Impact Factor
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Physiology of the TME and its...
Biomimetic platforms that...
Perspective and Conclusion
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(19):5259-5275. doi:10.7150/thno.29098 This issue Cite
Review
Chemoresistance of Cancer Cells: Requirements of Tumor Microenvironment-mimicking In Vitro Models in Anti-Cancer Drug Development
Yeonho Jo1,2, Nakwon Choi2,3, Kyobum Kim4, Hyung-Jun Koo5, Jonghoon Choi1 ![]() , Hong Nam Kim2,3
, Hong Nam Kim2,3 ![]()
1. School of Integrative Engineering, Chung-Ang University, Seoul 06974, Republic of Korea
2. Center for BioMicrosystems, Brain Science Institute, Korea Institute of Science and Technology (KIST), Seoul 02792, Republic of Korea
3. Division of Bio-Medical Science & Technology, KIST School, Korea University of Science and Technology, Seoul 02792, Republic of Korea
4. Division of Bioengineering, Incheon National University, Incheon, Republic of Korea
5. Department of Chemical and Biomolecular Engineering, Seoul National University of Science and Technology, Seoul, Republic of Korea
Received 2018-8-8; Accepted 2018-10-4; Published 2018-10-22
Abstract

For decades, scientists have been using two-dimensional cell culture platforms for high-throughput drug screening of anticancer drugs. Growing evidence indicates that the results of anti-cancer drug screening vary with the cell culture microenvironment, and this variation has been proposed as a reason for the high failure rate of clinical trials. Since the culture condition-dependent drug sensitivity of anti-cancer drugs may negatively impact the identification of clinically effective drug candidates, more reliable in vitro cancer platforms are urgently needed. In this review article, we provide an overview of how cell culture conditions can alter drug efficacy and highlight the importance of developing more reliable cancer drug testing platforms for use in the drug discovery process. The environmental factors that can alter drug delivery and efficacy are reviewed. Based on these observations of chemoresistant tumor physiology, we summarize the recent advances in the fabrication of in vitro cancer models and the model-dependent cytotoxicity of anti-cancer drugs, with a particular focus on engineered environmental factors in these platforms. It is believed that more physiologically relevant cancer models can revolutionize the drug discovery process.
Keywords: chemoresistance, tumor microenvironment, cancer cell, biomimetic, efficacy
Introduction
Since the launch of the “war on cancer” in 1971 in the United States, many cancer biologists and pharmaceutical companies have been striving to conquer cancer[1]. Cancer biologists have identified the molecular mechanisms underlying the characteristic behaviors of cancer cells including the extensive proliferation, degradation of the surrounding extracellular matrix, migration and even vascularization[2, 3]. Based on the identified signaling pathways and biomolecular factors, pharmaceutical companies have developed various anti-cancer drugs termed 'magic bullets,' which can specifically inhibit the actions of their molecular targets. Although a few drugs have demonstrated promising ability, such as those for the treatment of metastatic melanoma[4, 5], many of the drugs do not satisfy the expectations of conquering cancer. As proposed by Hanahan, this low efficacy of targeted cancer chemotherapy is because of the “adaptive and evasive resistance strategies developed by cancers under attack”[1]. One of the important factors is that cancer cells change their responsiveness to drugs by changing their interaction with the surroundings. Earlier studies focused mainly on the cancer cell itself rather than on the interactions between the cancer cells and their surroundings. However, the role of tumor microenvironment (TME) in tumor progression and drug efficacy has recently attracted much attention[6, 7].
TME includes, in addition to the cancer cells, the physical, chemical, and biological components around the cancer cells, such as the extracellular matrix (ECM), interstitial flow, stromal cells, immune cells, vascular networks, and biochemical concentration gradients (Fig. 1A). Recent studies have indicated that the TME can significantly affect cancer progression including malignancy, invasion, and metastasis[8, 9]. Frequently, researchers overlook the environmental factors and utilize traditional two-dimensional culture techniques because of the ease of handling and the possibility of a large number of parallel analyses[10-12]. However, the results of efficacy and toxicity assays using traditional cell culture models (e.g., monolayer cell culture) exhibit significant differences from those of animal studies and human trials. This is one of the reasons for the failure of many drug candidates in clinical trials[13, 14] and is partly due to the limited utility of 2D culture models for drug screening because of the absence of the complex microenvironment of native tumors[15]. Two-dimensional culture methods are not able to replicate the cell-cell and cell-extracellular environment interactions, which are important factors in vivo (Fig. 1B). These interactions are responsible for cell differentiation, proliferation, vitality, expression of genes and proteins, drug metabolism, and other cellular functions[16-18]. In addition, the modes of cell division and adhesion are restricted under 2D conditions. These features affect the organization of the intracellular structures and cell signaling[19, 20]. Finally, unlike natural in vivo tumors, 2D cultured cells in a monolayer have unlimited access to oxygen, nutrients, and signaling molecules from the culture medium[16].
The differences between the native tumor microenvironment (TME) and the conventional in vitro cancer models in terms of the recapitulation of physiological factors. (A) The physiological conditions within the native TME. (B) The features of the conventional 2D or plastic dish-based cancer models. Because the conventional cancer models do not reflect the important environmental cues observed in the TME, the behaviors and responses of cancer cells in vivo cannot be fully recapitulated in the experimental conditions. In particular, tests of the efficacy or cytotoxicity of anticancer drugs frequently show misleading drug screening results, increasing the time and cost of drug discovery.
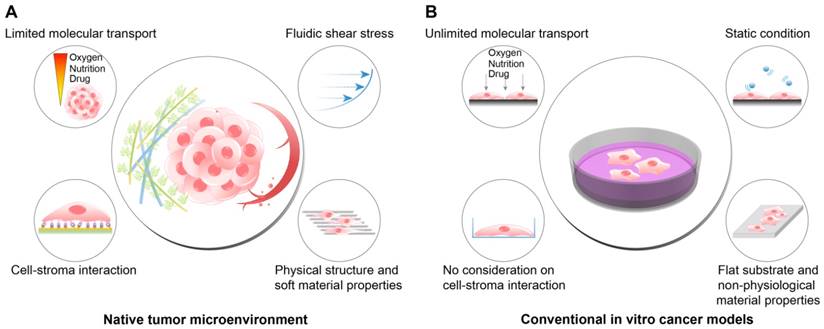
These environmental factors are significantly different in 2D cultures compared to those in the in vivo tumors and can skew the experimental results[21]. Clinically efficacious drug candidates might be eliminated during early screening, and compounds with lower or no clinical efficacy might progress into clinical trials, resulting in increased developmental cost and time. It is therefore necessary to develop physiologically relevant in vitro cancer models to better predict the efficacy and toxicity of anti-cancer drugs[22-24]. Several techniques have been developed to overcome the limitations of traditional 2D cell culture models and allow the experimental models to mimic the in vivo microenvironment more closely. These techniques replicates the physiological features of the TME such as cell-cell interactions, fluidic shear stress, and cell-ECM interactions. This review discusses how the efficacy or the toxicity of anti-cancer drug candidates can be changed by altering the cell culture conditions. For this purpose, we first discuss the physiological characteristics of the TME with a particular focus on the interaction between the TME components and cancer cells. The review will then describe the efforts for the development of biomimetic cell culture platforms, which can replicate the features of tumor physiology. Finally, this review will discuss the difference in the efficacy of anti-cancer drug candidates depending on the in vitro models used, which underscore the importance of reliable drug screening platforms.
Physiology of the TME and its effect on drug delivery and efficacy
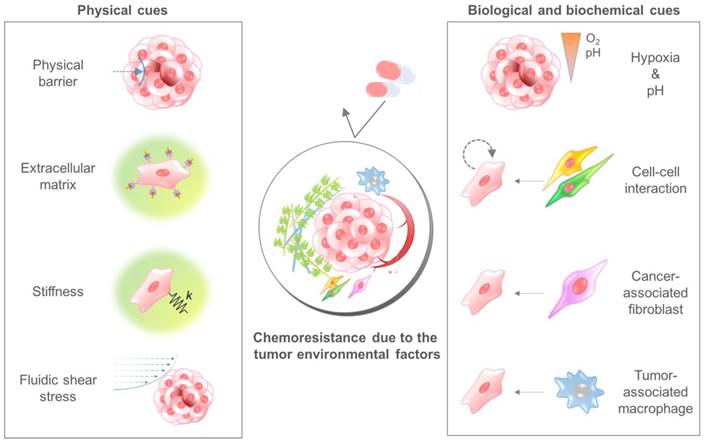
The TME comprises multiple cellular and noncellular components organized in a three-dimensional form[25, 26]. The representative TME factors that can affect the chemosensitivity of cancer cells are summarized in Table 1. Various TME factors are classified into two categories, physical and biological/biochemical cues, and their roles in drug delivery and efficacy are summarized in the next sections (Fig. 2).
The tumor environmental factors that affect the efficacy of anti-cancer drugs
| Tumor microenvironmental factors | Mechanism | Results | Ref. |
|---|---|---|---|
| Physical cues | |||
| Physical barrier | Limited penetration of drugs or drug carriers | Unable to deliver drugs into the core of the tumor mass | [31, 32] |
| Changed interstitial fluid flow | Outward fluid flux from the tumor mass to the surroundings | [34] | |
| ECM | Cell adhesion mediated drug resistance (CAM-DR) | Activation of anti-apoptotic signals by integrin-mediated ligand-receptor binding | [40, 77] |
| Porosity | Diffusion-limited molecular transport | [55] | |
| Stiffness | Matrix stiffness-induced mechanotransduction | Matrix stiffness-mediated induction of mechanotransduction pathways such as YAP and TAZ | [53] |
| Fluidic shear stress | Flow-mediated activation of autocrine signaling (IGF-1R pathway) | Increased IGF-1 release in response to increasing fluidic shear stress (feed-forward loop) | [161] |
| Caspase pathway-dependent receptor-mediated apoptosis (tumor necrosis factor apoptosis-inducing ligand, TRAIL) | TRAIL-induced apoptosis observed only under the fluidic shear stress condition | [157] | |
| PI3K/Akt signaling and microRNA-199-3p | Chemoresistance to cisplatin and paclitaxel under the fluidic shear stress condition | [156] | |
| Biological and biochemical cues | |||
| Hypoxia | Quiescence of cancer cells (nonproliferating or slow cell cycle) | Decreased cell death against anti-proliferating agents | [65] |
| HIF-1 mediated enhancement of drug efflux | Decreased intracellular concentration of drugs | [61, 62] | |
| HIF-1 mediated enhancement of antiapoptotic signals | Avoiding necrotic or apoptotic cell death | [63] | |
| pH | P-glycoprotein-mediated drug efflux | Enhanced activity of the drug efflux pump in the acidic microenvironment | [73] |
| Ion trapping | Reduced cell permeability of positively ionized weak base drugs in the acidic environment | [72] | |
| Chronic exposure to acid pH | Increased expression of heat shock protein HSP27 levels in tumor cells causing chemoresistance to cisplatin | [74] | |
| Cell-cell interaction | Cytokines secreted by nearby cells | Autocrine and paracrine-mediated activation of antiapoptosis signaling | [76] |
| Heterocellular interaction (stromal cell-cancer cell) | Trogocytosis-mediated chemoresistance | [75] | |
| Cancer-associated fibroblast (CAF) | Cytokines secreted by CAFs | Chemoresistance of cancer cells by CAF-secreted cytokines such as interleukins, CCL1, and SDF-1 | [79-83] |
| Exosome-mediated miRNA delivery from CAFs to cancer cells | Acquired chemoresistance via transferred miRNA such as miR-155, 100, 222, 30a, and 146a | [85-87] | |
| Changed metabolism of CAFs by effector T-cells | Abrogated stroma-mediated chemoresistance in cancer cells | [84] | |
| Tumor-associated macrophage (TAM) | Secretion of cytokines by TAM in an M2 polarization state | Activation of anti-apoptotic signals in the cancer cells | [92-95] |
The tumor microenvironmental factors that cause chemoresistance of cancer cells. Physical cues include the physical barrier, binding to the extracellular matrix component, stiffness-induced mechanotransduction, and fluidic shear stress. Biological and biochemical cues include hypoxia, low pH, cell-cell interaction, cancer-associated fibroblasts, and tumor-associated macrophages. Because each cue induces the chemoresistance of cancer cells through different mechanisms, a combinatorial consideration of those factors using innovative in vitro cancer models is required to identify the exact efficacy of anticancer drugs.
