Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
1. Introduction
2. Structure of V-ATPase
3. Subunits of V-ATPase and bone...
4. Functions of V-ATPase...
5. Complex contributions of...
6. Future applications of...
Related materials and methods
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(19):5379-5399. doi:10.7150/thno.28391 This issue Cite
Review
V-ATPases and osteoclasts: ambiguous future of V-ATPases inhibitors in osteoporosis
Xiaohong Duan1 ![]() , Shaoqing Yang1, Lei Zhang2, Tielin Yang3
, Shaoqing Yang1, Lei Zhang2, Tielin Yang3
1. State Key Laboratory of Military Stomatology, National Clinical Research Center for Oral Diseases, Department of Oral Biology, Clinic of Oral Rare and Genetic Diseases, School of Stomatology, the Fourth Military Medical University, 145 West Changle Road, Xi'an 710032, P. R. China.
2. Center for Genetic Epidemiology and Genomics, School of Public Health, Medical College of Soochow University, 199 Renai Road, Suzhou, Jiangsu, P. R. China.
3. Key Laboratory of Biomedical Information Engineering of Ministry of Education, and Institute of Molecular Genetics, School of Life Science and Technology, Xi'an Jiaotong University, 28 West Xianning Road, Xi'an 710049, People's Republic of China.
Received 2018-7-9; Accepted 2018-10-10; Published 2018-10-26
Abstract

Vacuolar ATPases (V-ATPases) play a critical role in regulating extracellular acidification of osteoclasts and bone resorption. The deficiencies of subunit a3 and d2 of V-ATPases result in increased bone density in humans and mice. One of the traditional drug design strategies in treating osteoporosis is the use of subunit a3 inhibitor. Recent findings connect subunits H and G1 with decreased bone density. Given the controversial effects of ATPase subunits on bone density, there is a critical need to review the subunits of V-ATPase in osteoclasts and their functions in regulating osteoclasts and bone remodeling. In this review, we comprehensively address the following areas: information about all V-ATPase subunits and their isoforms; summary of V-ATPase subunits associated with human genetic diseases; V-ATPase subunits and osteopetrosis/osteoporosis; screening of all V-ATPase subunits variants in GEFOS data and in-house data; spectrum of V-ATPase subunits during osteoclastogenesis; direct and indirect roles of subunits of V-ATPases in osteoclasts; V-ATPase-associated signaling pathways in osteoclasts; interactions among V-ATPase subunits in osteoclasts; osteoclast-specific V-ATPase inhibitors; perspective of future inhibitors or activators targeting V-ATPase subunits in the treatment of osteoporosis.
Keywords: V-ATPase, osteoclasts, osteoporosis, osteopetrosis, pH, inhibitor, signaling pathways
1. Introduction
Vacuolar ATPases (V-ATPases) are protein complexes that couple ATP hydrolysis to proton transport in intracellular compartments or across the plasma membrane. V-ATPases are important in maintaining the acidic environment of intracellular organelles, including secretory granules, endosomes, and lysosomes. The acidic intracellular environment is necessary for protein sorting, zymogen activation, and receptor-mediated endocytosis [1]. V-ATPases also control the extracellular acidification of osteoclasts, which is a key factor for bone resorption [2]. The V-ATPases-related regulation of extracellular acidification also exists in other tissues or cells such as kidney and metastatic cells [3-5].
V-ATPases are ubiquitously expressed in a variety of cell types and are considered the essential “housekeeping” enzymes in all eukaryotic cells; however, the specific functions of V-ATPases vary from cell to cell. Most cells only express a low level of V-ATPases to carry out housekeeping functions, but other cells, like osteoclasts, have abundant V-ATPases that control extracellular acidification and finally affect bone resorption and bone remodeling. In this review, we will describe the V-ATPases-involved bone phenotypes, the functions of V-ATPase subunits in osteoclasts, as well as the inhibitors targeting V-ATPases in the treatment of bone diseases.
2. Structure of V-ATPase
The mammalian V-ATPase proton pump is composed of the peripheral V1 component and membrane-bound V0 component with at least 13 subunits [6]. The V1 component drives ATP hydrolysis to energize and initiate the rotation of V0 domain, which includes eight subunits, A through H. V0 domain utilizes the energy generated by V1 domain to translocate protons across the membrane and includes a through e subunits. V-ATPases also have two accessory subunits, AP1 and AP2 [1, 7-10]. Some V-ATPase subunits have multiple isoforms, which are expressed or function in a tissue-specific manner [9, 11-13] (Table 1). N-glycosylation and other processes are crucial for the formation of subunit isoforms and protein stability [14, 15].
Subunits of human V-ATPases
| Location | Name | Isoform (s) | Official Symbol | Location (GRCh38.p7) | mRNA and Protein (s) | Alias | ||
|---|---|---|---|---|---|---|---|---|
| Gene | Protein | |||||||
| V1 | A | N/A | ATP6V1A | Chromosome 3, NC_000003.12 (113747019,113812058) | NM_001690.3 → NP_001681.2 | ATP6V1A, ARCL2D, HO68, VA68 | ATP6V1A, ARCL2D, HO68, VA68 | |
| B | B1 | ATP6V1B1 | Chromosome 2, NC_000002.12 (70935868,70965431) | kidney isoform: NM_001692.3 → NP_001683.2 | ATP6V1B1, ATP6B1 | ATP6V1B1, ATP6B1 | ||
| B2 | ATP6V1B2 | Chromosome 8, NC_000008.11 (20197193,20226852) | brain isoform: NM_001693.3 → NP_001684.2 | ATP6V1B2, HO57 | ATP6V1B2, HO57 | |||
| C | C1 | ATP6V1C1 | Chromosome 8, NC_000008.11 (103021020,103073057) | NM_001695.4 → NP_001686.1 | ATP6V1C1, ATP6C, ATP6D | ATP6V1C1, ATP6C, ATP6D | ||
| C2 | ATP6V1C2 | Chromosome 2, NC_000002.12 (10720973,10785110) | isoform a: NM_001039362.1 → NP_001034451.1 | ATP6V1C2, ATP6C2 | ATP6V1C2, ATP6C2 | |||
| isoform b: NM_144583.3 → NP_653184.2 | ||||||||
| D | D | ATP6V1D | Chromosome 14, NC_000014.9 (67337864, 67360003, complement) | NM_015994.3 → NP_057078.1 | ATP6V1D, ATP6M | ATP6V1D, ATP6M | ||
| E | E1 | ATP6V1E1 | Chromosome 22, NC_000022.11 (17592136, 17628822, complement) | isoform a: NM_001696.3 → NP_001687.1 | ATP6E, ATP6E2, ATP6V1E, ATP6V1E1 | ATP6E, ATP6E2, ATP6V1E, ATP6V1E1 | ||
| isoform b: NM_001039366.1 → NP_001034455.1 | ||||||||
| isoform c: NM_001039367.1 → NP_001034456.1 | ||||||||
| E2 | ATP6V1E2 | Chromosome 2, NC_000002.12 (46511835, 46542557, complement) | NM_001318063.1 → NP_001304992.1 | ATP6V1E2, ATP6E1, ATP6EL2 | ATP6V1E2, ATP6E1, ATP6EL2 | |||
| F | F | ATP6V1F | Chromosome 7, NC_000007.14 (128862803,128865849) | isoform 1: NM_004231.3 → NP_004222.2 | ATP6V1F, ATP6S14 | ATP6V1F, ATP6S14 | ||
| isoform 2: NM_001198909.1 → NP_001185838.1 | ||||||||
| G | G1 | ATP6V1G1 | Chromosome 9, NC_000009.12 (114587714,114598872) | NM_004888.3 → NP_004879.1 | ATP6V1G1, ATP6G, ATP6G1, ATP6GL, ATP6J | ATP6V1G1, ATP6G, ATP6G1, ATP6GL, ATP6J | ||
| G2 | ATP6V1G2 | Chromosome 6, NC_000006.12 (31544451,31546848, complement) | isoform a (longest): NM_130463.3 → NP_569730.1 | ATP6V1G2, ATP6G, ATP6G2 | ATP6V1G2, ATP6G, ATP6G2 | |||
| isoform b: NM_138282.2 → NP_612139.1 | ||||||||
| isoform c: NM_001204078.1 → NP_001191007.1 | ||||||||
| G3 | ATP6V1G3 | Chromosome 1, NC_000001.11 (198523222,198540945, complement) | isoform a: NM_133262.2 → NP_573569.1 | ATP6V1G3, ATP6G3 | ATP6V1G3, ATP6G3 | |||
| isoform b: NM_133326.1 → NP_579872.1 | ||||||||
| isoform c: NM_001320218.1 → NP_001307147.1 | ||||||||
| H | H | ATP6V1H | Chromosome 8, NC_000008.11 (53715543, 53843311, complement) | isoform 1: NM_015941.3 → NP_057025.2 NM_213620.2 → NP_998785.1. Two variants encode the same isoform 1 | ATP6V1H, CGI-11, SFD | ATP6V1H, CGI-11, SFD | ||
| isoform 2: NM_213619.2 → NP_998784.1 | ||||||||
| V0 | a | a1 | ATP6V0A1 | Chromosome 17, NC_000017.11 (42458844, 42522579) | isoform a : NM_001130020.1 → NP_001123492.1 | ATP6V0A1, ATP6N1, ATP6N1A | ATP6V0A1, ATP6N1, ATP6N1A | |
| isoform b: NM_001130021.1 → NP_001123493.1 | ||||||||
| isoform c: NM_005177.3 → NP_005168.2 | ||||||||
| a2 | ATP6V0A2 | Chromosome 12, NC_000012.12 (123712318,123761755) | NM_012463.3 → NP_036595.2 | ATP6V0A2, ARCL, ARCL2A, ATP6A2 | ATP6V0A2, ARCL, ARCL2A, ATP6A2 | |||
| a3 | TCIRG1 | Chromosome 11, NC_000011.10 (68038995, 68053846) | isoform a (OC116): NM_006019.3 → NP_006010.2 | TCIRG1, ATP6V0A3, Atp6i | TCIRG1, ATP6V0A3, Atp6i | |||
| isoform b (TCIR7): NM_006053.3 → NP_006044.1 | TIRC7 | TIRC7 | ||||||
| isoform c: NM_001351059.1 → NP_001337988.1 | ||||||||
| a4 | ATP6V0A4 | Chromosome 7, NC_000007.14 (138706294,138799839, complement) | NM_020632.2 → NP_065683.2 NM_130840.2 → NP_570855.2 NM_130841.2 → NP_570856.2 Three variants encode the same protein. | ATP6V0A4, ATP6N1B | ATP6V0A4, ATP6N1B | |||
| c | c | ATP6V0C | Chromosome 16, NC_000016.10 (2513726, 2520223) | NM_001198569.1 → NP_001185498.1 NM_001694.3→ NP_001685.1 Two variants encode the same protein. | ATP6V0C, ATP6L, ATP6C | ATP6V0C, ATP6L, ATP6C | ||
| b | b | ATP6V0B | Chromosome 1, NC_000001.11 (43974648, 43978300) | isoform 1: NM_004047.4 → NP_004038.1 | ATP6F, ATP6V0B | ATP6F, ATP6V0B | ||
| isoform 2: NM_001039457.2 → NP_001034546.1 | ||||||||
| isoform 3: NM_001294333.1 → NP_001281262.1 | ||||||||
| d | d1 | ATP6V0D1 | Chromosome 16, NC_000016.10 (67438014, 67481186, complement) | NM_004691.4 → NP_004682.2 | ATP6V0D1, ATP6D | ATP6V0D1, ATP6D | ||
| d2 | ATP6V0D2 | Chromosome 8, NC_000008.11 (86098910, 86154225) | NM_152565.1 → NP_689778.1 | ATP6V0D2, ATP6D2 | ATP6V0D2, ATP6D2 | |||
| e | e1 | ATP6V0E1 | Chromosome 5, NC_000005.10 (172983760,173034897) | NM_003945.3 → NP_003936.1 | ATP6V0E1, ATP6H | ATP6V0E1, ATP6H | ||
| e2 | ATP6V0E2 | Chromosome 7, NC_000007.14 (149872968,149880713) | isoform 1: NM_145230.3 → NP_660265.2 | ATP6V0E2 | ATP6V0E2 | |||
| isoform 2: NM_001100592.2 → NP_001094062.1 | ||||||||
| isoform 3: NM_001289990.1 → NP_001276919.1 | ||||||||
| Accessory | AP1 | AP1 | ATP6AP1 | Chromosome X, NC_000023.11 (154428632, 154436517) | NM_001183.5 → NP_001174.2 | ATP6AP1, ATP6IP1, ATP6S1, Ac45 | ATP6AP1, ATP6IP1, ATP6S1, Ac45 | |
| AP2 | AP2 | ATP6AP2 | Chromosome X, NC_000023.11 (40580964, 40606637) | NM_005765.2 → NP_005756.2 | ATP6AP2, APT6M8-9, ATP6IP2, ATP6M8-9, PRR, RENR | ATP6AP2, APT6M8-9, ATP6IP2, ATP6M8-9, PRR, RENR | ||
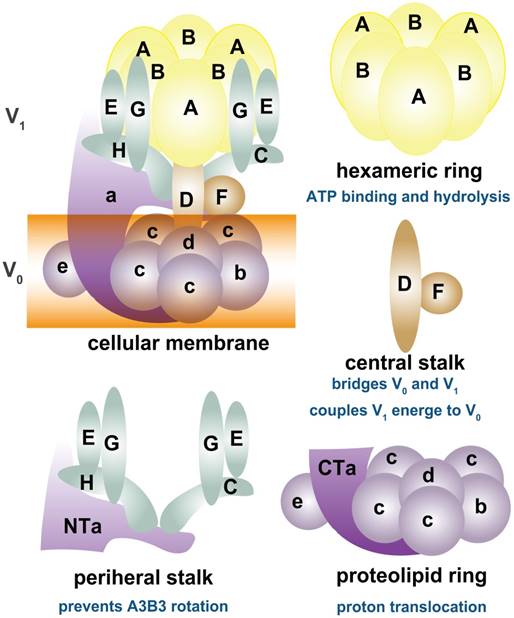
As shown in Figure 1, depending on ATP hydrolysis and reversible assembly process, the V-ATPase structure can be further divided into four parts: hexameric ring, central stalk, peripheral stalk, and proteolipid ring. The subunits A and B in the V1 component have three copies and they form an A3B3 hexameric ring in which ATP hydrolysis occurs. Three ATP catalytic sites are at the interfaces between subunits A and B [1]. Subunit A provides most of the residues for ATP binding. Other ATP binding sites may be located in subunit B [16-19]. The central stalk including subunit D and F bridges and stabilizes the interaction between V1 and V0 domains and couples the energy released from A3B3 to proton translocation in V0 [20, 21]. Subunit F is also crucial in ATP hydrolysis. The peripheral stalk, which contains three copies of subunits E and G, one or two copies of H [22], one copy of C, and the N-terminal domain of subunit a of the V0 domain, functions as a stator and tethers the A3B3 hexameric ring to subunit a for the next rotational catalysis. The central rotor and peripheral stalk maintain the stability of V-ATPase complex during catalytic rotation and proton translocation [23-25]. Finally, subunit c and b in V0 form a proteolipid ring-like structure in the membrane layer [10, 26, 27]. Subunit d functions as a “boxing glove” on the top of the proteolipid ring, interacts with subunit a, and provides a connection between the central stalk of V1 and the proteolipid ring of V0 [1, 28, 29]. The proton transportation in V-ATPase relies much on the C-terminal domain of subunit a within the proteolipid subunits, in which the hemichannels allow protons to enter and leave the membrane [30-34]. The rotation of the central rotor initiates a series of proton translocations from subunit a to subunit c [35-37] (Figure 1). Subunits a3, d, A, C, and D are related to the coupling efficiency of ATP hydrolysis to proton transport, which alternatively regulates V-ATPase activity [38-42]. Besides ATP hydrolysis and proton transport, the reversible dissociation of V1 and V0 complexes also regulates V-ATPase functions [1, 43].
Schematic map of V-ATPase. The mammalian V-ATPase proton pump is composed of peripheral V1 and membrane-bound V0. The V1 components include A-H subunits, whereas V0 includes a~e subunits. ATP hydrolysis and binding occur in the A3B3 hexameric ring. The central stalk D and F bridges and stabilizes V1/ V0 interaction and couples the energy released from A3B3 to proton translocation in V0. The peripheral stalks E, G, H, C, and N-terminal of subunit a (NTa) function as stators and tether the A3B3 hexameric ring to subunit a. The proton transport relies on the C-terminal of subunit a (CTa) and proteolipid-containing b, c, d, and e.
3. Subunits of V-ATPase and bone diseases
V-ATPase complex plays a significant role in biological and physiological processes. Mutations in the coding genes and non-coding regions of V-ATPase subunits cause various syndromes [44, 45]. The subunits of V-ATPase are ubiquitously expressed, and some of them have tissue or cell-specific distributions. Thus, the phenotypes of V-ATPase-related human diseases vary from the nervous system, kidney, and skin to skeletal system and many other tissues. Some subunits might contribute to common polygenic diseases, such as cancer and diabetes (Table 2). The two most striking and entirely distinct types of bone diseases that involve V-ATPases are osteopetrosis and osteoporosis.
