes [18-22] can be introduced to specifically identify the modified T cells in vivo. This technique is deemed safe when engineered T cells are injected at the tumor site of patients with malignant glioma [22]. However, this approach is not possible for endogenous T cells activated therapeutically by monoclonal antibodies such as anti-PD-1, anti-PD-L1, or anti-CTLA-4. Direct imaging of T cells, therefore, remains the most comprehensive method in the context of a variety of immunotherapies to evaluate efficacy and immune-mediated toxicity. Subpopulation-specific markers such as CD4 and CD8, as well as the TCR and CD3, have been proposed as targets and tested using various tracer molecules in murine models of immunotherapy [23-31]. Recently, a clinical trial has been initiated targeting CD8+ cells by 89Zr-labeled IAB22M2C in patients with solid tumors and Hodgkin lymphoma (NCT03107663) for which no modulating effects on adoptively transferred T cells have been observed using a T-cell engraftment model [32]. We have previously described a human TCR with a murinized domain (TCRmu) tracking specifically genetically modified T cells but not detecting immune modulated endogenous T cells [33,34]. Of note, the full antibody directed against the murinized domain showed a functional impact on T cells in vitro and in vivo, whereas the derived F(ab´)2 was largely inert [33]. Others have also observed impairment of T-cell functions when specific T-cell imaging technologies targeting CD4 or the TCR are applied [28,29], although in most studies, functionality testing remains insufficiently investigated.
T cells represent the main pharmacodynamically active partners in diverse immunotherapies. However, the effects may be mediated by different T-cell subpopulations [35-41]. With the aim to develop an approach that can be used in different settings, we selected antibodies targeting antigens broadly expressed on T cells and focused on CD2, CD3, and CD7. CD3 is a TCR co-receptor involved in T-cell stimulation and was previously chosen as target structure for T-cell imaging [30,31]. CD2 is expressed on T cells, NK cells, and thymocytes where it plays a role in T-cell adhesion and activation via CD58 [42-46]. Furthermore, the expression of CD2 has been described to highly correlate with cytolytic activity in tumors [47], making this target especially interesting to track effector T cells. CD7 is a member of the immunoglobulin superfamily, although there is limited information about this target so far. It is a known marker for mature T cells and NK cells but also for early-stage hematopoietic precursor cells [48,49].
Murine anti-human antibodies targeting these antigens were selected and tested for their potential as immune-imaging tracers to monitor T cells. The most promising antibody clones maintaining functional antitumor efficacy of targeted T cells in vitro were further investigated in vivo with respective 89Zr-labeled F(ab´)2 using a previously described mouse model of adoptive T-cell transfer [33].
Methods
Primary material and cell lines
Peripheral blood mononuclear cells (PBMC) were isolated via density gradient centrifugation from blood donated by healthy volunteers according to the standards of the local ethical board and the Declaration of Helsinki. Isolation, stimulation, and cultivation of cells were performed as previously described [33,34,50]. PBMC were nonspecifically stimulated with IL-2 (50 U/mL; PeproTech, USA) and OKT3 (30 ng/mL; BioLegend, San Diego, CA) and cultivated in RPMI supplemented with 5% human serum, 5% fetal calf serum, penicillin/streptomycin (100 U/mL), 10 mM non-essential amino acids, 2 mM L-glutamine, 1 mM sodium pyruvate, 10 mM HEPES, and recombinant human IL-7/IL-15 (5 ng/mL each). Human CD8+ central memory T cells (TCM) were isolated from PBMC via CD45RA-CD4-CD62L+ cell isolation using magnetic beads (Miltenyi Biotech, Bergisch Gladbach, Germany) and stimulated with human T-cell activating CD3/CD28 dynabeads (Thermo Fisher Scientific, Waltham, MA) and IL-2 (30 U/mL) according to manufacturer's recommendations.
The following cell lines were used: human acute leukemia cell line ML2 (The CABRI consortium, received in 2004), IL-15-producing NSO cells (provided by S. R. Riddell in 2011; [51]), OKT11 (anti-CD2) hybridoma (P3X63Ag8, Sigma-Aldrich, St.Louis, MO, in 2016), and T3-3A1 (anti-CD7) hybridoma (HB-2, ATCC, Manassas,VI, in 2016).
ML2 cells were retrovirally transduced with genes coding for HLA-B*07:02 (B7) or HLA-B*15:01 (B15) linked to enhanced green fluorescent protein (GFP). The stable ML2-derived cell lines ML2-B7GFP (ML2-B7) and ML2-B15GFP (ML2-B15) were generated by sorting and single cell cloning, respectively [33].
TCM were transduced retrovirally with the previously described MPO5-specific TCR 2.5D6 linked to red fluorescent protein (TCR2.5D6iRFP TCM) [33,50] or iRFP only (iRFP TCM). In addition to microscopic analysis, the cell lines were regularly tested for mycoplasma infection (VenorGeM Mycoplasma Detection Kit, Minerva Biolabs, Berlin, Germany) and cell line-specific surface markers.
Antibodies and F(ab´)2, blocking and Kd determination
For the clones OKT11 (anti-CD2) and T3-3A1 (anti-CD7), antibodies were isolated from the supernatant of the hybridoma cells using Protein A-Sepharose (GE Healthcare, Chicago, IL) and F(ab´)2 was produced by pepsin digestion using F(ab´)2-Preparation Kit (Thermo Fisher Scientific, Waltham, MA), as described elsewhere [33,34]. Protein purity was assessed by size-exclusion high-performance liquid chromatography (SE-HPLC; BioSep™ SEC-s3000 LC Column, Phenomenex, Torrance, CA) and binding function was confirmed by enzyme-linked immunosorbent assay (ELISA). In addition, the first in vitro experiments were performed with anti-CD3 antibodies of the clones BC3 (BioLegend, San Diego, CA), VIT3b (kindly provided by Institute of Immunology, Medical University Vienna), and OKT3 (BioLegend, San Diego, CA), as well as another anti-CD7 antibody (clone 4H9; Caprico Biotechnology, Norcross, GA). Anti-CD3 antibody OKT3 (Thermo Fisher Scientific, Waltham, MA) served as positive control [52,53], whereas mouse IgG1, IgG2a, and IgG2b isotype antibodies (Thermo Fisher Scientific, Waltham, MA) served as negative control.
To determine specific binding of T3-3A1 (anti-CD7) IgG1 and IgG2a antibody, CD7 blocking analysis of PBMC-derived T cells was performed as follows. Cells were stained with T3-3A1 (anti-CD7) antibody with and without pre-incubation of a polyclonal sheep anti-human CD7 antibody (R&D Systems, Minneapolis, MN). Afterwards, cells were washed and stained with either anti-mouse-IgG1 or anti-mouse-IgG2a antibody (BD Biosciences, San Jose, CA). Subsequently, cells were analyzed by flow cytometry. Specific binding of the sheep anti-human CD7 antibody was confirmed by staining with an anti-sheep-IgG antibody (clone A756; Thermo Fisher Scientific, Waltham, MA), followed by flow cytometric analysis.
To determine the dissociation constant (Kd), TCM were incubated with various concentrations of Pacific-Blue (PacBl)-labeled antibodies or F(ab´)2 (Antibody Labeling Kit, Invitrogen, Thermo Fisher Scientific, Waltham, MA) and analyzed by flow cytometry. The Kd was calculated by nonlinear regression analysis of plotted mean fluorescence intensity (MFI) values of 7-AAD- cells versus applied antibody concentrations.
Flow cytometric analysis
For flow cytometric analysis, the following antibodies were used: anti-human CD3 (clone UCHT1), anti-human CD3 (clone HIT3a), anti-human CD45 (clone J.33), anti-human CD56 (clone B159), anti-human CD4 (clone RPA-T4), anti-human CD8 (clone RPA-T8), anti-human CD62L (clone DREG-56), anti-human CD45RA (clone HI100), anti-human CD45RO (clone UCHL1), anti-human CD20 (clone 2H7), anti-human CD14 (clone M5E2), anti-human CD33 (clone WM53; all BD Biosciences, San Jose, CA), anti-human CD2 (RPA-2.10), anti-human CD127 (clone A019D5; both BioLegend, San Diego, CA), anti-human CD5 (clone BL1a), anti-human CD7 (clone 8H8.1; both Beckman Coulter, Brea, CA), anti-human CD56 (clone CMSSB), anti-human CD25 (clone BC96; both Thermo Fisher Scientific, Waltham, MA), and isotype controls (clones MOPC-21 and X40). Dead cells were identified with 7-aminoactinomycine (7-AAD; Sigma-Aldrich, St.Louis, MO) and samples were analyzed using LSRII (BD Biosciences, San Jose, CA) flow cytometer. Data were evaluated by FlowJoSoftware7.6.5 (FlowJo, LLC; Ashland, OR). Similarly, different blood cells and T-cell subpopulations were analyzed for CD2, CD7, and CD3.
Quantification of surface expression on specifically activated TCM
The surface expression of the target antigens CD2 and CD7 on specifically activated TCR2.5D6iRFP TCM was investigated during co-incubation with ML2-B7 tumor cells by flow cytometry. Measured geometric mean (GM) of surface markers was related to GM of quantification beads (BD Biosciences, San Jose, CA), and labeling efficacy of the antibodies was determined via nanophotometer (Implen, Munich, Germany). As controls, iRFP TCM were co-cultured with ML2-B7 and TCR2.5D6iRFP TCM with ML2-B15 tumor cells.
Internalization analysis
Antibodies and F(ab´)2 (100 nM) were added to 1×106 PBMC-derived T cells/mL to determine internalization as described by Li and colleagues [54]. After the residual antibodies were removed from the surface by acidic wash, T cells were stained for the respective surface molecules and 7-AAD and analyzed by flow cytometry. Internalization was determined by normalization of MFI of protein applied to non-pretreated cells.
In vitro testing of apoptosis, proliferation, and IFNγ production
PBMC-derived T cells (1×106/mL) were incubated with antibodies or F(ab´)2 at several concentrations. Analysis of apoptosis was performed after 18 h of incubation as previously described [33]. Harvested cells were washed with PBS and stained for 10 min at 25 °C with 3 µL of annexin V-APC (BD Bioscience, San Jose, CA) and 0.5 µg of 7-AAD. The reaction was stopped by addition of 100 µL binding buffer (0.01 M HEPES, 0.14 M NaCl, and 2.5 mM CaCl2; pH 7.4), and the samples were analyzed immediately by flow cytometry. Apoptotic cells were calculated as the percentage of either annexin V or 7-AAD positive cells of all cells under exclusion of debris.
