Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results
Discussion
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(14):3841-3855. doi:10.7150/thno.25784 This issue Cite
Research Paper
HSP40 co-chaperone protein Tid1 suppresses metastasis of head and neck cancer by inhibiting Galectin-7-TCF3-MMP9 axis signaling
Yu-Syuan Chen1,*, Ching-Wen Chang1,*, Yeou-Guang Tsay2, Liu-Ying Huang1, Yi-Chen Wu1, Li-Hao Cheng1, Cheng-Chieh Yang3,4, Cheng-Hsien Wu4, Wan-Huai Teo1, Kai-Feng Hung3,5, Chih-Yang Huang6,7,8, Te-Chang Lee9, Jeng-Fan Lo1,3,6,10,11,12 ![]()
1. Institute of Oral Biology, National Yang-Ming University, Taipei, Taiwan
2. Institute of Biochemistry and Molecular Biology, National Yang-Ming University, Taipei, Taiwan
3. Department of Dentistry, School of Dentistry, National Yang-Ming University, Taipei, Taiwan
4. Department of Stomatology, Taipei Veterans General Hospital, Taipei, Taiwan
5. Department of Medical Research and Education, Taipei Veterans General Hospital, Taipei, Taiwan
6. Graduate Institute of Chinese Medical Science and Institute of Medical Science, China Medical University, Taichung, Taiwan
7. Institute of Basic Medical Science, China Medical University, Taichung, Taiwan
8. Department of Health and Nutrition Biotechnology, Asia University, Taichung, Taiwan
9. Institute of Biomedical Sciences, Academia Sinica, Taipei, Taiwan
10. Genome Research Center, National Yang-Ming University, Taipei, Taiwan
11. Department of Dentistry, Taipei Veterans General Hospital, Taipei, Taiwan
12. Cancer Progression Center of Excellence, National Yang-Ming University, Taipei, Taiwan
* These authors contributed equally to this work.
Received 2018-2-27; Accepted 2018-5-18; Published 2018-6-13
Abstract

Human tumorous imaginal disc (Tid1), a DnaJ co-chaperone protein, is classified as a tumor suppressor. Previously, we demonstrated that Tid1 reduces head and neck squamous cell carcinoma (HNSCC) malignancy. However, the molecular details of Tid1-mediated anti-metastasis remain elusive.
Methods: We used affinity chromatography and systemic mass spectrometry to identify Tid1-interacting client proteins. Immunohistochemical staining of Tid1 in HNSCC patient tissues was examined to evaluate the association between the expression profile of Tid1-interacting client proteins with pathologic features and prognosis. The roles of Tid1-interacting client proteins in metastasis were validated both in vitro and in vivo. The interacting partner and downstream target of Tid1-interacting client protein were determined.
Results: Herein, we first revealed that Galectin-7 was one of the Tid1-interacting client proteins. An inverse association of protein expression profile between Tid1 and Galectin-7 was determined in HNSCC patients. Low Tid1 and high Galectin-7 expression predicted poor overall survival in HNSCC. Furthermore, Tid1 abolished the nuclear translocation of Galectin-7 and suppressed Galectin-7-induced tumorigenesis and metastasis. Keratinocyte-specific Tid1-deficient mice with 4-nitroquinoline-1-oxide (4NQO) treatment exhibited increased protein levels of Galectin-7 and had a poor survival rate. Tid1 interacted with Galectin-7 through its N-linked glycosylation to promote Tid1-mediated ubiquitination and proteasomal degradation of Galectin-7. Additionally, Galectin-7 played a critical role in promoting tumorigenesis and metastatic progression by enhancing the transcriptional activity of TCF3 transcription factor through elevating MMP-9 expression.
Conclusions: Overall, future treatments through activating Tid1 expression or inversely repressing the oncogenic function of Galectin-7 may exhibit great potential in targeting HNSCC progression.
Keywords: HSP40, tumor metastasis, MMP-9, Tid1, Galectin-7, head and neck squamous cell carcinoma (HNSCC)
Introduction
Head and neck squamous cell carcinoma (HNSCC) is one of the most common cancers worldwide with fewer than half of HNSCC patients surviving beyond five years [1]. Poor prognosis of HNSCC is predominantly attributed to local and distant metastasis [2]. Understanding the molecular basis by which metastasis of HNSCC is promoted may contribute to the development of a better treatment for decreasing the mortality of this cancer.
Molecular chaperones and co-chaperones play critical roles in maintaining substrate protein homeostasis through control of the folding, stability, and function of many proteins also known as “client proteins” [3, 4]. The molecular chaperone heat shock protein (HSP) 70 and its co-chaperone partner HSP40 is one major pair of chaperone and co-chaperone involved in preventing aggregation of misfolded proteins [4, 5]. In addition to their role in maintaining protein homeostasis, they are also important in tumor malignancy progression and metastasis [5]. However, the role of HSP40 in cancer still remains elusive.
Tid1, the human homolog of the Drosophila lethal tumorous imaginal disc Tid56 protein, acts as a Hsp40 co-chaperone of Hsp70. Tid1 binds to Hsp70 through its conserved DnaJ domain to regulate specificity and activity of their substrate proteins [12, 13]. Mutations in the DnaJ domain abolish part of Tid1-mediated cellular activities [14, 15]. Moreover, Tid1 is involved in multiple intracellular signaling pathways such as cell survival, cell proliferation, and apoptosis. It has also been demonstrated that Tid1 deficiency causes embryo lethality in mice [16]. In addition, Tid1 deficiency blocks early stage development of T cells [17]. Recently, we have shown that transgenic mice with muscle-specific Tid1 deletion display muscular dystrophic phenotype [18]. These results support the physiological roles and functions of Tid1 in cell survival and proliferation, and also provide the first evidence that co-chaperone protein is required for T cell and muscle development.
In addition to its involvement in the development of certain cells, we have also demonstrated the role of Tid1 as a tumor suppressor in HNSCC as well as in breast cancer [14, 19]. Although how Tid1 inhibits tumor growth is not completely understood, it is likely that the tumor-suppressing effect of Tid1 is mediated by its client/substrate/interacting proteins. The client proteins of Tid1, such as human papillomavirus E7 oncoprotein, ErbB-2, and EGFR protein, have been identified in a number of studies [19-21]. Consistently, we have shown that the protein level of EGFR in HNSCC and lung cancer, as well as that of ErbB2 in breast cancer, are down-regulated by Tid1 via ubiquitination-mediated degradation [15, 20, 22]. Taken together, these observations suggest that Tid1 plays a role in suppression of cancer development. Nevertheless, the molecular mechanisms mediated by Tid1 to suppress metastasis of HNSCC, however, are not yet well investigated.
Glycosylation is one of the most important and frequent post-translational protein modifications and occurs in about half of eukaryotic proteome [23]. N-linked glycosylation is one of the major types of protein glycosylation, which occurs in asparagine side-chains. Deregulation of protein glycosylation has contributed to several human diseases, including cancer [24], Alzheimer's disease [25] and diabetes [26]. It has been reported that mitochondrial proteins have glycosylated isoforms to regulate their functions within mitochondria or at extra-mitochondrial locations [1-3]. Since Tid1 proteins are mainly located in mitochondria, it would be of interest to determine how a mitochondrial protein such as Tid1 can be glycosylated as well as whether glycosylated Tid1 plays tumor suppressing roles during tumorigenesis.
Tid1 has two alternative splicing isoforms, Tid1 long form (Tid1-L) and Tid1 short form (Tid1-S). In this study, we first applied affinity chromatography and proteomic analyses to identify the interacting proteins of Tid1-L and Tid1-S, respectively. In this study, we provide an insight of how Tid1 modulates the client proteins to suppress HNSCC growth. Further research focusing on enhancement of Tid1 may potentially shed light on a better treatment for decreasing mortality of HNSCC.
Methods
Patient population and immunohistochemistry
Between 2004 and 2006, 56 patients with operable head and neck cancer, without histories of radiation therapy or chemotherapy, underwent surgical treatment at Taipei Veterans General Hospital (Taipei, Taiwan) and their tumor tissues were collected. This study was approved by the institutional review board and ethics committee of Taipei Veterans General Hospital and National Yang Ming University (IRB No.1000075). The deparaffinization, rehydration, antigen retrieval, antibody hybridization, visualization and grading of the collected tumor sections were performed as described previously [22].
Generation of K5-Tid1flox/flox mice and tumor induction
Keratinocytes-specific Tid1-deficient mice were generated by breeding mice bearing a loxP-flanked Tid1 gene (Tid1flox/flox) with K5-Cre mice (generously provided by Dr. Chun-Ming Chen) [16-18, 27]. The mice were injected intraperitoneally every other day for one week, starting at the age of 4 weeks, with either corn oil or 4-hydroxytamoxifen (4-5 mg/kg body weight) [27]. 4NQO (4-nitroquinoline-1-oxide; 100 mg/mL) was added to drinking water for 16 weeks [28]. Mice were sacrificed at the time points when body weight loss exceeded 30% of the initial weight, or at the defined time points [28]. All the experimental procedures regarding animal handling were approved by the Institutional Animal Care and Use Committee (IACUC) of the National Yang-Ming University (IACUC No. 991235, 1021279 and 1070402). [For animal studies, approval must be obtained from the appropriate animal care committee for compliance with the National Institutes of Health for use of laboratory animals or equivalent.]
