Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results
Discussion
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(16):4345-4358. doi:10.7150/thno.26862 This issue Cite
Research Paper
Transcriptomic signature associated with carcinogenesis and aggressiveness of papillary thyroid carcinoma
Huajing Teng1,#, ![]() , Fengbiao Mao1,#, Jialong Liang1, Meiying Xue1,2, Wenqing Wei1,2, Xianfeng Li1, Kun Zhang3, Dongdong Feng4, Baoguo Liu4, Zhongsheng Sun1,
, Fengbiao Mao1,#, Jialong Liang1, Meiying Xue1,2, Wenqing Wei1,2, Xianfeng Li1, Kun Zhang3, Dongdong Feng4, Baoguo Liu4, Zhongsheng Sun1, ![]()
1. Beijing Institutes of Life Science, Chinese Academy of Sciences, Beijing, China
2. University of Chinese Academy of Sciences, Beijing, China.
3. Institute of Genomic Medicine, Wenzhou Medical University, Wenzhou, China
4. Department of Head and Neck Surgery, Key Laboratory of Carcinogenesis and Translational Research (Ministry of Education), Peking University Cancer Hospital and Institute, Beijing, China.
#Authors contributed equally to this work.
Received 2018-4-24; Accepted 2018-7-16; Published 2018-7-30
Abstract

Papillary thyroid carcinoma (PTC) is the fastest-growing disease caused by numerous molecular alterations in addition to previously reported DNA mutations. There is a compelling need to identify novel transcriptomic alterations that are associated with the pathogenesis of PTC with potential diagnostic and prognostic implications.
Methods: We gathered and compared 242 expression profiles between paired PTC and adjacent normal tissues and identified and validated the coding and long non-coding RNAs (lncRNAs) associated with the extrathyroidal extension (ETE) of 655 PTC patients in two independent cohorts, followed by predicting their interactions with drugs. Co-expression, RNA interaction, Kaplan-Meier survival and multivariate Cox proportional regression analyses were performed to identify dysregulated lncRNAs and genes that correlated with clinical outcomes of PTC. Alternative splicing (AS), RNA circularization, and editing were also compared between transcriptomes to expand the repertoire of molecular alterations in PTC.
Results: Numerous genes related to cellular microenvironment and steroid hormone response were associated with the ETE of PTC. Drug susceptibility predictions of the expression signature revealed two highly ranked compounds, 6-bromoindirubin-3'-oxime and lovastatin. Co-expression and RNA interaction analysis revealed the essential role of lncRNAs in PTC pathogenesis by modulating extracellular matrix and cell adhesion. Eight genes and two novel lncRNAs were identified that correlated with the aggressive nature and disease-free survival of PTC. Furthermore, this study provided the transcriptome-wide landscape of circRNAs in PTC and uncovered dissimilar expression profiles among circRNAs originating from the same host gene, suggesting the functional complexity of circRNAs in PTC carcinogenesis. The newly identified AS events in the SERPINA1 and FN1 genes may improve the sensitivity and specificity of these diagnostic biomarkers.
Conclusions: Our study uncovered a comprehensive transcriptomic signature associated with the carcinogenesis and aggressive behavior of PTC, as well as presents a catalog of 10 potential biomarkers, which would facilitate PTC prognosis and development of new therapeutic strategies for this cancer.
Keywords: papillary thyroid carcinoma, long non-coding RNAs, circular RNAs, alternative splicing, RNA editing, drug susceptibility
Introduction
The incidence of thyroid cancer has tripled over the past three decades, attributed to the rapid rise of the papillary subtype [1-3]. Large-scale genomic characterization of papillary thyroid carcinoma (PTC) uncovered the critical role of genetic alterations in the tumorigenesis of this disease. Somatic mutations and gene fusions, which activate the mitogen-activated protein kinase (MAPK) and phosphoinositide 3-kinase AKT (PI3K-AKT) pathways, were identified as the primary molecular aberrations for PTC [4-9]. However, the carcinogenesis of PTC is a complex biological process characterized by various molecular abnormalities, and the reason for its high prevalence remains poorly understood. Extrathyroidal extension (ETE) is a prognostic factor that has been strongly associated with recurrence and survival in PTC patients [10, 11], but the molecular mechanisms underlying the progression of ETE remain mostly uncharacterized, limiting the ability to predict and treat patients with high recurrence risk [11, 12]. Recently, mutations in thyroglobulin (TG), not exclusive to mutations in the RAS-MAP kinase pathway, were associated with significantly worse clinical outcomes, suggesting their pathogenic role beyond driving initial oncogenesis [13]. Thus, for diagnosis and prognosis, there remains a compelling need to decode novel molecular targets and/or processes that underlie the pathogenesis and progression of PTC [14-18].
RNAs, the direct output of the genetic information that is encoded by genomes, play essential roles in cellular functions, and their regulation can diversify the genetic output. For example, the function of non-coding RNAs is indispensable for fine-tuning the genetic network, and its dysregulation is tightly associated with the pathogenesis of cancers [19-21]. In comparison with the well-defined role of microRNAs in the pathogenesis of thyroid cancer [22], the role of long noncoding RNAs (lncRNAs) in this process is still largely uncharacterized. The aberrant expression of several lncRNAs in PTC indicates their contribution to carcinogenesis [23-27]; however, key targets of these lncRNAs and their clinical roles remain to be elucidated. Emerging evidence indicates that several other processes, such as alternative splicing (AS), RNA editing, and RNA circularization, expand the complexity of the transcriptome, and provide a repertoire of candidate genes contributing to the pathogenesis of cancers [28, 29]. So far, little information is available on the transcriptomic signatures of AS and circular RNAs (circRNAs) in thyroid cancer. Although an overall increase in the A-to-I editing level has been detected in multiple cancer types including thyroid cancers [28], the specific sites that were altered in cancer tissues and their pathogenic role remain elusive.
Next-generation sequencing permits the investigation of an entire transcriptome, including alterations of coding/noncoding RNA expression, AS, and editing with unprecedented resolution and throughput. In this study, we collected 242 paired PTC and adjacent normal tissues, analyzed their expression profiles, and provided a comprehensive transcriptomic signature of lncRNAs, circRNAs, mRNAs, AS, and RNA editing associated with the carcinogenesis and aggressive behavior of PTC. More importantly, our study identified a catalog of 10 mRNAs and lncRNAs with prognostic significance for PTC. This study provides new insight into the pathogenesis and aggressiveness of PTC, which may facilitate the prognosis and development of new therapeutic strategies for this disease.
Methods
Patients and samples
PTC and adjacent normal tissues were obtained from 11 patients undergoing total thyroidectomy at the Head and Neck Surgery Department of the Beijing Cancer Hospital. The clinical characteristics of the patients are listed in Table S1-2. All tumor samples were reviewed by two experienced pathologists, who confirmed the diagnosis of PTC and ensured that the cancer foci contained more than 60% tumor cellularity. The TNM classification of American Joint Committee on Cancer (AJCC) ranging from stages I to IV, according to the primary tumor (T), regional lymph nodes (N), and distant metastasis (M), was used for PTC staging [30]. ETE, the risk factor strongly associated with recurrence and aggressive features of PTC [10, 31], was used to describe the aggressiveness of PTC. Three different degrees of ETE, none, minimal (extension to the sternothyroid muscle or perithyroidal soft tissue) and gross (extension to the subcutaneous soft tissue, larynx, trachea, esophagus, or recurrent laryngeal nerve), were defined according to the range of extension in the AJCC TNM system. The samples were snap-frozen in liquid nitrogen and stored at -80 °C until used for analysis of differential expression, circRNAs, AS, and RNA-editing after RNA sequencing. The study was approved by the Ethical Review Board of the hospital, and clinical data were collected after patients provided informed consent.