Subunits of V-ATPase and phenotypes in humans and animals.
| Gene Name | Phenotype MIM Number | Human Disease Data | Mouse/Zebrafish Data |
|---|---|---|---|
| ATP6V0A1 | N/A | N/A | Zebrafish: abnormalities in endosomes, autophagosomes, and phagolysosomes, as well as the migration of neural crest cells [46, 47]. |
| ATP6V0A2 | 219200 | Cutis laxa, autosomal recessive, type II A (ARCL-2A)[45, 48-51] | N/A |
| 278250 | Wrinkly skin syndrome [48-52] | N/A | |
| TCIRG1 | 259700 | Osteopetrosis, autosomal recessive 1 [44, 53, 54] | Mouse: hypocalcemia and osteopetrorickets[55-57] |
| ATP6V0A4 | 602722 | Renal tubular acidosis, distal, autosomal recessive [58, 59] | Mouse: distal renal tubular acidosis with hearing loss, severe metabolic acidosis, hypokalemia, early nephrocalcinosis, and bone loss [60, 61]. |
| ATP6V0B | N/A | N/A | Zebrafish: abnormal integument colorless, retina degeneration, and eye discoloration [62] |
| ATP6V0C | N/A | Eye development and maintenance [63]; glial cell death/cancer/dopamine release/neurodegenerative disease [64, 65] | Zebrafish: abnormalities in head size, surface structure quality, fin malformation, pigment cell quality, brain necrosis, retinal pigmented epithelium quality, melanocyte quality, pectoral fin quality, nervous system quality [63, 66, 67]. |
| ATP6V0D1 | N/A | N/A | Zebrafish: manifestation in animal organ development, eye development, multicellular organism development, pigmentation, sensory organ development [63, 68, 69]. |
| ATP6V0D2 | N/A | N/A | Mouse: increased bone intensity [70, 71]. |
| ATP6V0E1 | N/A | N/A | N/A |
| ATP6V0E2 | N/A | N/A Restricted tissue distribution in kidney and brain [72] | N/A |
| ATP6V1A | 617403 | Autosomal recessive cutis laxa type IID[73] | Zebrafish: several abnormalities including suppression of acid-secretion from skin, growth retardation, trunk deformation [74]. |
| ATP6V1B1 | 267300 | Renal tubular acidosis with deafness [58, 75] | Mouse: acidosis, tubular, renal, with progressive nerve deafness [76] |
| ATP6V1B2 | 124480 | Deafness, congenital, with onychodystrophy, autosomal dominant [77] | Mouse: hearing loss [77] |
| 616455 | Zimmermann-Laband syndrome [78] | ||
| N/A | Depression and hippocampal neurocognitive deficits [79] | ||
| ATP6V1C1 | N/A | N/A | N/A |
| ATP6V1C2 | 609946 | Deafness, autosomal recessive 47; DFNB47 [80] | N/A |
| ATP6V1D | N/A | N/A Cell division [81] | N/A |
| ATP6V1E1 | 617403 | Autosomal recessive cutis laxa type IID (ARCL2D)[73] | Zebrafish: abnormal ventral fin [82] |
| ATP6V1E2 | N/A | N/A Acrosome acidification [83] | N/A |
| ATP6V1F | N/A | Zebrafish: oculocutaneous albinos, defects in melanosomes and retinal pigmented epithelium [63] | |
| ATP6V1G1 | N/A | Bone loss [84] | N/A |
| ATP6V1G2 | N/A | N/A | Mouse: no obvious phenotype due to compensating increased G1 level [85]. |
| ATP6V1G3 | N/A | N/A | N/A |
| ATP6V1H | N/A | Bone loss [86, 87] | Mouse and zebrafish: bone loss [86, 87] |
N/A: no available data.
3.1 Subunits of V-ATPase and osteopetrosis
Osteopetrosis is a rare bone disorder with increased bone density. In the OMIM catalogue, autosomal recessive osteopetrosis is divided into seven subtypes (osteopetrosis, autosomal recessive 1~7, OPTB1~7; OMIM 259700, 259710, 259730, 611490, 259720, 611497and 612301) while autosomal dominant osteopetrosis is divided into two types (osteopetrosis, autosomal dominant 1~2, OPTA1~2; OMIM166600, 607634). The clinical phenotypes of osteopetrosis vary considerably from the early onset life-threatening severe cases to mild cases in which the patients usually do not realize their conditions. Generally, osteopetrosis is clinically divided into three groups, i.e., infantile malignant autosomal recessive osteopetrosis (ARO), intermediate autosomal recessive osteopetrosis (IARO) and autosomal dominant osteopetrosis (ADO II). ARO has a fatal outcome within the first decade of life.
T cell immune regulator 1(TCIRG1) encodes subunit a3 of V-ATPase and its mutations are the primary cause of autosomal recessive osteopetrosis [44, 53, 88] and infantile malignant osteopetrosis [54, 89]. The mutations in TCIRG1 underlie 50% of ARO patients [53]. The TCIRG1 variants include deletions, insertions, nonsense substitutions, and splice site mutations, may cause severe abnormalities in the protein product and likely represent null alleles [44, 53, 88]. Besides ARO, TCIRG1 mutations are also related to autosomal dominant severe congenital neutropenia [90]. In animal studies, mice deficient in Tcirg1 (Atp6i) show severe osteopetrosis. Atp6i-/- osteoclast-like cells lose the function of extracellular acidification but retain intracellular lysosomal proton pump activity [57]. Deletion of the 5-prime portion of Tcirg1 gene in mice causes hypocalcemia and osteopetrorickets phenotype with high bone mass [91]. Transgenic mice carrying a dominant missense mutation (R740S) in Tcirg1 gene also exhibit high bone density without affected osteoblast parameters [55].
Besides subunit a3, so far, no other V-ATPase subunits have been reported to be involved in osteopetrosis. Only Atp6v0d2-deficient mice show increased bone mass. Subunit d2 has been suggested to play important roles in coupling proton transport and ATP hydrolysis as well as the assembly of ATPase complexes [39, 40]. Subunit d2 is also involved in the regulation of osteoclast function and bone formation. Although mutations in the human ATP6V0D2 gene have not been reported in osteopetrosis, Atp6v0d2 gene-knockout mice have increased bone density and defective osteoclasts because of the requirement for fusion of preosteoclasts resulting in osteopetrosis [70, 71, 92]. ATP6V0D2 has recently been identified as a novel chondrocyte hypertrophy-associated gene [93].
3.2 Subunits of V-ATPase and osteoporosis or bone loss
Osteoporosis is a common metabolic bone disease that is characterized by reduced bone mineral density (BMD) and increased risk of osteoporotic fractures. In particular, genes involved in the functions of osteoclasts have been associated with the risk of osteoporosis [94-96].
H subunit is a small subunit of V-ATPases that connects the V1 and V0 domains. We previously reported that partial loss of ATP6V1H function resulted in osteoporosis/osteopenia in a population of 1625 Han Chinese as well as in an Italian pedigree [86, 87]. Atp6v1h+/- knockout mice generated by the CRISPR/Cas9 technique had decreased bone remodeling and a net bone matrix loss. Similarly, Atp6v1h+/- osteoclasts showed impaired bone formation and resorption activity. The increased intracellular pH of Atp6v1h+/- osteoclasts downregulated TGF-β1 activation, thereby reducing induction of osteoblast formation [86]. In a CRISPR/Cas9 zebrafish model, atp6v1h deficiency also caused bone loss [86, 87]. In another bivariate GWAS study, ATP6V1G1 was implicated as a novel pleiotropic gene affecting human BMD [84]. The above controversial effects of V-ATPase subunits on BMD suggest that the deficiency of V-ATPase subunits might not always lead to increased bone mass.
3.3 Other subunits of V-ATPase related to BMD
3.3.1 Genetic factors for osteoporosis (GEFOS) information
GEFOS Consortium is a large international collaboration of groups studying the genetics of osteoporosis using the meta-analysis of GWAS data with high-density SNP arrays. The BMD of GEFOS was measured at the femoral neck and lumbar spine using dual-energy X-ray absorptiometry in 32,961 subjects (http://www.gefos.org/?q=content/data-release) [97]. We screened the variants of V-ATPase subunits in GEFOS data and in-house data to find whether more subunits are involved in BMD regulation. Based on our VEGAS analysis results of 2012 GEFOS-released data [98, 99], ATP6V1A, ATP6V0A1, ATP6V1E2, ATP6V0A4, ATP6V1F, ATP6V1G2 and ATP6V1G3 are related to BMD (Table 3). Further analysis showed that SNPs in other subunits of V-ATPase are also associated with BMD (Table S1). A detailed analysis of the GWAS data of 1627 Han Chinese [100, 101] revealed that other SNPs of V-ATPase subunits might be related to BMD (Table S2).
Association of V-ATPase subunits and bone mass in GEFOS.
| FNK* P value | SPN* P value | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Chr | Gene name | nSNPs | Start Position | Stop Position | Male | Female | Total | Male | Female | Total |
| 3 | ATP6V1A | 30 | 113465865 | 113530905 | 0.00596 | 0.00142 | 2.00×10-5 | 0.08991 | 0.195804 | 0.023298 |
| 2 | ATPV1B1 | 29 | 71162997 | 71192561 | 0.921079 | 0.204795 | 0.17182817 | 0.533467 | 0.410589 | 0.433566 |
| 8 | ATP6V1B2 | 20 | 20054703 | 20079207 | 0.415584 | 0.675325 | 0.60539461 | 0.662338 | 0.908092 | 0.705295 |
| 8 | ATP6V1C1 | 53 | 104033247 | 104085285 | 0.638362 | 0.917083 | 0.94705295 | 0.413586 | 0.497502 | 0.434565 |
| 2 | ATP6V1C2 | 31 | 10861774 | 10925236 | 0.125874 | 0.381618 | 0.15284715 | 0.416583 | 0.579421 | 0.686314 |
| 14 | ATP6V1D | 17 | 67804580 | 67826720 | 0.411588 | 0.955045 | 0.77922078 | 0.386613 | 0.644356 | 0.234765 |
| 22 | ATP6V1E1 | 44 | 18074902 | 18111588 | 0.195804 | 0.375624 | 0.32467532 | 0.651349 | 0.25974 | 0.084915 |
| 2 | ATP6V1E2 | 10 | 46738985 | 46747096 | 0.282717 | 0.100899 | 0.01359864 | 0.882118 | 0.021498 | 0.203796 |
| 7 | ATP6V1F | 3 | 128502856 | 128505903 | 0.713287 | 0.40959 | 0.49350649 | 0.564436 | 0.031597 | 0.456543 |
| 9 | ATP6V1G1 | 8 | 117349993 | 117361152 | 0.96004 | 0.812188 | 0.75624376 | 0.390609 | 0.121878 | 0.172827 |
| 6 | ATP6V1G2 | 5 | 31512227 | 31514625 | 0.724276 | 0.145854 | 0.16683317 | 0.323676 | 0.361638 | 0.325674 |
| 1 | ATP6V1G3 | 12 | 198492351 | 198510075 | 0.163836 | 0.017698 | 0.08691309 | 0.466533 | 0.42957 | 0.155844 |
| 8 | ATP6V1H | 62 | 54628102 | 54755871 | 0.088911 | 0.301698 | 0.13386613 | 0.999001 | 0.528472 | 0.358641 |
| 17 | ATP6V0A1 | 12 | 40610861 | 40674597 | 0.448551 | 0.175824 | 0.10589411 | 0.164835 | 0.0061 | 0.022598 |
| 12 | ATP6V0A2 | 35 | 124196864 | 124246301 | 0.797203 | 0.382617 | 0.71628372 | 0.717283 | 0.103896 | 0.462537 |
| 7 | ATP6V0A4 | 86 | 138391038 | 138482941 | 0.025597 | 0.944056 | 0.84615385 | 0.034497 | 0.536464 | 0.614386 |
| 1 | ATP6V0B | 4 | 44440601 | 44443972 | 0.94006 | 0.769231 | 0.83616384 | 0.458541 | 0.364635 | 0.508492 |
| 16 | ATP6V0D1 | 12 | 67471916 | 67515089 | 0.664336 | 0.846154 | 0.71128871 | 0.427572 | 0.55045 | 0.460539 |
| 8 | ATP6V0D2 | 93 | 87111138 | 87166454 | 0.204795 | 0.874126 | 0.87112887 | 0.904096 | 0.30969 | 0.361638 |
| 5 | ATP6V0E1 | 21 | 172410762 | 172461900 | 0.183816 | 0.326673 | 0.16883117 | 0.708292 | 0.183816 | 0.438561 |
| 7 | ATP6V0E2 | 3 | 149570056 | 149577801 | 0.657343 | 0.667333 | 0.60639361 | 0.288711 | 0.913087 | 0.85015 |
#TCIRG1 (ATP6V0A3) and ATP6V0C genes were not included in the analysis because of the insufficient SNPs in the GEFOS data base. FNK: femoral neck; SPN: lumbar spine; nSNP: number of SNP. Bold font shows the genes with a significant P value (P<0.05).
3.3.2 Subunits of V-ATPase possibly related to BMD
Not much information is available on the location and functions of the above BMD-related subunits (A, E2, G2, G3, a1 and a4) of V-ATPase in osteoclasts.
Subunit A: The mRNA level of Atp6v1a has been reported in rat osteoclasts and may respond to fluid shear stress changes [102]. No other information about subunit A in osteoclastic function is available. The mutations in ATP6V1A gene caused autosomal recessive cutis laxa type IID (ARCL2D) [73]. Subunit A interacts with the N terminal of Wolfram syndrome 1 (WFS1) protein in human embryonic kidney (HEK) 293 cells and human neuroblastoma cells, which might be important both for pump assembly in the endoplasmic reticulum (ER) and for granular acidification [103]. ATP6V1A also controls the extracellular acidification of intercalated cells in kidney, and its phosphorylation is regulated by the metabolic sensor AMP-activated protein kinase (AMPK) at Ser 384 [104]. Subunit A has also been detected in intracellular structures such as trans-Golgi network (TGN) of principal cells and narrow/clear cells in the epididymis and vas deferens [105]. The morpholinos against atp6v1a in zebrafish result in several abnormalities including suppression of acid-secretion from the skin, growth retardation, trunk deformation, and loss of internal Ca2+ and Na2+[74].
Subunit E2: Unlike testis-specific subunit E1, subunit E2, the isoform of E1 shows a ubiquitous distribution [83, 106]. Subunit E2 was found to be present in the perinuclear compartments of spermatocytes and rat epididymis [83, 105].
Subunit F: The mRNA level of subunit F has been reported in human prostate carcinoma cells and biopsy specimens from the antral mucosa [107, 108]. A zebrafish atp6v1f model was used to study its role during eye development. Atp6v1f mutant zebrafish showed oculocutaneous albinos and defects in melanosomes and retinal pigmented epithelium [63].
Subunit G2: Subunit G2 has a brain-specific distribution [109]. ATP6V1G2 was found to be associated with myocardial infarction according to analysis of SNPs and transcriptome sequencing [110, 111]. The mRNA of ATP6V1G2 was detected in lipopolysaccharide-stimulated human macrophages [112] and human neuroblastoma cell line SH-SY5Y [113].
Subunit G3: Subunit G3 is expressed in kidney and rat epididymis [105, 109]. Its protein is used as a novel immunohistochemical marker for differentiating subtypes of chromophobe renal cell carcinoma (RCC) including clear cell, papillary RCCs [114, 115], and osteosarcoma [116].
Subunits a1 and a4: Subunit a has four isoforms, a1-a4, among which subunit a3 plays an essential role in osteoclasts and bone density. Subunit a4 is expressed in α intercalated cells in both human and mouse kidney [117] as well as inner ear, olfactory epithelium, the uterus of pregnant animals, embryonic visceral yolk sac, prostatic alveoli, ampullary glands, epididymis, and vas deferens [118]. It is also associated with autosomal recessive distal renal tubular acidosis [58, 59, 109]. Atp6v0a4-knockout mice showed distal renal tubular acidosis with hearing loss, severe metabolic acidosis, hypokalemia, early nephrocalcinosis, and bone loss [60, 61]. Mutations in ATP6V0A4 were associated with atypical progressive sensorineural hearing loss in a Chinese patient with distal renal tubular acidosis [119]. A zebrafish study showed that rbc3a and atp6v0a1 promote endosomal maturation to coordinate Wnt signaling in neural crest [46].
