Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
General aspects regarding AMPK
Regulation of AMPK in ischemic...
Therapeutic research
Prospects
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(16):4535-4551. doi:10.7150/thno.25674 This issue Cite
Review
AMPK: Potential Therapeutic Target for Ischemic Stroke
Shuai Jiang1,2*, Tian Li3*, Ting Ji1*, Wei Yi4, Zhi Yang3, Simeng Wang5, Yang Yang1 ![]() , Chunhu Gu4
, Chunhu Gu4 ![]()
1. Key Laboratory of Resource Biology and Biotechnology in Western China, Ministry of Education. Faculty of Life Sciences, Northwest University, 229 Taibai North Road, Xi'an 710069, China
2. Department of Aerospace Medicine, The Fourth Military Medical University, 169 Changle West Road, Xi'an 710032, China
3. Department of Biomedical Engineering, The Fourth Military Medical University, 169 Changle West Road, Xi'an 710032, China
4. Department of Cardiovascular Surgery, Xijing Hospital, The Fourth Military Medical University, 127 Changle West Road, Xi'an 710032, China
5. Center for Human Nutrition, University of Texas Southwestern Medical Center, 5323 Harry Hines Blvd, Dallas, TX 75390, USA
*These authors contributed equally to this work.
Received 2018-2-22; Accepted 2018-7-16; Published 2018-8-10
Abstract

5'-AMP-activated protein kinase (AMPK), a member of the serine/threonine (Ser/Thr) kinase group, is universally distributed in various cells and organs. It is a significant endogenous defensive molecule that responds to harmful stimuli, such as cerebral ischemia, cerebral hemorrhage, and, neurodegenerative diseases (NDD). Cerebral ischemia, which results from insufficient blood flow or the blockage of blood vessels, is a major cause of ischemic stroke. Ischemic stroke has received increased attention due to its '3H' effects, namely high mortality, high morbidity, and high disability. Numerous studies have revealed that activation of AMPK plays a protective role in the brain, whereas its action in ischemic stroke remains elusive and poorly understood. Based on existing evidence, we introduce the basic structure, upstream regulators, and biological roles of AMPK. Second, we analyze the relationship between AMPK and the neurovascular unit (NVU). Third, the actions of AMPK in different phases of ischemia and current therapeutic methods are discussed. Finally, we evaluate existing controversy and provide a detailed analysis, followed by ethical issues, potential directions, and further prospects of AMPK. The information complied here may aid in clinical and basic research of AMPK, which may be a potent drug candidate for ischemic stroke treatment in the future.
Keywords: 5'-AMP-activated protein kinase, Ischemic stroke, Metformin, Energy metabolism, Autophagy
Introduction
The brain is an organ that serves as the central nervous system among all vertebrate. Stroke is caused by occulusion of cerebral vessels or angiorhagia [1-4]. It is reported that stroke is the primary cause of disability and affects approximately 795,000 people annually. Stroke is characterized by '3H', namely high mortality, high morbidity, and high disability, it causes immense health and economic burdens to families and society [5]. Stroke may contribute to brain dysfunction due to a disturbance in the cerebral blood supply, which is caused by occlusion of vessels due to a blood clot or a bleeding vessel [6]. Mild or chronic cerebral ischemia might be nonfatal and cause mild somatic symptoms, whereas acute stroke may cause hemiplegia, cardiac dysfunction, and even death. Ischemic stroke is more common than hemorrhagic stroke in all cases, we pay attention to the former throughout this review.
Previous studies have demonstrated that the dysfunction of energy metabolism plays a significant role in ischemic stroke. The underlying mechanism appears to involve an energy sensor called AMPK, a member of the serine/threonine (Ser/Thr) kinase group [7-11]. The historical background of AMPK may date back to two independent works by Gibon and Carbson in 1973. Later, it was extracted and sequenced by Carling et al until 1994 and was identified as a peripheral 'energy sensor' under conditions of energy deprivation, ischemic stress, and heavy exercise [12]. Studies have demonstrated that AMPK plays an important role in various organs, including the brain [13], heart [14], liver [15], kidney [16], gastrointestinal tract [17], lung [18], and pancreas [19]. AMPK, which functions as the cellular energy sensor, can resist cerebral ischemic injury by switching on catabolic pathways and switching off ATP-consuming processes [20, 21]. Over the past 40 years, the annual number of publications on AMPK has surged from several to nearly 2000. Our laboratory has discovered that AMPK is a protective molecule in myocardial ischemia [22], vasorelaxation [23], angiogenesis [24], fluid shear stress [25, 26], and diabetic cardiac injury[27]. A positive effect of AMPK has been reviewed in the heart [28], liver [29], and kidney [30], etc., whereas its relationship with ischemic stroke has not been well discussed. Thus, further understanding of AMPK and ischemic stroke may aid in experimental studies and clinical therapeutics.
Herein, we first introduce the background of AMPK and ischemic stroke, followed by a review of its upstream agonists and antagonists. Second, we summarize the regulatory mechanism of AMPK in ischemic stroke, including oxidative stress, autophagy, apoptosis, mitochondrial dysfunction, glutamate excitotoxicity, neuroinflammation, and angiogenesis. AMPK stands at the crossroads of diverse regulatory mechanism to main energy homeostasis during stroke. Third, therapeutic methods and benefits beyond ischemia are discussed. Finally, we bring attention to various controversial opinions, ethical issues, and potential directions. This review highlights recent advances and provides a comprehensive picture of AMPK, which may be helpful in drug design and clinical therapy for treatment of ischemic stroke.
General aspects regarding AMPK
Structure of AMPK
AMPK is a heterotrimeric complex distributed in different tissues and organs denoted α1, α2, β1, β2, γ1, γ2, and γ3 subunits [31]. As a result, there are 7 isoforms in eukaryotic cells, which allow for the expression of 12 different AMPK complexes, such as α2β1γ1 [32] and α1β1γ1 [33].
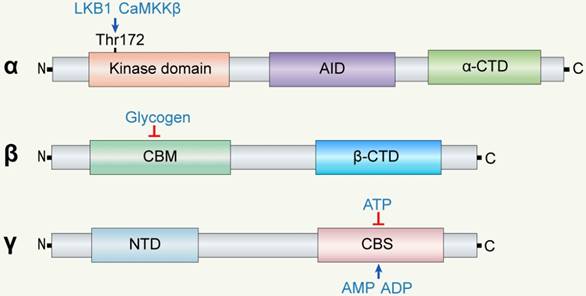
The α subunit is composed of a kinase domain, an auto-inhibitor domain (AID), and an α-subunit carboxy-terminal domain (α-CTD) from the N-terminus to the C-terminus. Phosphorylation of Thr172 at the α subunit increases AMPK activity by 2-3 orders of magnitude [34, 35]. AMP binding promotes phosphorylation by LKB1. Scott et al discovered that AMP and A769662 cause synergistic allosteric activation even when using kinase that is not phosphorylated on Thr172 [36]. The AID lowers AMPK activity in the absence of AMP, which is a crucial component of AMPK regulation independent of Thr172 phosphorylation.
The β subunit comprises a carbohydrate-binding module (CBM) and a β-CTD. The β-CTD functions as a scaffold, interacting with the α-CTD of the α subunit and the N-terminal region (NTR) of the γ subunits. Excess accumulated glycogen can interact with CBM and inhibit the activity of AMPK, while energy deprivation might activate AMPK especially in conditions of cerebral ischemia.
The γ subunit has four tandem repeats of cystathionine β synthase (CBS) and an NTR. The four CBS domains (CBS1 to CBS4) form two Bateman domains that are able to bind to AMP, ADP, or ATP. Mutations in these domains contribute to some diseases, such as PRKAG2 cardiomyopathy [37]. The NTR is connected to the β subunit to maintain the stability of AMPK. Overall, the main function of the γ subunit is promoting the allosteric activation of AMPK.
Expression and localization of AMPK
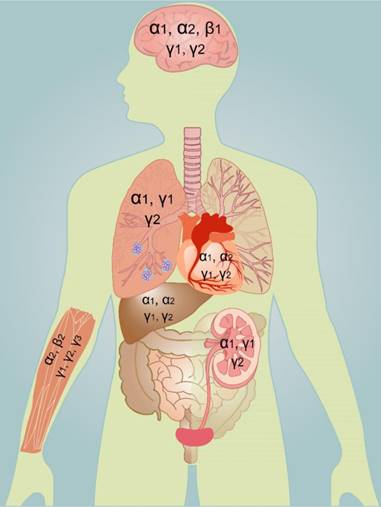
AMPK is widely but distinctly distributed in different tissues and organs, which might explain the susceptibility of different organs to ischemic stimuli. The early work of Turnley and colleagues reported a systematic study on AMPK that provides a comprehensive understanding of the distribution of AMPK in specific types of cells [20]. The α1 subunit mainly exists in the brain, heart, lung, kidney, and liver. The α2 subunit mainly exists in the liver, heart, skeletal muscle, and cerebral neurons. The α1 subunit is present in the cytoplasm whereas the α2 subunit is present in both the cytoplasm and nucleus [38]. The α2 subunit can regulate transcription in the nucleus, which allows neurons that contain more AMPK α2 subunit to regulate energy metabolism and to survive metabolic stress during ischemia. The β1 subunit has a low content in skeletal muscle, while the β2 subunit is highly expressed in skeletal muscle. The γ3 subunit is almost exclusively restricted to fast twitch skeletal muscle [39], whereas the γ1 and subunit are widely expressed in almost all tissues. The γ2 subunit is not expressed in high concentration in all tissues [20].
AMPK is expressed in the central nervous system (CNS) from at least the time of neuroepithelial formation. Gao and colleagues detected the expression of AMPK in the adult brain in 1995 [40]. The brain mRNA levels of the α2 subunit are increased from the 10th to 14th day of the embryonic period, whereas the α1, β1, and γ1 subunits are expressed consistently at all ages. Moreover, the α2 catalytic subunit is highly expressed in neurons and activated astrocytes, whereas the α1 catalytic subunit exhibits only low expression in neuropil. Some neurons, such as granule cells of the olfactory bulb, do not express detectable levels of the β subunit [20]. The disparate expression and nuclear localization of the α, β, and γ subunits suggest specific functions and physiological roles of AMPK in different nerve cells.
Upstream regulators of AMPK
Upstream kinases/activators
In the brain, ischemia is one of the most important forms of energy deprivation. ATP is the primary energy source for various biological actions, and the production and utilization of ATP are highly related to cerebral homeostasis. In mammals, AMPK is activated by increases in AMP/ATP or ADP/ATP ratios under energy stress [41, 42]. The increase in AMP is always much greater than the increase in ADP or decrease in ATP, which suggests that AMP might be the primary regulatory signal to energy consumption [43]. Therefore, AMPK acts as an early sensor of energy deprivation for maintaining metabolic homeostasis.
In addition to AMP, other upstream kinases/activators play significant roles in the activation of AMPK, such as liver kinase B1 (LKB1) [44, 45], Ca2+/calmodulin-dependent protein kinase β (CaMKKβ), 5-aminoimidazole-4-carboxyamide ribonucleoside (AICAR), and antimycin A. In middle cerebral artery occlusion (MCAO) mice [46], Li et al detected that the activity of LKB1 is significantly increased compared to controls, concomitant with increased expression of AMPK [47]. Studies have revealed that LKB1 can phosphorylate AMPK under energy deprivation, and this process may be facilitated by adapter proteins that are able to recruit LKB1 and AMPK on the two-dimensional surfaces of lysosomes [35, 48]. Zhang's group reported that the late lysosome v-ATPase-Regulator complex may activate the LKB1/AMPK pathway under glucose starvation, thereby providing a switch from catabolism to anabolism [35]. CaMKKβ functions as AMPK kinase in the hypothalamus to control food intake, indicating this pathway's contribution to energy balance of whole body [49]. Calcium activates CaMKKβ to increase T172 phosphorylation. One group study reported that both AMP and ADP promote T172 phosphorylation via CaMKKβ [41], while another study reported AMP not ADP regulates activation via LKB1, but not CaMKKβ [38].