The clinical data of 12 paired PTC and noncancerous tissues (Table S1-3) and 24 transcriptome datasets of total RNA were downloaded from ArrayExpress (https://www.ebi.ac.uk/arrayexpress/experiments/E-GEOD-83520/) [23]. We also downloaded the clinical data of 125 PTC patients (Table S1-4) and 165 transcriptome data including 80 paired cancerous and noncancerous transcriptomes from the European Nucleotide Archive database with accession number PRJEB11591 (http://www.ebi.ac.uk/ena/data/view/PRJEB11591) [16]. The clinical data of 58 PTC patients, each containing both tumor and normal tissues (Table S1-5) and their matched 116 RNA-seq read count data were downloaded from the TCGA THCA cohort (https://portal.gdc.cancer.gov).
cDNA library preparation and sequencing
Total RNA was isolated using an RNeasy Mini Kit (Qiagen, Shanghai, China). The RNA samples with an RNA Integrity Number (RIN) greater than eight, as assessed by Agilent 2100 Bioanalyzer (Agilent, Santa Clara, CA, USA), were used for RNA library construction. cDNA libraries were prepared according to the manufacturer's protocol (Illumina, San Diego, CA, USA). Each library was sequenced using the Illumina HiSeq™ 2000 platform and generated 100 bp paired-end (PE) reads. The raw sequencing datasets were deposited in the Sequence Read Archive of NCBI (http://www.ncbi.nlm.nih.gov/sra) under accession number PRJNA431763.
Identification of differentially expressed genes (DEGs) and lncRNAs
After removing adapters and low-quality bases, sequencing reads of 211 transcriptomes from 148 PTC patients in our collected cohorts (Table S1-1) were aligned to the human reference genome (UCSC hg19) using STAR [32] with default parameters. Next, HTSeq [33] was used to count the reads according to the hg19 coordinates of protein-coding genes and lncRNAs in GENCODE (V22), and differential expression between the paired normal and cancer samples was identified using the Bioconductor package edgeR [34] with a cut-off of 5% false discovery rate (FDR). To identify the prominently altered genes and RNAs, the differentially expressed lncRNAs and genes with greater than two-fold expression changes were considered. Hierarchical clustering of the prominent altered gene and lncRNA expression profiles were performed using Cluster 3.0 (http://bonsai.hgc.jp/~mdehoon/software/cluster/). After taking into consideration the differences between tumor and adjacent normal tissues, an ANOVA-like test for differential expression across different degrees of ETE in our collected and TCGA THCA cohorts was performed independently using edgeR [34] to identify lncRNAs and genes associated with the progression of PTC. The gene expression profiles in response to ETE were compared with drug response signatures listed in the Connectivity Map (CMAP) build 02 (Broad Institute) [35]. A protein-protein interaction network of DEGs associated with PTC aggressiveness was also constructed based on text mining and the experimentally determined protein-protein interaction database of String [36].
Co-expression analysis of prominently altered lncRNAs and genes
We used the list of differentially expressed lncRNAs to analyze their correlation with the DEGs detected in PTC tissues. The expression level of lncRNAs and genes in each tumor or normal tissue was used to calculate the Pearson correlation coefficient and probability. Subsequently, gene pairs with Pearson correlation coefficient > 0.7 and P < 0.05 were selected and constructed into a co-expression network using Cytoscape. To further identify the interaction partners, lncRNA-mRNA interactions were predicted for each lncRNA based on the seed-and-extension approach and RNA secondary structure energy model using Riblast according to default parameters [37].
Survival analysis of dysregulated lncRNAs and genes
We downloaded RNA-seq normalized data for 493 THCA patients with clinical data from TCGA (https://portal.gdc.cancer.gov) (Table S2). Kaplan-Meier survival analysis, univariate and multivariate Cox proportional regression analyses were employed with the R survival package to analyze the association between the expression of a specific molecule and clinical outcome. Age, gender, TNM staging, mutation of BRAFV600E, histological subtypes (classical, follicular, and tall cell), and history of other malignancies were taken into account in the multivariate Cox regression analysis. The information on Cox proportional hazard ratios and the 95% confidence interval were also obtained.
circRNA analysis
Sequencing reads of paired PTC and noncancerous tissues in our collected cohorts were aligned to a human reference genome (UCSC hg19) using BWA-MEM [38] with default parameters. Subsequently, circRNAs were identified using CIRI_v2.0.6 [39, 40] with at least two supporting reads. Differentially expressed circRNAs between normal and tumor tissues were further identified using CircTest [41]. Regulatory elements on circRNA, including microRNA response elements and RNA-binding protein binding sites, were predicted using CircView [42].
Detection of AS events
rMATS uses a modified generalized linear mixed model to detect differential AS events from RNA-seq data with replicates [43]. We used rMATS to detect five types of AS events [43], including skipped exon (SE), retained intron (RI), mutually exclusive exons (MXE) and alternative 5′ and 3′ splice sites (A5SS, A3SS), from the RNA-seq data.
Motif enrichment analysis
We used rMAPs [44] to identify binding sites of RNA-binding proteins that were significantly enriched in differential exon skipping events between cancer and normal tissues. Motif occurrences were scanned separately in exons or 250 bp upstream or downstream intronic sequences, and alternative exons without splicing changes (rMATS FDR>50%, maxPSI>15%, and minPSI<85%) were used as a control. P-values and FDR for motif enrichment were calculated for each motif after counting the number of occurrences in the differentially spliced exons and the control exons.
RNA-editing analysis
After aligning the sequencing reads to the human reference genome using STAR [32], processes, including duplicate removal, reassign mapping qualities, and base quality recalibration, were performed using PICARD (http://picard.sourceforge.net) and the Genome Analysis Toolkit (GATK). Single nucleotide variants (SNV) were called using GATK HaplotypeCaller. RNA editing sites were identified using GIREMI [45] with the obtained SNV as the training set. RNA editing sets were compared with the known RNA-edited sites available from public database, Rigorously Annotated Database of A-to-I RNA Editing (RADAR) [46]. The overall editing levels across all edited sites were compared between normal and tumor tissues using Wilcoxon rank-sum test. For each of the sites, differences in editing level between paired PTC and adjacent normal tissues were calculated using Fisher's exact test followed by 5% FDR multiple-testing correction.
Gene set enrichment analysis
Functional enrichment analysis of the genes was performed on Cytoscape plugin BINGO [47].
Results
Differentially expressed genes (DEGs) and lncRNAs associated with carcinogenesis and aggressive disposition of PTC
PTC was reported to have a low frequency of somatic mutations and low intratumor heterogeneity [8]. To identify the transcriptome-wide alterations of PTC, differential expression analysis of lncRNAs and coding genes was first performed by comparing 242 expression profiles between paired PTC and adjacent normal tissues. Surprisingly, 52.73% (10,427/19,776) of the coding genes and 32.83% (5,192/15,817) of the lncRNAs exhibited differential expression (FDR<0.05) between tumor and normal tissues, indicating the widespread alterations in both lncRNA and mRNA expression in the cancer known to have a low frequency of mutations. When a greater than two-fold change in the expression levels was considered significant, 1,437 lncRNAs (464 upregulated and 973 downregulated) and 1,582 coding genes (672 upregulated and 910 downregulated) with differential expression were identified (Figure 1A-B). Correlation (R2) of the expression differences in PTC versus normal tissues for 38 DEGs between our study and a previous meta-analysis [48] was 0.9352 (Figure S1), suggesting a high degree of reliability of the DEG analysis. Hierarchical clustering analysis using the identified DEGs and lncRNAs revealed that expression levels of these entities could define the majority of the malignant and adjacent benign thyroid tissues (Figure 1A-B).
