Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results
Discussion
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(14):3808-3823. doi:10.7150/thno.25255 This issue Cite
Research Paper
Self-assembled dual fluorescence nanoparticles for CD44-targeted delivery of anti-miR-27a in liver cancer theranostics
Xiao Zheng1, Fan Zhang1, Yawei Zhao1, Jing Zhang1, Jianati Dawulieti1, Yue Pan1, Lianzhi Cui4, Madi Sun1, Dan Shao1,3 ![]() , Mingqiang Li3, Kan He1, Ming Zhang1, Jing Li1
, Mingqiang Li3, Kan He1, Ming Zhang1, Jing Li1 ![]() , Li Chen1,2
, Li Chen1,2 ![]()
1. Department of Pharmacology, Nanomedicine Engineering Laboratory of Jilin Province, College of Basic Medical Sciences, Jilin University, Changchun 130021, China
2. School of Nursing, Jilin University, Changchun 130021, China
3. Department of Biomedical Engineering, Columbia University, New York, NY 10027, USA
4. Clinical Laboratory, Jilin Cancer Hospital, Changchun 130012, China
Received 2018-1-31; Accepted 2018-5-8; Published 2018-6-13
Abstract

Despite the vital role miRNA-27a plays in driving the development and progress of liver cancer, miRNA-based inhibition therapy is hampered due to its undesired degradation and off-target effects. Herein, a multifunctional nanoparticle for noninvasive tracking of targeted delivery of anti-miR-27a oligonucleotides against liver cancer was constructed.
Methods: Dual-fluorescent conjugates (QD-HA-PEI) were first fabricated through crosslinking hyaluronic acid (HA), polyethyleneimine (PEI) and near-infrared (NIR) fluorescent quantum dots (QDs) via a facile one-pot approach. Antisense oligonucleotide was then encapsulated by QD-HA-PEI to form anti-miR-27a/QD-HA-PEI via electrostatic interactions. Targeting, biodistribution, bioimaging, in vitro cytotoxicity and in vivo anti-tumor effects were evaluated and the underlying mechanism was studied.
Results: The NIR fluorescence of anti-miR-27a/QD-HA-PEI could be employed to monitor CD44 receptor-targeted cellular uptake and tumor accumulation. Importantly, the intrinsic fluorescence of anti-miR-27a/QD-HA-PEI remained in the “ON” state in extracellular or blood environment, but switched to the “OFF” state in the intracellular environment, indicating pH-responsive oligonucleotide release. Furthermore, anti-miR-27a/QD-HA-PEI exhibited effective and selective anti-cancer effects in vitro and in vivo with fewer side effects via the direct down-regulation of oncogenic transcription factors FOXO1 and PPAR-γ.
Conclusion: Our findings validate the dual-fluorescent nanoparticles as delivery vectors of therapeutic miRNA, capable of simultaneous tumor imaging and tracking of miRNA-based modulation therapy, thereby providing an efficient and safe approach for liver cancer theranostics.
Keywords: Dual fluorescence, HA-PEI, anti-miR-27a, theranostics, liver cancer
Introduction
Despite the significant progress that has been made towards therapeutic approaches, including liver transplantation, surgical resection, thermal ablation and chemotherapy, unsatisfactory response continuously poses a tremendous challenge to effective liver cancer therapy. [1-3] Among all the efforts that have been made, RNA interference (RNAi) technology has emerged as a nonnegligible tool in liver cancer treatment due to its specific silencing effect of oncological targets. [4-6] microRNA (miRNA) is a class of small, endogenous noncoding RNAs (containing 18-25 nucleotides) that functions as negative gene regulators at the post-transcriptional level. [7-11] The discovery of miRNAs and their multiple roles in the development and progression of liver cancer has provided a comprehensive picture of cellular regulation with the potential for generating new RNAi-based therapeutic approaches. [12-20] Recent evidence has shown that the dysregulation of many miRNAs including let-7a, miR-16, miR-25, miR-27a, miR-34a, miR-107, etc., could play a crucial role in accelerating the deterioration of liver cancer. [15-18] Among the known miRNAs, miR-27a was found to be highly enriched in both serum and tumor tissue of liver cancer patients. It could promote liver tumorigenesis by directly down-regulating transcription factors including FOXO-1 and PPAR-γ, and subsequently inhibiting the expressions of their downstream targets such as p21, p27, Bax and caspase-3, ultimately promoting proliferation and reducing apoptosis of liver cancer cells. [19, 20]
Although remarkable advances have been made in miRNA-based inhibition therapies, critical barriers remain to be overcome before their transition to clinical application. [21, 22] On the one hand, synthetic naked miRNAs are unstable in nuclease-rich serum and plasma environments, resulting in low therapeutic efficacy. [23, 24] On the other hand, excessive accumulation of exogenous miRNA therapeutics in the non-malignant tissues might interfere with normal cell activities, causing impaired specificity and off-target side effects. [25, 26] Therefore, an ideal miRNA delivery system must be developed to achieve satisfying performance. The system should meet the following characteristics: (1) reduce miRNA degradation and prolong in vivo circulation, (2) target liver cancer cells in a highly selective manner and facilitate intracellular uptake, (3) allow for tailored release profiles in response to micro-environmental stimulus and promote endosome release/escape of the payload miRNAs, (4) noninvasively monitor drug delivery and release in a real-time manner and simultaneously image the tumor site.
