Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(17):4824-4836. doi:10.7150/thno.24747 This issue Cite
Research Paper
Neurogliovascular dysfunction in a model of repeated traumatic brain injury
Conner Adams1,2 ![]() , Paolo Bazzigaluppi1,2,3, Tina L. Beckett2, Jossana Bishay1,2, Iliya Weisspapir2, Adrienne Dorr2, James R. Mester1,2, Joe Steinman1,4, Lydiane Hirschler5,6,7, Jan M. Warnking5,7, Emmanuel L. Barbier5,7, JoAnne McLaurin2,8, John G. Sled1,4, Bojana Stefanovic1,2
, Paolo Bazzigaluppi1,2,3, Tina L. Beckett2, Jossana Bishay1,2, Iliya Weisspapir2, Adrienne Dorr2, James R. Mester1,2, Joe Steinman1,4, Lydiane Hirschler5,6,7, Jan M. Warnking5,7, Emmanuel L. Barbier5,7, JoAnne McLaurin2,8, John G. Sled1,4, Bojana Stefanovic1,2
1. University of Toronto, Department of Medical Biophysics, Toronto, Ontario, Canada
2. Sunnybrook Research Institute, Toronto, Ontario, Canada
3. Fundamental Neurobiology, Krembil Research Institute, University Health Network, M5T 2S8 Toronto, Ontario, Canada
4. Mouse Imaging Centre, The Hospital for Sick Children, Toronto, Ontario, Canada
5. Université Grenoble Alpes, Grenoble Institut des Neurosciences, Grenoble, France
6. Bruker Biospin MRI, Ettlingen, Germany
7. Inserm, U1216, Grenoble, France
8. University of Toronto, Department of Laboratory Medicine and Pathobiology, Toronto, Ontario Canada
Received 2018-1-4; Accepted 2018-8-2; Published 2018-9-9
Abstract

Traumatic brain injury (TBI) research has focused on moderate to severe injuries as their outcomes are significantly worse than those of a mild TBI (mTBI). However, recent epidemiological evidence has indicated that a series of even mild TBIs greatly increases the risk of neurodegenerative and psychiatric disorders. Neuropathological studies of repeated TBI have identified changes in neuronal ionic concentrations, axonal injury, and cytoskeletal damage as important determinants of later life neurological and mood compromise; yet, there is a paucity of data on the contribution of neurogliovascular dysfunction to the progression of repeated TBI and alterations of brain function in the intervening period.
Methods: Here, we established a mouse model of repeated TBI induced via three electromagnetically actuated impacts delivered to the intact skull at three-day intervals and determined the long-term deficits in neurogliovascular functioning in Thy1-ChR2 mice. Two weeks post the third impact, cerebral blood flow and cerebrovascular reactivity were measured with arterial spin labelling magnetic resonance imaging. Neuronal function was investigated through bilateral intracranial electrophysiological responses to optogenetic photostimulation. Vascular density of the site of impacts was measured with in vivo two photon fluorescence microscopy. Pathological analysis of neuronal survival and astrogliosis was performed via NeuN and GFAP immunofluorescence.
Results: Cerebral blood flow and cerebrovascular reactivity were decreased by 50±16% and 70±20%, respectively, in the TBI cohort relative to sham-treated animals. Concomitantly, electrophysiological recordings revealed a 97±1% attenuation in peri-contusional neuronal reactivity relative to sham. Peri-contusional vascular volume was increased by 33±2% relative to sham-treated mice. Pathological analysis of the peri-contusional cortex demonstrated astrogliosis, but no changes in neuronal survival.
Conclusion: This work provides the first in-situ characterization of the long-term deficits of the neurogliovascular unit following repeated TBI. The findings will help guide the development of diagnostic markers as well as therapeutics targeting neurogliovascular dysfunction.
Keywords: repeated TBI, neurovascular, optogenetics, closed head injury
Introduction
Traumatic brain injury (TBI) is a widespread condition, with a global incidence greater than 6 for every 1000 people [1]. The severe injuries are often fatal and even moderate ones have permanent effects on cognition, memory, and mood disorders. In contrast, a mild TBI (mTBI) may simply cause brief confusion or cognitive impairment, but its prevalence is much higher than that of moderate and severe injuries with some estimates suggesting that up to 90% of TBI cases involve mild injuries [1]. While immediate sequelae of mTBI are often transient, there is mounting evidence of an association between mTBI and late-onset neurodegenerative and mood disorders [2-4]. This risk is amplified in the case of repeated head trauma, which may lead to chronic traumatic encephalopathy—a progressive neurodegenerative disease involving cognitive decline—as well as neuropsychiatric conditions including depression and suicidal tendencies [5,6]. Although the determinants of long-term effects of repeated mTBI are not well understood, the epidemiological evidence of long term dysfunction following multiple mTBI has prompted many organizations to implement “three-strike” rules mandating that those who suffer a third mTBI be removed from the environment in which the injury occurred [7]. Notwithstanding, hitherto, studies of repeated mTBI have focused on acute pathology. To address this gap, we set out to determine changes in brain function elicited by a series of mild TBI, which may underlie the increased risk of later life neurodegenerative and mood disturbances.
Assessments of the neurogliovascular unit following repeated mTBI have been limited to histological investigations, with a paucity of data on in situ brain function. Functional imaging has, however, been key in our understanding of neuropathological progression and disease monitoring in stroke, Alzheimer's disease, and moderate to severe TBI [8]. In particular, cerebrovascular reactivity (CVR), the brain's ability to elevate blood flow above baseline, is compromised following moderate to severe TBI [9] and has been identified as critical in the ensuing pathological progression and functional decline [10]. Making inferences about mTBI based on preclinical assessments of CVR following moderate to severe TBI may be difficult due to the great disparity in the degree of functional impairment between moderate to severe and mild TBI, as well as the supralinear effects of the repeated mild insults. These considerations have prompted the development of preclinical models of mild TBI and repeated mild TBI: their histopathological characterization indicated that mild TBI, when repeated, results in compromised blood-brain-barrier, pyramidal neuron death, astrocytic activation, and recruitment of macrophages/ microglia to the site of the injury [11-15]. Furthermore, behavioural assessments of repeated mTBI mice have revealed cognitive and motor deficits greater than those predicted from summation of compromise seen in mice challenged with a single mild TBI [16]. Despite the progress made in characterizing repeated mild TBI using histopathology and behavioural testing, functional assessments of the neurogliovascular unit following repeated mTBI are lacking [17].
In this study, we report on the first in situ functional assessments of the neuronal and cerebrovascular functions in the chronic phase of a series of mild impacts. Using a repeated closed head injuries (CHIs) model comprising one closed skull strike delivered every three days for a total of three impacts, we quantified the resting cerebral blood flow (CBF) and the CVR via pseudo-continuous arterial spin labelling (pCASL) MRI two weeks after the final impact of the repeated impact paradigm. Neuronal network functioning was assessed using electrophysiological recordings of evoked potentials in response to photostimulation of channelrhodopsin (ChR2)-positive cortical pyramidal neurons. Results of in situ assays were then interpreted alongside immunohistochemical measurements of neuronal, glial, and vascular state. These mice exhibited cerebrovascular dysfunction as indicated by reduced CBF and CVR, as well as remodelling of cerebrovasculature observed via in vivo two-photon fluorescence microscopy. Furthermore, the amplitude of evoked neuronal response to optogenetic photostimulation was reduced, despite no significant changes in neuronal density on NeuN, neurofilament, and MAP2 immunofluorescence. Altogether, our results reveal pronounced neurogliovascular impairments two weeks following a series of three mild impacts.