For analysis of proliferative capacity, PBMC-derived T cells, activated with the anti-CD3 antibody OKT3 (30 ng/mL) and IL-2 (50 U/mL) for 2-3 days, were stained with CellTrace Violet (CTV) using the CTV Cell Proliferation Kit (Thermo Fisher Scientific, Waltham, MA) according to the manufacturer's information. Briefly, PBMC-derived T cells (1×106/mL) were incubated with 5 µM of CTV staining solution for 10 min at 37 °C. Afterwards, cells were washed with ice cold PBS containing 2% FCS and resuspended in prewarmed T-cell medium containing IL-7 and IL-15 (5 ng/mL each). Labeling efficiency was determined by flow cytometry. Afterwards, 5×104 stained cells were incubated with the antibodies and F(ab´)2 and analyzed at different time points by flow cytometry evaluating CTV-MFI of all 7-AAD- cells. Supernatants derived from co-incubation assays were analyzed for IFNγ by ELISA according to the manufacturer's instructions (BD Bioscience, San Jose, CA) as previously described [50].
Analysis of cytotoxic potential of T cells in vitro
Cytotoxic efficacy of specifically activated T cells in the presence of antibodies and F(ab´)2 was investigated by co-cultivation of TCR2.5D6iRFP TCM and ML2-B7 tumor cells. Cells were stained with 7-AAD and analyzed by flow cytometry in the presence of AccuCheck Counting Beads (Thermo Fisher Scientific, Waltham, MA). The total number of 7-AAD-GFP+ tumor cells/µL was calculated according to the manufacturer's instructions. The product of detected cells and the amount of beads/µL was related to the total number of beads detected. IFNγ secretion in the supernatant was determined by ELISA.
In vivo studies
F(ab´)2 of the most promising antibodies were tested in animal experiments following approval by the local government using a previously described mouse tumor model with NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG mice) (Figure S1A-B) [33,34]. Briefly, ML2-B7 and ML2-B15 control tumor cells were injected subcutaneously into each flank of NSG mice. After seven days, the animals underwent total body irradiation with 0.5-1 Gy followed by intravenous injection of 2×106 TCR2.5D6iRFP TCM the next day. iRFP TCM or PBS injected mice served as controls. After 3 days, mice received either non-radiolabeled (200 µL PBS) or 89Zr-radiolabeled F(ab´)2 (20 µg in 200 µL PBS) intravenously. Size of tumors was determined daily by caliper measurement. For the in vivo functionality experiment, mice were injected intraperitoneally with irradiated (80 Gy) NSO-IL15 cells twice per week starting on the day of T-cell injection.
Preparation of 89Zr-labeled F(ab´)2
Conjugation and radiolabeling of F(ab´)2 were performed as previously described [33,34]. For radiolabeling of F(ab')2, the p-isothiocanatobenzyl derivate of desferrioxamine (DFO-Bz-NCS, Macrocyclics, Inc., Richardson, TX) served as a bifunctional chelator for 89Zr. Chelator and F(ab´)2 were compounded in a molar ratio of 3:1, followed by purification of the immunoconjugate by size-exclusion chromatography (Sephadex G-25 M, PD10 column, GE Healthcare, Chicago, IL) and determination of the conjugation yield by a nanophotometer. The radiolabeling was performed according to a protocol of Perk and colleagues [55] with slight modifications as described elsewhere [33,34]. For the in vivo studies, animals were injected intravenously with 0.7-1.5 MBq (10-20 µg) of 89Zr-radiolabeled F(ab´)2 in 0.25 M sodium acetate/gentisic acid 5 mg/mL buffer solution (pH 5.5).
PET/CT imaging
48 h after the injection of 89Zr-OKT11 (anti-CD2)-F(ab´)2 or 89Zr-T3-3A1 (anti-CD7)-F(ab´)2, mice were anesthetized and imaging was performed with the Inveon small animal PET/CT scanner (Siemens, Knoxville, TN) as previously described [33,34]. The CT scan was followed by 20-min static PET acquisition. A modified Feldcamp algorithm was used for reconstruction of CT images and 3-dimensional ordered subsets expectation maximum algorithm (OSEM3D/MAP) with a pixel size of 0.77 mm for the reconstruction of PET images. Fusion images were then created using the Inveon Research Workplace (Siemens, Knoxville, TN). Data were normalized and corrected for random, dead time, and decay with no correction for attenuation or scatter. To quantify tracer uptake, regions of interest were drawn based on the CT image and the activity accumulation referred as percentage of injected dose per gram of tissue (%ID/g, 1 cc = 1 g). Pictures are shown as 3D-maximum intensity projections (MIP) of co-registered PET/CT images.
Ex vivo analysis
After imaging, the animals were killed, tumors and tissues were weighted, and activity in respective tissues was measured by the gamma counter 2480Wizard2 (PerkinElmer, Waltham, MA). The percentage of injected dose per gram of tissue (%ID/g) was calculated and depicted as ratio to blood uptake. Tumors and organs were homogenized, erythrocytes were lysed (ACK lysis buffer, Thermo Fisher Scientific, Waltham, MA), and cells were stained and analyzed by flow cytometry for human T cells (CD45+CD3+GFP-7-AAD- or CD45+CD5+GFP-7-AAD-), TCR2.5D6iRFP TCM (CD45+CD3+iRFP+GFP-7-AAD-), and tumor cells (CD45+GFP+7-AAD-). The number of cells was expressed in relation to all 7-AAD- cells.
Histology and immunohistochemistry
On the day of imaging, tumor tissues were collected for immunohistochemistry. These mouse tissues were fixed in 4% neutral-buffered formalin solution for 48 h, dehydrated under standard conditions (ASP300S, Leica Biosystems, Wetzlar, Germany), and embedded in paraffin. Serial 2-µm sections prepared with a rotary microtome (HM355S, Thermo Fisher Scientific, Waltham, MA) were collected and subjected to histological and immunohistochemical analysis.
Hematoxylin-eosin (H.-E.) staining was performed on deparaffinized sections with eosin and Mayer's haemalaun according to a standard protocol (data not shown).
Immunohistochemistry was performed using a Bond RXm system (Leica Biosystems, Wetzlar, Germany) with the anti-human CD2 (clone EPR6451, dilution 1:250, Abcam, Cambridge, UK) and anti-human CD7 (clone EPR4242, dilution 1:100, Abcam, Cambridge, UK) or for a double staining anti-CD3 (clone SP7, dilution 1:250, Zytomed Systems, Berlin, Germany) and anti-cleaved caspase 3 (clone 5A1, dilution 1:300, Cell Signaling, Danvers, USA). Briefly, slides were deparaffinized using deparaffinization solution, pretreated with Epitope retrieval solution 1 (corresponding to citrate buffer, pH 6) for 30 min. Antibody binding was detected with a polymer refine detection kit without post primary reagent and visualized with DAB as a dark brown precipitate. The double staining of anti-CD3/anti-cleaved caspase 3 was performed as described by sequential staining procedure. Briefly, after application of the anti-CD3 antibody and visualization with DAB, anti-cleaved caspase3 antibody was applied and visualized with Fast Red as chromogen.
Counterstaining was done with hematoxylin. In addition, we performed immunohistochemistry with a BenchMark XT automated stainer (Ventana, Tucson, AZ) for anti-human CD3 antibody (clone MRQ-39, Sigma-Aldrich), using the ultraVIEW DAB Detection Kit (all reagents from Ventana, Tucson, AZ). Briefly, the tissue sections were deparaffinized with EZ Prep at 75-76 °C, heat pretreated in Cell Conditioning 1 for antigen retrieval at 76-100 °C, and then incubated with the primary antibody diluted in antibody diluent 1:500 for 32 min at 37 °C after inactivation of the endogenous peroxidase using UV-inhibitor for 4 min at 37 °C. The slides were incubated with a HRP Universal Multimer for 8 min. Antibody binding was detected using DAB as chromogen and counterstained with hematoxylin for 10 min with subsequent bluing in bluing reagent for 10 min.
Slides were then dehydrated manually by alcohol washes of increasing concentration (70%, 96%, and 100%) and xylene and coverslipped using Pertex® mounting medium (Histolab, Goeteborg, Sweden). The stained slides were evaluated by an experienced certified pathologist (K.S.) using a BX53 stereomicroscope (Olympus, Shinjuku, Japan) and scanned using a slide scanner (AT-2, Leica Biosystems, Wetzlar, Germany). Representative images were collected using the AperioImagescope software (version 12.3, Leica Biosystems, Wetzlar, Germany).
Statistical analysis
Mean ± standard deviation (±SD) and unpaired t-tests were calculated using GraphPad Prism software version 5.01 (San Diego, CA). Normal distribution of data was confirmed by probability plots using MiniTab trial software (State Collage, PE). P-values of <0.05 were set as statistically significant and indicated in figure legends.
Results
CD2 and CD7 are highly expressed on different T-cell subsets and upregulated on specifically activated T cells
Focusing on a specific pan T-cell marker, we investigated CD2, CD3, and CD7 as potential target antigens for T-cell tracking regarding their suitability for diagnostic immuno-PET. First, expression of these antigens was analyzed on different T-cell populations (Figure 1A, Figure S2A-B and Table S1-S2), as well as various other blood cell components such as B cells, NK cells, and monocytes (Figure S3). CD2 was highly expressed on TCM. CD7 and CD3 showed especially high expression on TN (Figure 1A). Although the expression pattern of the target structures was similar for different donors, the MFI on T-cell subpopulations varied (data not shown).