To date, numerous delivery systems including cationic polymers, polysaccharides, and liposomes have shown great promise in the protection of miRNA from degradation and have realized targeted cancer gene therapy. [27-35] Among all these polyionic nanosystems, hyaluronic acid-polyethyleneimine conjugate (HA-PEI) has been widely studied for nuclear acid delivery. [32-35] In this system, HA acts as a targeting component due to its high affinity for CD44 receptor, which is a cell adhesion membrane glycoprotein overexpressed on the surface of many types of liver cancer cells, while PEI has a high capacity for nuclear acid complexation and facilitates endosomal escape through the proton sponge effect. [34] Besides integrating each advantage, HA-PEI exhibits biodegradable, biocompatible, and non-immunogenic properties for the systemic administration of miRNA. [35] Moreover, the active residues of HA-PEI allow its easy modification with fluorescent probes that permit real-time tracking of the miRNA delivery process in vivo and monitoring of miRNA release in vitro. Hence, the combination of imaging nanoprobes with miRNA in HA-PEI cationic polymer is an ideal nanosystem to achieve efficient and safe liver cancer theranostics.
In the present study, a bifluorescent conjugate was firstly fabricated using hyaluronic acid, polyethyleneimine and quantum dots (QDs) through a facile one-pot approach, then negatively charged anti-miR-27a were encapsulated to form anti-miR-27a/QD-HA-PEI nanoparticle for near-infrared (NIR) fluorescence imaging and miRNA-based modulation therapy against liver cancer (Scheme 1). The physicochemical properties of anti-miR-27a/QD-HA-PEI including hydrodynamic size, zeta potential, optical properties, and stability were characterized. Importantly, the targeted cellular uptake of nanoparticles could be monitored by the QDs' NIR fluorescence, while the “ON-OFF” intrinsic fluorescence of the HA-PEI conjugate might be employed to track the release behavior of anti-miR-27a. The anti-cancer efficacy, miR-27a modulation effects, and molecular mechanisms of anti-miR-27a/QD-HA-PEI were investigated through both in vitro and in vivo models. The bioimaging, biodistribution and biocompatibility of anti-miR-27a/QD-HA-PEI after systemic administration were additionally studied. We suggest that bifluorescent anti-miR-27a/QD-HA-PEI has great potential for applications in liver cancer theranostics.
Schematic illustration of the fabrication of anti-miR-27a/QD-HA-PEI for simultaneous bioimaging and tracking of miRNA-based modulation therapy of liver cancer.