Cell lines
The human embryonic kidney cell line 293T was originally from ATCC and maintained under recommended culture conditions. The human tongue carcinoma cell line SAS was originally from the Japanese Collection of Research Bioresources (Tokyo, Japan) [29]. Furthermore, we have successfully established the metastatic cell lines SASM-1 and SASM-5, which were derived from metastatic lung tumors generated by sequential tail vein injection and harvesting of the metastatic counterpart [30]. All of these cell lines were grown in DMEM medium containing 10% FBS and 1% L-glutamine. The OECM-1 cells were provided by Dr. Ching-Liang Meng (National Defense Medical College, Taipei, Taiwan) and cultured in RPMI 1640 medium containing 10% FBS. The OC-3 cell line was a gift from Dr. Shu-Chun Lin (National Yang-Ming University) and cultured in mixed K-SFM-DMEM medium (2:1 v/v) supplemented with 10% FBS.
Statistical analysis
The statistical package for social sciences software (version 24; SPSS, Inc., Chicago, IL) was used for statistical analysis. The data are presented as mean ± SD of one experiment performed in triplicate. Continuous variables between groups were compared by the independent Student's t-test or ANOVA. P < 0.05 was considered a significant difference for all the tests.
Results
Galectin-7 is a Tid1 client protein
To determine the molecular mechanisms by which Tid1, a co-chaperone DnaJ protein, acts as a tumor suppressor during tumorigenesis, we applied combined screening methodologies with affinity chromatography and mass spectrometry analysis to identify Tid1-interacting client proteins (Figure S1A-C). As hypothesized, the Tid1 client proteins are transferred to Hsp70 upon the binding of Tid1 with Hsp70 through its DnaJ domain [12, 13]. Thus, to identify Tid1 client proteins that are only associated with Tid1 but not Hsp70, we generated mutant Tid1 lacking the functional DnaJ domain by site-directed mutagenesis of HPD loop of DnaJ domain (HPD mutant; H466Q, P467N, and D468A) (Figure S1A). The Tid1 client proteins would not be transferred to Hsp70 but are retained on the mutant Tid1. By doing so, we could eliminate the Tid1 non-specific binding proteins through interacting directly with Hsp70. In addition, we also wanted to identify the interacting proteins of the two isoforms of Tid1. The protocol to identify the Tid1 client proteins is described in Figure S1B. As shown in Figure S1C, we observed the differential patterns of client proteins interacting with Tid1 long form wild-type (Tid1-L-wt), Tid1 long form mutant (Tid1-L-mut), Tid1 short form wild-type (Tid1-S-wt) and Tid1 short form mutant (Tid1-S-mut).
Recent studies have shown that two alternative splicing isoforms of Tid1 may have different expression levels and functions [22, 31-33]. Especially, Tid1-L functions as a tumor suppressor in various cancers, including HNSCC and non-small cell lung cancer (NSCLC) [15, 20]. Therefore, we further focused on characterizing the Tid1-L client proteins that are associated with the tumor suppressor function of Tid1. By analyzing the public database, we found that the top Tid1-L-mut interaction protein, differentially expressed between tumor and non-tumor, was Galectin-7 (Figure S1D). Galectin-7 is a member of β-galactoside-binding lectins family containing one or more conserved carbohydrate-recognition domains (CRDs) that bind to N-linked or O-linked sugars of glycoproteins [6]. In addition, Galectin‑7 is overexpressed in breast, ovarian cancer, and HNSCC and has been implicated in promoting metastasis and inducing chemoresistance of breast cancer [9-11]. Thus, the level of Galectin-7 may be associated with HNSCC progression and metastasis.
Inverse protein expression of Tid1 and Galectin-7 in HNSCC is associated with lymph node metastasis and poor prognosis
Recently, we have demonstrated that the expression of Tid1 is inversely related to tumor stage in HNSCC patients [15]. However, the role of Galectin-7 in Tid1-mediated tumor suppression in HNSCC patients needs to be determined. For this purpose, the expressions of Tid1 and Galectin-7 proteins were examined by immunohistochemistry analysis of 56 HNSCC tissue sections. The HNSCC tissues that exhibited strong staining of Tid1 were mostly weakly stained for Galectin-7, whereas HNSCC tissues showing weak Tid1 staining were more likely to exhibit strong Galectin-7 staining (Figure 1A, Table 2 and Figure S2A-B). In addition, Kaplan-Meier analysis demonstrated that HNSCC patients with a low level of Tid1 or a high level of Galectin-7 had lymph node metastasis and reduced survival (Table 1 and Figure 1A, C). Moreover, the majority of nuclear Galectin-7 were observed in HNSCC tissue that exhibited weak to moderate Tid1 staining; conversely, a high proportion of cytoplasmic Galectin-7 was found in HNSCC showing strong positivity of Tid1 staining (Figure 1A and Table 2). Meanwhile, as expected, patients whose HNSCC tissue staining exhibited nuclear Galectin-7 staining had a poor survival compared to patients with cytoplasmic staining of Galectin-7 (Figure 1C). To further determine the negative role of Tid1 in the modulation of Galectin-7 protein during head and neck cancer progression, keratinocyte-specific Tid1-deficient mice were generated by crossing Tid1 floxed mice (Tid1flox/flox) generated in our laboratory with tamoxifen-inducible cre-expressing transgenic mice driven by the K5 promoter [16-18, 27]. 4NQO was added in the drinking water to induce tumors on the tongue surface of mice [28]. 4NQO-treated mice with deleted Tid1 gene exhibited increased protein levels of Galectin-7 and had a poor survival rate (Figure 1D and Figure S2C). These results were inconsistent with our clinical findings and support that Tid1 negatively regulates the expression of Galectin-7 and its nuclear translocation. Therefore, the expression of Galectin-7 in HNSCC is indeed inversely correlated with Tid1, and the contribution of Tid1 to suppression of HNSCC is likely mediated by inhibition of Galectin-7 protein and its nuclear translocation.
Predictive survival of HNSCC patients based on the expression profile of Tid1 and Galectin-7. (A) Immunohistochemical staining (IHC) of Tid1 and Galectin-7 on HNSCC patients' tumor tissues with/without lymph node metastasis. Black arrows indicating the positive nuclear staining of Galectin-7. (B) Kaplan-Meier analysis of overall survival according to the expression of the Tid1 score (left up panel), Galectin-7 score (middle panels) and multivariate analysis (right panel). (C) Kaplan-Meier analysis of overall survival according to Galectin-7 subcellular localization. (D) Galectin-7 expression in the cutaneous epithelium of Tid1f/f mice without 4NQO treatment, and of Tid1f/f and K5-Tid1f/f mice treated with 4NQO, respectively, was stained by immunohistochemistry.
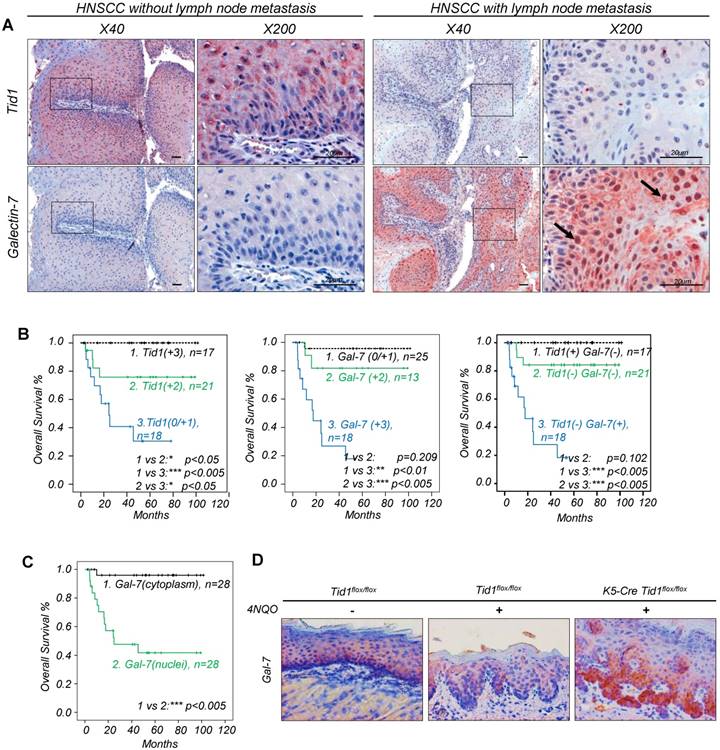
Fisher's exact test for the association between Tid1 and Galectin-7 expression
| Tid1 expression (patients n=56) | p Value | ||||
|---|---|---|---|---|---|
| Weak positive (0/1+) | Moderate positive (2+) | Strong positive (3+) | |||
| Gal-7 expression | Weak positive (0/1+) | 1 (1.8%) | 9 (16.1%) | 15 (26.8%) | ***<0.001 |
| Moderate positive (2+) | 2 (3.6%) | 9 (16.1%) | 2 (3.6%) | ||
| Strong positive (3+) | 15 (26.8%) | 3 (5.4%) | 0 (0%) | ||
Characteristics of 56 patients with HNSCC with Tid1 or Galectin-7 expressiona
| Variables | Tid1 Highb | Tid1 Lowc | P-value | Galectin-7 Highd | Galectin-7 Lowe (n=25) | P-value |
|---|---|---|---|---|---|---|
| (n=38) | (n=18) | (n=31) | ||||
| Age | ||||||
| ≧54 | 20 | 8 | 18 | 10 | ||
| <54 | 18 | 10 | 0.775 | 13 | 15 | 0.282 |
| Overall stage | ||||||
| Pre-cancer-II | 8 | 1 | 3 | 6 | ||
| III-IV | 30 | 17 | 0.245 | 28 | 19 | 0.272 |
| T-stage | ||||||
| T1-T2 | 8 | 3 | 4 | 7 | ||
| T3-T4 | 30 | 15 | 0.047f | 27 | 18 | 0.069 |
| Lymph node metastasis | ||||||
| N=0 | 31 | 8 | 16 | 23 | ||
| N≧0 | 7 | 10 | 0.004f | 15 | 2 | 0.003f |
| Differentiation | ||||||
| Well | 26 | 11 | 17 | 20 | ||
| Moderate to Poor | 12 | 7 | 0.763 | 14 | 5 | 0.09 |
| Recurrence | ||||||
| Yes | 8 | 8 | 12 | 4 | ||
| No | 30 | 10 | 0.112 | 19 | 21 | 0.079 |
aBy Fisher test.