Physical cues
The physical components of the TME can affect the sensitivity of tumor cells to drugs. Physical cues that can affect drug delivery and efficacy include high cell density, fluid pressure, the extracellular matrix, and stiffness.
Drugs are generally delivered to the tumor mass through interstitial spaces via diffusion and pressure-driven circulation. During the delivery process, drugs encounter physical and biochemical barriers. Sutherland et al. demonstrated that the efficacy of anti-cancer drugs was reduced when the cells were cultured in 3D spheroids rather than in 2D monolayer cultures due to the limited transport of drugs through the densely packed cells[27]. Therefore, a majority of the anticancer drugs exhibit limited penetration into solid tumors and reduced toxicity[28-32]. For example, the organization of the surrounding ECM increases the interstitial fluid pressure because the expansion of the tumor mass strongly affects the efficiency of drug delivery[33]. The rapid growth of the tumor mass induces outward fluid flux from the tumor mass to the surroundings and hinders drug delivery towards the neoplasms[34].
Chemoresistance mediated by receptor-ligand interaction of the cancer cell with the surroundings is termed cell adhesion-mediated drug resistance (CAM-DR)[35]. Cancer cells can interact with stromal cells in their proximity[36] or with the extracellular matrix to which they adhere, such as collagen, laminin, and fibronectin[33]. When a cancer cell (either hematopoietic or epithelial) comes into intimate contact with the stromal cells or ECM components via integrins, adhesion induces not only the quiescent state but also the production of pro- and anti-apoptotic molecules[37, 38]. Therefore, cancer cells can acquire chemoresistance via their interaction with stromal cells or with the ECM[39]. For example, the acute myelogenous leukemia (AML) cell lines, when cultured on fibronectin-coated substrates, exhibit increased chemoresistance to treatment with daunorubicin (DNR) and arabinoside (AraC); integrin α4β1 is important for this chemoresistance[40]. It has also been shown that integrin α4β1-mediated chemoresistance is closely related to patient outcome, showing a higher relapse rate and a decreased survival rate in the case of patients expressing high levels of integrin α4β1. Since the mechanism underlying cell-cell and cell-ECM interaction-mediated chemoresistance is not associated with transcriptional regulation[37, 41, 42], drug sensitivity can be restored when the microenvironmental factors are removed[43].
The composition of the ECM can also significantly affect chemoresistance. Previous studies on native tumor microenvironments have indicated that type I collagen, a major component of the tumor stroma, can promote the invasive progression of cancer[44]. Furthermore, the correlation between the chemoresistance and the abundance of the tumor stroma of pancreatic cancer has also been considered a therapeutic target to overcome the limitations of conventional chemotherapy[45, 46]. In this context, Cramer et al. demonstrated that the ECM-component-mediated epithelial-to-mesenchymal transition is an important factor in chemoresistance[47]. In this study, the tumor spheroids were embedded in type I collagen-rich or laminin-rich bulk ECM matrix, and the behaviors of the cancer cells were monitored. Interestingly, the cancer cells embedded in a spheroid form within the type I collagen matrix showed significant invasion into the surrounding matrix, similar to individual cells showing epithelial-to-mesenchymal transition, whereas the spheroids embedded in a laminin-rich matrix showed no such dissemination. In addition, upon oxaliplatin chemotherapy, the invading populations of cancer cells (observed only in the collagen I matrix) showed significant chemoresistance, while the tumor spheroids in both matrices were reduced in size, demonstrating tumor spheroid-specific chemosensitivity.
The effect of matrix stiffness on the malignancy of cancer cells can be evaluated by using stiffness-tunable hydrogels[48, 49]. Previous studies have indicated that the stroma around the native tumor becomes highly fibrotic and stiffen, especially for pancreatic ductal adenocarcinoma[50, 51], and the altered ECM mechanics is related to the malignancy of tumor[48]. Furthermore, matrix stiffness has been also known to induce chemoresistance to paclitaxel in hepatocellular carcinoma [52]. Rice et al. investigated the effect of matrix stiffness on the chemoresistance with a particular focus on the matrix stiffness-induced epithelial-mesenchymal transition (EMT) and the subsequent acquisition of chemoresistance [53, 54].
The 3D environment in which cancer cells are surrounded by the ECM has different drug delivery conditions compared to the widely used 2D plastic dish. In the case of the 2D plastic dish, drugs, oxygen, and nutrients can access the cells unlimitedly; therefore, the diffusion-limited or perfusion-controlled delivery mechanism observed in vivo cannot be recapitulated. According to a previous study, the cytotoxic effect of the anti-cancer drug paclitaxel was lower in cells grown in the 3D hydrogel environment (40-60% survival rate) compared to that in cells grown in the 2D plastic dish (20% survival rate) due to the limited access of drugs to the cancer cells in the former[55].
Biological and biochemical cues
The uncontrolled growth of cancer cells generates densely packed cell spheroids. In this explosive growth stage, the high rate of metabolism of cancer cells and the limited availability of oxygen results in a concentration gradient of oxygen along the depth of the tumor mass[56, 57]. The presence of hypoxia in tumors has been reported to increase the expression of the genes encoding P-glycoprotein and dihydrofolate reductase, which induce resistance to substrates of P-glycoprotein and folate antagonists, respectively[58-60]. Furthermore, increased expression of hypoxia-inducible factor 1 (HIF-1) stimulates cancer cells and promotes drug efflux out of the cancer cells by the overexpression of P-glycoprotein and the multidrug resistance (MDR1) gene[61, 62]. The increased encoding of HIF-1 in cancer cells is also known to enhance the anti-apoptotic signals of cancer cells, and thus the survival rate of cancer cells increases upon exposure to chemotherapeutic agents[63]. For example, a hypoxic condition promotes the production of growth factors such as platelet-derived growth factor B (PDGF-B), transforming growth factor β (TGF-β), and epidermal growth factor (EGF) and increases the survival rate of cancer cells[63, 64]. The quiescence of cancer cells due to the low level of oxygen tension can limit the cytotoxic effect of the drugs targeting rapidly proliferating cancer cells. Such an effect is especially important in the submarginal layer of the tumor volume in which the cancer cells neither proliferate nor die, showing a quiescent metabolic state unless another stimulation is applied[65]. Many anti-cancer drugs are designed to generate free radicals, which are harmful to DNA in the presence of oxygen. The free radicals accept electrons from biologic sources and then transfer the electrons to oxygen[66]. However, in a hypoxic environment such as the deeper regions of the tumor mass, the cytotoxicity of anti-cancer drugs whose activity is mediated by free radicals is inevitably weakened[67].
In addition to the limited availability of oxygen, the hypoxic regions of tumors are also likely to have limited access to nutrients such as glucose and essential amino acids. For this reason, tumor cells in the inner regions of the tumor mass use glycolysis to generate energy for survival and proliferation, leading to the production of CO2 and carbonic acid[68]. The accumulation of these acidic products of metabolism results in low interstitial pH, which is one of the characteristics of solid tumors[69, 70]. The pH in the TME influences the efficacy of anti-cancer drugs. Drug molecules diffuse passively into the cell in an uncharged form. In the acidic extracellular pH, weakly basic drugs such as doxorubicin, mitoxantrone, vincristine, and vinblastine, are prone to protonation. Since the cell membrane is negatively charged, positively charged drugs suffer from lower cellular uptake than those in the neutral state of charge, which is known as 'ion trapping' [69, 71, 72]. As well as the inefficient drug delivery into the cancer cells, low pH promotes P-glycoprotein-mediated drugs efflux and reduce the intracellular drug concentration[73]. In addition, the chronic exposure to acidic pH increases the level of intracellular proteins such as heat shock protein HSP27, causing chemoresistance to chemotherapeutics[74].
Intercellular interactions can also enhance the chemoresistance of the cells[36]. According to earlier studies, the stromal cells near the cancer cells play supportive roles in the survival of cancer cells and the acquisition of chemoresistance via several mechanisms. For example, the stromal cells can enhance the chemoresistance via direct cell-cell interactions such as crosstalk or oncologic trogocytosis[75], and the autocrine- and paracrine-mediated activation of anti-apoptosis signaling increases chemoresistance[76]. The stromal cells near the cancer cells or surrounding ECM can activate anti-apoptotic signaling via the binding of integrin receptors of cancer cells and the corresponding ligands provided by the stromal cells or the ECM components[40, 77].
The coculture of cancer cells with stromal cells also induces cytokine-mediated chemoresistance[78]. For example, cancer cells acquired chemoresistance when cocultured with cancer-associated fibroblasts (CAFs) via CAF-secreted chemokines such as interleukin-6 (IL-6), interleukin-11 (IL-11), interleukin-17 (IL-17), chemokine C-C motif ligand-1 (CCL1), and stromal cell-derived factor 1 (SDF-1)[79-83]. Recent studies have also proposed that effector T-cells can abrogate such CAF-induced chemoresistance by modulating the metabolism of CAFs[84].
Exosomes secreted by cancer cells or nearby CAF have also been known to mediate cancer resistance. Among the components loaded in the exosomes, the encapsulated miRNAs (e.g., miR-155, miR-100, miR-222, miR-30a, and miR-146a), which are produced by chemotherapy-treated cancer cells and CAFs, play significant roles in the acquisition of chemoresistance[85-87]. For instance, the miR-155 transferred between gemcitabine-treated pancreatic cancer cells promotes chemoresistance by the detoxification of reactive oxygen species and miR-155-mediated suppression of the gemcitabine-metabolizing enzyme[88].
Tumor-associated macrophages (TAMs) are observed in tumor tissues with high density, but their clinical implications are different depending on the organs: TAMs in colorectal cancer show an anti-tumor (M1) phenotype while most of the TAMs in other organs display pro-tumor and immunosuppressive properties (M2)[89-91]. Importantly, in TAMs with pro-tumor properties, numerous TAM-secreted cytokines including IL-6, signal transducer and activator of transcription 3 (STAT3), and cathepsin B and S have been known to induce the chemoresistance of tumors by activating the anti-apoptotic programs of tumor cells[92-95]. Recent studies have indicated that the polarization of macrophages is a crucial determinant of the drug response, especially through the activation of the cytotoxic response of T cells[96]. For instance, docetaxel, partly induces antitumor activity by depleting immunosuppressive TAMs and by activating anti-tumoral monocytes[97]. These results indicate that the reprogramming of the polarization states of TAMs might be an efficient way to resolve the chemoresistance of cancer.
As discussed above, the physical and biological/biochemical cues in the TME weaken the cytotoxic effects of anti-cancer drugs. Therefore, it is obvious that the traditional monolayer cell culture model does not represent the complex microenvironment of the tumor. The following section discusses how the engineered cell culture models, with their ability to reproduce the surrounding environment including the physical, biological/biochemical cues, can address these issues[98-103].
Biomimetic platforms that replicate the physiology of the TME
In this section, recent approaches that mimic the TME using the engineered platforms are overviewed. The following examples controlled various factors of the TME such as aggregation of cancer cells, cell-cell interaction via direct contact or cytokine-mediated crosstalk, fluidic shear stress, and structure and composition of biomaterials. These platforms successfully mimicked the TME factors in the engineered cancer models, and demonstrated more in vivo-like cancer cell growth, cytokine release, cell-cell interaction and drug delivery. Cancer cells usually showed chemoresistance against the anticancer drugs in the TME-mimetic cancer platforms, suggesting the importance of physiologically relevant cancer models in the testing of the anticancer drug efficacy. In the following sections, the examples of the engineered cancer models will be reviewed.