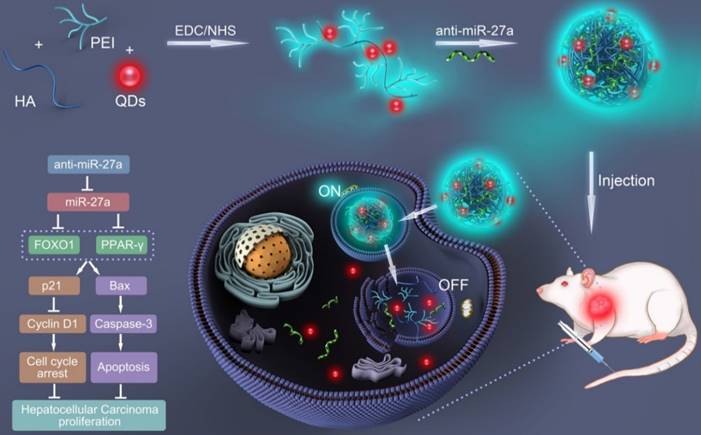
Methods
Materials
Sodium hyaluronate (HA, 10 kDa) was purchased from Bloomage Freda Biopharma Co., LTD. Polyethyleneimine (PEI, linear, 25 kDa) was obtained from Polysciences Inc. Qdot™ 800 ITK™ Amino (PEG) Quantum Dots were purchased from Thermo-Fisher Scientific Corp. Inhibitor of miR-27a (anti-miR-27a) and cy3-labeled RNA were purchased from Gene Pharma Co., Shanghai. 1-Ethyl-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC), N-hydroxysuccinimide (NHS) and sulforhodamine B were obtained from Sigma-Aldrich. RPMI-1640 medium was obtained from GIBCO. Fetal bovine serum (FBS), penicillin, streptomycin, assay kits for cell cycle and cell apoptosis were from the Beyotime Institute of Biotechnology (Jiangsu, China). Antibodies for western blotting were purchased from Abcam. Assay kits for alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase (ALP), blood urea nitrogen (BUN), creatinine (CRE) and total triglycerides (TG) were purchased from Nanjing Jiancheng Bioengineering Institute. Formamide and ethyl alcohol were obtained from Beijing Chemical Works. All reagents were commercially available products with analytical grade purity and were used without further purification.
Synthesis of anti-miR-27a/QD-HA-PEI
QD- and PEI-conjugated HA based on a modified EDC/NHS reaction was carried out according to a previous report. [32,36-37] In brief, sodium hyaluronate (10KD, 10 μmol) was dissolved in 10 mL of dry formamide in a glass weighing bottle by warming up the reaction container to 50 ℃. After obtaining a clear solution, the reaction mixture was cooled down to room temperature, and then 10 mg of EDC and NHS were added to the solution. After activating for 30 min, 10 μL of QDs (0.32 μmol/mL) that were dissolved in formamide in advance was added to the reaction mixture. This step was repeated 10 times and the interval was 10 min; 100 μL of QDs was added to the mixture in total (0.032 μmol). Then 30 mg of PEI (25 kDa, 12 μmol) was added to the reaction mixture. Soon the solution turned hazy and became clear in 20 min. The reaction was stirred overnight using a magnetic stirrer. The resulting solution was added to a large excess of EtOH (250 mL) to precipitate the polymer. The precipitate was centrifuged and collected and the washings were discarded. The above step of EtOH precipitation and washing was repeated thrice to purify the polymer. Finally, the precipitate polymer (QD-HA-PEI) was re-dissolved in deionized water and lyophilized using a freeze dryer (Labconco FreeZone 2.5). Then QD-HA-PEI powder was dissolved into saline together with anti-miR-27a, vortexed for 1 min and incubated for 15 min at room temperature to form miRNA-loaded nanoparticles (anti-miR-27a/QD-HA-PEI).
Characterization
The morphologies of the NPs were characterized using a JEM-2100F transmission electron microscope (JEOL, Ltd., Japan) and a scanning electron microscope (SEM, FEI quanta 200F). Fluorescence spectroscopy was performed using a Shimadzu RF-5301 PC spectrophotometer. NMR spectra were obtained on an AVANCEIII500 (500 MHz) from Bruker. Fourier transform infrared (FTIR) spectra were collected with a Nicolet AVATAR 360 FTIR instrument. The size distribution and zeta potential of the NPs in cell culture media (DMEM containing 10% FBS) were characterized using a Nano-ZS 90 Nanosizer (Malvern Instruments Ltd., Worcestershire, UK).