bHigh, representative HNSCC with intense Tid1 immunoreactivity (+2 and +3).
cLow, representative HNSCC showing negative expression and almost absent Tid1 immunoreactivity (0/+1).
dHigh, representative HNSCC with intense Galectin-7 immunoreactivity (+2 and +3).
eLow, representative HNSCC showing negative expression and almost absent Galectin-7 immunoreactivity (0/+1).
fP<0.05
Down-regulation of Galectin-7 inhibits tumorigenic potential in metastatic HNSCC cell lines
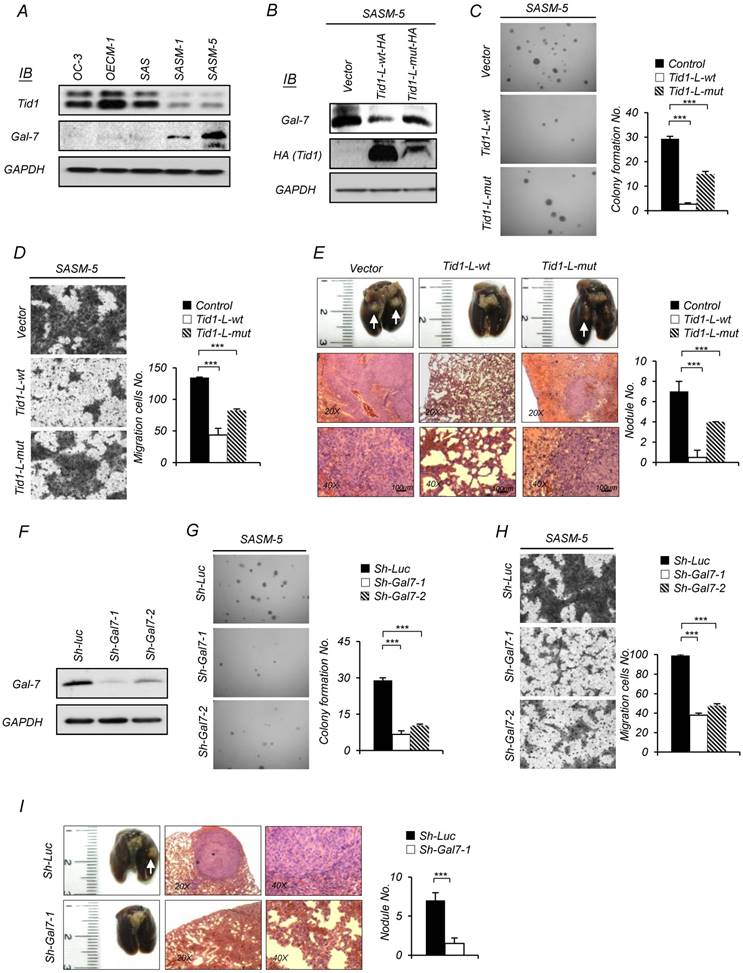
We first evaluated the endogenous expression pattern of both Tid1 and Galectin-7 proteins in established HNSCC cells, including the metastases-derived cell lines [34, 35]. As shown in Figure 2A, Galectin-7 was significantly up-regulated in highly metastatic lines (SASM-1 and SASM-5) but not in their parent cells (SAS) with limited metastatic potential, or two different HNSCC lines (OC-3 and OECM-1) that exhibit robust expression of the endogenous Tid1 protein. The inverse correlation of Tid1 and Galectin-7 expression was further validated by the findings that overexpression of Tid1-L-wt, but not Tid1-L-mut, in SASM-5 cells significantly down-regulated the level of endogenous Galectin-7 (Figure 2B). These findings suggest that Galectin-7 may enhance metastatic potential. Consistently, we showed that both the in vitro anchorage-independent growth and migration abilities, as well as the in vivo metastatic ability, were inhibited by overexpression of Tid1-L-wt (Figure 2C-E) or depletion of endogenous Galectin-7 expression by small hairpin RNAi (Sh-Gal7-1 and Sh-Gal7-2) in SASM-5 cells (Figure 2F-I). Overall, these results suggest that Galectin-7 is required to promote the malignant phenotype of HNSCC, which may be negatively regulated by Tid1.
Tid1 suppresses tumorigenesis and metastasis of HNSCC by downregulating Galectin-7 expression. (A) Expression of endogenous Tid1 and Galectin-7 protein of five HNSCCs (OC3, OECM1, SAS, SASM-1, and SASM-5) was examined by immunoblot assay using an antibody against Tid1. (B-E) SASM-5 cells were transfected with control, Tid1-L-wt-HA, or Tid1-L-mut-HA plasmids, respectively. (B) Whole cell protein lysates from the transfected cells were collected and examined for the endogenous amount of Gal-7 by immunoblotting assay with an antibody against Gal-7. The amount of GAPDH was served as a loading control. In vitro malignancy of the transfected cells was examined by (C) anchorage-independent growth and (D) Transwell® migration ability, respectively. (E) Representative images of collected lungs (top panel) and tissue sections stained with hematoxylin and eosin (H&E; middle and bottom panel) from nude mice with tail vein injection of transfected cells. White arrows indicating metastatic tumor nodules. Quantification of the tumor nodules in the lungs of nude mice by gross examination (right panel). (F-I) SASM-5 cells were transduced with sh-Gla7-1 or sh-Gal7-2 shRNA lentivirus to downregulate Gal-7 expression (sh-Luc was used as a control). (F) Knockdown efficiency of Gal-7 protein was confirmed by immunoblotting. Representative images of anchorage-independent growth assay (G) and Transwell® migration assays (H) of Gal-7-knockdown (sh-Gla7-1 or sh-Gal7-2) SASM-5 cells and control (sh-Luc) SASM-5 cells, in which the cells were stained with crystal violet. (I) Representative images of collected lungs (left panel) and tissue sections stained with H&E (middle and right panels) from nude mice after tail vein injection with Gal-7 knockdown (sh-Gla7-1) or control (sh-Luc) SASM-5 cells, respectively. Quantification of the tumor nodule in the lungs of nude mice by gross examination (right panel). The histograms shown are the mean ± SD from three independent experiments and analyzed by Student's t-test (∗p < 0.01; ∗∗p < 0.005).
Tid1 suppresses tumorigenicity through inhibition of Galectin-7 functions in HNSCC
To further investigate the inhibitory role of Tid1 in regulating Galectin-7 protein level, we performed transient co-transfection with increasing amounts of Tid1 DNA plasmid and constant amounts of Galectin-7 DNA plasmid in 293T cells. By immunoblot analyses, a declined protein level of Galectin-7 was observed along with an increased expression of Tid1-L-wt but not Tid1-L-mut proteins (Figure S3A). Next, we co-expressed Tid1-L-wt or Tid1-L-mut with Galectin-7 to test whether Tid1 could suppress the Galectin-7-induced tumorigenicity in HNSCCs. We found that the ectopic expression of Galectin-7 in SAS cells led to increases in anchorage-independent growth, cell migration and invasion ability, which was abolished upon further ectopic co-expression of Tid1-wt but not Tid1-mut proteins (Figure S3B-D). Additionally, in SAS cells, the protein level of Galectin-7 was markedly increased by knockdown of Tid1 along with the ectopic co-expression of Galectin-7; simultaneously, the invasion capacity was also promoted (Figure S3E-F). Together, these data suggest that Tid1 plays a role in negative regulation of head and neck cancer cell malignancy through suppression of Galectin-7 protein.