Methods
Animals
Thirty-six 9-week-old transgenic Thy1-ChR2 mice underwent either repeated CHI (n=19, 10 male, 9 female) or sham preparation (n=17, 9 male, 8 female). This age is considered equivalent to puberty/young adulthood in humans [18]. Repeated TBI was induced via three electromagnetically actuated impacts delivered at 3-day intervals to the skull region corresponding to the forelimb region in the primary somatosensory cortex (S1FL). Sham-treated mice underwent the same surgical procedures with no impacts. To assess cerebrovascular dysfunction in the chronic phase of injury, pCASL imaging (n=8) was used to measure resting CBF and CVR to hypercapnic challenge. The brains of a subset of these mice (n=5) were then harvested for pathology. A cohort of mice underwent in vivo two-photon fluorescence microscopy to measure peri-contusional vascularity (n=4). Neuronal function was probed via electrophysiological recordings of evoked responses to optogenetic photostimulation (n=4). The final cohort sizes in each assay are summarized in Table 1.
Cohort sizes after attrition.
| Group | CBF MRI | CVR MRI | IHC total | Overlap between MRI and IHC | Electrophysiology | 2PFM |
|---|---|---|---|---|---|---|
| Repeated CHI (n=19) | 5 | 6 | 5 | 2 | 4 | 4 |
| Sham (n=17) | 5 | 5 | 5 | 4 | 4 | 4 |
2PFM: 2-photon fluorescence microscopy; CBF: cerebral blood flow; CHI: closed head injury; CVR: cerebrovascular reactivity; IHC: immunohistochemistry; MRI: magnetic resonance imaging
Repeated CHI induction
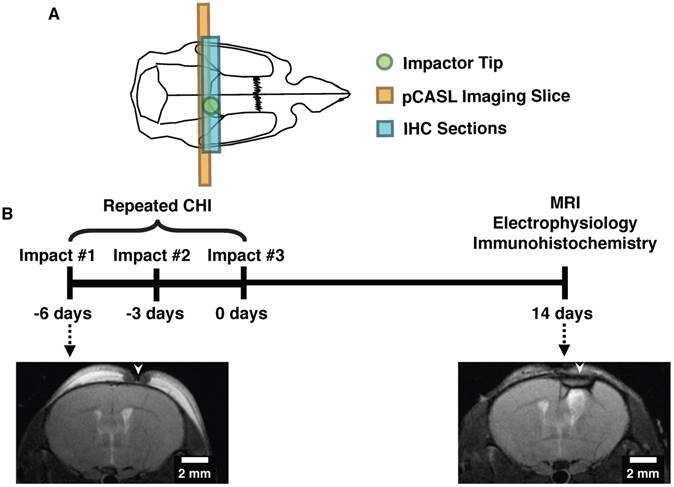
All animal experiments were approved by the Sunnybrook Research Institute (SRI) Animal Care and Use Committee. Transgenic Thy1-ChR2-YFP mice (stock no. 007612, strain B6.Cg-Tg (Thy1-COP4/EYFP)18Gfng/J from Jackson Labs, Bar Harbor, ME; bred in-house) were used in this study (YFP expression in a sample animal is shown in Figure S1). These mice have been bred to express channelrhodopsin-, a light-gated cation channel that is non-selective towards H+, Na+, K+, and CA2+, in cortical pyramidal neurons, enabling optical control of neuronal activation [19,20]. Induction of repeated mTBI or sham preparation was achieved via three individual closed head injuries with a three-day inter-impact interval (Figure 1). For each CHI, mice were anesthetized with isoflurane (5% induction, 2% maintenance), administered 5 mg/kg Xylocaine (Astrazeneca, Mississauga, Canada) and 5 mg/kg Baytril (Rompun, Bayer Inc.) subcutaneously, placed on a feedback-controlled heating pad, and fixed on a stereotactic surgical frame. Under aseptic conditions, a midline excision was then performed to expose the skull surface, and the forelimb region of the primary somatosensory cortex was located stereotactically (1.5 mm mediolateral from midline on the right-hand side, 0.5 mm anterior of bregma) [21]. The impactor was positioned to deliver an impact perpendicular to the skull surface, and a single impact was delivered at 2 m/s, to a depth of 1000 μm below the skull surface, with a dwell time of 200 ms, using a 1.5 mm diameter impactor tip (Leica Impact One stereotaxic actuator, Leica Biosystems, Wetzlar, Germany). These procedures were performed in sham treated mice as well, but the impact was not delivered. The midline incision was then sutured closed, isoflurane discontinued, and the mice recovered under a heating lamp. This surgery was repeated a further two times at 3-day intervals.
Experimental design and timeline for induction of repeated mTBI and readouts. (A) Diagram showing the location of the closed head injuries (green circle), pCASL imaging slice (orange rectangle), and region characterized by immunohistochemistry (blue rectangle) on a mouse skull diagram. (B) Repeated mTBI consisted of three closed head injuries (CHIs) delivered to the skull surface overlying S1FL. Impacts were delivered at 2 m/s, with a dwell time of 200 ms, to a depth of 1 mm, and using a 1.5 mm impactor tip. Each impact was separated by a 3-day inter-impact interval. Two weeks following the final CHI, mice underwent either electrophysiology or MRI followed by immunohistochemistry. Sham-treated mice underwent the same timeline and surgical procedures, but no impacts. T2-weighted images of structural damage at two timepoints of the study are shown on the bottom from two mice with alterations that are representative of each timepoint. The left image was acquired 24 h after a single impact, whereas the right image was taken two weeks after the last impact. White arrows indicate the locations to which impacts were delivered.
Imaging
Two weeks following the third CHI, mice underwent either MRI or electrophysiology. For imaging procedures, mice were anesthetized under isoflurane (5% induction, 1% for imaging), underwent orotracheal intubation with a 22-gauge catheter, and were mechanically ventilated (SAR 830/P, CWE Inc.). Stable physiological state was ensured by monitoring heart rate and pO2 via pulse oximetry (MouseOx Plus, STARR Life Sciences Group), temperature via rectal probe, and end-tidal CO2 via a rodent capnograph (microCapStar End-Tidal CO2 Analyzer, CWE Inc.). Mice were immobilized via an incisor bar and ear bars.
Magnetic resonance imaging
Imaging was performed on a 7 T preclinical horizontal MRI system (Bruker, BioSpec, Ettlingen, Germany). A birdcage body coil was used for excitation and a 20 mm diameter loop coil for reception. Mice were immobilized via an incisor bar and ear bars, and positioned such that the brain region of interest was located at isocenter.
To assess structural damage associated with TBI, thirteen coronal T2-weighted rapid acquisition with refocused echoes (RARE) images were acquired (TR/TE = 2500/33 ms, 62.5 μm nominal in-plane resolution, 0.7 mm slice thickness, and RARE factor = 8) [22]. From these anatomical images, a slice containing no hypointense regions was selected for subsequent pCASL experiments.
pCASL imaging was performed using an unbalanced labelling scheme (4 μT pulses with Gav/Gmax = 10/45 mT/mm, Hanning-shaped pulses of duration 400 μs, 800 μs pulse interval, and with interpulse phase changes to optimize pCASL signal) [23]. Adiabatic labelling pulses were applied through a plane positioned perpendicular to the common carotid artery, 13 mm posterior to the imaging slice. Single slice, single shot, echo planar images (EPI) were then collected with TR/TE = 3500/11 ms, 1 mm coronal slice thickness, and 250 μm nominal in-plane resolution. Quantification of resting perfusion was achieved via multi-post label delay (PLD) pCASL imaging; thirty-two labelled-unlabelled EPI pairs were acquired for each of nine PLDs (0, 50, 150, 250, 350, 500, 750, 1000, and 2000 ms in randomized order). Voxelwise resting CBF and arterial transit time (ATT) were then computed in absolute units by modelling the mean pCASL signal vs. PLD, using an in-house Python implementation (Python Software Foundation, https://www.python.org/) of the single-compartment model [24].