Determination of RNA loading and protection
To determine the encapsulation capability of QD-HA-PEI, samples of different polymer-to-miRNA weight ratios, ranging from 1:1 to 9:1, were prepared and then run on Agarose gel (0.5%) according to a previous report [38]. To visualize miRNAs, a substitute for EB called Goodview nucleic acid stain (SBS Genetech Co., ltd) was used. The encapsulation ability was assessed after imaging the gel under UV light. In the protection assay, anti-miR-27a/QD-HA-PEI was incubated with RNase A at 37 °C for 30 min, and then EDTA solution was added to denature RNase. 10 μL of each sample were mixed and vortexed with 10 μL of 2% polyacrylic acid (PAA), since PAA would compete with the anionic polymer and release the miRNA, which then appears as a free band in the gel. Finally, 20 μL of each sample was run on agarose gel. Naked miRNA was used as a control in both experiments.
Cell culture and cellular imaging
The cell lines used in this study, including human hepatocellular carcinoma cell line (HepG2), human hepatic embryo cell line (HL-7702), and mouse fibroblasts NIH-3T3, were obtained from ATCC. Cells were maintained at 37 ℃ and 5% CO2 in RPMI-1640 supplemented with 10% (v/v) heat-inactivated fetal bovine serum, penicillin (100 U/mL), and streptomycin (100 mg/mL). To determine the cellular entry of the designed nanoparticle and its targeting feature, negative control (N.C.) miRNA encapsulated in QD-HA-PEI was incubated with cells for 3 h. In parallel, a competition group of each cell line was made. The cell culture medium in these groups was replaced with serum-free media containing excess free HA at a concentration of 10 mg/mL for 1 h to block CD44 receptors before incubation with the N.C. /QD-HA-PEI for 3 h under identical conditions. All sets of cells were washed with PBS, then stained with Hoechst 33258 and visualized under a confocal fluorescence microscope (CLSM, Olympus FV1000). To further quantify the cellular uptake efficiency, the same N.C. miRNA/QD-HA-PEI-treated cells were washed, trypsinized and resuspended for flow cytometry (FACS, Becton-Dickinson Biosciences, Drive Franklin Lakes, U.S.).
Determination of pH-dependent stability and RNA release
To further evaluate if the stability of QD-HA-PEI was related to pH, cy3-labelled miRNA-loaded QD-HA-PEI was first mixed with PBS of different pH (pH=7.4 and pH=5.5) for 24 h and solution from each group at the time points of 0, 1, 3, 6, 12, 18, and 24 h was collected. Photostability was determined by the fluorescence intensity of HA-PEI, while miRNA release behavior was assessed by RT-PCR. Then 2 sets of HepG2 cells were incubated with cy3-labelled miRNA-loaded QD-HA-PEI for 1 h and 6 h, respectively, then stained with LysoTracker blue and visualized under the CLSM. To further quantify the fluorescence intensity, the same cy3-labelled miRNA/QD-HA-PEI-treated cells were washed, trypsinized and resuspended for FACS.
Evaluation on in vitro anti-cancer effect
HepG2 cells were first incubated with QD-HA-PEI to determine whether this vector was safe or not. Cells were seeded in 96-well plates at a density of 5000 cells per well and then treated with QD-HA-PEI, of which the maximum concentration was 200 μg/mL, for 24 h. SRB assay was performed for cytotoxicity assessment. [39-41] Then both cell lines were treated with free anti-miR-27a and anti-miR-27a-loaded QD-HA-PEI. The maximum concentration of RNA used was 200 nM for 24 h. Since the polymer-to-miRNA weight ratio was set to 9:1, the concentrations of QD-HA-PEI were 11.8 μg/mL and 14.4 μg/mL, respectively.