Fisher's exact test for the association between Tid1 expression and Galectin-7 subcellular localization.
| Tid1 expression (patients n=56) | p Value | ||||
|---|---|---|---|---|---|
| Weak positive (0/1+) | Moderate positive (2+) | Strong positive (3+) | |||
| Gal-7 localization | Cytoplasm | 3 (5.4%) | 9 (16.1%) | 16 (28.6%) | ***<0.001 |
| Nucleus | 15 (26.8%) | 12 (21.0%) | 1 (1.8%) | ||
Tid1 N-glycosylation mediates the interaction of Tid1 and Galectin-7
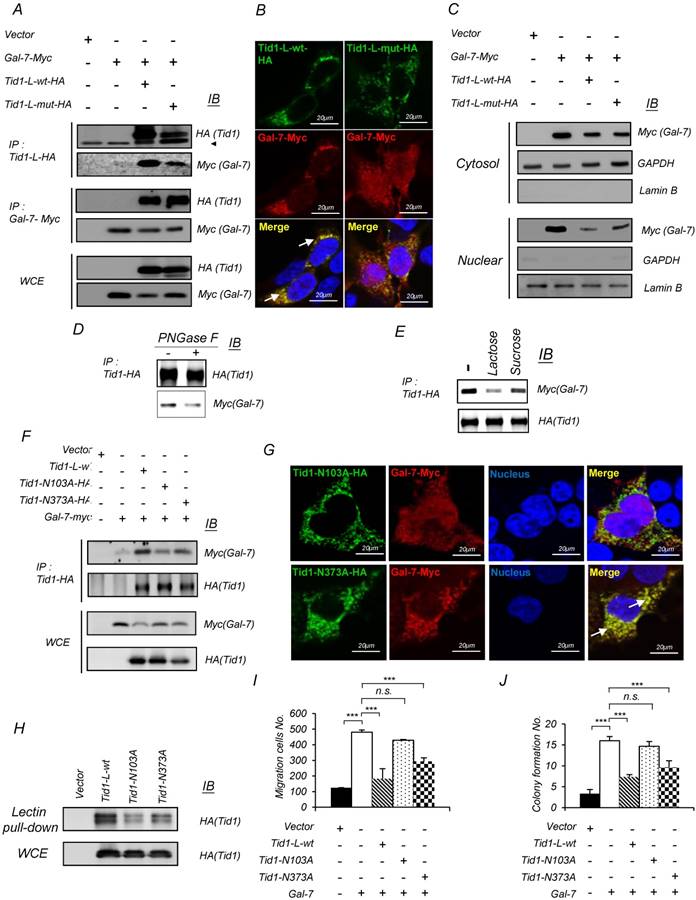
The interaction between Tid1-L-wt or Tid1-L-mut and Galectin-7 was further validated with the demonstration that HA-tagged Tid1-L-wt or Tid1-L-mut was co-immunoprecipitated with Myc-tagged Galectin-7 and vice versa with HA-tagged Tid1-L in 293T cells, co-transfected with HA-tagged Tid1-L-wt (or Tid1-L-mut) and Myc-tagged Galectin-7 plasmids (Figure 3A). Interestingly, we found that the protein level of exogenous Galectin-7 was reduced in Tid1-wt transfected cells in comparison with that in Tid1-mut transfected cells (Figure 3A). Consistent with the findings of co-immunoprecipitation, confocal immunofluorescence analysis showed that Galectin-7 was more significantly co-localized with Tid1-L-wt than Tid1-L-mut (Figure 3B). Moreover, Tid1-L-mut-transfected cells exhibited increased nuclear localization of Galectin-7 compared to Tid1-L-wt-transfected cells (Figure 3C). Previous studies show that the carbohydrate branches of N-glycan possess a high affinity to Galectins [36]. Therefore, we speculate that the interaction between Tid1 and Galectin-7 can be bridged by N-linked glycosylation of Tid1. Herein, we found that treatment with peptide N-Glycosidase F (PNGase F) diminished the Tid1-Galectin-7 interaction (Figure 3D). In addition, it has been shown that lactose binds to the carbohydrate-recognition domain (CRD) of Galectin to block protein-protein interaction [36]. By co-immunoprecipitation assay, we also demonstrated that lactose inhibited the interaction between Tid1 and Galectin-7 (Figure 3E). With the aid of software prediction (NetNGlyc 1.0 Server; http://www.cbs.dtu.dk/services/NetNGlyc/), we noticed that there are two putative N-linked glycosylation sites (103N and 373N) in Tid1 protein. Cell lines expressing the mutant Tid1 proteins (N103A and N373A) containing mutation sites (replacement of arginine to alanine) for N-linked glycosylation were generated, respectively (Figure S4). Compared to the Tid1-L-wt protein, cells expressing the mutant Tid1 (N103A) displayed reduced interaction with Galectin-7 and sustained more Galectin-7 proteins (Figure 3F). Further, we found diminished co-localization of mutant Tid1-N103A and Galectin-7 but not that of mutant Tid1-N373A and Galectin-7 by confocal imaging analysis (Figure 3G). Additionally, cells expressing Tid1-N103A protein also displayed increased nuclear localization of Galectin-7, in comparison with cells expressing the mutant Tid1-N373A proteins (Figure 3G). By using lectin-pull down assay, we also recovered the least Tid1-N103A protein (Figure 3H). Lastly, the inhibitory effect of Tid1 on Galectin-7-enhanced anchorage-independent growth and migration ability of cells expressing Tid1-N103A proteins was attenuated by disrupting the interaction between mutant Tid1 and Galectin-7 (Figure 3I-J). Together, our results suggest that the N-glycosylation at the 103 arginine residue of Tid1 protein is important for Tid1 and Galectin-7 interaction and has a profound effect on Tid1-mediated tumor suppression.
Tid1 interacts with Galectin-7 through N-linked glycosylation to mediate Galectin-7-induced malignancy in HNSCC cells. (A) 293T cells were transfected with Myc-tagged Galectin-7 alone or together with either Tid1-L-wt-HA or Tid1-L-mut-HA plasmids. Total protein lysates of the transfected cells were precipitated (IP) with either anti-HA (Tid1) or anti-Myc (Gal-7) antibody followed by immunoblotting (IB) with the indicated antibodies. (B) Immunofluorescence staining showing the subcellular localization of Gal-7 (Red) in 293T cells expressing Tid1-L-wt, or Tid1-L-mut, respectively. Co-localization of Tid1 and Gal-7 (yellow) was detected in the cytosol, as indicated by white arrows. (C) Subcellular localization of Myc (Gal-7), actin and Lamin B proteins in the nuclear and cytosolic fractions, prepared from 293T cells transfected with indicated plasmids, was determined. Actin was detectable mainly in the cytoplasmic extract, whereas Lamin B was mainly visible in the nuclear extract. (D) 293T cells were transfected with Tid1-L-wt-HA plasmids. Protein lysates of the transfected cells were treated without or with PNGase F, followed by immunoprecipitation with an anti-HA antibody, then, immunoblotted with antibody against HA or Myc, respectively. (E) 293T cells were co-transfected with Tid1-L-wt-HA and Gal-7-myc. Protein lysates of the transfected cells were incubated with lactose or sucrose, respectively, followed by immunoprecipitation with anti-HA, then, immunoblotted with antibody against HA and Myc. (F) Various plasmids of Tid1-L N-link glycosylation site mutants (Tid1N102A and Tid1N372A) were co-transfected with Gal-7-Myc into 293T cells. Protein lysates of the transfected cells were immunoprecipitated with anti-HA, and immunoblotted with antibody against HA and Myc antibodies, respectively. (G) Confocal fluorescence microscopy showed the intracellular localization of Tid1N102A or Tid1N372A plus Gal-7-Myc detected by double staining with antibodies against HA and Myc tag. Co-localization of Tid1 and Gal-7 is indicated with white arrows. (H) 293T cells were transfected with the HA-tagged WT Tid1 (Tid1-L-wt-HA) or Tid1-L mutants (Tid1N102A and Tid1N372A) plasmid and then pulled down by PNA agarose beads. The pulled-down proteins were separated by SDS-PAGE and analyzed by IB with anti-HA antibody. SAS cells were transfected with various combinations of the indicated plasmids. The transfected cells were examined for Transwell® migration ability (I), and anchorage-independent growth (J). The histograms shown are the mean ± SD from three independent experiments and analyzed by Student's t-test (∗p < 0.01; ∗∗p < 0.005).