Transcriptional patterns and protein-protein interactions of differentially expressed genes. Hierarchical clustering of differentially expressed genes (DEGs) (A) and lncRNAs (B) define the majority of the malignant and adjacent benign tissues of 121 papillary thyroid carcinoma patients. Protein-protein interaction network (C) and functional enrichment (D) of DEGs associated with the extrathyroidal extension (ETE). Nodes with dark green shading indicate genes response to hormone stimulus.
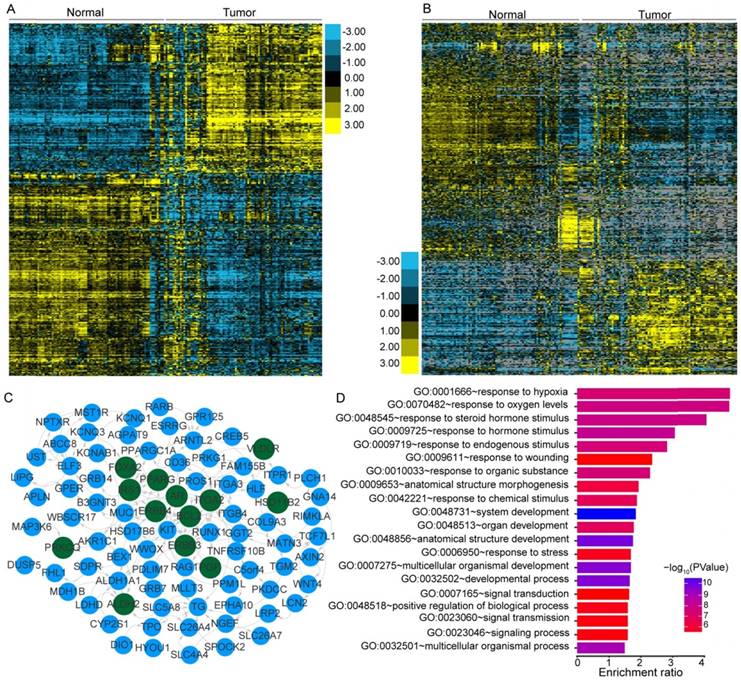
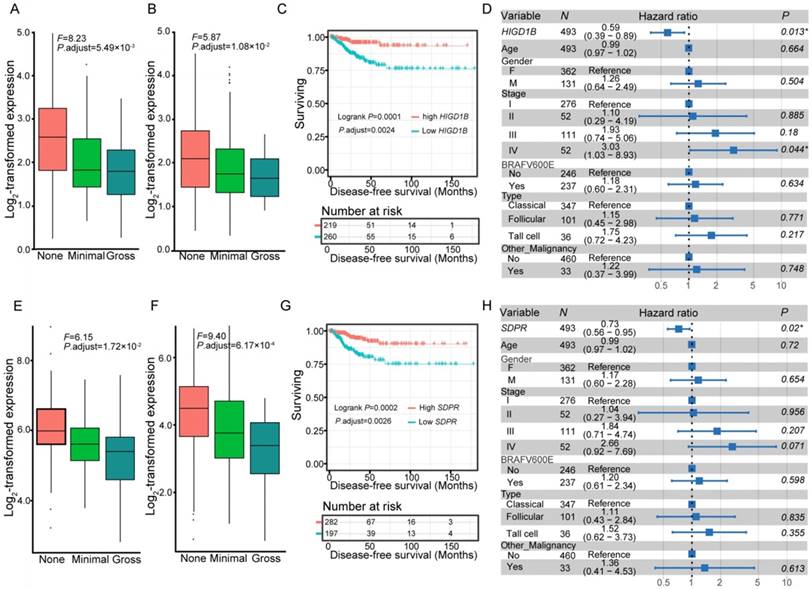
We further compared the transcriptomes of different degrees (none, minimal, and gross) of ETE to identify transcripts associated with the aggressive features of PTC. We obtained 264 DEGs and 95 differentially expressed lncRNAs across various degrees of ETE after considering the differences between tumor and adjacent normal tissues (Table S3), and found that 35 genes and 7 lncRNAs were differentially expressed between all analyzed groups. The correlation between expression levels of these identified transcripts and ETE was validated in PTC data from the TCGA cohort (Table S3). After Kaplan-Meier survival, univariate and multivariate Cox regression analyses, a total of 8 genes were predicted to be correlated with the disease-free survival of PTC patients (Table S4), thus expanding the number of prognostic biomarkers in this disease. For example, decreased mRNA expression of HIGD1B and SDPR was observed in minimal and gross ETE tumor tissues in both cohorts (Figure 2A-B, E-F), which was significantly associated with worse disease-free survival of PTC patients after adjusting for multiple testing (Figure 2C, G) and multiple clinical factors including age, gender, TNM staging, mutation of BRAFV600E, histological subtypes, and history of other malignancies (Figure 2D, H). In addition to several tumor microenvironment-related categories, such as “proteinaceous extracellular matrix,” and “blood vessel morphogenesis”, we found two other categories enriched in the identified gene set associated with ETE, “response to oxygen levels” and “response to steroid hormone stimulus” (Figure 1D), indicating their essential role in the aggressive behavior of PTC. Comparative analysis between the gene expression profiles in response to ETE and drug response signatures listed in the CMAP database [35] identified 6-bromoindirubin-3'-oxime and lovastatin. 6-bromoindirubin-3'-oxime, a pan-JAK inhibitor to induce apoptosis of human melanoma cells [49], was found to be the highly ranked compound with antagonistic effects on genes associated with ETE (enrichment score: -0.811; Table S5). The compound ranked second is lovastatin, a cholesterol-lowering drug with dual effects on the proliferation of anaplastic thyroid cancer [50], which could induce the biological state encoded in the signature associated with ETE (enrichment score: 0.925; Table S5). The identification of these compounds might be beneficial for PTC patients with high risk of progression.
Expression and survival of different genes. Expression of HIGD1B (A-B) and SDPR (E-F) under different degrees of extrathyroidal extension (ETE) in our collected (A, E) and the THCA (B, F) cohorts. Subjects from the THCA cohort were stratified according to the expression of HIGD1B (C) and SDPR (G). Multivariate Cox regression analysis of HIGD1B (D) and SDPR (H) after taking into account age, gender, TNM stage, BRAFV600E mutation, histological subtypes, and history of other malignancies.
