Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(21):5960-5971. doi:10.7150/thno.28960 This issue Cite
Research Paper
Islet α-cell Inflammation Induced By NF-κB inducing kinase (NIK) Leads to Hypoglycemia, Pancreatitis, Growth Retardation, and Postnatal Death in Mice
Xinzhi Li1,2, Linna Jia1, Xiaoyue Chen1, Ying Dong1, Xiaomeng Ren1, Yuefan Dong1, Ying Chen1, Liwei Xie6, Ming Liu4, Chiyo Shiota5, George K. Gittes5, Liangyou Rui3, Zheng Chen2 ![]()
1. Key Laboratory of Molecular Epigenetics of the Ministry of Education (MOE), School of Life Sciences, Northeast Normal University, Changchun, 130024, China
2. HIT Center for Life Sciences, School of Life Science and Technology, Harbin Institute of Technology, Harbin 150080, China
3. Department of Molecular & Integrative Physiology, University of Michigan Medical School, Ann Arbor, Michigan, USA
4. Department of endocrinology and metabolism, Tianjin Medical University General Hospital, Tianjin, China
5. Division of Pediatric Surgery, Department of Surgery, Children's Hospital of Pittsburgh of UPMC, University of Pittsburgh School of Medicine, Pittsburgh, USA
6. State Key Laboratory of Applied Microbiology Southern China, Guangdong Provincial Key Laboratory of Microbial Culture Collection and Application, Guangdong Open Laboratory of Applied Microbiology, Guangdong Institute of Microbiology, Guangzhou 510070, China.
Received 2018-8-2; Accepted 2018-10-4; Published 2018-11-13
Abstract

Islet α-cell dysfunction has been shown to contribute to type 2 diabetes; however, whether islet α-cell inflammation is involved in the occurrence of pancreatitis is largely unknown. The aims of this study were to investigate how NF-κB inducing kinase (NIK) regulates pancreatic α-cell function, both in vitro and in vivo, and to assess how islet α-cell inflammation induced by NIK affects the development of pancreatitis.
Methods: We utilized adenovirus-mediated NIK overexpression, ELISA, qPCR, RNA-seq, and Western blot analyses to study the role of NIK in islet α cells in vitro. Islet α-cell-specific NIK overexpressing (α-NIK-OE) mice were generated, and pancreatic α/β-cell function and the occurrence of pancreatitis in these mice were assessed via ELISA, qPCR, and immunohistochemical analyses.
Results: The LTβR/noncanonical NF-κB signaling pathway is present in islet α cells. Overexpression of NIK in αTC1-6 cells induces inflammation and cell death, contributing to a decrease in the expression and secretion of glucagon. Additionally, α-cell specific overexpression of NIK (α-NIK-OE) results in α-cell death, lower serum glucagon levels, and hypoglycemia in mice. Strikingly, α-NIK-OE mice also display a reduced β-cell mass, growth retardation, pancreatitis, and postnatal death.
Conclusions: Islet α-cell specific overexpression of NIK results in islet α-cell dysfunction and causes islet β-cell death and pancreatitis, which are most likely due to paracrine secretion of cytokines and chemokines from islet α cells, thus leading to hypoglycemia, growth retardation, and postnatal death in mice.
Keywords: islet α cell, NF-κB inducing kinase, inflammation, pancreatitis, chemokine
Introduction
The pancreas contains endocrine islets and exocrine acinar cells, which produce hormones and digestive enzymes that are key regulators of glucose metabolism and food digestion. Islet β-cell damage can lead to diabetes, while injury of acinar cells can cause pancreatitis, with inflammation being one of primary factors involve in the development of both diabetes and pancreatitis [1, 2]. Islet β-cell inflammation has been shown to contribute to pancreatitis [3], but whether or not islet α-cell inflammation can lead to pancreatitis is largely unknown.
NF-κB-inducing kinase (NIK) is a widely expressed Ser/Thr kinase that functions as a central mediator of noncanonical NF-κB activation [4]; however, its expression is quite low in quiescent cells due to TRAF2/TRAF3/cIAP1/2-mediated ubiquitination and degradation [4]. In the presence of specific cytokines, such as BAFF, CD40L, LTβ, and RANKL, receptors for these cytokines are activated, which promotes the degradation of TRAF2/TRAF3/cIAP1/ 2, leading to the stabilization and activation of NIK. In turn, NIK phosphorylates and activates IKK α [4, 5], and activated IKKα phosphorylates p100 (the inactive full-length NF-κB2), promoting its proteolytic cleavage to produce p52 (the active form of NF-κB2) [6]. Several studies have shown that NIK can regulate T/B-cell development [7, 8], and influence liver fibrosis [9, 10], and metabolic syndrome in mice [11]; however, the role of NIK in islet α cells is not completely understood. Whether islet α-cell inflammation induced by NIK can contribute to pancreatitis is also largely unknown.
In this study, we demonstrate that overexpression of NIK in islet α cells decreases glucagon secretion and induces inflammation and cell death both in vitro and in vivo. This further promotes the cell death of islet β cells and pancreatic acinar cells via increased paracrine secretion of inflammatory cytokines and chemokines, thus leading to hypoglycemia, growth retardation, pancreatitis, and postnatal death in mice.
Materials and Methods
Animal experiments
Animal experiments were carried out in strict accordance with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health and were approved by the Animal Experimental Ethics Committee of Northeast Normal University and Harbin Institute of Technology. STOP-NIK and Glucagon-cre mice (C57BL/6 background) were described previously [9, 10, 12, 13]. STOP-NIK mice were crossed with glucagon-cre mice to generate islet α-cell-specific NIK overexpression (α-NIK-OE: STOP-NIK+/-; Glucagon- Cre+/-) mice. Control mice were their littermates (Genotype: STOP-NIK+/-). ROSA26-EYFP reporter mice were purchased from Shanghai Biomodel Organism Science & Technology Development Co., Ltd. Mice were housed on a 12-h light/12-h dark cycle, and were fed with a normal chow and free access to water. Male littermates were used for experiments. Blood glucose levels were measured as desscribed previsouly [10]. Blood samples were collected from orbital sinus. Serum glucagon and insulin levels were measured using glucagon ELISA kits (DGCG0, R&D Systems) and insulin ELISA kits (EZRMI-13K, Millipore Corporation), respectively. Serum amylase activity was measured using α-Amylase assay kits (C016-1, Nanjing Jiancheng Bioengineering Institute). Pancreatic trypsin activity was measured using Trypsin ELISA kits (D59091, Immuno-Biological Laboratories Co., Ltd.) following the manufacturer's recommended procedure.