For assessment of CVR to 10% CO2 in the inspired gas mixture, pCASL imaging was performed as above (with PLD = 250 ms, TR = 2500 ms) during four separate one-minute hypercapnic challenges (10% CO2, 69% N2, 21% O2 delivered via programmable gas mixer, CWE inc., Boston, MA, USA) separated by four minutes of medical air breathing (0% CO2, 79% N2, 21% O2). Data were motion-corrected (AFNI, Analysis of Functional Neuroimages, 2dImReg), masked to isolate cortical grey matter, and blurred within the mask (AFNI, 3dBlurInMask, 0.5 mm FWHM). The motion-corrected, blurred data was then fit using a General Linear Model (AFNI, 3dDeconvolve), regressed against subject-specific, within-mask spatially averaged signal time courses of responses to CO2. Data were then masked for significance (excluding voxels with p>0.05), and clusterized using a cluster size of 5 (pre-determined from AFNI's 3dClustSim to give false discovery rate of 0.05).
Electrophysiology
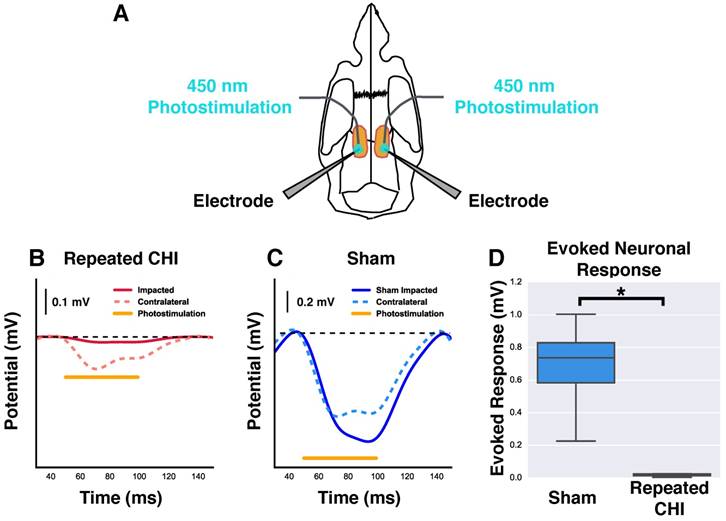
For electrophysiological experiments, bilateral craniotomies (~5 mm diameter) were performed using a high-speed micro-drill (Foredom Electric Co., Bethel Connecticut, USA). One craniotomy was centered around the site at which the impacts had been delivered, while the second was located symmetrically on the contralateral side. In each cranial window, an electrode was placed near the site of the impacts or the contralateral location via micromanipulators. Extracellular potential recordings of neuronal activity were acquired at ~10 kHz, amplified, and recorded using a HEKA EPC-10 (Heka Instruments inc., MA, USA). To measure evoked neuronal responses to optogenetic stimulation, an optical fibre (200 μm diameter, 0.27 numerical aperture) coupled to a 450 nm laser diode (Doric Lenses inc., Quebec, Canada) was positioned over the tip of the electrode in either the impacted, or contralateral cortex. Optical stimulation was delivered at ~30 mW/mm2, using 50 ms pulses at 10 Hz, for a total of 10 s.
Time-course data was bandpass filtered between 1 and 300 Hz to remove baseline drift and noise, and notch filtered (55-65 Hz stopband) to remove 60 Hz electrical noise. The amplitude of evoked neuronal responses was computed as the mean amplitude of change in potential from baseline resulting from photostimulation.
Histopathology
Immediately following imaging, mice were given a high dose of ketamine-xylazine anesthetic (100/10 mg/kg, Vetalar, Boehringer Ingelheim Vetmedica). Peripheral blood was flushed with PBS containing 0.1% heparin, followed by fixation with 4% paraformaldehyde (PFA). Brains were collected, stored overnight in 4% PFA, then cryoprotected with 30% sucrose in PBS until the time of sectioning. Brains were sectioned coronally via sliding freezing microtome (40 µm; SM2010 R Sliding Microtome, Leica Biosystems, Concord, ON). Sections were collected within the region of the CHIs, with the most anterior section at Bregma +1.5 mm and the most posterior section at Bregma -1.7 mm, with an inter-section spacing of 300-500 μm. We mounted 5 slices per slide and stained (with each stain) 1 slide per animal. The slices were stained for GFAP (an astrocytic marker; 1:1000, DAKO #Z0334), NeuN (a neuronal marker; 1:500, EMD Millipore #ABN90) MAP2 (1:250, EMD Millipore #MAB3418), and Neurofilament (1:500, BioLegend # SMI312). GFAP sections were incubated with secondary antibody conjugated to AlexaFluor-647 (1:500, Invitrogen #A31573). Similarly, NeuN sections were incubated with secondary antibody conjugated to AlexaFluor-647 (1:200, EMD Millipore #AP193SA6). Stained sections were then mounted on VistaVision HistoBond slides (VWR, Mississauga, ON, Canada) using polyvinyl-alcohol mounting medium with DABCO (PVA-DABCO, Sigma-Aldrich), and sealed with a coverslip.
All slides were imaged using a Zeiss Apotome 2 Microscope with StereoInvestigator Software (MBF Biosciences) under a 10x objective, with NA = 0.17 (Zeiss, Oberkochen, Germany). In total we analyzed 5 coronal sections per animal, per stain. The region analyzed spanned from ~0.2 mm below cortical surface to ~1.2 mm below cortical surface, corresponding to cortical layers II through VI (given 1.4 mm cortical thickness in this coronal slab). Image analysis was performed using ImageJ following previous work [25]. Briefly, images underwent edge detection, background subtraction, and binarization via ImageJ's MakeBinary algorithm. The binarization of the images sets all pixels below a certain threshold to zero, and all others to one. For each stain, a different threshold was applied and held constant between animals. Cortical regions of interest were then selected near the site of the impact (or sham impact, for sham-treated animals), and the percentage area occupied by NeuN, Neurofilament, MAP2, or GFAP was computed. ROIs were defined within the region of the insets shown so as to comprise the annular region from one to two times the semi-minor and semi-major axes of the elliptical contusion region segmented based on local contrast on NeuN staining, as demonstrated in Figure S2.
In-vivo two-photon fluorescence microscopy
Following standard protocols, 2 weeks following the final impact (or sham surgery), mice were implanted with an ~2-3 mm diameter cranial window centered over the location of impacts. To allow vascular visualization, a 70 kDa Texas Red dextran (Invitrogen, USA) dissolved in PBS (8.33 mg/mL) was administered via the tail vein (25 mg/kg). The cerebrovasculature was imaged at 900 nm on a two-photon fluorescence microscope (FV-1000MPE, Olympus, Japan) using a 25x, 1.05 water immersion lens (Olympus, Japan). The red channel data were segmented and the percent vascular occupancy (intravascular volume normalized by the total cortical volume) was estimated in the imaged volume of each animal, from 50 μm to 150 μm below the cortical surface.
Statistical analyses
To enable sensitive assessment of the focal cerebrovascular damage caused by TBI, a logistic regression classifier was trained to differentiate between healthy and unhealthy tissue voxels. For each animal, we defined a training data set consisting of manually selected voxels representing either healthy or pathological levels of resting CBF or cerebrovascular reactivity to CO2. Voxels classified as having a greater than 65% probability of belonging to the peri-contusional tissue were then clusterized using AFNI's 3dmerge and included in the analysis (See Figure S3 for representative ROIs).