AICAR, the first discovered pharmacologic activator of AMPK, is an adenosine-regulating agent that was approved by the Food and Drug Administration (FDA), named Acadesine [50]. AICAR can be phosphorylated into the AMP analog “ZMP” and enhance the activity of AMPK, which may prolong the survival of cultured rat embryonic hippocampal neurons subjected to oxygen glucose deprivation (OGD), a classical model used to mimic ischemia in vitro [51]. Inhibition of AMPK by Compound C (a classical AMPK antagonist) exacerbates OGD-induced neuronal injury, while treatment with AICAR reverses the effects above [52]. Antimycin A, a mitochondrial respiratory inhibitor in oxidative phosphorylation, is able to maintain intracellular pH homeostasis of the hippocampal neurons by recruiting Na+/H+ exchanger 5 (NHE5) to the cytomembrane, whereas these effects are effectively eliminated by Compound C, the NHE inhibitor HOE694, or AMPK mutation [53, 54]. Together, upstream agonists/activators are necessary for the activation of AMPK and to guarantee its protective effects during metabolic stress.
Antagonists
Compounds C and C75 are two important antagonists of AMPK. Zhou and colleagues studied more than 10,000 hydrones and identified that Compound C is a reversible inhibitor of AMPK under various levels of ATP [55]. Initially, studies on Compound C were still limited to in vitro experiments of the CNS, despite in vivo experiments began to be carried out a few years ago [47, 56]. Zhu et al discovered that inhibition of AMPK by Compound C reduces the viability and inhibits the energy metabolism of cultured primary cortical neurons under physiological conditions [57]. Moreover, atorvastatin treatment enhances the phosphorylation of AMPK and the expression of vascular endothelial growth factor (VEGF) in cultured human umbilical vein endothelial cells (HUVEC), whereas these effects are abolished by Compound C [34]. The inhibition of AMPK by Compound C was also reported by Li's [58] and Izumi's groups [59].
C75 is a synthetic FA synthase inhibitor or carnitine palmitoyltransferase (CPT-1) stimulator that indirectly restrains the phosphorylation of AMPK [60]. C75 rapidly reduces the level of the phosphorylated AMPK α subunit (pAMPKα) in the hypothalamus, even in fasted mice with elevated hypothalamic pAMPKα levels. C75 reduces the levels of pAMPK α and phosphorylated cAMP response element-binding protein (pCREB) in the arcuate nucleus neurons of the hypothalamus [61]. Moreover, Landree and colleagues discovered that C75 can decrease food intake and induce weight loss by inhibiting AMPK in primary cultures of cortical neurons [62]. McCullough and colleagues reported that the suppression of AMPK by C75 plays a role in not only the ischemic area but also the non-ischemic area. The inhibition of AMPK by C75 in the non-ischemic area is delayed and secondary to the ischemic area [56]. It was reported that C75 injections in mice induce rapid reduction in phosphorylated-AMPK (pAMPK) levels [61] and ATP levels in cultured hypothalamic neurons [63]. Strictly speaking, compound C and antimycin A cannot be regarded as specific inhibitors of AMPK, indicating the pleiotropic effects of them should be observed.
Regulation of energy metabolism in the brain
The concentration of ATP within most eukaryotic cells is kept at a considerably constant level, despite wide fluctuations in the demand for ATP. In order to achieve this goal, cellular changes in ATP levels are required to be monitored for restoring ATP levels to main energy homeostasis. Of all systems, AMPK stands at the crossroads of various metabolic processes and is involved in the metabolic changes that are closely related to cell survival, growth, aging, and cell death [64]. A number of studies have indicated that AMPK is able to protect against ischemic stroke [65, 66], hemorrhage stroke [67], NDD [68], and brain tumors [69], suggesting that AMPK is a protective regulator in cerebral diseases. The brain constitutes only 2% of the body's weight but utilizes half of the total glucose supply, which may be attributed to the fact that neurons are the most metabolically demanding cells in humans[70]. Neurons manifest as high energy metabolism and conduct numerous energy-demanding processes, including maintaining ion gradients across membrane, firing action potentials, and producing other forms of bioelectricity. Neurons are able to synthesize glycogen, whereas astrocytes are one of the only 'warehouses' of glycogen, which allows glycogen to transfer into astrocytes [71]. Unlike cardiomyocytes, neurons utilize only glucose and poorly store energy, which makes neurons more vulnerable to cerebral ischemia that might cause irreversible injury to the cerebrum. Under harmful stimuli, neurons can be activated and have a higher expression of AMPK for glucose uptake and utilization. Therefore, sufficient and stable AMPK content is necessary to maintain the life and activity of neurons.
In the brain, glutamate is the primary neurotransmitter regulating most synaptic transmission associated with AMPK signaling [72]. Stimulation of glutamate by reduced cellular ATP contributes to compensatory increases in AMPK in cerebellar granule neurons [73]. Glutamate excitation leads to an increase in GLUT3 that persists for hours, concomitant with enhanced expression of AMPK, whereas knockdown or pharmacological inhibition of AMPK may contribute to a reduction of GLUT3 and glucose utilization [72]. Taken together, AMPK acts as a nuclear regulator of energy metabolism and plays a pivotal role in cerebral energy homeostasis (Fig 1).
Structure of AMPK subunits. AMPK is a heterotrimeric complex of α, β, and γ subunits. The main functions of α, β, and γ subunits are catalysis, regulation, and conjunction, respectively. The α subunit is composed of a kinase domain, an auto-inhibitor domain (AID), and a α-subunit carboxy-terminal domain (α-CTD). The β subunit consists of β-CTD and CBM and functions as the 'bridge' between the α and γ subunits via β-CTD. The γ subunit has an N-terminal domain (NTD) and four tandem repeats of cystathionine β synthase (CBS) to form Bateman domains.
Regulation of AMPK in ischemic stroke
Oxidative stress
Oxidative stress is a pathological process manifesting overproduction of reactive oxygen species (ROS) and reactive nitrogen species (RNS) under harmful stimuli [74-79]. These alterations result in an imbalance of antioxidants and ROS/RNS and cause immense injury to neurons. The brain is a highly energy-consuming organ that almost utilizes 50% of the total energy. However, the energy storage of brain can sustain only a few minutes without energy influx, which determines the high liability of the brain to ischemic stimuli.
Compelling evidence has suggested that oxidative stress involving AMPK plays a pivotal role in the brain. Trans-caryophyllene (TC), a bicyclic sesquiterpene, is a selective agonist of type 2 cannabinoid receptor (CB2R). Choi and colleagues studied cortical neuron/glia-mixed cultures from Sprague-Dawley rats and found that aftertreatment of TC enhances AMPK/cAMP response element-binding protein (CREB) signaling and reduces OGD/reoxygenation-evoked neuronal oxidative stress. Exposure to hypoxia results in a significant increase in ROS that is closely related to neurological pathologies. Extracellular superoxide dismutase transgenic mice exposed to hypoxia for 10 days display increased pAMPK activity and decreased malondialdehyde (MDA) and lactic acid levels compared to the controls. This revealed that activation of cortical CB2R can ameliorate oxidative stress via activating AMPK/CREB signaling [80]. Pathophysiologically, quiescent astrocytes convert to the activated state and protect neurons from harmful stimuli, therefore rapidly alleviating injury to NVUs. AICAR, a cell-permeable activator of AMPK, significantly promotes the production of ketone bodies in rat cortical astrocytes under hypoxic conditions, thereby attenuating the oxidative injury of NVUs [81]. These data demonstrate that AMPK has potential prophylactic roles in diseases with chronic ischemia/hypoxia-induced cerebral injury, suggesting that AMPK is a pivotal and protective molecule in cerebral ischemic/hypoxic diseases.
Autophagy
Energy deprivation secondary to cerebral ischemia often leads to cellular autophagy, a conservation process that produces energy by degrading and recycling cellular constituents. Autophagy is a dynamic intracellular process consisting of five steps: autophagy induction, vesicle formation, autophagosome fusion, lysosome fusion, and autophagosome degradation [82-87].
Pre-activation of AMPK has been proven to promote neurological autophagy and to ameliorate ischemic injury [88, 89]. Cerebral ischemic preconditioning (IPC), consisting of transient episodes of subthreshold ischemic insult, induces a low level of energy stress and increases cerebral tolerance to subsequent fatal ischemic exposure [90]. Cerebral IPC can activate AMPK and induce autophagy, accompanied by reductions in cell apoptosis, infract volume, and neurological deficits during ischemic stroke. Furthermore, rats injected with a single dose of Compound C 5 min before cerebral IPC display suppressed autophagy and increased infarct size, suggesting that AMPK-dependent autophagy contributes to neuroprotection [90]. Metformin, discovered in 1922, is the first-line oral medication for the treatment of type 2 diabetes [91, 92]. Acute metformin preconditioning induces autophagy by activating AMPK and confers neuroprotection against subsequent cerebral ischemia. Jiang and colleagues discovered that metformin significantly reduces the infarct volume and neurological deficits 96 h after ischemia, implying that the neuroprotection of metformin could last for at least 4 days, whereas this process can be inhibited by pretreatment with Compound C. Notably, they also discovered that metformin does not significantly affect some physiological parameters (including PaCO2, PaO2, blood pressure, and blood glucose levels), further supporting the pharmacological value of AMPK. This suggests that pre-activation of autophagy by AMPK is protective against cerebral ischemia [93]. Kallikrein, a subgroup of serine proteases, protects SH-SY5Y neurons against OGD-induced injury via autophagy induction via AMPK/tuberous sclerosis complex (TSC)/mTOR pathway. However, inhibition of AMPK by Compound C prevents the phosphorylation of AMPK and inhibits autophagy. Meanwhile, the knockdown of AMPK α1 by lentivirus achieves the same effects [94]. Notably, several studies have demonstrated that metformin is not a direct activator of AMPK, and has effects that are AMPK-independent [95, 96], suggesting that we shall equitably think of the effects of metformin-induced AMPK activation.
Apoptosis
Apoptosis is a process of programmed cell death that occurs in multicellular organisms. It is a dynamic process of blebbing, cell shrinkage, nuclear fragmentation, chromatin condensation, chromosomal DNA fragmentation, and global mRNA decay [97-101]. Apoptosis represents the execution of an ATP-dependent death program, which is often initiated by death ligand/death receptor interactions, such as Fas ligand and Bax/Bcl-2 [102]. Neuron apoptosis might be induced and even worsen under pathological situations, including ischemia and hyperglycemia [103].
Neuron apoptosis is usually secondary to ischemic injury and serves as an index of ischemic extent, whereas this process may be ameliorated by AMPK. IPC can activate AMPK and ameliorate cell apoptosis in the peri-infarct region, as evidenced by the reduced percentage of TUNEL-positive cells, suggesting the ability of AMPK to reduce apoptosis in ischemic stroke [90]. Acute preconditioning of metformin ameliorates neuron apoptosis via activating AMPK, corresponding to a reduced percentage of TUNEL-positive cells and endoplasmic reticulum stress (ERS) during subsequent ischemic stimuli [93, 104]. Chikusetsu Saponin Iva preconditioning activates AMPK/GSK-3β pathway and inhibits apoptosis of PC12 cells, as evidenced by reduced amounts of caspase-3 and Bax/Bcl-2. Activated AMPK and suppressed apoptosis also reduce infarct size and improve neurological outcomes after ischemia reperfusion injury (IRI) in MCAO mice. This indicates that Chikusetsu Saponin Iva protects against cerebral ischemia through the AMPK/GSK-3β-restrained apoptosis pathway [105]. Moreover, Yang et al discovered that preconditioning of Apelin-13 promotes the phosphorylation of AMPK and suppresses apoptosis after cerebral IR, while Apelin-13 has minimal effect on attenuating apoptosis when Compound C is added. This study provides evidence that Apelin-13 upregulates the levels of AMPK to perform neuroprotective effects by restraining apoptosis [106]. Estrogen deficiency induced by ovariectomy aggravates brain infarction in experimentally induced cerebral ischemic rats, while estrogen pretreatment reduces ischemia-induced cerebral injuries. In OGD neurons, aftertreatment of estrogen promotes AMPK activation and suppresses apoptosis through estrogen receptor α while neuroprotection of estrogen is prevented by AMPK inhibition [107]. These evidences suggest that inhibiting apoptosis by AMPK is a potentially valuable therapeutic strategy for ischemic stroke in the future.