3.4 Identification of subunits of V-ATPase involved in osteoclast formation
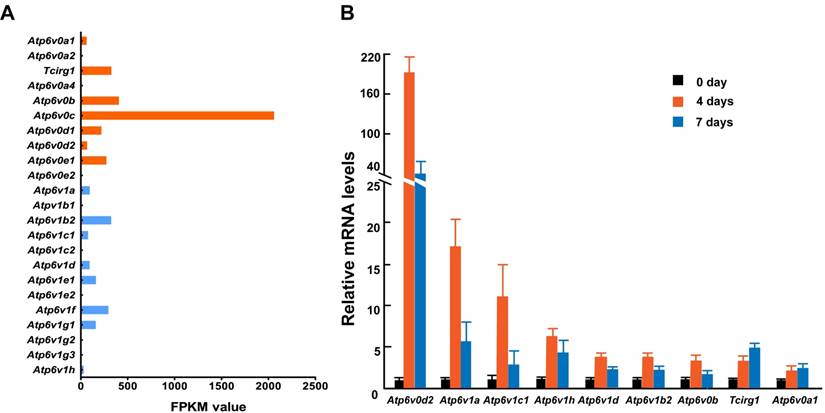
To further identify the relationship between osteoclasts and the subunits described above, we primary cultured mouse osteoclasts with induction by macrophage colony-stimulating factor (M-CSF) and receptor activator of nuclear factor kappa-B ligand (RANKL) and analyzed the mRNA levels of 13 V-ATPase subunits using RNA sequencing and Q-PCR. While we could not detect the expression of all subunit isoforms, at least one isoform was expressed representing each of the 13 V-ATPase subunits either by FPKM analysis or by Q-PCR analysis. Among the 15 expressed isoforms, nine (60%) showed an increased level of mRNA during osteoclastogenesis. Tcirg1 and Atp6v0a1 showed a time-dependent increase, and the other seven genes showed a sharp increase on day 4 and then dropped down on day 7, suggesting that subunits b, d2, A, B2, C1, D, and H contribute more to the early maturation stage of osteoclasts, and subunit a1 and a3 might play more important roles in late osteoclast maturation (Figure 2). Both RNA sequencing and Q-PCR analysis showed that the mRNA levels of subunits a4, e2, B1, C2, E2, G2, and G3 are quite low or undetectable in mature osteoclasts. In the above GEFOS analysis, ATP6V0A4, ATP6V1G2, and ATP6V1G3 are probably related to bone density. The low mRNA levels of a4, G2, and G3 in the mature mouse osteoclasts suggest that these subunits might affect bone density by their interactions with other subunits of osteoclasts or through a more complicated and systematic pathway instead of the osteoclast-specific pathway.
mRNA expression of V-ATPase subunits in mouse osteoclasts. Mouse osteoclasts were primary cultured from bone marrow cells by inducing with M-CSF (50ng/mL) and RANKL (100ng/mL). Total mRNA of cells was extracted at 0, 4, and 7 days of induction. (A) RNA sequencing results. The average FPKM value was obtained from cells induced by M-CSF/RANKL for 7 days; n=3. (B) Q-PCR results from one representative experiment. ≥2-fold change was regarded as statistically significant (P<0.05). Among the 15 expressed isoforms, nine showed a statistically significant increased level of mRNA during osteoclastogenesis.Tcirg1 and Atp6v0a1 showed time-dependent increases. Seven other isoforms showed a greater increase on day 4 than on day 7. Other isoforms with smaller than 2-fold change are not presented in the figure. Error bars represent standard deviation. Each experiment was repeated at least three times.
4. Functions of V-ATPase subunits in osteoclasts
The controversial effects of various subunits of V-ATPase on bone phenotypes imply their complicated biological functions in vivo and in vitro. While analyzing the bone phenotypes of various V-ATP subunits, we need to be cognizant of whether the subunits are specifically expressed in osteoclasts, the pathway(s) by which the subunits affect osteoclast function, and whether it is a common phenomenon or unique to a particular subunit. At present, there are only limited human or animal data on the relationships between subunits a3, d2, B2 [120, 121], C1[122-126], and H and osteoclast functions. It is, therefore, difficult to conclude that a particular subunit is specifically expressed in osteoclasts and only regulates osteoclastic function. As per our mRNA screening of ATPase subunits in mouse osteoclasts, more subunits, such as b, c, e1, A, D, E1, F and G1, have potential functions in osteoclasts. Thus, it is necessary to consider the direct or indirect functions of V-ATPase subunits in osteoclasts and non-osteoclasts while exploring the mechanisms of V-ATPase-regulated bone phenotypes.
4.1 Direct roles
4.1.1 Regulation of extracellular acidification
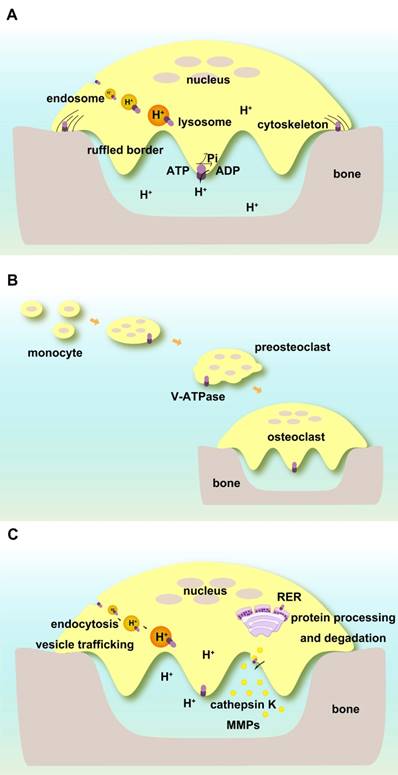
V-ATPases complexes containing subunits a3, d2, and C1 are present on the ruffled borders of osteoclasts to maintain the acidic extracellular environment [127]. Subunit a3 of V-ATPase controls the extracellular acidification of bone resorptive lacunae [57]. Atp6v0d2 is highly expressed in mature mouse osteoclasts, and its depletion abolishes their extracellular acidification [71]. Subunit C1 is also highly expressed in osteoclasts and interacts with the a3 subunit on the ruffled borders. Deficiency of C1 subunit causes severely impaired osteoclast acidification activity and bone resorption [122] (Figure 3A).
Functions of V-ATPases in osteoclasts. (A) Direct regulation of extracellular and intracellular pH. V-ATPases on ruffled borders maintain the acidic extracellular environment for bone resorption. V-ATPases in endosomes and lysosomes keep pH homeostasis of intracellular compartments. V-ATPases are associated with cytoskeletal F-actin. (B) V-ATPases are required for the fusion of preosteoclasts. (C) Indirect roles in osteoclasts. V-ATPases regulate endocytosis, vesicle trafficking, protein processing, and degradation as well as enzyme secretion and enzymatic activities.
4.1.2 Maintenance of pH homeostasis of intracellular compartments
V-ATPases exist within intracellular compartments including endosomes and lysosomes. They control the acidification of these vesicles and regulate membrane trafficking processes such as receptor-mediated endocytosis, intracellular trafficking of lysosomal enzymes from Golgi complexes to lysosomes, protein processing and degradation, and transportation of small molecules and ions. The low pH in the intracellular compartments enables endocytosis of ligands or other proteins including low-density lipoprotein (LDL) and their dissociation from their receptors [128, 129]. ATP6V1H is localized in osteoclasts, and its deficiency causes an increase in intracellular pH and inhibition of the formation and function of osteoclasts such as bone resorption [86] (Figure 3A).
V-ATPase complex also facilitates the entry of many viruses, and endosomal acidification enables fusion of viruses with infected cells [130, 131]. Subunit H is a representative example that helps HIV internalization [132]. During infection of group A rotaviruses (RVAs), the outer capsid proteins of the RVA strain bind to cell surface receptors and phosphorylate PI3K, Akt, and ERK, which, in turn, directly interact with subunit E to acidify late endosomes for uncoating of RVAs [133]. Also, ATP6V0C is related to human cytomegalovirus (HCMV) [134].
4.1.3 Assembly of cytoskeletal F-actin
Subunit C1 regulates cytoskeletal F-actin assembly during osteoclast activation [123-125] and the reversible dissociation of V-ATPases [126]. Local treatment of AAV-shRNA-Atp6v1c1 attenuates the bone erosion and inflammation caused by periodontitis. Atp6v1c1 silencing severely impairs osteoclast acidification and bone resorption as well as F-actin ring formation, whereas cell differentiation does not appear to be affected [135] (Figure 3A).
Subunit B2, and not the B1 isoform, is located in intracellular vesicles and on ruffled membranes of osteoclasts [120, 121]. Phosphatidylinositol 3-kinase (PI 3-kinase) is involved with the association of B2 and F-actin that is important for recruitment of V-ATPase complexes to the osteoclasts' ruffled border during polarization and bone resorption [136-139]. Alteration of B2 and F-actin association by the mutated binding site does not influence V-ATPase assembly or ATP hydrolysis [138, 139].
Golgi-associated V-ATPase activity relies on actin and the Golgi pH. Actin depolymerization promotes dissociation of V1 and V0 domains followed by translocation of subunit B2 from Golgi membranes to the cytosol. Actin may regulate Golgi pH homeostasis, which can maintain the coupling of V1-V0 domains of V-ATPase through the binding of microfilaments to subunits B and C [140].
4.1.4 Fusion of preosteoclasts
Subunit d2 is required for the fusion of preosteoclasts. Atp6v0d2-deficient mice have defective osteoclasts and increased bone mass [70, 71, 92] (Figure 3B).
4.2 Indirect roles
4.2.1 Effects on endocytosis and vesicle trafficking
V-ATPase complex functions as a proton pump and acidifies endosomes and lysosomes. V-ATPase-specific inhibitors or acidotropic agents may inhibit endosomal acidification, thus affecting endocytosis, vesicle trafficking, and the processing of secretory and lysosomal proteins [1, 141]. Atp6v0c-mutant mice show acidification defects in endocytosis and Golgi complex that affect the development of embryonic and extraembryonic tissues [142]. Subunit c and a2 interact with two small GTPases, Arf6 and ARNO, respectively; inhibition of these interactions causes an alteration in endocytosis. Recruitment of Arf6 and ARNO from the cytosol to endosomal membranes causes intra-endosomal acidification [143]. In yeast, regulator of the H+-ATPase of vacuoles and endosomes (RAVE) is essential for the reversible assembly of V-ATPase. RAVE complex consists of Rav1p, Rav2p, and Skp1p, which play an essential role in RAVE regulation of V-ATPase activity [144]. It is unclear whether V-ATPases promote membrane fusion in the endocytic and exocytic pathways independent of their acidification functions [145].
4.2.2 Effects on endoplasmic reticulum (ER)
Besides endosomes and lysosomes, V-ATPases are also localized in the ER. The functions of V-ATPases in the ER are related to protein processing and degradation. Mutation analysis shows that R444L causes subunit a3 to be stuck in the ER instead of lysosomes. The oligosaccharide moiety of the mutant protein is misprocessed, and its degradation is through the ER-associated degradation pathway. R445L mutated protein is also degraded quickly in differentiated osteoclasts due to itsaltered protein conformation [146]. Archazolid, a V-ATPase inhibitor, affects the secretion of cytokines TNF-α, interleukin-6, and -8, and causes accumulation of these cytokines at the ER [147].
4.2.3 Effects of V-ATPases on enzyme activity
There are several reports on V-ATPases and cathepsins. Cathepsin K (CTSK), an important enzyme for osteoclasts to resorb bone matrix, requires V-ATPase to keep an acidic environment for its normal activity [148]. Archazolid induces secretion of the pro-forms of cathepsin B and D. By inhibiting mannose-6-phosphate receptor-dependent trafficking, Archazolid abrogates the cathepsin B maturation process, reduces the intracellular mature cathepsin B protein abundance, and decreases cathepsin B activity [149].
Other enzymes related to V-ATPases include members of the matrix metalloproteinase (MMP) family. We recently observed that deficiency of subunit H affected MMP9 and MMP13 in the bone system [87]. In pancreatic cancer cells, V-ATPase colocalizes with cortactin, an F-actin-stabilizing protein, which helps in the release of MMPs. V-ATPase selectively modulates specific MMPs such as MMP9 and MMP2, which might be linked to an invasive cancer phenotype [150]. Treatment with concanamycin or Atp6v1e shRNA affects MMP9 and MMP2 activities. Targeting inhibition of a2 subunit suppresses the activities of MMP9 and MMP2 in ovarian cancer cells and plays a critical role in tumor invasion [151] (Figure 3C).
4.3 Signaling pathways in osteoclasts involving V-ATPases
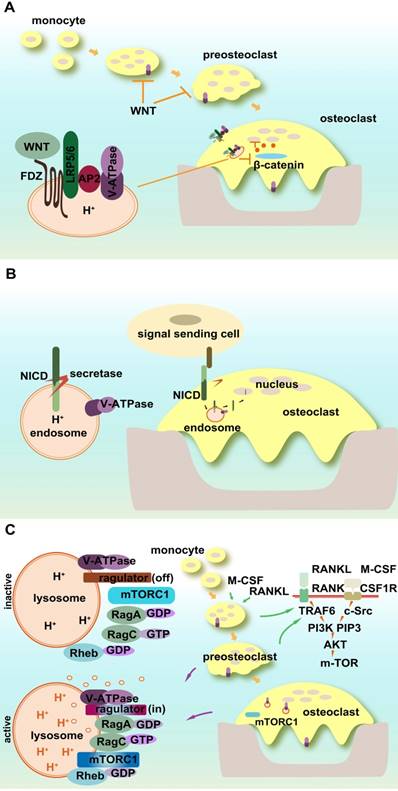
Cellular signaling pathways play critical roles in cell-cell and cell-environment interactions and maintaining extrinsic and intrinsic homeostasis. The important association between V-ATPases and cellular signaling has been documented in cancer cell biology to affect apoptosis, tumor cell invasion, migration, and metastasis [128, 152]. Recent findings have shown that V-ATPases also affect cellular signaling pathways in osteoclasts and bone metabolism [86] (Figure 4A-C).
Signaling pathways in osteoclasts involving V-ATPases. (A) WNT/β-catenin signaling pathway. V-ATPases influence WNT signaling through its trafficking and activation pathways. The accessory subunit of V-ATPase AP2 (PRR) activates WNT receptor. (B) Notch signaling. Acidic endosomes enable S3 cleavage through secretase and NICD release in a V-ATPase-dependent manner. (C) mTOR signaling. V-ATPases sense the signals of amino acid sufficiency and transfer it to mTORC1. V-ATPases also control the off/on switch of the V-ATPase-regulator complex.
4.3.1 WNT/β-catenin signaling pathway
WNT signaling is initiated by the binding of WNT ligand to its receptor Frizzled (FZ), forming a complex with a specific co-receptor low-density lipoprotein receptor-related protein 5 or 6 (LRP5/6). WNT signaling includes β-catenin-dependent canonical and β-catenin-independent noncanonical signaling pathways. WNT signalings, such as Wnt3a and LRP5/6, have been shown to influence bone mass [153, 154] or inhibit osteoclast differentiation by activating canonical and noncanonical cAMP/PKA pathways [153-159]. Lack of LRP5/6 may impair osteoclast progenitor proliferation [155].
V-ATPases influence WNT signaling through its trafficking and activation pathway including blocking the phosphorylation of its receptor and its internalization after ligand binding. In addition, localization and activation of the WNT receptor require an adaptor (Pro) renin receptor (PRR), one of the accessory subunits of the V-ATPase complex. WNT secretion also requires its binding to the carrier protein Wntless (WLS), and V-ATPase-controlled vacuolar acidification facilitates the release of palmitoylated WNT3A from WLS in secretory vesicles [160].
4.3.2 Notch signaling pathway
Notch signaling connects the communication between signal-sending and signal-receiving cells. As a single pass transmembrane receptor, Notch receptor includes extracellular and intracellular domains (NICD). The ligand binding to the extracellular domain of the Notch receptor activates its successive proteolytic cleavage and releases NICD, which then translocates to the nucleus and alters gene transcription, cell identity, and growth. Notch signaling affects osteoclastic differentiation, maturation, and its resorption activity. Both osteoclast precursors and bone stromal cells are regulated by Notch signaling [161-168]. Notch2 in osteoclasts controls bone resorption via PYK2-c-Src-microtubule signaling pathway [162].
The proper processing, trafficking, and activation of Notch signaling require V-ATPase participation [169]. The acidic environment of early endosomes enables S3 cleavage through secretase and NICD release in a V-ATPase-dependent manner [170]. Mutant V-ATPase affects the internalization of Notch signals and causes its accumulation in lysosomes as well as a substantial loss in the physiological and ectopic Notch activation of endosomes [170]. Rabconnectin-3 α and β (Rbcn-3A and B) regulate the V-ATPase proton pump and mutations in Rbcn-3A and Rbcn-3B cause defects in endocytic trafficking and the accumulation of Notch in late endosomal compartments in Drosophila [171].
4.3.3 mTOR signaling pathway
The mechanistic target of rapamycin (mTOR), the former mammalian target of rapamycin, is a member of phosphatidylinositol 3-kinase-related kinase family. mTORC1 is a central node of cellular signaling, and its activity is influenced by multiple factors such as growth factors, stress, energy status, and cellular amino acid levels, whereas mTORC2 is involved in survival signaling. The inhibition of V-ATPases impairs lysosomal acidification and alters lysosomal amino acid efflux, which also shows an mTOR-dependent regulation mechanism [172].