Molecular mechanisms of anti-cancer effect
HepG2 cells were first seeded in 6-well plates at a density of 50000 cells per well and then treated with saline, QD-HA-PEI (3.6 μg/mL), anti-miR-27a (50 nM) and anti-miR-27a/QD-HA-PEI for 24 h. Effects on cell cycle and apoptosis were determined by FACS, and the procedures were conducted according to the instruction of each assay kit. Briefly, to determine cell cycle, cells were harvested and adjusted to 106 cells per sample. Then cells were fixed with 70% cold ethanol for 1 h at room temperature. After that, cells were washed with PBS twice and resuspended with 0.5 mL PI/RNase Staining Solution. Next the suspension was gently vortexed and incubated for 30 min at room temperature, protected from light, and finally analyzed by FACS. To determine apoptosis, cells were harvested, washed with cold PBS and resuspended in 1x Binding Buffer. Then the suspensions were centrifuged for 10 min at 300 ×g to remove the Binding Buffer from the cell pellet. After that, cells were resuspended in 1x Binding Buffer and adjusted to 106 cells/mL. 100 μL of cells (105 cells) were added to each labeled tube. Next 5 μL of Annexin V-FITC was added to appropriate tubes. Tubes were gently vortexed and incubated for 10 min at room temperature, protected from light. Then 5 μL of PI was added and incubated for 5 min at room temperature, protected from light. Finally, samples were analyzed by FACS within 1 h. Then total RNAs of each group were extracted, and the miR-27a expression was determined by RT-PCR. Proteins of the above-mentioned groups were also extracted, and the expressions of FOXO-1, PPAR-γ, p21, Bax, cyclin D1 and caspase-3 were assessed by western blot. The primers and antibodies were selected according to our previous reports. [19, 20]
Animals and in vivo bioimaging and biodistribution
All animal experimental protocols were approved by the Ethics Committee for the Use of Experimental Animals of Jilin University. The tumor models for this study were developed in nude mice obtained from Weitonglihua Co., Beijing. 6-week-old male nude mice were injected subcutaneously (s.c.) with 2×106 HepG2 cells under the right shoulder. Tumor size was measured twice a week to monitor tumor growth. HepG2 tumor-bearing mice were injected intravenously (i.v.) with anti-miR-27a/QD-HA-PEI containing 20 nmol/kg of miRNA and visualized using an NIR Imaging System at 3 h and 24 h after injection. To evaluate its targeting feature, HepG2 tumor-bearing mice were pre-treated with free HA (10 mg/kg) 3 h before administration of anti-miR-27a/QD-HA-PEI. After 24 h of injection, all mice (n=6) were sacrificed and heart, lung, liver, spleen, left kidney and tumor were collected and imaged. NIR fluorescence intensity at the ROI is expressed as the mean ± SD for three mice in each group.
In vivo anti-cancer efficacy and systemic safety evaluation
HepG2 tumor-bearing nude mice were randomly divided into 3 groups (n=6) and were intravenously (i.v.) injected with saline, free anti-miR-27a (1 mg/kg) and anti-miR-27a (1 mg/kg)-loaded QD-HA-PEI when the tumor volume reached 50-100 mm3, respectively. Mice weight was measured every third day while tumor sizes were measured every fifth day. The estimated tumor volume (mm3) was calculated based on the formula length × width2 × 0.52. All mice were sacrificed at the 40th day. Tumors were measured, weighed, and then partly embedded in paraffin before being subjected to TUNEL and Ki67 staining. The miR-27a, FOXO1 and PPAR-γ expression in tumor were detected by RT-PCR and western blot, respectively. Blood was collected, and serum was separated for assessing alkaline aminotransferase (ALT), aspartate phosphatase (AST), alkaline phosphatase (ALP), blood urea nitrogen (BUN), creatinine (CRE), and triglyceride (TG) levels. Heart, lung, liver, spleen and left kidney of each mouse were conserved for H/E staining.
Statistical analysis
All experiments were performed at least three times and expressed as mean ± SD. Statistical significance (p< 0.05) was evaluated using Student's t-test when only two groups were compared. If more than two groups were compared, evaluation of significance was performed using one-way ANOVA analysis of variance, followed by Bonferroni's post hoc test.