Mitochondrial dysfunction
Mitochondria, also referred to as the 'cellular power centers', are semiautonomous, self-reproducing organelles found in the cytoplasm of most eukaryotes [108, 109]. Mitochondria can generate high quantities of ATP for various biological actions in both physiological and pathological conditions [110]. Mitochondrial dysfunction is the insufficiency and dysregulation of mitochondria, usually caused by harmful or extreme conditions, including cerebral ischemia and hypoxia [111, 112].
AMPK is also able to ameliorate cerebral ischemic/hypoxic injury via the improvement of mitochondrial dysfunction. Recent studies reported that AMPK can signal to the mitochondrial fission factor (MFF) and induce rapid mitochondrial fragmentation, thereby initiating the biogenesis of new mitochondria and alleviating mitochondrial dysfunction under serious energy crises [113]. Activation of CB2R decreases mitochondrial depolarization and maintains normal mitochondrial membrane potential caused by OGD/R in mixed culture of cortical neurons/glia, as evidenced by the reduced release of mitochondrial proteins and cytochrome c in rat cortical neuron/glia-mixed cultures [80]. Ashabi and colleagues discovered that pretreatment of metformin for 2 weeks enhances the activity of AMPK. They further demonstrated that AMPK induces neuronal mitochondrial biogenesis in ischemic rats via proliferator-activated receptor gamma coactivator-1α (PGC-1α) signaling. However, inhibition of AMPK by Compound C reverses such protective effects and exacerbates the levels of dysfunction [66, 114]. Downregulation of Sestrin2 inhibits neuronal mitochondrial biogenesis by downregulating AMPK/PGC-1α pathway following transient MCAO, which correlates with reduced expression of NRF-1 and TFAM. However, treatment of AICAR activates AMPK and enhances mitochondrial biogenesis, thereby alleviating ischemic injury [115]. Taken together, these studies demonstrate that AMPK is a pivotal molecule against mitochondrial dysfunction through maintaining mitochondrial biogenesis and normal membrane potential.
Glutamate excitotoxicity
Glutamate, the derivative of glutamic acid, is the most common excitatory neurotransmitter in the CNS and plays a pivotal role in neuronal activity [116]. Glutamate mediates synaptic transmission and is necessary for neuronal growth, maturation, and synaptic plasticity. Cerebral ischemia induces excessive release of extra-synaptic glutamate and the activation of glutamate receptors [117], the process of which is also known as glutamate excitotoxicity. It may trigger disproportional calcium and sodium into neurons and serious neuronal death [118].
Compelling evidence has exhibited that activation of AMPK is able to protect neurons from glutamate excitotoxicity [119]. Curcumin, a natural polyphenolic compound from Curcuma longa, has been proven to exhibit beneficial effects on neuronal protection by activating AMPK. Li and colleagues reported that pretreatment of curcumin-induced production of AMPK protects neurons from glutamate neurotoxicity in the hippocampus [120]. GABA(B) receptors are heterodimeric G protein-coupled receptors composed of R1 and R2 subunits. AMPK binds directly to GABA(B) receptors, phosphorylates site S783 of the R2 subunits and suppresses the release of glutamate, which enhances the coupling of GABA(B) receptors to G-protein-gated inward rectifier K+ (GIRK) channels, promoting energy storage and neuronal survival after hypoxic injury. This reveals that AMPK may decrease synaptic activity and limit neuronal excitotoxic injury to conserve cellular energy levels [121]. Intriguingly, it is worth noting that injury-induced phosphorylation of GABA was observed mainly in the hippocampal region, which usually disaccords with the location of MCAO [88]. These findings suggest a probable link between glutamate excitotoxicity and AMPK involving neuroprotection, which paves a new avenue for targeting AMPK in the treatment of cerebral ischemia.
Neuroinflammation
Neuroinflammation is inflammation of the nervous tissue, which might be initiated in response to infection, toxic metabolites, or autoimmunity [122, 123]. It is characterized by activated microglia and astrocytes that express inflammatory mediators, chemokines, and complement proteins in the CNS [124, 125]. Neuroinflammation remains the primary cause of morbidity and mortality in stroke-induced secondary brain injury [126]. The CNS is an immunologically privileged site because the blood-brain barrier (BBB) can block peripheral immune cells. Under ischemic stroke, circulating peripheral immune cells may surpass BBB and encounter neurons and glial cells, which express major histocompatibility complex (MHC) molecules for protection. This response is concomitant with the further migration of leukocytes through the BBB and may lead to injury of the cerebrum.
Ischemic stroke may initiate a series of cellular responses that include the activation of resident glial cells and the recruitment of peripheral immune cells, thereby contributing to central neuroinflammation and injury. Metformin can promote the phosphorylation of AMPK and alleviate neutrophil infiltration, as evidenced by inhibited activities of nuclear factor-κB (NF-κB), IL-1β, IL-6, and tumor necrosis factor-α (TNF-α) after MCAO. Furthermore, activated AMPK by metformin aftertreatment alleviates OGD-induced expression of intercellular adhesion molecular (ICAM)-1, whereas this effect is reversed by the administration of Compound C [127]. Pretreatment of metformin activates AMPK, reduces cellular levels of NF-κB, TNF-α, and cyclooxygenase-2 but increases heme oxygenase-1 (HO-1) and glutathione, which accelerates anti-inflammatory pathways and inhibits neurological inflammatory responses after transient global cerebral ischemia [65]. Aftertreatment of sinomenine inhibits the activation of NLRP3 inflammasome and release of neuronal inflammatory cytokines (IL-1β, IL-6, IL-18, and TNF-α) in C57BL/6 mice after MCAO. Further, the suppressive effect of sinomenine on NLRP3 inflammasome is blocked by an AMPK inhibitor, Compound C. This finding demonstrates that sinomenine activates AMPK and exerts a neuroprotective effect in ischemic stroke by inhibiting NLRP3 inflammasome [128]. These findings demonstrate that AMPK is a strong protective molecule against cerebral ischemia by attenuating neuroinflammation (Fig 2).
Expression of different subunits of AMPK in human organs. AMPK is widely but distinctly distributed in different tissues and organs. The brain mainly contains the α1, α2, β1, γ1, and γ2 subunits. The heart and liver mainly contain the α1, α2, γ1, and γ2 subunits. The lung and kidney mainly contain the α1, γ1, and γ2 subunits. The skeletal muscle mainly contains the α2, β2, γ1, γ2, and γ3 subunits.
Endothelial cells and angiogenesis
Endothelial cells are epithelium that line the interior surface of blood vessels and lymphatic vessels. Among them, endothelial cells that line vessel walls are called vascular endothelial cells. Clinically, stroke patients with a higher density of blood vessels usually have less morbidity and longer survival, indicating that angiogenesis is important for neurological repair in the ischemic region [129]. Venna and colleague used C57BL/6N male mice subjected to 60-min MCAO and discovered that aftertreatment of metformin significantly promotes the expression of VEGF, concomitant with decreased infarct size and glial scaring. When tested in AMPKα2 knockout mice, they found that metformin did not have the same beneficial angiogenic effects, suggesting that metformin-induced protective effects are mediated by AMPK. Notably, they also reported that a 30-day treatment of metformin may improve neurological outcomes, further supporting the pharmacological value of metformin in ischemic stroke [130]. Jin et al reported that chronic metformin treatment after stroke activates AMPK and significantly enhances angiogenesis, as revealed by elevated vessel density and increased CD31, the endothelial marker, in the ischemic striatum [131]. Therefore, AMPK has an important role in upregulating angiogenic factor VEGF and promoting angiogenesis to ameliorate ischemic injury.
Roles of AMPK in ischemic stroke. A. AMPK inhibits neuroinflammation in ischemic stroke B. AMPK suppresses the levels of oxidative stress in ischemic stroke. C. AMPK promotes autophagy against ischemic stroke. D. AMPK restrains glutamate release and excitotoxicity. E. AMPK ameliorates mitochondrial dysfunction against ischemic stroke. F. AMPK inhibits apoptosis in ischemic stroke. In short, AMPK helps to main the energy homeostasis via regulating the mechanisms above.
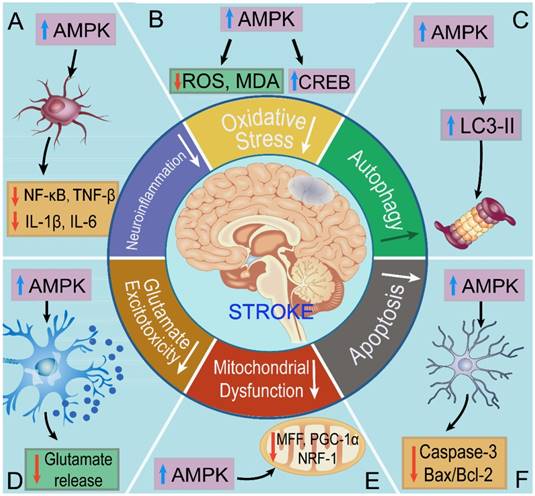
To sum up, we discussed the regulatory mechanisms of AMPK in ischemic stroke, including oxidative stress, autophagy, apoptosis, mitochondrial dysfunction, glutamate excitotoxicity, neuroinflammation, and angiogenesis. AMPK stands at the crossroads of diverse regulatory mechanism to main energy homeostasis during stroke. The findings above demonstrate that pretreatment of estrogen (by inhibiting apoptosis) [107], metformin (by promoting mitochondrial biogenesis and inhibiting neuroinflammation) [66], curcumin (by inhibiting glutamate excitotoxicity) [120], may ameliorates the ischemic or hypoxic symptoms. Moreover, aftertreatment of estrogen (by inhibiting apoptosis) [107], trans-caryophyllene (by inhibiting oxidative stress) [80], metformin (by inhibiting neuroinflammation) [127], and sinomenine (by inhibiting neuroinflammation) also helps to resist against stroke in vivo and in vitro [128]. Moreover, AMPK attenuates ischemic injury in the neurovascular unit, such as neurons, astrocytes, microglial cells, to maintain the neurological homeostasis.
Therapeutic research
A number of studies have introduced several therapeutic strategies for ischemic stroke, such as medical treatment, physical therapy, and receptor-targeted therapy. These treatments all induce their neuroprotective effects on ischemic stroke through AMPK signaling.
Pharmacotherapy
Pharmacotherapy, also referred to as medical treatment, is remarkably distinguished from surgical therapy, radiotherapy, and physical therapy. Due to its approval and preference by patients for thousands of years, it has become the most classical and popular treatment. Ashabi's study revealed that induction of 30 min global cerebral IR leads to significant neuron death, whereas pretreatment with metformin significantly promotes mitochondrial biogenesis and alleviates apoptotic cell death and ischemia-related neurodegeneration in the ischemic hippocampus [66]. Resveratrol is a small natural phytoalexin that plays a defensive role against IRI. Wang and colleagues reported that administration of resveratrol activates AMPK, significantly reducing infarct volumes and protecting neurons from death in recurrent ischemic stroke models, concomitant with improved neurological scores and behavioral deficits [132]. Moreover, resveratrol can activate the cAMP/AMPK/SIRT1 pathway by inhibiting phosphodiesterases and alleviating injury following IR, as evidenced by reduced infarct volumes and improved neurological deficits [133]. Atorvastatin, a lipid-lowering agent, is a powerful AMPK activator [134]. Chaturvedi and colleagues enrolled 4731 patients with a recent stroke or transient ischemic attack and discovered that atorvastatin significantly reduces low-density lipoprotein (LDL) and improves the post-stroke outcomes, suggesting the ability of atorvastatin in stroke treatment [135, 136].