A novel polypeptide translated by long non-coding RNAs exists in the late endosome/lysosome and interacts with the lysosomal V-ATPase, which may negatively regulate mTORC1 activation by amino acid stimulation, rather than by growth factors [173, 174]. mTOR activity is essential for osteoclastic function and bone metabolism [175-177]. It regulates osteoclast formation by modulating the C/EBP-β isoform ratio [177]. mTOR and AMPK function as the nutrient and energy sensors of osteoclasts and regulate osteoclastogenesis [176].
mTOR1 activity is dependent on V-ATPase function. V-ATPases sense the signal of amino acid sufficiency and transfer it to mTORC1. V-ATPases also control the off/on switch of the V-ATPase-Ragulator complex, which plays a vital role in the activation of AMPK in lysosomes [128]. Amino acid starvation strengthens the association of Ragulator with the V1 segment of V-ATPase instead of V0 [178]. mTOR undergoes both lysosomal and proteasomal degradation, which follows the guanine nucleotide exchange of Rag small GTP-binding protein activated by V-ATPase [179, 180]. In osteoclasts, mTOR activation and deactivation rely on the lysosomal environment. The direct evidence for the interaction between V-ATPase and mTOR1 in osteoclasts is that R740S mutation in subunit a3 alters mTOR1 expression and activity in osteoclasts [175].
4.3.4 GTP-binding protein-coupled receptors (GPCR) signaling pathway
GPCRs belong to a large family with seven transmembrane domains and constitute extracellular sensing components and intracellular signal transduction cascade. The representative GPCRs that regulate osteoclasts and bone metabolism include the parathyroid hormone receptor (PTHR) [181, 182].
V-ATPases regulate GPCRs through the modulation of intracellular pH. Acidification of intracellular components induces dissociation of PTH from PTHR and stimulates the signaling of activated receptors and their recycling [128]. PTH also induces the phosphorylation of subunit a via cAMP-dependent protein kinase (PKA), which positively regulates the catalytic activity of the pump [179]. In addition, V-ATPase-mediated endosomal acidification provides negative feedback on PTH signaling. Normal pH enables binding of arrestin, and lower endosomal pH causes enhanced retromer binding and turns off PTH receptor signaling [183].
5. Complex contributions of V-ATPase subunits to bone phenotypes
5.1 Interactions between V-ATPase subunits
The direct interactions between various subunits of V-ATPase have been studied for the past decades to clarify the assembly and mechanisms of V-ATPase complex using bioinformatic model systems, mutation analysis, binding experiments, etc. In the model of the V1 component of Thermus thermophilus V-ATPase, interactions between A3B3 and D/F subcomplexes were observed, and asymmetry was realized by rigid-body rearrangements of the relative position between A and B subunits [184].
Based on hybridization or pull-down analysis, subunit a3 is connected with subunits d2 or B2. The connection between a3 and B2 was found to be quite important for the trafficking of ruffled border V-ATPase in activated osteoclasts [136] and this interaction was affected by benzohydrazide derivative IPI and KM91104 [3,4-dihydroxy-N-(2-hydroxybenzylidene) benzohydrazide] [185]. According to acryo-EM reconstruction model of yeast V-ATPase, when the V1 segment was released from V0, the N-terminal cytoplasmic domain of subunit a changed its conformation and bound to the rotor subunit d [186]. N-termini of subunits a3 and d2 had high-affinity interactions as found by glutathione-S-transferase (GST) pull-down assay, and were co-expressed in mammalian cells [71]. Luteolin inhibited V-ATPase activity by interfering with a3-d2 interaction without affecting the transcription or protein levels of these subunits [187-190], and Janus kinase (JAK) signaling pathway was involved in this process [191].
Using an RNA sequencing technique, the mRNA response of all V-ATPase subunits was observed in haploinsufficiency mouse osteoclasts (Atp6v1h+/-), and some subunits showed increased expression (Table 4). Subunit d2 showed a two-fold increase in expression, whereas other subunits spatially close to subunit H, such as subunits B2, C1, D, A, and G1, exhibited a varied level of increased expression to compensate for the subunit H haploinsufficiency. Subunit H has been regarded as a functional bridge between V1 and V0 and its absence resulted in an inactive V1/V0 complex by reducing communication between the two components [192]. Thus, subunit H might function as a negative regulator of osteoclastic formation and function, and the upregulated mRNA levels of the other V-ATPase subunits might not definitely lead to increased V-ATPase function.
Comparison of mRNA levels of V-ATPase subunits in mouse osteoclasts.a
| Gene Name | Length | Atp6v1h+/+ Expression | Atp6v1h+/-Expression | log2 Fold Change (Atp6v1h+/-/Atp6v1h+/+) | Padj | Up/ Down b | P value |
|---|---|---|---|---|---|---|---|
| Atp6v1b2 | 2742 | 23955.49 | 42023.38 | 0.810836 | 1.24×10-6 | - | 2.02×10-8 |
| Atp6v0d2 | 2518 | 4611.703 | 9576.583 | 1.054211 | 0.00014 | Up | 5.98×10-6 |
| Atp6v1c1 | 2117 | 4933.716 | 6962.68 | 0.496968 | 0.000454 | - | 2.47×10-5 |
| Atp6ap2 | 2376 | 4700.42 | 7147.419 | 0.604633 | 0.008669 | - | 0.00096 |
| Atp6v1d | 1410 | 4022.583 | 5443.382 | 0.436381 | 0.029975 | - | 0.004732 |
| Atp6v1a | 3959 | 14607.46 | 19902.6 | 0.446251 | 0.036436 | - | 0.006087 |
| Atp6v1g1 | 1109 | 4890.781 | 6122.023 | 0.323943 | 0.039252 | - | 0.006736 |
| Atp6v0b | 996 | 11082.52 | 14157.32 | 0.353262 | 0.04682 | - | 0.008451 |
| Atp6v0e | 800 | 5985.754 | 7517.969 | 0.32881 | 0.096098 | - | 0.021782 |
| Atp6v1e1 | 1219 | 5479.907 | 6871.928 | 0.326563 | 0.09754 | - | 0.022242 |
| Tcirg1 | 2719 | 25921.58 | 31894.51 | 0.299154 | 0.101496 | - | 0.023429 |
| Atp6v0c | 1150 | 61788.39 | 77716.21 | 0.33088 | 0.137199 | - | 0.035293 |
| Atp6v1f | 635 | 4880.556 | 5955.098 | 0.28708 | 0.208681 | - | 0.063749 |
| Atp6v1g2 | 1632 | 59.3336 | 42.74398 | -0.47313 | 0.412445 | - | 0.169455 |
| Atp6v0a1 | 3997 | 8717.704 | 6991.545 | -0.31834 | 0.53171 | - | 0.249876 |
| Atp6v0d1 | 1617 | 10210.97 | 8998.879 | -0.1823 | 0.612442 | - | 0.31818 |
| Atp6v0a2 | 5357 | 2573.471 | 2373.729 | -0.11656 | 0.657228 | - | 0.356739 |
| Atp6ap1 | 2189 | 22026.99 | 19970.68 | -0.14139 | 0.658755 | - | 0.358188 |
| Atp6v0e2 | 1849 | 5.238299 | 3.990053 | -0.39269 | 0.816266 | - | 0.549267 |
a All data were generated by our group. Osteoclasts were primary cultured from wild-type and Atp6v1h+/-mice as previously reported. Total mRNA of cells was extracted after induction by M-CSF and RANKL for 7 days [86]. High-quality RNA was obtained and RNA sequencing analysis was performed. All coding genes of V-ATPase subunits were compared between two groups. N=3.
b Bold: Up / Down: log2-fold change >1 or -1 and P value <0.05; Bold font shows the mRNA changes with a significant P value (P < 0.05).
5.2 Interactions between osteoclasts and osteoblasts
Although there is no direct evidence for V-ATPases in osteoblasts, increasing data imply some functions of V-ATPases in osteoblasts and bone formation. For example, deletion of the 5-prime portion of the Tcirg1 gene (subunit a3) in mice caused hypocalcemia and osteopetrorickets phenotype combined with decreased bone formation [91]. Atp6v0d2-deficient mice had enhanced bone formation and osteopetrosis [70, 71, 92]. Atp6v1h+/- mice showed a decrease in cartilage and bone formation with reduced bone formation rate and mineral apposition rate. The numbers of osteoblasts, the area of osteoblast surfaces as well as the osteoblast ALP level were reduced in Atp6v1h+/- mice [86]. ATP6V1H was recently detected in mouse bone marrow stromal cells and deficiency of ATP6V1H impaired their osteogenic differentiation and enhanced adiogenic differentiation[193].
5.3 Systemic factors
A variety of systemic factors may affect bone metabolism, including growth factors and hormones. Due to the ubiquitous distribution of V-ATPases, various subunits of V-ATPase may affect bone phenotypes in a general and complex fashion.
5.3.1 Immune factors
Subunit a3 has two transcript variants, the longer isoform OC116 and the shorter isoform TIRC7. The short one uses a transcription start site within the exon 5 of the longer variant (OC116) and includes the downstream intron as part of its 5' UTR. TIRC7 is expressed in T lymphocytes and is essential for normal T cell activation. OC116 isoform encodes osteoclast-specific subunit a3, while TIRC7 mRNA is expressed by alloactivated T lymphocytes as a T cell inhibitory receptor [194, 195]. Six new alternative splice events in TCIRG1 were observed in 28 human tissues, implying that more functions of TCIRG1 might exist besides immune response and bone resorption [196].
5.3.2 Insulin and other hormone factors
Insulin activates ERK1/2 MAP kinase of osteoclasts and induces the expression of NFATc1 and Atp6v0d2. The insulin-induced expression of Atp6v0d2 was blocked by the ERK1/2 inhibitor or knockdown of insulin receptor [197]. The acidifying secretory vesicles in B-cells of pancreatic islets are the major site of proinsulin to insulin conversion. Subunit a3 is expressed in the endomembrane of secretory vesicles of normal islet B-cells but is absent in insulinoma cells, suggesting that subunit a3 may have a profound effect on the efficiency of proteolytic cleavage of proinsulin [198].
A mouse model with a null mutation at the subunit a3 locus exhibited a reduced level of insulin without changing the processing of insulin. In this respect, subunit a3 was believed to regulate the exocytosis process of insulin secretion [59]. V-ATPase functioned as a sensor of cytosolic pH, and was required for full activation of the cAMP-dependent PKA pathway in response to glucose in the Min6 β-cell line and contributed to insulin secretion [199]. Bafilomycin exposure also inactivated the insulin/IGF signaling pathway intermediate FOXO1 and increased the insulin content in neonatal islets [200].
The accessory subunits of V-ATPase such as AP1 (Ac45) and AP2 (PRR) are related to insulin. Ac45 was highly expressed in Langerhans cells of islets. Downregulation of Ac45 reduced insulin secretion and proinsulin II processing [201]. AP2 was abundant in islets including both α and β cells and modulated both glucagon-like peptide-1 receptor (GLP1R) and insulin processing to affect insulin secretion [202]. AP2 (PRR), another accessory protein of V-ATPase, was expressed in pituitary adenoma cells and regulated growth hormone (GH) release via V-ATPase-induced cellular acidification [203]. The PRR blocker could reduce body weight and fat mass and improve insulin sensitivity in high-fat-fed mice. Knocking out the PRR gene of adipose tissue also prevented weight gain and insulin resistance [204].
5.3.3 Growth factors
V-ATPases and specifically its subunits B2, c, and E were found to be involved in TGF-β1-mediated epithelial-to-mesenchymal transition (EMT) in rat proximal tubular epithelial cells (NRK52E) [205]. Since TGF-β1 activation is pH sensitive [206, 207] and TGF-β1 regulates the functions of osteoclasts and osteoblasts [208, 209], changes in the intracellular pH of Atp6v1h +/- osteoclasts altered the level and activity of TGF-β1, which further affected osteoblasts and osteoclasts [86]. Atp6v0a2-knockout mice showed delayed mammary morphogenesis associated with aberrant activation of Notch and TGF-β pathways [210].
The low pH in intracellular components was also found to be responsible for ligand dissociation and receptor trafficking of epidermal growth factor receptors (EGFRs) and insulin receptor. There were differential routings of internalized EGFRs induced by EGF, TGF α, and the superagonist EGF-TGF alpha chimera E4T. Bafilomycin treatment blocked EGFR but not c-Cbl degradation [211].
6. Future applications of V-ATPase inhibitors in osteoporosis
Osteoporosis is characterized by relatively increased bone resorption, and current osteoporosis therapeutics are mainly based on antiresorptive treatment [212, 213]. The anti-resorptive molecules antagonize integrin or inhibit Src tyrosine kinase, V-ATPases, chloride channels or cathepsin K. Bisphosphonates and Denosumab, the antibody against RANKL, are widely recommended as the first-line antiresorptive therapy [214]. Some of these have disadvantages such as coupling of inhibition of bone resorption and bone formation. Thus, the specificity of the antiresorptive therapy is essential.
As reviewed above, bone resorption requires V-ATPases activity and localization of some subunits of V-ATPase in osteoclasts. Successive generations of antiresorptive drugs based on V-ATPase complex or a specific subunit are becoming increasingly attractive. However, future efforts should focus on reducing side effects, minimizing the frequency of dosing, and increasing efficacy to halt osteoporotic bone loss [215-220].
6.1 V-ATPase inhibitors as anti-resorptives
6.1.1 Non-osteoclast-specific inhibitors of V-ATPase
The representative inhibitors of V-ATPase include bafilomycin A1 and B1, concanamycin A, and plecomacrolides. These highly specific V-ATPase inhibitors have been widely used in experimental cell biology for decades. Besides these inhibitors, macrolactone, archazolid, benzolactoneenamide and apicularen type inhibitors have also been shown to bind to the V0 component of V-ATPase. The agents are linked to the holoenzyme at or near the a/c subunit interface [213, 221, 222]. Many V-ATPase inhibitors show anti-tumor effects by targeting EMT or changing intracellular pH [223-226]. Other new inhibitors including FK506 might be possible new therapies for treating neurodegenerative diseases [227].
6.1.2 Osteoclasts-specific inhibitors of V-ATPase
The above inhibitors have broad effects and, therefore, many other osteoclasts-specific inhibitors of V-ATPases have become more attractive for osteoporosis treatment in recent decades [185, 191]. SB242784 is one of the attractive inhibitors with high potency and selectivity for the osteoclast V-ATPase [228]. Other inhibitors targeting osteoclastic V-ATPases include FR167356, FR202126, FR177995, Diphyllin, saliphenylhalamide (saliPhe) and Iejimalides (IEJLs) [229-232]. These inhibitors are summarized as follows (Table 5).
Osteoclast-specific V-ATPase inhibitors.
| Name | Class of Derivatives | Targeting Sites | Selectivity | In vivo | In vitro | Other Effects |
|---|---|---|---|---|---|---|
| SB242784 | indole derivative from bafilomycin | c, a, or V0 domain | high | Inhibited retinoid-induced hypercalcemia in thyroparathyroidectomized rats Prevented bone loss in ovariectomized rats | Inhibited V-ATPase activity in chicken osteoclasts (IC50 of 29 nM), human osteoclastoma (IC50 of 22 nM) and human osteoclastic bone resorption (IC50 of 3.4 nM) | |
| Iejimalides | 24-membered ring macrolides | V0 domain | low | N/A | Irreversibly inhibited V-ATPase-mediated intracellular acidification in osteoclasts with potent cytotoxicity | Anti-tumor activity in vivo and in vitro Inhibited V-ATPase-mediated intracellular acidification in yeast cells |
| FR167356 | benzamide | unknown | high selectivity in inhibiting osteoclast plasma membrane V-ATPase | Reduced retinoic acid-induced hypercalcemia in thyroparathyroidectomized rats Prevented bone loss in ovariectomized rats | Inhibited plasma membrane V-ATPase complex (IC50 of 190 nM) and lysosomal V-ATPase activity | Blocked macrophage and kidney V-ATPase activity |
| FR202126 | benzamide | unknown | low | Reduced hypercalcemia induced by retinoic acid in thyroparathyroidectomized-OVX rats Prevented alveolar bone loss in experimental periodontitis in rats | Prevented bone resorption by murine osteoclasts (IC50 2.6-20 nM) | Unclear effects on other cell types expressing plasma membrane V-ATPases |
| FR177995 | benzamide | unknown | low | Reduced bone loss in adjuvant-induced model of arthritis in rats; attenuated inflammation and articular cartilage damage | Non-specific inhibition of lysosomal and endosomal V-ATPase activity in dendritic cells | |
| Diphyllin | natural lignin compound | unknown | low | N/A | Inhibited V-ATPase-mediated lysosomal and extracellular acidification (IC50 of 14 nM) Enhanced osteoclast number and survival without cytotoxic effects (up to 100 nM) | Anti-cancer, anti-inflammatory effects |
| KM91104 | Benzohydrazide derivative | a3-B2 interaction | medium | N/A | Inhibited osteoclast resorption with an IC50 of ∼1.2μM | |
| Enoxacin | fluoroquinolone antibiotic | actin binding site on B2 | low | Interfered with osteoclast formation and activity (IC50 of ∼10μM) | Phototoxicity, neurological problems, severe tendinitis, adverse immune activity, and renal failure | |
| Salicylihalamide A (saliA), saliphenylhalamide (saliPhe) | benzolactoneenamide family | V0 domain | low | Inhibited osteoclastic bone resorption and attenuated titanium particle-induced osteolysis in mice | Anti-tumor agent Treatment of urinary tract infections and gonorrhea |
SB242784: (2Z,4E)-5-(5,6-dichloro-2-indolyl)-2-methoxy-N-(1,2,2,6,6-pentamethylpiperidin-4-yl)-2,4-pentadienamide, a novel indole derivative optimized from bafilomycin, SB242784 exhibited inhibitory effect on bone resorption of chicken osteoclasts and human osteoclastoma in a low nanomolar range. Compared with the inhibition efficiency on V-ATPase complexes from other tissues including kidney, liver, spleen, stomach, brain, and/or endothelial cells, SB242784 showed a greater than 1000-fold selectivity for the osteoclast V-ATPase [228]. Administration of SB242748 for six months as a potential anti-osteoporosis therapy prevented bone loss in ovariectomized rats [228, 233].