Results
Characterization of anti-miR-27a/QD-HA-PEI
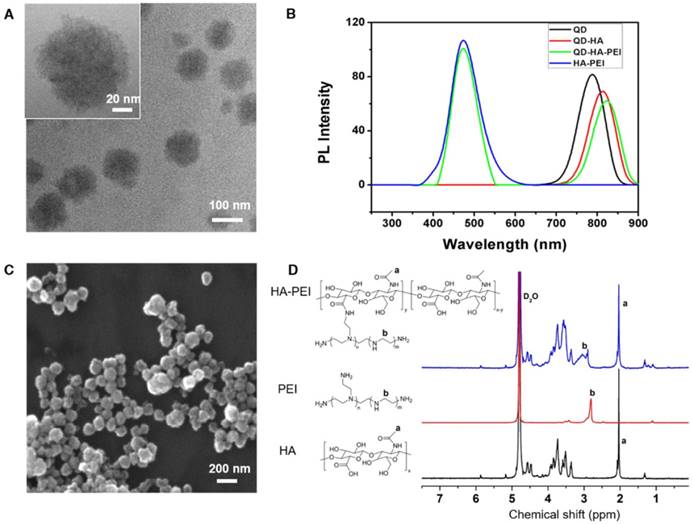
QD-HA-PEI conjugate was fabricated first and anti-miR-27a/QD-HA-PEI was self-assembled in the present study. As shown in Figure 1A-B, anti-miR-27a/QD-HA-PEI displayed a uniform spherical shape with sizes in the range of 70-100 nm. Hydrodynamic size and zeta-potential of anti-miR-27a/QD-HA-PEI were 120 ± 4.3 nm (PDI was 0.218) (Figure S1) and -11.6 ± 1.4 mV, respectively (Figure S2). The optical properties of anti-miR-27a/QD-HA-PEI are shown in Figure 1C and Figure S3. As expected, anti-miR-27a/QD-HA-PEI exhibited similar emission patterns as QDs, with major emission peaks at around 850 nm in the NIR region. To our surprise, there was a strong fluorescence emission peak at 460 nm, which was considered to originate from the amide bond formed between HA and PEI due to a similar peak in the emission spectrum of HA-PEI but not in that of QD-HA. The 1H-NMR spectrum in Figure 1D shows characteristic resonance peaks for the methyl group of HA at 1.94 ppm and the methylene groups of PEI at 2.40-3.30 ppm, demonstrating that PEI was successfully grafted onto the HA chains. As shown in Figure S4, QD-HA-PEI could efficiently bind anti-miR-27a, as evidenced by significant retardation of miRNA migration in gel electrophoresis at a w/w (QD-HA-PEI to RNA) ratio above 9:1. The RNA protection results indicated rapid degradation of free anti-miR-27a in RNase solution; whereas, QD-HA-PEI could approximately 100% protect anti-miR-27a from RNase-based degradation at the minimal w/w ratio of 9:1 (Figure S5). Consequently, we used this dose ratio in the following biological experiments. Moreover, anti-miR-27a/QD-HA-PEI possessed good colloidal stability and photostability in cell culture media for 24 h (Figure S6 and Figure S7), suggesting its considerable potential to be applied in vitro and in vivo.
Characterization of anti-miR-27a/QD-HA-PEI. (A) High-resolution TEM image of anti-miR-27a/QD-HA-PEI; scale bars are 100 nm and 20 nm (inset). (B) SEM image of anti-miR-27a/QD-HA-PEI; scale bar is 100 nm. (C) Emission spectra and (D) 1H-NMR spectra of anti-miR-27a/QD-HA-PEI.
CD44 receptor-mediated endocytosis of anti-miR-27a/QD-HA-PEI and “ON-OFF” fluorescence-based drug release tracking
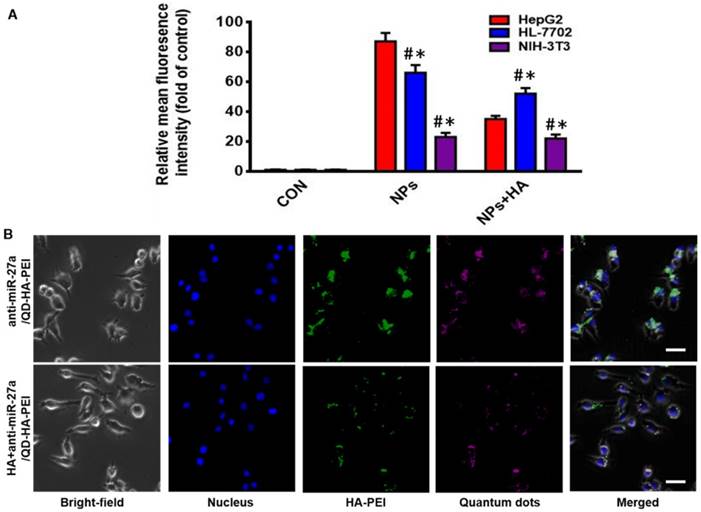
As one of the important tumor initiating cell markers in liver cancer, CD44 is a hyaluronic acid receptor and major cell surface glycoprotein, which plays a key role in cell adhesion, cell migration and invasion [42]. To determine the CD44 receptor-mediated endocytosis of anti-miR-27a/QD-HA-PEI, CD44 receptor positive liver cancer HepG2 cells, CD44 receptor negative NIH-3T3 cells and CD44 low-expression normal liver HL-7702 cells were selected for a comparative study. From the FACS analysis (Figure 2A), anti-miR-27a/QD-HA-PEI could be taken up by HepG2 cells with HA receptors more efficiently than NIH-3T3 or HL-7702 cells. It was also found that pre-treatment with free HA could significantly decrease anti-miR-27a/QD-HA-PEI ingestion in HepG2 cells rather than in NIH-3T3 or HL-7702 cells, which are lined with other receptors [43,44]. As shown in Figure 2B and Figure S8, we first concluded that endocytosis of the nanoparticles was induced by CD44 receptor and determined by its expression level. As expected, NIR fluorescence of QDs and intrinsic fluorescence of HA-PEI conjugate exhibited strong co-localization in the cytoplasm after 3 h of treatment, further indicating the conjugation of QD-HA-PEI.