Tid1 mediates ubiquitination of Galectin-7
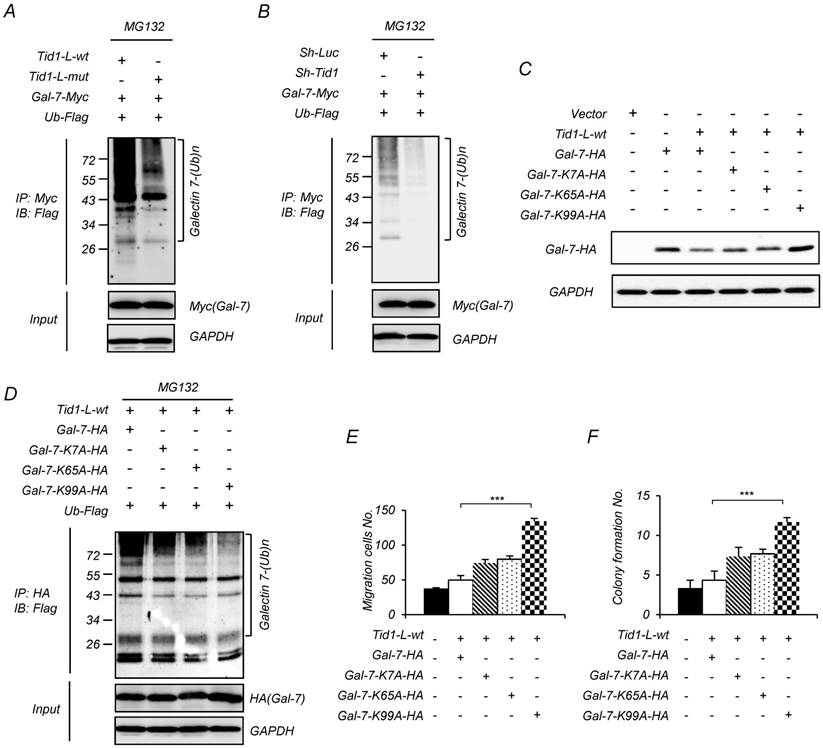
Tid1 could cause the instability of client proteins through ubiquitination followed by proteasome-mediated degradation [15, 19, 37]. Herein, we speculate that Tid1 would direct Galectin-7 degradation by the ubiquitin-proteasome pathway. The working hypothesis is that the Tid1-bound Galectin-7 would be delivered to Hsp70 through the consequential interaction between Tid1 (co-chaperone) and Hsp70 (chaperone), and would be subject to further 26S proteasome-mediated protein degradation. To test the hypothesis, we first co-immunoprecipitated the Tid1/Galectin-7 complex by using an antibody against Myc to bring down the C-terminal Myc-tagged Galectin-7. The antibody against Flag-tagged ubiquitin was used to detect the putative ubiquitinated Galectin-7 according to the molecular weight shift from 16 kDa to 26, 36 or 46 kDa dependent on the number of ubiquitin (1-3 ubiquitin addition; each Flag-ubiquitin is around 10 kDa). Compared with cells expressing Tid1-L-mut (without functional DnaJ domain), the transfected cell expressing Tid1-L-wt strongly displayed ubiquitinated Galectin-7 (Figure 4A). In addition, in cells with downregulation of Tid1 by shRNAi, we observed less ubiquitinated Galectin-7 (Figure 4B). To further support the idea that the anti-tumor function of Tid1 is associated with Galectin-7 ubiquitination, we generated three Galectin-7 mutants, of which the putative ubiquitination sites on lysine 7, lysine 65, or lysine 99 were separately substituted to alanine (G7K7A, G7K65A, and G7K99A), which were predicted by the UbPred software (www.ubpred.org). All constructs were validated by DNA sequencing (Figure S4). As expected, mutations of Galectin-7 at these putative ubiquitination sites (mutant G7K99A Galectin-7 in particular) reduced the downregulation of Galectin-7 in cells with overexpression of Tid1-L-wt (Figure 4C), likely because these mutants of Galectin-7 were less ubiquitinated (Figure 4D). These findings indeed support the hypothesis that Galectin-7 was regulated by Tid1-mediated ubiquitination. Similarly, cells expressing mutant Galectin-7 displayed increased migration and anchorage-independent growth abilities (Figure 4E-F). Hence, Tid1-mediated ubiquitination of Galectin-7 is likely the mechanism by which Tid1 down-regulates Galectin-7 and suppresses tumorigenicity.
Tid1 reduces the protein level of Galectin-7 by promoting ubiquitination, and the ubiquitination of Galectin-7 is required for inhibiting the malignancy of HNSCC cells. (A-B) 293T cells were transfected with various combinations of indicated plasmids. 24 h after transfection, the cells were treated with MG132 for 6 h. Protein lysates from the transfected cells were immunoprecipitated with anti-Myc (Gal-7), then, immunoblotted with antibody against Flag (Ubiquitin, Ub). (C) Various plasmids of Gal-7 ubiquitination site mutants (K7A-HA, K65A-HA, and K99A-HA) were transiently introduced into 293T cells. The expression of wild-type and mutant Gal-7 protein was determined by immunoblotting assays. (D) Various plasmids of Gal-7 mutants were co-transfected with Tid1-L-wt and Ub-Flag plasmids into 293T cells. Protein lysates from the transfected cells were immunoprecipitated with anti-HA (Gal-7) and immunoblotted with antibody against Flag (Ubiquitin). SAS cells were transfected with various combinations of indicated plasmids. Transwell® migration ability (E) and anchorage-independent growth (F) of the transfected cells were examined. The histograms shown are the mean ± SD from three independent experiments and analyzed by Student's t-test (∗p < 0.01; ∗∗p < 0.005).
Galectin-7 up-regulates MMP-9 expression through the increase of TCF3 transcriptional activity
It has been demonstrated that Galectin-7 promotes lymphoma metastasis by increasing the transcriptional level of MMP-9 [38]. Herein, we found that ectopic expression of Galectin-7 in SAS cells led to upregulation of the protein level and enzyme activity of MMP-9 (Figure 5A). However, the upregulation of MMP-9 was abolished upon co-ectopic expression with Tid1-wt but not Tid1-mut proteins (Figure 5A). In SASM-5 cells, which harboring abundant amount of endogenous Galectin-7 protein, the introduction of Tid1-wt diminished the protein level and enzyme activity of MMP-9 (Figure 5B). Additionally, Galectin-7 knockdown led to a reduction of the protein level and enzyme activity of MMP-9 in SASM-5 (Figure 5C).
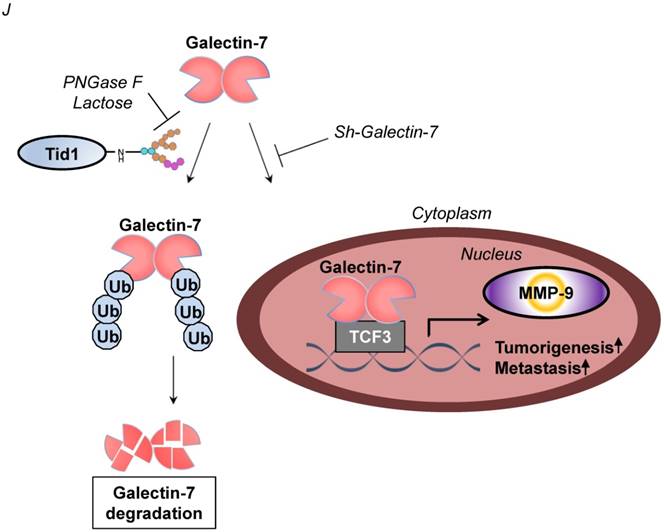
Tid1 suppresses Galectin-7-induced cis-activation of MMP-9 in HNSCC cells. (A) Gelatin zymogram assay (upper panel) and immunoblotting analysis (lower panel) were conducted on SAS cells transfected with control, Gal-7, Tid1-L-wt, or Tid1-L-mut plasmids as indicated. The conditioned media of transfected cells were collected and further used to measure the activity of MMP-9. Gelatin zymogram assay (upper panel) and immunoblotting analysis (lower panel) were conducted on SAS-M5 cells transfected with (B) control, Tid1-L-wt or Tid1-L-mut plasmids plus (C) sh-luc, sh-Gal-7-1 or sh-Gal-7-2 plasmids as indicated. The conditioned media of transfected cells were collected and further used to measure the activity of MMP-9. (D) Schematic depicting the transcriptional factor TCF-3 binding sites (E1: -66 to -71 bp; E2: -555 to -560 bp; E3: -1168 to -1173) within the MMP-9 promoter region. (E) ChIP assays were performed in 293T cells transfected with Gal-7-HA plasmid by using control anti-IgG or anti-HA antibodies. Precipitated genomic DNA fragments containing MMP-9 promoter region (Chip E1 primers: -169 to +106; Chip E2 primers: -727 to -501; Chip E3 primers: -1266 to -1054) were amplified by PCR primer sets. One percent of the prepared chromatin was examined by PCR to verify the presence of genomic DNA fragments as the loading control (Input). (F) MMP-9 promoter activity of 293T cells expressing Gal-7, Tid1-L-wt or Tid1-L-mut proteins was measured by luciferase reporter assays. (G) SAS cells were transiently transfected with Gal-7-HA plasmid. Protein lysates of the transfected cells were collected and immunoprecipitated (IP) with an antibody against HA (Gal-7), then, immunoblotted (IB) with either anti-HA or anti-TCF3 antibodies. (H) Immunofluorescence showing the staining of TCF3 (red) and Gal-7 (green) in SAS cells transfected with Gal-7 with Tid1-L-wt or Tid1-L-mut. Co-localization (yellow) of TCF3 and Gal-7 was detected in the nucleus. DAPI-stained nuclei are blue. (I) Pearson's correlation analyses were used to examine the mRNA expression profile of DNAJA3 (Tid1 gene name), MMP-9, TCF3, and LGALS7 (Gal-7 gene name) genes derived from the GEO datasets (GSE13601). The Pearson's correlation coefficients (r) and P values are displayed (n = 57). (J) Schematic depicting the negative regulation of Galectin-7 by Tid1 in HNSCC.