Tumor spheroids
Tumor spheroids have been one of the widely used 3D models for cancer study. Tumor spheroids can recapitulate the dense 3D structures of native tumors in terms of their shape and the characteristics of tumors such as hypoxia, limited molecular transport, and metabolic changes. From an engineering point of view, 3D tumor spheroids can be fabricated by facilitating cell-cell interactions via physical confinement. In this process, the cell-substrate interaction must be minimized with adequate surface coating or modification. Otherwise, the cells adhere to the substrate rather than form cell aggregates[104]. Various methods have been developed to generate tumor cell spheroids. The hanging drop method is a commonly used technique for generating cell aggregates[105]. A small aliquot (normally 10 to 30 μL) of the cell suspension is placed onto the lid of the culture plate, and the lid is subsequently inverted. The drop of cell suspension remains adhered to the lid, and thus the cell suspension makes a hanging drop. Cells accumulate at the bottom of the drop (liquid-air interface) and assemble into a 3D spheroid form[106]. However, the culture media cannot be exchanged in this method, and thus it is not suitable for long-term culture. The microwell technique was developed to address this shortcoming. Using low surface energy materials such as elastomer or coating the wells with cell-repellent materials such as poly (2-hydroxyethyl methacrylate) (pHEMA), the spontaneous formation of cell spheroids at the bottom of the microwell can be facilitated because the cell-cell interaction is stronger than the cell-substrate interaction[107, 108]. In contrast to the hanging drop method, this microwell system enables the changing of the media during incubation. For cell-cell communication, interspheroid interaction is available via paracrine signaling through the media[109]. A rotating wall vessel can be used to induce polarity or to apply fluidic shear stress to the cell spheroid[110-112].
According to previous studies, 3D tumor spheroid models showed higher resistance to drug candidates than 2D models. There are several straightforward explanations for the differential drug resistance observed between 2D and 3D cultures (Fig. 3A). First, the penetration of drugs into the 3D spheroid is difficult compared to that into cell monolayers due to the physical barrier of outer cell layers[113-115]. Most of the cytotoxic effects of the drugs can be observed in the outer layers of the cells in the spheroids. Second, drug sensitivity changes in response to acidity[116]. Earlier studies have suggested that weakly acidic drugs might have the advantage of enhanced permeability because of the slightly acidic extracellular pH[117-119]. Third, the quiescent zone in the tumor spheroid can alter the cytotoxic effect of drugs. Some anti-cancer drugs act by inducing DNA damage during cellular proliferation, ultimately leading to apoptosis. Therefore, these drugs are effective in rapidly proliferating cancer cells and are termed antimitotic chemotherapeutic agents[65]. However, due to the nonproliferating nature of cells in the quiescent zone, which is located between the core and outer layer, cells in the dormant stage remain protected. For this reason, tumor spheroids may shrink but are not completely killed. Therefore, the tumor can resume proliferation once the cells in the outer layer are dead[114, 120]. Fourth, the low oxygen level in the inner zone of the tumor mass induces the expression of HIF-1 in the cancer cells[121]. The increased level of HIF-1 has been known to be associated with chemoresistance in various cancer cells[122, 123].
The tumor spheroid as a three-dimensional model for recapitulating the environmental cues originating from the high density of cancer cells such as a physical barrier against drug delivery, the concentration gradient of oxygen and nutrition, and low pH in the core of the tumor mass. (A) Representative features of tumor spheroids in terms of the physiological similarity with the native tumor. (B-a) The response of prostate cancer cell (LNCaP) spheroids in the aggregated form against the anticancer drug docetaxel. (B-b) The difference in cell viability depending on the culture dimension. Reprinted with permission from Chambers et al.[124]. (C-a) The time-dependent penetration of doxorubicin into the tumor spheroids. Red: doxorubicin. (C-b) The size-dependent chemoresistance of human breast adenocarcinoma spheroids against doxorubicin. Reprinted with permission from Gong et al.[127].
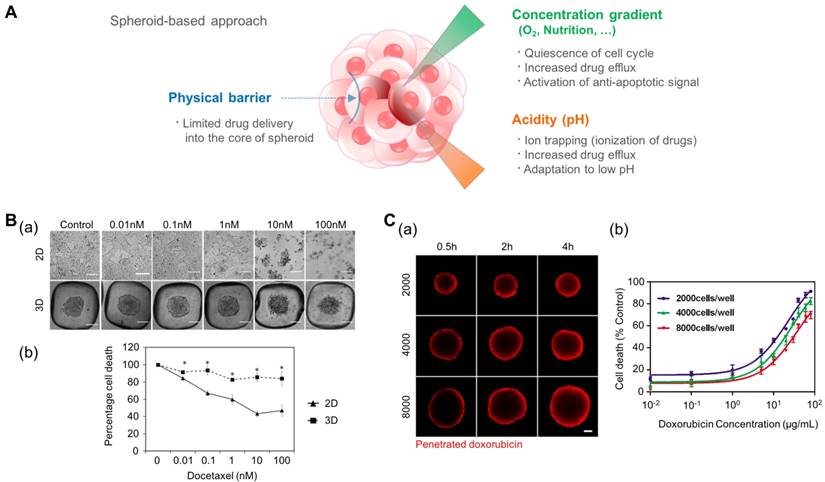
For these reasons, the spheroid model is promising in its ability to predict the cytotoxic effect of anti-cancer drugs. Chambers et al. developed a PDMS microwell system that produces cancer microaggregates (Fig. 3B)[124], which form through forced aggregation. Using this system, LNCaP cells (human prostate cancer cell line) were cultured for two days with and without the PDMS well and were treated with docetaxel for 72 h. LNCaP microaggregates showed higher resistance to docetaxel compared to cells grown as monolayers. Imamura et al. developed patient-derived xenografts (PDX) as dense 2D plates or 3D aggregated forms and observed their chemoresistance and proliferation profiles[125]. The cancer cells cultured in the aggregated form (3D) showed greater resistance to paclitaxel and doxorubicin and reduced expression of Ki-67 (a marker for proliferation) compared to the 2D cultured cells. From these results, the author concluded that the anti-apoptotic behavior of aggregated cancer cells is related to the dormant population of cancer cells. This reduced efficacy of chemotherapy in cancer spheroid models, compared to that of 2D models, was also observed in other cancer types such as leukemia and pancreatic cancer[120, 126].
In addition to differences between the monolayer (2D) and spheroidal (3D) culture environments, the size of the tumor spheroid is also an important issue. To address the size-dependent drug sensitivity of cancer cells, Gong et al. utilized agarose-based scaffolds, which have different sized microwells (Fig. 3C)[127]. They controlled the number of cancer cells in each microwell at 2,000, 4,000, and 8,000 cells per microwell, and controlled the size of the tumor spheroid in the range of 300-500 μm. They observed the penetration of doxorubicin by monitoring the fluorescence signals of cross-sectioned spheroids and found limited transport of doxorubicin into the spheroids with approximately 100 μm depth during a 4 h drug treatment. Therefore, the ratio of the drug-penetrated area was larger in the smaller tumor spheroids. When drug sensitivity is compared based on the size of the tumor spheroids, larger tumor spheroids demonstrate a higher drug resistance than smaller spheroids. Drug resistance observed in larger tumor spheroids is closely correlated with limited drug penetration into the large tumor spheroids. Because the anticancer drugs cannot be efficiently delivered into the core of the tumor mass, the development of drug carriers has been extensively studied. Tests of the drug delivery efficiency of carriers have shown that tumor spheroids are able to present a real-time monitoring window. For example, Albanese et al. demonstrated the size-dependent penetration of fluorescently labeled nanoparticles into tumor spheroids, showing more efficient delivery of smaller nanoparticles compared to larger nanoparticles into the tumor spheroids[128].
Microfluidic cell culture systems
Microfluidics refers to the technology that allows the manipulation of tiny amounts (10-9 to 10-18 liters) of fluids using channels with specific dimensions on the order of tens to hundreds of micrometers[129]. Because of the physical properties associated with the microscale, microfluidic devices offer several advantages for the study of cancer biology. With the help of microfabrication techniques, a confined region with a small volume can be fabricated such as multiple microchambers, which are interconnected with one another via microfluidic channels. These isolated microchambers can act as individual cell culture modules[130, 131]. Furthermore, the laminar flow of the medium, in parallel with largely diffusion-mediated molecular transport, enables the establishment of a stable biochemical gradient[132, 133]. Therefore, various types and concentrations of biochemical factors can be tested in a high-throughput manner. For example, Kim et al. fabricated a microfluidic device with individually addressable multiple microchambers to generate tumor spheroids and to analyze the dose-dependent effect of anti-cancer drugs on a chip platform[134]. MCF-7 human breast cancer cells were cultured in a 2D monolayer and 3D spheroid form and subsequently exposed to various concentrations of three anti-cancer drugs, mitomycin C, 5-fluorouracil, and doxorubicin. The cytotoxic effect was greater in the cells in the 2D monolayer form than in those in the 3D spheroid. This study shows that the microfluidic platform is useful not only for the mass production of cancer spheroids but also for high-throughput screening. The microfluidic platform can be used for the in situ monitoring of chemoresistant cancer cell behavior. Using a concentration gradient, the time-dependent emergence of chemoresistance can be observed. Han et al. fabricated an interconnected microchamber array, which was surrounded by two microfluidic channels[135]. By passing doxorubicin-containing medium and normal medium in two anti-parallel microchannels, they established a stable concentration gradient of the anti-cancer drug across the array of microchambers. The U87 glioblastoma cells displayed sensitivity against the anti-cancer drugs, as reflected in the reduced number of cell-occupied chambers in the early stages of exposure to the drug. However, the cells acquired chemoresistance after day 5, as demonstrated by the reoccupation of microchambers, even in the presence of anti-cancer drugs. Thus, the acquisition of chemoresistance could be directly visualized by monitoring the microchambers.
One of the advantages of the microfluidic system is the ability to reproduce the dynamic fluidic microenvironment that is critical for organ functionality and disease progression[136]. Although the effects of hemodynamic fluidic shear stress on different cell types, such as endothelial cells and smooth muscle cells, have been extensively studied[137-139], the effect of fluidic forces on cancer cells is not yet clearly understood because of the diversity in tissue origin and drug-dependent sensitization mechanisms[140]. Cancer cells experience two main types of fluid shear stress—one that results from the flow of blood in the vascular microenvironment and the other generated by interstitial flows in the TME[141, 142]. Interstitial flow is the slow stream of plasma passing through extracellular or intercellular spaces[143]. Although the velocity of the interstitial fluid flow may vary depending on the location in the body, it is generally ~0.1 dyn/cm2 around tumors[144-146]. Interstitial flow plays a crucial role in the growth and pathophysiology of cancer[147]. For example, the rapid increase in the volume of solid tumors induces outward flow, and this flow changes the drug delivery path, ultimately reducing the efficiency of drug delivery[148-152]. Furthermore, the flow reinforces the autocrine signaling in tumor cells, obtaining biochemical stimulation from the factors secreted by the cells themselves, and thereby stimulating directional invasion and migration[153, 154]. This interstitial flow-mediated directional cancer cell invasion is termed 'autologous chemotaxis'.
Drug screening results using microfluidic platforms have demonstrated that the fluidic condition can change the chemosensitivity of cancer cells[155]. Ip et al. demonstrated that shear stress enhances the chemoresistance of tumor spheroid cells (Fig. 4B)[156]. They cultured ovarian cancer cells in the spheroid form under a fluidic shear stress of 0.02 dyn/cm2 and treated the spheroids with cisplatin and paclitaxel. The cell viability under static conditions was low: 9.2% for cisplatin and 10.15% for paclitaxel. In contrast, under conditions of shear stress, tumor spheroids exhibited significantly higher chemoresistance with a 65.59% viability in the presence of cisplatin and a 69.9% viability in the presence of paclitaxel. From the changes in protein and miRNA levels and pharmacological inhibition studies, the authors proposed that the downregulation of miR-199a-3p and the consequent upregulation of phosphoinositide 3-kinase/Akt (PI3K/Akt) signaling as a potential mechanism for the fluidic condition-mediated chemoresistance. In contrast, Mitchell et al. reported that the chemo-resistant behavior can vary with the drug type even under the same fluidic conditions (Fig. 4C)[157]. When they treated colon and prostate cancer cells with tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), the cancer cells demonstrated dose- and time-dependent increases in chemoresistance. However, when they treated those cancer cells with doxorubicin, there was no difference between the chemo-sensitivity of the cells under static conditions and that of the cells under fluidic conditions. In this study, the authors proposed the death receptor trimerization and subsequent recruitment of TRAIL-induced apoptosis signaling as the potential mechanism for TRAIL-mediated chemoresistance under fluidic conditions.