Co-expression networks reveal the critical role of lncRNAs in PTC carcinogenesis
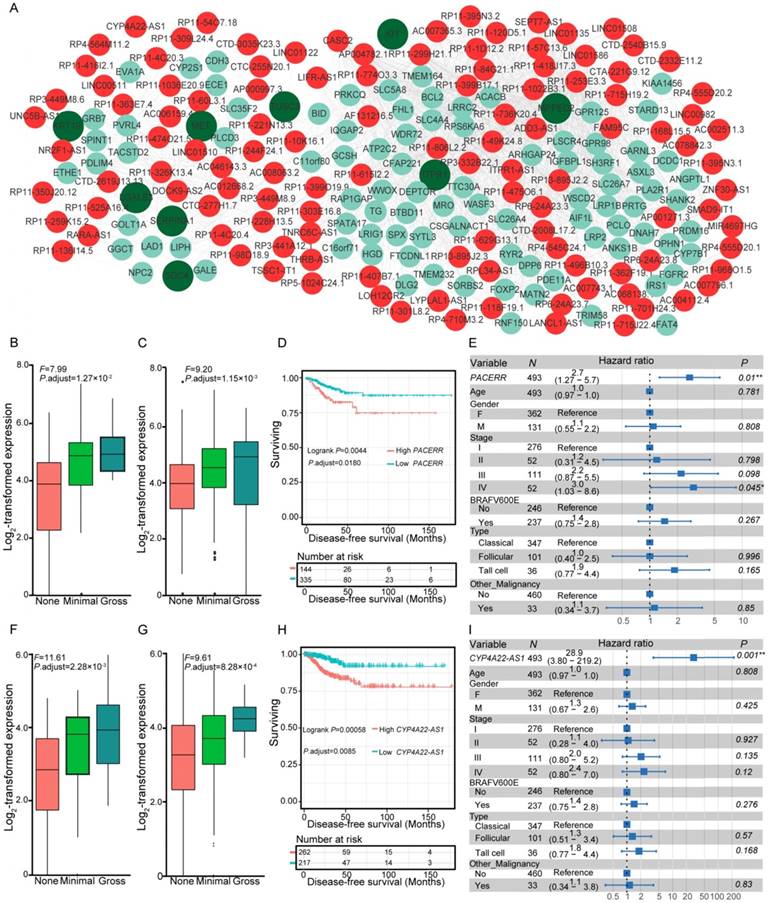
Gene expression can be organized into modules, which results in co-expression variations across conditions. We investigated the co-expression relationships between the differentially expressed lncRNAs and coding genes associated with the carcinogenesis of PTC. Following the identification of significantly co-expressed lncRNA-mRNA pairs and RNA-RNA interactions, we obtained several networks that consisted of 1,528 co-expression relationships between 349 lncRNAs and 491 mRNAs (Table S6). The maximum connected subgraph of the network contained 120 lncRNAs and 103 mRNAs (Figure 3A). Highly connected nodes were likely to be the hubs with more competing interactions. Higher degrees of interactions were observed in the lncRNA nodes than the coding-gene nodes (Figure 3A), and the top 10 high-degree nodes were all of the lncRNAs. We observed significantly enriched processes relevant for extracellular matrix microenvironments among the genes impacted by lncRNAs, including “cell adhesion”, “proteinaceous extracellular matrix”, and “transmembrane receptor protein tyrosine kinase activity”, which have been proposed to be important in the pathogenesis of thyroid cancer [48]. Some of the identified coding genes impacted by lncRNAs have already been described to be involved in thyroid cancer development, such as MET, KRT19, and SERPINA1 [48]. These results suggested the functional importance of lncRNAs in the PTC carcinogenesis.
Co-expression of differentially expressed genes (DEGs) and lncRNAs, and expression and survival of various lncRNAs. (A) Co-expression network of DEGs and lncRNAs. Nodes with red and cyan shading correspond to identified differentially expressed lncRNAs and DEGs, respectively. Nodes with dark green indicate pathogenesis-related genes of thyroid cancer [48]. Expression of PACERR (B-C) and CYP4A22-AS1 (F-G) under different degrees of extrathyroidal extension (ETE) in our collected (B, F) and the THCA (C, G) cohorts. Subjects from the THCA cohort were stratified according to the expression of PACERR (D) and CYP4A22-AS1 (H). Multivariate Cox regression analysis of PACERR (E) and CYP4A22-AS1 (I) after taking into account age, gender, TNM stage, BRAFV600E mutation, histological subtypes, and history of other malignancies.
Dysregulated lncRNAs are markers of aggressive behavior and survival in PTC
Co-expression networks suggested the essential role of lncRNAs in PTC pathogenesis, which prompted us to assess whether the expression levels of these co-expressed lncRNAs correlated with disease progression and survival of PTC patients. We examined the co-expression matrix and the repository of lncRNAs identified to be correlated with progression of ETE in the two independent cohorts (Table S3). After performing Kaplan-Meier survival analysis and taking into consideration the confounding clinical factors in multivariate Cox proportional hazards regression analysis, two dysregulated lncRNAs, PACERR (Figure 3B-E) and CYP4A22-AS1 (Figure 3F-I), were for the first time identified to be correlated with the disease-free survival of PTC patients (Table S7). Increased expression of PACERR (Figure 3B-C) and CYP4A22-AS1 (Figure 3F-G) was observed during the progression of ETE in two independent cohorts, and their increase was associated with worse disease-free survival of PTC patients in the Kaplan-Meier survival analysis after controlling for multiple hypothesis tests (Figure 3D, H). Furthermore, the association between the increased expression of the two lncRNAs and the disease-free survival of PTC patients remained statistically significant after accounting for age, gender, TNM staging, mutation of BRAFV600E, histological subtypes, and history of other malignancies in the multivariate Cox regression analysis (Figure 3E, I). Thus, these two dysregulated lncRNAs provide novel prognostic biomarkers for PTC.
Differentially expressed circRNAs between PTC and adjacent normal tissues
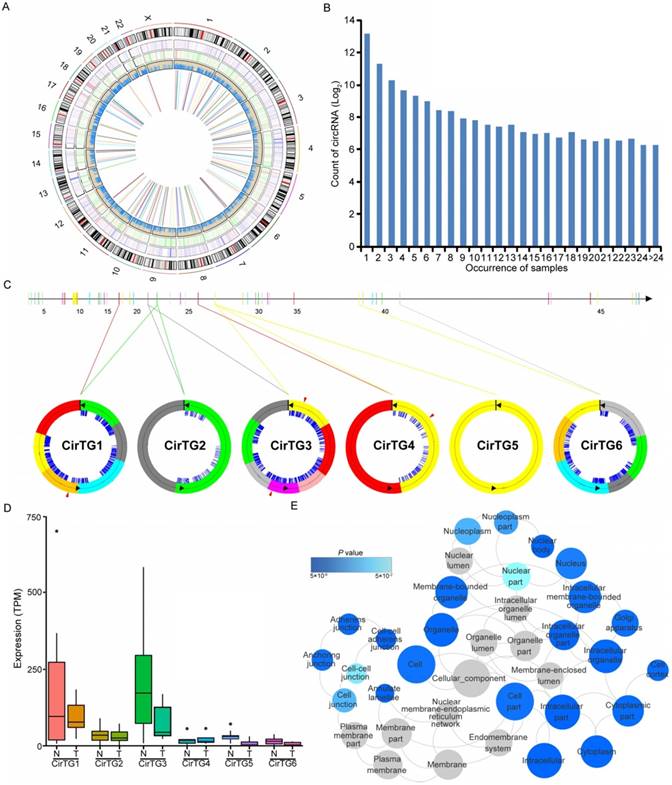
Increasing evidence has suggested the crucial functions of another class of noncoding RNAs, circRNAs, in regulating gene expression and tumorigenesis [51-54]. Our search for these noncoding RNAs identified in total 17,864 circRNAs in thyroid tissues, 19.29% of which were shared by more than five samples (Figure 4B). By comparing each of these circRNAs between normal and tumor tissues, we identified 146 differentially expressed circRNAs generated from 128 annotated host genes (Figure 4A and Table S8). We observed significantly enriched GO terms based on host genes of these circRNAs, including “intracellular part” and “cell-cell adherens junction”, which are functional processes that are reported to be relevant to the tumorigenesis of PTC [55]. Interestingly, some genes yielded multiple distinct circularized RNAs that were observed in our study. It is of note that six distinct circRNAs were observed in the TG gene, whose mutations were associated with significantly worse clinical outcomes of PTC [13] (Figure 4C). Some circRNAs had different acceptors but shared the same donor, indicating the existence of alternative splicing events (Figure 4C). An abundance of circRNA isoforms revealed dissimilar expression profiles among different circRNAs originating from the same host gene (Figure 4D). Multiple binding sites of miRNA and RNA binding proteins were predicted in the identified circRNAs (Figure 4C), which implied that these circRNAs could function as miRNA sponges to regulate mRNA expression [52, 53]. Collectively, the above results suggested the role of circRNAs in the carcinogenesis of PTC, and that the alternative back-splicing contributed to the diversity and functional complexity of PTC circRNAs.