First, we confirmed that all samples were normally distributed (with Shapiro-Wilk normality test) and checked for equal variances of populations being compared (F-test). If both conditions were satisfied, a linear mixed effects model (lme4 [26]) was used for comparison between TBI and sham groups. Alternatively, a Kruskal-Wallis test was used. We examined the effect of TBI on the amplitude of CBF, CVR, and ATT for voxels having been classified as belonging to the affected hemisphere, with the subject as a random effect. Linear mixed effects modelling is particularly well suited for this study as it provides robust maximum likelihood ratios in the presence of many observations for each subject and allows modeling of subject as a random effect. To obtain p-values, a likelihood ratio test between the model with TBI or sham as an effect against the model without TBI as an effect (i.e., the null model) was performed [27]. For immunohistochemical analyses, the optical density for each stain at the site of the impacts and contralaterally was computed as previously described, and a Kruskal-Wallis test was applied. For electrophysiological data, the mean evoked response amplitude was computed for pericontusional cortices and comparisons between repeated CHI mice and sham mice was performed using a Kruskal-Wallis test.
Results
The final cohort sizes along with incidence and type of surgical complications and attrition are summarized in Table 1 and Table 2. Both TBI and sham mice were maintained in a stable physiological state during imaging procedures, as summarized in Table 3. There were no significant differences between groups for any physiological parameters. The average lesion volume in the TBI cohort estimated from NeuN staining was 100 ± 10 μm3.
Surgical complications.
| Group | Ear bleeding (included in study) | Skull Fracture (excluded from study) | Animals sacrificed due to poor surgical recovery |
|---|---|---|---|
| Repeated CHI | 4 | 6 | 4 |
| Sham | 2 | 0 | 2 |
CHI: closed head injury
Physiological monitoring parameters during normocapnic mixture breathing.
| Group | Heart Rate (bpm) | Respiratory Rate (bpm) | SpO2 (%) | Temperature (°C) | End-tidal CO2 (mmHg) |
|---|---|---|---|---|---|
| Repeated CHI (n=3) | 460 ± 60 | 120 ± 1 | 95 ± 2 | 37 ± 1 | 35 ± 7 |
| Sham (n=3) | 470 ± 50 | 121 ± 1 | 96 ± 2 | 37 ± 1 | 35 ± 7 |
BPM: beats per minute; CHI: closed head injury; SpO2: blood oxygen saturation
Cerebrovascular dysfunction
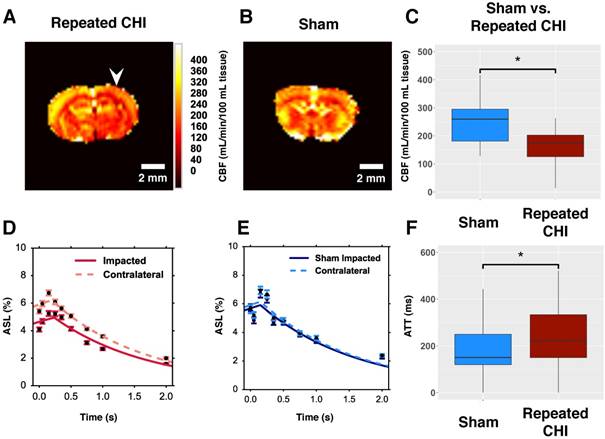
We hypothesized that TBI mice would exhibit persistent cerebrovascular dysfunction proximal to the site of the CHIs. We compared the CBF, as measured by multiple post label delay (mPLD) pCASL MRI, in TBI versus sham mice two weeks following the final impact of our repeated impact paradigm. TBI mice (n=5) showed reduced perfusion of peri-contusional cortex (140±30 mL/min/100 mL) relative to perfusion level in the corresponding brain region of sham-treated mice (280±30 mL/min/100 mL, n=5, χ2= 10.78, p=0.001) (Figure 2). Additionally, mPLD pCASL imaging allowed estimation of ATT—the time taken for labeled carotid arterial blood to reach imaged brain regions. ATT of TBI mice was longer, on average, than that of sham-treated mice (260±50 ms in TBI vs. 120±6 ms in sham treated mice, χ2= 6.53, p=0.01). In contrast to the peri-contusional tissue, CBF of the contralateral hemisphere (240±20 mL/min/100 mL, n=5) was not reduced relative to that of sham-treated mice (210±20 mL/min/100 mL, n=5, χ2= 2.75, p=0.1) nor was the ATT of the contralateral hemisphere reduced relative to that of sham-treated mice (108±20 ms in TBI vs. 150±20 ms in sham-treated mice, χ2= 3.33, p=0.07). These findings are indicative of impairment of the brain's ability to maintain circulation to peri-contusional tissue. Chronically reduced resting CBF may lead to irreversible selective neuronal loss [28].
Reduced resting cerebral blood flow and elongated arterial transit time. (A) CBF map for a representative repeated mTBI mouse computed voxel-wise from modelling pCASL signal as a function of PLD using a single compartment model. The white arrow indicates the location to which impacts were delivered. (B) CBF map for a sham-operated mouse. (C) The average CBF of peri-contusional brain (n=5) was reduced relative to the corresponding brain region of sham-treated mice (n=5). (D) ROI analysis of pCASL signal as a function of PLD for a representative repeated mTBI mouse. The data points represent the mean ASL signal across the ROI for a given PLD with error bars showing standard error of the mean across the ROI. The modeled time course of the peri-contusional ROI is shown as a solid line, while the contralateral tissue model is shown with a dotted line. (E) ROI analysis of pCASL signal as a function of PLD for a representative sham-operated mouse. This modeling allowed estimation of CBF and ATT in the peri-contusional hemisphere for each subject. (F) Similarly, peri-contusional ATT was elongated in repeated mTBI mice (n=5) relative to sham-treated mice (n=5).
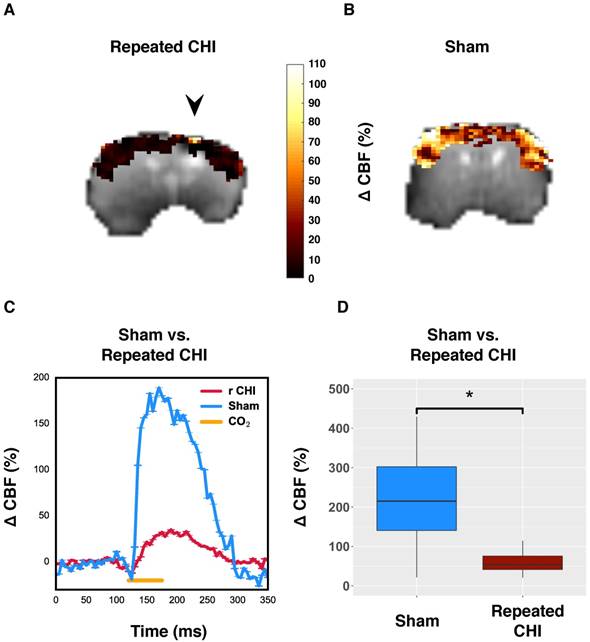
To further probe the cerebrovascular impairment, the brain's ability to increase blood flow, the CVR, was investigated. In TBI mice (n=6), the CO2-induced change in CBF of impacted tissue was 50±10%, compared to 250±30% in that of sham-treated mice (n=5) (χ2= 9.0, p=0.003) (Figure 3). Unlike resting brain hemodynamics, cerebrovascular reactivity was affected bilaterally two weeks following induction of injury: TBI mice had reduced contralateral CVR compared to sham-treated mice (45±7% TBI vs. 250±40% sham, χ2= 5.33, p=0.02). Cerebrovascular reactivity is a critical aspect of healthy brain function as the brain depends on tuning of blood flow to the local tissue metabolic needs. Impairment of CVR is known to lead to cognitive dysfunction and hence could contribute to the functional deficits seen in repeated TBI patients.