Engineered platforms that mimic the fluidic shear stress around native tumor cells. (A) Schematic illustration of tumor spheroids and adhered cancer cells in a 3D matrix or circulating tumor cells upon exposure to a physiologically relevant stream. (B-a) A photograph and schematic illustration of the tumor spheroid-embedding microfluidic device. (B-b) The viable cells (denoted as Annexin V-/PI- cells) after the treatment with anticancer drugs in the presence or absence of fluidic shear stress. Reprinted with permission from Ip et al.[156]. (C-a) The microscopic images of COLO 205 cells exposed to tumor necrosis factor apoptosis-inducing ligand (TRAIL) and fluidic shear stress. Scale bar = 30 μm. (C-b) Apoptosis of COLO 205 cells by TRAIL in the presence or absence of fluidic shear stress. The COLO 205 cells were treated with 0.1 μg/mL TRAIL before applying fluidic shear stress. *p<0.05. Reprinted with permission from Mitchell et al.[157]. (D-a) Fluorescence images of the sectioned scaffolds with Ewing sarcoma cells. Blue: DAPI. Scale bar = 200 μm. (D-b) The released amount of IGF-1 in response to fluidic shear stress (S: static, B-04: 0.04 mL/min, B-08: 0.08 mL/min, and B-40: 0.4 mL/min). After the treatment with the IGF-1R inhibitor (MK-0646), the release of IGF-1 was reduced in all fluidic conditions, whereas the addition of exogenous IGF-1 significantly promoted the amount of released IGF-1. Reprinted with permission from Santoro et al.[161].
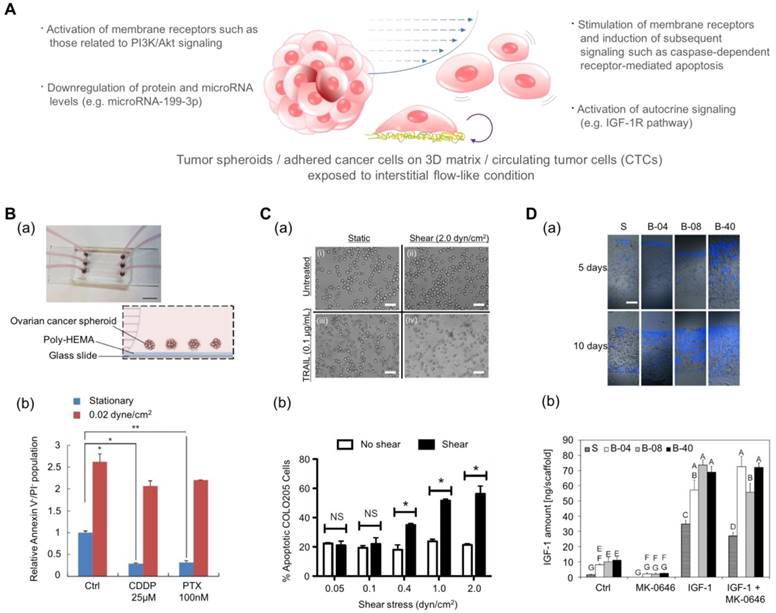
In addition to the stimulation of membrane receptors in cancer cells, the fluidic shear stress can also affect the autocrine signaling of cancer cells. In recent years, the production of growth factors and the self-response to these factor, known as autocrine signaling, have attracted much attention because autocrine signaling stimulates cancer cells to proliferate and migrate and to prevent apoptosis[158-160]. Santoro et al. cultured Ewing sarcoma cells on an electrospun poly(ε-caprolactone) scaffold and applied fluidic shear stress using a flow perfusion bioreactor (Fig. 4D)[161]. Under the fluidic condition, the cancer cells showed enhanced secretion of insulin-like growth factor-1 (IGF-1) in the presence of fluidic shear stress, and the amount of secreted IGF-1 was further increased by adding exogenous IGF-1 in the fluidic culture condition. These results indicate that the fluidic shear stress can promote Ewing sarcoma cells to form a positive feed-forward loop of IGF-1 signaling. By adding IGF-1 ligand (dalotuzumab, MK-0646), the feed-forward loop could be broken. Such results imply that the chemoresistance of an anti-cancer drug is not simply a function of the fluidic condition but can be the result of a complex interaction between various factors such as receptor expression, fluidic condition, and drug type.
Tumor angiogenesis, which drives de novo blood vessel formation towards the tumor, promotes the supply of oxygen and nutrition to the tumor. Since cancer cells can proliferate and even metastasize due to the existence of blood vessels, targeting tumor angiogenesis in conjunction with solid tumors has emerged as a methodology to treat cancer [162]. In this regard, the vascularized tumor model, which is prepared as a hydrogel-laden microfluidic system, is a good platform to screen the cytotoxic effect of anticancer drugs to both cancer and blood vessels. For example, Sobrino et al. have developed a vascularized tumor model by coculturing the cancer, endothelial, and stromal cells within interconnected microchambers for 7 days[163]. Since the endothelial cells spontaneously form perfusable vascular networks, which were connected to reservoirs, anti-cancer drugs could be delivered to the tumor through the vasculature. The authors introduced various anticancer drugs into the vascularized tumor-on-a-chip and screened their effect on the regression of the tumor or the blood vessels. This vascularized tumor-on-a-chip model can be extended to a 96-well compatible platform, and a library of anticancer drugs can be screened in a high throughput manner[164]. In addition, a dynamic microenvironment can be incorporated in the vascularized tumor models. A cyclically stretchable lung-on-a-chip platform can give valuable information about the importance of the lung-specific mechanical environment in the chemoresistance of lung cancer. Hassell et al. have fabricated a multilayered nonsmall-cell lung cancer model by culturing cancer cells and lung epithelial cells on one side of the porous elastomeric membrane and endothelial cells on the other side[165]. They found that the cyclic mechanical stretching not only suppresses cancer cell growth and invasion but also induces chemoresistance to tyrosine kinase inhibitor therapy. They found that the cyclic stretching-induced chemoresistance was attributed by the downregulation of epidermal growth factor receptor (EGFR) and changes in EGFR-related phosphorylation. This result implies that the tissue-specific microenvironment is an important factor for the identification of anticancer drug efficacy.
Polymeric scaffolds
The physical structure of the ECM in the TME plays a crucial role in regulating various cancer cell behaviors such as growth, proliferation, tumor invasion, transformation, and drug response in a number of cell types[48, 166-168]. The ECM can be described in a simplified way as fibrous physical structures and biochemical factors bound to the surface of fibers. Therefore, the structure and function of the ECM can be mimicked by fabricating an artificial matrix, which has similar physical structure, and subsequently coating it with biochemical factors observed in the native ECM[169-172]. In contrast to cells in two-dimensional culture systems, the cancer cells can form focal adhesions with a neighboring cell or ECM, but only on the surface of the artificial matrix[173]. Therefore, the distribution and clustering of focal adhesions are different from those on a plastic dish, resulting in a change in the signaling pathway of focal adhesion kinase and its downstream cascade[174, 175]. Therefore, cancer cells, as well as other stromal cell types, show different cell behaviors including polarization, directional migration, proliferation, and modified intracellular organization on an artificial matrix[176, 177]. Since the anisotropic topography can mimic the remodeled ECM bundles and microtracks in the TME, this model was used to study the mechanism underlying the invasion of cancer in response to geometrical signals[178-180]. Such multiscale topography can be fabricated with various top-down and bottom-up methods. The top-down strategy refers to techniques that create small features on bulk materials such as laser machining and photolithography. The bottom-up strategy describes the technologies that build small units into organized forms, such as electrospinning and particle lithography[181]. The details of these nanofabrication techniques can be found elsewhere[182-184].
In addition to inducing changes in the behavior of cancer cells, multiscale topography, as a biomimetic platform, can alter the cytotoxic profiles of anti-cancer drugs. Abdellatef et al. investigated the effect of surface topography on the cellular cytotoxicity and morphology of HepG2 cells[185]. They cultured HepG2 liver cancer cells on patterned TiO2 substrates and treated them with cisplatin. Even with a short exposure (8 h) to the drug, HepG2 cells on the patterned substrate displayed a significantly higher cytotoxic response than those on the flat surface. Tan et al. investigated the effect of cell-morphology-like topography on the response of cancer cells to anticancer drugs (Fig. 5B)[186]. Human endometrial cancer cells were incubated on substrates containing cell-like features that had been fabricated using a bioimprint methodology. The mold was prepared as a positive (convex) and a negative (concave) surface. When the cancer cells were treated for 48 h with commonly used drugs, paclitaxel and doxorubicin, the cells showed the most reduced growth rate on the negative imprint pattern. It is assumed that the results might be regulated by the balance of forces generated by the substrate topography via adhesion complexes and the metabolism induced by the drugs. However, the mechanism underlying the increased sensitivity is not yet clear.
Multiscale topography- and 3D scaffold-induced cancer cell behaviors that mimic the physical aspect of the cell-stroma interactions. (A) Schematic illustration of cancer cells (either spheroids or single cell) in response to multiscale topography and 3D scaffolds. (B-a) Preparation of the cell morphology-imprinted substrates. Two types of cell topography-replicated substrates (negative and positive) are prepared as well as a flat one for control. (B-b) The surface topography-dependent viability of the Ishikawa cells treated with paclitaxel and doxorubicin for 48 h. Cancer cells showed different sensitivities depending on the topography, showing the highest sensitivity in the negative topography. Reprinted with permission from Tan et al.[186]. (C-a) Scanning electron microscope images of Ewing sarcoma cells cultured on a 3D electrospun poly(ε-caprolactone) fiber matrix. Scale bar: upper row = 200 μm and lower row = 50 μm. (C-b) The response of Ewing sarcoma cells to doxorubicin. Percentage of cell viability for the 2D and 3D culture conditions, and percentage of tumor volume for in vivo as xenografts. *p<0.05 for 3D vs. 2D. Reprinted with permission from Fong et al.[188]. (D-a) The 3D scaffolds that can incorporate dissociated tumor cells and tumor spheroids. (D-b) Efficacy of doxorubicin against U251 cancer cells depending on the culture conditions. MS: 3D scaffolds with monolayer-cultured cells. SS: 3D scaffolds with spheroid-cultured cells. Reprinted with permission from Ho et al.[189]
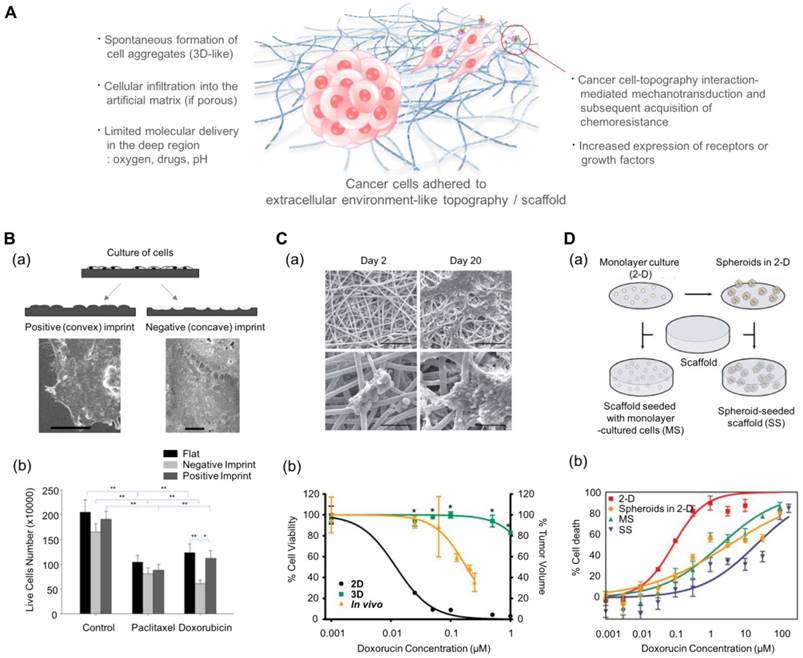
Kim et al. prepared nanogrooved structures with UV-curable polyurethane acrylate (PUA)[187]. HeLa cells and human mesenchymal cells (hMSCs) were cultured on these substrates for 24 h and treated with cisplatin. The cytotoxic effects of cisplatin were observed to be dose-dependent in both cell types. There was no significant difference between the behavior of the HeLa cells on the flat substrates and those on the nanopatterned substrates. In contrast, hMSCs showed higher cytotoxicity when cultured on the nanopatterns rather than on the flat substrate. Considering that tissue regeneration is mediated by stem cells, this high death rate of hMSCs during the treatment process can be a potentially important issue. Therefore, the parallel analyses of the cytotoxic effect on stem cells and cancer cells is potentially beneficial in the drug screening process.