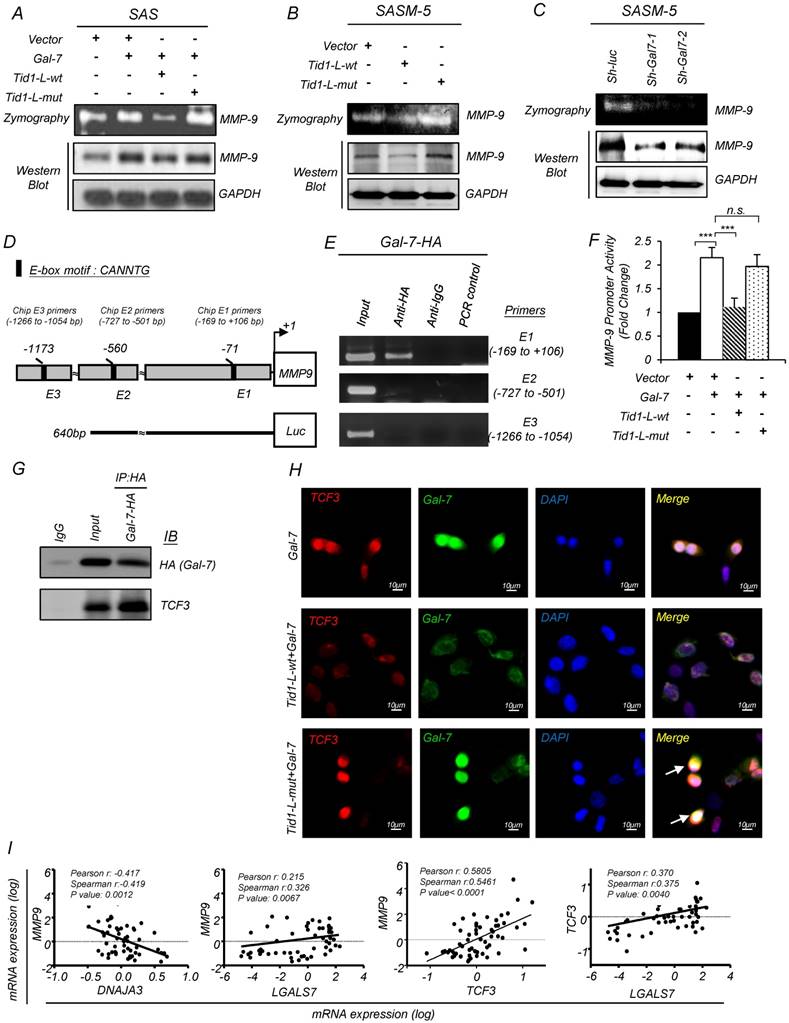
Galectin-7 could localize in the nucleus and contribute to transcriptional activation [39]. Next, we used the BioGRID database to find Galectin-7-interacting proteins, which might be involved in MMP-9 induction. The previous study showed that transcription factor 3 (TCF3) binds directly to the MMP-9 promoter [40]. Interestingly, we found that TCF3 interacts with Galectin-7 through a protein-protein interaction network (Figure S5A). Three E-box sequences (E1, E2, and E3) with a motif sequence CANNTG were identified in the MMP-9 promoter region by using the Integrated Genomics Viewer (IGV 2.3.61) (Figure 5D). To validate the association of Galectin-7 proteins with the promoter DNA region of MMP-9, we performed chromatin immunoprecipitation (ChIP) analysis targeting E-box sequences (E1, E2, and E3) with antibodies specifically against Galectin-7. We found that Galectin-7 was most strongly associated with the MMP-9 promoter region (-169 to +106 base pairs) (Figure 5E). Next, we performed reporter assays to understand whether the MMP-9 promoter can be activated by Galectin-7. This reporter construct contains the MMP-9 promoter (-640 to +133 base pairs) (Figure 5D). Ectopic expression of Galectin-7 up-regulated the MMP-9 promoter activity, which was abolished upon co-transfection with Tid1-wt plasmid but not Tid1-mut plasmid (Figure 5F). We also performed co-immunoprecipitation to validate the interaction between Galectin-7 and TCF3 (Figure 5G). Concordantly, immunofluorescence analysis showed that Galectin-7 co-localized with TCF3 in the nucleus; however, the above Galectin-7/TCF3 nuclear co-localization was abolished upon further ectopic expression of Tid1-wt but not Tid1-mut (Figure 5I and Figure S5B). We then compared the co-expression of MMP-9 with Tid1 (Gene Symbol: DNAJA3), Galectin-7 (Gene Symbol: LGALS7), and TCF3 in clinical patients by analyzing the GEO datasets (GSE13601). A negative correlation was observed between the expression of MMP-9 and Tid1 (Figure 5I). In contrast, a positive correlation was observed between the expression of MMP-9, Galectin-7, and TCF3 (Figure 5I). These findings suggest that Tid1 represses the oncogenic functions of Galectin-7 where Galectin-7 co-operates with TCF3 to upregulate the expression of MMP-9 by directly binding to the MMP-9 promoter (Figure 5J).
Discussion
Metastasis is known as a critical clinical parameter associated with poor overall survival among cancers including HNSCC [41, 42]. Poor prognoses of HNSCC patients have been associated with downregulation of Tid1 (Figure 1C and Table 1), yet the detailed molecular basis is not well understood [15]. In this study, we characterized the anti-metastatic role of Tid1 in HNSCC. We first identified Galectin-7 as one of the Tid1-L-interacting proteins. The interaction between Tid1-L and Galectin-7 not only abolished the nuclear translocation of Galectin-7 but also suppressed Galectin-7-induced tumorigenesis and metastasis. Notably, this interaction, which is mediated through Tid1 N-linked glycosylation, promotes the ubiquitination and proteasomal degradation of Galectin-7. In addition, we found that Galectin-7 plays a critical role in promoting tumorigenesis and metastasis by enhancing the transcriptional activity of TCF3 transcription factor to elevate MMP-9 expression. Consistent with our in vitro finding, we showed with HNSCC patient tumor sections that the level of Tid1 is inversely associated with that of Galectin-7. To our knowledge, this is the first study delineating the molecular mechanisms by which Tid1 negatively regulates Galectin-7 to suppress tumorigenesis of HNSCC.
Tid1, a DnaJ co-chaperone with a highly conserved J domain, is capable of binding to Hsp70, thus providing substrate specificity for this chaperone protein [13]. The formation of partner/DnaJ/Hsp70 protein complex facilitates protein folding [43], protein degradation, and assembly/disassembly of a multiprotein complex [44]. Recent studies suggest that the co-chaperones and regulatory function of Tid1 in suppressing tumor malignancy are possibly mediated by ubiquitination and consequent degradation of oncoproteins via HSP70-associated pathway [20, 22, 37]. Tid1 protein has two alternatively spliced forms in humans, Tid1-L and Tid1-S, which have different lengths and amino acid sequences in the C- termini. Additionally, Tid1-L has been shown to exhibit a superior cytosolic stability with enhanced ability to induce apoptosis, whereas Tid1-S has an increased rate of mitochondrial import and is able to suppress apoptosis [45, 46]. We have previously reported that Tid1-L can act as a tumor suppressor by enhancing EGFR ubiquitination and consequent degradation through interaction with HSP70 and HSP90 proteins in lung cancer [20]. Ahn et al. reported that Tid1-S can enhance mitochondrial translocation of mutant p53 to restore the apoptotic activity of mutant p53 in human breast cancer and glioma cells [33]. Additional support for the idea that these two proteins have distinct functions is our identification of the differential binding partners of Tid1-L or Tid1-S by affinity chromatography and proteomic analyses (Figure S1). However, the function of these client proteins as well as how these proteins are regulated by Tid1 mostly remain to be determined.
Galectin-7, a member of β-galactoside-binding lectins, binds to the glycans of glycoproteins with diverse biological functions [36]. Galectin-7 is highly expressed in aggressive cancers and has been associated with increased metastasis tendency and poor survival (Figure 1 and Table 1) [9, 38, 47]. Importantly, the augmented level of Galectin-7 is correlated with reduced expression of Tid1, particularly in tissue from cancer patients with lymph node metastasis, recurrence, and poor survival (Figure 1 and Table 1) [15, 20, 22]. Consistently, we also demonstrated both in vitro and in vivo that Tid1-L negatively regulates Galectin-7-induced malignant phenotypes (Figure 2 and Figure 3). Furthermore, we demonstrated that Tid1-L abrogates Galectin-7-induced tumorigenesis and metastatic progression by interacting with Galectin-7 through its N-linked glycosylation, thereby promoting degradation of ubiquitinated Galectin-7 (Figure 3 and Figure 4). However, several reports also suggest that Galectin-7 may have a distinct role in different types of cancer [6]. Previous studies have demonstrated that Galectin-7 negatively regulates tumor growth and exhibits pro-apoptotic function in gastric cancer cells and colon carcinoma cells [48, 49]. Nonetheless, Moisan et al. found that increased expression of Galectin-7 in cancer cells is associated with poor progression and an aggressive phenotype [50]. Moreover, overexpression of Galectin-7 enhances metastasis of cancer cells and resists apoptosis [9, 38, 47]. Therefore, cancer type-specific research investigating the role of Galectin-7 in cancer progression is necessary.