Circular RNAs (circRNAs) identified in papillary thyroid carcinoma (PTC). (A) Genome-wide distribution of differentially expressed genes, lncRNAs, RNA editing and circRNAs in thyroid cancer. Two circles adjacent to the karyotypes show lines representing the distribution of differentially expressed coding genes and lncRNAs. The innermost circle shows lines representing differentially expressed circRNAs. The next circle shows lines representing differentially edited sites. (B) The number of circRNAs shared by samples. (C) Six circRNAs identified in TG gene with binding sites of miRNAs (blue lines) and RNA binding proteins (red triangles). (D) Expression of circRNAs in thyroid cancer and paired normal tissues. (E) Functional enrichment of parental genes of differentially expressed circRNAs.
The landscape of AS in PTC
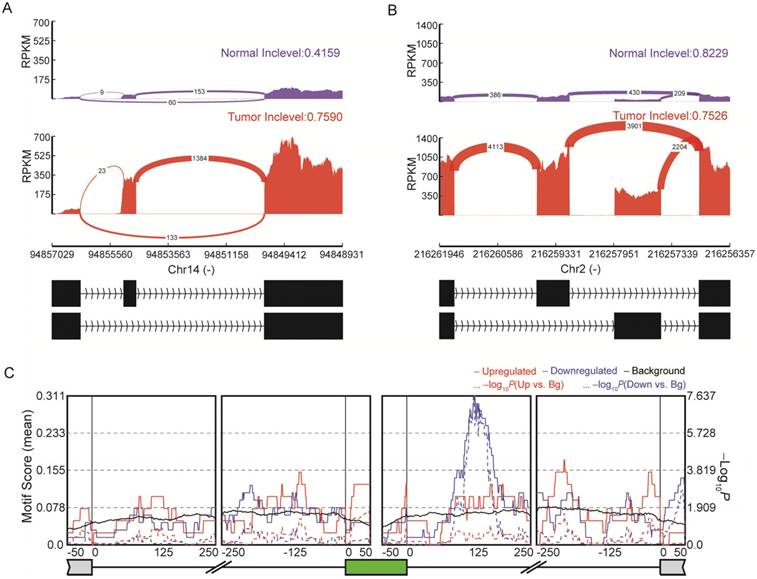
Emerging evidence indicates the important role of AS in the pathophysiology of many human cancers [56, 57]. We obtained a total of 107 highly reliable differential AS events between normal and tumor tissues (Figure 5 and Table S9), including 57 SE, six RI, 31 MXE, seven A5SS and six A3SS events. Among the genes with identified AS events, some had already been proven to be crucial for thyroid cancer development. For instance, SERPINA1 exhibited differential exon skipping between tumor and normal tissues (Figure 5A), and a mutually exclusive exon of the FN1 gene was shown with its flanking exons (Figure 5B). RNA binding proteins (RBPs) can regulate AS through binding to regulatory RNA sequence motifs in the exons and/or flanking intronic sequences of alternatively spliced exons [58]. Analyses of binding motifs in the exons and flanking intronic sequences of SE revealed 14 significantly enriched RBPs (Table S10). For example, IGF2BP3 binding sites were observed downstream of the included exons (Figure 5C). The newly identified AS events are expected to facilitate a better understanding of this disease.
Alternative splicing (AS) in papillary thyroid carcinoma (PTC). (A) SERPINA1 exhibits differential exon skipping between tissues of thyroid cancer and non-cancerous tissues. (B) Mutually exclusive exon of the FN1 gene is shown with its flanking exons. Histograms represent exon read density and arcs represent splice junctions with the number of reads mapped to the junctions indicated by the thickness of the arc. (C) Binding site analyses of RNA-binding proteins (RBPs) around the 57 skipped exon events reveal enrichment of IGF2BP3.
Differential RNA-editing pattern in PTC
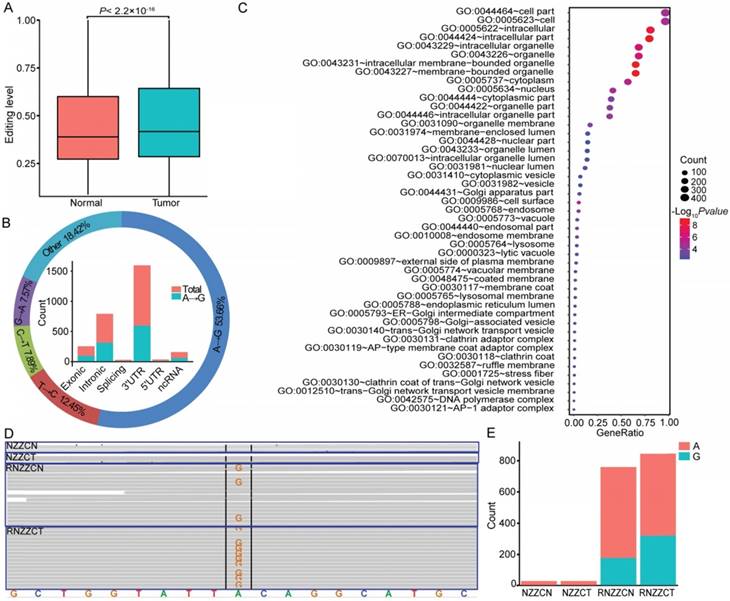
RNA editing may supplement alterations in genomic DNA to drive tumorigenesis [28, 59, 60]. In total, we detected 113,884 RNA editing sites in thyroid tissues, and approximately 71.02% (71,663 of 100,905) of A-to-I (G) editing sites were annotated in the RADAR [46]. To determine whether editing is altered in PTC, the mean editing levels across all edited sites were compared between normal and tumor tissues. An elevated RNA editing level was observed in tumor compared to normal tissues (Figure 6A). To identify specific sites that were altered in cancers, we next compared the editing level of each site between paired cancer and normal tissues. Overall, we detected 2,469 differentially edited sites in tumors compared with normal tissues, and there was a significant enrichment of the A-to-G variation (Figure 6B and Table S11). When considering the distribution of RNA editing variants in different genomic regions, we observed that 33.35% of the differentially edited sites resided in the 3′ untranslated regions (UTR) derived from 506 genes (Figure 6B). The most enriched functional categories for the differentially edited 3' UTR variants fell into “intracellular membrane-bounded organelle”, “cell surface”, and “posttranscriptional regulation of gene expression”, which are all core functional processes in the cell. RNA editing at 3' UTRs could regulate the stability of target transcripts by creating or eliminating miRNA targeting sites [61, 62]. We obtained 36 sites within 3' UTRs of 27 genes that showed altered complementary seed sites of miRNA targets. For example, in the 3' UTR of METTL7A, which is a tumor suppressor in some cancers, we found two A-to-G editing sites (chr12:51324436; chr12:51324506) in four samples (Figure 6D-E). The site (chr12:51324436) was a complementary seed site of multiple miRNAs, including miR-320, miR-4251, miR-4329, miR-4663, miR-6761-5p, miR-7160-5p, and miR-8077. Notably, miR-320a, one of the members of miR-320, was downregulated in thyroid cancer of the TCGA cohort, ovarian cancer [63], colon cancer [64], and medulloblastoma [65]. These data shed new light on the functions of RNA editing variants in the tumorigenesis of PTC.
RNA editing events in papillary thyroid carcinoma (PTC). (A) Editing level is elevated in thyroid cancer tissues compared to normal tissues. (B) The figure summarizes the results for different categories (outer circle) and genomic locations (inner histogram) of differential RNA editing variants. (C) Functional enrichment of genes with differentially edited 3' UTR variants. (D-E) Differential RNA editing site (chr12:51324506) identified in the genome (NZZCN, NZZCT) and transcriptome (RNZZCN, RNZZCT) of paired normal and thyroid cancer tissues.