CD2, CD3, and CD7 are expressed on all T-cell subsets and CD2 as well as CD7 show an upregulation on specifically activated T cells. (A) Analysis of CD2, CD3, and CD7 expression on the surface of different T-cell subsets from freshly isolated peripheral blood mononuclear cells (PBMC). Mean fluorescence intensity (MFI) of PE-labeled anti-CD2, anti-CD3, and anti-CD7 antibodies is shown normalized to PE-isotype control for cells pre-gated on CD4 or CD8. T-cell subpopulations include TCM (central memory T cells; CD45RA-CD62L+), TEFF (effector T cells; CD45RA+CD62L-), TEM (effector memory T cells; CD45RA-CD62L-), TN (naive T cells; CD45RA+CD62L+), and TReg (regulatory T cells; CD4+CD25+CD127low). Analysis of CD2 and CD7 on T-cell subpopulations of two other donors revealed the same pattern for each subset at other MFI values. Flow cytometry data for LSR II was evaluated using FlowJoSoftware7.6.5 and is shown for cells pre-gated on single and 7-AAD- cells. (B) CD2, CD3, and CD7 molecules on the surface of T cells and tumor cells (control). TCR2.5D6iRFP or iRFP TCM (both GFP-) were co-cultivated for 24 h with ML2-B7 or ML2-B15 (both GFP+) tumor cells as indicated and subsequently analyzed for surface expression with respective PE-labeled antibodies. For calculation of the number of surface molecules, detected geometric mean (GM) was related to GM of PE quantification beads and the labeling efficiency of antibodies was determined by nanophotometer.
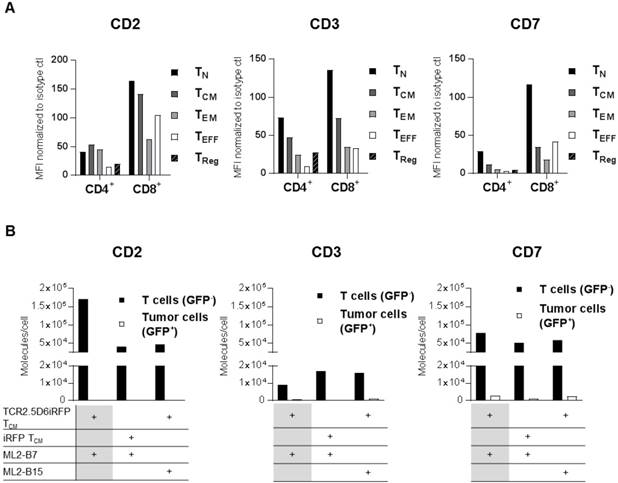
The expression of these markers on specifically activated T cells was determined by co-incubation of TCR2.5D6iRFP TCM or control iRFP TCM with ML2 tumor cells bearing either the restriction element HLA-B7 (ML2-B7) or the irrelevant HLA-B15 (ML2-B15) molecule. Analysis of CD2 and CD7 expression after co-cultivation with respective target cells demonstrated that specific activation of T cells led to strong induction of CD2 and moderate enhancement of CD7, whereas CD3 expression was reduced (Figure 1B).
Subsequently, we selected various antibody clones directed against these three target structures. OKT11 (anti-CD2), T3-3A1, and 4H9 (both anti-CD7), as well as OKT3, VIT3b, and BC3 (all anti-CD3) were tested for their effect on the viability of PBMC-derived T cells. These in vitro studies on non-activated T cells revealed induction of apoptosis for all selected anti-CD3 antibodies (Figure S4A), but for none of the anti-CD2 and anti-CD7 antibodies. IFNγ levels in supernatants corresponded with these findings, indicating T-cell stimulation by all anti-CD3 antibodies, whereas the IFNγ level after incubation with anti-CD2 and anti-CD7 antibodies was comparable to the isotype control (Figure S4B).
OKT11 (anti-CD2) and T3-3A1 (anti-CD7) IgG and F(ab´)2 show specific, high-affinity binding to PBMC-derived T cells resulting in partial internalization
Based on the in vitro results, we selected OKT11 (anti-CD2) antibody (OKT11 (anti-CD2)-IgG) and T3-3A1 (anti-CD7) antibody (T3-3A1 (anti-CD7)-IgG) to further characterize immunotracer candidates for in vivo T-cell imaging. The CD3-specific antibody OKT3 served as positive control. We isolated the full antibodies from respective hybridoma supernatants and F(ab´)2 were produced because of their general superiority with respect to in vivo pharmacokinetics. Purity was validated by size-exclusion HPLC and binding was verified by ELISA. Although isolated OKT11 (anti-CD2)-IgG showed one peak reflecting IgG1 (Figure S5A-B), we detected two isotype subclasses, IgG1 and IgG2a, for T3-3A1 (anti-CD7)-IgG (Figure S5A-B). Blocking experiments using flow cytometry confirmed the specific detection of human CD7 by both isolated isotype subclasses (Figure S5C).
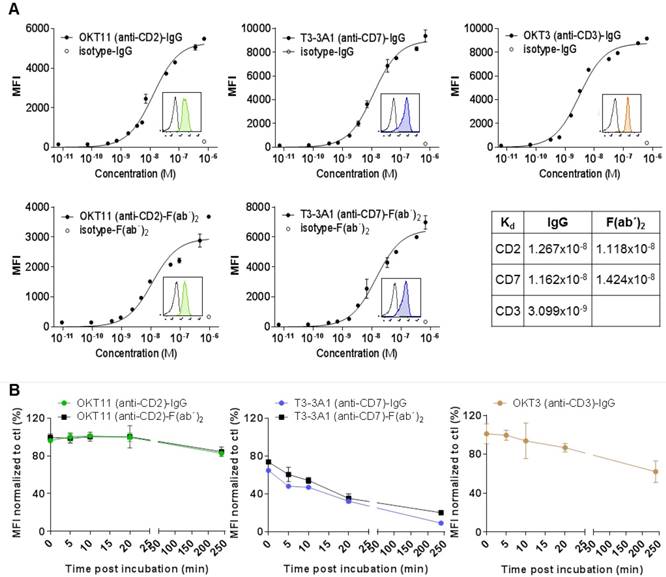
Flow cytometry analysis of IgG and respective F(ab´)2 showed specific binding to PBMC-derived T cells with a Kd in the range of 10-8 M (Figure 2A). We observed internalization after stimulation especially for T3-3A1 (anti-CD7) IgG and F(ab´)2. In contrast, OKT11 (anti-CD2) IgG and F(ab´)2 showed only slight internalization of the receptor molecule (Figure 2B).
OKT11 (anti-CD2) and T3-3A1 (anti-CD7) IgG and F(ab´)2 show high specificity and affinity to selected antigens on T cells, with highest internalization effects observed for CD7 binding. (A) Affinity binding curves of OKT11 (anti-CD2) and T3-3A1 (anti-CD7) IgG and F(ab´)2 to PBMC-derived T cells at different concentrations. OKT3 (anti-CD3)-IgG was used as a positive control. Flow cytometric data (performed in triplicates) are depicted as semi-logarithmic plots in comparison to the isotype. Each value is shown as mean ± standard deviation (SD). Histograms within the graphs show specific binding of tested PacBlue-labeled IgG and F(ab´)2 (colored filled histograms) to PBMC-derived T cells at a concentration of 100 nM in comparison to respective PacBlue-isotype controls (gray histogram). Constant of dissociation (Kd) values are depicted in the table. (B) Internalization of IgG and F(ab´)2 after incubation with PBMC-derived T cells. MFI values of CD2, CD7 and CD3 staining with respective PE-labeled antibodies at various time points after application of indicated IgG or F(ab´)2 were normalized to MFI of control sample without addition of proteins beforehand. The experiment was performed in duplicates and results are shown as mean ± SD (n = 3).
OKT11 (anti-CD2) and T3-3A1 (anti-CD7) IgG and F(ab´)2 do not affect viability, IFNγ production, and proliferative capacity of PBMC-derived T cells
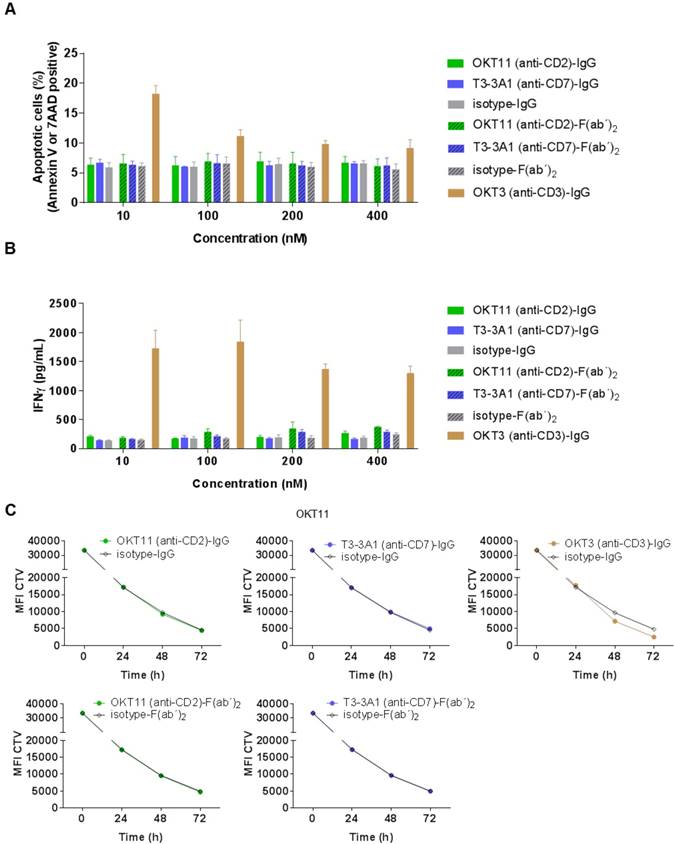
After determining general characteristics of the selected IgG and F(ab´)2, we investigated their functional effect on non-activated PBMC-derived T cells. Different concentrations of OKT11 (anti-CD2) and T3-3A1 (anti-CD7) IgG and F(ab´)2 revealed no enhancement of apoptosis on PBMC-derived T cells (Figure 3A). Similarly, the corresponding IFNγ levels showed no difference to respective isotype controls independent from concentration (Figure 3B) or different time points (Figure S6). Additionally, proliferative capacity of PBMC-derived T cells in the presence of the tested proteins did not differ from the isotype control (Figure 3C), whereas addition of OKT3 (anti-CD3)-IgG led to enhanced proliferation (Figure 3C). Potential dilution effects deriving from the IgG1/IgG2a mixture of the T3-3A1 (anti-CD7) IgG or F(ab´)2 were excluded by determination of proliferation capacity and IFNγ secretion with two-fold concentrations showing comparable results (Figure S7A-C).