Engineered 3D scaffolds can serve as a useful platform for anticancer drugs. From the structural point of view, the 3D scaffolds present penetrable pore networks in which cells can migrate and aggregate and thus present a more dynamic microenvironment for the cancer cells. For example, cancer cells form clusters on the scaffolds[188] or invade into the porous networks in a prolonged culture[161]. In addition, the amount of oxygen through the porous networks per unit time is limited, possessing potentials in the recapitulating in vivo-like concentration gradient. In collaboration with the limited molecular transport and the extensive consumption of oxygen by the cancer cells, the hypoxic and acidic microenvironment can spontaneously form, allowing a TME-like microenvironment[189]. In the polymeric scaffolds, cancer cells also receive mechanotransductional inputs via integrin-mediated surface adhesion[190].
Electrospun multiscale matrices can be used as a useful platform in the studies of three-dimensional cancer cell-matrix interactions. For example, Fong et al. cultured Ewing sarcoma tumor cells on electrospun poly(ε-caprolactone) scaffolds. These tumor cells spontaneously formed tumor cell clusters and showed enhanced resistance to doxorubicin compared to those cultured in a 2D monolayer (Fig. 5C)[188]. Furthermore, the Ewing sarcoma cells cultured on the electrospun nanofiber matrix showed enhanced expression of insulin-like growth factor 1 receptor, c-Kit, and Her2 compared to the conventional 2D cultured Ewing sarcoma cells.
Such effect of cell-structure interaction can be evaluated by using 3D porous scaffolds. Ho et al. prepared the collagen-coated poly(lactic-co-glycolic acid) (PLGA) scaffolds to compare the effect of culture dimension (2D vs. 3D) (Fig. 5D)[189] and prepared dissociated cancer cells and cancer spheroids to compare the effects of cell aggregation. They prepared four cases of in vitro cancer models depending on the aggregated form (single cell vs. spheroid) and the existence of scaffolds: (i) monolayer cancer cells (2D), (ii) tumor spheroids in 2D conditions, (iii) individual cancer cells embedded in 3D scaffolds, and (iv) tumor spheroids embedded in 3D scaffolds. The 3D scaffold-embedded single cancer cells and spheroids showed higher drug resistance than those cultured on 2D scaffolds and even those in the 3D cultures, and the cancer spheroids demonstrated higher drug resistance than cancer cells seeded at the single cell level. The authors found that the enhanced drug resistance observed in the presence of scaffolds might be induced by the low pH-mediated glycolysis and higher production of angiogenic factors due to the acidic environment.
Perspective and Conclusion
Out of every few thousands of drug candidates screened and tested, very few drugs are approved for clinical use. Even if a drug candidate passes the preclinical studies, it is frequently dropped from the clinical trials because of unexpected side effects or toxicity[191]. There might be multiple reasons for the low approval rate of drugs; however, scientists and bioengineers believe that this high failure rate is primarily caused by the use of less reliable testing platforms for screening the drug candidates. As shown by numerous studies, the effect of the anti-cancer drug can differ depending on the environment in which the cancer cells reside: 2D vs. 3D, static vs. fluidic, noncoated vs. matrix-rich, and smooth vs. textured.
In recent years, mouse models have been predominantly used in anticancer drug discovery due to their close resemblance to human tumors. The xenograft mouse model is the most commonly used preclinical model for evaluating the efficacy of anticancer drugs. Xenograft models are established by implanting human cancer cells into heterogeneous mouse tissues (such as human brain cancer cells to subcutaneous tissue, which is termed heterotopic) or into the same tissue origin (such as the implantation of human brain cancer cells to the mouse brain, which is termed orthotopic)[192]. Although this in vivo xenograft model has revolutionized the drug discovery process, it does not reflect the human tumor microenvironment such as the immune response because of the utilization of immunocompromised mice[193]. A more advanced model, patient-derived tumor xenograft (PDX), has been developed[194]. The PDX model utilizes patient-derived primary cancer cells when establishing in vivo models; therefore, this model better reflects patient-specific drug responses, showing the possibility to be used in personalized medicine[195]. However, the PDX model also has several unresolved limitations such as a time-consuming process and high cost[163]. To overcome the limitations of in vivo mouse models, researchers in biomedical engineering have been working on the development of more in vivo-like models. By developing more physiologically relevant models and using them in the drug discovery process, drug screening speed and efficiency will increase, and developmental costs will decrease.
There are additional issues in the development of physiologically relevant in vitro platforms. First, high-throughput screening ability is required for testing large numbers of drug candidates. In the initial stages of drug screening, different concentrations of thousands of drug candidates are tested on various cancer cell types with different genetic mutations. Thus, the number of combinations is so large that high-throughput screening becomes a critical need. To address this issue, pharmaceutical companies have been employing well plate-based assays using 96- or 384-well plates; however, these platforms only provide a two-dimensional, static environment. Recently developed technologies such as patterned spheroid arrays[196] are believed to resolve this high-throughput screening issue without sacrificing the physiological features of the tumor microenvironment.
The second issue is the development of precision medicine or patient-specific drugs. Recent strategies of pharmaceutical companies include targeting specific genetic mutations, signaling pathways, cytokines, and enzymes with target-specific antibodies. Since the therapeutic effect of a single drug or drug cocktail depends on the individual patient characteristics, the identification of an effective agent in an in vitro model prepared using patient-derived cells is a promising approach for the development of targeted treatments. The organ-on-a-chip, an organoid made of patient-derived iPS, and PDX models can be useful for this strategy. However, in some models, the time required to establish a platform is so long that a more innovative approach with reduced process time is critical for the effective treatment of patients.
Third, the acquisition of patient-derived cells and the establishment of reliable cell lines are also important issues in the development of physiologically relevant in vitro models[197]. To address the advancement of antibody-based chemotherapy, cancer cells expressing specific ligands and activated signaling pathways are needed to test the efficacy of newly developed drugs. However, the limited availability of patient-derived cells for use in precision medicine and the stable cell lines for high-throughput screening hinders the development of in vitro models. In addition to that of cancer cells, the availability of patient-derived stromal cells is also essential because of their largely unknown regulatory mechanisms[198].
Finally, including multiple environmental factors in a single platform is also an unmet need. Although the effects of individual factors, such as ECM, flow, pH, and hypoxia, have been investigated, their synergistic role remains largely unknown. In stem cell research, stem cell differentiation is synergistically enhanced when multiple cues are provided simultaneously[199, 200]. These results imply that the tumor cells might also be synergistically affected by the environmental factors because of their inherent complexity. Therefore, such synergistic effect of multiple environmental factors on cancer progression and chemoresistance needs to be investigated. In particular, the interaction between cancer and stromal cells is largely unknown because of the limited number of available cell sources and analysis techniques. It is expected that more reliable in vitro cancer models will be developed in the future because of the advancement of engineering techniques and the abundance of cell sources. These advanced platforms can revolutionize the drug discovery process.
In this article, we have summarized the recent advances in the development of drug screening models, specifically those used to study the efficacy of anti-cancer drug candidates. We first gave an overview of the characteristics of the tumor microenvironment in terms of physical and biochemical properties including hypoxia, pH, interstitial flow, cell-cell and cell-matrix interactions. Next, we discussed engineered platforms that attempted to reproduce these environmental factors. Finally, the changes in the cytotoxicity of anti-cancer drugs in response to the engineered environmental factors were summarized. Several studies comparing the cytotoxicity of different anti-cancer drugs have shown that drug efficacy is strongly influenced by the culture conditions of the cancer cells. However, the current technologies do not fully mimic the complex microenvironment of the tumor because of the limited availability of fabrication techniques, biomaterials, and cell sources. If the in vitro cancer models can mimic the in vivo physiology more closely, the efficacy and toxicity of drug candidates can be gauged accurately.
Acknowledgements
This work was supported by the Nano•Material Technology Development Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (No. 2017M3A7B8061942) and by the National Research Foundation of Korea (NRF) grant funded by the Korea Government (MSIT) (No. 2018R1A2A3075013). This research was also supported by the KIST Institutional Program (2E27910).
Conflict of Interest
The authors declare no conflict of interest.
References
1. Hanahan D. Rethinking the war on cancer. Lancet. 2014;383:558-63
2. Friedl P, Gilmour D. Collective cell migration in morphogenesis, regeneration and cancer. Nat Rev Mol Cell Bio. 2009;10:445-57
3. Chung AS, Lee J, Ferrara N. Targeting the tumour vasculature: insights from physiological angiogenesis. Nat Rev Cancer. 2010;10:505-14
4. Rosenberg SA. Raising the Bar: The Curative Potential of Human Cancer Immunotherapy. Sci Transl Med. 2012;4:127ps8
5. Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM. et al. Nivolumab plus Ipilimumab in Advanced Melanoma. New Engl J Med. 2013;369:122-33
6. Pattabiraman DR, Weinberg RA. Tackling the cancer stem cells - what challenges do they pose? Nat Rev Drug Discov. 2014;13:497-512