For cerulein-induced acute pancreatitis, 9-week old male C57BL/6 mice were intraperitoneally injected with 50 μg/kg cerulein (Sigma-Aldrich, St. Louis, MO) in saline every hour for a total of seven injections. Mice were sacrificed at 12 h time point, and pancreases were fixed with 4% paraformaldehyde and subjected to immunostaining assays.
Pancreatic islet and acinar cell isolation
Male mice were euthanized. Pancreases were cut into small pieces, and digested with 1 mg/mL collagenase P (Roche Diagnostics) in Hanks' balanced salt solution (HBSS) as shown previously [14]. Pancreatic islets and acinar cells were hand-picked.
Transient transfection and luciferase assays
HEK293 cells were divided equally in a 24-well plate and cultured overnight. The cells were cotransfected with mouse glucagon promoter (-1000-0 bp) luciferase reporter plasmid with NIK or p52 at different doses (0, 100, 200, 400 ng) for 24 h. The cells were then harvested in reporter lysis buffer (Promega, Madison, WI, USA). Luciferase activity was measured and normalized to β-Gal activity as shown previously [10].
Cell culture, adenoviral infection, and low glucose-stimulated glucagon secretion (LGSGS)
αTC1-6 cells (a mouse pancreatic alpha cell line) were cultured at 37°C in 5% CO2 in DMEM supplemented with 100 units ml-1 penicillin, 100 units ml-1 streptomycin, and 10% FBS. INS-1 832/13 cells (a rat insulinoma cell line) were cultured at 37°C and 5% CO2 in RPMI-1640 medium supplemented with 10% FBS and 50 mM β-mercaptoethanol as shown previously [15, 16]. β-Gal, NIK, and NIK(KA) adenovirus were described before [10, 17]. αTC1-6 cells were infected with β-Gal and NIK adenovirus for 48 h and subjected to MTT and TUNEL assays. For LGSGS assay, αTC1-6 cells were infected with β-Gal and NIK adenovirus for 16 h, and these cells were incubated at 37°C in 200 μL of HBSS (pH 7.4) containing 25 mM or 1 mM glucose for 1 h. Medium was collected to measure LGSGS. Cells were then harvested in a lysis buffer, and protein concentrations were measured. The cell extracts were then mixed with acid-ethanol (1.5% HCl in 70% EtOH) and were used to measure glucagon content. Glucagon secretion was normalized to protein levels.
Immunoblotting
αTC1-6 cells were harvested in a lysis buffer (50 mM Tris HCl, pH 7.5, 1.0% NP-40, 150 mM NaCl, 2 mM EGTA, 1 mM Na3VO4, 100 mM NaF, 10 mM Na4P2O7, 1 mM PMSF, 10 μg/mL aprotinin, 10 μg/mL leupeptin). Cell extracts were immunoblotted with the indicated antibodies and were visualized using the ECL. Antibody dilution ratios were as follows: Flag (F1804, Sigma, 1:5000 dilution), NF-κB2 (4882, Cell Signaling Technology, 1:2000 dilution), Tubulin (sc5286, Santa Cruz, 1:5000 dilution).
Quantitative real-time PCR (qPCR) analysis
αTC1-6 cells were infected with β-Gal, NIK and NIK(KA) adenovirus for 16 h. Total RNAs were extracted using TriPure Isolation Reagent (Roche, Mannheim, Germany), and the first-strand cDNAs were synthesized using random primers and M-MLV reverse transcriptase (Promega, Madison, WI) as shown before [10]. RNA abundance was measured using ABsolute qPCR SYBR Mix (Roche, Mannheim, Germany) and Roche LightCycler 480 real-time PCR system (Roche, Mannheim, Germany). The expression of individual genes was normalized to the expression of 36B4, a house-keeping gene. Primers for real time qPCR were listed in Table S1.
Treatment of INS-1 832/13 cells with conditioned medium from αTC1-6 cell
αTC1-6 cells were infected with β-Gal and NIK adenovirus for 18 h, and then the cells were grown in fresh serum-free DMEM for 24 h. Conditioned media from NIK-overexpressing αTC1-6 cells (NIKCM) and control cells (βGalCM) were then collected. Half of NIKCM was neutralized with antibodies (Proteintech) against TNFα, CCL2 and CCL5, denoted as NIKCM+Ab. INS-1 832/13 cells were treated with these conditioned media for 16 h. MTT and TUNEL assays were performed as shown previously [15]. TNFα, CCL2, and CCL5 protein levels in these conditioned media and αTC1-6 cells were measured by ELISA kits (Proteintech). For signal detection, Streptavidin-HRP was added, followed by tetramethyl-benzidine (TMB) reagent. Solution containing sulfuric acid was used to stop color development and the color intensity which was proportional to the quantity of bound protein was measurable at 450 nm.
Immunostaining and TUNEL assays
Pancreata were fixed in 4% paraformaldehyde for 3 h and then in 30% sucrose overnight. Frozen pancreatic sections (5-8 μm) were stained with the indicated antibodies. For the measurement of islet glucagon and insulin positive area, pancreatic sections were immunostained with anti-glucagon and anti-insulin antibodies. Islet glucagon and insulin positive areas were measured using Image J software, and normalized to total pancreatic section area as described before [15]. Antibody dilution ratios were as follows: insulin (A0564, Dako, 1:1000 dilution); glucagon (G2654, Sigma, 1:1000 dilution); and Ki67 (VP-RM04, Vector Laboratories, 1:100 dilution); F4/80 (14-4801-82, eBiosience, 1:200 dilution); LTβR (20331-1-AP, Proteintech, 1:200 dilution); NF-κB2 (4882, Cell Signaling Technology, 1:100 dilution); CCL2 (66272-1-Ig, Proteintech, 1:100 dilution). Frozen pancreatic sections, αTC1-6, and INS-1 832/13 cells were fixed with 4% paraformaldehyde and subjected to TUNEL assays using cell death detection kits (Roche Diagnostics) following the manufacturer's recommended procedure. The samples were stained with DAPI to visualize total cells. Pancreatic sections were also immunostained with anti-glucagon antibody to identify islet α cells or with anti-insulin antibody to identify islet β cells. TUNEL-positive cells were counted and normalized to total islet cell number.