FR167356, FR202126, and FR177995: This group of inhibitors exhibited a higher inhibitory effect on plasma membrane V-ATPase than on the lysosomal V-ATPase in osteoclasts and inhibited bone resorption in a dose-dependent manner. FR167356 also inhibited the V-ATPase complex of the kidney with similar efficacy [229].
Diphyllin: This compound has been shown to inhibit human osteoclastic bone resorption by inhibiting acid influx and lysosomal acidification compared with bafilomycin A1 [230].
Salicylihalamide A (SaliA): SaliA belongs to the benzolactoneenamide family of V-ATPase inhibitors and may act on the V0 domain via a mechanism different from the classical macrolides such as bafilomycin and concanamycin [234]. A phenyl derivative of saliA, saliphenylhalamide (saliPhe), effectively inhibited osteoclastic bone resorption in a titanium particle-induced osteolysis mouse model [231].
IEJLs: These 24-membered ring macrolides were previously used in anti-tumor research and inhibited V-ATPase-mediated intracellular acidification in osteoclasts [232]. Other reports found that Iejimalides A~D also inhibited lysosomal V-ATPase activity and induced S-phase arrest and apoptosis in MCF-7 cells and HeLa cells without inhibiting actin polymerization [235, 236].
Artemisia capillaris: The anti-osteoporotic activity of Artemisia capillaris has recently been reported. Its extracts diminished osteoclast differentiation and bone resorption, attenuated acidification, and reduced tumor necrosis factor receptor-associated factor 6 (TRAF6) expression and its association with V-ATPase [237].
6.2 Targeting sites of V-ATPase inhibitors in anti-resorptives
Subunit B2 of V-ATPase binds to F-actin with a profiling pocket-like structure. The inhibitor Enoxacin, a fluoroquinolone antibiotic, may target the interaction of F-actin and subunit B2 together with Binhib16 [216, 238, 239]. Some inhibitors target the interactions of V-ATPase subunits. The interaction between subunit a3 and B2 is crucial to the trafficking of ruffled border V-ATPase in activated osteoclasts that enables osteoclasts to keep acidified lacunae [136, 138, 213]. KM91104 could affect the interaction without changing cell viability or RANKL-mediated osteoclast differentiation [185, 219]. Luteolin, a naturally occurring flavonoid, could inhibit V-ATPase against a3-d2 interaction and reduce bone resorption without affecting the levels of these subunits and V-ATPase assembly [187-190]. The inhibition of bone resorption did not affect osteoclastic actin ring formation and cellular viability [191].
6.3 Prospects of V-ATPase inhibitors as anti-resorptives
We have an in-depth understanding of the complicated effects of V-ATPase complex in the bone system from the literature, human GWAS data, and mouse data from our own research. Defects in subunits a3 and d2 result in increased bone density. Many V-ATPase inhibitors are designed against these two subunits to reduce the osteoclast bone resorptive activity. Although subunit H stimulates the formation and resorptive activity of osteoclasts in vitro, its deficiency in vivo results in bone loss [86]. Thus, the traditional concepts based on the anti-resorptive V-ATPase inhibitors might not be adequate, and the directions of drug design need to be adjusted. The design of future anti-resorptive drugs should take a more comprehensive and multifaceted approach rather than focusing on a specific subunit of V-ATPase given the versatile functions of ATPases in osteoclasts as well as other cell types.
First, it is important to take into consideration the interactions among various subunits of V-ATPase that have direct physical interactions with a3-d2 and a3-B2. Meanwhile, the transcript levels of some V-ATPase subunits is changed with reduced levels of subunit H. However, the nature of these interactions between subunit H and those subunits is not clear and probably involves both direct and indirect contacts. Thus, a comprehensive understanding of the network of V-ATPase subunits will help us predict the net effect of V-ATPase in osteoclasts.
Second, the emphasis so far has been on the location of osteoclastic V-ATPases on ruffled borders and their function of extracellular acidification. In this review, we describe possible versatile locations and functions of intracellular V-ATPase via direct or non-direct involvements. As is evident by our Q-PCR analysis, the mRNA levels of most V-ATPase subunits are increased during osteoclastogenesis. The locations of these subunits in osteoclasts are not clear and may be either on the ruffled borders or in the intracellular components including endosomes, lysosomes, and ER. Most importantly, the functions of intracellular V-ATPases in osteoclasts may vary from pH regulation and endocytosis to protein recycling, as well as involvement in several signaling pathways such as Wnt, Notch, mTOR and GPCR pathways, which are essential for the development of bone and osteoclasts.
Third, the precise functions of most subunits of V-ATPase in osteoclasts are not well understood yet. For example, besides influencing osteoclast function, subunit d2 affects bone mass by regulating osteoblasts. Subunit H targets the interaction between osteoclasts and osteoblasts through the TGF-β1 pathway. GEFOS analysis data imply that some subunits of V-ATPase are related to bone density without being present in osteoclasts.
In summary, in the future, direct or indirect functions of V-ATPases in osteoblasts as well as other cell types related to the bone system should be explored. Furthermore, the effects of V-ATPase inhibitors not only on osteoblasts but also on other cell types should be taken into consideration. It is of note that expressions of Tcirg1, Atp6v0d2, and Atp6v1b2 were recently detected in atherosclerotic lesions in mice, which might act against plaque calcification [240], providing an interesting perspective for the potential use of V-ATPase inhibitors in patients suffering from atherosclerosis and osteoporosis simultaneously. We believe that a new balance should be established between finding osteoclast-specific V-ATPase inhibitors or activators and considering their possible effects on other cells and tissues as well as their secondary effects on osteoclasts.
Related materials and methods
1. GEFOS and VEGAS analysis
GEFOS 2012 meta-analysis data including 32,961 subjects from 17 studies (http://www.gefos.org/?q=content/data-release) were used in our study [99]. SNPs in the genomic DNA of all V-ATPase subunits and the 50kb upstream and downstream region of certain genes were selected and analyzed with VEGAS methods [98]. The overall impacts of SNPs of a specific gene, as well as a single SNP, were compared.
2. In-house analysis
1627 Chinese volunteers were recruited from Midwestern Chinese Han adults living in Xi'an and Changsha cities. The analytical methods have been described previously [86, 100, 101]. Five groups were generated based on the Z-score; thresholds were -2, -1, 1, 2. All SNPs of V-ATPase subunits were selected and compared among groups (Table S2).
3. Osteoclasts culture and mRNA detection
At present, the locations and functions of V-ATPase subunits in osteoclasts have only been verified in a few subunits. To investigate whether more V-ATPase subunits are involved in osteoclastogenesis, osteoclasts were cultured, and Q-PCR was used to detect the mRNA levels of all V-ATPase subunits. The details are as follows: Bone marrow cells were separated from the femur and tibia of 6-week-old C57BL/6 mice and were treated with red blood cell lysis buffer (Solarbio, China) for 5 min. Subsequently, the cells were cultured in the presence of M-CSF (50ng/mL) (R&D, USA) for 24 hand then treated with M-CSF (50ng/mL) and RANKL (100ng/mL)(R&D, USA) for 4 or 7 days [86]. Total mRNA of cells at 0d, 4d, and 7d were extracted using the E.Z.N.A. total RNA kit (Omega, China). The PrimeScriptTM RT Reagent Kit was used to synthesize cDNA (TaKaRa, Japan). Realtime PCR was performed to analyze the mRNA levels of V-ATPase subunits using SYBR® Premix Ex TagTM (TaKaRa) and ABI 7500 real-time PCR system (Applied Biosystems) [241]. The primers of all subunits of V-ATPase are listed in Table S3.
4. RNA sequencing
Atp6v1h knockout mice were generated using the CRISPR/Cas9 system. Osteoclasts were cultured as described above from wild-type and heterozygous mice (Atp6v1h+/-). All animals were treated in accordance with the ethical guidelines of the School of Stomatology, the Fourth Military Medical University (Xi'an, China). Total mRNA of cells was extracted after induction with M-CSF and RANKL for 7 days [86]. High-quality RNA from each sample was combined into a single large pool to maximize the diversity of transcriptional units. The RNA library was constructed using Illumina's TruSeq RNA Sample Preparation Kit (Illumina Inc, San Diego, CA, USA). The integrity of the RNA library was evaluated using Agilent 2100 Bioanalyzer (Agilent RNA 6000 Nano Kit). The amplified flowcell was used for paired-end sequencing on BGISEQ-500. After cleaning and quality checks, clean reads were generated. All coding genes of V-ATPase subunits were used to compare wild-type and heterozygous mice.
Supplementary Material
Supplementary tables.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81470728, 81800787).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Forgac M. Vacuolar ATPases: rotary proton pumps in physiology and pathophysiology. Nat Rev Mol Cell Biol. 2007;8:917-29
2. Sun-Wada GH, Wada Y, Futai M. Vacuolar H+ pumping ATPases in luminal acidic organelles and extracellular compartments: common rotational mechanism and diverse physiological roles. J Bioenerg Biomembr. 2003;35:347-58
3. Gluck S, Nelson R. The role of the V-ATPase in renal epithelial H+ transport. J Exp Biol. 1992;172:205-18
4. Pathare G, Dhayat NA, Mohebbi N, Wagner CA, Bobulescu IA, Moe OW. et al. Changes in V-ATPase subunits of human urinary exosomes reflect the renal response to acute acid/alkali loading and the defects in distal renal tubular acidosis. Kidney Int. 2018;93(4):871-80
5. Lozupone F, Borghi M, Marzoli F, Azzarito T, Matarrese P, Iessi E. et al. TM9SF4 is a novel V-ATPase-interacting protein that modulates tumor pH alterations associated with drug resistance and invasiveness of colon cancer cells. Oncogene. 2015;34:5163-74
6. Mazhab-Jafari MT, Rohou A, Schmidt C, Bueler SA, Benlekbir S, Robinson CV. et al. Atomic model for the membrane-embedded VO motor of a eukaryotic V-ATPase. Nature. 2016;539:118-22
7. Nishi T, Forgac M. The vacuolar (H+)-ATPases-nature's most versatile proton pumps. Nat Rev Mol Cell Biol. 2002;3:94-103
8. Jefferies KC, Cipriano DJ, Forgac M. Function, structure and regulation of the vacuolar (H+)-ATPases. Arch Biochem Biophys. 2008;476:33-42
9. Toei M, Saum R, Forgac M. Regulation and isoform function of the V-ATPases. Biochemistry. 2010;49:4715-23
10. Supek F, Supekova L, Mandiyan S, Pan YC, Nelson H, Nelson N. A novel accessory subunit for vacuolar H(+)-ATPase from chromaffin granules. J Biol Chem. 1994;269:24102-6
11. Sun-Wada GH, Yoshimizu T, Imai-Senga Y, Wada Y, Futai M. Diversity of mouse proton-translocating ATPase: presence of multiple isoforms of the C, d and G subunits. Gene. 2003;302:147-53
12. Sun-Wada GH, Murata Y, Namba M, Yamamoto A, Wada Y, Futai M. Mouse proton pump ATPase C subunit isoforms (C2-a and C2-b) specifically expressed in kidney and lung. J Biol Chem. 2003;278:44843-51
13. Zhao W, Zhang Y, Yang S, Hao Y, Wang Z, Duan X. Analysis of two transcript isoforms of vacuolar ATPase subunit H in mouse and zebrafish. Gene. 2018;638:66-75
14. Esmail S, Kartner N, Yao Y, Kim JW, Reithmeier RAF, Manolson MF. N-linked glycosylation of a subunit isoforms is critical for vertebrate vacuolar H(+) -ATPase (V-ATPase) biosynthesis. J Cell Biochem. 2018;119:861-75
15. Esmail S, Yao Y, Kartner N, Li J, Reithmeier RA, Manolson MF. N-linked glycosylation is required for vacuolar H(+) -ATPase (V-ATPase) a4 subunit stability, assembly, and cell surface expression. J Cell Biochem. 2016;117:2757-68
16. Liu Q, Kane PM, Newman PR, Forgac M. Site-directed mutagenesis of the yeast V-ATPase B subunit (Vma2p). J Biol Chem. 1996;271:2018-22
17. Liu Q, Leng XH, Newman PR, Vasilyeva E, Kane PM, Forgac M. Site-directed mutagenesis of the yeast V-ATPase A subunit. J Biol Chem. 1997;272:11750-6
18. MacLeod KJ, Vasilyeva E, Baleja JD, Forgac M. Mutational analysis of the nucleotide binding sites of the yeast vacuolar proton-translocating ATPase. J Biol Chem. 1998;273:150-6
19. Maher MJ, Akimoto S, Iwata M, Nagata K, Hori Y, Yoshida M. et al. Crystal structure of A3B3 complex of V-ATPase from Thermus thermophilus. EMBO J. 2009;28:3771-9
20. Tomashek JJ, Graham LA, Hutchins MU, Stevens TH, Klionsky DJ. V1-situated stalk subunits of the yeast vacuolar proton-translocating ATPase. J Biol Chem. 1997;272:26787-93
21. Tomashek JJ, Garrison BS, Klionsky DJ. Reconstitution in vitro of the V1 complex from the yeast vacuolar proton-translocating ATPase. Assembly recapitulates mechanism. J Biol Chem. 1997;272:16618-23
22. Ohira M, Smardon AM, Charsky CM, Liu J, Tarsio M, Kane PM. The E and G subunits of the yeast V-ATPase interact tightly and are both present at more than one copy per V1 complex. J Biol Chem. 2006;281:22752-60
23. Fethiere J, Venzke D, Madden DR, Bottcher B. Peripheral stator of the yeast V-ATPase: stoichiometry and specificity of interaction between the EG complex and subunits C and H. Biochemistry. 2005;44:15906-14
24. Hildenbrand ZL, Molugu SK, Stock D, Bernal RA. The C-H peripheral stalk base: a novel component in V1-ATPase assembly. PLoS One. 2010;5:e12588
25. Kitagawa N, Mazon H, Heck AJ, Wilkens S. Stoichiometry of the peripheral stalk subunits E and G of yeast V1-ATPase determined by mass spectrometry. J Biol Chem. 2008;283:3329-37