7. Joyce JA. Therapeutic targeting of the tumor microenvironment. Cancer Cell. 2005;7:513-20
8. Mbeunkui F, Johann DJ. Cancer and the tumor microenvironment: a review of an essential relationship. Cancer Chemoth Pharm. 2009;63:571-82
9. Whiteside TL. The tumor microenvironment and its role in promoting tumor growth. Oncogene. 2008;27:5904-12
10. Stringfellow DA, Gray BW, Lauerman LH, Thomson MS, Rhodes PJ, Bird RC. Monolayer culture of cells originating from a preimplantation bovine embryo. In Vitro Cell Dev Biol. 1987;23:750-4
11. Ivanov I, Taherzadeh D, Nunziante M. 2D cell culture can approximate 3D behavior: Applications for screening individual agents and drug combinations. Mol Cancer Ther. 2013;12:DOI 10.1158/535-7163.Targ-13-A206
12. Sun T, Jackson S, Haycock JW, MacNeil S. Culture of skin cells in 3D rather than 2D improves their ability to survive exposure to cytotoxic agents. J Biotechnol. 2006;122:372-81
13. El-Ali J, Sorger PK, Jensen KF. Cells on chips. Nature. 2006;442:403-11
14. Tan JL, Tien J, Pirone DM, Gray DS, Bhadriraju K, Chen CS. Cells lying on a bed of microneedles: an approach to isolate mechanical force. Proc Natl Acad Sci USA. 2003;100:1484-9
15. Shamir ER, Ewald AJ. Three-dimensional organotypic culture: experimental models of mammalian biology and disease. Nat Rev Mol Cell Biol. 2014;15:647-64
16. Pampaloni F, Reynaud EG, Stelzer EHK. The third dimension bridges the gap between cell culture and live tissue. Nat Rev Mol Cell Bio. 2007;8:839-45
17. Hickman JA, Graeser R, de Hoogt R, Vidic S, Brito C, Gutekunst M. et al. Three-dimensional models of cancer for pharmacology and cancer cell biology: capturing tumor complexity in vitro/ex vivo. Biotechnol J. 2014;9:1115-28
18. Baker BM, Chen CS. Deconstructing the third dimension: how 3D culture microenvironments alter cellular cues. J Cell Sci. 2012;125:3015-24
19. Petersen OW, Ronnovjessen L, Howlett AR, Bissell MJ. Interaction with Basement-Membrane Serves to Rapidly Distinguish Growth and Differentiation Pattern of Normal and Malignant Human Breast Epithelial-Cells. P Natl Acad Sci USA. 1992;89:9064-8
20. Vondermark K, Gauss V, Vondermark H, Muller P. Relationship between Cell-Shape and Type of Collagen Synthesized as Chondrocytes Lose Their Cartilage Phenotype in Culture. Nature. 1977;267:531-2
21. Hutmacher DW. Biomaterials offer cancer research the third dimension. Nat Mater. 2010;9:90-3
22. Chen CS. 3D Biomimetic Cultures: The Next Platform for Cell Biology. Trends Cell Biol. 2016;26:798-800
23. Niu N, Wang L. In vitro human cell line models to predict clinical response to anticancer drugs. Pharmacogenomics. 2015;16:273-85
24. Campbell RM, Tummino PJ. Cancer epigenetics drug discovery and development: the challenge of hitting the mark. J Clin Invest. 2014;124:64-9
25. Li HC, Fan XL, Houghton J. Tumor microenvironment: The role of the tumor stroma in cancer. J Cell Biochem. 2007;101:805-15
26. Finger EC, Giaccia AJ. Hypoxia, inflammation, and the tumor microenvironment in metastatic disease. Cancer Metast Rev. 2010;29:285-93
27. Sutherland RM, Eddy HA, Bareham B, Reich K, Vanantwerp D. Resistance to adriamycin in multicellular spheroids. Int J Radiat Oncol Biol Phys. 1979;5:1225-30
28. Holle AW, Young JL, Spatz JP. In vitro cancer cell-ECM interactions inform in vivo cancer treatment. Adv Drug Deliv Rev. 2016;97:270-9
29. Jang SH, Wientjes MG, Lu D, Au JL. Drug delivery and transport to solid tumors. Pharm Res. 2003;20:1337-50
30. Kyle AH, Huxham LA, Yeoman DM, Minchinton AI. Limited tissue penetration of taxanes: A mechanism for resistance in solid tumors. Clin Cancer Res. 2007;13:2804-10
31. Tannock IF, Lee CM, Tunggal JK, Cowan DSM, Egorin MJ. Limited penetration of anticancer drugs through tumor tissue: A potential cause of resistance of solid tumors to chemotherapy. Clin Cancer Res. 2002;8:878-84
32. Minchinton AI, Tannock IF. Drug penetration in solid tumours. Nat Rev Cancer. 2006;6:583-92
33. Correia AL, Bissell MJ. The tumor microenvironment is a dominant force in multidrug resistance. Drug Resist Update. 2012;15:39-49
34. Jain RK. Delivery of molecular and cellular medicine to solid tumors. Adv Drug Deliver Rev. 2001;46:149-68
35. Meads MB, Gatenby RA, Dalton WS. Environment-mediated drug resistance: a major contributor to minimal residual disease. Nat Rev Cancer. 2009;9:665-74
36. Castells M, Thibault B, Delord JP, Couderc B. Implication of tumor microenvironment in chemoresistance: tumor-associated stromal cells protect tumor cells from cell death. Int J Mol Sci. 2012;13:9545-71
37. Hazlehurst LA, Argilagos RF, Dalton WS. beta 1 integrin mediated adhesion increases Bim protein degradation and contributes to drug resistance in leukaemia cells. Brit J Haematol. 2007;136:269-75
38. Hazlehurst LA, Damiano JS, Buyuksal I, Pledger WJ, Dalton WS. Adhesion to fibronectin via beta1 integrins regulates p27kip1 levels and contributes to cell adhesion mediated drug resistance (CAM-DR). Oncogene. 2000;19:4319-27
39. Landowski TH, Olashaw NE, Agrawal D, Dalton WS. Cell adhesion-mediated drug resistance (CAM-DR) is associated with activation of NF-kappa B (RelB/p50) in myeloma cells. Oncogene. 2003;22:2417-21
40. Matsunaga T, Takemoto N, Sato T, Takimoto R, Tanaka I, Fujimi A. et al. Interaction between leukemic-cell VLA-4 and stromal fibronectin is a decisive factor for minimal residual disease of acute myelogenous leukemia. Nat Med. 2003;9:1158-65
41. Shain KH, Landowski TH, Dalton WS. Adhesion-mediated intracellular redistribution of c-Fas-associated death domain-like IL-1-converting enzyme-like inhibitory protein-long confers resistance to CD95-induced apoptosis in hematopoietic cancer cell lines. J Immunol. 2002;168:2544-53
42. Lwin T, Hazlehurst LA, Dessureault S, Lai R, Bai WL, Sotomayor E. et al. Cell adhesion induces p27Kip1-associated cell-cycle arrest through down-regulation of the SCFSkp2 ubiquitin ligase pathway in mantle-cell and other non-Hodgkin B-cell lymphomas. Blood. 2007;110:1631-8
43. Meads MB, Hazlehurst LA, Dalton WS. The bone marrow microenvironment as a tumor sanctuary and contributor to drug resistance. Clin Cancer Res. 2008;14:2519-26
44. Shintani Y, Hollingsworth MA, Wheelock MJ, Johnson KR. Collagen I promotes metastasis in pancreatic cancer by activating c-Jun NH(2)-terminal kinase 1 and up-regulating N-cadherin expression. Cancer Res. 2006;66:11745-53
45. Gore J, Korc M. Pancreatic Cancer Stroma: Friend or Foe? Cancer Cell. 2014;25:711-2
46. Ozdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng XF, Wu CC, Simpson TR. et al. Depletion of Carcinoma-Associated Fibroblasts and Fibrosis Induces Immunosuppression and Accelerates Pancreas Cancer with Reduced Survival. Cancer Cell. 2014;25:719-34
47. Cramer GM, Jones DP, El-Hamidi H, Celli JP. ECM Composition and Rheology Regulate Growth, Motility, and Response to Photodynamic Therapy in 3D Models of Pancreatic Ductal Adenocarcinoma. Mol Cancer Res. 2017;15:15-25
48. Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A. et al. Tensional homeostasis and the malignant phenotype. Cancer Cell. 2005;8:241-54
49. Liang Y, Jeong J, DeVolder RJ, Cha C, Wang F, Tong YW. et al. A cell-instructive hydrogel to regulate malignancy of 3D tumor spheroids with matrix rigidity. Biomaterials. 2011;32:9308-15
50. Erkan M, Hausmann S, Michalski CW, Fingerle AA, Dobritz M, Kleeff J. et al. The role of stroma in pancreatic cancer: diagnostic and therapeutic implications. Nat Rev Gastro Hepat. 2012;9:454-67
51. Whatcott CJ, Diep CH, Jiang P, Watanabe A, LoBello J, Sima C. et al. Desmoplasia in Primary Tumors and Metastatic Lesions of Pancreatic Cancer. Clin Cancer Res. 2015;21:3561-8
52. Liu C, Liu Y, Xie HG, Zhao S, Xu XX, Fan LX. et al. Role of three-dimensional matrix stiffness in regulating the chemoresistance of hepatocellular carcinoma cells. Biotechnol Appl Bioc. 2015;62:556-62
53. Rice AJ, Cortes E, Lachowski D, Cheung BCH, Karim SA, Morton JP. et al. Matrix stiffness induces epithelial-mesenchymal transition and promotes chemoresistance in pancreatic cancer cells. Oncogenesis. 2017;6:e352
54. Yang Q, Huang J, Wu Q, Cai Y, Zhu L, Lu X. et al. Acquisition of epithelial-mesenchymal transition is associated with Skp2 expression in paclitaxel-resistant breast cancer cells. Brit J Cancer. 2014;110:1958-67
55. Loessner D, Stok KS, Lutolf MP, Hutmacher DW, Clements JA, Rizzi SC. Bioengineered 3D platform to explore cell-ECM interactions and drug resistance of epithelial ovarian cancer cells. Biomaterials. 2010;31:8494-506
56. Vaupel P, Mayer A. Hypoxia in cancer: significance and impact on clinical outcome. Cancer Metast Rev. 2007;26:225-39
57. Brown JM, William WR. Exploiting tumour hypoxia in cancer treatment. Nat Rev Cancer. 2004;4:437-47
58. Rice GC, Hoy C, Schimke RT. Transient Hypoxia Enhances the Frequency of Dihydrofolate-Reductase Gene Amplification in Chinese-Hamster Ovary Cells. P Natl Acad Sci USA. 1986;83:5978-82
59. Rice GC, Ling V, Schimke RT. Frequencies of independent and simultaneous selection of Chinese hamster cells for methotrexate and doxorubicin (adriamycin) resistance. Proc Natl Acad Sci USA. 1987;84:9261-4
60. Comerford KM, Wallace TJ, Karhausen J, Louis NA, Montalto MC, Colgan SP. Hypoxia-inducible factor-1-dependent regulation of the multidrug resistance (MDR1) gene. Cancer Res. 2002;62:3387-94
61. Gottesman MM. Mechanisms of cancer drug resistance. Annu Rev Med. 2002;53:615-27
62. Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: Role of ATP-dependent transporters. Nat Rev Cancer. 2002;2:48-58
63. Rohwer N, Cramer T. Hypoxia-mediated drug resistance: Novel insights on the functional interaction of HIFs and cell death pathways. Drug Resist Update. 2011;14:191-201
64. Tredan O, Galmarini CM, Patel K, Tannock IF. Drug resistance and the solid tumor microenvironment. J Natl Cancer I. 2007;99:1441-54
65. Mitchison TJ. The proliferation rate paradox in antimitotic chemotherapy. Mol Biol Cell. 2012;23:1-6
66. Kovacic P, Osuna JA Jr. Mechanisms of anti-cancer agents: emphasis on oxidative stress and electron transfer. Curr Pharm Des. 2000;6:277-309
67. Kennedy KA. Hypoxic Cells as Specific Drug Targets for Chemotherapy. Anti-Cancer Drug Des. 1987;2:181-94
68. Warburg O. On the origin of cancer cells. Science. 1956;123:309-14
69. Tannock IF, Rotin D. Acid Ph in Tumors and Its Potential for Therapeutic Exploitation. Cancer Res. 1989;49:4373-84
70. Helmlinger G, Yuan F, Dellian M, Jain RK. Interstitial pH and pO2 gradients in solid tumors in vivo: high-resolution measurements reveal a lack of correlation. Nat Med. 1997;3:177-82
71. Gerweck LE, Vijayappa S, Kozin S. Tumor pH controls the in vivo efficacy of weak acid and base chemotherapeutics. Mol Cancer Ther. 2006;5:1275-9
72. Raghunand N, Gillies RJ. pH and drug resistance in tumors. Drug Resist Update. 2000;3:39-47