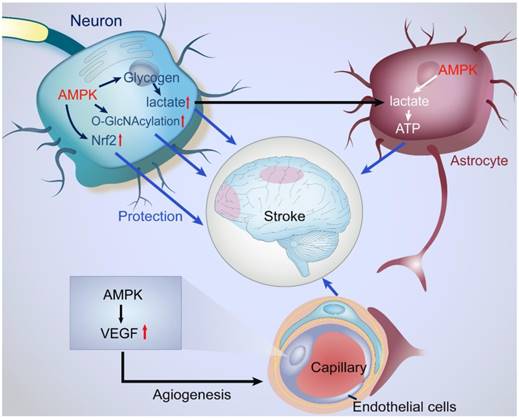
Protective actions of AMPK in the NVU under ischemic stroke. NVU is composed of neurons, astrocytes, endothelial cells, microvessels, basal membrane, and ECM. Under ischemic condition, activated AMPK initiates a series of protective mechanisms in the NVU. AMPK enhances the catabolism of glycogen and glucose to produce ATP in astrocytes while astrocytes do not utilize the metabolite of glucose, lactate. The remaining lactate is transported into neurons for supplying ATP. In neurons, AMPK promotes the O-GlcNAcylation of intracellular proteins against ischemic stimuli. In endothelial cells, AMPK upregulates the levels of VEGF, enhance angiogenesis, and eventually improves microcirculation.
Thrombolytic therapy is another classical method for treating ischemic stroke, especially acute cerebral infarction. Tang et al reported that salvianolic acid, an AMPK agonist and a new drug for ischemic stroke treatment, can alleviate neuron death in the penumbra, reduce infarction volume, and improve neurological scores and outcomes. Notably, they discovered that administrating salvianolic acid 2 minutes before reperfusion is a promising time point for thrombolysis therapy, suggesting the possibility of salvianolic acid as an adjuvant thrombolytic drug [137]. Tissue-type plasminogen activator (tPA) is a thrombolytic drug that activates AMPK. Wu and colleagues discovered that the intravenous administration of recombinant tPA after tMCAO induces AMPK activation in the synaptic space and glucose uptake in the ischemic brain, as revealed by positron emission tomography (PET) studies and biochemical analyses with synaptoneurosomes [138]. This reveals that treatment with tPA activates AMPK signaling in the synaptic space and enhances cerebral tolerance to ischemic metabolic stress. However, the time of thrombolysis is short and usually within 6 h, which limits its utilization in stroke patients.
Physical therapy
Electroacupuncture, also known as acuelectrostimulation pretreatment, is a special form of acupuncture that has shown therapeutic effects in animal experiments and stroke patients [139]. In 2003, Xiong and colleagues proposed that electroacupuncture preconditioning is able to induce cerebral ischemic tolerance [140]. Electroacupuncture, which combines the traditional advantages of acupuncture treatment and acuelectrostimulation, has great advantages, as it can be readily controlled, standardized, and objectively measured [141]. Electroacupuncture preconditioning significantly enhances the expressions of AMPKα and pAMPKα, concomitant with reduced TUNEL-positive cells and attenuated neuron apoptosis after stroke. However, intraperitoneal injections of Compound C suppress this phenomenon and exacerbate ischemic injury. This finding suggests that electroacupuncture preconditioning alleviates cerebral ischemic injury via AMPK signaling [142].
Exercise can be viewed as a chronic metabolic stress that leads to an increase in regional cerebral blood flow and metabolic functioning [143]. Adult male Sprague-Dawley rats were subjected to 1-3 weeks of exercise and displayed high expression of AMPK, concomitant with decreased infarct volume and enhanced glucose uptake and ATP production after stroke. Notably, they discovered that exercise attenuates neurological deficits and improves neurological outcomes in the long-term [144]. This study revealed that an increased expression of AMPK following exercise may enhance cerebral ischemic tolerance. Collectively, physical therapy targeting AMPK is an important type of non-pharmacotherapy compliant to stroke patients.
Receptor-targeted therapy
N-methyl-D-aspartic acid (NMDA), first synthesized in the 1960s, is a water-soluble synthetic substance and amino acid derivative. Its receptor has been widely thought to promote survival in the CNS [145]. Upregulation of NMDA receptor activity contributes to sustained AMPK activation and increased glucose uptake by facilitating the transportation of GLUT4 to the plasma membrane. In contrast, ketamine, an antagonist of NMDA receptors, decreases the activity of AMPK and the content of GLUT4 in the plasma membrane following cerebral ischemia [146]. As discussed in Section 4.1, trans-caryophyllene is a CB2R-selective agonist and an AMPK activator. In vitro, trans-caryophyllene ameliorates neuron death under the condition of OGD/R, which is correlated with reduced mitochondrial dysfunction and intracellular oxidative stress. In vivo, post-ischemic treatment with TC decreases cerebral infarct size and edema and increases CREB and brain-derived neurotrophic factor, while a selective AMPK inhibitor abolishes the neuroprotective effects [80]. These findings reveal that AMPK is a potent drug candidate of receptor-targeted therapy for cerebral ischemia (Table 1).
Therapeutic research of AMPK in ischemia stroke
| Models/Subjects | Therapeutics | Methods | Effects | Evidence | Reference |
|---|---|---|---|---|---|
| Young male Wister rats | Pharmacotherapy | Pretreatment of metformin for two weeks before global cerebral IR | Metformin significantly promotes mitochondrial biogenesis, alleviates apoptotic cell death and ischemia-related neurodegeneration in the ischemic hippocampus | Elevated levels of AMPK and decreased levels of Bax/Bcl-2, caspase-3, and PARP | [66] |
| Old male Wistar rats | Pharmacotherapy | Pretreatment of resveratrol for 3 days before MCAO | Resveratrol activates AMPK and reduces infarct volume | Improved neurological scores and decreased neuron death | [132] |
| Old male Sprague-Dawley rats | Pharmacotherapy | Pretreatment of resveratrol for 5 days before the MCAO | Resveratrol reduces infarct volume, improves neurological deficits and injury following IR. | Activated cAMP/AMPK/SIRT1 pathway and reduced phosphodiesterases | [133] |
| cMale Sprague-Dawley rats | Pharmacotherapy | Administration of TSI for 2 min before reperfusion via femoral vein injection | TSI reduces infarction volume, and improves neurological scores after thrombolysis therapy | Increased levels of AMPK, NADPH, and PKC, and decreased levels of ROS | [137] |
| Male wild-type C57BL/6J mice | Pharmacotherapy | Intravenous administration of recombinant tPA after tMCAO | Recombinant tPA induces glucose uptake against ischemic injury | Increased expression of GLUT around synaptic space | [138] |
| Wild-type C57BL/6J | Electroacupuncture | Preconditioning for 5 days before ischemia | Electroacupuncture attenuates neuronal apoptosis and ischemic injury after stroke | Reduced number of TUNEL-positive cells | [142] |
| Adult male Sprague-Dawley rats | Exercise | For 1-3 weeks before ischemia | Decreased infarct volume and neurological deficits, and improved neurological outcome | Enhanced glucose uptake and production of ATP after stroke | [144] |
| Adult male Sprague-Dawley rats | Receptor-targeted therapy | NMDA receptor activation under ischemic condition | Sustained AMPK activation and increased glucose uptake | Facilitating the transportation of GLUT4 to the plasma membrane | [146] |
| Rat cortical neurons/glia mixed cultures | Receptor-targeted therapy | TC was applied immediately before initiation of OGD | Ameliorated neuron death, decreased cerebral infarct volume and edema | Reduced mitochondrial dysfunction and intracellular oxidative stress | [80] |
Prospects
Equitable evaluation of AMPK
Existing contradictions
Although numerous studies have shown that AMPK is a protective molecule in ischemic stroke, we would like to discuss existing discrepancies to provide comprehensive information to readers. Some studies have revealed that restrained AMPK is capable of exerting protective effects. Nam et al discovered that increased AMPK immunoreactivity may enhance the ischemic injury in the hippocampal CA1 region 1-2 days after IR, whereas Compound C reverses the effects above, correlated with decreased reactive gliosis, ATP depletion, and lactate accumulation, suggesting that inhibition of AMPK has protective effects against ischemic stroke [147]. Suppression of AMPK by Compound C reduces infarct size and improves neurological outcome at 24 h after reperfusion, which is correlated with restrained release of microglial pro-inflammatory factors [148]. Hypothermia is one of the few therapies that has been translated into the clinic for global cerebral ischemia [149]. Hypothermia-related inhibition of pAMPK reduces infarct size, edema, and cerebral metabolic rate after MCAO, and this protection is enhanced after administration of Compound C. This study provides evidence that hypothermia exerts its neuroprotective effects by inhibiting AMPK in stroke [150].
Activated AMPK or elevated expressions of AMPK have been shown to exert harmful effects. One high dose of metformin remarkably increases the levels of pAMPK and lactic acid, promotes the consumption of energy, and exacerbates cerebral metabolic crisis and neuron death [47, 56]. Moreover, long-term treatment with metformin for treatment of stroke may increase the deposition of β-amyloid, which possibly results from chronic metformin treatment [151]. These findings all suggest that AMPK is harmful, and its downregulation might be a potent therapy for stroke.
Additionally, some studies maintain that AMPK is a double-edged sword in ischemic injury. AMPK overexpression downregulates the levels of CREB by promoting the SIRT1/miR-134 pathway. However, phosphorylated CREB is decreased in AMPK knockout primary hippocampal neurons. Therefore, AMPK plays a dual role in the regulation of CREB in OGD models, exerting a negative effect on total CREB expression by elevating the levels of SIRT1/miR-134 and a positive effect by phosphorylating CREB [152]. In primary neurons of hypoxia/ischemia, AMPK activation results in elevated cell death, whereas inhibition of AMPK may protect neurons from harmful stimuli, implicating that activated AMPK is involved in the injury process. Conversely, activation of AMPK prior to injury enhances the tolerance of neurons to subsequent insult. These data reveal that the dual roles of AMPK likely depend on whether it is activated prior to or during the ischemic/hypoxic insult [153]. In summary, AMPK indeed has dual actions on ischemic stroke, and its internal role needs further research.
Equitable evaluation
Since early research in the 1980s, the enthusiasm and efforts dedicated to studying AMPK have continuously increased. AMPK can restrain oxidative stress, apoptosis, mitochondrial dysfunction, glutamate excitotoxicity and neuroinflammation and promote autophagy, thereby promoting glucose uptake and energy metabolism. Enhanced glucose uptake replenishes cellular energy deprivation and ameliorates metabolic crises during and after ischemia. We reckoned that during IPC and the post-stroke chronic convalescent period, elevated AMPK is helpful for repairing brain tissue and metabolic homeostasis by producing more ATP, thus enhancing the processes of angiogenesis and neurogenesis. However, elevated AMPK is harmful and consumes excessive energy during acute cerebral ischemia, which may exacerbate cerebral injury and lead to off-target effects during the midanaphase of acute cerebral ischemia [73, 154].
The question remains how numerous studies have reached opposite conclusions. It is possible that these differences may result from different ischemic degrees, durations, and models and different cell types [155, 156]. The inhibition of AMPK contributes to 'neuronal hibernation' during acute ischemia and provides continuous neuroprotective effects, just as hypothermia can decrease energy requirements and attenuate metabolic crises. Activated AMPK promotes the expression of GLUT and the translocation of abundant glucose to neurons, which may result in post-ischemic hyperglycemia and subsequent brain injury, including cellular edema, apoptosis, and even dysfunction of the BBB. These alterations can promote the production of ROS in endothelial cells, destroy vessel walls, and ultimately enhance hemorrhagic risk after cerebral infarct [73, 154]. Simulating in vitro neuronal metabolism is difficult and differs from in vivo metabolism. Neurons are post-mitotic cells and are intolerant to cellular stress due to poor energy stores, especially without the supporting glial cells and vasculature that characterize the in vivo state. Moreover, the culture media of neurons typically contains 25 mmol/L glucose, which is much higher than physiological levels (between 0.82 and 2.4 mmol/L) [156].
For example, Li et al reported that the timing, duration, and amount of AMPK activation are key factors in determining the ultimate effects of AMPK in ischemic brains, including pre-ischemia and post-ischemia, acute versus chronic medication, and normal expression versus over-expression of AMPK [157]. They discovered that acute administration of metformin (3d) exerts deleterious actions, while chronic administration (3w) reverses these effects and exerts a neuroprotective role in ischemic stroke. Iwabuchi and colleagues systematically reviewed the complex influence of AMPK on cerebral ischemic tolerance. They reported that inhibition of AMPK during preconditioning initially suppresses the upregulation of GLUT3 but subsequently induces rapid upregulation of GLUT3, thereby leading to a biphasic effect [158]. Collectively, most studies suggest that AMPK is a protective molecule, although few studies maintain opposing opinions.