RNA-seq
αTC1-6 cells were infected with β-Gal, NIK and NIK(KA) adenovirus for 16 h. Total RNAs were extracted using TriPure Isolation Reagent (Roche, Mannheim, Germany). mRNA profiles of β-Gal-, NIK-, and NIK(KA)-overexpressing αTC1-6 cells were generated by deep sequencing, in a single test, using Illumina. RNA sequencing libraries were generated using NEBNext® Ultra™ RNA Library Prep Kit for Illumina® (NEB, USA) following manufacturer's recommendations and library quality was assessed on the Agilent Bioanalyzer 2100 system. The mRNA library was sequenced on an Illumina Hiseq platform and 125 bp/150 bp paired-end reads were generated. Paired-end clean reads were aligned to the mouse reference genome (Ensemble_GRCm38.89) with TopHat (version 2.0.12), and the aligned reads were used to quantify mRNA expression by using HTSeq-count (version 0.6.1).Differential expression analysis of two conditions was performed using the DEGSeq R package (1.20.0). The P values were adjusted using the Benjamini & Hochberg method. Corrected P-value of 0.005 and log2 (Fold change) of 1 were set as the threshold for significantly differential expression. Gene Ontology (GO) enrichment analysis of differentially expressed genes was implemented by the GOseq R package, in which gene length bias was corrected. GO terms with corrected P value less than 0.05 were considered significantly enriched by differential expressed genes. KEGG is a database resource for understanding high-level functions and utilities of the biological system, such as the cell, the organism and the ecosystem, from molecular-level information, especially large-scale molecular datasets generated by genome sequencing and other high-through put experimental technologies (http://www.genome.jp/kegg/). We used KOBAS software to test the statistical enrichment of differential expression genes in KEGG pathways. RNA-seq data that support the findings of this study have been deposited in GEO under accession code GSE112511.
Statistical Analysis
Data were presented as mean ±SEM. Differences between groups were analyzed by two-tailed Student's t tests. P< 0.05 was considered statistically significant.
Results
The LTβR/noncanonical NF-κB signaling pathway is present in pancreatic islet α cells
It has been shown that noncanonical NF-κB signaling can be stimulated by the activation of BAFF-R, CD40, LTβR, or RANK. To determine the relative expression levels of these four receptors, we performed qPCR assays in αTC1-6 cells (a mouse pancreatic α cell line). As shown in Figure 1A, LTβR was highly expressed in αTC1-6 cells; however, BAFF-R, CD40, and RANK were barely detectable (Figure 1A). Compared with pancreatic acinar cells, αTC1-6 cells and pancreatic islets displayed higher levels of LTβR (Figure 1B and 1C). Consistent with these observations, immunofluorescence data indicated that LTβR was highly expressed in pancreatic islet α cells (Figure 1C). We noticed that insulin positive cells also displayed high levels of LTβR (Figure S1). Additionally, treatment with an anti-LTβR antibody resulted in increased p52 protein levels in αTC1-6 cells (Figure 1D). LIGHT, a ligand for LTβR, was able to increase p52 protein level (Figure 2A) and suppress low glucose-stimulated glucagon secretion (LGSGS) in αTC1-6 cells (Figure 2B). LIGHT also induced α-cell death (Figure 2C). These data demonstrated that the components of the LTβR/noncanonical NF-κB signaling pathway are present in pancreatic α cells, which may regulate islet α cell function.
LTβR/noncanonicalNF-κB2 signaling pathway is present in islet α cells. A, The relative expression of BAFF-R, CD40, LTβR, and RANK were measured by qPCR (left) and semi-quantitative PCR (right) in αTC1-6 cells (n = 6/group). B, The relative expression of LTβR was measured by qPCR in αTC1-6 cells, pancreatic acinar cells, and pancreatic islets (n = 6/group). C, Immunostaining of LTβR and glucagon in the section of pancreas. D, αTC1-6 cells were treated with anti-LTβR antibody at different concentrations (0, 2, 10 μg/mL) for 2 h, and p52 protein levels were measured by Western blot. **, p < 0.01.
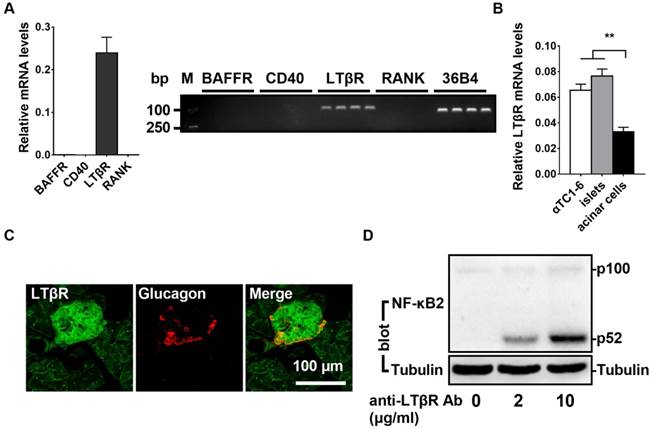
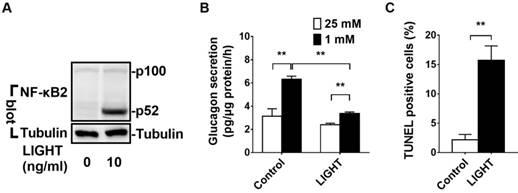
LIGHT activates noncanonical NF-κB2 signaling pathway, impairs glucagon secretion and increases cell death in αTC1-6 cells. A, αTC1-6 cells were treated with LIGHT at 0 and 10 ng/mL for 2 h. p100 and p52 protein levels were measured by Western blot. B-C, αTC1-6 cells were treated with LIGHT at 0 and 10 ng/mL for 16 h. Low glucose (1 mM) stimulated glucagon secretion (LGSGS) was measured by ELISA (B), and TUNEL-positive cells were measured (C). n = 3-4. **, p < 0.01.