Reduced cerebrovascular reactivity to 10% inhaled CO2. (A) Map of percent change in CBF due to hypercapnic challenge in a representative repeated mTBI mouse, shown overlaid on the corresponding echo planar image with the site of impacts indicated by an arrow. (B) pCASL reactivity map for a representative sham-operated mouse. (C) Time-courses of mean response to CO2 for sham-treated and repeated mTBI mice. (D) Percent change in peri-contusional CBF in response to hypercapnia for representative sham and repeated mTBI mice. Injured mice showed reduced reactivity to hypercapnia (n=6) relative to sham-treated mice (n=5).
Reduced evoked neuronal responses to optogenetic photostimulation
We next investigated whether this cerebrovascular dysfunction was accompanied by neuronal function deficits. The amplitude of evoked neuronal responses to photostimulation of peri-contusional tissue of TBI mice (n=4) was reduced relative to that of sham-treated mice (n=4) (0.02±0.01 mV TBI vs. 0.7±0.2 mV sham-treated, χ2= 5.33, p=0.02). Additionally, the evoked response amplitude of contralateral hemisphere (0.2±0.1 mV) was reduced by 67±19% relative to that of sham-treated mice (0.6±0.1 mV, χ2=4.08, p=0.04). The signals detected with these extracellular recordings are an integration of neuronal depolarizations that occur proximal to the electrodes; reduction in the amplitude of evoked responses suggests fewer viable neurons and/or impaired functioning of the surviving neurons.
Astrogliosis and cerebrovascular volume increase with preserved neuronal density
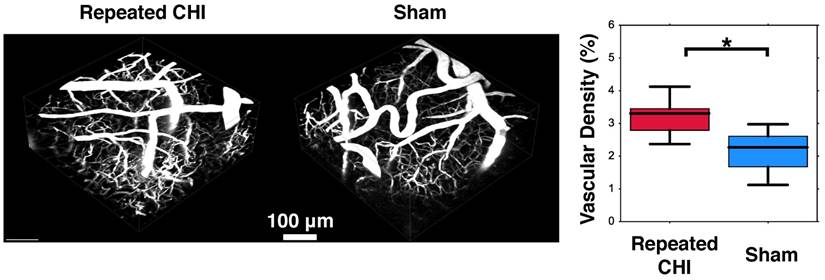
Impairments of the cerebrovascular circulation as well as altered neuronal function may have produced an imbalance between the metabolic demands of the tissue and the brain's ability to deliver these metabolites. This mismatch has been identified as key in the pathological progression of moderate to severe TBI. To contextualize the observed dysfunction of neuronal and cerebrovascular networks, we undertook morphological examination of key components of the neurogliovascular unit. Relative to sham-treated animals, the TBI mice showed increased vascular density on in vivo 2-photon fluorescence microscopy in the cortical region surrounding the impacts' site relative to that in the homologous region in sham mice (3.2±0.4% TBI, 2.4±0.6% sham, p=0.02, Figure 6). Increased peri-contusional vascular density was accompanied by an increase in GFAP immunoreactivity (29±1% for TBI mice, 1.5±0.2% for sham-treated mice, p = 0.009) (Figure 5). GFAP-positive cells at the site of the impact had the characteristic star-shaped appearance of astrocytes, whereas an approximately normal proportion of GFAP-positive cells were present contralaterally to the injured site (4±2% for TBI vs. 1.7±0.5% for sham-treated mice, p=0.17).
Angiogenesis. Two weeks following the final closed head injury (CHI) or sham preparation, mice underwent in-vivo 2-photon fluorescence microscopy (2PFM). An intravenous injection of red dextran die was administered to highlight the vasculature, and 3D images of the peri-contusional tissue were collected. These are shown for the CHI (left) and sham (middle) treated mice. Statistical comparison of these two groups revealed elevated cerebrovascular density of CHI mice (right).
Reduction of neuronal response to photostimulation. (A) Schematic showing the orientation and placement of recording electrodes and optical fibers for electrophysiological experiments. Mice underwent bilateral craniotomy at the site of injury and in the homologous location contralaterally, allowing access for extracellular recording and photostimulation in both hemispheres. (B-C) Change in extracellular potential due to photostimulation (shown as orange bar) for representative repeated CHI and sham-treated mice. (B) Repeated CHI mice showed greatly reduced (n=4) reactivity to photostimulation in peri-contusional (solid red line) relative to contralateral tissue (dotted red line). (C) The peri-contusional cortical responses in sham mice were greatly reduced relative to those of sham-treated mice (n=4) (bottom middle, blue lines). (D) Box plot comparison of evoked response amplitude of sham-treated mice (blue) and repeated mTBI mice (red).
Astrogliosis and neuronal survival. Brains were stained for GFAP (top) and NeuN (bottom). The left column shows slices from representative repeated CHI mice, while the middle column shows slices from sham-treated animals. Insets show the general cortical area below the skull location that was impacted, not the ROIs analyzed (the latter are displayed in Figure S2). For each stain, a box and whisker plot comparing optical density of peri-contusional tissue of repeated CHI (n=5, GFAP and NeuN) vs. sham-treated mice (n=5, GFAP and NeuN) is shown (right column). Top: GFAP stains of representative repeated mTBI (left) and sham (right) mice. GFAP fluorescence was elevated peri-contusionally relative to sham treated mice. No difference was observed in optical density of the peri-contusional tissue of NeuN-stained sections.
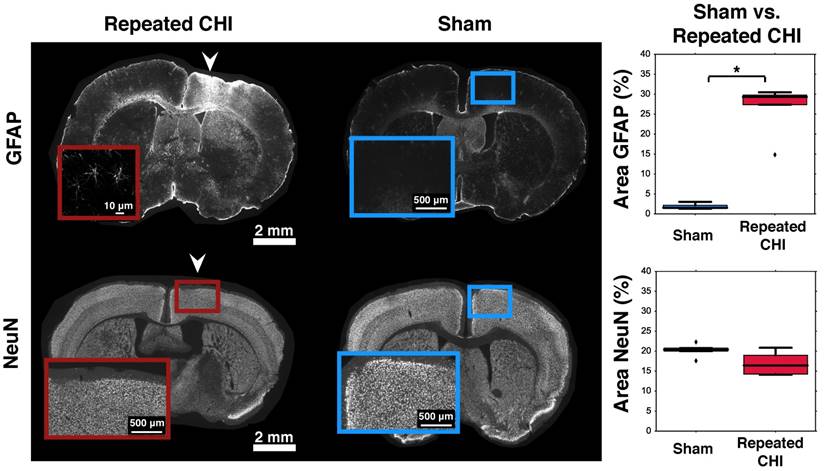
To provide context to the electrophysiological recordings, we characterized neuronal density of pericontusional and contralateral cortex. The neuronal density of TBI animals was not different from that of sham-treated mice in either region. We observed no significant changes in NeuN, MAP2, or neurofilament immunofluorescence in the perilesional cortex (NeuN: 16±2% for TBI vs. 20.4±0.4% for sham-treated mice, p = 0.11; MAP2: 77±9% for TBI vs. 58±2% for sham-treated mice, p = 0.08; and neurofilament: 77±33% for TBI mice vs. 58±23% for sham-treated mice, p = 0.1). No differences were observed in the contralateral cortex either (NeuN: 22±1% for TBI vs. 19±1% for sham-treated mice, p = 0.1; MAP2: 63±7% for TBI vs. 46±3% for sham-treated mice, p = 0.2; and neurofilament: 25±3% for TBI vs. 28±2% for sham-treated mice, p = 0.8).