It has been known that Hsp70 regulates substrate protein stability through co-chaperone interactions, consequently promoting ubiquitination-mediated protein degradation [19]. In our previous study, we demonstrated that Tid1 enhances EGFR ubiquitination through Hsp70 complex and downregulates EGFR signaling in HNSCC [15]. In this study, we find that Galectin-7 ubiquitination could be enhanced by Tid1-L-wt rather than Tid1-L-mut (Figure 4A). The Galectin-7 ubiquitination site mutant is more stable than the wild-type Galectin-7 (Figure 4A-D), and thus displays a potent activity for inhibiting cancer malignancy (Figure 4E-F). In a previous study, Galectin-7 ubiquitination was identified by a large-scale analysis of the human ubiquitin-related proteome [51]. Together, these data suggest Tid1 with E3 ubiquitin ligase activity could bridge Hsp70-mediated Galectin-7 ubiquitination for degradation of Galectin-7.
Galectin-7 has been shown to be secreted extracellularly as a lectin but has been proposed to possess intracellular functions [49, 52]. Nuclear staining of Galectin-7 has been observed in different cancer cell lines and tissues [9, 11]. Demers and colleagues reported that ectopic expression of galectin-7 promotes the aggressiveness of cancer cells by increasing the transcript level of MMP-9 [38, 47]. They further found that knockdown of galectin-7 lead to reduced MMP-9 expression in lymphoma cells [47]. Accordingly, these findings suggest that Galectin-7 affects gene expression through transcriptional regulation. However, the target genes of Galectin-7 are thus far unknown, and the specific mechanism by which Galectin-7 is translocated to the nucleus remains unclarified. In the present study, we demonstrated that expression of Tid1-L-wt significantly decreased the nuclear accumulation of Galectin-7 compared with expression of Tid1-L-mut (Figure 3B-C). Furthermore, Galectin-7 increased the transcriptional activity of TCF3 transcription factor and subsequently elevated MMP-9 expression (Figure 5). Interestingly, it is evident that Galectin-3 increases the transcriptional activity of TCF4 [53]. Based on these findings, we proposed that differential galectin expression may differentially activate distinct transcription factors to modulate downstream target genes. Meanwhile, we also found that higher nuclear staining of Galectin-7 was associated with lower Tid1 expression and poor survival in the HNSCC patients (Figure 1C, E). These findings suggest that Tid1 may play a negative role in Galectin-7-associated cancer progression and metastasis by downregulating MMP activation through inhibition of TCF3 activity.
Previous studies have shown that Galectins bind to complex N-glycans to regulate cell biological functions [36]. Further, N-linked glycans can have a direct effect on the protein folding process and protein degradation [53]. Nevertheless, N- and O-glycans modulate Galectin-1 to reduce the binding of galectin-1 to other glycoproteins [54]. Herein, we found that the interaction between Tid1 and Galectin-7 was bridged by N-linked glycosylated Tid1 (Figure 3). Moreover, we found that Galectin-7 and Tid1-L-wt were mostly co-localized, whereas, N-linked glycosylation mutant of Tid1 (N103A) reduced the interaction between Galectin-7 and Lectin (Figure 3F, H). Additionally, N-linked glycosylation mutant of Tid1 (N103A) enhanced the metastatic ability of SAS cells compared to Tid1-L-wt group (Figure 3I). We, therefore, speculate that N-linked glycosylation of Tid1 is required to interact with Galectin-7 to downregulate Galectin-7; consequently, the downregulation of Galectin-7 by Tid1 can attenuate cancer progression and metastasis.
Collectively, our data demonstrate the crucial role of glycosylated Tid1 in its interaction with Galectin-7 to attenuate tumorigenicity and metastasis of HNSCC (Figure 5J). Additionally, the inhibition of metastasis mediated by Tid1 to promote Galectin-7 degradation and reduce nuclear accumulation of Galectin-7 may imply that Galectin-7 could be a possible therapeutic target for future HNSCC treatment.
Abbreviations
4NQO: 4-nitroquinoline-1-oxide; ChIP: chromatin immunoprecipitation; CRDs: carbohydrate-recognition domains; HNSCC: head and neck squamous cell carcinoma; PNGase F: N-Glycosidase F; TCF3: transcription factor 3; Tid1-L: Tid1 long form; Tid1-S: Tid1 short form; Tid1flox/flox: loxP-flanked Tid1 gene; Tid1-L-wt: Tid1 long form wild-type; Tid1-L-mut : Tid1 long form mutant; Tid1-S-wt: Tid1 short form wild-type; Tid1-S-mut: Tid1 short form mutant.
Acknowledgements
We thank the Instrumentation Resource Center and Proteomics Research Center at National Yang-Ming University for technical support. We also thank the technical services provided by the Transgenic Mouse Model Core Facility of the National Core Facility Program for Biotechnology, National Science Council and the Gene Knockout Mouse Core Laboratory of National Taiwan University Center of Genomic Medicine. This study was supported by research grants from the Ministry of Science and Technology of Taiwan (NSC102-2314-B-010-046, MOST103-2320-B-010-028, MOST104-2314-B-010-047-MY3 and MOST105-2320-B-010-025-MY3) and National Yang-Ming University (Ministry of Education, “Aim for the Top University Plan: 104AC-T204, 105AC-T203, and 106AC-T302”; “Higher Education Sprout Project:107CRC-T402”).
Supplementary Material
Supplementary materials and methods and figures.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA: a cancer journal for clinicians. 2015;65:5-29
2. Doci CL, Mikelis CM, Lionakis MS, Molinolo AA, Gutkind JS. Genetic identification of SEMA3F as an antilymphangiogenic metastasis suppressor gene in head and neck squamous carcinoma. Cancer research. 2015;75:2937-48
3. Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nat Rev Cancer. 2005;5:761-72
4. Sterrenberg JN, Blatch GL, Edkins AL. Human DNAJ in cancer and stem cells. Cancer Lett. 2011;312:129-42
5. Boudesco C, Cause S, Jego G, Garrido C. Hsp70: A cancer target inside and outside the cell. Methods Mol Biol. 2018;1709:371-96
6. Kaur M, Kaur T, Kamboj SS, Singh J. Roles of galectin-7 in cancer. Asian Pac J Cancer Prev. 2016;17:455-61
7. Wang J, Liu Y, Yang Y, Xu Z, Zhang G, Liu Z. et al. High expression of galectin-7 associates with poor overall survival in patients with non-metastatic clear-cell renal cell carcinoma. Oncotarget. 2016;7:41986-95
8. Tsai CJ, Sulman EP, Eifel PJ, Jhingran A, Allen PK, Deavers MT. et al. Galectin-7 levels predict radiation response in squamous cell carcinoma of the cervix. Gynecologic oncology. 2013;131:645-9
9. Demers M, Rose AA, Grosset AA, Biron-Pain K, Gaboury L, Siegel PM. et al. Overexpression of galectin-7, a myoepithelial cell marker, enhances spontaneous metastasis of breast cancer cells. Am J Pathol. 2010;176:3023-31
10. Kim HJ, Jeon HK, Lee JK, Sung CO, Do IG, Choi CH. et al. Clinical significance of galectin-7 in epithelial ovarian cancer. Anticancer research. 2013;33:1555-61
11. Zhu X, Ding M, Yu ML, Feng MX, Tan LJ, Zhao FK. Identification of galectin-7 as a potential biomarker for esophageal squamous cell carcinoma by proteomic analysis. BMC cancer. 2010;10:290
12. Lu B, Garrido N, Spelbrink JN, Suzuki CK. Tid1 isoforms are mitochondrial DnaJ-like chaperones with unique carboxyl termini that determine cytosolic fate. J Biol Chem. 2006;281:13150-8
13. Silver PA, Way JC. Eukaryotic DnaJ homologs and the specificity of Hsp70 activity. Cell. 1993;74:5-6
14. Kim SW, Hayashi M, Lo JF, Fearns C, Xiang R, Lazennec G. et al. Tid1 negatively regulates the migratory potential of cancer cells by inhibiting the production of interleukin-8. Cancer Res. 2005;65:8784-91
15. Chen CY, Chiou SH, Huang CY, Jan CI, Lin SC, Hu WY. et al. Tid1 functions as a tumour suppressor in head and neck squamous cell carcinoma. J Pathol. 2009;219:347-55
16. Lo JF, Hayashi M, Woo-Kim S, Tian B, Huang JF, Fearns C. et al. Tid1, a cochaperone of the heat shock 70 protein and the mammalian counterpart of the Drosophila tumor suppressor l(2)tid, is critical for early embryonic development and cell survival. Molecular and cellular biology. 2004;24:2226-36