26. Arai H, Terres G, Pink S, Forgac M. Topography and subunit stoichiometry of the coated vesicle proton pump. J Biol Chem. 1988;263:8796-802
27. Wang Y, Cipriano DJ, Forgac M. Arrangement of subunits in the proteolipid ring of the V-ATPase. J Biol Chem. 2007;282:34058-65
28. Thaker YR, Roessle M, Gruber G. The boxing glove shape of subunit d of the yeast V-ATPase in solution and the importance of disulfide formation for folding of this protein. J Bioenerg Biomembr. 2007;39:275-89
29. Wilkens S, Forgac M. Three-dimensional structure of the vacuolar ATPase proton channel by electron microscopy. J Biol Chem. 2001;276:44064-8
30. Cain CC, Sipe DM, Murphy RF. Regulation of endocytic pH by the Na+,K+-ATPase in living cells. Proc Natl Acad Sci U S A. 1989;86:544-8
31. Eya S, Maeda M, Futai M. Role of the carboxyl terminal region of H(+)-ATPase (F0F1) a subunit from Escherichia coli. Arch Biochem Biophys. 1991;284:71-7
32. Fillingame RH, Dmitriev OY. Structural model of the transmembrane Fo rotary sector of H+-transporting ATP synthase derived by solution NMR and intersubunit cross-linking in situ. Biochim Biophys Acta. 2002;1565:232-45
33. Kawasaki-Nishi S, Nishi T, Forgac M. Arg-735 of the 100-kDa subunit a of the yeast V-ATPase is essential for proton translocation. Proc Natl Acad Sci U S A. 2001;98:12397-402
34. Kawasaki-Nishi S, Bowers K, Nishi T, Forgac M, Stevens TH. The amino-terminal domain of the vacuolar proton-translocating ATPase a subunit controls targeting and in vivo dissociation, and the carboxyl-terminal domain affects coupling of proton transport and ATP hydrolysis. J Biol Chem. 2001;276:47411-20
35. Yokoyama K, Nakano M, Imamura H, Yoshida M, Tamakoshi M. Rotation of the proteolipid ring in the V-ATPase. J Biol Chem. 2003;278:24255-8
36. Hirata R, Graham LA, Takatsuki A, Stevens TH, Anraku Y. VMA11 and VMA16 encode second and third proteolipid subunits of the Saccharomyces cerevisiae vacuolar membrane H+-ATPase. J Biol Chem. 1997;272:4795-803
37. Flannery AR, Graham LA, Stevens TH. Topological characterization of the c, c', and c" subunits of the vacuolar ATPase from the yeast Saccharomyces cerevisiae. J Biol Chem. 2004;279:39856-62
38. Curtis KK, Francis SA, Oluwatosin Y, Kane PM. Mutational analysis of the subunit C (Vma5p) of the yeast vacuolar H+-ATPase. J Biol Chem. 2002;277:8979-88
39. Nishi T, Kawasaki-Nishi S, Forgac M. Expression and function of the mouse V-ATPase d subunit isoforms. J Biol Chem. 2003;278:46396-402
40. Owegi MA, Pappas DL, Finch MW Jr, Bilbo SA, Resendiz CA, Jacquemin LJ. et al. Identification of a domain in the V0 subunit d that is critical for coupling of the yeast vacuolar proton-translocating ATPase. J Biol Chem. 2006;281:30001-14
41. Shao E, Nishi T, Kawasaki-Nishi S, Forgac M. Mutational analysis of the non-homologous region of subunit A of the yeast V-ATPase. J Biol Chem. 2003;278:12985-91
42. Shao E, Forgac M. Involvement of the nonhomologous region of subunit A of the yeast V-ATPase in coupling and in vivo dissociation. J Biol Chem. 2004;279:48663-70
43. McGuire C, Stransky L, Cotter K, Forgac M. Regulation of V-ATPase activity. Front Biosci (Landmark Ed). 2017;22:609-22
44. Susani L, Pangrazio A, Sobacchi C, Taranta A, Mortier G, Savarirayan R. et al. TCIRG1-dependent recessive osteopetrosis: mutation analysis, functional identification of the splicing defects, and in vitro rescue by U1 snRNA. Hum Mutat. 2004;24:225-35
45. Esmail S, Kartner N, Yao Y, Kim JW, Reithmeier RAF, Manolson MF. Molecular mechanisms of cutis laxa and distal renal tubular acidosis-causing mutations in V-ATPase a subunits, ATP6V0A2 and ATP6V0A4. J Biol Chem. 2018;293(8):2787-800
46. Tuttle AM, Hoffman TL, Schilling TF. Rabconnectin-3a regulates vesicle endocytosis and canonical Wnt signaling in zebrafish neural crest migration. PLoS Biol. 2014;12:e1001852
47. Peri F, Nusslein-Volhard C. Live imaging of neuronal degradation by microglia reveals a role for v0-ATPase a1 in phagosomal fusion in vivo. Cell. 2008;133:916-27
48. Fischer B, Dimopoulou A, Egerer J, Gardeitchik T, Kidd A, Jost D. et al. Further characterization of ATP6V0A2-related autosomal recessive cutis laxa. Hum Genet. 2012;131:1761-73
49. Hucthagowder V, Morava E, Kornak U, Lefeber DJ, Fischer B, Dimopoulou A. et al. Loss-of-function mutations in ATP6V0A2 impair vesicular trafficking, tropoelastin secretion and cell survival. Hum Mol Genet. 2009;18:2149-65
50. Kornak U, Reynders E, Dimopoulou A, van Reeuwijk J, Fischer B, Rajab A. et al. Impaired glycosylation and cutis laxa caused by mutations in the vesicular H+-ATPase subunit ATP6V0A2. Nat Genet. 2008;40:32-4
51. Van Maldergem L, Dobyns W, Kornak U. ATP6V0A2-related cutis laxa. In: (ed.) Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH. et al. GeneReviews(R). Seattle (WA). 1993-2018 [Updated 2015 Feb 12](Open)
52. Rajab A, Kornak U, Budde BS, Hoffmann K, Jaeken J, Nurnberg P. et al. Geroderma osteodysplasticum hereditaria and wrinkly skin syndrome in 22 patients from Oman. Am J Med Genet A. 2008;146A:965-76
53. Sobacchi C, Frattini A, Orchard P, Porras O, Tezcan I, Andolina M. et al. The mutational spectrum of human malignant autosomal recessive osteopetrosis. Hum Mol Genet. 2001;10:1767-73
54. Kornak U, Schulz A, Friedrich W, Uhlhaas S, Kremens B, Voit T. et al. Mutations in the a3 subunit of the vacuolar H(+)-ATPase cause infantile malignant osteopetrosis. Hum Mol Genet. 2000;9:2059-63
55. Ochotny N, Flenniken AM, Owen C, Voronov I, Zirngibl RA, Osborne LR. et al. The V-ATPase a3 subunit mutation R740S is dominant negative and results in osteopetrosis in mice. J Bone Miner Res. 2011;26:1484-93
56. Deng W, Stashenko P, Chen W, Liang Y, Shimizu K, Li YP. Characterization of mouse Atp6i gene, the gene promoter, and the gene expression. J Bone Miner Res. 2001;16:1136-46
57. Li YP, Chen W, Liang Y, Li E, Stashenko P. Atp6i-deficient mice exhibit severe osteopetrosis due to loss of osteoclast-mediated extracellular acidification. Nat Genet. 1999;23:447-51
58. Stover EH, Borthwick KJ, Bavalia C, Eady N, Fritz DM, Rungroj N. et al. Novel ATP6V1B1 and ATP6V0A4 mutations in autosomal recessive distal renal tubular acidosis with new evidence for hearing loss. J Med Genet. 2002;39:796-803
59. Smith AN, Skaug J, Choate KA, Nayir A, Bakkaloglu A, Ozen S. et al. Mutations in ATP6N1B, encoding a new kidney vacuolar proton pump 116-kD subunit, cause recessive distal renal tubular acidosis with preserved hearing. Nat Genet. 2000;26:71-5
60. Norgett EE, Golder ZJ, Lorente-Canovas B, Ingham N, Steel KP, Karet Frankl FE. Atp6v0a4 knockout mouse is a model of distal renal tubular acidosis with hearing loss, with additional extrarenal phenotype. Proc Natl Acad Sci U S A. 2012;109:13775-80
61. Stehberger PA, Schulz N, Finberg KE, Karet FE, Giebisch G, Lifton RP. et al. Localization and regulation of the ATP6V0A4 (a4) vacuolar H+-ATPase subunit defective in an inherited form of distal renal tubular acidosis. J Am Soc Nephrol. 2003;14:3027-38
62. Lee J, Willer JR, Willer GB, Smith K, Gregg RG, Gross JM. Zebrafish blowout provides genetic evidence for Patched1-mediated negative regulation of Hedgehog signaling within the proximal optic vesicle of the vertebrate eye. Dev Biol. 2008;319:10-22
63. Nuckels RJ, Ng A, Darland T, Gross JM. The vacuolar-ATPase complex regulates retinoblast proliferation and survival, photoreceptor morphogenesis, and pigmentation in the zebrafish eye. Invest Ophthalmol Vis Sci. 2009;50:893-905
64. Mangieri LR, Mader BJ, Thomas CE, Taylor CA, Luker AM, Tse TE. et al. ATP6V0C knockdown in neuroblastoma cells alters autophagy-lysosome pathway function and metabolism of proteins that accumulate in neurodegenerative disease. PLoS One. 2014;9:e93257
65. Jin D, Muramatsu S, Shimizu N, Yokoyama S, Hirai H, Yamada K. et al. Dopamine release via the vacuolar ATPase V0 sector c-subunit, confirmed in N18 neuroblastoma cells, results in behavioral recovery in hemiparkinsonian mice. Neurochem Int. 2012;61:907-12
66. Chung AY, Kim MJ, Kim D, Bang S, Hwang SW, Lim CS. et al. Neuron-specific expression of atp6v0c2 in zebrafish CNS. Dev Dyn. 2010;239:2501-8
67. Kishi S, Bayliss PE, Uchiyama J, Koshimizu E, Qi J, Nanjappa P. et al. The identification of zebrafish mutants showing alterations in senescence-associated biomarkers. PLoS Genet. 2008;4:e1000152
68. Madsen EC, Gitlin JD. Zebrafish mutants calamity and catastrophe define critical pathways of gene-nutrient interactions in developmental copper metabolism. PLoS Genet. 2008;4:e1000261
69. Ramos-Balderas JL, Carrillo-Rosas S, Guzman A, Navarro RE, Maldonado E. The zebrafish mutants for the V-ATPase subunits d, ac45, E, H and c and their variable pigment dilution phenotype. BMC Res Notes. 2013;6:39
70. Lee SH, Rho J, Jeong D, Sul JY, Kim T, Kim N. et al. v-ATPase V0 subunit d2-deficient mice exhibit impaired osteoclast fusion and increased bone formation. Nat Med. 2006;12:1403-9
71. Wu H, Xu G, Li YP. Atp6v0d2 is an essential component of the osteoclast-specific proton pump that mediates extracellular acidification in bone resorption. J Bone Miner Res. 2009;24:871-85
72. Blake-Palmer KG, Su Y, Smith AN, Karet FE. Molecular cloning and characterization of a novel form of the human vacuolar H+-ATPase e-subunit: an essential proton pump component. Gene. 2007;393:94-100
73. Van Damme T, Gardeitchik T, Mohamed M, Guerrero-Castillo S, Freisinger P, Guillemyn B. et al. Mutations in ATP6V1E1 or ATP6V1A cause autosomal-recessive cutis laxa. Am J Hum Genet. 2017;100:216-27
74. Horng JL, Lin LY, Huang CJ, Katoh F, Kaneko T, Hwang PP. Knockdown of V-ATPase subunit A (atp6v1a) impairs acid secretion and ion balance in zebrafish (Danio rerio). Am J Physiol Regul Integr Comp Physiol. 2007;292:R2068-76
75. Vargas-Poussou R, Houillier P, Le Pottier N, Strompf L, Loirat C, Baudouin V. et al. Genetic investigation of autosomal recessive distal renal tubular acidosis: evidence for early sensorineural hearing loss associated with mutations in the ATP6V0A4 gene. J Am Soc Nephrol. 2006;17:1437-43
76. Paunescu TG, Russo LM, Da Silva N, Kovacikova J, Mohebbi N, Van Hoek AN. et al. Compensatory membrane expression of the V-ATPase B2 subunit isoform in renal medullary intercalated cells of B1-deficient mice. Am J Physiol Renal Physiol. 2007;293:F1915-26
77. Yuan Y, Zhang J, Chang Q, Zeng J, Xin F, Wang J. et al. De novo mutation in ATP6V1B2 impairs lysosome acidification and causes dominant deafness-onychodystrophy syndrome. Cell Res. 2014;24:1370-3
78. Kortum F, Caputo V, Bauer CK, Stella L, Ciolfi A, Alawi M. et al. Mutations in KCNH1 and ATP6V1B2 cause Zimmermann-Laband syndrome. Nat Genet. 2015;47:661-7
79. Gonda X, Eszlari N, Anderson IM, Deakin JF, Bagdy G, Juhasz G. Association of ATP6V1B2 rs1106634 with lifetime risk of depression and hippocampal neurocognitive deficits: possible novel mechanisms in the etiopathology of depression. Transl Psychiatry. 2016;6:e945
80. Hassan MJ, Santos RL, Rafiq MA, Chahrour MH, Pham TL, Wajid M. et al. A novel autosomal recessive non-syndromic hearing impairment locus (DFNB47) maps to chromosome 2p25.1-p24.3. Hum Genet. 2006;118:605-10
81. Kittler R, Putz G, Pelletier L, Poser I, Heninger AK, Drechsel D. et al. An endoribonuclease-prepared siRNA screen in human cells identifies genes essential for cell division. Nature. 2004;432:1036-40
82. Monteiro J, Aires R, Becker JD, Jacinto A, Certal AC, Rodriguez-Leon J. V-ATPase proton pumping activity is required for adult zebrafish appendage regeneration. PLoS One. 2014;9:e92594
83. Sun-Wada GH, Imai-Senga Y, Yamamoto A, Murata Y, Hirata T, Wada Y. et al. A proton pump ATPase with testis-specific E1-subunit isoform required for acrosome acidification. J Biol Chem. 2002;277:18098-105
84. Tan LJ, Wang ZE, Wu KH, Chen XD, Zhu H, Lu S. et al. Bivariate genome-wide association study implicates ATP6V1G1 as a novel pleiotropic locus underlying osteoporosis and age at menarche. J Clin Endocrinol Metab. 2015;100:E1457-66
85. Kawamura N, Sun-Wada GH, Wada Y. Loss of G2 subunit of vacuolar-type proton transporting ATPase leads to G1 subunit upregulation in the brain. Sci Rep. 2015;5:14027
86. Duan X, Liu J, Zheng X, Wang Z, Zhang Y, Hao Y. et al. Deficiency of ATP6V1H causes bone loss by inhibiting bone resorption and bone formation through the TGF-beta1 pathway. Theranostics. 2016;6:2183-95
87. Zhang Y, Huang H, Zhao G, Yokoyama T, Vega H, Huang Y. et al. ATP6V1H deficiency impairs bone development through activation of MMP9 and MMP13. PLoS Genet. 2017;13:e1006481
88. Scimeca JC, Quincey D, Parrinello H, Romatet D, Grosgeorge J, Gaudray P. et al. Novel mutations in the TCIRG1 gene encoding the a3 subunit of the vacuolar proton pump in patients affected by infantile malignant osteopetrosis. Hum Mutat. 2003;21:151-7
89. Frattini A, Orchard PJ, Sobacchi C, Giliani S, Abinun M, Mattsson JP. et al. Defects in TCIRG1 subunit of the vacuolar proton pump are responsible for a subset of human autosomal recessive osteopetrosis. Nat Genet. 2000;25:343-6
90. Makaryan V, Rosenthal EA, Bolyard AA, Kelley ML, Below JE, Bamshad MJ. et al. TCIRG1-associated congenital neutropenia. Hum Mutat. 2014;35:824-7
91. Scimeca JC, Franchi A, Trojani C, Parrinello H, Grosgeorge J, Robert C. et al. The gene encoding the mouse homologue of the human osteoclast-specific 116-kDa V-ATPase subunit bears a deletion in osteosclerotic (oc/oc) mutants. Bone. 2000;26:207-13
92. Kim K, Lee SH, Ha Kim J, Choi Y, Kim N. NFATc1 induces osteoclast fusion via up-regulation of Atp6v0d2 and the dendritic cell-specific transmembrane protein (DC-STAMP). Mol Endocrinol. 2008;22:176-85
93. Ayodele BA, Mirams M, Pagel CN, Mackie EJ. The vacuolar H(+) ATPase V0 subunit d2 is associated with chondrocyte hypertrophy and supports chondrocyte differentiation. Bone Rep. 2017;7:98-107