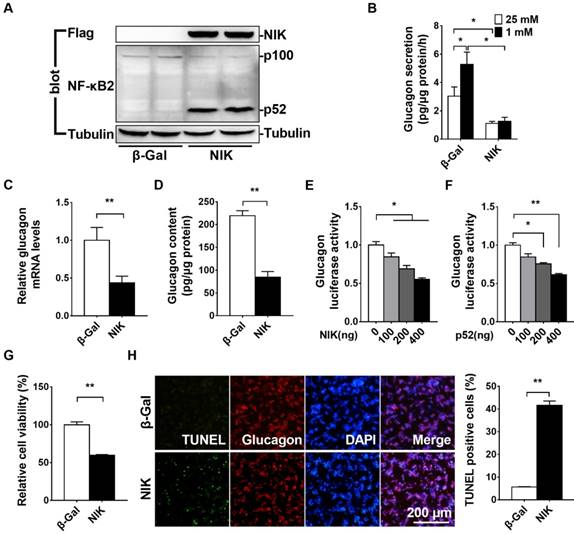
NIK overexpression impairs glucagon secretion and increases cell death in αTC1-6 cells
NIK is the key activator of the noncanonical NF-κB signaling pathway [4]. To determine whether NIK can regulate islet α cell function, we overexpressed NIK in αTC1-6 cells by infecting them with an ad-NIK adenovirus and then assessed glucagon secretion and cell viability. NIK and p52 protein levels were very high in ad-NIK infected αTC1-6 cells (Figure 3A). As shown in Figure 3B, LGSGS was present in the β-Gal control group, which was completely blocked in NIK overexpressing αTC1-6 cells. Moreover, glucagon mRNA and protein levels were significantly decreased in αTC1-6 cells after activation of NIK signaling (Figure 3C and 3D). NIK and p52 dose-dependently inhibited glucagon promoter luciferase activity (Figure 3E and 3F). These data suggested that NIK/p52 directly inhibited glucagon gene transcription. Additionally, cell viability was significantly decreased and the number of TUNEL-positive cells was dramatically increased in NIK-overexpressing αTC1-6 cells (Figure 3G and 3H), which also contributed to the observed impairment in LGSGS and reduced glucagon expression.
NIK overexpression impairs glucagon secretion and increases cell death in αTC1-6 cells. αTC1-6 cells were infected with β-Gal and NIK adenovirus for 16 h. A, NIK, p52, and Tubulin protein levels were measured by Western blot. B, Low glucose (1 mM) stimulated glucagon secretion (LGSGS) was measured by ELISA (n = 3-4). C, Glucagon mRNA was measured by RT-qPCR (n = 5). D, Glucagon content was measured by ELISA (n = 4). E-F, Glucagon promoter luciferase reporter and NIK/p52 plasmids were cotransfected into HEK293 cells for 24 h. Luciferase activity was then measured and normalized to β-Gal activity (n = 6). G-H, Cell viability (MTT) and the number of TUNEL-positive cells were measured after virus infection for 48 h (n = 5-6). *, p< 0.05. **, p < 0.01.
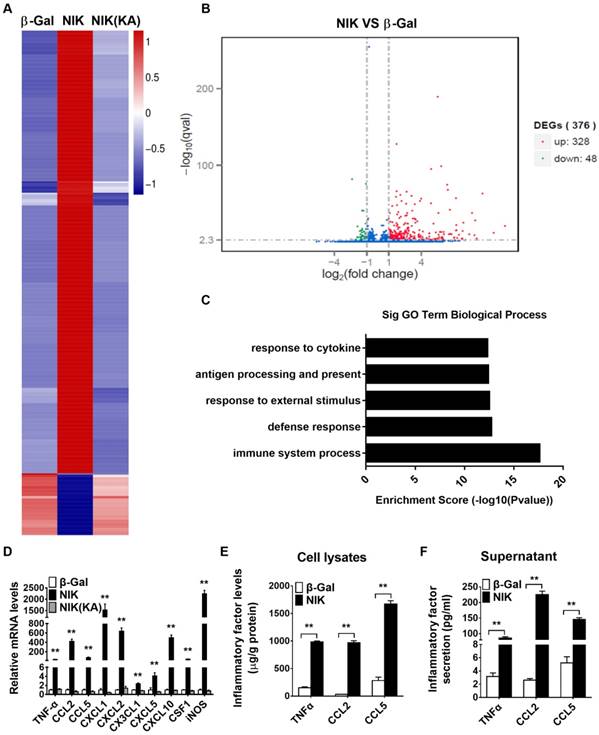
NIK overexpression triggers an immune response, inflammation, and chemokine secretion in αTC1-6 cells
To further determine the molecular mechanisms involved in the NIK regulation of α cell function, we performed RNA-seq analysis in β-Gal-overexpressing (control), NIK-overexpressing, and NIK(KA) (the dominate negative form of NIK)-overexpressing αTC1-6 cells. As shown in Figure 4A and 4D, NIK(KA)-overexpressing αTC1-6 cells displayed a very similar gene expression pattern with β-Gal control. However, 328 genes were upregulated and 48 genes were downregulated following NIK overexpression (Figure 4B). GO analysis indicated that the upregulated genes were primarily related to the immune response (Figure 4C). KEGG pathway enrichment analysis showed that immune signaling pathways, including the TNF, NF-κB, and chemokine signaling pathways, were significantly activated by NIK overexpression in αTC1-6 cells (Figure S2A), while genes related to metabolism-associated signaling pathways were significantly decreased (Figure S2B). Among the upregulated genes, the expression of the highest-ranked cytokines and chemokines were confirmed by qPCR (Figure 4D) and ELISA (Figure 4E). CXCL1 and iNOS mRNA levels were increased by more than 1500-fold, and CCL2, CXCL2, and CXCL10 mRNA levels were increased by more than 400-fold (Figure 4D). TNFα, CCL5, CXCL3, CXCL5, and CSF1 mRNA levels were also significantly increased (Figure 4D). Moreover, results of ELISA clearly showed that TNFα, CCL2, and CCL5 were secreted from αTC1-6 cells infected with ad-NIK (Figure 4F). These data indicated that NIK overexpression impairs glucagon secretion and induces cell death due to the activation of an immune response and inflammation in αTC1-6 cells.
NIK overexpression triggers an immune response, inflammation, and chemokine secretion in αTC1-6 cells. αTC1-6 cells were infected with β-Gal, NIK and NIK(KA) adenovirus for 16 h. RNA-seq analysis in both β-Gal-overexpressing (control), NIK-overexpressing, and NIK(KA)-overexpressing αTC1-6 cells. A, Heat map derived from RNA-Seq expression data ofthe differentially expressed genes (DEGs). B, DEGs (NIK VS β-Gal). C, Top GO biological process terms enriched in upregulated genes in NIK-overexpressing αTC1-6 cells compared with β-Gal control. D, Relative TNFα, CCL2, CCL5, CSCL1, CXCL2, CX3CL1, CXCL5, CXCL10, CSF1, and iNOS mRNA levels were measured by RT-qPCR (n = 6). E-F, TNFα, CCL2, and CCL5 protein levels in cell lysis and supernatant were measured by ELISA (n = 5-8). *, p < 0.05. **, p < 0.01.