Thrombolytic management and clinical research
As discussed above, stroke is a major global disease that threatens human health. It is characterized by high mortality and disability, and there are no ideal therapeutic methods. Currently, thrombolysis via the AMPK pathway is almost the only available treatment for acute ischemic stroke [159]. The ideal thrombolytic time of ischemic stroke is 3-6 hours, which is also known as the window phase. During this period, energy deprivation is the 'blasting fuse' that leads to a series of secondary injuries, including oxidative stress, acidosis, apoptosis, and excitotoxicity, and thus proper thrombolysis during this period is effective and might reduce post-stroke complications. However, only 3%-5% of stroke patients can receive thrombolytic therapy as a result of the strict time window, low precaution consciousness of cerebrovascular accidents, and reperfusion injury after thrombolysis [160]. Thereby, clinical and basic research require additional efforts to solve this problem.
Clinically, anesthetists and surgeons are committed to cerebral resuscitation and protection during operations [161]. Cerebral injury during the perioperative period is a significant adverse event that results from the relative cerebral ischemia, usually including coronary artery bypass grafts (CABG) and intracranial tumor surgeries [162]. Glucose-insulin-potassium (GIK) is an important method for upregulating the levels of AMPK. In a previous study, 217 patients who underwent aortic valve replacement were assigned to GIK or placebo over a 4-year period. The result revealed that GIK treatment induces a substantial increase in pAMPK and its downstream molecule Akt compared to controls, concomitant with a significant reduction in the incidence of low cardiac output [163]. According to the conditions of systemic circulation, the blood flow of the internal carotid artery and the vertebral artery depends largely on cardiac output, and thus, we presume that GIK infusion during the perioperative period upregulates the levels of AMPK, which is helpful for restoring brain blood flow and ameliorating cerebral injury. Moreover, Kim et al recruited 1609 subjects (aged from 60 to 80) and performed a multivariate logistic regression analyses. They discovered that the polymorphism of PRKAG2-26C/T gene has a significant association with cognitive impairment in stroke patients (OR, 1.6; 95% CI, 1.1—2.3) [164], suggesting that AMPK has a role not only in metabolic function but also in cognitive function of post-stroke patients. Notably, Campbell's group recruited 70 patients and randomly assigned patients into two groups. The results showed that patients who undergo stent retriever thrombectomy and receive alteplase (an AMPK activator) display improved reperfusion, early neurologic recovery, and improved functional outcomes compared to those who receive alteplase alone. Of the experimental group, 89% of patients exhibited restored the cerebral perfusion compared to 34% of patients in the control group. In addition, 71% of experimental patients achieved functional independence compared to 40% of control patients [165]. This trial demonstrated that an integrated combination of thrombolysis and interventional therapy may achieve profit maximization for ischemic stroke rather than AMPK-related thrombolysis alone.
Ethical issues
Additionally, medical ethical issues concerning treating stroke patients via AMPK are issues that cannot be neglected. Medical decision-making in stroke patients can be complex and often involves ethical challenges, from the selection of thrombolytic drugs to therapeutic methods [166]. Over the past few years, several predictive models have been proposed and are conducive for estimating the outcomes of ischemic stroke, which may affect the choice of the therapeutic method in patients [167, 168]. For patients with cerebrovascular stenosis, simple medical treatment with AMPK-related thrombolytic drugs (alteplase) may restore blood flow and ameliorate neurological impairments [165]. However, thrombolytic therapy is unavailable in some stroke patients who should have received cerebrovascular interventional therapy. Previous cardiogenic stroke patients can take thrombolytic drugs orally to prevent a recurrent attack, whereas such drugs increase the possibility of acute hemorrhage, and effective treatments to remedy injury are lacking [169]. Thus, the selection of the ideal treatment is difficult and is usually decided when patients are unconsciousness, which may leave a series of ethical issues. Moreover, previous studies have shown that exercise can ameliorate ischemic symptoms via AMPK signaling [144]. Thus, we suggest that post-stroke rehabilitation training activates the AMPK pathway and may replace drugs to some extent, which also represents an ethical concern regarding promoting the improvement of the locomotor system and cerebral function of stroke patients.
Potential directions
So far as the status, stroke is still the fifth leading cause of death but the first cause of disability among all diseases, which produces in the United States and globally. Annually, about 795,000 people experience ischemic or hemorrhagic stroke. Approximately 610,000 of them are first events and 185,000 are recurrent events in the United States [170].
Metabolic therapy for treatment of ischemic stroke via AMPK signaling is almost the most important finding and of great potential for future research. The greatest discovery in this field may be the establishment of the MCAO model and the application of thrombolytic drugs. The MCAO model includes a relatively simple extracranial microsurgical dissection procedure that interrupts cerebral blood flow, which paves a way for subsequent scientific research. Clinically, both statins and metformin are used clinically, which is helpful to high-risk population and reduces the stroke incidence [135, 171]. Thrombolytic therapy is applied in acute cerebral ischemia, especially acute cerebral infarction. However, there still exist some problems for the treatment of AMPK. First, whether AMPK plays a protective role in ischemic stroke is elusive, and some reports have identified negative effects [147-149]. Second, only a few of the AMPK activators have been applied in the clinic, and there are very few clinical trials of AMPK. Third, pre-stroke patients are almost asymptomatic, and most strokes are acute and may contribute to secondary injury. Finally, the thrombolytic time of stroke is short, and there are no sufficient effective therapeutic methods based on AMPK signaling after the window phase. New directions of AMPK studies should 1) evaluate the actions of AMPK in acute/chronic cerebral ischemia, or cerebral hemorrhage, 2) choose a more selective AMPK activator, 3) target NVUs rather than the neuron alone in ischemic stroke, 4) provide more reliable clinical evidence (e.g. randomized controlled trials) based on AMPK signaling, and 5) explore a cerebral drug delivery system involving AMPK signaling [172].
This review is committed to describing the beneficial roles of AMPK following ischemic stroke. AMPK is a Ser/Thr kinase that maintains cerebral metabolic homeostasis by enhancing catabolic pathways and restraining ATP-consuming processes. Initially, we provide a general introduction of AMPK and ischemic stroke, including descriptions of its structure, expression, localization, and upstream regulators. The high expression of AMPK in the cerebrum determines its protective biological actions under ischemic stimuli, such as inhibiting oxidative stress, apoptosis, mitochondrial dysfunction, glutamate excitotoxicity, and neuroinflammation and promoting autophagy. The NVU is the basic elementary unit of the brain tissues, and therefore, targeting NVUs via AMPK may induce better effects than targeting neurons alone. Next, we introduce the roles of AMPK in different phases, in commonly used animal models, and in various therapeutic methods, including medical treatment, receptor-targeted therapy, and physical therapy. However, the results of some studies question the protective role of AMPK in ischemic stroke, and we have discussed these discrepancies in detail. Moreover, additional benefits of AMPK beyond those for cerebral ischemic stroke have also been found for other cerebral diseases, such as neurological improvements and neurogenesis, the attenuation cerebral hemorrhage, benefits in NDD, and amelioration of neural tumors. Lastly, we discuss controversial opinions, ethical issues, and potential directions of future AMPK studies based on the existing evidence. Although some scholars suggest that activating AMPK is harmful and can aggravate ischemic injury, most studies demonstrate that it is a protective molecule in ischemic stroke. However, few studies have identified the role of AMPK in the clinic, which leads to an incomplete picture of AMPK. Therefore, more clinical studies are required to elucidate the roles of AMPK, which may aid in the prevention and treatment of stroke in the future.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81500263), China Postdoctoral Science Foundation (2016T90973 and 2015M572681), and Natural Science Foundation of Shaanxi Province (S2018-JC-YB-1960).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Barra de la Tremblaye P, Plamondon H. Alterations in the corticotropin-releasing hormone (CRH) neurocircuitry: Insights into post stroke functional impairments. Front Neuroendocrinol. 2016;42:53-75
2. Holmes MM. Social regulation of adult neurogenesis: A comparative approach. Front Neuroendocrinol. 2016;41:59-70
3. Dumais KM, Veenema AH. Vasopressin and oxytocin receptor systems in the brain: Sex differences and sex-specific regulation of social behavior. Front Neuroendocrinol. 2016;40:1-23
4. Gammie SC, Driessen TM, Zhao C, Saul MC, Eisinger BE. Genetic and neuroendocrine regulation of the postpartum brain. Front Neuroendocrinol. 2016;42:1-17
5. Benjamin EJ, Blaha MJ, Chiuve SE, Cushman M, Das SR, Deo R. et al. Heart Disease and Stroke Statistics-2017 Update: A Report From the American Heart Association. Circulation. 2017;135:e146-e603
6. Lv J, Hu W, Yang Z, Li T, Jiang S, Ma Z. et al. Focusing on claudin-5: A promising candidate in the regulation of BBB to treat ischemic stroke. Prog Neurobiol. 2018;161:79-96
7. Li T, Jiang S, Yang Z, Ma Z, Yi W, Wang D. et al. Targeting the energy guardian AMPK: another avenue for treating cardiomyopathy? Cell Mol Life Sci. 2017;74:1413-29
8. Liang Z, Li T, Jiang S, Xu J, Di W, Yang Z. et al. AMPK: a novel target for treating hepatic fibrosis. Oncotarget. 2017;8:62780-92
9. Jiang S, Li T, Yang Z, Yi W, Di S, Sun Y. et al. AMPK orchestrates an elaborate cascade protecting tissue from fibrosis and aging. Ageing Res Rev. 2017;38:18-27
10. Salminen A, Kaarniranta K, Kauppinen A. Age-related changes in AMPK activation: Role for AMPK phosphatases and inhibitory phosphorylation by upstream signaling pathways. Ageing Res Rev. 2016;28:15-26