CD44-targeted cellular uptake of anti-miR-27a/QD-HA-PEI. (A) Quantitative analysis of the internalization of anti-miR-27a/QD-HA-PEI in HepG2, HL-7702 and NIH-3T3 cells after 3 h of exposure through FACS. These data represent three separate experiments and are presented as the mean values ± SD. *P < 0.05 versus the NPs group, #P < 0.05 versus the HepG2 group. (B) CLSM images of HepG2 cells after 3 h of incubation; the scale bars represent 10 μm.
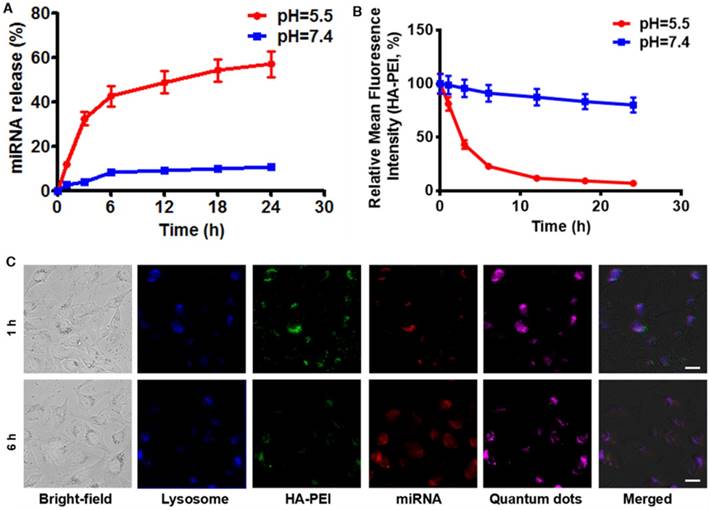
To investigate whether anti-miR-27a/QD-HA-PEI is pH-sensitive, we first measured its RNA release profile at different levels of pH (Figure 3A). About 50% release of anti-miR-27a was observed at pH 5.5, while less than 10% of anti-miR-27a was released at pH 7.4. The release of anti-miR-27a could reach a plateau within 6 h, which may be due to degradation of the HA-PEI backbone and destruction of the integrity of the spherical structure under acidic conditions. As expected, the intrinsic fluorescence of the HA-PEI backbone was quenched at pH 5.5 and reduced along with incubation time, while the NIR fluorescence of the QDs only changed a little over time (Figure 3B and Figure S9). It is worth noting that there was probably a negative correlation between miRNA release and fluorescence quenching of HA-PEI. We further investigated whether the dual-fluorescence nanosystem could be employed to monitor intracellular pH-triggered miRNA release by detecting the fluorescence quenching of HA-PEI. HepG2 cells were incubated with anti-miR-27a/QD-HA-PEI for different lengths of time. As shown in Figure 3C, the co-localization of QD, HA-PEI, RNA and lysosome was observed after 1 h of incubation. Importantly, a time-course enhancement of RNA fluorescence in the cancer cells clearly indicated the pH-controlled drug release. In the meantime, gradual fluorescence quenching of HA-PEI backbone was observed in lysosomes, while there was no significant QD fluorescence loss during the incubation. Correspondingly, FACS results demonstrated the decrease of HA-PEI fluorescence signal and stability of QD fluorescence signal over time (Figure S10).