Discussion
PTC is the fastest-growing disease caused by a series of molecular alterations [2, 3, 8]. Previous studies attempting to identify molecular alterations underpinning PTC have focused primarily on characterizing aberrations at the DNA level, such as mutations and structural variations [8, 13]. Besides identifying variations at the genetic level, a detailed transcriptomic characterization is expected to provide a better understanding of this disease, as well as facilitate the prognosis and development of new therapeutic strategies. Here we report a comprehensive transcriptome-wide analysis of the noncoding RNAs and mRNAs, AS, and RNA editing, revealing a clinically significant relationship between mRNAs/lncRNAs and PTC prognosis.
Genes associated with the progression of PTC
Although several genes have been implicated in PTC carcinogenesis, the molecular markers associated with the progression of this disease remain largely unknown, which limits early diagnosis and treatment of patients with high recurrence risk [66]. ETE, a prognostic factor strongly associated with recurrence and aggressive features of PTC [10, 31], was used to assess the progression of PTC in our study. We obtained and validated 264 genes associated with ETE of PTC in two independent cohorts, and identified 8 genes correlated with the disease-free survival of PTC patients after adjusting for multiple clinical factors, thus expanding the repertoire of prognostic biomarkers in this disease. Some of them have been reported to be associated with survival of other types of cancers. For example, SDPR could function as a metastasis suppressor in breast cancer by promoting apoptosis, and its loss of expression correlated with significantly reduced distant-metastasis-free and relapse-free survival in breast cancer patients [67]. It has been demonstrated that the expression of GALNT9, another gene identified in our survival analysis, was associated with high overall survival in neuroblastoma patients, independent of the standard risk-stratification covariates [68]. The enrichment of the genes associated with ETE of PTC revealed the pivotal role of the cellular microenvironment in the progression of this disease. Interestingly, we found that some of the genes were related to steroid hormone response, indicating the essential role of this function in PTC progression. Further comparison between the expression profiles in response to ETE and drug response signatures in the CMAP database [35] revealed two highly ranked compounds, 6-bromoindirubin-3'-oxime and lovastatin. As an inhibitor of 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase, lovastatin can inhibit the conversion of mevalonate from HMG-CoA. It was previously reported that it could suppress invasiveness of anaplastic thyroid cancer cells by inhibiting Rho geranylgeranylation and RhoA/ROCK signaling [69]. Recently, dual effects for lovastatin in anaplastic thyroid cancer, inducing neoplastic proliferation at low doses but antineoplastic effects at high doses, have been reported [50]. A positive correlation between signatures associated with ETE and effect of lovastatin (10 μM) was observed in our study. These results suggested a need for the careful evaluation of dose effects before treatment of thyroid cancer patients with lovastatin. In another example, 6-bromoindirubin-3'-oxime, a pan-JAK inhibitor that could induce apoptosis in human melanoma cells [49], was found to have antagonistic effects on genes associated with ETE. Thus, it is possible that these compounds provide an alternative strategy for treatment of patients with a high recurrence risk of PTC.
Dysregulation of non-coding RNAs associated with aggressive behavior of PTC
Non-coding RNAs can have regulatory functions to diversify the genetic output and, due to their high stability and association with the pathogenesis of human cancers, are promising diagnostic and prognostic biomarkers [19-21]. They may also function to amplify the malignancy and progression of cancer induced by other genomic alterations [70]. Although several lncRNAs are aberrantly expressed in PTC [23-27], their key targets have not yet been identified. In this study, our co-expression network analysis of DEGs and differentially expressed lncRNAs suggested the essential role of lncRNAs in PTC pathogenesis by modulating extracellular matrix and cell adhesion. The survival analysis identified two co-expressed lncRNAs, PACERR and CYP4A22-AS1, which were associated with disease-free survival of thyroid cancer patients after considering the confounding clinical factors including age, gender, TNM staging, mutation of BRAFV600E, histological subtypes, and history of other malignancies. It has been reported that PACERR could activate the expression of COX-2, a gene whose deregulated expression is linked to progression and outcome of several types of cancers, by occluding repressive NF-kB complexes, and play a role in inflammation and cancer [71]. Our study also identified the association of CYP4A22-AS1 with the survival of PTC patients. The expression of CYP4A22-AS1 has been reported to be enriched in nuclear fractions of several cancer cell lines including K562, HelaS3, and HepG2 [72], and its expression levels could discriminate clear cell renal cell carcinoma from normal renal tissues [73]. These two dysregulated lncRNAs can potentially be used as novel prognostic biomarkers in PTC.
AS and RBPs involved in the carcinogenesis and development of PTC
AS events have been linked to the pathophysiology of many human cancers [56, 57]. This study has cataloged numerous AS events in thyroid cancer. The expression of SERPINA1 could be used to identify PTC against benign nodules with 90% accuracy [74]. Most encouragingly, in our study, we detected differential exon skipping of SERPINA1 between tumor and normal tissues. Overexpression of the FN1 gene was regarded as an important determinant of thyroid cancer aggressiveness [75]. A mutually exclusive exon of this gene was observed in our study. The newly discovered AS events of SERPINA1 and FN1 here have the potential to improve the sensitivity and specificity of these biomarkers. Analysis of binding motifs in the exons and flanking intronic sequences of SE revealed 14 significantly enriched RBPs, including IGF2BP3 and PCBP2. In thyroid tumors, IGF2BP3 could activate IGF2 translation and IGF1 receptor signaling via PI3K and MAPK cascades, as well as promote cell proliferation, invasion, and transformation [76]. It is speculated that the alternatively spliced transcripts regulated by RBPs are potentially involved in the carcinogenesis and development of PTC, although further studies are warranted to define the binding activity of these RBPs.
In conclusion, we have provided a comprehensive transcriptomic signature associated with the carcinogenesis and aggressiveness of PTC, and present a catalog of 10 lncRNAs and mRNAs for PTC prognosis. The findings in this study provide new insights into the pathogenesis of PTC, and are expected to facilitate the prognosis and development of new therapeutic strategies for the disease.
Abbreviations
PTC: papillary thyroid carcinoma; lncRNAs: long non-coding RNAs; ETE: extrathyroidal extension; AS: alternative splicing; MAPK: mitogen-activated protein kinase; PI3K-AKT: phosphoinositide 3-kinase AKT; TG: thyroglobulin; circRNAs: circular RNAs; AJCC: American Joint Committee on Cancer; FDR: false discovery rate; SE: skipped exon; RI: retained intron; MXE: mutually exclusive exons; A5SS: alternative 5′ splice sites; A3SS: alternative 3′ splice sites; DEGs: Differentially expressed genes; RBPs: RNA binding proteins; UTR: untranslated regions.
Acknowledgements
This work was supported by the National Key Research and Development Program of China (2016YFC0900400) and the National Natural Science Foundation of China (81672649).
Supplementary Material
Supplementary figure 1.
Supplementary tables 1-2.
Supplementary tables 3-8.
Supplementary table 9.
Supplementary table 10.