11. Phillipson OT. Alpha-synuclein, epigenetics, mitochondria, metabolism, calcium traffic, & circadian dysfunction in Parkinson's disease. An integrated strategy for management. Ageing Res Rev. 2017;40:149-67
12. Carling D, Aguan K, Woods A, Verhoeven AJ, Beri RK, Brennan CH. et al. Mammalian AMP-activated protein kinase is homologous to yeast and plant protein kinases involved in the regulation of carbon metabolism. J Biol Chem. 1994;269:11442-8
13. Amato S, Liu X, Zheng B, Cantley L, Rakic P, Man HY. AMP-activated protein kinase regulates neuronal polarization by interfering with PI 3-kinase localization. Science. 2011;332:247-51
14. Miller EJ, Li J, Leng L, McDonald C, Atsumi T, Bucala R. et al. Macrophage migration inhibitory factor stimulates AMP-activated protein kinase in the ischaemic heart. Nature. 2008;451:578-82
15. Duca FA, Cote CD, Rasmussen BA, Zadeh-Tahmasebi M, Rutter GA, Filippi BM. et al. Metformin activates a duodenal Ampk-dependent pathway to lower hepatic glucose production in rats. Nat Med. 2015;21:506-11
16. Zhao J, Miyamoto S, You YH, Sharma K. AMP-activated protein kinase (AMPK) activation inhibits nuclear translocation of Smad4 in mesangial cells and diabetic kidneys. Am J Physiol Renal Physiol. 2015;308:F1167-77
17. Muanprasat C, Wongkrasant P, Satitsri S, Moonwiriyakit A, Pongkorpsakol P, Mattaveewong T. et al. Activation of AMPK by chitosan oligosaccharide in intestinal epithelial cells: Mechanism of action and potential applications in intestinal disorders. Biochem Pharmacol. 2015;96:225-36
18. Zhao S, Wu J, Tang Q, Zheng F, Yang L, Chen Y. et al. Chinese herbal medicine Xiaoji decoction inhibited growth of lung cancer cells through AMPKalpha-mediated inhibition of Sp1 and DNA methyltransferase 1. J Ethnopharmacol. 2016;181:172-81
19. Kone M, Pullen TJ, Sun G, Ibberson M, Martinez-Sanchez A, Sayers S. et al. LKB1 and AMPK differentially regulate pancreatic beta-cell identity. FASEB J. 2014;28:4972-85
20. Turnley AM, Stapleton D, Mann RJ, Witters LA, Kemp BE, Bartlett PF. Cellular distribution and developmental expression of AMP-activated protein kinase isoforms in mouse central nervous system. J Neurochem. 1999;72:1707-16
21. Gadalla AE, Pearson T, Currie AJ, Dale N, Hawley SA, Sheehan M. et al. AICA riboside both activates AMP-activated protein kinase and competes with adenosine for the nucleoside transporter in the CA1 region of the rat hippocampus. J Neurochem. 2004;88:1272-82
22. Sun Y, Yi W, Yuan Y, Lau WB, Yi D, Wang X. et al. C1q/tumor necrosis factor-related protein-9, a novel adipocyte-derived cytokine, attenuates adverse remodeling in the ischemic mouse heart via protein kinase A activation. Circulation. 2013;128:S113-20
23. Yi W, Sun Y, Yuan Y, Lau WB, Zheng Q, Wang X. et al. C1q/tumor necrosis factor-related protein-3, a newly identified adipokine, is a novel antiapoptotic, proangiogenic, and cardioprotective molecule in the ischemic mouse heart. Circulation. 2012;125:3159-69
24. Zheng Q, Yuan Y, Yi W, Lau WB, Wang Y, Wang X. et al. C1q/TNF-related proteins, a family of novel adipokines, induce vascular relaxation through the adiponectin receptor-1/AMPK/eNOS/nitric oxide signaling pathway. Arterioscler Thromb Vasc Biol. 2011;31:2616-23
25. Yang Y, Fan C, Deng C, Zhao L, Hu W, Di S. et al. Melatonin reverses flow shear stress-induced injury in bone marrow mesenchymal stem cells via activation of AMP-activated protein kinase signaling. J Pineal Res. 2016;60:228-41
26. Zhao L, Fan C, Zhang Y, Yang Y, Wang D, Deng C. et al. Adiponectin enhances bone marrow mesenchymal stem cell resistance to flow shear stress through AMP-activated protein kinase signaling. Sci Rep. 2016;6:28752
27. Yuan Y, Lau WB, Su H, Sun Y, Yi W, Du Y. et al. C1q-TNF-related protein-9, a novel cardioprotetcive cardiokine, requires proteolytic cleavage to generate a biologically active globular domain isoform. Am J Physiol Endocrinol Metab. 2015;308:E891-8
28. Zaha VG, Young LH. AMP-activated protein kinase regulation and biological actions in the heart. Circ Res. 2012;111:800-14
29. Yang YM, Han CY, Kim YJ, Kim SG. AMPK-associated signaling to bridge the gap between fuel metabolism and hepatocyte viability. World J Gastroenterol. 2010;16:3731-42
30. Hallows KR, Mount PF, Pastor-Soler NM, Power DA. Role of the energy sensor AMP-activated protein kinase in renal physiology and disease. Am J Physiol Renal Physiol. 2010;298:F1067-77
31. Carling D, Thornton C, Woods A, Sanders MJ. AMP-activated protein kinase: new regulation, new roles? Biochem J. 2012;445:11-27
32. Xiao B, Sanders MJ, Carmena D, Bright NJ, Haire LF, Underwood E. et al. Structural basis of AMPK regulation by small molecule activators. Nat Commun. 2013;4:3017
33. Calabrese MF, Rajamohan F, Harris MS, Caspers NL, Magyar R, Withka JM. et al. Structural basis for AMPK activation: natural and synthetic ligands regulate kinase activity from opposite poles by different molecular mechanisms. Structure. 2014;22:1161-72
34. Suter M, Riek U, Tuerk R, Schlattner U, Wallimann T, Neumann D. Dissecting the role of 5'-AMP for allosteric stimulation, activation, and deactivation of AMP-activated protein kinase. J Biol Chem. 2006;281:32207-16
35. Zhang CS, Jiang B, Li M, Zhu M, Peng Y, Zhang YL. et al. The lysosomal v-ATPase-Ragulator complex is a common activator for AMPK and mTORC1, acting as a switch between catabolism and anabolism. Cell Metab. 2014;20:526-40
36. Scott JW, Ling N, Issa SM, Dite TA, O'Brien MT, Chen ZP. et al. Small molecule drug A-769662 and AMP synergistically activate naive AMPK independent of upstream kinase signaling. Chem Biol. 2014;21:619-27
37. Li T, Jiang S, Yang Z, Ma Z, Yi W, Wang D. et al. Targeting the energy guardian AMPK: another avenue for treating cardiomyopathy?. Cell Mol Life Sci. 2016
38. Salt I, Celler JW, Hawley SA, Prescott A, Woods A, Carling D. et al. AMP-activated protein kinase: greater AMP dependence, and preferential nuclear localization, of complexes containing the alpha2 isoform. Biochem J. 1998;334( Pt 1):177-87
39. Cheung PC, Salt IP, Davies SP, Hardie DG, Carling D. Characterization of AMP-activated protein kinase gamma-subunit isoforms and their role in AMP binding. Biochem J. 2000:346 Pt 3: 659-69
40. Gao G, Widmer J, Stapleton D, Teh T, Cox T, Kemp BE. et al. Catalytic subunits of the porcine and rat 5'-AMP-activated protein kinase are members of the SNF1 protein kinase family. Biochim Biophys Acta. 1995;1266:73-82
41. Oakhill JS, Steel R, Chen ZP, Scott JW, Ling N, Tam S. et al. AMPK is a direct adenylate charge-regulated protein kinase. Science. 2011;332:1433-5
42. Lewinska A, Adamczyk-Grochala J, Deregowska A, Wnuk M. Sulforaphane-Induced Cell Cycle Arrest and Senescence are accompanied by DNA Hypomethylation and Changes in microRNA Profile in Breast Cancer Cells. Theranostics. 2017;7:3461-77
43. Gowans GJ, Hawley SA, Ross FA, Hardie DG. AMP is a true physiological regulator of AMP-activated protein kinase by both allosteric activation and enhancing net phosphorylation. Cell Metab. 2013;18:556-66
44. Li C, Sun W, Gu C, Yang Z, Quan N, Yang J. et al. Targeting ALDH2 for Therapeutic Interventions in Chronic Pain-Related Myocardial Ischemic Susceptibility. Theranostics. 2018;8:1027-41
45. Zeng J, Liu W, Fan YZ, He DL, Li L. PrLZ increases prostate cancer docetaxel resistance by inhibiting LKB1/AMPK-mediated autophagy. Theranostics. 2018;8:109-23
46. Wiesmann M, Zinnhardt B, Reinhardt D, Eligehausen S, Wachsmuth L, Hermann S. et al. A specific dietary intervention to restore brain structure and function after ischemic stroke. Theranostics. 2017;7:493-512
47. Li J, Zeng Z, Viollet B, Ronnett GV, McCullough LD. Neuroprotective effects of adenosine monophosphate-activated protein kinase inhibition and gene deletion in stroke. Stroke. 2007;38:2992-9
48. Zhang YL, Guo H, Zhang CS, Lin SY, Yin Z, Peng Y. et al. AMP as a low-energy charge signal autonomously initiates assembly of AXIN-AMPK-LKB1 complex for AMPK activation. Cell Metab. 2013;18:546-55
49. Anderson KA, Ribar TJ, Lin F, Noeldner PK, Green MF, Muehlbauer MJ. et al. Hypothalamic CaMKK2 contributes to the regulation of energy balance. Cell Metab. 2008;7:377-88
50. Mangano DT. Effects of acadesine on myocardial infarction, stroke, and death following surgery. A meta-analysis of the 5 international randomized trials. The Multicenter Study of Perioperative Ischemia (McSPI) Research Group. JAMA. 1997;277:325-32
51. Culmsee C, Monnig J, Kemp BE, Mattson MP. AMP-activated protein kinase is highly expressed in neurons in the developing rat brain and promotes neuronal survival following glucose deprivation. J Mol Neurosci. 2001;17:45-58
52. Wang P, Xu TY, Guan YF, Tian WW, Viollet B, Rui YC. et al. Nicotinamide phosphoribosyltransferase protects against ischemic stroke through SIRT1-dependent adenosine monophosphate-activated kinase pathway. Ann Neurol. 2011;69:360-74
53. Jinadasa T, Szabo EZ, Numat M, Orlowski J. Activation of AMP-activated protein kinase regulates hippocampal neuronal pH by recruiting Na(+)/H(+) exchanger NHE5 to the cell surface. J Biol Chem. 2014;289:20879-97
54. Wang LT, Chen BL, Wu CT, Huang KH, Chiang CK, Hwa Liu S. Protective role of AMP-activated protein kinase-evoked autophagy on an in vitro model of ischemia/reperfusion-induced renal tubular cell injury. PLoS One. 2013;8:e79814
55. Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J. et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167-74
56. McCullough LD, Zeng Z, Li H, Landree LE, McFadden J, Ronnett GV. Pharmacological inhibition of AMP-activated protein kinase provides neuroprotection in stroke. J Biol Chem. 2005;280:20493-502
57. Zhu J, Aja S, Kim EK, Park MJ, Ramamurthy S, Jia J. et al. Physiological oxygen level is critical for modeling neuronal metabolism in vitro. J Neurosci Res. 2012;90:422-34
58. Li X, Han Y, Pang W, Li C, Xie X, Shyy JY. et al. AMP-activated protein kinase promotes the differentiation of endothelial progenitor cells. Arterioscler Thromb Vasc Biol. 2008;28:1789-95
59. Lopez-Lopez C, Dietrich MO, Metzger F, Loetscher H, Torres-Aleman I. Disturbed cross talk between insulin-like growth factor I and AMP-activated protein kinase as a possible cause of vascular dysfunction in the amyloid precursor protein/presenilin 2 mouse model of Alzheimer's disease. J Neurosci. 2007;27:824-31
60. Li J, McCullough LD. Effects of AMP-activated protein kinase in cerebral ischemia. J Cereb Blood Flow Metab. 2010;30:480-92
61. Kim EK, Miller I, Aja S, Landree LE, Pinn M, McFadden J. et al. C75, a fatty acid synthase inhibitor, reduces food intake via hypothalamic AMP-activated protein kinase. J Biol Chem. 2004;279:19970-6
62. Landree LE, Hanlon AL, Strong DW, Rumbaugh G, Miller IM, Thupari JN. et al. C75, a fatty acid synthase inhibitor, modulates AMP-activated protein kinase to alter neuronal energy metabolism. J Biol Chem. 2004;279:3817-27