Islet α-cell specific overexpression of NIK (α-NIK-OE) causes postnatal death, lower body weight, hypoglycemia, and impaired islet structure and function in mice
To examine the role of NIK in pancreatic α cells in vivo, we generated α-cell specific NIK overexpression (α-NIK-OE) mice by crossing STOP-NIK mice with glucagon-cre transgenic mice. The specificity of the glucagon-promoter driven Cre recombinase (glucagon-cre) was confirmed by crossing ROSA26-EYFP reporter mice with glucagon-cre mice and immunostaining for EYFP in pancreatic islets, hypothalamus, and intestine cells. As shown in Figure 5A, more than 95% of glucagon-positive cells expressed EYFP and none of insulin positive cells expressed EYFP, suggesting a high specificity of glucagon-cre expressed in islet α cells. We noticed that very few (about 0.55%) YFP-positive cells expressed somatostatin (Figure 5A). We also observed YFP signal in a very small amount of intestine L cells and neurons in the brain, which is consistent with previous observations [13].
Islet α-cell-specific overexpression of NIK causes postnatal death, lower body weight, hypoglycemia, and impaired islet structure and function in mice. A, Co-immunostaining of YFP and glucagon or YFP and insulin in pancreatic sections of Glucagon-cre/ROSA26-YFP mice. B, Pancreatic islets were isolated from α-NIK-OE mice and control littermates at 27 days age. NIK mRNA levels were measured by RT-qPCR and normalized to 36B4 levels (n = 4/group). C, Representative α-NIK-OE mice and control littermates at 23 days age. D, Growth curve of α-NIK-OE mice and control littermates (n = 7-14/group). E, Survival curves (n = 29/group). F, Fed blood glucose levels (n = 9-14/group). G-H, Serum glucagon and insulin levels at 20-28 days age (n = 10-15/group). I-J, Pancreas glucagon and insulin contents at 20-28 days age (n = 9-12/group). K, Islet number/section (n = 6/group). L, Representative islet from α-NIK-OE mice and control littermates was immunostained with insulin and glucagon. M-N, Relative glucagon- and insulin-positive areas. O, Ratio of islet glucagon /insulin-positive areas in α-NIK-OE mice and control littermates (n = 5-6/group).*, p < 0.05. **, p < 0.01.
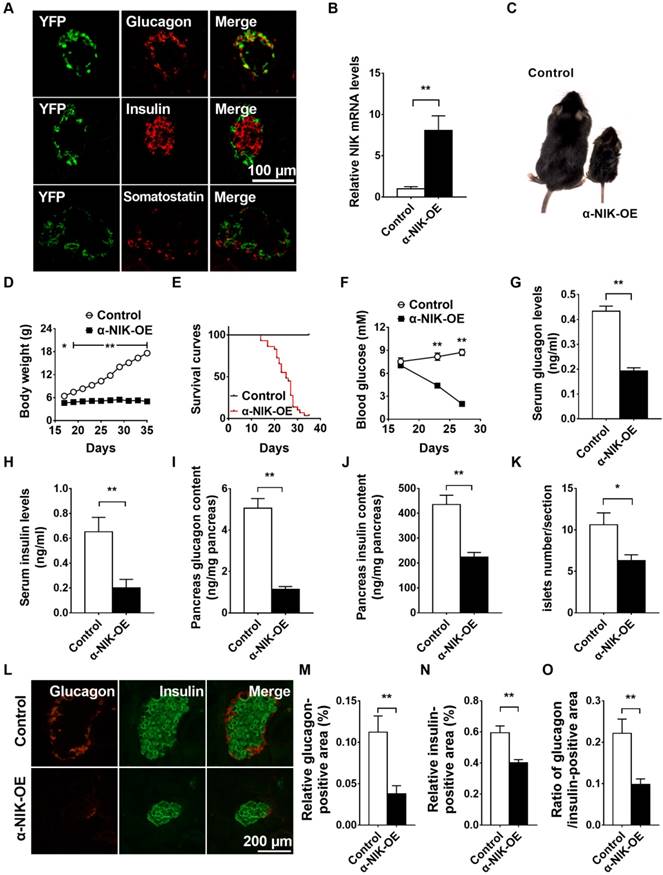
NIK mRNA levels were increased by ∼8-fold in pancreatic islets of α-NIK-OE mice (Figure 5B). α-NIK-OE mice did not gain any body weight (Figure 5C and 5D), and died within 40 days after birth (Figure 5E), and the fed blood glucose levels in α-NIK-OE mice gradually decreased after birth (Figure 5F). As glucagon and insulin are two key hormones that tightly control blood glucose levels, we measured the serum glucagon and insulin levels in α-NIK-OE and control mice. As shown in Figure 5G, the serum glucagon levels were significantly reduced by 55.6% in α-NIK-OE mice. Surprisingly, the serum insulin levels were also dramatically decreased in these mice (Figure 5H). Accordingly, pancreatic glucagon and insulin contents were significantly decreased in α-NIK-OE mice relative to the control mice (Figure 5I and 5J).
To determine whether the reduced glucagon and insulin levels in α-NIK-OE mice were caused by a reduction in islet insulin- and glucagon-positive areas, pancreatic sections from α-NIK-OE and control mice were stained using antibodies against insulin and glucagon. Compared with the control mice, islet number, the size of individual islet and total islet area were significantly decreased in α-NIK-OE mice (Figure 5K and 5L). The relative glucagon-positive area was also lower in α-NIK-OE mice (Figure 5L and 5M), and the relative insulin-positive area was also lower in these mice (Figure 5L and 5N). The ratio of islet glucagon/insulin-positive area was significantly reduced in α-NIK-OE mice (Figure 5O), indicating that the reduction of islet α cell area was substantially more than that in the control mice.