Supplementary table 11.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Burgess JR, Tucker P. Incidence trends for papillary thyroid carcinoma and their correlation with thyroid surgery and thyroid fine-needle aspirate cytology. Thyroid. 2006;16:47-53
2. Davies L, Welch HG. Increasing incidence of thyroid cancer in the United States, 1973-2002. Jama. 2006;295:2164-7
3. Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64:9-29
4. Liang J, Cai W, Feng D, Teng H, Mao F, Jiang Y. et al. Genetic landscape of papillary thyroid carcinoma in the Chinese population. J Pathol. 2018;244:215-26
5. Nair A, Lemery SJ, Yang J, Marathe A, Zhao L, Zhao H. et al. FDA approval summary: lenvatinib for progressive, radio-iodine-refractory differentiated thyroid cancer. Clin Cancer Res. 2015;21:5205-8
6. Brose MS, Nutting CM, Jarzab B, Elisei R, Siena S, Bastholt L. et al. Sorafenib in radioactive iodine-refractory, locally advanced or metastatic differentiated thyroid cancer: a randomised, double-blind, phase 3 trial. Lancet. 2014;384:319-28
7. Haraldsdottir S, Shah MH. New era for treatment in differentiated thyroid cancer. Lancet. 2014;384:286-8
8. Network TCGAR. Integrated genomic characterization of papillary thyroid carcinoma. Cell. 2014;159:676-90
9. Liu R, Zhang T, Zhu G, Xing M. Regulation of mutant TERT by BRAF V600E/MAP kinase pathway through FOS/GABP in human cancer. Nat Commun. 2018;9:579
10. Xing M. Molecular pathogenesis and mechanisms of thyroid cancer. Nat Rev Cancer. 2013;13:184-99
11. Radowsky JS, Howard RS, Burch HB, Stojadinovic A. Impact of degree of extrathyroidal extension of disease on papillary thyroid cancer outcome. Thyroid. 2014;24:241-4
12. Londero SC, Krogdahl A, Bastholt L, Overgaard J, Pedersen HB, Hahn CH. et al. Papillary thyroid carcinoma in Denmark, 1996-2008: outcome and evaluation of established prognostic scoring systems in a prospective national cohort. Thyroid. 2015;25:78-84
13. Siraj AK, Masoodi T, Bu R, Beg S, Al-Sobhi SS, Al-Dayel F. et al. Genomic profiling of thyroid cancer reveals a role for thyroglobulin in metastasis. Am J Hum Genet. 2016;98:1170-80
14. Nikiforov YE, Yip L, Nikiforova MN. New strategies in diagnosing cancer in thyroid nodules: impact of molecular markers. Clin Cancer Res. 2013;19:2283-8
15. Lu Z, Sheng J, Zhang Y, Deng J, Li Y, Lu A. et al. Clonality analysis of multifocal papillary thyroid carcinoma by using genetic profiles. J Pathol. 2016;239:72-83
16. Yoo SK, Lee S, Kim SJ, Jee HG, Kim BA, Cho H. et al. Comprehensive analysis of the transcriptional and mutational landscape of follicular and papillary thyroid cancers. PLoS Genet. 2016;12:e1006239
17. Son HY, Hwangbo Y, Yoo SK, Im SW, Yang SD, Kwak SJ. et al. Genome-wide association and expression quantitative trait loci studies identify multiple susceptibility loci for thyroid cancer. Nat Commun. 2017;8:15966
18. Ye L, Zhou X, Huang F, Wang W, Qi Y, Xu H. et al. The genetic landscape of benign thyroid nodules revealed by whole exome and transcriptome sequencing. Nat Commun. 2017;8:15533
19. Gong J, Li Y, Liu CJ, Xiang Y, Li C, Ye Y. et al. A Pan-cancer analysis of the expression and clinical relevance of small nucleolar RNAs in human cancer. Cell Rep. 2017;21:1968-81
20. Rupaimoole R, Calin GA, Lopez-Berestein G, Sood AK. miRNA deregulation in cancer cells and the tumor microenvironment. Cancer Discov. 2016;6:235-46
21. Esteller M. Non-coding RNAs in human disease. Nat Rev Genet. 2011;12:861-74
22. Pallante P, Battista S, Pierantoni GM, Fusco A. Deregulation of microRNA expression in thyroid neoplasias. Nat Rev Endocrinol. 2014;10:88-101
23. Liyanarachchi S, Li W, Yan P, Bundschuh R, Brock P, Senter L. et al. Genome-wide expression screening discloses long noncoding RNAs involved in thyroid carcinogenesis. J Clin Endocrinol Metab. 2016;101:4005-13
24. Yang M, Tian J, Guo X, Yang Y, Guan R, Qiu M. et al. Long noncoding RNA are aberrantly expressed in human papillary thyroid carcinoma. Oncol Lett. 2016;12:544-52
25. Kim D, Lee WK, Jeong S, Seol MY, Kim H, Kim KS. et al. Upregulation of long noncoding RNA LOC100507661 promotes tumor aggressiveness in thyroid cancer. Mol Cell Endocrinol. 2016;431:36-45
26. He H, Nagy R, Liyanarachchi S, Jiao H, Li W, Suster S. et al. A susceptibility locus for papillary thyroid carcinoma on chromosome 8q24. Cancer Res. 2009;69:625-31
27. Lei H, Gao Y, Xu X. LncRNA TUG1 influences papillary thyroid cancer cell proliferation, migration and EMT formation through targeting miR-145. Acta Biochim Biophys Sin (Shanghai). 2017;49:588-97
28. Paz-Yaacov N, Bazak L, Buchumenski I, Porath HT, Danan-Gotthold M, Knisbacher BA. et al. Elevated RNA editing activity is a major contributor to transcriptomic diversity in tumors. Cell Rep. 2015;13:267-76
29. Mayr C, Bartel DP. Widespread shortening of 3'UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell. 2009;138:673-84
30. Lang BH, Chow SM, Lo CY, Law SC, Lam KY. Staging systems for papillary thyroid carcinoma: a study of 2 tertiary referral centers. Ann Surg. 2007;246:114-21
31. Xing M, Clark D, Guan H, Ji M, Dackiw A, Carson KA. et al. BRAF mutation testing of thyroid fine-needle aspiration biopsy specimens for preoperative risk stratification in papillary thyroid cancer. J Clin Oncol. 2009;27:2977-82
32. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2014;29:15-21
33. Anders S, Pyl PT, Huber W. HTSeq-a Python framework to work with high-throughput sequencing data. Bioinformatics. 2014;31:166-9
34. Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139-40
35. Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ. et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006;313:1929-35
36. Szklarczyk D, Morris JH, Cook H, Kuhn M, Wyder S, Simonovic M. et al. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017;45:D362-D8
37. Fukunaga T, Hamada M. RIblast: an ultrafast RNA-RNA interaction prediction system based on a seed-and-extension approach. Bioinformatics. 2017;33:2666-74
38. Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv. 2013:1303 3997
39. Gao Y, Wang J, Zhao F. CIRI: an efficient and unbiased algorithm for de novo circular RNA identification. Genome Biol. 2015;16:4
40. Gao Y, Zhang J, Zhao F. Circular RNA identification based on multiple seed matching. Brief Bioinform. 2017 DOI: 10.1093/bib/bbx014
41. Cheng J, Metge F, Dieterich C. Specific identification and quantification of circular RNAs from sequencing data. Bioinformatics. 2016;32:1094-6
42. Feng J, Xiang Y, Xia S, Liu H, Wang J, Ozguc FM. et al. CircView: a visualization and exploration tool for circular RNAs. Brief Bioinform. 2017 DOI: 10.1093/bib/bbx070
43. Shen S, Park JW, Lu ZX, Lin L, Henry MD, Wu YN. et al. rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc Natl Acad Sci USA. 2014;111:E5593-601