63. Andersson U, Filipsson K, Abbott CR, Woods A, Smith K, Bloom SR. et al. AMP-activated protein kinase plays a role in the control of food intake. J Biol Chem. 2004;279:12005-8
64. Carling D. AMPK signalling in health and disease. Curr Opin Cell Biol. 2017;45:31-7
65. Ashabi G, Khalaj L, Khodagholi F, Goudarzvand M, Sarkaki A. Pre-treatment with metformin activates Nrf2 antioxidant pathways and inhibits inflammatory responses through induction of AMPK after transient global cerebral ischemia. Metab Brain Dis. 2015;30:747-54
66. Ashabi G, Khodagholi F, Khalaj L, Goudarzvand M, Nasiri M. Activation of AMP-activated protein kinase by metformin protects against global cerebral ischemia in male rats: interference of AMPK/PGC-1alpha pathway. Metab Brain Dis. 2014;29:47-58
67. Zhou Y-Y, Li Y, Jiang W-Q, Zhou L-F. MAPK/JNK signalling: a potential autophagy regulation pathway. Bioscience Reports. 2015:35
68. Vazquez-Manrique RP, Farina F, Cambon K, Dolores Sequedo M, Parker AJ, Millan JM. et al. AMPK activation protects from neuronal dysfunction and vulnerability across nematode, cellular and mouse models of Huntington's disease. Hum Mol Genet. 2016;25:1043-58
69. Kim Y, Powathil G, Kang H, Trucu D, Kim H, Lawler S. et al. Strategies of eradicating glioma cells: a multi-scale mathematical model with MiR-451-AMPK-mTOR control. PLoS One. 2015;10:e0114370
70. Leuner B, Sabihi S. The birth of new neurons in the maternal brain: Hormonal regulation and functional implications. Front Neuroendocrinol. 2016;41:99-113
71. Vilchez D, Ros S, Cifuentes D, Pujadas L, Valles J, Garcia-Fojeda B. et al. Mechanism suppressing glycogen synthesis in neurons and its demise in progressive myoclonus epilepsy. Nat Neurosci. 2007;10:1407-13
72. Amato S, Man HY. Bioenergy sensing in the brain: the role of AMP-activated protein kinase in neuronal metabolism, development and neurological diseases. Cell Cycle. 2011;10:3452-60
73. Weisova P, Concannon CG, Devocelle M, Prehn JH, Ward MW. Regulation of glucose transporter 3 surface expression by the AMP-activated protein kinase mediates tolerance to glutamate excitation in neurons. J Neurosci. 2009;29:2997-3008
74. Jiang T, Sun Q, Chen S. Oxidative stress: A major pathogenesis and potential therapeutic target of antioxidative agents in Parkinson's disease and Alzheimer's disease. Prog Neurobiol. 2016;147:1-19
75. Li T, Jiang S, Han M, Yang Z, Lv J, Deng C. et al. Exogenous melatonin as a treatment for secondary sleep disorders: A systematic review and meta-analysis. Front Neuroendocrinol. 2018
76. Sanchez-Flores M, Marcos-Perez D, Costa S, Teixeira JP, Bonassi S, Pasaro E. et al. Oxidative stress, genomic features and DNA repair in frail elderly: A systematic review. Ageing Res Rev. 2017;37:1-15
77. Lephart ED. Skin aging and oxidative stress: Equol's anti-aging effects via biochemical and molecular mechanisms. Ageing Res Rev. 2016;31:36-54
78. Ipson BR, Fisher AL. Roles of the tyrosine isomers meta-tyrosine and ortho-tyrosine in oxidative stress. Ageing Res Rev. 2016;27:93-107
79. Ahmed Z, Shah ZH, Rehman HM, Shahzad K, Daur I, Elfeel A. et al. Genomics: A Hallmark to Monitor Molecular and Biochemical Processes Leading Toward a Better Perceptive of Seed Aging and ex-situ Conservation. Curr Issues Mol Biol. 2017;22:89-112
80. Choi IY, Ju C, Anthony Jalin AM, Lee da I, Prather PL, Kim WK. Activation of cannabinoid CB2 receptor-mediated AMPK/CREB pathway reduces cerebral ischemic injury. Am J Pathol. 2013;182:928-39
81. Blazquez C, Woods A, de Ceballos ML, Carling D, Guzman M. The AMP-activated protein kinase is involved in the regulation of ketone body production by astrocytes. J Neurochem. 1999;73:1674-82
82. Auriel E, Bornstein NM. Neuroprotection in acute ischemic stroke-current status. J Cell Mol Med. 2010;14:2200-2
83. Moloudizargari M, Asghari MH, Ghobadi E, Fallah M, Rasouli S, Abdollahi M. Autophagy, its mechanisms and regulation: Implications in neurodegenerative diseases. Ageing Res Rev. 2017;40:64-74
84. Shah SZA, Zhao D, Hussain T, Yang L. Role of the AMPK pathway in promoting autophagic flux via modulating mitochondrial dynamics in neurodegenerative diseases: Insight into prion diseases. Ageing Res Rev. 2017;40:51-63
85. Hyttinen JMT, Blasiak J, Niittykoski M, Kinnunen K, Kauppinen A, Salminen A. et al. DNA damage response and autophagy in the degeneration of retinal pigment epithelial cells-Implications for age-related macular degeneration (AMD). Ageing Res Rev. 2017;36:64-77
86. Liang M, Habib Z, Sakamoto K, Chen X, Cao G. Mycobacteria and Autophagy: Many Questions and Few Answers. Curr Issues Mol Biol. 2017;21:63-72
87. Drake LE, Springer MZ, Poole LP, Kim CJ, Macleod KF. Expanding perspectives on the significance of mitophagy in cancer. Semin Cancer Biol. 2017;47:110-24
88. Manwani B, McCullough LD. Function of the master energy regulator adenosine monophosphate-activated protein kinase in stroke. J Neurosci Res. 2013;91:1018-29
89. Sheng R, Zhang LS, Han R, Liu XQ, Gao B, Qin ZH. Autophagy activation is associated with neuroprotection in a rat model of focal cerebral ischemic preconditioning. Autophagy. 2010;6:482-94
90. Jiang T, Yu JT, Zhu XC, Zhang QQ, Tan MS, Cao L. et al. Ischemic preconditioning provides neuroprotection by induction of AMP-activated protein kinase-dependent autophagy in a rat model of ischemic stroke. Mol Neurobiol. 2015;51:220-9
91. Maruthur NM, Tseng E, Hutfless S, Wilson LM, Suarez-Cuervo C, Berger Z. et al. Diabetes Medications as Monotherapy or Metformin-Based Combination Therapy for Type 2 Diabetes: A Systematic Review and Meta-analysis. Ann Intern Med. 2016;164:740-51
92. Arbelaez-Quintero I, Palacios M. To Use or Not to Use Metformin in Cerebral Ischemia: A Review of the Application of Metformin in Stroke Rodents. Stroke Res Treat. 2017;2017:9756429
93. Jiang T, Yu JT, Zhu XC, Wang HF, Tan MS, Cao L. et al. Acute metformin preconditioning confers neuroprotection against focal cerebral ischaemia by pre-activation of AMPK-dependent autophagy. Br J Pharmacol. 2014;171:3146-57
94. Liu Y, Lu Z, Cui M, Yang Q, Tang Y, Dong Q. Tissue kallikrein protects SH-SY5Y neuronal cells against oxygen and glucose deprivation-induced injury through bradykinin B2 receptor-dependent regulation of autophagy induction. J Neurochem. 2016;139:208-20
95. Kalender A, Selvaraj A, Kim SY, Gulati P, Brule S, Viollet B. et al. Metformin, independent of AMPK, inhibits mTORC1 in a rag GTPase-dependent manner. Cell Metab. 2010;11:390-401
96. Rena G, Pearson ER, Sakamoto K. Molecular mechanism of action of metformin: old or new insights? Diabetologia. 2013;56:1898-906
97. Gusev GP, Govekar R, Gadewal N, Agalakova NI. Understanding quasi-apoptosis of the most numerous enucleated components of blood needs detailed molecular autopsy. Ageing Res Rev. 2017;35:46-62
98. Birkinshaw RW, Czabotar PE. The BCL-2 family of proteins and mitochondrial outer membrane permeabilisation. Semin Cell Dev Biol. 2017;72:152-62
99. Curti V, Di Lorenzo A, Dacrema M, Xiao J, Nabavi SM, Daglia M. In vitro polyphenol effects on apoptosis: An update of literature data. Semin Cancer Biol. 2017;46:119-31
100. Diwanji N, Bergmann A. An unexpected friend - ROS in apoptosis-induced compensatory proliferation: Implications for regeneration and cancer. Semin Cell Dev Biol. 2018;80:74-82
101. Ma Z, Xin Z, Hu W, Jiang S, Yang Z, Yan X. et al. Forkhead box O proteins: Crucial regulators of cancer EMT. Semin Cancer Biol. 2018;50:21-31
102. King TD, Song L, Jope RS. AMP-activated protein kinase (AMPK) activating agents cause dephosphorylation of Akt and glycogen synthase kinase-3. Biochem Pharmacol. 2006;71:1637-47
103. Horike N, Sakoda H, Kushiyama A, Ono H, Fujishiro M, Kamata H. et al. AMP-activated protein kinase activation increases phosphorylation of glycogen synthase kinase 3beta and thereby reduces cAMP-responsive element transcriptional activity and phosphoenolpyruvate carboxykinase C gene expression in the liver. J Biol Chem. 2008;283:33902-10
104. Li T, Jiang S, Lu C, Hu W, Ji T, Han M. et al. Snapshots: Endoplasmic Reticulum Stress in Lipid Metabolism and Cardiovascular Disease. Curr Issues Mol Biol. 2018;28:14-28
105. Duan J, Yin Y, Cui J, Yan J, Zhu Y, Guan Y. et al. Chikusetsu Saponin IVa Ameliorates Cerebral Ischemia Reperfusion Injury in Diabetic Mice via Adiponectin-Mediated AMPK/GSK-3beta Pathway In Vivo and In Vitro. Mol Neurobiol. 2016;53:728-43
106. Yang Y, Zhang XJ, Li LT, Cui HY, Zhang C, Zhu CH. et al. Apelin-13 protects against apoptosis by activating AMP-activated protein kinase pathway in ischemia stroke. Peptides. 2016;75:96-100
107. Guo JM, Shu H, Wang L, Xu JJ, Niu XC, Zhang L. SIRT1-dependent AMPK pathway in the protection of estrogen against ischemic brain injury. CNS Neurosci Ther. 2017;23:360-9
108. Kim HJ, Maiti P, Barrientos A. Mitochondrial ribosomes in cancer. Semin Cancer Biol. 2017;47:67-81
109. St John JC. Mitochondrial DNA copy number and replication in reprogramming and differentiation. Semin Cell Dev Biol. 2016;52:93-101
110. Ham PB 3rd, Raju R. Mitochondrial function in hypoxic ischemic injury and influence of aging. Prog Neurobiol. 2016
111. Sacca SC, Gandolfi S, Bagnis A, Manni G, Damonte G, Traverso CE. et al. From DNA damage to functional changes of the trabecular meshwork in aging and glaucoma. Ageing Res Rev. 2016;29:26-41
112. DeBalsi KL, Hoff KE, Copeland WC. Role of the mitochondrial DNA replication machinery in mitochondrial DNA mutagenesis, aging and age-related diseases. Ageing Res Rev. 2017;33:89-104
113. Toyama EQ, Herzig S, Courchet J, Lewis TL Jr, Loson OC, Hellberg K. et al. Metabolism. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science. 2016;351:275-81
114. Lv J, Jiang S, Yang Z, Hu W, Wang Z, Li T. et al. PGC-1alpha sparks the fire of neuroprotection against neurodegenerative disorders. Ageing Res Rev. 2018;44:8-21
115. Li L, Xiao L, Hou Y, He Q, Zhu J, Li Y. et al. Sestrin2 Silencing Exacerbates Cerebral Ischemia/Reperfusion Injury by Decreasing Mitochondrial Biogenesis through the AMPK/PGC-1alpha Pathway in Rats. Sci Rep. 2016;6:30272
116. Meyerson JR, Kumar J, Chittori S, Rao P, Pierson J, Bartesaghi A. et al. Structural mechanism of glutamate receptor activation and desensitization. Nature. 2014;514:328-34
117. Soria FN, Perez-Samartin A, Martin A, Gona KB, Llop J, Szczupak B. et al. Extrasynaptic glutamate release through cystine/glutamate antiporter contributes to ischemic damage. J Clin Invest. 2014;124:3645-55
118. Lai TW, Zhang S, Wang YT. Excitotoxicity and stroke: identifying novel targets for neuroprotection. Prog Neurobiol. 2014;115:157-88
119. Connolly NM, Dussmann H, Anilkumar U, Huber HJ, Prehn JH. Single-cell imaging of bioenergetic responses to neuronal excitotoxicity and oxygen and glucose deprivation. J Neurosci. 2014;34:10192-205