73. Goda K, Balkay L, Marian T, Tron L, Aszalos A, Szabo G. Intracellular pH does not affect drug extrusion by P-glycoprotein. J Photoch Photobio B. 1996;34:177-82
74. Wachsberger PR, Landry J, Storck C, Davis K, OHara MD, Owen CS. et al. Mammalian cells adapted to growth at pH 6 center dot 7 have elevated HSP27 levels and are resistant to cisplatin. Int J Hyperther. 1997;13:251-5
75. Rafii A, Mirshahi P, Poupot M, Faussat AM, Simon A, Ducros E. et al. Oncologic Trogocytosis of an Original Stromal Cells Induces Chemoresistance of Ovarian Tumours. Plos One. 2008;3:e3894
76. Jones VS, Huang RY, Chen LP, Chen ZS, Fu LW, Huang RP. Cytokines in cancer drug resistance: Cues to new therapeutic strategies. Bba-Rev Cancer. 2016;1865:255-65
77. Kijima T, Maulik G, Ma PC, Madhiwala P, Schaefer E, Salgia R. Fibronectin enhances viability and alters cytoskeletal functions (with effects on the phosphatidylinositol 3-kinase pathway) in small cell lung cancer. J Cell Mol Med. 2003;7:157-64
78. Lee JR, J.-L. Development of an in vitro cell-sheet cancer model for chemotherapeutic screening. Theranostics. 2018;8:3964-73
79. Pasquier J, Gosset M, Geyl C, Hoarau-Vechot J, Chevrot A, Pocard M. et al. CCL2/CCL5 secreted by the stroma induce IL-6/PYK2 dependent chemoresistance in ovarian cancer. Mol Cancer. 2018;17:47
80. Tao LL, Huang GC, Wang R, Pan Y, He ZY, Chu XY. et al. Cancer-associated fibroblasts treated with cisplatin facilitates chemoresistance of lung adenocarcinoma through IL-11/IL-11R/STAT3 signaling pathway. Sci Rep-Uk. 2016;6:38408
81. Lotti F, Jarrar AM, Pai RK, Hitomi M, Lathia J, Mace A. et al. Chemotherapy activates cancer-associated fibroblasts to maintain colorectal cancer-initiating cells by IL-17A. J Exp Med. 2013;210:2851-72
82. Li ZQ, Chan KY, Qi YF, Lu LL, Ning F, Wu ML. et al. Participation of CCL1 in Snail-Positive Fibroblasts in Colorectal Cancer Contribute to 5-Fluorouracil/Paclitaxel Chemoresistance. Cancer Res Treat. 2018;50:894-907
83. Li JM, Guan J, Long XP, Wang Y, Xiang XD. mir-1-mediated paracrine effect of cancer-associated fibroblasts on lung cancer cell proliferation and chemoresistance. Oncol Rep. 2016;35:3523-31
84. Wang WM, Kryczek I, Dostal L, Lin H, Tan LJ, Zhao LL. et al. Effector T Cells Abrogate Stroma-Mediated Chemoresistance in Ovarian Cancer. Cell. 2016;165:1092-105
85. Santos JC, Lima ND, Sarian LO, Matheu A, Ribeiro ML, Derchain SFM. Exosome-mediated breast cancer chemoresistance via miR-155 transfer. Sci Rep-Uk. 2018;8:829
86. Chen WX, Liu XM, Lv MM, Chen L, Zhao JH, Zhong SL. et al. Exosomes from Drug-Resistant Breast Cancer Cells Transmit Chemoresistance by a Horizontal Transfer of MicroRNAs. Plos One. 2014;9:e95240
87. Richards KE, Zeleniak AE, Fishel ML, Wu J, Littlepage LE, Hill R. Cancer-associated fibroblast exosomes regulate survival and proliferation of pancreatic cancer cells. Oncogene. 2017;36:1770-8
88. Patel GK, Khan MA, Bhardwaj A, Srivastava SK, Zubair H, Patton MC. et al. Exosomes confer chemoresistance to pancreatic cancer cells by promoting ROS detoxification and miR-155-mediated suppression of key gemcitabine-metabolising enzyme, DCK. Brit J Cancer. 2017;116:609-19
89. Ruffell B, Coussens LM. Macrophages and Therapeutic Resistance in Cancer. Cancer Cell. 2015;27:462-72
90. Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. 2010;11:889-96
91. Ong SM, Tan YC, Beretta O, Jiang DS, Yeap WH, Tai JJY. et al. Macrophages in human colorectal cancer are pro-inflammatory and prime T cells towards an anti-tumour type-1 inflammatory response. Eur J Immunol. 2012;42:89-100
92. Shree T, Olson OC, Elie BT, Kester JC, Garfall AL, Simpson K. et al. Macrophages and cathepsin proteases blunt chemotherapeutic response in breast cancer. Gene Dev. 2011;25:2465-79
93. Mitchem JB, Brennan DJ, Knolhoff BL, Belt BA, Zhu Y, Sanford DE. et al. Targeting Tumor-Infiltrating Macrophages Decreases Tumor-Initiating Cells, Relieves Immunosuppression, and Improves Chemotherapeutic Responses. Cancer Res. 2013;73:1128-41
94. Jinushi M, Chiba S, Yoshiyama H, Masutomi K, Kinoshita I, Dosaka-Akita H. et al. Tumor-associated macrophages regulate tumorigenicity and anticancer drug responses of cancer stem/initiating cells. P Natl Acad Sci USA. 2011;108:12425-30
95. Yu H, Lee H, Herrmann A, Buettner R, Jove R. Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat Rev Cancer. 2014;14:736-46
96. De Palma M, Lewis CE. Macrophage Regulation of Tumor Responses to Anticancer Therapies. Cancer Cell. 2013;23:277-86
97. Kodumudi KN, Woan K, Gilvary DL, Sahakian E, Wei S, Djeu JY. A Novel Chemoimmunomodulating Property of Docetaxel: Suppression of Myeloid-Derived Suppressor Cells in Tumor Bearers. Clin Cancer Res. 2010;16:4583-94
98. Tredan O, Galmarini CM, Patel K, Tannock IF. Drug resistance and the solid tumor microenvironment. J Natl Cancer Inst. 2007;99:1441-54
99. Mitsiades CS, Mitsiades NS, Munshi NC, Richardson PG, Anderson KC. The role of the bone microenvironment in the pathophysiology and therapeutic management of multiple myeloma: Interplay of growth factors, their receptors and stromal interactions. Eur J Cancer. 2006;42:1564-73
100. Vincent T, Mechti N. Extracellular matrix in bone marrow can mediate drug resistance in myeloma. Leukemia Lymphoma. 2005;46:803-11
101. Sloan EK, Pouliot N, Stanley KL, Chia J, Moseley JM, Hards DK. et al. Tumor-specific expression of alpha v beta 3 integrin promotes spontaneous metastasis of breast cancer to bone. Breast Cancer Res. 2006;8:R20
102. Chrenek MA, Wong P, Weaver VM. Tumour-stromal interactions - Integrins and cell adhesions as modulators of mammary cell survival and transformation. Breast Cancer Res. 2001;3:224-9
103. Damiano JS, Dalton WS. Integrin-mediated drug resistance in multiple myeloma. Leukemia Lymphoma. 2000;38:71-81
104. Folkman J, Moscona A. Role of Cell-Shape in Growth-Control. Nature. 1978;273:345-9
105. Keller GM. In vitro differentiation of embryonic stem cells. Curr Opin Cell Biol. 1995;7:862-9
106. Breslin S, O'Driscoll L. Three-dimensional cell culture: the missing link in drug discovery. Drug Discov Today. 2013;18:240-9
107. Mohr JC, de Pablo JJ, Palecek SP. 3-D microwell culture of human embryonic stem cells. Biomaterials. 2006;27:6032-42
108. Ivascu A, Kubbies M. Rapid generation of single-tumor spheroids for high-throughput cell function and toxicity analysis. Journal of Biomolecular Screening. 2006;11:922-32
109. Wang Y, Kim MH, Tabaei SR, Park JH, Na K, Chung S. et al. Spheroid Formation of Hepatocarcinoma Cells in Microwells: Experiments and Monte Carlo Simulations. PLoS One. 2016;11:e0161915
110. Barrila J, Radtke AL, Crabbe A, Sarker SF, Herbst-Kralovetz MM, Ott CM. et al. Organotypic 3D cell culture models: using the rotating wall vessel to study host-pathogen interactions. Nat Rev Microbiol. 2010;8:791-801
111. Hammond TG, Hammond JM. Optimized suspension culture: the rotating-wall vessel. Am J Physiol Renal Physiol. 2001;281:F12-25
112. Unsworth BR, Lelkes PI. Growing tissues in microgravity. Nat Med. 1998;4:901-7
113. Perche F, Torchilin VP. Cancer cell spheroids as a model to evaluate chemotherapy protocols. Cancer Biol Ther. 2012;13:1205-13
114. Chitcholtan K, Asselin E, Parent S, Sykes PH, Evans JJ. Differences in growth properties of endometrial cancer in three dimensional (3D) culture and 2D cell monolayer. Exp Cell Res. 2013;319:75-87
115. Chitcholtan K, Sykes PH, Evans JJ. The resistance of intracellular mediators to doxorubicin and cisplatin are distinct in 3D and 2D endometrial cancer. J Transl Med. 2012;10:38
116. Lovitt CJ, Shelper TB, Avery VM. Advanced cell culture techniques for cancer drug discovery. Biology (Basel). 2014;3:345-67
117. Gerweck LE, Seetharaman K. Cellular pH gradient in tumor versus normal tissue: Potential exploitation for the treatment of cancer. Cancer Res. 1996;56:1194-8
118. Gerweck LE. Tumor pH: Implications for treatment and novel drug design. Semin Radiat Oncol. 1998;8:176-82
119. Mikkelsen RB, Asher C, Hicks T. Extracellular pH, transmembrane distribution and cytotoxicity of chlorambucil. Biochem Pharmacol. 1985;34:2531-4
120. Aljitawi OS, Li D, Xiao Y, Zhang D, Ramachandran K, Stehno-Bittel L. et al. A novel three-dimensional stromal-based model for in vitro chemotherapy sensitivity testing of leukemia cells. Leuk Lymphoma. 2014;55:378-91
121. Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006;3:187-97
122. Liu L, Ning X, Sun L, Zhang H, Shi Y, Guo C. et al. Hypoxia-inducible factor-1 alpha contributes to hypoxia-induced chemoresistance in gastric cancer. Cancer Sci. 2008;99:121-8
123. Wilson WR, Hay MP. Targeting hypoxia in cancer therapy. Nat Rev Cancer. 2011;11:393-410
124. Chambers KF, Mosaad EMO, Russell PJ, Clements JA, Doran MR. 3D Cultures of Prostate Cancer Cells Cultured in a Novel High-Throughput Culture Platform Are More Resistant to Chemotherapeutics Compared to Cells Cultured in Monolayer. Plos One. 2014;9:e111029
125. Imamura Y, Mukohara T, Shimono Y, Funakoshi Y, Chayahara N, Toyoda M. et al. Comparison of 2D-and 3D-culture models as drug-testing platforms in breast cancer. Oncol Rep. 2015;33:1837-43
126. Longati P, Jia X, Eimer J, Wagman A, Witt MR, Rehnmark S. et al. 3D pancreatic carcinoma spheroids induce a matrix-rich, chemoresistant phenotype offering a better model for drug testing. BMC Cancer. 2013;13:95
127. Gong X, Lin C, Cheng J, Su JS, Zhao H, Liu TL. et al. Generation of Multicellular Tumor Spheroids with Microwell-Based Agarose Scaffolds for Drug Testing. Plos One. 2015;10:e0130348
128. Albanese A, Lam AK, Sykes EA, Rocheleau JV, Chan WCW. Tumour-on-a-chip provides an optical window into nanoparticle tissue transport. Nat Commun. 2013;4:2718
129. Whitesides GM. The origins and the future of microfluidics. Nature. 2006;442:368-73
130. Hung PJ, Lee PJ, Sabounchi P, Lin R, Lee LP. Continuous perfusion microfluidic cell culture array for high-throughput cell-based assays. Biotechnol Bioeng. 2005;89:1-8
131. Wu MH, Huang SB, Lee GB. Microfluidic cell culture systems for drug research. Lab Chip. 2010;10:939-56
132. Jeon NL, Dertinger SKW, Chiu DT, Choi IS, Stroock AD, Whitesides GM. Generation of solution and surface gradients using microfluidic systems. Langmuir. 2000;16:8311-6
133. Kim S, Kim HJ, Jeon NL. Biological applications of microfluidic gradient devices. Integr Biol. 2010;2:584-603
134. Kim C, Bang JH, Kim YE, Lee SH, Kang JY. On-chip anticancer drug test of regular tumor spheroids formed in microwells by a distributive microchannel network. Lab Chip. 2012;12:4135-42
135. Han J, Jun Y, Kim SH, Hoang HH, Jung Y, Kim S. et al. Rapid emergence and mechanisms of resistance by U87 glioblastoma cells to doxorubicin in an in vitro tumor microfluidic ecology. Proc Natl Acad Sci U S A. 2016;113:14283-8
136. Huh D, Hamilton GA, Ingber DE. From 3D cell culture to organs-on-chips. Trends Cell Biol. 2011;21:745-54
137. Moazzam F, DeLano FA, Zweifach BW, Schmid-Schonbein GW. The leukocyte response to fluid stress. Proc Natl Acad Sci USA. 1997;94:5338-43