OKT11 (anti-CD2) and T3-3A1 (anti-CD7) IgG and F(ab´)2 show no functional T-cell impairment during short-term co-incubation. (A-B) PBMC-derived T cells were incubated for 18 h at 37 °C with various concentrations of IgG and F(ab´)2 in triplicates. Respective isotypes served as negative control, whereas OKT3 was used as positive control. Mean ± SD is depicted (n = 3). (A) Percentage of apoptotic PBMC-derived T cells after co-incubation at indicated protein concentrations. Values are based on flow cytometry analysis identifying annexin V-APC or 7-AAD positive cells of all cells detected. (B) Corresponding IFNγ release of PBMC-derived T cells in supernatants after incubation with indicated concentrations of IgG or F(ab´)2 determined by ELISA. (C) CTV-labeled PBMC-derived T cells were incubated with 100 nM of the indicated antibodies or F(ab´)2 at 37 °C over a period of 3 days. Incubation with isotypes served as negative control and OKT3 as positive control. Decrease of MFI of CTV indicates T-cell proliferation in response to indicated IgG or F(ab´)2 depicted as mean ± SD from triplicates (n = 3).
T3-3A1 (anti-CD7) does not impair functionality of specifically activated TCR2.5D6iRFP TCM, whereas co-incubation with OKT11 (anti-CD2) results in slightly modulated T-cell function
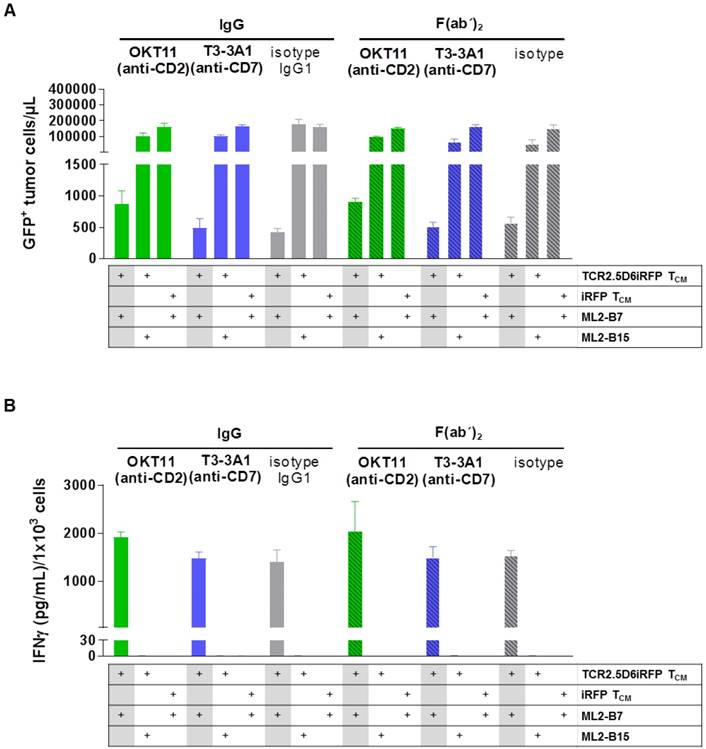
We then investigated the influence of the selected antibodies and F(ab´)2 on specific cytotoxicity of T cells against tumor cells. IgG and F(ab´)2 were added to a co-culture of TCR2.5D6iRFP TCM and GFP+ ML2-B7 tumor cells. iRFP TCM and GFP+ ML2-B15 tumor cells served as controls. The percentage of tumor cells was strongly reduced for TCR2.D6iRFP TCM incubated with isotype IgG and F(ab´)2, indicating a strong cytotoxic activity of specifically activated T cells (Figure 4A). Incubation with T3-3A1 (anti-CD7) IgG or F(ab´)2 showed comparable results. In case of OKT11 (anti-CD2) IgG and F(ab´)2, the percentage of tumor cells was slightly enhanced, suggesting an impairment of cytotoxic T-cell activity (Figure 4A). In addition, specifically activated TCR2.5D6iRFP TCM showed slightly higher IFNγ levels in supernatants for OKT11 (anti-CD2) IgG and F(ab´)2 compared to the isotype in contrast to T3-3A1 (anti-CD7) IgG and F(ab´)2 (Figure 4B). Taken together, statistically significant differences were observed in single but not all experiments for full IgG and F(ab´)2.
T3-3A1 (anti-CD7) IgG and F(ab´)2 have no impact on the cytotoxic efficacy of specifically activated TCR2.5D6iRFP TCM in contrast to co-incubation with OKT11 (anti-CD2) resulting in minor changes in tumor cells killing and IFNγ secretion. TCR2.5D6iRFP TCM were co-cultured with tumor cells bearing either the respective restriction element HLA-B7 or the irrelevant HLA-B15 for 24 h at 37 °C. As control, iRFP TCM were co-incubated with ML2-B7 tumor cells. Each condition is indicated below the graph. After incubation of the co-culture with respective IgG or F(ab´)2, cells were harvested for flow cytometric analysis and IFNɣ secretion in supernatants was analyzed by ELISA. The experiment was performed in triplicates and results are shown as mean ± SD (n = 3). One representative experiment out of three is depicted. Statistically significant differences were observed in single but not all experiments for full IgG and F(ab´)2. (A) GFP+ tumor cells per microliter calculated from flow cytometric analysis. (B) Corresponding IFNγ cytokine secretion in co-culture supernatant.
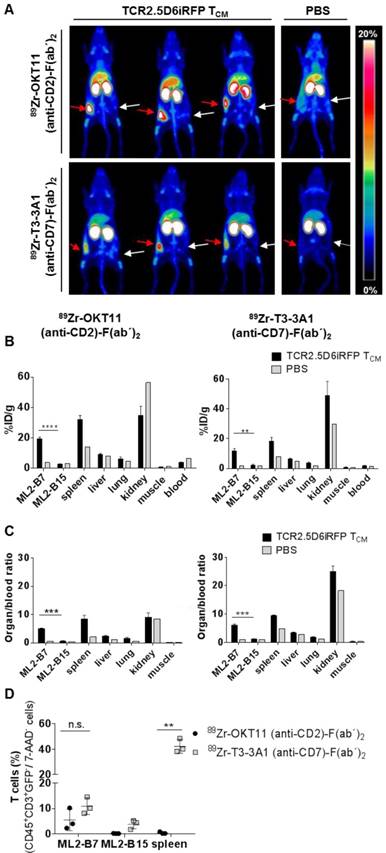
In vivo imaging using 89Zr-labeled OKT11 (anti-CD2)-F(ab´)2 and T3-3A1 (anti-CD7)-F(ab´)2 showed a distinct signal of T cells at the tumor site
Based on the lack or only minor functional impairment of T cells after co-incubation with OKT11 (anti-CD2) and T3-3A1 (anti-CD7) IgG and F(ab´)2, we tested the potential of 89Zr-labeled F(ab´)2 to track T cells in vivo in a previously described myeloid sarcoma model of adoptive T-cell transfer (Figure S1A; [33, 34]). PET images showed a distinct signal of T cells at the site of the MHC-matched antigen-expressing tumor for both tracers, whereas no signal was detected in the MHC-mismatched antigen-expressing control tumor (Figure 5A). Biodistribution analyses of isolated organs confirmed the imaging results revealing enhanced tracer uptake especially in the ML2-B7 tumor and in the spleen for 89Zr-OKT11 (anti-CD2)-F(ab´)2 and 89Zr-T3-3A1 (anti-CD7)-F(ab´)2 (Figure 5B and Table S3-S4). The organ-to-blood ratios were comparable for both tracers (Figure 5C). For the control tumor ML2-B15, radioactive uptake was not enhanced at the same level as in mice that had not received T cells (Figure 5B-C). Of note, flow cytometric analysis of selected organs showed the presence of T cells in ML2-B7 tumor and spleen of animals injected with 89Zr-T3-3A1 (anti-CD7)-F(ab´)2 as expected (Figure 5D), whereas T cells were quantitatively reduced especially in the spleen of animals injected with 89Zr-OKT11 (anti-CD2)-F(ab´)2 (Figure 5D).
In vivo imaging of T cells at the specific tumor site by immuno-PET using 89Zr-OKT11 (anti-CD2)-F(ab´)2 and 89Zr-T3-3A1 (anti-CD7)-F(ab´)2. (A) 3D-maximum intensity projection (MIP) pictures of PET/CT imaging of mice. Mice bearing ML2-B7 or ML2-B15 tumors that received TCR2.5D6iRFP TCM or PBS and 89Zr-labeled F(ab´)2 as indicated. Red arrows point at the ML2-B7 tumor site recognized by TCR2.5D6iRFP TCM and white arrows mark the ML2-B15 control tumor site. Animals injected with PBS served as negative controls. The scale bar shows 0-20% of percentage injected dose per gram (%ID/g). (B) Biodistribution analysis based on activity accumulation measurement by a gamma counter of the depicted organs for animals that received 89Zr-OKT11 (anti-CD2)-F(ab´)2 (left panel) or 89Zr-T3-3A1 (anti-CD7)-F(ab´)2 (right panel). %ID/g is depicted as mean ± SD for mice injected with TCR2.5D6iRFP TCM (n = 3 each) or PBS (n = 1 each) as control for various organs as indicated. Unpaired t-test: ** p < 0.01 and **** p < 0.0001. (C) Respective organ-to-blood ratio from %ID/g values shown as mean ± SD for 89Zr-OKT11 (anti-CD2)-F(ab´)2 (left panel) and 89Zr-T3-3A1 (anti-CD7)-F(ab´)2 (right panel). Unpaired t-test: *** p < 0.001. (D) Percentage of T cells detected by flow cytometry in tumors and spleen for animals injected with TCR2.5D6iRFP TCM and either 89Zr-OKT11 (anti-CD2)-F(ab´)2 or 89Zr-T3-3A1 (anti-CD7)-F(ab´)2. Unpaired t-test: ** p < 0.01.