120. Li Y, Li J, Li S, Li Y, Wang X, Liu B. et al. Curcumin attenuates glutamate neurotoxicity in the hippocampus by suppression of ER stress-associated TXNIP/NLRP3 inflammasome activation in a manner dependent on AMPK. Toxicol Appl Pharmacol. 2015;286:53-63
121. Kuramoto N, Wilkins ME, Fairfax BP, Revilla-Sanchez R, Terunuma M, Tamaki K. et al. Phospho-dependent functional modulation of GABA(B) receptors by the metabolic sensor AMP-dependent protein kinase. Neuron. 2007;53:233-47
122. Rahimifard M, Maqbool F, Moeini-Nodeh S, Niaz K, Abdollahi M, Braidy N. et al. Targeting the TLR4 signaling pathway by polyphenols: A novel therapeutic strategy for neuroinflammation. Ageing Res Rev. 2017;36:11-9
123. Song Z, Albers HE. Cross-talk among oxytocin and arginine-vasopressin receptors: Relevance for basic and clinical studies of the brain and periphery. Front Neuroendocrinol. 2017
124. Ransohoff RM, Perry VH. Microglial physiology: unique stimuli, specialized responses. Annu Rev Immunol. 2009;27:119-45
125. Xiong XY, Liu L, Yang QW. Functions and mechanisms of microglia/macrophages in neuroinflammation and neurogenesis after stroke. Prog Neurobiol. 2016;142:23-44
126. Chen YJ, Nguyen HM, Maezawa I, Grossinger EM, Garing AL, Kohler R. et al. The potassium channel KCa3.1 constitutes a pharmacological target for neuroinflammation associated with ischemia/reperfusion stroke. J Cereb Blood Flow Metab. 2016;36:2146-61
127. Liu Y, Tang G, Li Y, Wang Y, Chen X, Gu X. et al. Metformin attenuates blood-brain barrier disruption in mice following middle cerebral artery occlusion. J Neuroinflammation. 2014;11:177
128. Qiu J, Wang M, Zhang J, Cai Q, Lu D, Li Y. et al. The neuroprotection of Sinomenine against ischemic stroke in mice by suppressing NLRP3 inflammasome via AMPK signaling. Int Immunopharmacol. 2016;40:492-500
129. Krupinski J, Kaluza J, Kumar P, Wang M, Kumar S. Prognostic value of blood vessel density in ischaemic stroke. Lancet. 1993;342:742
130. Venna VR, Li J, Hammond MD, Mancini NS, McCullough LD. Chronic metformin treatment improves post-stroke angiogenesis and recovery after experimental stroke. Eur J Neurosci. 2014;39:2129-38
131. Jin Q, Cheng J, Liu Y, Wu J, Wang X, Wei S. et al. Improvement of functional recovery by chronic metformin treatment is associated with enhanced alternative activation of microglia/macrophages and increased angiogenesis and neurogenesis following experimental stroke. Brain Behav Immun. 2014;40:131-42
132. Wang LM, Wang YJ, Cui M, Luo WJ, Wang XJ, Barber PA. et al. A dietary polyphenol resveratrol acts to provide neuroprotection in recurrent stroke models by regulating AMPK and SIRT1 signaling, thereby reducing energy requirements during ischemia. Eur J Neurosci. 2013;37:1669-81
133. Wan D, Zhou Y, Wang K, Hou Y, Hou R, Ye X. Resveratrol provides neuroprotection by inhibiting phosphodiesterases and regulating the cAMP/AMPK/SIRT1 pathway after stroke in rats. Brain Res Bull. 2016;121:255-62
134. Ohira M, Endo K, Saiki A, Miyashita Y, Terai K, Murano T. et al. Atorvastatin and pitavastatin enhance lipoprotein lipase production in L6 skeletal muscle cells through activation of adenosine monophosphate-activated protein kinase. Metabolism. 2012;61:1452-60
135. Chaturvedi S, Zivin J, Breazna A, Amarenco P, Callahan A, Goldstein LB. et al. Effect of atorvastatin in elderly patients with a recent stroke or transient ischemic attack. Neurology. 2009;72:688-94
136. Mascitelli L, Pezzetta F. Statin therapy after stroke or transient ischemic attack. N Engl J Med. 2006;355:2369-70 author reply 70-1
137. Tang H, Pan CS, Mao XW, Liu YY, Yan L, Zhou CM. et al. Role of NADPH oxidase in total salvianolic acid injection attenuating ischemia-reperfusion impaired cerebral microcirculation and neurons: implication of AMPK/Akt/PKC. Microcirculation. 2014;21:615-27
138. Wu F, Nicholson AD, Haile WB, Torre E, An J, Chen C. et al. Tissue-type plasminogen activator mediates neuronal detection and adaptation to metabolic stress. J Cereb Blood Flow Metab. 2013;33:1761-9
139. Si QM, Wu GC, Cao XD. Effects of electroacupuncture on acute cerebral infarction. Acupunct Electrother Res. 1998;23:117-24
140. Xiong L, Lu Z, Hou L, Zheng H, Zhu Z, Wang Q. et al. Pretreatment with repeated electroacupuncture attenuates transient focal cerebral ischemic injury in rats. Chin Med J (Engl). 2003;116:108-11
141. Tominaga A, Ishizaki N, Naruse Y, Kitakoji H, Yamamura Y. Repeated application of low-frequency electroacupuncture improves high-fructose diet-induced insulin resistance in rats. Acupunct Med. 2011;29:276-83
142. Ran QQ, Chen HL, Liu YL, Yu HX, Shi F, Wang MS. Electroacupuncture preconditioning attenuates ischemic brain injury by activation of the adenosine monophosphate-activated protein kinase signaling pathway. Neural Regen Res. 2015;10:1069-75
143. Ogoh S, Ainslie PN. Cerebral blood flow during exercise: mechanisms of regulation. J Appl Physiol (1985). 2009;107:1370-80
144. Dornbos D 3rd, Zwagerman N, Guo M, Ding JY, Peng C, Esmail F. et al. Preischemic exercise reduces brain damage by ameliorating metabolic disorder in ischemia/reperfusion injury. J Neurosci Res. 2013;91:818-27
145. Papadia S, Hardingham GE. The dichotomy of NMDA receptor signaling. Neuroscientist. 2007;13:572-9
146. Li M, Zhao J, Hu Y, Lu H, Guo J. Oxygen free radicals regulate energy metabolism via AMPK pathway following cerebral ischemia. Neurol Res. 2010;32:779-84
147. Nam HG, Kim W, Yoo DY, Choi JH, Won MH, Hwang IK. et al. Chronological changes and effects of AMP-activated kinase in the hippocampal CA1 region after transient forebrain ischemia in gerbils. Neurol Res. 2013;35:395-405
148. Ma Y, Bu J, Dang H, Sha J, Jing Y, Shan-jiang AI. et al. Inhibition of adenosine monophosphate-activated protein kinase reduces glial cell-mediated inflammation and induces the expression of Cx43 in astroglias after cerebral ischemia. Brain Res. 2015;1605:1-11
149. van der Worp HB, Sena ES, Donnan GA, Howells DW, Macleod MR. Hypothermia in animal models of acute ischaemic stroke: a systematic review and meta-analysis. Brain. 2007;130:3063-74
150. Li J, Benashski S, McCullough LD. Post-stroke hypothermia provides neuroprotection through inhibition of AMP-activated protein kinase. J Neurotrauma. 2011;28:1281-8
151. Greco SJ, Sarkar S, Johnston JM, Tezapsidis N. Leptin regulates tau phosphorylation and amyloid through AMPK in neuronal cells. Biochem Biophys Res Commun. 2009;380:98-104
152. Huang W, Cao J, Liu X, Meng F, Li M, Chen B. et al. AMPK Plays a Dual Role in Regulation of CREB/BDNF Pathway in Mouse Primary Hippocampal Cells. J Mol Neurosci. 2015;56:782-8
153. Rousset CI, Leiper FC, Kichev A, Gressens P, Carling D, Hagberg H. et al. A dual role for AMP-activated protein kinase (AMPK) during neonatal hypoxic-ischaemic brain injury in mice. J Neurochem. 2015;133:242-52
154. Saposnik G, Young B, Silver B, Di Legge S, Webster F, Beletsky V. et al. Lack of improvement in patients with acute stroke after treatment with thrombolytic therapy: predictors and association with outcome. JAMA. 2004;292:1839-44
155. Kleman AM, Yuan JY, Aja S, Ronnett GV, Landree LE. Physiological glucose is critical for optimized neuronal viability and AMPK responsiveness in vitro. J Neurosci Methods. 2008;167:292-301
156. Ronnett GV, Ramamurthy S, Kleman AM, Landree LE, Aja S. AMPK in the brain: its roles in energy balance and neuroprotection. J Neurochem. 2009;109(Suppl 1):17-23
157. Li J, Benashski SE, Venna VR, McCullough LD. Effects of metformin in experimental stroke. Stroke. 2010;41:2645-52
158. Iwabuchi S, Kawahara K, Harata NC. Effects of pharmacological inhibition of AMP-activated protein kinase on GLUT3 expression and the development of ischemic tolerance in astrocytes. Neurosci Res. 2014;84:68-71
159. Adams HP Jr, Adams RJ, Brott T, del Zoppo GJ, Furlan A, Goldstein LB. et al. Guidelines for the early management of patients with ischemic stroke: A scientific statement from the Stroke Council of the American Stroke Association. Stroke. 2003;34:1056-83
160. Wang X, Tsuji K, Lee SR, Ning M, Furie KL, Buchan AM. et al. Mechanisms of hemorrhagic transformation after tissue plasminogen activator reperfusion therapy for ischemic stroke. Stroke. 2004;35:2726-30
161. Desmurget M, Bonnetblanc F, Duffau H. Contrasting acute and slow-growing lesions: a new door to brain plasticity. Brain. 2007;130:898-914
162. Chakkarapani E, Dingley J, Liu X, Hoque N, Aquilina K, Porter H. et al. Xenon enhances hypothermic neuroprotection in asphyxiated newborn pigs. Ann Neurol. 2010;68:330-41
163. Howell NJ, Ashrafian H, Drury NE, Ranasinghe AM, Contractor H, Isackson H. et al. Glucose-insulin-potassium reduces the incidence of low cardiac output episodes after aortic valve replacement for aortic stenosis in patients with left ventricular hypertrophy: results from the Hypertrophy, Insulin, Glucose, and Electrolytes (HINGE) trial. Circulation. 2011;123:170-7
164. Kim E, Lee SH, Lee KS, Cheong HK, Namkoong K, Hong CH. et al. AMPK gamma2 subunit gene PRKAG2 polymorphism associated with cognitive impairment as well as diabetes in old age. Psychoneuroendocrinology. 2012;37:358-65
165. Campbell BC, Mitchell PJ, Kleinig TJ, Dewey HM, Churilov L, Yassi N. et al. Endovascular therapy for ischemic stroke with perfusion-imaging selection. N Engl J Med. 2015;372:1009-18
166. Kelly AG, Sahin B, Holloway RG. Ethical considerations in stroke patients. Curr Opin Neurol. 2014;27:61-5
167. Gill TM. The central role of prognosis in clinical decision making. JAMA. 2012;307:199-200
168. Rabinstein A, Rundek T. Prediction of outcome after ischemic stroke: the value of clinical scores. Neurology. 2013;80:15-6
169. Connolly SJ, Ezekowitz MD, Yusuf S, Eikelboom J, Oldgren J, Parekh A. et al. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med. 2009;361:1139-51
170. Writing Group M, Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ. et al. Heart Disease and Stroke Statistics-2016 Update: A Report From the American Heart Association. Circulation. 2016;133:e38-60
171. Selvin E, Bolen S, Yeh HC, Wiley C, Wilson LM, Marinopoulos SS. et al. Cardiovascular outcomes in trials of oral diabetes medications: a systematic review. Arch Intern Med. 2008;168:2070-80
172. Dong X. Current Strategies for Brain Drug Delivery. Theranostics. 2018;8:1481-93
Author contact
![]() Corresponding authors: Yang Yang MD., PhD. and Chunhu Gu MD., PhD., Key Laboratory of Resource Biology and Biotechnology in Western China, Ministry of Education, Faculty of Life Sciences, Northwest University, 229 Taibai North Road, Xi'an 710069, China. Telephone: +86 13379217366; Email address: yang200214yycom (Yang Yang) and chunhuguxijingcom (Chunhu Gu)
Corresponding authors: Yang Yang MD., PhD. and Chunhu Gu MD., PhD., Key Laboratory of Resource Biology and Biotechnology in Western China, Ministry of Education, Faculty of Life Sciences, Northwest University, 229 Taibai North Road, Xi'an 710069, China. Telephone: +86 13379217366; Email address: yang200214yycom (Yang Yang) and chunhuguxijingcom (Chunhu Gu)