17. Lo JF, Zhou H, Fearns C, Reisfeld RA, Yang Y, Lee JD. Tid1 is required for T cell transition from double-negative 3 to double-positive stages. J Immunol. 2005;174:6105-12
18. Cheng LH, Hung KF, Lee TC, Huang CY, Chiu WT, Lo JF. et al. Mitochondrial co-chaperone protein Tid1 is required for energy homeostasis during skeletal myogenesis. Stem Cell Res Ther. 2016;7:185
19. Kim SW, Chao TH, Xiang R, Lo JF, Campbell MJ, Fearns C. et al. Tid1, the human homologue of a Drosophila tumor suppressor, reduces the malignant activity of ErbB-2 in carcinoma cells. Cancer Res. 2004;64:7732-9
20. Chen CY, Jan CI, Lo JF, Yang SC, Chang YL, Pan SH. et al. Tid1-L inhibits EGFR signaling in lung adenocarcinoma by enhancing EGFR ubiquitinylation and degradation. Cancer Res. 2013;73:4009-19
21. Schilling B, De-Medina T, Syken J, Vidal M, Munger K. A novel human DnaJ protein, hTid-1, a homolog of the Drosophila tumor suppressor protein Tid56, can interact with the human papillomavirus type 16 E7 oncoprotein. Virology. 1998;247:74-85
22. Jan CI, Yu CC, Hung MC, Harn HJ, Nieh S, Lee HS. et al. Tid1, CHIP and ErbB2 interactions and their prognostic implications for breast cancer patients. J Pathol. 2011;225:424-37
23. Stowell SR, Ju T, Cummings RD. Protein glycosylation in cancer. Annu Rev Pathol. 2015;10:473-510
24. Pinho SS, Reis CA. Glycosylation in cancer: mechanisms and clinical implications. Nature reviews Cancer. 2015;15:540-55
25. Yu H, Yang C, Chen S, Huang Y, Liu C, Liu J. et al. Comparison of the glycopattern alterations of mitochondrial proteins in cerebral cortex between rat Alzheimer's disease and the cerebral ischemia model. Sci Rep. 2017;7:39948
26. Ohtsubo K, Takamatsu S, Minowa MT, Yoshida A, Takeuchi M, Marth JD. Dietary and genetic control of glucose transporter 2 glycosylation promotes insulin secretion in suppressing diabetes. Cell. 2005;123:1307-21
27. Liang CC, You LR, Chang JL, Tsai TF, Chen CM. Transgenic mice exhibiting inducible and spontaneous Cre activities driven by a bovine keratin 5 promoter that can be used for the conditional analysis of basal epithelial cells in multiple organs. J. Biomed Sci. 2009;16:2
28. Chen YF, Yang CC, Kao SY, Liu CJ, Lin SC, Chang KW. MicroRNA-211 enhances the oncogenicity of carcinogen-induced oral carcinoma by repressing TCF12 and increasing antioxidant activity. Cancer Res. 2016;76:4872-86
29. Okumura K, Konishi A, Tanaka M, Kanazawa M, Kogawa K, Niitsu Y. Establishment of high- and low-invasion clones derived for a human tongue squamous-cell carcinoma cell line SAS. J. Cancer Res Clin Oncol. 1996;122:243-8
30. Zhu H, Wu TC, Chen WQ, Zhou LJ, Wu Y, Zeng L. et al. Roles of galectin-7 and S100A9 in cervical squamous carcinoma: Clinicopathological and in vitro evidence. Int J Cancer. 2013;132:1051-9
31. Wang TH, Lin YH, Yang SC, Chang PC, Wang TC, Chen CY. Tid1-S regulates the mitochondrial localization of EGFR in non-small cell lung carcinoma. Oncogenesis. 2017;6:e361
32. Chen CY, Jan CI, Pi WC, Wang WL, Yang PC, Wang TH. et al. Heterogeneous nuclear ribonucleoproteins A1 and A2 modulate expression of Tid1 isoforms and EGFR signaling in non-small cell lung cancer. Oncotarget. 2016;7:16760-72
33. Ahn BY, Trinh DL, Zajchowski LD, Lee B, Elwi AN, Kim SW. Tid1 is a new regulator of p53 mitochondrial translocation and apoptosis in cancer. Oncogene. 2010;29:1155-66
34. Chen YS, Huang WL, Chang SH, Chang KW, Kao SY, Lo JF. et al. Enhanced filopodium formation and stem-like phenotypes in a novel metastatic head and neck cancer cell model. Oncol Rep. 2013;30:2829-37
35. Chen CY, Chiou SH, Huang CY, Jan CI, Lin SC, Tsai ML. et al. Distinct population of highly malignant cells in a head and neck squamous cell carcinoma cell line established by xenograft model. J Biomed Sci. 2009;16:100
36. Liu FT, Rabinovich GA. Galectins as modulators of tumour progression. Nature reviews Cancer. 2005;5:29-41
37. Bae MK, Jeong JW, Kim SH, Kim SY, Kang HJ, Kim DM. et al. Tid-1 interacts with the von Hippel-Lindau protein and modulates angiogenesis by destabilization of HIF-1alpha. Cancer Res. 2005;65:2520-5
38. Demers M, Magnaldo T, St-Pierre Y. A novel function for galectin-7: promoting tumorigenesis by up-regulating MMP-9 gene expression. Cancer Res. 2005;65:5205-10
39. Inagaki Y, Higashi K, Kushida M, Hong YY, Nakao S, Higashiyama R. et al. Hepatocyte growth factor suppresses profibrogenic signal transduction via nuclear export of Smad3 with galectin-7. Gastroenterology. 2008;134:1180-90
40. Zhang L, Xiong W, Xiong Y, Liu H, Li N, Du Y. et al. Intracellular wnt/beta-catenin signaling underlying 17beta-estradiol-induced matrix metalloproteinase 9 expression in human endometriosis. Biol Reprod. 2016;94:70
41. Goldson TM, Han Y, Knight KB, Weiss HL, Resto VA. Clinicopathological predictors of lymphatic metastasis in HNSCC: implications for molecular mechanisms of metastatic disease. J Exp Ther Oncol. 2010;8:211-21
42. Garcia J, Lopez M, Lopez L, Bague S, Granell E, Quer M. et al. Validation of the pathological classification of lymph node metastasis for head and neck tumors according to the 8th edition of the TNM Classification of Malignant Tumors. Oral Oncol. 2017;70:29-33
43. Suzuki H, Ikeda A, Tsuchimoto S, Adachi K, Noguchi A, Fukumori Y. et al. Synergistic binding of DnaJ and DnaK chaperones to heat shock transcription factor sigma32 ensures its characteristic high metabolic instability: implications for heat shock protein 70 (Hsp70)-Hsp40 mode of function. J Biol Chem. 2012;287:19275-83
44. Cyr DM, Langer T, Douglas MG. DnaJ-like proteins: molecular chaperones and specific regulators of Hsp70. Trends Biochem Sci. 1994;19:176-81
45. Ng AC, Baird SD, Screaton RA. Essential role of TID1 in maintaining mitochondrial membrane potential homogeneity and mitochondrial DNA integrity. Mol Cell Biol. 2014;34:1427-37
46. Syken J, De-Medina T, Munger K. TID1, a human homolog of the Drosophila tumor suppressor l(2)tid, encodes two mitochondrial modulators of apoptosis with opposing functions. Proc Natl Acad Sci U S A. 1999;96:8499-504
47. Demers M, Biron-Pain K, Hebert J, Lamarre A, Magnaldo T, St-Pierre Y. Galectin-7 in lymphoma: elevated expression in human lymphoid malignancies and decreased lymphoma dissemination by antisense strategies in experimental model. Cancer research. 2007;67:2824-9
48. Kopitz J, Andre S, von Reitzenstein C, Versluis K, Kaltner H, Pieters RJ. et al. Homodimeric galectin-7 (p53-induced gene 1) is a negative growth regulator for human neuroblastoma cells. Oncogene. 2003;22:6277-88
49. Kuwabara I, Kuwabara Y, Yang RY, Schuler M, Green DR, Zuraw BL. et al. Galectin-7 (PIG1) exhibits pro-apoptotic function through JNK activation and mitochondrial cytochrome c release. J Biol Chem. 2002;277:3487-97
50. Moisan S, Demers M, Mercier J, Magnaldo T, Potworowski EF, St-Pierre Y. Upregulation of galectin-7 in murine lymphoma cells is associated with progression toward an aggressive phenotype. Leukemia. 2003;17:751-9
51. Matsumoto M, Hatakeyama S, Oyamada K, Oda Y, Nishimura T, Nakayama KI. Large-scale analysis of the human ubiquitin-related proteome. Proteomics. 2005;5:4145-51
52. Villeneuve C, Baricault L, Canelle L, Barboule N, Racca C, Monsarrat B. et al. Mitochondrial proteomic approach reveals galectin-7 as a novel BCL-2 binding protein in human cells. Mol Biol Cell. 2011;22:999-1013
53. Vladoiu MC, Labrie M, St-Pierre Y. Intracellular galectins in cancer cells: potential new targets for therapy (Review). Int J Oncol. 2014;44:1001-14
54. Earl LA, Bi S, Baum LG. N- and O-glycans modulate galectin-1 binding, CD45 signaling, and T cell death. J Biol Chem. 2010;285:2232-44
Author contact
![]() Corresponding author: Jeng-Fan Lo, Institute of Oral Biology, National Yang-Ming University, No. 155, Section 2, Li-Nong Street, Taipei, 11221, Taiwan, ROC. Telephone: 886-2-28267222; Fax: 886-2-28264053; E-mail: jfloedu.tw.
Corresponding author: Jeng-Fan Lo, Institute of Oral Biology, National Yang-Ming University, No. 155, Section 2, Li-Nong Street, Taipei, 11221, Taiwan, ROC. Telephone: 886-2-28267222; Fax: 886-2-28264053; E-mail: jfloedu.tw.