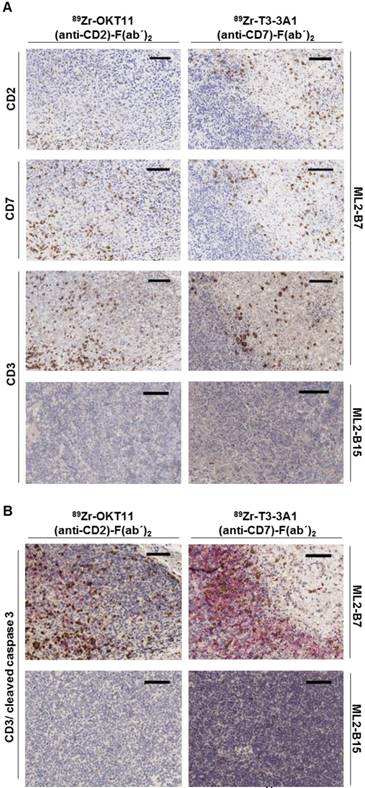
Ex vivo tissue staining shows T cells expressing CD2 and CD7 at the specifically detected tumor site but reveals less effective tumor-cell cytotoxic activity in samples derived from OKT11 (anti-CD2)-F(ab´)2 in contrast to T3-3A1 (anti-CD7)-F(ab´)2-injected mice
The presence of T cells expressing CD2 and CD7 in ML2-B7 tumor tissue harvested from mice on the imaging day was confirmed by immunohistochemistry. We detected CD2 and CD7 expression comparable to conventional CD3 on T cells within the ML2-B7 tumor from mice injected with 89Zr-OKT11 (anti-CD2)-F(ab´)2 and 89Zr-T3-3A1 (anti-CD7)-F(ab´)2, respectively (Figure 6A). In ML2-B15 tumors, only single T cells were detectable as expected. We additionally performed staining for cleaved caspase 3 to investigate T-cell functionality in vivo. We observed substantially reduced cleaved caspase 3 positive tumor cells adjacent to infiltrating T cells in 89Zr-OKT11 (anti-CD2)-F(ab´)2 compared to 89Zr-T3-3A1 (anti-CD7)-F(ab´)2 injected mice, indicating an impaired T-cell function in vivo (Figure 6B).
Immunohistochemistry confirmed the presence of CD2+ and CD7+ T cells in the ML2-B7 tumor specifically recognized by TCR2.5D6 transduced TCM. Tumor tissue from 89Zr-OKT11 (anti-CD2)-F(ab´)2 (left panel) or 89Zr-T3-3A1 (anti-CD7)-F(ab´)2 (right panel) injected mice was harvested after imaging. The bars in the upper right corner represent 100 µm. (A) ML2-B7 tumor slides stained for CD2, CD7, and CD3, as well as ML2-B15 tumor slides stained for CD3 as control. (B) ML2-B7 and ML2-B15 tumor slides stained for CD3 (brown)/ cleaved caspase 3 (red).
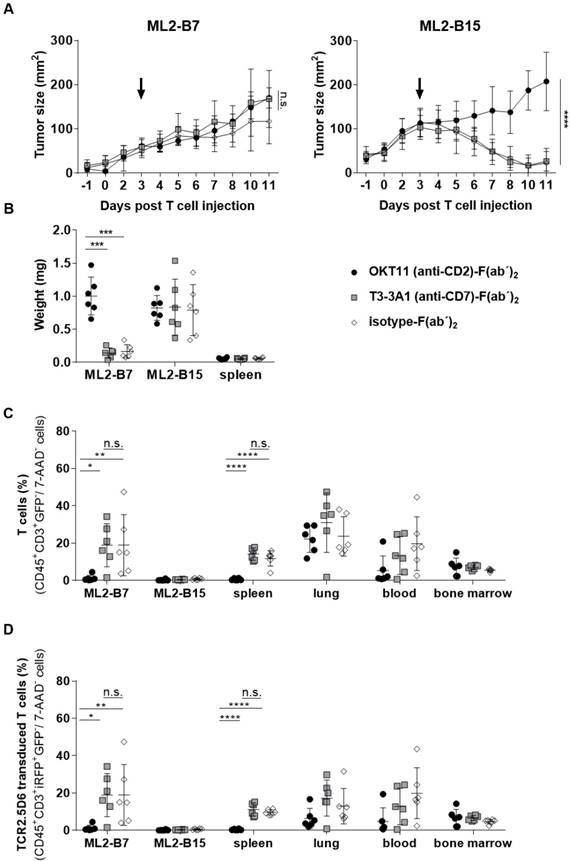
Animals injected with T3-3A1 (anti-CD7)-F(ab´)2 successfully reject ML2-B7 tumors, whereas OKT11 (anti-CD2)-F(ab´)2 administration results in failure of tumor rejection
We then investigated the long-term effects of respective F(ab´)2 administration on T-cell functionality in vivo. Therefore, we analyzed ML2-B7 tumor growth within NSG mice that had been co-injected with TCR2.5D6iRFP TCM and, analogous to the imaging setting described above, the same dose of OKT11 (anti-CD2)-F(ab´)2, T3-3A1 (anti-CD7)-F(ab´)2, or isotype-F(ab´)2 as control (Figure S1B). After an initial growth phase, T cells of mice injected with T3-3A1 (anti-CD7)-F(ab´)2 and isotype-F(ab´)2 were able to reject MHC-matched tumors efficiently. In contrast, the administration of OKT11 (anti-CD2)-F(ab´)2 led to failure of tumor rejection (Figure 7A). ML2-B15 control tumors were not rejected independent of the F(ab´)2 treatment. Eleven days after TCM injection, the tumor weight from OKT11 (anti-CD2)-F(ab´)2-treated mice was significantly higher than that of mice receiving T3-3A1 (anti-CD7)-F(ab´)2 or isotype-F(ab´)2 (Figure 7B). These results correspond to flow cytometry data showing a strong reduction of T cells within the ML2-B7 tumor 11 days after TCM transfer for the group injected with OKT11 (anti-CD2)-F(ab´)2 (Figure 7C). This was similarly observed in the spleen, and by trend in the blood. Of note, we have not observed depletion of T cells within the lungs and bone marrow in animals injected with OKT11 (anti-CD2)-F(ab´)2 (Figure 7C). Equivalent results were obtained investigating specific TCR2.5D6iRFP TCM in OKT11 (anti-CD2)-F(ab´)2 injected animals (Figure 7D).
Injection of OKT11 (anti-CD2)-F(ab´)2, but not T3-3A1 (anti-CD7)-F(ab´)2, impaired T-cell function in vivo. After injection of ML2-B7 tumor cells into the right flank and ML2-B15 tumor cells into the left flank, as well as total body irradiation, mice received 2×107 TCR2.5D6iRFP TCM. Comparable to the imaging experiments, three days after TCM injection, mice received 20 µg of OKT11 (anti-CD2)-F(ab´)2, T3-3A1 (anti-CD7)-F(ab´)2, or isotype-F(ab´)2 (n = 6 for each group). Animals were sacrificed on day 11 after T-cell administration (Figure S1B). Unpaired t-test: * p < 0.05, ** p < 0.01, *** p < 0.001, and **** p < 0.0001. (A) Tumor growth kinetics of ML2-B7 (left side) and ML2-B15 control tumors (right side) for mice injected as indicated. The arrow indicates the time point of respective F(ab´)2 injection. Tumor size is shown in mm2 as mean ± SD over different days post intravenous TCM injection. (B) Weight of tumors and spleen 11 days after TCM injection for mice receiving different F(ab´)2 constructs as indicated. The weight of the organs is shown in milligram as mean ± SD. (C) Percentage of human T cells in indicated organs analyzed by flow cytometry on day 11 post TCM injection. The percentage of CD45+GFP-CD3+ cells of all 7-AAD- cells is depicted as mean ± SD for the indicated groups. (D) The percentage of TCR2.5D6iRFP+ TCM of all 7-AAD- cells in indicated organs is shown as mean ± SD.
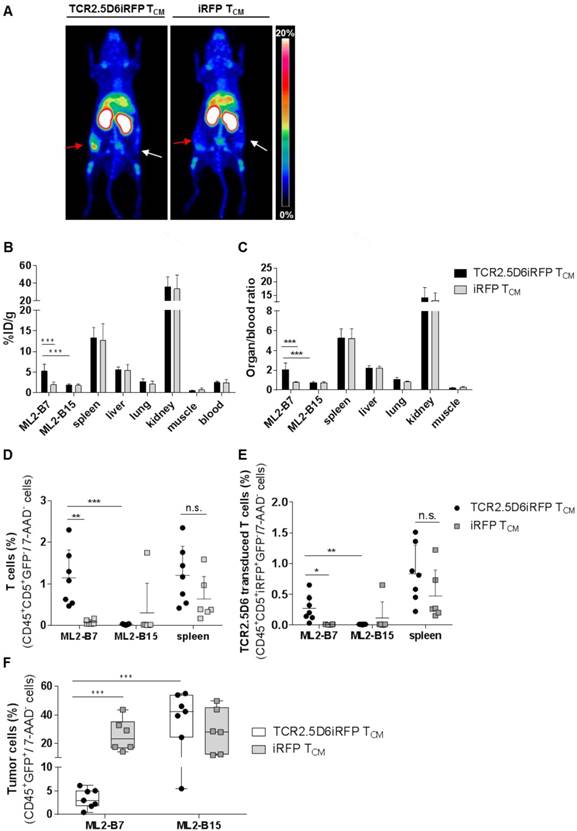
In vivo imaging with 89Zr-T3-3A1 (anti-CD7)-F(ab´)2 detects tumor-reactive T cells specifically at the tumor site
Because CD7 represents a pan T-cell marker expressed also on non-stimulated T cells, T3-3A1 (anti-CD7)-F(ab´)2 may also bind to nonspecific T cells in vivo. We therefore explored how reliably tumor-specific T cells can be detected in comparison to non-tumor-specific T cells in vivo. TCR2.5D6iRFP TCM and iRFP TCM were injected into tumor-bearing mice (Figure S1A). After the application of 89Zr-T3-3A1 (anti-CD7)-F(ab´)2 and PET imaging, we detected a strong signal at the ML2-B7 tumor site in mice receiving tumor-specific TCR2.5D6iRFP TCM, but not in mice receiving nonspecific iRFP TCM (Figure 8A). Ex vivo biodistribution analysis confirmed these imaging results, showing higher 89Zr-uptake in the ML2-B7 tumor in animals receiving TCR2.5D6iRFP TCM and an enhanced organ-to-blood ratio compared to the control group (Figure 8B-C). Flow cytometric analysis validated these data. T cells, in general, and TCR2.5D6iRFP T cells were enhanced in ML2-B7 tumors of TCR2.5D6iRFP TCM-treated mice (Figure 8D-E), corresponding to a reduction of GFP+ ML2-B7 tumor cells (Figure 8F).