94. Raisz LG. Pathogenesis of osteoporosis: concepts, conflicts, and prospects. J Clin Invest. 115: 3318-25.
95. Peacock M, Turner CH, Econs MJ, Foroud T. Genetics of Osteoporosis. Endocr Rev. 2002;23:303-26
96. Yang S, Duan X. Epigenetics, bone remodeling and osteoporosis. Curr Stem Cell Res Ther. 2018;13(2):101-109
97. Veldhuis-Vlug AG, Oei L, Souverein PC, Tanck MW, Rivadeneira F, Zillikens MC. et al. Association of polymorphisms in the beta-2 adrenergic receptor gene with fracture risk and bone mineral density. Osteoporos Int. 2015;26:2019-27
98. Liu JZ, McRae AF, Nyholt DR, Medland SE, Wray NR, Brown KM. et al. A versatile gene-based test for genome-wide association studies. Am J Hum Genet. 2010;87:139-45
99. Estrada K, Styrkarsdottir U, Evangelou E, Hsu YH, Duncan EL, Ntzani EE. et al. Genome-wide meta-analysis identifies 56 bone mineral density loci and reveals 14 loci associated with risk of fracture. Nat Genet. 2012;44:491-501
100. Yang TL, Guo Y, Liu YJ, Shen H, Liu YZ, Lei SF. et al. Genetic variants in the SOX6 gene are associated with bone mineral density in both Caucasian and Chinese populations. Osteoporos Int. 2012;23:781-7
101. Yang TL, Guo Y, Li J, Zhang L, Shen H, Li SM. et al. Gene-gene interaction between RBMS3 and ZNF516 influences bone mineral density. J Bone Miner Res. 2013;28:828-37
102. Guo D, Zhang Q, Li J, Liang X, Chen Y, Wang H. et al. Fluid shear stress changes cell morphology and regulates the expression of ATP6V1A and TCIRG1 mRNA in rat osteoclasts. Mol Med Rep. 2010;3:173-8
103. Gharanei S, Zatyka M, Astuti D, Fenton J, Sik A, Nagy Z. et al. Vacuolar-type H+-ATPase V1A subunit is a molecular partner of Wolfram syndrome 1 (WFS1) protein, which regulates its expression and stability. Hum Mol Genet. 2013;22:203-17
104. Alzamora R, Al-Bataineh MM, Liu W, Gong F, Li H, Thali RF. et al. AMP-activated protein kinase regulates the vacuolar H+-ATPase via direct phosphorylation of the A subunit (ATP6V1A) in the kidney. Am J Physiol Renal Physiol. 2013;305:F943-56
105. Pietrement C, Sun-Wada GH, Silva ND, McKee M, Marshansky V, Brown D. et al. Distinct expression patterns of different subunit isoforms of the V-ATPase in the rat epididymis. Biol Reprod. 2006;74:185-94
106. Imai-Senga Y, Sun-Wada GH, Wada Y, Futai M. A human gene, ATP6E1, encoding a testis-specific isoform of H(+)-ATPase subunit E. Gene. 2002;289:7-12
107. Grzmil M, Voigt S, Thelen P, Hemmerlein B, Helmke K, Burfeind P. Up-regulated expression of the MAT-8 gene in prostate cancer and its siRNA-mediated inhibition of expression induces a decrease in proliferation of human prostate carcinoma cells. Int J Oncol. 2004;24:97-105
108. Wang SY, Shen XY, Wu CY, Pan F, Shen YY, Sheng HH. et al. Analysis of whole genomic expression profiles of Helicobacter pylori related chronic atrophic gastritis with IL-1B-31CC/-511TT genotypes. J Dig Dis. 2009;10:99-106
109. Smith AN, Borthwick KJ, Karet FE. Molecular cloning and characterization of novel tissue-specific isoforms of the human vacuolar H(+)-ATPase C, G and d subunits, and their evaluation in autosomal recessive distal renal tubular acidosis. Gene. 2002;297:169-77
110. Iida A, Ozaki K, Ohnishi Y, Tanaka T, Nakamura Y. Identification of 46 novel SNPs in the 130-kb region containing a myocardial infarction susceptibility gene on chromosomal band 6p21. J Hum Genet. 2003;48:476-9
111. Eicher JD, Wakabayashi Y, Vitseva O, Esa N, Yang Y, Zhu J. et al. Characterization of the platelet transcriptome by RNA sequencing in patients with acute myocardial infarction. Platelets. 2016;27:230-9
112. Mewar D, Marinou I, Lee ME, Timms JM, Kilding R, Teare MD. et al. Haplotype-specific gene expression profiles in a telomeric major histocompatibility complex gene cluster and susceptibility to autoimmune diseases. Genes Immun. 2006;7:625-31
113. Romero A, Ramos E, Ares I, Castellano V, Martinez M, Martinez-Larranaga MR. et al. Oxidative stress and gene expression profiling of cell death pathways in alpha-cypermethrin-treated SH-SY5Y cells. Arch Toxicol. 2017;91:2151-64
114. Zodro E, Jaroszewski M, Ida A, Wrzesinski T, Kwias Z, Bluyssen H. et al. FUT11 as a potential biomarker of clear cell renal cell carcinoma progression based on meta-analysis of gene expression data. Tumour Biol. 2014;35:2607-17
115. Shinmura K, Igarashi H, Kato H, Koda K, Ogawa H, Takahashi S. et al. BSND and ATP6V1G3: novel immunohistochemical markers for chromophobe renal cell carcinoma. Medicine (Baltimore). 2015;94:e989
116. Liu ZZ, Cui ST, Tang B, Wang ZZ, Luan ZX. Identification of key biomarkers involved in osteosarcoma using altered modules. Genet Mol Res. 2016;15(3):10.4238 /gmr.15038277 (Open)
117. Su Y, Zhou A, Al-Lamki RS, Karet FE. The a-subunit of the V-type H+-ATPase interacts with phosphofructokinase-1 in humans. J Biol Chem. 2003;278:20013-8
118. Golder ZJ, Karet Frankl FE. Extra-renal locations of the a4 subunit of H(+)ATPase. BMC Cell Biol. 2016;17:27
119. Li X, Chai Y, Tao Z, Li L, Huang Z, Li Y. et al. Novel mutations in ATP6V0A4 are associated with atypical progressive sensorineural hearing loss in a Chinese patient with distal renal tubular acidosis. Int J Pediatr Otorhinolaryngol. 2012;76:152-4
120. Lee BS, Holliday LS, Ojikutu B, Krits I, Gluck SL. Osteoclasts express the B2 isoform of vacuolar H(+)-ATPase intracellularly and on their plasma membranes. Am J Physiol. 1996;270:C382-8
121. Bartkiewicz M, Hernando N, Reddy SV, Roodman GD, Baron R. Characterization of the osteoclast vacuolar H(+)-ATPase B-subunit. Gene. 1995;160:157-64
122. Feng S, Deng L, Chen W, Shao J, Xu G, Li YP. Atp6v1c1 is an essential component of the osteoclast proton pump and in F-actin ring formation in osteoclasts. Biochem J. 2009;417:195-203
123. Lee BS, Gluck SL, Holliday LS. Interaction between vacuolar H(+)-ATPase and microfilaments during osteoclast activation. J Biol Chem. 1999;274:29164-71
124. Vitavska O, Merzendorfer H, Wieczorek H. The V-ATPase subunit C binds to polymeric F-actin as well as to monomeric G-actin and induces cross-linking of actin filaments. J Biol Chem. 2005;280:1070-6
125. Vitavska O, Wieczorek H, Merzendorfer H. A novel role for subunit C in mediating binding of the H+-V-ATPase to the actin cytoskeleton. J Biol Chem. 2003;278:18499-505
126. Perez-Sayans M, Suarez-Penaranda JM, Barros-Angueira F, Diz PG, Gandara-Rey JM, Garcia-Garcia A. An update in the structure, function, and regulation of V-ATPases: the role of the C subunit. Braz J Biol. 2012;72:189-98
127. Hemken P, Guo XL, Wang ZQ, Zhang K, Gluck S. Immunologic evidence that vacuolar H+ ATPases with heterogeneous forms of Mr = 31,000 subunit have different membrane distributions in mammalian kidney. J Biol Chem. 1992;267:9948-57
128. Stransky L, Cotter K, Forgac M. The Function of V-ATPases in Cancer. Physiol Rev. 2016;96:1071-91
129. Bouche V, Espinosa AP, Leone L, Sardiello M, Ballabio A, Botas J. Drosophila Mitf regulates the V-ATPase and the lysosomal-autophagic pathway. Autophagy. 2016;12:484-98
130. Hunt SR, Hernandez R, Brown DT. Role of the vacuolar-ATPase in Sindbis virus infection. J Virol. 2011;85:1257-66
131. Perreira JM, Aker AM, Savidis G, Chin CR, McDougall WM, Portmann JM. et al. RNASEK is a V-ATPase-associated factor required for endocytosis and the replication of rhinovirus, influenza A virus, and dengue virus. Cell Rep. 2015;12:850-63
132. Lu X, Yu H, Liu SH, Brodsky FM, Peterlin BM. Interactions between HIV1 Nef and vacuolar ATPase facilitate the internalization of CD4. Immunity. 1998;8:647-56
133. Soliman M, Seo JY, Kim DS, Kim JY, Park JG, Alfajaro MM. et al. Activation of PI3K, Akt, and ERK during early rotavirus infection leads to V-ATPase-dependent endosomal acidification required for uncoating. PLoS Pathog. 2018;14:e1006820
134. Pavelin J, McCormick D, Chiweshe S, Ramachandran S, Lin YT, Grey F. Cellular v-ATPase is required for virion assembly compartment formation in human cytomegalovirus infection. Open Biol. 2017:7 160298 (Open)
135. Li S, Hao L, Wang L, Lu Y, Li Q, Zhu Z. et al. Targeting Atp6v1c1 prevents inflammation and bone erosion caused by periodontitis and reveals its critical function in osteoimmunology. PLoS One. 2015;10:e0134903
136. Holliday LS, Lu M, Lee BS, Nelson RD, Solivan S, Zhang L. et al. The amino-terminal domain of the B subunit of vacuolar H+-ATPase contains a filamentous actin binding site. J Biol Chem. 2000;275:32331-7
137. Nakamura I, Takahashi N, Udagawa N, Moriyama Y, Kurokawa T, Jimi E. et al. Lack of vacuolar proton ATPase association with the cytoskeleton in osteoclasts of osteosclerotic (oc/oc) mice. FEBS Lett. 1997;401:207-12
138. Chen SH, Bubb MR, Yarmola EG, Zuo J, Jiang J, Lee BS. et al. Vacuolar H+-ATPase binding to microfilaments: regulation in response to phosphatidylinositol 3-kinase activity and detailed characterization of the actin-binding site in subunit B. J Biol Chem. 2004;279:7988-98
139. Zuo J, Jiang J, Chen SH, Vergara S, Gong Y, Xue J. et al. Actin binding activity of subunit B of vacuolar H+-ATPase is involved in its targeting to ruffled membranes of osteoclasts. J Bone Miner Res. 2006;21:714-21
140. Serra-Peinado C, Sicart A, Llopis J, Egea G. Actin filaments are involved in the coupling of V0-V1 domains of vacuolar H+-ATPase at the Golgi complex. J Biol Chem. 2016;291:7286-99
141. Forgac M. Structure and properties of the clathrin-coated vesicle and yeast vacuolar V-ATPases. J Bioenerg Biomembr. 1999;31:57-65
142. Sun-Wada G, Murata Y, Yamamoto A, Kanazawa H, Wada Y, Futai M. Acidic endomembrane organelles are required for mouse postimplantation development. Dev Biol. 2000;228:315-25
143. Hurtado-Lorenzo A, Skinner M, El Annan J, Futai M, Sun-Wada GH, Bourgoin S. et al. V-ATPase interacts with ARNO and Arf6 in early endosomes and regulates the protein degradative pathway. Nat Cell Biol. 2006;8:124-36
144. Zhang Z, Wang X, Gao T, Gu C, Sun F, Yu L. et al. Characterization of the complex involved in regulating V-ATPase activity of the vacuolar and endosomal membrane. J Bioenerg Biomembr. 2017;49:347-55
145. Kissing S, Hermsen C, Repnik U, Nesset CK, von Bargen K, Griffiths G. et al. Vacuolar ATPase in phagosome-lysosome fusion. J Biol Chem. 2015;290:14166-80
146. Bhargava A, Voronov I, Wang Y, Glogauer M, Kartner N, Manolson MF. Osteopetrosis mutation R444L causes endoplasmic reticulum retention and misprocessing of vacuolar H+-ATPase a3 subunit. J Biol Chem. 2012;287:26829-39
147. Scherer O, Steinmetz H, Kaether C, Weinigel C, Barz D, Kleinert H. et al. Targeting V-ATPase in primary human monocytes by archazolid potently represses the classical secretion of cytokines due to accumulation at the endoplasmic reticulum. Biochem Pharmacol. 2014;91:490-500
148. Xue Y, Wang L, Xia D, Li Q, Gao S, Dong M. et al. Dental abnormalities caused by novel compound heterozygous CTSK mutations. J Dent Res. 2015;94:674-81
149. Kubisch R, Frohlich T, Arnold GJ, Schreiner L, von Schwarzenberg K, Roidl A. et al. V-ATPase inhibition by archazolid leads to lysosomal dysfunction resulting in impaired cathepsin B activation in vivo. Int J Cancer. 2014;134:2478-88
150. Chung C, Mader CC, Schmitz JC, Atladottir J, Fitchev P, Cornwell ML. et al. The vacuolar-ATPase modulates matrix metalloproteinase isoforms in human pancreatic cancer. Lab Invest. 2011;91:732-43
151. Kulshrestha A, Katara GK, Ibrahim S, Pamarthy S, Jaiswal MK, Gilman Sachs A. et al. Vacuolar ATPase 'a2' isoform exhibits distinct cell surface accumulation and modulates matrix metalloproteinase activity in ovarian cancer. Oncotarget. 2015;6:3797-810
152. Cotter K, Stransky L, McGuire C, Forgac M. Recent insights into the structure, regulation, and function of the V-ATPases. Trends Biochem Sci. 2015;40:611-22
153. Kobayashi Y, Uehara S, Koide M, Takahashi N. The regulation of osteoclast differentiation by Wnt signals. Bonekey Rep. 2015;4:713
154. Rawadi G, Roman-Roman S. Wnt signalling pathway: a new target for the treatment of osteoporosis. Expert Opin Ther Targets. 2005;9:1063-77
155. Weivoda MM, Ruan M, Hachfeld CM, Pederson L, Howe A, Davey RA. et al. Wnt signaling inhibits osteoclast differentiation by activating canonical and noncanonical cAMP/PKA pathways. J Bone Miner Res. 2016;31:65-75
156. Liedert A, Rontgen V, Schinke T, Benisch P, Ebert R, Jakob F. et al. Osteoblast-specific Krm2 overexpression and Lrp5 deficiency have different effects on fracture healing in mice. PLoS One. 2014;9:e103250
157. Qiang YW, Chen Y, Brown N, Hu B, Epstein J, Barlogie B. et al. Characterization of Wnt/beta-catenin signalling in osteoclasts in multiple myeloma. Br J Haematol. 2010;148:726-38
158. Bodine PV, Komm BS. Wnt signaling and osteoblastogenesis. Rev Endocr Metab Disord. 2006;7:33-9
159. Abe E, Marians RC, Yu W, Wu XB, Ando T, Li Y. et al. TSH is a negative regulator of skeletal remodeling. Cell. 2003;115:151-62
160. Coombs GS, Yu J, Canning CA, Veltri CA, Covey TM, Cheong JK. et al. WLS-dependent secretion of WNT3A requires Ser209 acylation and vacuolar acidification. J Cell Sci. 2010;123:3357-67
161. Kaur G, Ahn J, Hankenson KD, Ashley JW. Stimulation of notch signaling in mouse osteoclast precursors. J Vis Exp. 2017;120:e55234 (Open)
162. Jin WJ, Kim B, Kim JW, Kim HH, Ha H, Lee ZH. Notch2 signaling promotes osteoclast resorption via activation of PYK2. Cell Signal. 2016;28:357-65
163. Ashley JW, Ahn J, Hankenson KD. Notch signaling promotes osteoclast maturation and resorptive activity. J Cell Biochem. 2015;116:2598-609
164. Colombo M, Thummler K, Mirandola L, Garavelli S, Todoerti K, Apicella L. et al. Notch signaling drives multiple myeloma induced osteoclastogenesis. Oncotarget. 2014;5:10393-406
165. Duan L, Ren Y. Role of notch signaling in osteoimmunology—from the standpoint of osteoclast differentiation. Eur J Orthod. 2013;35:175-82
166. Duan L, de Vos P, Fan M, Ren Y. Notch is activated in RANKL-induced osteoclast differentiation and resorption. Front Biosci. 2008;13:7064-71
167. Bai S, Kopan R, Zou W, Hilton MJ, Ong CT, Long F. et al. NOTCH1 regulates osteoclastogenesis directly in osteoclast precursors and indirectly via osteoblast lineage cells. J Biol Chem. 2008;283:6509-18
168. Yamada T, Yamazaki H, Yamane T, Yoshino M, Okuyama H, Tsuneto M. et al. Regulation of osteoclast development by Notch signaling directed to osteoclast precursors and through stromal cells. Blood. 2003;101:2227-34
169. Pamarthy S, Kulshrestha A, Katara GK, Beaman KD. The curious case of vacuolar ATPase: regulation of signaling pathways. Mol Cancer. 2018;17:41
170. Vaccari T, Duchi S, Cortese K, Tacchetti C, Bilder D. The vacuolar ATPase is required for physiological as well as pathological activation of the Notch receptor. Development. 2010;137:1825-32