Discussion
This study provides the first in vivo measures of neurovascular functioning in chronic repeated TBI, induced via serial closed head injuries. The model was optimized so as to potentiate clinical relevance by limiting the amount of injury induced by each individual closed head injury, i.e., by rendering the individual impacts mild (single impact being negative on neuroimaging). Mice were young adults (9 weeks old) at the time of first impact; in the human population, young adults suffer a disproportionate number of mild head injuries through participation in athletics and military service [18,29]. Of note, however, rodent and human ages should not be conflated due to the different time scales of important complex biological processes [18,29]. They suggest that rodents may reach sexual maturity around 50 days of age, but do not enter adulthood until 210 days; hence, the mice in this study are sexually mature but not yet adults. Furthermore, while some sources report sex-based differences of biological responses to brain trauma [30-32], recent work from Tucker et al. suggests that behavioural outcomes between female and male C57BL/6J mice post TBI are equivalent [33]. Although an approximately equal number of male and female mice were included in both sham and repeated CHI groups in the present study, the sizes of cohorts precluded analysis of the sex effects; future studies with larger cohort sizes are required to investigate the effects of sex on neurovascular function. The 3-day inter-impact interval was motivated by prior reports of a “window of vulnerability,” whereby greatest behavioural deficits are seen in mice impacted every 3 days [34]. Similar “windows of vulnerability” have been observed in the clinic as well: in the most extreme cases, a second impact can be fatal [35]. In recognition of noxiousness of repeated injuries, at least three days is typically required for military servicemen to return to duty following an mTBI [7]. In light of the “three strike rule,” we delivered a total of three impacts. T2-weighted anatomical images of the site of the impact acquired at 24 h after a single CHI in a subset of mice confirmed an absence of significant structural damage following a single insult (Figure 1), as would be expected of a mild TBI in the clinic [36]. However, two weeks following the final impact the same anatomical imaging revealed hypointense regions at the site of the impacts, suggesting that individual mild insults delivered in series appear similar to a single moderate brain injury on neuroimaging. T2 hypointensities suggest microhemorrhages. In support of this notion, previous work on repeated controlled cortical impacts (two impacts delivered to opposite hemispheres at 3 and 7 day inter-impact-intervals) in rats reported extravascular iron deposition near lesions composed of blood [37]. Iron depositions result in T2 tashortening, potentially explaining the hypointensities seen on T2-weighted imaging here [38]. Sham-treated animals showed no contrast on T2-weighted imaging.
Alterations of cerebral blood flow resulting from experimental repeated TBI are poorly understood, as investigations have been limited by modalities that lack topographical information [39]. Here, we have utilized pCASL to map resting cerebral blood flow in absolute units [24]. Two weeks post final impact, we observed a reduction of resting cerebral blood flow at the site of the impacts that was not present either contralaterally or in sham-treated animals. Reduced cerebral blood flow may be the result of biomechanical damage to vessels caused by the initial impact [40]. Cortical swelling and the ensuing elevation in intracranial pressure may also have contributed to reduced resting cerebral blood flow [41]. In vivo two-photon fluorescence microscopy indicated increased peri-contusional vascular volume. This may have resulted either from angiogenesis with partial pruning and/or from dilation of immature vessels due to the lack of vascular smooth muscle cells in their walls [42]. Notwithstanding, cerebrovascular segmentation of three-dimensional in vivo imaging data acquisitions should be undertaken in future work to understand the details of cerebrovascular remodeling. An increase of cerebral blood volume concomitant to decreased cerebral blood flow may have produced a decrease of blood velocity and a decreased efficiency of metabolite delivery by the circulation, as seen in moderate to severe TBI [40,43].
Cerebrovascular dysfunction was further reflected in arterial transit time changes. Mice that underwent repeated TBI had longer arterial transit time in damaged tissue relative to sham-treated mice. In the context of the single-compartment model of arterial spin labelling signal, arterial transit time is a measure of the superposition of individual vessel arrival times in a given voxel. The observed lengthening of arterial transit time could manifest, as a result, increased dispersion of microvascular route lengths and microvascular reactivity, which have been previously reported in models of moderate TBI [44,45]. The aforementioned blood velocity decrease surrounding the site of the injury could have also contributed to the observed elongation of arterial transit time. Hayward et al., observed acute reduction of vessel density followed by a chronic stage increase in vessel density on rat endothelial cell antibody-1 immunohistochemistry in a lateral fluid percussive model of moderate TBI in rats [44]. Ostergaard et al. proposed that moderate TBI could lead to an increase in heterogeneity of capillary transit times [45]. In vivo two-photon fluorescence microscopy in our study indeed indicated increased vessel density near the site of the injury in repeated TBI-treated mice relative to shams: the reduced contractility of immature new vessels may have contributed to increases in heterogeneity of vascular transit times [42]. Although a number of prior studies have reported angiogenesis and vasculogenesis in single impact TBI models [44,46-48], this is the first report of cerebrovascular remodeling and arterial transit time elongation in a model of repeated TBI.
The observed deficits in cerebrovascular reactivity to hypercapnia likely resulted from incomplete pruning of new vessels in combination with mechanical damage to the existing vessels. While clinical repeated TBI is known to impair cerebrovascular reactivity, this had not previously been observed in experimental repeated TBI [49,50]. When challenged with an increase of CO2 in the inspired gas mixture, repeated TBI mice exhibited depressed cerebral blood flow response relative to sham-operated mice. Chronically depressed cerebrovascular reactivity has the potential to worsen functional outcomes. Pharmacological impairment of the coupling between neuronal activity and CBF in mice has been shown to result in cognitive impairments [51]. Here, we postulate that cerebrovascular reactivity impairment was due to the newly sprouted vessels seen on in vivo two-photon fluorescence microscopy that were not yet functionally mature, hence having thin vessel walls [52] and limited endothelium-associated smooth muscle cells [42]. As the maturation of these vessels is believed to be influenced by hemodynamic and metabolic stimuli, exercise and increased activity may be beneficial whereas rest and lack of external stimulus may be counter-indicated in the chronic phase of repeated mild TBI. Future work is needed to establish whether such interventions are required for optimization of long-term cerebrovascular functioning. Of note, an elevated peri-contusional vascular density and altered topology of the vessel network have also been seen in animal models of moderate to severe TBI [44,46]: this remodelling has been suggested to underlie changes in the neurovascular unit functioning [53].
In addition to compromised cerebrovascular function, we observed an increase in GFAP fluorescence. Enhancement of GFAP has been previously reported in the acute phase of repeated TBI as part of the neuroinflammatory cascade [54]. When short on glucose, the astrocytes are unable to provide adequate support to neurons, further endangering neuronal survival [55]. Enhanced GFAP fluorescence is sometimes also associated with astrocytic swelling, which has been observed to reduce capillary lumen and in turn reduce cerebral blood flow and microvascular hematocrit [56].
A dearth of available metabolites may also lead to neuronal dysfunction and loss [28], motivating us to examine neuronal survival and functioning. Neuronal dysfunction was manifested as bilaterally reduced amplitude of evoked responses to photoactivation of channelrhodopsin. Electrophysiological responses of cortical pyramidal neurons of Thy1-ChR2 mice to photostimulation have been shown to be comparable to those evoked by peripheral functional stimulation, such as forelimb stimulation [57]. Further, an earlier study in a model of moderate TBI reported decreased electrophysiological responses to contralateral forepaw stimulation two weeks following a single controlled cortical impact [58]. Photostimulation of opsin-expressing cells affords two important advantages over peripheral stimulation in that it enables finer spatial targeting of the neuronal perturbation in relation to the site of the injury as well as cell-type specific activation. Given that there was no statistically significant effect of the serial impacts on neuronal survival (on NeuN, MAP2, and neurofilament immunofluorescence), the reduction of evoked response amplitude may be a result of impaired responses on the individual unit level. Mild impact may cause subtle dendritic and axonal damage, thus altering connections between cortical neurons: fewer recruited neurons following photostimulation could thus also have contributed to the observed decrease in evoked response amplitude [59]. To our knowledge, ours is the first study to employ optogenetic photostimulation as a readout of neuronal function following TBI.