T-cell tracking by immuno-PET using 89Zr-T3-3A1 (anti-CD7)-F(ab´)2 reveals prominent signals corresponding to accumulation of specific tumor infiltrating lymphocytes at the tumor site. Mice were treated following the imaging protocol depicted in Figure S1A. (A) Representative 3D-MIP PET images of ML2-B7 and ML2-B15 tumor-bearing mice injected with either TCR2.5D6iRFP TCM (n = 7) or iRFP TCM (n = 6). Animals received 89Zr-T3-3A1 (anti-CD7)-F(ab´)2 intravenously and PET imaging was performed 48 h post injection. Red arrows point at ML2-B7 tumor site, and white arrows mark ML2-B15 control tumor site. The scale bar represents 0-20% ID/g. (B) Ex vivo biodistribution analysis of different organs analyzed on the day of imaging. Mean ± SD of %ID/g is shown for different organs from mice receiving either TCR2.5D6iRFP or iRFP TCM. Unpaired t-test: *** p < 0.001. (C) Respective organ-to-blood ratio for TCR2.5D6iRFP or iRFP TCM treated mice. Unpaired t-test: *** p < 0.001. (D) Flow cytometric analysis of T cells detected in the ML2-B7, ML2-B15 control tumor, or the spleen. Mean ± SD of percentage of CD45+CD5+GFP- T cells of 7-AAD- cells is shown for the two groups. Unpaired t-test: ** p < 0.01 and *** p < 0.001. (E) Percentages of TCR2.5D6iRFP transduced T cells of all viable cells analyzed by flow cytometric analysis of respective organs. Mean ± SD is depicted. Unpaired t-test: * p < 0.05 and ** p < 0.01 (F) Percentages of GFP+ tumor cells of all viable cells in ML2-B7 tumors and ML2-B15 control tumors evaluated by flow cytometry. Mean ± SD is shown. Unpaired t-test: *** p < 0.001.
Discussion
Immunotherapies have recently demonstrated a high efficacy in various malignant diseases, and the variety of treatment strategies and distinguishable response patterns is constantly extending. In contrast to many conventional targeted or chemotherapies, overall survival is considered a far more important endpoint than progression-free survival, indicating the value of potentially improved long-term survival of patients treated with immunotherapies [56]. Imaging technologies represent one of the most important surrogate markers in oncology. 18FDG-PET/CT, routinely used to evaluate response to cancer treatment, can detect immunotherapeutic responses including early ones due to its high resolution [15,16]. However, the broad spectrum of responses to immunotherapies, including pseudo-progression is often difficult to assess [12-14]. Furthermore, distinction between primary and secondary immune resistance at later time points can currently only be assessed following disease progression and biopsy. Thus, novel imaging technologies are essential for better understanding, as well as early modification and adjustment of therapies, especially in tumor entities harboring more therapeutic options.
Several novel technologies have been tested in preclinical and in a few clinical settings with respect to response assessment of immunotherapeutic treatments. Adoptive transfer of T cells genetically modified by CAR or TCR facilitates simultaneous integration of reporter genes to track transferred T cells by addition of respective tracer substrates within in vivo mice studies [18-21] and clinically [22]. Similarly, TCR-transgenic T cells with a murinized part of the TCRβ chain are detectable by a respective antibody and F(ab´)2 [33,34]. However, these approaches fail to evaluate immunotherapies inducing endogenous immune responses, such as immune checkpoint modulators or vaccination approaches [1,57,58]. To detect T cells directly at the tumor site, T-cell-associated markers as cell-derived secreted granules, such as granzyme B, have been targeted [59], or T-cell-selective [18F]fluoro-9-β-D-arabinofuranosylguanine ([18F]F-AraG) has been applied [60]. Granzyme B-based PET imaging allowed differentiation between responders and non-responders in a mouse model of checkpoint modulation [59], but detailed functional analyses are missing, although antibodies against other T-cell-secreted granules such as TNFα are known to possess potent immunosuppressive activity [61]. [18F]F-AraG was shown to detect activated T cells in an acute graft-versus-host disease model [60]. However, beside high liver background, in vitro or in vivo experiments investigating T-cell function following tracer application have not been published so far.
The most direct approach to visualize T cells in the tumor environment would be targeting specific surface markers on T cells. Several groups have recently investigated those as potential targets for antibody-derived tracers and immuno-imaging technologies, showing their suitability as surrogate markers to distinguish between responders and non-responders in the course of immunotherapy [24-31]. A more detailed functional characterization has been performed for an anti-murine CD4 cys-diabody by Freise and colleagues who investigated CD4 expression and proliferation of T cells at two different concentrations, revealing a dose-dependent decrease in CD4 expression and inhibition of proliferation [28]. Recently, the application of a pan T-cell marker, an anti-murine CD3 antibody, was reported to modulate the distribution of T-cell subsets in vivo, reducing CD4+ T cells and stimulating CD8+ TCM and TEFF[31]. Although long-term effects were not investigated, the study further emphasizes the relevance of functional analyses during the clinical translation of imaging technologies.
In this study, we performed an in-depth analysis for several pan T-cell-specific antibodies with the aim to identify one that does not modulate T-cell-related immune responses and would be suitable for diagnostic immuno-imaging with respect to immunotherapies potentially affecting different T-cell subpopulations [35-41]. Primarily, antibodies specific for CD2, CD3, and CD7 were selected. CD3 is a fundamental co-stimulator involved in TCR signaling and full anti-CD3 antibodies tested here in the in vitro experiments all demonstrated an unspecific modulation of T-cell function as previously reported [52,53,62,63]. Moreover, bivalent and monovalent anti-CD3 F(ab) constructs have been demonstrated to modulate T-cell function in vivo [64-66]. This target was, therefore, not further investigated. In contrast, targeting of CD2 and CD7 showed no major functional modulation in vitro, except for a slight impact on specifically activated T cells, which was observed for anti-CD2 targeting. Nevertheless, we continued investigating OKT11 (anti-CD2) and T3-3A1 (anti-CD7) IgG and F(ab´)2 as potential imaging tracers in vivo.
CD2, expressed on T cells, NK cells, and thymocytes, plays a role in T-cell adhesion via CD58 and T-cell signaling [42-46]. CD2 expression highly correlates with cytolytic activity in tumors in the context of immune checkpoint modulation [47], making this target especially interesting to track effector T cells. Three immunoglobulin-like domains are described as target structures for murine antibodies. While T11.1 is mainly involved in the interaction with CD58, T11.3 is only accessible upon activation [67,68]. Single targeting of T11.1 or T11.2 by antibodies is supposed to be non-mitogenic, whereas a co-application with an anti-T11.3 antibody results in T-cell stimulation [68]. OKT11 (anti-CD2)-IgG, binding the T11.1 epitope, was then selected for our experiments [69]. Previously, a functional impact of anti-CD2 targeting on T cells including OKT11 had been reported; however, these effects seem to be dose dependent and occur in a specific timely relation to T-cell priming [70-73]. In our experiments, we observed a high binding of this antibody to specifically activated T cells and TCM. Although apoptosis, proliferation, and IFNγ response of labeled T cells were not affected in short-term T-cell assays using nonspecifically stimulated T cells in vitro, specific tumor lysis and IFNγ secretion were slightly modulated. In contrast, cytotoxic reactivity and tumor rejection capacity were strongly affected in our in vivo model, allowing tumor growth similar to the positive control tumors lacking the defined HLA-restriction element. Additionally, T cells were eliminated in tumors and the spleen of these animals. These results may be explained by a direct cytotoxic effect of OKT11 upon TCR activation as previously described for anti-CD2-specific antibodies including OKT11 full antibody [70,73]. Thus, T-cell antigens involved in T-cell signaling or function should be employed with caution for diagnostic imaging purposes. In addition, our study demonstrates the importance of testing tracer candidates in a well-characterized model of tumor rejection in vivo prior to clinical translation.
Our data indicate favorable properties of CD7 as target structure for T-cell imaging. CD7, also a member of the immunoglobulin superfamily, is a marker of early-stage lymphocytes and hematopoietic precursor cells, as well as mature T cells and NK cells [48]. Furthermore, CD7 has shown an enhanced expression on naive and memory cells [49]. We observed an enhanced expression of CD7 after specific TCR stimulation and our T3-3A1 (anti-CD7)-F(ab´)2 tracer provided an effective and selective imaging of CD7+ T cells at the tumor site picturing the accumulation of specific T cells within the tumor. Although the function of this molecule is not well understood, it seems less essential for T-cell function as CD7-deficient animals present no major T-cell dysfunction [74,75]. In fact, CD7-binding did not impair T-cell function in vitro and did not impact tumor rejection in our in vivo model. Modification of de novo induction of antigen-specific cytotoxic immune responses, as observed in CD7-deficient animals [75], is likely not relevant for transient CD7 immunotracking. Because our tumor model uses NSG mice lacking any human immune cells, further in vivo testing is required such as investigating the detrimental effects of CD7 targeting in an animal model with an established human immune system. However, CD7 targeting has already been performed clinically using anti-CD7 immunotoxins in patients with T-cell lymphoma [76]. The immunotoxin showed some efficacy and safety with vascular leak syndrome, representing the main dose limiting toxicity likely associated to tumor lysis which would not be relevant in a diagnostic setting. These data indicate that targeting of CD7 is feasible. Moreover, CD7-directed CARs have been developed to treat T-cell lymphoma and T-cell acute lymphoblastic leukemia with promising preclinical data in gene-edited CD7- T cells supporting the therapeutic potential for CD7 targeting in T-cell malignancies [77]. Thus, anti-CD7 antibody constructs may have theranostic value if labeled with respective radionuclides.
Further development of this diagnostic approach is needed for clinical application. We used F(ab´)2 fragments in our experiments to exclude any potential Fc-mediated functional T-cell modulation. Based on our well-established model, we used 89Zr for labeling of F(ab´)2 fragments for the preclinical investigation of the novel approaches [33,34]. 89Zr is certainly not the ideal isotope to be used, because of the difference between the half-life of 89Zr (78.4 h; [78]) and the biological blood half-life of F(ab´)2 (24-48 h; [79]). However, due to an Eβ+max of 0.9 MeV and intrinsic spatial resolution loss of 1 mm, 89Zr emits a suitable PET signal at the time point selected for the presented study [78]. In addition, the maximum energy emitted by 89Zr (902 KeV) implies that the travel distance of an emitted positron in tissue is relatively short before annihilation, thus providing high-resolution images suitable for small target structure distribution investigation, such as tumor infiltrating T cells, as shown previously [33,34]. Furthermore, the use of 89Zr to radiolabel F(ab´)2 fragments [80] and even smaller molecules such as cys-diabodies [28] is reported to provide good contrast images. Using our tracer, we observed extensive uptake of 89Zr-OKT11 (anti-CD2)-F(ab´)2 and 89Zr-T3-3A1 (anti-CD7)-F(ab´)2 in the kidneys. For the clinical translation of this imaging method, the co-administration of cationic amino acid solutions or basic polypeptides might be necessary to reduce renal uptake and to avoid abnormal radioactive exposure of the kidneys [81].