138. Civelek M, Ainslie K, Garanich JS, Tarbell JM. Smooth muscle cells contract in response to fluid flow via a Ca2+-independent signaling mechanism. J Appl Physiol. 2002;93:1907-17
139. Li YS, Haga JH, Chien S. Molecular basis of the effects of shear stress on vascular endothelial cells. J Biomech. 2005;38:1949-71
140. Mitchell MJ, King MR. Computational and experimental models of cancer cell response to fluid shear stress. Front Oncol. 2013;3:44
141. Michor F, Liphardt J, Ferrari M, Widom J. What does physics have to do with cancer? Nat Rev Cancer. 2011;11:657-70
142. Swartz MA, Lund AW. Lymphatic and interstitial flow in the tumour microenvironment: linking mechanobiology with immunity. Nat Rev Cancer. 2012;12:210-9
143. Munson JM, Shieh AC. Interstitial fluid flow in cancer: implications for disease progression and treatment. Cancer Manag Res. 2014;6:317-28
144. Schmid-Schonbein GW. Microlymphatics and lymph flow. Physiol Rev. 1990;70:987-1028
145. Pedersen JA, Boschetti F, Swartz MA. Effects of extracellular fiber architecture on cell membrane shear stress in a 3D fibrous matrix. J Biomech. 2007;40:1484-92
146. Tarbell JM, Shi ZD. Effect of the glycocalyx layer on transmission of interstitial flow shear stress to embedded cells. Biomech Model Mechanobiol. 2013;12:111-21
147. Chary SR, Jain RK. Direct measurement of interstitial convection and diffusion of albumin in normal and neoplastic tissues by fluorescence photobleaching. Proc Natl Acad Sci USA. 1989;86:5385-9
148. Boucher Y, Jain RK. Microvascular Pressure Is the Principal Driving Force for Interstitial Hypertension in Solid Tumors - Implications for Vascular Collapse. Faseb J. 1993;7:5110-4
149. Boucher Y, Leunig M, Jain RK. Tumor angiogenesis and interstitial hypertension. Cancer Res. 1996;56:4264-6
150. Boucher Y, Baxter LT, Jain RK. Interstitial pressure gradients in tissue-isolated and subcutaneous tumors: implications for therapy. Cancer Res. 1990;50:4478-84
151. Netti PA, Baxter LT, Boucher Y, Skalak R, Jain RK. Time-Dependent Behavior of Interstitial Fluid Pressure in Solid Tumors - Implications for Drug-Delivery. Cancer Res. 1995;55:5451-8
152. Lunt SJ, Fyles A, Hill RP, Milosevic M. Interstitial fluid pressure in tumors: therapeutic barrier and biomarker of angiogenesis. Future Oncol. 2008;4:793-802
153. Shields JD, Fleury ME, Yong C, Tomei AA, Randolph GJ, Swartz MA. Autologous chemotaxis as a mechanism of tumor cell homing to lymphatics via interstitial flow and autocrine CCR7 signaling. Cancer Cell. 2007;11:526-38
154. Polacheck WJ, Charest JL, Kamm RD. Interstitial flow influences direction of tumor cell migration through competing mechanisms. P Natl Acad Sci USA. 2011;108:11115-20
155. Pandya HJ, Dhingra K, Prabhakar D, Chandrasekar V, Natarajan SK, Vasan AS. et al. A microfluidic platform for drug screening in a 3D cancer microenvironment. Biosens Bioelectron. 2017;94:632-42
156. Ip CK, Li SS, Tang MY, Sy SK, Ren Y, Shum HC. et al. Stemness and chemoresistance in epithelial ovarian carcinoma cells under shear stress. Sci Rep. 2016;6:26788
157. Mitchell MJ, King MR. Fluid Shear Stress Sensitizes Cancer Cells to Receptor-Mediated Apoptosis via Trimeric Death Receptors. New J Phys. 2013;15:015008
158. Sporn MB, Roberts AB. Autocrine growth factors and cancer. Nature. 1985;313:745-7
159. Scheel C, Eaton EN, Li SH, Chaffer CL, Reinhardt F, Kah KJ. et al. Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell. 2011;145:926-40
160. Lee HJ, Diaz MF, Price KM, Ozuna JA, Zhang S, Sevick-Muraca EM. et al. Fluid shear stress activates YAP1 to promote cancer cell motility. Nat Commun. 2017;8:14122
161. Santoro M, Lamhamedi-Cherradi SE, Menegaz BA, Ludwig JA, Mikos AG. Flow perfusion effects on three-dimensional culture and drug sensitivity of Ewing sarcoma. Proc Natl Acad Sci USA. 2015;112:10304-9
162. Zhao YJ, Adjei AA. Targeting Angiogenesis in Cancer Therapy: Moving Beyond Vascular Endothelial Growth Factor. Oncologist. 2015;20:660-73
163. Sobrino A, Phan DTT, Datta R, Wang XL, Hachey SJ, Romero-Lopez M. et al. 3D microtumors in vitro supported by perfused vascular networks. Sci Rep-Uk. 2016;6:31589
164. Phan DTT, Wang XL, Craver BM, Sobrino A, Zhao D, Chen JC. et al. A vascularized and perfused organ-on-a-chip platform for large-scale drug screening applications. Lab on a Chip. 2017;17:511-20
165. Hassell BA, Goyal G, Lee E, Sontheimer-Phelps A, Levy O, Chen CS. et al. Human Organ Chip Models Recapitulate Orthotopic Lung Cancer Growth, Therapeutic Responses, and Tumor Dormancy In Vitro. Cell Rep. 2017;21:508-16
166. Carloni V, Luong TV, Rombouts K. Hepatic stellate cells and extracellular matrix in hepatocellular carcinoma: more complicated than ever. Liver Int. 2014;34:834-43
167. Nguyen-Ngoc KV, Cheung KJ, Brenot A, Shamir ER, Gray RS, Hines WC. et al. ECM microenvironment regulates collective migration and local dissemination in normal and malignant mammary epithelium. Proc Natl Acad Sci USA. 2012;109:E2595-604
168. Goetz JG, Minguet S, Navarro-Lerida I, Lazcano JJ, Samaniego R, Calvo E. et al. Biomechanical remodeling of the microenvironment by stromal caveolin-1 favors tumor invasion and metastasis. Cell. 2011;146:148-63
169. Huh D, Torisawa YS, Hamilton GA, Kim HJ, Ingber DE. Microengineered physiological biomimicry: organs-on-chips. Lab Chip. 2012;12:2156-64
170. Jang KJ, Mehr AP, Hamilton GA, McPartlin LA, Chung S, Suh KY. et al. Human kidney proximal tubule-on-a-chip for drug transport and nephrotoxicity assessment. Integr Biol. 2013;5:1119-29
171. Kim HN, Jiao A, Hwang NS, Kim MS, Kang DH, Kim DH. et al. Nanotopography-guided tissue engineering and regenerative medicine. Adv Drug Deliv Rev. 2013;65:536-58
172. Puppi D, Zhang X, Yang L, Chiellini F, Sun X, Chiellini E. Nano/microfibrous polymeric constructs loaded with bioactive agents and designed for tissue engineering applications: a review. J Biomed Mater Res B Appl Biomater. 2014;102:1562-79
173. Kim HN, Hong Y, Kim MS, Kim SM, Suh KY. Effect of orientation and density of nanotopography in dermal wound healing. Biomaterials. 2012;33:8782-92
174. Kim DH, Provenzano PP, Smith CL, Levchenko A. Matrix nanotopography as a regulator of cell function. J Cell Biol. 2012;197:351-60
175. Biggs MJP, Richards RG, Dalby MJ. Nanotopographical modification: a regulator of cellular function through focal adhesions. Nanomed-Nanotechnol. 2010;6:619-33
176. Beliveau A, Thomas G, Gong J, Wen Q, Jain A. Aligned Nanotopography Promotes a Migratory State in Glioblastoma Multiforme Tumor Cells. Sci Rep. 2016;6:26143
177. Stevens MM, George JH. Exploring and engineering the cell surface interface. Science. 2005;310:1135-8
178. Smith CL, Kilic O, Schiapparelli P, Guerrero-Cazares H, Kim DH, Sedora-Roman NI. et al. Migration Phenotype of Brain-Cancer Cells Predicts Patient Outcomes. Cell Rep. 2016;15:2616-24
179. Shin JY, Kim HN, Bhang SH, Yoon JK, Suh KY, Jeon NL. et al. Topography-Guided Control of Local Migratory Behaviors and Protein Expression of Cancer Cells. Adv Healthc Mater. 2017;6:1700155
180. Chaudhuri PK, Pan CQ, Low BC, Lim CT. Topography induces differential sensitivity on cancer cell proliferation via Rho-ROCK-Myosin contractility. Sci Rep-Uk. 2016;6:19672
181. Yang L, Liu H, Lin Y. Biomaterial nanotopography-mediated cell responses: experiment and modeling. Int J Smart Nano Mater. 2014;5:227-56
182. Bettinger CJ, Langer R, Borenstein JT. Engineering Substrate Topography at the Micro- and Nanoscale to Control Cell Function. Angew Chem Int Edit. 2009;48:5406-15
183. Kim DH, Lee H, Lee YK, Nam JM, Levchenko A. Biomimetic Nanopatterns as Enabling Tools for Analysis and Control of Live Cells. Adv Mater. 2010;22:4551-66
184. Kim HN, Kang DH, Kim MS, Jiao A, Kim DH, Suh KY. Patterning Methods for Polymers in Cell and Tissue Engineering. Annals of Biomedical Engineering. 2012;40:1339-55
185. Abdellatef SA, Tange R, Sato T, Ohi A, Nabatame T, Taniguchi A. Nanostructures Control the Hepatocellular Responses to a Cytotoxic Agent "Cisplatin". Biomed Res Int. 2015;2015:925319
186. Tan LH, Sykes PH, Alkaisi MM, Evans JJ. Cell-like features imprinted in the physical nano- and micro-topography of the environment modify the responses to anti-cancer drugs of endometrial cancer cells. Biofabrication. 2017;9:015017
187. Kim J, Kim YJ, Bae WG, Jang KJ, Lim KT, Choung PH. et al. Topographical extracellular matrix cues on anticancer drug-induced cytotoxicity in stem cells. J Biomed Mater Res B Appl Biomater. 2015;103:1320-7
188. Fong ELS, Lamhamedi-Cherradi SE, Burdett E, Ramamoorthy V, Lazar AJ, Kasper FK. et al. Modeling Ewing sarcoma tumors in vitro with 3D scaffolds. P Natl Acad Sci USA. 2013;110:6500-5
189. Ho WJ, Pham EA, Kim JW, Ng CW, Kim JH, Kamei DT. et al. Incorporation of multicellular spheroids into 3-D polymeric scaffolds provides an improved tumor model for screening anticancer drugs. Cancer Sci. 2010;101:2637-43
190. Chin LK, Xia YT, Discher DE, Janmey PA. Mechanotransduction in cancer. Curr Opin Chem Eng. 2016;11:77-84
191. Hait WN. Anticancer drug development: the grand challenges. Nat Rev Drug Discov. 2010;9:253-4
192. Kim MP, Evans DB, Wang HM, Abbruzzese JL, Fleming JB, Gallick GE. Generation of orthotopic and heterotopic human pancreatic cancer xenografts in immunodeficient mice. Nat Protoc. 2009;4:1670-80
193. Day CP, Merlino G, Van Dyke T. Preclinical Mouse Cancer Models: A Maze of Opportunities and Challenges. Cell. 2015;163:39-53
194. Tentler JJ, Tan AC, Weekes CD, Jimeno A, Leong S, Pitts TM. et al. Patient-derived tumour xenografts as models for oncology drug development. Nat Rev Clin Oncol. 2012;9:338-50
195. Lai YX, Wei XR, Lin SH, Qin L, Cheng L, Li P. Current status and perspectives of patient-derived xenograft models in cancer research. J Hematol Oncol. 2017;10:106
196. Tung YC, Hsiao AY, Allen SG, Torisawa YS, Ho M, Takayama S. High-throughput 3D spheroid culture and drug testing using a 384 hanging drop array. Analyst. 2011;136:473-8
197. Gao H, Korn JM, Ferretti S, Monahan JE, Wang YZ, Singh M. et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat Med. 2015;21:1318-25
198. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19:1423-37
199. Park J, Bauer S, Pittrof A, Killian MS, Schmuki P, von der Mark K. Synergistic Control of Mesenchymal Stem Cell Differentiation by Nanoscale Surface Geometry and Immobilized Growth Factors on TiO2 Nanotubes. Small. 2012;8:98-107
200. Kim J, Kim HN, Lim KT, Kim Y, Pandey S, Garg P. et al. Synergistic effects of nanotopography and co-culture with endothelial cells on osteogenesis of mesenchymal stem cells. Biomaterials. 2013;34:7257-68
Author contact
![]() Corresponding authors: Dr. Hong Nam Kim, Email: hongnam.kimre.kr; Prof. Jonghoon Choi, Email: nanomedac.kr
Corresponding authors: Dr. Hong Nam Kim, Email: hongnam.kimre.kr; Prof. Jonghoon Choi, Email: nanomedac.kr