44. Park JW, Jung S, Rouchka EC, Tseng YT, Xing Y. rMAPS: RNA map analysis and plotting server for alternative exon regulation. Nucleic Acids Res. 2016;44:W333-8
45. Zhang Q, Xiao X. Genome sequence-independent identification of RNA editing sites. Nat Methods. 2015;12:347-50
46. Ramaswami G, Li JB. RADAR: a rigorously annotated database of A-to-I RNA editing. Nucleic Acids Res. 2014;42:D109-13
47. Maere S, Heymans K, Kuiper M. BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics. 2005;21:3448-9
48. Griffith OL, Melck A, Jones SJ, Wiseman SM. Meta-analysis and meta-review of thyroid cancer gene expression profiling studies identifies important diagnostic biomarkers. J Clin Oncol. 2006;24:5043-51
49. Liu L, Nam S, Tian Y, Yang F, Wu J, Wang Y. et al. 6-Bromoindirubin-3'-oxime inhibits JAK/STAT3 signaling and induces apoptosis of human melanoma cells. Cancer Res. 2011;71:3972-9
50. Cai J, Wei J, Schrott V, Zhao J, Bullock G, Zhao Y. Induction of deubiquitinating enzyme USP50 during erythropoiesis and its potential role in the regulation of Ku70 stability. J Investig Med. 2018;66:1-6
51. Gao Y, Zhao F. Computational Strategies for Exploring Circular RNAs. Trends Genet. 2018 DOI: 10.1016/j.tig.2017.12.016
52. Li Y, Zheng F, Xiao X, Xie F, Tao D, Huang C. et al. CircHIPK3 sponges miR-558 to suppress heparanase expression in bladder cancer cells. EMBO Rep. 2017;18:1646-59
53. Zheng Q, Bao C, Guo W, Li S, Chen J, Chen B. et al. Circular RNA profiling reveals an abundant circHIPK3 that regulates cell growth by sponging multiple miRNAs. Nat Commun. 2016;7:11215
54. Yang Y, Gao X, Zhang M, Yan S, Sun C, Xiao F. et al. Novel Role of FBXW7 Circular RNA in Repressing Glioma Tumorigenesis. J Natl Cancer Inst. 2017:110
55. Hess J, Thomas G, Braselmann H, Bauer V, Bogdanova T, Wienberg J. et al. Gain of chromosome band 7q11 in papillary thyroid carcinomas of young patients is associated with exposure to low-dose irradiation. Proc Natl Acad Sci USA. 2011;108:9595-600
56. Lu ZX, Huang Q, Park JW, Shen S, Lin L, Tokheim CJ. et al. Transcriptome-wide landscape of pre-mRNA alternative splicing associated with metastatic colonization. Mol Cancer Res. 2015;13:305-18
57. Zhao Q, Caballero OL, Davis ID, Jonasch E, Tamboli P, Yung WK. et al. Tumor-specific isoform switch of the fibroblast growth factor receptor 2 underlies the mesenchymal and malignant phenotypes of clear cell renal cell carcinomas. Clin Cancer Res. 2013;19:2460-72
58. Fu XD, Ares M Jr. Context-dependent control of alternative splicing by RNA-binding proteins. Nat Rev Genet. 2014;15:689-701
59. Han L, Diao L, Yu S, Xu X, Li J, Zhang R. et al. The Genomic Landscape and Clinical Relevance of A-to-I RNA Editing in Human Cancers. Cancer Cell. 2015;28:515-28
60. Xu X, Wang Y, Liang H. The role of A-to-I RNA editing in cancer development. Curr Opin Genet Dev. 2017;48:51-6
61. Borchert GM, Gilmore BL, Spengler RM, Xing Y, Lanier W, Bhattacharya D. et al. Adenosine deamination in human transcripts generates novel microRNA binding sites. Hum Mol Genet. 2009;18:4801-7
62. Wang Q, Hui H, Guo Z, Zhang W, Hu Y, He T. et al. ADAR1 regulates ARHGAP26 gene expression through RNA editing by disrupting miR-30b-3p and miR-573 binding. RNA. 2013;19:1525-36
63. Shahab SW, Matyunina LV, Mezencev R, Walker LD, Bowen NJ, Benigno BB. et al. Evidence for the complexity of microRNA-mediated regulation in ovarian cancer: a systems approach. PLoS One. 2011;6:e22508
64. Zhang JX, Song W, Chen ZH, Wei JH, Liao YJ, Lei J. et al. Prognostic and predictive value of a microRNA signature in stage II colon cancer: a microRNA expression analysis. Lancet Oncol. 2013;14:1295-306
65. Ferretti E, De Smaele E, Po A, Di Marcotullio L, Tosi E, Espinola MS. et al. MicroRNA profiling in human medulloblastoma. Int J Cancer. 2009;124:568-77
66. Schlumberger MJ. Papillary and follicular thyroid carcinoma. N Engl J Med. 1998;338:297-306
67. Ozturk S, Papageorgis P, Wong CK, Lambert AW, Abdolmaleky HM, Thiagalingam A. et al. SDPR functions as a metastasis suppressor in breast cancer by promoting apoptosis. Proc Natl Acad Sci USA. 2016;113:638-43
68. Berois N, Gattolliat CH, Barrios E, Capandeguy L, Douc-Rasy S, Valteau-Couanet D. et al. GALNT9 gene expression is a prognostic marker in neuroblastoma patients. Clin Chem. 2013;59:225-33
69. Zhong WB, Liang YC, Wang CY, Chang TC, Lee WS. Lovastatin suppresses invasiveness of anaplastic thyroid cancer cells by inhibiting Rho geranylgeranylation and RhoA/ROCK signaling. Endocr Relat Cancer. 2005;12:615-29
70. Goedert L, Placa JR, Fuziwara CS, Machado MCR, Placa DR, Almeida PP. et al. Identification of long noncoding RNAs deregulated in papillary thyroid cancer and correlated with BRAF(V600E) mutation by bioinformatics integrative analysis. Sci Rep. 2017;7:1662
71. Krawczyk M, Emerson BM. p50-associated COX-2 extragenic RNA (PACER) activates COX-2 gene expression by occluding repressive NF-kappaB complexes. Elife. 2014;3:e01776
72. Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H. et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 2012;22:1775-89
73. Ellinger J, Alam J, Rothenburg J, Deng M, Schmidt D, Syring I. et al. The long non-coding RNA lnc-ZNF180-2 is a prognostic biomarker in patients with clear cell renal cell carcinoma. Am J Cancer Res. 2015;5:2799-807
74. Vierlinger K, Mansfeld MH, Koperek O, Nohammer C, Kaserer K, Leisch F. Identification of SERPINA1 as single marker for papillary thyroid carcinoma through microarray meta analysis and quantification of its discriminatory power in independent validation. BMC Med Genomics. 2011;4:30
75. Sponziello M, Rosignolo F, Celano M, Maggisano V, Pecce V, De Rose RF. et al. Fibronectin-1 expression is increased in aggressive thyroid cancer and favors the migration and invasion of cancer cells. Mol Cell Endocrinol. 2016;431:123-32
76. Panebianco F, Kelly LM, Liu P, Zhong S, Dacic S, Wang X. et al. THADA fusion is a mechanism of IGF2BP3 activation and IGF1R signaling in thyroid cancer. Proc Natl Acad Sci USA. 2017;114:2307-12
Author contact
![]() Corresponding authors: Zhongsheng Sun, Beijing Institutes of Life Science, Chinese Academy of Sciences, Beichen West Road, Chao Yang District, Beijing 100101, China. Tel.: +86 10 64864959; Fax: +86 10 84504120; E-mail: sunzsac.cn and Huajing Teng, Beijing Institutes of Life Science, Chinese Academy of Sciences, Beichen West Road, Chao Yang District, Beijing 100101, China. Tel.: +86 10 64887229; Fax: +86 10 84504120; E-mail: tenghjac.cn
Corresponding authors: Zhongsheng Sun, Beijing Institutes of Life Science, Chinese Academy of Sciences, Beichen West Road, Chao Yang District, Beijing 100101, China. Tel.: +86 10 64864959; Fax: +86 10 84504120; E-mail: sunzsac.cn and Huajing Teng, Beijing Institutes of Life Science, Chinese Academy of Sciences, Beichen West Road, Chao Yang District, Beijing 100101, China. Tel.: +86 10 64887229; Fax: +86 10 84504120; E-mail: tenghjac.cn