Our target-oriented approach identified a suitable T-cell tracer applicable for diagnostic T-cell imaging without impacting T-cell function. Based on our results, immunotracers using these targets can be further optimized; for instance, by using monovalent antibody-derived constructs. When a target and an optimized construct are identified, a suitable radioisotope should be selected, and the novel tracer will need to be again tested for specificity, feasibility, and safety including detailed dosimetry in the subsequent clinical translation process [82].
Conclusions
In conclusion, we hereby show that in-depth functional analyses of T cells are essential to safely develop clinically relevant T-cell tracers for diagnostic imaging. We reveal noninvasive CD7 immuno-imaging as a highly promising strategy to track T-cell responses in the context of diverse immunotherapies in vivo, with additional theranostic potential in T-cell-mediated diseases.
Supplementary Material
Supplementary figures and tables.
Abbreviations
7-AAD: 7-aminoactinomycine; CAR: chimeric antigen receptor; CT: computed tomography; CTV: CellTrace Violet; ELISA: enzyme-linked immunesorbent assay; [18F]F-AraG: [18F]fluoro-9-β-D-arabinofuranosylguanine; 18F-FDG-PET: 18F-fluorodeoxyglucose-positron-emission-tomography; GFP: enhanced green fluorescent protein; GM: geometric mean; iRFP TCM: iRFP transduced TCM; Kd: dissociation constant; MFI: mean fluorescence intensity; MIP: maximum intensity projections; ML2-B7: HLA-B*07:02; ML2-B15: HLA-B*15:01; NSG mice: NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ mice; OD: optical density; PacBl: Pacific-Blue; PBMC: peripheral blood mononuclear cells; RECIST: response evaluation criteria in solid tumors; SD: standard deviation; SE-HPLC: size-exclusion high-performance liquid chromatography; TCM: central memory T cells; TEFF: effector T cells; TN: naive T cells; TReg: regulatory T cells; TCR: T-cell receptor; TCR2.5D6iRFP TCM: 2.5D6 linked to red fluorescent protein transduced TCM; TCRmu: TCR with a murinized domain; 89Zr: zirconium-89.
Acknowledgements
This work was supported by a grant from the Deutsche Forschungsgemeinschaft (SFB824/C10) (A.M.K., C.D.). The authors thank Francesco De Rose for support in labeling, Stephanie Rämisch, Sibylle Reder, and Markus Mittelhäuser for excellent other technical support and Johannes Stöckl (Institute of Immunology, Medical University Vienna) for providing the VIT3b (anti-CD3)-IgG.
Contributions
K.E.M. and S.M. performed in vitro experiments; K.E.M., S.M., and D.G. performed in vivo experiments; N.Y. and L.R. did radiolabeling and HPLC analysis; K.S. designed, performed and analyzed histology and immunohistochemistry; H.O.B., S.A., and E.B. supported in vivo studies; R.W. designed the in vitro apoptosis assay; A.S. performed immunohistochemistry staining; F.B., W.W., W.W., and M.S. reviewed the manuscript; K.E.M., S.M., and A.M.K. designed studies; K.E.M., S.M., N.Y., E.B., S.A., C.A., and A.M.K. analyzed and interpreted experimental data; K.E. M., S.M., and A.M.K. made the figures; K.E.M., S.M., C.A., and A.M.K. wrote the manuscript; F.B., W.W., W.W., M.S., C.D., and A.M.K. contributed with administrative duties, database construction, technical and material support; A.M.K. and C.D. supervised the study.
Competing Interests
M.S. received a commercial research grant from Siemens Medical Research and speakers´ bureau honoraria from Siemens Lunch Symposium, and he has ownership interest (including patents) in Siemens. The other authors have no competing interest to declare.
References
1. Larkin J, Chiarion-Sileni V, Gonzalez R. et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373:23-34
2. Borghaei H, Paz-Ares L, Horn L. et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N Engl J Med. 2015;373:1627-39
3. Grupp SA, Kalos M, Barrett D. et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368:1509-18
4. Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365:725-33
5. Morgan RA, Dudley ME, Wunderlich JR. et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314:126-9
6. Robbins PF, Kassim SH, Tran TL. et al. A pilot trial using lymphocytes genetically engineered with an NY-ESO-1-reactive T-cell receptor: long-term follow-up and correlates with response. Clin Cancer Res. 2015;21:1019-27
7. Audehm S, Krackhardt AM. Specific adoptive cellular immunotherapy in allogeneic stem cell transplantation. Oncol Res and Treat. 2017;40:691-6
8. Johnson LA, Morgan RA, Dudley ME. et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood. 2009;114:535-46
9. Schwartz LH, Litiere S, de Vries E. et al. RECIST 1.1-Update and clarification: From the RECIST committee. Eur J Cancer. 2016;62:132-7
10. Wolchok JD, Hoos A, O'Day S. et al. Guidelines for the evaluation of immune therapy activity in solid tumors: immune-related response criteria. Clin Cancer Res. 2009;15:7412-20
11. Seymour L, Bogaerts J, Perrone A. et al. iRECIST: guidelines for response criteria for use in trials testing immunotherapeutics. Lancet Oncol. 2017;18:e143-52
12. Wong ANM, McArthur GA, Hofman MS, Hicks RJ. The advantages and challenges of using FDG PET/CT for response assessment in melanoma in the era of targeted agents and immunotherapy. Eur J Nucl Med Mol Imaging. 2017;44:67-77
13. Cho SY, Lipson EJ, Im HJ. et al. Prediction of response to immune checkpoint inhibitor therapy using early-time-point (18)F-FDG PET/CT imaging in patients with advanced melanoma. J Nucl Med. 2017;58:1421-8
14. Cheson BD, Ansell S, Schwartz L. et al. Refinement of the Lugano Classification lymphoma response criteria in the era of immunomodulatory therapy. Blood. 2016;128:2489-96
15. Dimitrakopoulou-Strauss A. Monitoring of patients with metastatic melanoma treated with immune checkpoint inhibitors using PET-CT. Cancer Immunol Immunother. 2018 [Epub ahead of print]
16. Seith F, Forschner A, Schmidt H. et al. 18F-FDG-PET detects complete response to PD1-therapy in melanoma patients two weeks after therapy start. Eur J Nucl Med Mol Imaging. 2018;45:95-101
17. von Eyben FE, Baumann GS, Baum RP. PSMA diagnostics and treatments of prostate cancer become mature. Clin Transl Imaging. 2018;6:145-8
18. Dobrenkov K, Olszewska M, Likar Y. et al. Monitoring the efficacy of adoptively transferred prostate cancer-targeted human T lymphocytes with PET and bioluminescence imaging. J Nucl Med. 2008;49:1162-70
19. Vedvyas Y, Shevlin E, Zaman M. et al. Longitudinal PET imaging demonstrates biphasic CAR T cell responses in survivors. JCI Insight. 2016;1:e90064
20. Najjar AM, Manuri PR, Olivares S. et al. Imaging of sleeping beauty-modified CD19-specific T cells expressing HSV1-thymidine kinase by positron emission tomography. Mol Imaging Biol. 2016;18:838-48
21. McCracken MN, Vatakis DN, Dixit D, McLaughlin J, Zack JA, Witte ON. Noninvasive detection of tumor-infiltrating T cells by PET reporter imaging. J Clin Invest. 2015;125:1815-26
22. Keu KV, Witney TH, Yaghoubi S. et al. Reporter gene imaging of targeted T cell immunotherapy in recurrent glioma. Sci Transl Med. 2017:9
23. Kanwar B, Gao DW, Hwang AB. et al. In vivo imaging of mucosal CD4+ T cells using single photon emission computed tomography in a murine model of colitis. J Immunol Methods. 2008;329:21-30
24. Tavare R, Escuin-Ordinas H, Mok S. et al. An effective immuno-PET imaging method to monitor CD8-dependent responses to immunotherapy. Cancer Res. 2016;76:73-82
25. Tavare R, McCracken MN, Zettlitz KA. et al. Engineered antibody fragments for immuno-PET imaging of endogenous CD8+ T cells in vivo. Proc Natl Acad Sci U S A. 2014;111:1108-13
26. Tavare R, McCracken MN, Zettlitz KA. et al. Immuno-PET of murine T cell reconstitution postadoptive stem cell transplantation using anti-CD4 and anti-CD8 cys-diabodies. J Nucl Med. 2015;56:1258-64
27. Rashidian M, Ingram JR, Dougan M. et al. Predicting the response to CTLA-4 blockade by longitudinal noninvasive monitoring of CD8 T cells. J Exp Med. 2017;214:2243-55
28. Freise AC, Zettlitz KA, Salazar FB, Lu X, Tavare R, Wu AM. ImmunoPET imaging of murine CD4+ T cells using anti-CD4 Cys-Diabody: Effects of protein dose on T cell function and imaging. Mol Imaging Biol. 2017;19:599-609
29. Griessinger CM, Maurer A, Kesenheimer C. et al. 64Cu antibody-targeting of the T-cell receptor and subsequent internalization enables in vivo tracking of lymphocytes by PET. Proc Natl Acad Sci U S A. 2015;112:1161-6
30. Larimer BM, Wehrenberg-Klee E, Caraballo A, Mahmood U. Quantitative CD3 PET imaging predicts tumor growth response to anti-CTLA-4 therapy. J Nucl Med. 2016;57:1607-11
31. Beckford Vera DR, Smith CC, Bixby LM. et al. Immuno-PET imaging of tumor-infiltrating lymphocytes using zirconium-89 radiolabeled anti-CD3 antibody in immune-competent mice bearing syngeneic tumors. PLoS One. 2018;13:e0193832