While the observed increases in cerebrovascular density and GFAP immunoreactivity were limited to the peri-contusional cortex, impairments of cerebrovascular and neuronal reactivity were observed bilaterally, although the peri-contusional effects were much stronger than the contralateral ones. Given the density of transcallosal connections between homologous regions of the cortex, focal damage at the site of the injury may well have affected the contralateral hemisphere. Global reduction of cerebrovascular reactivity could result from increased intracranial pressure or be secondary to global decreases in neuronal activity and thus their signaling to the vessels. As the neuronal and cerebrovascular networks are closely coupled functionally, impairments of one could affect the other, and vice versa.
In conclusion, this study provides initial characterization of neurogliovascular dysfunction two weeks following repeated TBI. Our repeated closed head injury model reveals that repeated impacts-induced injury comprises sustained peri-contusional decreases in cerebral blood flow, elongation of arterial transit time, increase in cerebrovascular density, and bilaterally reduced reactivity to hypercapnia, in addition to astrogliosis. Furthermore, we present the first measures of impaired neuronal excitability (in response to opsin photoactivation) not only peri-contusionally, but also contralaterally, two weeks following repeated TBI. Our findings indicate that a series of individually mild impacts can trigger pathological changes on par with those seen post moderate TBI, and call for development of therapeutic strategies for a series of mild injuries.
Abbreviations
2PFM: two-photon fluorescence microscopy; AFNI: Analysis of Functional Neuroimages; ASL: arterial spin labelling; ATT: arterial transit time; CBF: cerebral blood flow; CHI: closed head injury; ChR2: channelrhodopsin; CVR: cerebrovascular reactivity; EPI: echo planar images; GFAP: glial fibrillary acidic protein; IHC: immunohistochemistry; mPLD: multiple post label delay; MRI: magnetic resonance imaging; mTBI: mild traumatic brain injury; PFA: paraformaldehyde; pCASL: pseudo-continuous arterial spin labelling; PLD: post label delay; PVA-DABCO: polyvinyl-alcohol mounting medium with DABCO; RARE: rapid acquisition with refocused echoes; ROI: region of interest; TBI: traumatic brain injury.
Supplementary Material
Supplementary figures.
Acknowledgements
This work was funded by the Canadian Institute of Health Research (FRN 148521).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Cassidy JD, Carroll LJ, Peloso PM, Borg J, von Holst H, Holm L. et al. Incidence, risk factors and prevention of mild traumatic brain injury: results of the WHO Collaborating Centre Task Force on Mild Traumatic Brain Injury. J Rehabil Med. 2004;43(Suppl):28-60
2. Bryant R. Post-traumatic stress disorder vs traumatic brain injury. Dialogues Clin Neurosci. 2011;13:251-262
3. Van Den Heuvel C, Thornton E, Vink R. Traumatic brain injury and Alzheimer's disease: a review. Prog Brain Res. 2007;161:303-316
4. Lee Y-K, Hou S-W, Lee C-C, Hsu C-Y, Huang Y-S, Su Y-C. Increased risk of dementia in patients with mild traumatic brain injury: a nationwide cohort study. PLoS One. 2013;8:e62422
5. McKee AC, Cantu RC, Nowinski CJ, Hedley-Whyte ET, Gavett BE, Budson AE. et al. Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J Neuropathol Exp Neurol. 2009;68:709-735
6. McKee AC, Stern RA, Nowinski CJ, Stein TD, Alvarez VE, Daneshvar DH. et al. The spectrum of disease in chronic traumatic encephalopathy. Brain. 2013;136:43-64
7. DoD Policy Guidance for Management of Mild Traumatic Brain Injury. 28 September 2012. https://www.hsdl.org/?view&did=722999
8. Weiller C, May A, Sach M, Buhmann C, Rijntjes M. Role of functional imaging in neurological disorders. J Magn Reson Imaging. 2006;23:840-850
9. Amyot F, Kenney K, Moore C, Harber M, Turtzo LC, Shenouda CN. et al. Imaging of cerebrovascular function in chronic traumatic brain injury. J Neurotrauma. 2017;5(10):1116-1123
10. Kenney K, Amyot F, Haber M, Pronger A, Bogoslovsky T, Moore C. et al. Cerebral vascular injury in traumatic brain injury. Exp Neurol. 2016:275 Pt 3: 353-366
11. Mouzon BC, Bachmeier C, Ferro A, Ojo J-O, Crynen G, Acker CM. et al. Chronic neuropathological and neurobehavioral changes in a repetitive mild traumatic brain injury model. Ann Neurol. 2014;75:241-254
12. Tzekov R, Quezada A, Gautier M, Biggins D, Frances C, Mouzon B. et al. Repetitive mild traumatic brain injury causes optic nerve and retinal damage in a mouse model. J Neuropathol Exp Neurol. 2014;73:345-361
13. Yang Z, Wang P, Morgan D, Lin D, Pan J, Lin F. et al. Temporal MRI characterization, neurobiochemical and neurobehavioral changes in a mouse repetitive concussive head injury model. Sci Rep. 2015;5:11178
14. Laurer HL, Bareyre FM, Lee VM, Trojanowski JQ, Longhi L, Hoover R. et al. Mild head injury increasing the brain's vulnerability to a second concussive impact. J Neurosurg. 2001;95:859-870
15. Ojo J-O, Mouzon B, Greenberg MB, Bachmeier C, Mullan M, Crawford F. Repetitive mild traumatic brain injury augments tau pathology and glial activation in aged hTau mice. J Neuropathol Exp Neurol. 2013;72:137-151
16. Mouzon B, Chaytow H, Crynen G, Bachmeier C, Stewart J, Mullan M. et al. Repetitive mild traumatic brain injury in a mouse model produces learning and memory deficits accompanied by histological changes. J Neurotrauma. 2012;29:2761-2773
17. Fehily B, Fitzgerald M. Repeated mild traumatic brain injury: potential mechanisms of damage. Cell Transplant. 2017;26:1131-1155
18. Agoston DV. How to translate time? The temporal aspect of human and rodent biology. Front Neurol. 2017;8:92
19. Nagel G, Szellas T, Huhn W, Kateriya S, Adeishvili N, Berthold P. et al. Channelrhodopsin-2, a directly light-gated cation-selective membrane channel. Proceedings of the National Academy of Sciences. 2003;100:13940-13945
20. Arenkiel BR, Peca J, Davison IG, Feliciano C, Deisseroth K, Augustine GJ. et al. In vivo light-induced activation of neural circuitry in transgenic mice expressing channelrhodopsin-2. Neuron. 2007;54:205-218
21. Hawrylycz M, Ng L, Feng D, Sunkin S, Szafer A, Dang C. The Allen brain atlas. Springer handbook of bio-/neuroinformatics. Berlin, Heidelberg: Springer. 2014:1111-1126
22. Hennig J, Nauerth A, Friedburg H. RARE imaging: A fast imaging method for clinical MR. Magn Reson Med. 1986;3:823-833
23. Hirschler L, Debacker CS, Voiron J, Köhler S, Warnking JM, Barbier EL. Interpulse phase corrections for unbalanced pseudo-continuous arterial spin labeling at high magnetic field. Magn Reson Med. 2017;79(3):1314-1324
24. Parkes LM, Tofts PS. Improved accuracy of human cerebral blood perfusion measurements using arterial spin labeling: accounting for capillary water permeability. Magn Reson Med. 2002;48:27-41