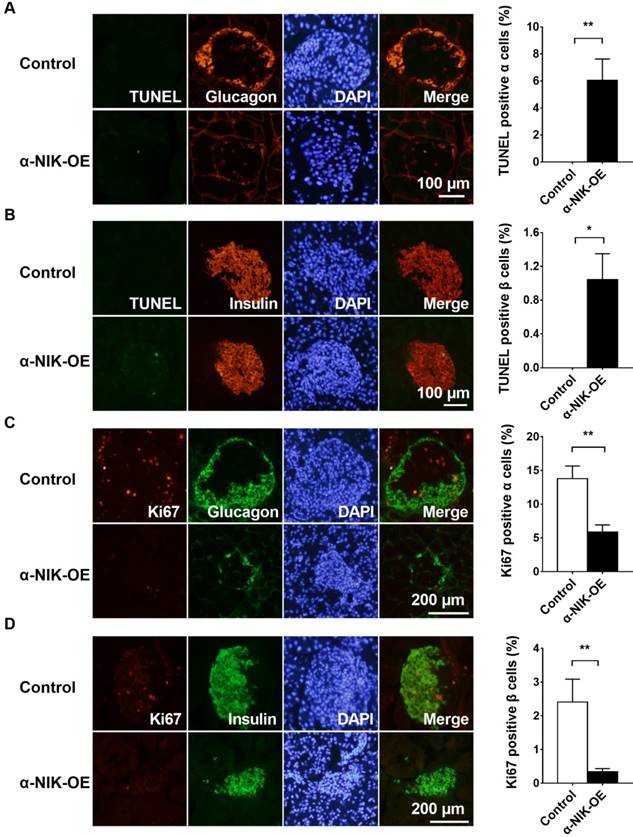
To determine whether cell death contributed to the observed reduction in glucagon- or insulin-positive areas in α-NIK-OE mice, we measured islet α- and β-cell apoptosis via TUNEL staining. Pancreatic sections were co-immunostained with anti-glucagon and anti-insulin antibodies to visualize α and β cells, respectively. The number of TUNEL-positive α or β cells was substantially more in α-NIK-OE mice than that in control mice (Figure 6A and 6B). We also examined islet α- and β-cell proliferation by immunostaining pancreatic sections with an anti-Ki67 antibody, and islets from α-NIK-OE mice demonstrated markedly fewer Ki67-positive α and β cells (Figure 6C and 6D). Together, these data suggested that increased apoptosis and decreased proliferation of α and β cells contributed to the reduced islet mass and the decreased serum glucagon and insulin levels observed in α-NIK-OE mice.
Islet α-cell-specific overexpression of NIK increases cell death and decreased cell proliferation in both islet α and β cells. A-B, Islet α- and β-cell apoptosis in α-NIK-OE mice and control littermates at 17-27 days age was measured by TUNEL assays. C-D, Islet α- and β-cell proliferation was measured by ki67 staining. Pancreatic sections were also immunostained with anti-glucagon antibody to identify islet α cells or with anti-insulin antibody to identify islet β cells (n = 5-6/group). *, p < 0.05. **, p < 0.01.
α-Cell-specific overexpression of NIK induces both α-cell death and β-cell death, indicating a crosstalk between α and β cells. To test this hypothesis, we collected conditioned media from NIK-overexpressing αTC1-6 cells (NIKCM) and control cells (βGalCM), and then treated INS-1 832/13 cells with these conditioned media. As shown in Figure S3A, cell viability was significantly decreased in INS-1 832/13 cells treated with NIKCM compared with the βGalCM group. Consistent with these observations, the presence of TUNEL-positive cells was dramatically increased in NIKCM-treated INS-1 832/13 cells (Figure S3B), which was most likely due to high levels of TNFα, CCL2, and CCL5 in NIKCM (Figure 4F). A combination of TNFα, CCL2 and CCL5 was able to induce β-cell death (Figure S3C). After neutralization with antibodies against TNFα, CCL2 and CCL5, NIKCM was unable to induce β-cell death (Figure S3A and S3B). These data suggested that activation of NIK in α cells resulted in β-cell death via paracrine secretion of cytokines and chemokines.
In addition to the role of islets in the regulation of blood glucose levels, the liver is sensitive to increased glucagon levels secreted by islet α cells in response to hypoglycemia. To address the cause of the hypoglycemia in α-NIK-OE mice, we measured the expression of the gluconeogenesis related genes G6pase and PEPCK using qPCR. The expression levels of G6pase and PEPCK were relatively high in α-NIK-OE mice as compared to those in control mice (Figure S4A), which may be a compensatory response to the hypoglycemia and lower serum glucagon levels present in α-NIK-OE mice. Importantly, normal liver structure was observed in α-NIK-OE mice (Figure S4B). These data indicated that lower serum glucagon levels could be the primary cause of hypoglycemia in α-NIK-OE mice.