32. Olafsen T, Torgov M, Zhang GG. et al. Pet imaging of cytotoxic human T cells using an 89Zr-labeled anti-CD8 minibody. J Immunother Cancer. 2015;3:P388
33. Mall S, Yusufi N, Wagner R. et al. Immuno-PET imaging of engineered human T cells in tumors. Cancer Res. 2016;76:4113-23
34. Yusufi N, Mall S, Bianchi HO. et al. In-depth characterization of a TCR-specific tracer for sensitive detection of tumor-directed transgenic T cells by immuno-PET. Theranostics. 2017;7:2402-16
35. Kreiter S, Vormehr M, van de Roemer N. et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature. 2015;520:692-6
36. Davids MS, Kim HT, Bachireddy P. et al. Ipilimumab for patients with relapse after allogeneic transplantation. N Engl J Med. 2016;375:143-53
37. Hoepner S, Loh JM, Riccadonna C. et al. Synergy between CD8 T cells and Th1 or Th2 polarised CD4 T cells for adoptive immunotherapy of brain tumours. PLoS One. 2013;8:e63933
38. Church SE, Jensen SM, Antony PA, Restifo NP, Fox BA. Tumor-specific CD4+ T cells maintain effector and memory tumor-specific CD8+ T cells. Eur J Immunol. 2014;44:69-79
39. Sommermeyer D, Hudecek M, Kosasih PL. et al. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia. 2016;30:492-500
40. Mattes J, Hulett M, Xie W. et al. Immunotherapy of cytotoxic T cell-resistant tumors by T helper 2 cells: an eotaxin and STAT6-dependent process. J Exp Med. 2003;197:387-93
41. Trzonkowski P, Bieniaszewska M, Juscinska J. et al. First-in-man clinical results of the treatment of patients with graft versus host disease with human ex vivo expanded CD4+CD25+CD127- T regulatory cells. Clin Immunol. 2009;133:22-6
42. Bierer BE, Barbosa J, Herrmann S, Burakoff SJ. Interaction of CD2 with its ligand, LFA-3, in human T cell proliferation. J Immunol. 1988;140:3358-63
43. Hahn WC, Burakoff SJ, Bierer BE. Signal transduction pathways involved in T cell receptor-induced regulation of CD2 avidity for CD58. J Immunol. 1993;150:2607-19
44. Hunig T, Tiefenthaler G, Meyer zum Buschenfelde KH, Meuer SC. Alternative pathway activation of T cells by binding of CD2 to its cell-surface ligand. Nature. 1987;326:298-301
45. Howard FD, Ledbetter JA, Wong J, Bieber CP, Stinson EB, Herzenberg LA. A human T lymphocyte differentiation marker defined by monoclonal antibodies that block E-rosette formation. J Immunol. 1981;126:2117-22
46. Moingeon P, Chang HC, Wallner BP, Stebbins C, Frey AZ, Reinherz EL. CD2-mediated adhesion facilitates T lymphocyte antigen recognition function. Nature. 1989;339:312-4
47. Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 2015;160:48-61
48. Stillwell R, Bierer BE. T cell signal transduction and the role of CD7 in costimulation. Immunol Res. 2001;24:31-52
49. Aandahl EM, Sandberg JK, Beckerman KP, Tasken K, Moretto WJ, Nixon DF. CD7 is a differentiation marker that identifies multiple CD8 T cell effector subsets. J Immunol. 2003;170:2349-55
50. Klar R, Schober S, Rami M. et al. Therapeutic targeting of naturally presented myeloperoxidase-derived HLA peptide ligands on myeloid leukemia cells by TCR-transgenic T cells. Leukemia. 2014;28:2355-66
51. Berger C, Jensen MC, Lansdorp PM, Gough M, Elliott C, Riddell SR. Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. J Clin Invest. 2008;118:294-305
52. Van Wauwe JP, De Mey JR, Goossens JG. OKT3: a monoclonal anti-human T lymphocyte antibody with potent mitogenic properties. J Immunol. 1980;124:2708-13
53. Chang TW, Testa D, Kung PC, Perry L, Dreskin HJ, Goldstein G. Cellular origin and interactions involved in gamma-interferon production induced by OKt3 monoclonal antibody. J Immunol. 1982;128:585-9
54. Li N, Hill KS, Elferink LA. Analysis of receptor tyrosine kinase internalization using flow cytometry. Methods Mol Biol. 2008;457:305-17
55. Perk LR, Vosjan MJ, Visser GW. et al. p-Isothiocyanatobenzyl-desferrioxamine: a new bifunctional chelate for facile radiolabeling of monoclonal antibodies with zirconium-89 for immuno-PET imaging. Eur J Nucl Med Mol Imaging. 2010;37:250-9
56. Schadendorf D, Hodi FS, Robert C. et al. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J Clin Oncol. 2015;33:1889-94
57. Hodi FS, O'Day SJ, McDermott DF. et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711-23
58. Weber JS, D'Angelo SP, Minor D. et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015;16:375-84
59. Larimer BM, Wehrenberg-Klee E, Dubois F. et al. Granzyme B PET imaging as a predictive biomarker of immunotherapy response. Cancer Res. 2017;77:2318-27
60. Ronald JA, Kim BS, Gowrishankar G. et al. A PET imaging strategy to visualize activated T cells in acute graft-versus-host disease elicited by allogenic hematopoietic cell transplant. Cancer Res. 2017;77:2893-902
61. Shealy DJ, Visvanathan S. Anti-TNF antibodies: lessons from the past, roadmap for the future. Handb Exp Pharmacol. 2008:101-29
62. Holter W, Majdic O, Stockinger H, Knapp W. Analysis of T cell activation with a non-mitogenic anti CD3 antibody and the phorbol ester TPA. Clin Exp Immunol. 1985;62:600-6
63. Anasetti C, Martin PJ, Storb R. et al. Treatment of acute graft-versus-host disease with a nonmitogenic anti-CD3 monoclonal antibody. Transplantation. 1992;54:844-51
64. Kohm AP, Williams JS, Bickford AL. et al. Treatment with nonmitogenic anti-CD3 monoclonal antibody induces CD4+ T cell unresponsiveness and functional reversal of established experimental autoimmune encephalomyelitis. J Immunol. 2005;174:4525-34
65. Sarangi PP, Kim B, Rouse BT. Non-mitogenic anti-CD3F(ab')2 monoclonal antibody: a novel approach to control herpetic stromal keratitis. Invest Ophthalmol Vis Sci. 2008;49:5425-33
66. Juraske C, Wipa P, Morath A. et al. Anti-CD3 Fab fragments enhance tumor killing by human gammadelta T cells independent of Nck recruitment to the gammadelta T cell antigen receptor. Front Immunol. 2018;9:1579
67. Bierer BE, Hahn WC. T cell adhesion, avidity regulation and signaling: a molecular analysis of CD2. Semin Immunol. 1993;5:249-61
68. Meuer SC, Hussey RE, Fabbi M. et al. An alternative pathway of T-cell activation: a functional role for the 50 kd T11 sheep erythrocyte receptor protein. Cell. 1984;36:897-906
69. Verbi W, Greaves MF, Schneider C. et al. Monoclonal antibodies OKT 11 and OKT 11A have pan-T reactivity and block sheep erythrocyte "receptors". Eur J Immunol. 1982;12:81-6
70. Gückel B, Berek C, Lutz M, Altevogt P, Schirrmacher V, Kyewski BA. Anti-CD2 antibodies induce T cell unresponsiveness in vivo. J Exp Med. 1991;174:957-67
71. Xu Y, Kolber-Simonds D, Hope JA. et al. The anti-CD2 monoclonal antibody BTI-322 generates unresponsiveness by activation-associated T cell depletion. Clin Exp Immunol. 2004;138:476-83
72. Dumont C, Deas O, Mollereau B. et al. Potent apoptotic signaling and subsequent unresponsiveness induced by a single CD2 mAb (BTI-322) in activated human peripheral T cells. J Immunol. 1998;160:3797-804
73. Van Wauwe J, Goossens J, Decock W, Kung P, Goldstein G. Suppression of human T-cell mitogenesis and E-rosette formation by the monoclonal antibody OKT11A. Immunology. 1981;44:865-71
74. Bonilla FA, Kokron CM, Swinton P, Geha RS. Targeted gene disruption of murine CD7. Int Immunol. 1997;9:1875-83
75. Lee DM, Staats HF, Sundy JS. et al. Immunologic characterization of CD7-deficient mice. J Immunol. 1998;160:5749-56
76. Frankel AE, Laver JH, Willingham MC, Burns LJ, Kersey JH, Vallera DA. Therapy of patients with T-cell lymphomas and leukemias using an anti-CD7 monoclonal antibody-ricin A chain immunotoxin. Leuk Lymphoma. 1997;26:287-98
77. Gomes-Silva D, Srinivasan M, Sharma S. et al. CD7-edited T cells expressing a CD7-specific CAR for the therapy of T-cell malignancies. Blood. 2017;130:285-96
78. Zhang Y, Hong H, Cai W. PET tracers based on Zirconium-89. Curr Radiopharm. 2011;4:131-9
79. Goldenberg DM. Targeted therapy of cancer with radiolabeled antibodies. J Nucl Med. 2002;43:693-713
80. Sham JG, Kievit FM, Grierson JR. et al. Glypican-3-targeting F(ab')2 for 89Zr PET of hepatocellular carcinoma. J Nucl Med. 2014;55:2032-7
81. Behr TM, Goldenberg DM, Becker W. Reducing the renal uptake of radiolabeled antibody fragments and peptides for diagnosis and therapy: present status, future prospects and limitations. Eur J Nucl Med. 1998;25:201-12
82. Bailey DL, Pichler BJ, Guckel B. et al. Combined PET/MRI: Global warming-summary report of the 6th international workshop on PET/MRI, March 27-29, 2017, Tübingen, Germany. Mol Imaging Biol. 2018;20:4-20
Author contact
![]() Corresponding author: Angela M. Krackhardt, Klinik und Poliklinik für Innere Medizin III, Klinikum rechts der Isar, Technische Universität München, Munich, Germany. E-mail: angela.krackhardtde Phone: +49 (0) 89-4140-4124 Fax: +49 (0) 89-4140-4879
Corresponding author: Angela M. Krackhardt, Klinik und Poliklinik für Innere Medizin III, Klinikum rechts der Isar, Technische Universität München, Munich, Germany. E-mail: angela.krackhardtde Phone: +49 (0) 89-4140-4124 Fax: +49 (0) 89-4140-4879