25. Lake EMR, Mester J, Thomason LA, Adams C, Bazzigaluppi P, Koletar M. et al. Modulation of the peri-infarct neurogliovascular function by delayed COX-1 inhibition. J Magn Reson Imaging. 2017;46:505-517
26. Bates D, Mächler M, Bolker B, Walker S. Fitting linear mixed-effects models using lme4. J Stat Softw. 2015;67:1-48
27. Bolker BM, Brooks ME, Clark CJ, Geange SW, Poulsen JR, Stevens MHH. et al. Generalized linear mixed models: a practical guide for ecology and evolution. Trends Ecol Evol. 2009;24:127-135
28. Baron J-C, Yamauchi H, Fujioka M, Endres M. Selective neuronal loss in ischemic stroke and cerebrovascular disease. J Cereb Blood Flow Metab. 2014;34:2-18
29. Scopaz KA, Hatzenbuehler JR. Risk modifiers for concussion and prolonged recovery. Sports Health. 2013;5:537-541
30. Ferguson SA, Mouzon BC, Lynch C, Lungmus C, Morin A, Crynen G. et al. Negative impact of female sex on outcomes from repetitive mild traumatic brain injury in hTau mice is age dependent: A chronic effects of neurotrauma consortium study. Front Aging Neurosci. 2017;9:416
31. Velosky AG, Tucker LB, Fu AH, Liu J, McCabe JT. Cognitive performance of male and female C57BL/6J mice after repetitive concussive brain injuries. Behav Brain Res. 2017;324:115-124
32. Hsu H-L, Chen DY-T, Tseng Y-C, Kuo Y-S, Huang Y-L, Chiu W-T. et al. Sex differences in working memory after mild traumatic brain injury: A functional MR imaging study. Radiology. 2015;276:828-835
33. Tucker LB, Fu AH, McCabe JT. Performance of male and female C57BL/6J mice on motor and cognitive tasks commonly used in pre-clinical traumatic brain injury research. J Neurotrauma. 2016;33:880-894
34. Longhi L, Saatman KE, Fujimoto S, Raghupathi R, Meaney DF, Davis J. et al. Temporal window of vulnerability to repetitive experimental concussive brain injury. Neurosurgery. 2005;56:364-74 discussion 364-74
35. Bey T, Ostick B. Second impact syndrome. West J Emerg Med. 2009;10:6-10
36. Mittl RL, Grossman RI, Hiehle JF, Hurst RW, Kauder DR, Gennarelli TA. et al. Prevalence of MR evidence of diffuse axonal injury in patients with mild head injury and normal head CT findings. AJNR Am J Neuroradiol. 1994;15:1583-1589
37. Donovan V, Bianchi A, Hartman R, Bhanu B, Carson MJ, Obenaus A. Computational analysis reveals increased blood deposition following repeated mild traumatic brain injury. Neuroimage Clin. 2012;1:18-28
38. Wu G, Xi G, Hua Y, Sagher O. T2* magnetic resonance imaging sequences reflect brain tissue iron deposition following intracerebral hemorrhage. Transl Stroke Res. 2010;1:31-34
39. Hylin MJ, Orsi SA, Rozas NS, Hill JL, Zhao J, Redell JB. et al. Repeated mild closed head injury impairs short-term visuospatial memory and complex learning. J Neurotrauma. 2013;30:716-726
40. Meaney DF, Smith DH. Biomechanics of concussion. Clin Sports Med. 2011;30:19-31
41. Unterberg AW, Stover J, Kress B, Kiening KL. Edema and brain trauma. Neuroscience. 2004;129:1021-1029
42. Carmeliet P. Angiogenesis in health and disease. Nat Med. 2003;9:653-660
43. Giza CC, Hovda DA. The new neurometabolic cascade of concussion. Neurosurgery. 2014;75(Suppl 4):S24-33
44. Hayward NMEA, Tuunanen PI, Immonen R, Ndode-Ekane XE, Pitkänen A, Gröhn O. Magnetic resonance imaging of regional hemodynamic and cerebrovascular recovery after lateral fluid-percussion brain injury in rats. J Cereb Blood Flow Metab. 2011;31:166-177
45. Østergaard L, Engedal TS, Aamand R, Mikkelsen R, Iversen NK, Anzabi M. et al. Capillary transit time heterogeneity and flow-metabolism coupling after traumatic brain injury. J Cereb Blood Flow Metab. 2014;34:1585-1598
46. Obenaus A, Ng M, Orantes AM, Kinney-Lang E, Rashid F, Hamer M. et al. Traumatic brain injury results in acute rarefication of the vascular network. Sci Rep. 2017;7:239
47. Guo X, Liu L, Zhang M, Bergeron A, Cui Z, Dong J-F. et al. Correlation of CD34+ cells with tissue angiogenesis after traumatic brain injury in a rat model. J Neurotrauma. 2009;26:1337-1344
48. Morgan R, Kreipke CW, Roberts G, Bagchi M, Rafols JA. Neovascularization following traumatic brain injury: possible evidence for both angiogenesis and vasculogenesis. Neurol Res. 2007;29:375-381
49. Mutch WAC, Ellis MJ, Ryner LN, Ruth Graham M, Dufault B, Gregson B. et al. Brain magnetic resonance imaging CO2 stress testing in adolescent postconcussion syndrome. J Neurosurg. 2016;125:648-660
50. Ellis MJ, Ryner LN, Sobczyk O, Fierstra J, Mikulis DJ, Fisher JA. et al. Neuroimaging assessment of cerebrovascular reactivity in concussion: Current concepts, methodological considerations, and review of the literature. Front Neurol. 2016;7:61
51. Tarantini S, Hertelendy P, Tucsek Z, Valcarcel-Ares MN, Smith N, Menyhart A. et al. Pharmacologically-induced neurovascular uncoupling is associated with cognitive impairment in mice. J Cereb Blood Flow Metab. 2015;35:1871-1881
52. Pries AR, Reglin B, Secomb TW. Remodeling of blood vessels: responses of diameter and wall thickness to hemodynamic and metabolic stimuli. Hypertension. 2005;46:725-731
53. Jullienne A, Obenaus A, Ichkova A, Savona-Baron C, Pearce WJ, Badaut J. Chronic cerebrovascular dysfunction after traumatic brain injury. J Neurosci Res. 2016;94:609-622
54. Bolton AN, Saatman KE. Regional neurodegeneration and gliosis are amplified by mild traumatic brain injury repeated at 24-hour intervals. J Neuropathol Exp Neurol. 2014;73:933-947
55. Lee C-Y, Dallérac G, Ezan P, Anderova M, Rouach N. Glucose tightly controls morphological and functional properties of astrocytes. Front Aging Neurosci. 2016;8:82
56. Bouzat P, Millet A, Boue Y, Pernet-Gallay K, Trouve-Buisson T, Gaide-Chevronnay L. et al. Changes in brain tissue oxygenation after treatment of diffuse traumatic brain injury by erythropoietin. Crit Care Med. 2013;41:1316-1324
57. Vazquez AL, Fukuda M, Crowley JC, Kim S-G. Neural and hemodynamic responses elicited by forelimb- and photo-stimulation in channelrhodopsin-2 mice: insights into the hemodynamic point spread function. Cereb Cortex. 2014;24:2908-2919
58. Li N, Yang Y, Glover DP, Zhang J, Saraswati M, Robertson C. et al. Evidence for impaired plasticity after traumatic brain injury in the developing brain. J Neurotrauma. 2014;31:395-403
59. Sword J, Masuda T, Croom D, Kirov SA. Evolution of neuronal and astroglial disruption in the peri-contusional cortex of mice revealed by in vivo two-photon imaging. Brain. 2013;136:1446-1461
Author contact
![]() Corresponding author: Conner Adams, Conner.adamsutoronto.ca. Sunnybrook Health Sciences Centre, 2075 Bayview Ave., S636, Toronto, ON, M4N 3M5
Corresponding author: Conner Adams, Conner.adamsutoronto.ca. Sunnybrook Health Sciences Centre, 2075 Bayview Ave., S636, Toronto, ON, M4N 3M5