α-Cell-specific overexpression of NIK induces pancreatitis
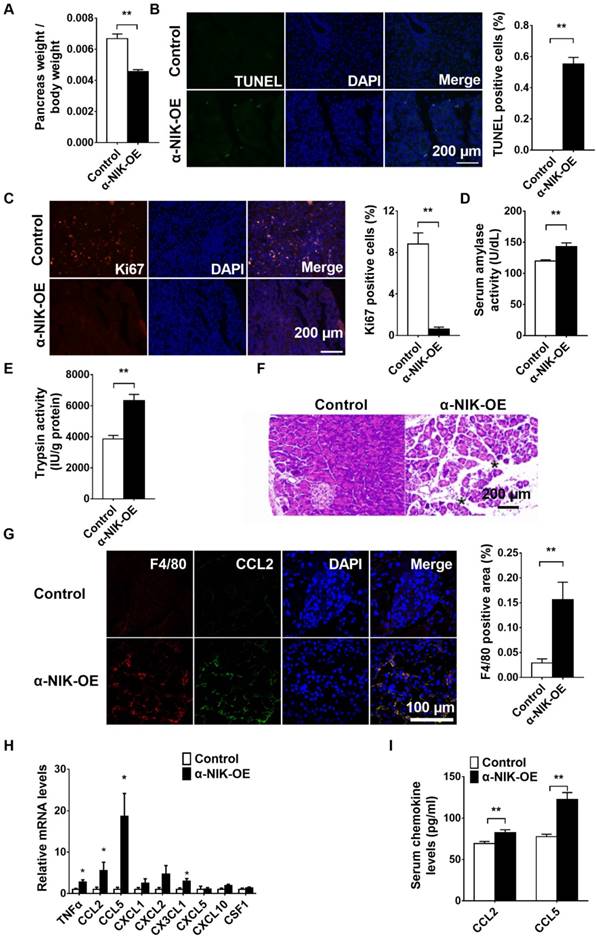
Several reports have shown that glucagon deficiency in mice only slightly decreases blood glucose levels without affecting growth or postnatal survival [18, 19], which indicates that other factors in addition to islet dysfunction contribute to the growth retardation and postnatal death observed in α-NIK-OE mice. Nuclear NF-κB2 is increased in pancreatic islet α cells in acute pancreatitis mouse model (Figure S5), indicating that NIK/p52 activation in islet α cells may be associated with pancreatitis. To assess the function of the pancreas, the size of the pancreas was first compared between α-NIK-OE and control mice. The pancreas-to-body-weight ratio was significantly decreased by 31.7% in α-NIK-OE mice relative to the control mice (Figure 7A). To check whether the smaller pancreas in these mice was due to increased apoptosis and/or decreased cell proliferation of acinar cells, pancreas sections were stained with TUNEL and Ki67. Figure 7B and 7C clearly showed that the number of TUNEL-positive cells was significantly increased while the presence of Ki67-positive cells was dramatically reduced in α-NIK-OE mice. We further checked the presence of pancreatitis markers in α-NIK-OE mice. The level of amylase was significantly increased in α-NIK-OE mice (Figure 7D), indicating tissue damage within the pancreas. Activation of trypsin is considered a key event in the onset of pancreatitis; as such, we next measured trypsin activity in the pancreas of α-NIK-OE mice. Figure 7E clearly showed that trypsin activity was significantly increased in α-NIK-OE mice. Moreover, H&E staining of pancreas sections showed that α-NIK-OE mice developed significantly more edema (Figure 7F). The relative F4/80-positive area was dramatically increased in the pancreas of α-NIK-OE mice (Figure 7G), indicating the infiltration of inflammatory cells. Moreover, the expression of inflammatory cytokine (TNFα) and chemokines (CCL2, CCL5, CXCL2, and CX3CL1) was dramatically increased in α-NIK-OE mice (Figure 7H). Immunostaining data showed that CCL2 colocalized with F4/80 (Figure 7G), indicating that increased chemokines were most likely from infiltrated F4/80-positive cells. Furthermore, CCL2 and F4/80 were also localized in islets of α-NIK-OE mice (Figure S6A and S6B), suggesting that α-NIK-OE mice also developed insulitis. Consistent with these observations, serum CCL2 and CCL5 levels were also increased in α-NIK-OE mice (Figure 7I), and a combination of TNFα, CCL2 and CCL5 was able to induce acinar cell death (Figure S7). However, we did not observe any obvious inflammation in the small intestines of α-NIK-OE mice (Figure S8). Taken together, these data demonstrated that islet α-cell-specific overexpression of NIK induces islet cell dysfunction and pancreatitis, leading to hypoglycemia, growth retardation, and postnatal death in mice.
Islet α-cell-specific overexpression of NIK induces pancreatitis. The pancreas of α-NIK-OE mice and control littermates at 20-28 days age were analyzed. A, Pancreas weight/body weight (n = 9/group). B-C, TUNEL and ki67 staining of pancreatic sections in α-NIK-OE mice and control littermates (n = 5-6/group). D, Serum amylase levels (n = 7/group). E, Pancreatic trypsin activity (n = 8/group). F, Representative H&E staining of pancreatic sections in α-NIK-OE mice and control littermates. Note the increased edema (black stars) in α-NIK-OE mice. G, F4/80 and CCL2 were co-immunostained in the pancreatic sections of α-NIK-OE mice and control littermates (n = 5-6/group). H, Pancreas mRNA abundance was measured by RT-qPCR and normalized to 36B4 levels (n = 9-12/group). I, Serum chemokine (CCL2 and CCL5) levels were measured by ELISA (n = 8-10/group). *, p < 0.05. **, p < 0.01.
Discussion
In this study, we investigated the role of NIK in the regulation of islet α cells and showed that islet α-cell-specific overexpression of NIK induces islet α-cell dysfunction. Importantly, islet α-cell-specific overexpression of NIK results in islet β-cell death and pancreatitis, which is most likely due to paracrine secretion of cytokines and chemokines from the affected islet α cells. Ultimately, this islet α-cell dysfunction leads to hypoglycemia, growth retardation, and postnatal death in these mice. These observations indicate that islet α-cell inflammation induced by NIK is directly linked to pancreatitis.
In support of these conclusions, we first showed that the LTβR/noncanonical NF-κB signaling pathway can be activated in pancreatic α cells. Stimulation of LTβR by an anti-LTβR antibody or LIGHT is able to activate the noncanonical NF-κB signaling pathway in islet α cells. As NIK is the key activator of the noncanonical NF-κB signaling pathway [4], overexpression of NIK in islet α cells results in impaired glucagon secretion, both in vitro and in vivo, which is at least partially due to decreased glucagon expression. Both NIK and p52 could dose-dependently inhibit glucagon promoter Luciferase activity, indicating that NIK/p52 directly suppresses glucagon gene transcription. Overexpression of NIK also causes islet α-cell death and reduced cell proliferation, leading to a decreased islet α-cell mass, further contributing to decreased serum glucagon levels in α-NIK-OE mice. Insufficient secretion of glucagon, thus, contributes to the observed hypoglycemia in these mice.
In addition to islet α-cell phenotypes, we also observed islet β-cell phenotypes in α-NIK-OE mice. Serum insulin levels and the islet β-cell mass were significantly decreased in α-NIK-OE mice due to increased islet β-cell death and decreased islet β-cell proliferation. This crosstalk between islet α and β cells is further supported by the results of our conditioned medium experiments. NIK overexpression in islet α cell induces inflammatory cytokine and chemokine expression, which are then secreted from islet α cells and act in a paracrine manner on islet β cells, causing islet β-cell death and reduced β-cell proliferation. Finally, these results in lower serum insulin levels, which may contribute to the growth retardation observed in α-NIK-OE mice.
Because glucagon deficiency and islet-cell dysfunction do not cause postnatal death in mice [18, 19], we further assessed the function of the pancreas, liver, and intestine of α-NIK-OE mice. It was observed that only pancreas develops severe pathological phenotypes (pancreatitis). This is the first evidence that islet α-cell inflammation can cause pancreatitis. Consistent with the association between islet β-cell stress in type 2 diabetes mellitus and the development of pancreatitis [3], islet α cells overexpressing NIK releases inflammatory cytokines and chemokines, which may further damage acinar cells and recruit macrophage into the pancreas, inducing inflammation and pancreatitis. Subsequently, hypoglycemia and pancreatitis ultimately result in growth retardation and postnatal death in α-NIK-OE mice.