171. Yan Y, Denef N, Schupbach T. The vacuolar proton pump, V-ATPase, is required for notch signaling and endosomal trafficking in Drosophila. Dev Cell. 2009;17:387-402
172. Abu-Remaileh M, Wyant GA, Kim C, Laqtom NN, Abbasi M, Chan SH. et al. Lysosomal metabolomics reveals V-ATPase- and mTOR-dependent regulation of amino acid efflux from lysosomes. Science. 2017;358:807-13
173. Matsumoto A, Pasut A, Matsumoto M, Yamashita R, Fung J, Monteleone E. et al. mTORC1 and muscle regeneration are regulated by the LINC00961-encoded SPAR polypeptide. Nature. 2017;541:228-32
174. Stransky LA, Forgac M. Amino acid availability modulates vacuolar H+-ATPase assembly. J Biol Chem. 2015;290:27360-9
175. Hu Y, Carraro-Lacroix LR, Wang A, Owen C, Bajenova E, Corey PN. et al. Lysosomal pH plays a key role in regulation of mTOR activity in osteoclasts. J Cell Biochem. 2016;117:413-25
176. Indo Y, Takeshita S, Ishii KA, Hoshii T, Aburatani H, Hirao A. et al. Metabolic regulation of osteoclast differentiation and function. J Bone Miner Res. 2013;28:2392-9
177. Smink JJ, Begay V, Schoenmaker T, Sterneck E, de Vries TJ, Leutz A. Transcription factor C/EBPbeta isoform ratio regulates osteoclastogenesis through MafB. EMBO J. 2009;28:1769-81
178. Zoncu R, Bar-Peled L, Efeyan A, Wang S, Sancak Y, Sabatini DM. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science. 2011;334:678-83
179. Sun-Wada GH, Wada Y. Role of vacuolar-type proton ATPase in signal transduction. Biochim Biophys Acta. 2015;1847:1166-72
180. Sigismund S, Confalonieri S, Ciliberto A, Polo S, Scita G, Di Fiore PP. Endocytosis and signaling: cell logistics shape the eukaryotic cell plan. Physiol Rev. 2012;92:273-366
181. Nevius E, Pinho F, Dhodapkar M, Jin H, Nadrah K, Horowitz MC. et al. Oxysterols and EBI2 promote osteoclast precursor migration to bone surfaces and regulate bone mass homeostasis. J Exp Med. 2015;212:1931-46
182. Menon P, Yin G, Smolock EM, Zuscik MJ, Yan C, Berk BC. GPCR kinase 2 interacting protein 1 (GIT1) regulates osteoclast function and bone mass. J Cell Physiol. 2010;225:777-85
183. Gidon A, Al-Bataineh MM, Jean-Alphonse FG, Stevenson HP, Watanabe T, Louet C. et al. Endosomal GPCR signaling turned off by negative feedback actions of PKA and v-ATPase. Nat Chem Biol. 2014;10:707-9
184. Nagamatsu Y, Takeda K, Kuranaga T, Numoto N, Miki K. Origin of asymmetry at the intersubunit interfaces of V1-ATPase from Thermus thermophilus. J Mol Biol. 2013;425:2699-708
185. Kartner N, Yao Y, Li K, Crasto GJ, Datti A, Manolson MF. Inhibition of osteoclast bone resorption by disrupting vacuolar H+-ATPase a3-B2 subunit interaction. J Biol Chem. 2010;285:37476-90
186. Stam NJ, Wilkens S. Structure of the lipid nanodisc-reconstituted vacuolar ATPase proton channel: definition of the interaction of rotor and stator and implications for enzyme regulation by reversible dissoication. J Biol Chem. 2017;292:1749-61
187. Shin DK, Kim MH, Lee SH, Kim TH, Kim SY. Inhibitory effects of luteolin on titanium particle-induced osteolysis in a mouse model. Acta Biomater. 2012;8:3524-31
188. Kim TH, Jung JW, Ha BG, Hong JM, Park EK, Kim HJ. et al. The effects of luteolin on osteoclast differentiation, function in vitro and ovariectomy-induced bone loss. J Nutr Biochem. 2011;22:8-15
189. Lee JW, Ahn JY, Hasegawa S, Cha BY, Yonezawa T, Nagai K. et al. Inhibitory effect of luteolin on osteoclast differentiation and function. Cytotechnology. 2009;61:125-34
190. Zhang J, Ahn MJ, Sun QS, Kim KY, Hwang YH, Ryu JM. et al. Inhibitors of bone resorption from Halenia corniculata. Arch Pharm Res. 2008;31:850-5
191. Crasto GJ, Kartner N, Yao Y, Li K, Bullock L, Datti A. et al. Luteolin inhibition of V-ATPase a3-d2 interaction decreases osteoclast resorptive activity. J Cell Biochem. 2013;114:929-41
192. Liu M, Tarsio M, Charsky CM, Kane PM. Structural and functional separation of the N- and C-terminal domains of the yeast V-ATPase subunit H. J Biol Chem. 2005;280:36978-85
193. Li L, Yang S, Zhang Y, Ji D, Jin Z, Duan X. ATP6V1H regulates the growth and differentiation of bone marrow stromal cells. Biochem Biophys Res Commun. 2018;502:84-90
194. Heinemann T, Bulwin GC, Randall J, Schnieders B, Sandhoff K, Volk HD. et al. Genomic organization of the gene coding for TIRC7, a novel membrane protein essential for T cell activation. Genomics. 1999;57:398-406
195. Utku N, Heinemann T, Tullius SG, Bulwin GC, Beinke S, Blumberg RS. et al. Prevention of acute allograft rejection by antibody targeting of TIRC7, a novel T cell membrane protein. Immunity. 1998;9:509-18
196. Smirnova AS, Morgun A, Shulzhenko N, Silva ID, Gerbase-DeLima M. Identification of new alternative splice events in the TCIRG1 gene in different human tissues. Biochem Biophys Res Commun. 2005;330:943-9
197. Oh JH, Lee JY, Joung SH, Oh YT, Kim HS, Lee NK. Insulin enhances RANKL-induced osteoclastogenesis via ERK1/2 activation and induction of NFATc1 and Atp6v0d2. Cell Signal. 2015;27:2325-31
198. Ohta T, Yamamoto M, Numata M, Iseki S, Kitagawa H, Kayahara M. et al. Differential expression of vacuolar-type H+-ATPase between normal human pancreatic islet B-cells and insulinoma cells. Int J Oncol. 1997;11:597-601
199. Dechant R, Binda M, Lee SS, Pelet S, Winderickx J, Peter M. Cytosolic pH is a second messenger for glucose and regulates the PKA pathway through V-ATPase. EMBO J. 2010;29:2515-26
200. Hettiarachchi KD, Zimmet PZ, Danial NN, Myers MA. Transplacental exposure to the vacuolar-ATPase inhibitor bafilomycin disrupts survival signaling in beta cells and delays neonatal remodeling of the endocrine pancreas. Exp Toxicol Pathol. 2008;60:295-306
201. Louagie E, Taylor NA, Flamez D, Roebroek AJ, Bright NA, Meulemans S. et al. Role of furin in granular acidification in the endocrine pancreas: identification of the V-ATPase subunit Ac45 as a candidate substrate. Proc Natl Acad Sci U S A. 2008;105:12319-24
202. Dai FF, Bhattacharjee A, Liu Y, Batchuluun B, Zhang M, Wang XS. et al. A novel GLP1 receptor interacting protein ATP6ap2 regulates insulin secretion in pancreatic beta cells. J Biol Chem. 2015;290:25045-61
203. Tani Y, Yamada S, Inoshita N, Hirata Y, Shichiri M. Regulation of growth hormone secretion by (pro)renin receptor. Sci Rep. 2015;5:10878
204. Shamansurova Z, Tan P, Ahmed B, Pepin E, Seda O, Lavoie JL. Adipose tissue (P)RR regulates insulin sensitivity, fat mass and body weight. Mol Metab. 2016;5:959-69
205. Cao X, Yang Q, Qin J, Zhao S, Li X, Fan J. et al. V-ATPase promotes transforming growth factor-beta-induced epithelial-mesenchymal transition of rat proximal tubular epithelial cells. Am J Physiol Renal Physiol. 2012;302:F1121-32
206. Pircher R, Jullien P, Lawrence DA. Beta-transforming growth factor is stored in human blood platelets as a latent high molecular weight complex. Biochem Biophys Res Commun. 1986;136:30-7
207. Miyazono K, Hellman U, Wernstedt C, Heldin CH. Latent high molecular weight complex of transforming growth factor beta 1. Purification from human platelets and structural characterization. J Biol Chem. 1988;263:6407-15
208. Tang Y, Wu X, Lei W, Pang L, Wan C, Shi Z. et al. TGF-[beta]1-induced migration of bone mesenchymal stem cells couples bone resorption with formation. Nat Med. 2009;15:757-65
209. Zhen G, Wen C, Jia X, Li Y, Crane JL, Mears SC. et al. Inhibition of TGF-[beta] signaling in mesenchymal stem cells of subchondral bone attenuates osteoarthritis. Nat Med. 2013;19:704-12
210. Pamarthy S, Mao L, Katara GK, Fleetwood S, Kulshreshta A, Gilman-Sachs A. et al. The V-ATPase a2 isoform controls mammary gland development through Notch and TGF-beta signaling. Cell Death Dis. 2016;7:e2443
211. Alwan HA, van Zoelen EJ, van Leeuwen JE. Ligand-induced lysosomal epidermal growth factor receptor (EGFR) degradation is preceded by proteasome-dependent EGFR de-ubiquitination. J Biol Chem. 2003;278:35781-90
212. Ng KW. Future developments in osteoporosis therapy. Endocr Metab Immune Disord Drug Targets. 2009;9:371-84
213. Kartner N, Manolson MF. Novel techniques in the development of osteoporosis drug therapy: the osteoclast ruffled-border vacuolar H(+)-ATPase as an emerging target. Expert Opin Drug Discov. 2014;9:505-22
214. Chen JS, Sambrook PN. Antiresorptive therapies for osteoporosis: a clinical overview. Nat Rev Endocrinol. 2011;8:81-91
215. Thudium CS, Jensen VK, Karsdal MA, Henriksen K. Disruption of the V-ATPase functionality as a way to uncouple bone formation and resorption - a novel target for treatment of osteoporosis. Curr Protein Pept Sci. 2012;13:141-51
216. Toro EJ, Ostrov DA, Wronski TJ, Holliday LS. Rational identification of enoxacin as a novel V-ATPase-directed osteoclast inhibitor. Curr Protein Pept Sci. 2012;13:180-91
217. Yao G, Feng H, Cai Y, Qi W, Kong K. Characterization of vacuolar-ATPase and selective inhibition of vacuolar-H(+)-ATPase in osteoclasts. Biochem Biophys Res Commun. 2007;357:821-7
218. Farina C, Gagliardi S. Selective inhibition of osteoclast vacuolar H(+)-ATPase. Curr Pharm Des. 2002;8:2033-48
219. Qin A, Cheng TS, Pavlos NJ, Lin Z, Dai KR, Zheng MH. V-ATPases in osteoclasts: structure, function and potential inhibitors of bone resorption. Int J Biochem Cell Biol. 2012;44:1422-35
220. Xu J, Cheng T, Feng HT, Pavlos NJ, Zheng MH. Structure and function of V-ATPases in osteoclasts: potential therapeutic targets for the treatment of osteolysis. Histol Histopathol. 2007;22:443-54
221. Osteresch C, Bender T, Grond S, von Zezschwitz P, Kunze B, Jansen R. et al. The binding site of the V-ATPase inhibitor apicularen is in the vicinity of those for bafilomycin and archazolid. J Biol Chem. 2012;287:31866-76
222. Huss M, Sasse F, Kunze B, Jansen R, Steinmetz H, Ingenhorst G. et al. Archazolid and apicularen: novel specific V-ATPase inhibitors. BMC Biochem. 2005;6:13
223. Kim YS, Jin HO, Hong SE, Song JY, Hwang CS, Park IC. Silencing of secretory clusterin sensitizes NSCLC cells to V-ATPase inhibitors by downregulating survivin. Biochem Biophys Res Commun. 2018;495:2004-9
224. Costa GA, de Souza SB, da Silva Teixeira LR, Okorokov LA, Arnholdt ACV, Okorokova-Facanha AL. et al. Tumor cell cholesterol depletion and V-ATPase inhibition as an inhibitory mechanism to prevent cell migration and invasiveness in melanoma. Biochim Biophys Acta. 2018;1862:684-91
225. Chen H, Liu P, Zhang T, Gao Y, Zhang Y, Shen X. et al. Effects of diphyllin as a novel V-ATPase inhibitor on TE-1 and ECA-109 cells. Oncol Rep. 2018;39:921-8
226. Merk H, Messer P, Ardelt MA, Lamb DC, Zahler S, Muller R. et al. Inhibition of the V-ATPase by archazolid A: anew strategy to inhibit EMT. Mol Cancer Ther. 2017;16:2329-39
227. Kim D, Hwang HY, Kim JY, Lee JY, Yoo JS, Marko-Varga G. et al. FK506, an immunosuppressive drug, induces autophagy by binding to the V-ATPase catalytic subunit A in neuronal cells. J Proteome Res. 2017;16:55-64
228. Visentin L, Dodds RA, Valente M, Misiano P, Bradbeer JN, Oneta S. et al. A selective inhibitor of the osteoclastic V-H(+)-ATPase prevents bone loss in both thyroparathyroidectomized and ovariectomized rats. J Clin Invest. 2000;106:309-18
229. Niikura K, Takano M, Sawada M. A novel inhibitor of vacuolar ATPase, FR167356, which can discriminate between osteoclast vacuolar ATPase and lysosomal vacuolar ATPase. Br J Pharmacol. 2004;142:558-66
230. Sorensen MG, Henriksen K, Neutzsky-Wulff AV, Dziegiel MH, Karsdal MA. Diphyllin, a novel and naturally potent V-ATPase inhibitor, abrogates acidification of the osteoclastic resorption lacunae and bone resorption. J Bone Miner Res. 2007;22:1640-8
231. Lebreton S, Jaunbergs J, Roth MG, Ferguson DA, De Brabander JK. Evaluating the potential of vacuolar ATPase inhibitors as anticancer agents and multigram synthesis of the potent salicylihalamide analog saliphenylhalamide. Bioorg Med Chem Lett. 2008;18:5879-83
232. Kazami S, Muroi M, Kawatani M, Kubota T, Usui T, Kobayashi J. et al. Iejimalides show anti-osteoclast activity via V-ATPase inhibition. Biosci Biotechnol Biochem. 2006;70:1364-70
233. Price PA, June HH, Buckley JR, Williamson MK. SB 242784, a selective inhibitor of the osteoclastic V-H+ATPase, inhibits arterial calcification in the rat. Circ Res. 2002;91:547-52
234. Xie XS, Padron D, Liao X, Wang J, Roth MG, De Brabander JK. Salicylihalamide A inhibits the V0 sector of the V-ATPase through a mechanism distinct from bafilomycin A1. J Biol Chem. 2004;279:19755-63
235. Kazami S, Takaine M, Itoh H, Kubota T, Kobayashi J, Usui T. Iejimalide C is a potent V-ATPase inhibitor, and induces actin disorganization. Biol Pharm Bull. 2014;37:1944-7
236. McHenry P, Wang WL, Devitt E, Kluesner N, Davisson VJ, McKee E. et al. Iejimalides A and B inhibit lysosomal vacuolar H+-ATPase (V-ATPase) activity and induce S-phase arrest and apoptosis in MCF-7 cells. J Cell Biochem. 2010;109:634-42
237. Lee SH, Lee JY, Kwon YI, Jang HD. Anti-osteoclastic activity of Artemisia capillaris Thunb. extract depends upon attenuation of osteoclast differentiation and bone resorption-associated acidification due to chlorogenic acid, hyperoside, and scoparone. Int J Mol Sci. 2017;18(2):pii E322 (Open)
238. Ostrov DA, Magis AT, Wronski TJ, Chan EK, Toro EJ, Donatelli RE. et al. Identification of enoxacin as an inhibitor of osteoclast formation and bone resorption by structure-based virtual screening. J Med Chem. 2009;52:5144-51
239. Toro EJ, Zuo J, Ostrov DA, Catalfamo D, Bradaschia-Correa V, Arana-Chavez V. et al. Enoxacin directly inhibits osteoclastogenesis without inducing apoptosis. J Biol Chem. 2012;287:17894-904
240. Wierer M, Prestel M, Schiller HB, Yan G, Schaab C, Azghandi S. et al. Compartment-resolved proteomic analysis of mouse aorta during atherosclerotic plaque formation reveals osteoclast-specific protein expression. Mol Cell Proteomics. 2018;17:321-34
241. Wang H, Wang R, Wang Z, Liu Q, Mao Y, Duan X. ClC-3 chloride channel functions as a mechanically sensitive channel in osteoblasts. Biochem Cell Biol. 2015;93:558-65
Author contact
![]() Corresponding author: Xiaohong Duan, Department of Oral Biology, Clinic of Oral Rare and Genetic Diseases, School of Stomatology, the Fourth Military Medical University, 145 West Changle Road, Xi'an 710032, P. R. China. Tel: 86-29-84776169 Fax: 86-29-84776169 E-mail: xhduanedu.cn
Corresponding author: Xiaohong Duan, Department of Oral Biology, Clinic of Oral Rare and Genetic Diseases, School of Stomatology, the Fourth Military Medical University, 145 West Changle Road, Xi'an 710032, P. R. China. Tel: 86-29-84776169 Fax: 86-29-84776169 E-mail: xhduanedu.cn