In conclusion, we have uncovered a role for NIK in the regulation of islet α cells and found that islet α-cell-specific overexpression of NIK both induces islet α cell dysfunction and causes islet β cell death resulting in pancreatitis, which is most likely due to paracrine secretion of cytokines and chemokines from islet α cells, resulting in hypoglycemia, growth retardation, and postnatal death in these mice. These data suggest for the first time a relationship between islet α-cell inflammation and the development of pancreatitis.
Abbreviations
NF-κB inducing kinase: NIK, α-cell specific overexpressing of NIK: α-NIK-OE, Hanks' balanced salt solution: HBSS, low glucose-stimulated glucagon secretion: LGSGS, conditioned medium from NIK-overexpressing αTC1-6 cells: NIKCM, conditioned medium from βGal-overexpressing αTC1-6 cells: βGalCM, Quantitative real time PCR: qPCR.
Acknowledgements
This study was supported by the National Natural Science Foundation of China Grant (31500957 and 31671225 to Z.C.; 81570699 and 81620108004 to M.L.), the Fok Ying Tong Education Foundation (151022), the Jilin Science and Technology Development Program (20160101204JC and 20170519009JH), Changbai Mountain Scholars Program of The People's Government of Jilin Province (2013046).
Author contributions
Z.C. conceived and designed the project, acquired research data, and wrote the manuscript. X.L., L.J., X.C., Y.D., X.R., Y.D., and Y.C. acquired research data. C.S and GK. G. provided Glucagon-cre mice and reviewed the manuscript. L.R. provided STOP-NIK mice and reviewed the manuscript. M.L. and L.X. contributed to helpful discussion and reviewed the manuscript.
Supplementary Material
Supplementary figures and tables.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Imai Y, Dobrian AD, Morris MA, Nadler JL. Islet inflammation: A unifying target for diabetes treatment? Trends in endocrinology and metabolism: TEM. 2013;24:351-360
2. Gukovsky I, Li N, Todoric J. et al. Inflammation, Autophagy, and Obesity: Common Features in the Pathogenesis of Pancreatitis and Pancreatic Cancer. Gastroenterology. 2013;144:1199-1209.e1194
3. Schludi B, Moin ASM, Montemurro C. et al. Islet inflammation and ductal proliferation may be linked to increased pancreatitis risk in type 2 diabetes. JCI Insight. 2017;2:e92282
4. Sun SC. Non-canonical NF-kappaB signaling pathway. Cell Res. 2011;21:71-85
5. Cildir G, Low KC, Tergaonkar V. Noncanonical NF-κB Signaling in Health and Disease. Trends in Molecular Medicine. 2016;22:414-429
6. Xiao G, Fong A, Sun S-C. Induction of p100 Processing by NF-κB-inducing Kinase Involves Docking IκB Kinase α (IKKα) to p100 and IKKα-mediated Phosphorylation. Journal of Biological Chemistry. 2004;279:30099-30105
7. Mair F, Joller S, Hoeppli R. et al. The NFκB-inducing kinase is essential for the developmental programming of skin-resident and IL-17-producing γδ T cells. eLife. 2015;4:e10087
8. Hahn M, Macht A, Waisman A, Hövelmeyer N. NF-κB-inducing kinase is essential for B-cell maintenance in mice. European Journal of Immunology. 2016;46:732-741
9. Shen H, Sheng L, Chen Z. et al. Mouse hepatocyte overexpression of NF-kappaB-inducing kinase (NIK) triggers fatal macrophage-dependent liver injury and fibrosis. Hepatology. 2014;60:2065-2076
10. Ren X, Li X, Jia L. et al. A small-molecule inhibitor of NF-κB-inducing kinase (NIK) protects liver from toxin-induced inflammation, oxidative stress, and injury. The FASEB Journal. 2017;31:711-718
11. Sheng L, Zhou Y, Chen Z. et al. NF-kappaB-inducing kinase (NIK) promotes hyperglycemia and glucose intolerance in obesity by augmenting glucagon action. Nat Med. 2012;18:943-949
12. Sasaki Y, Calado DP, Derudder E. et al. NIK overexpression amplifies, whereas ablation of its TRAF3-binding domain replaces BAFF:BAFF-R-mediated survival signals in B cells. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:10883-10888
13. Shiota C, Prasadan K, Guo P. et al. α-Cells are dispensable in postnatal morphogenesis and maturation of mouse pancreatic islets. American Journal of Physiology-Endocrinology and Metabolism. 2013;305:E1030-E1040
14. Chen Z, Morris DL, Jiang L. et al. SH2B1 in β-Cells Promotes Insulin Expression and Glucose Metabolism in Mice. Molecular Endocrinology. 2014;28:696-705
15. Chen Z, Morris DL, Jiang L. et al. SH2B1 in β-Cells Regulates Glucose Metabolism by Promoting β-Cell Survival and Islet Expansion. Diabetes. 2014;63:585-595
16. Hohmeier HE, Mulder H, Chen G. et al. Isolation of INS-1-derived cell lines with robust ATP-sensitive K+ channel-dependent and -independent glucose-stimulated insulin secretion. Diabetes. 2000;49:424-430
17. de Leon-Boenig G, Bowman Krista K, Feng Jianwen A. et al. The Crystal Structure of the Catalytic Domain of the NF-κB Inducing Kinase Reveals a Narrow but Flexible Active Site. Structure. 2012;20:1704-1714
18. Hayashi Y. Metabolic impact of glucagon deficiency. Diabetes, Obesity and Metabolism. 2011;13:151-157
19. Hancock AS, Du A, Liu J. et al. Glucagon Deficiency Reduces Hepatic Glucose Production and Improves Glucose Tolerance In Adult Mice. Molecular Endocrinology. 2010;24:1605-1614
Author contact
![]() Corresponding author: Zheng Chen, Ph.D. HIT Center for Life Sciences, School of Life Science and Technology, Harbin Institute of Technology, Harbin 150080, China, TEL: +86 45186402029 E-mail: chenzhengedu.cn
Corresponding author: Zheng Chen, Ph.D. HIT Center for Life Sciences, School of Life Science and Technology, Harbin Institute of Technology, Harbin 150080, China, TEL: +86 45186402029 E-mail: chenzhengedu.cn