RNA release behavior and “ON-OFF” fluorescence switching of anti-miR-27a/QD-HA-PEI. (A) RNA release profile and (B) normalized fluorescence intensity of anti-miR-27a/QD-HA-PEI under different pH conditions. (C) CLSM images of HepG2 cells after 1 or 6 h of incubation; the scale bars represent 10 μm.
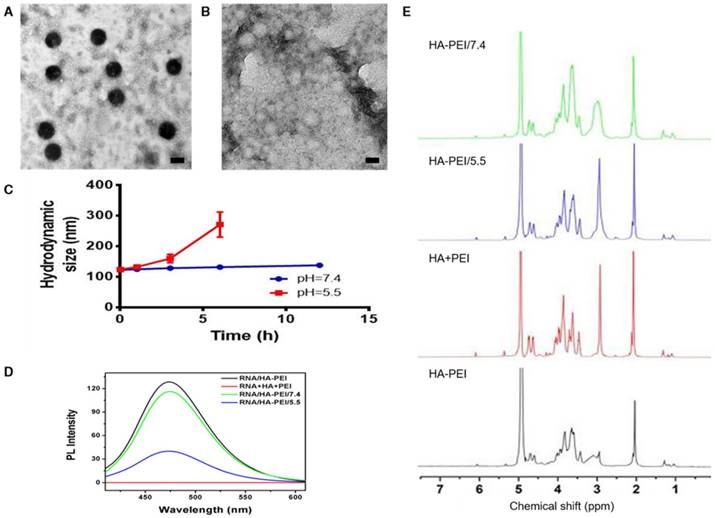
Investigation on the relationship between “ON/OFF” effect and structural change under different pH conditions
To verify our assumption that the emission peak at 460 nm was the result of amide bonds formed between HA and PEI, RNA/HA-PEI NPs were synthesized and incubated in neutral (pH=7.4) and acidic (pH=5.5) PBS for 12 h. As shown in Figure 4A-B, RNA/HA-PEI kept their spherical morphology in the neutral environment, while they degraded into membrane-like fragments in the acidic environment; these observations were collectively confirmed by the hydrodynamic size changes (Figure 4C). What's more, the intensity of the emission peak at 460 nm also significantly decreased in acidic PBS (Figure 4D). After comparing the results in Figure 1C and Figure 4D, it is obvious that RNA contributed nothing to this emission peak. We then concluded from these results that a stable connection between HA and PEI is essential for the existence of the 460 nm fluorescence peak. To further prove our point, 1H-NMR was performed. As shown in Figure 4E, characteristic resonance peaks for the methyl group of HA at 1.94 ppm and the methylene groups of PEI at 2.40-3.30 ppm were observed in the spectrum of free HA and PEI mixture. As expected, the spectrum of HA-PEI/5.5 was quite similar to that of HA and PEI mixture, indicating most of the amide bonds in HA-PEI hydrolyzed in acidic PBS, while only a small part of HA-PEI hydrolyzed in neutral PBS. In conclusion, hydrolyzation of amide bonds in HA-PEI under acidic conditions not only promotes the destruction of its spherical morphology, but also leads to the quenching of its intrinsic fluorescence at 460 nm. This feature was further named an “ON/OFF” effect and applied to monitor RNA release in vitro.
pH-dependent degradation behavior of anti-miR-27a/QD-HA-PEI. Changes of anti-miR-27a/QD-HA-PEI under different pH conditions. (A) TEM image of anti-miR-27a/QD-HA-PEI incubated in PBS (pH=7.4) for 12 h; scale bar is 100 nm. (B) TEM image of anti-miR-27a/QD-HA-PEI incubated in PBS (pH=5.5) for 12 h; scale bar is 100 nm. (C) Hydrodynamic size changes of anti-miR-27a/QD-HA-PEI in PBS (pH=5.5, red and pH=7.4, blue). (D) Emission spectra and (E) 1H-NMR results of HA-PEI, HA+PEI, HA-PEI in PBS (pH=5.5, blue and pH=7.4, green).
CD44-targeted anti-cancer efficacy, miR-27a modulation effects and molecular mechanisms of anti-miR-27a/QD-HA-PEI
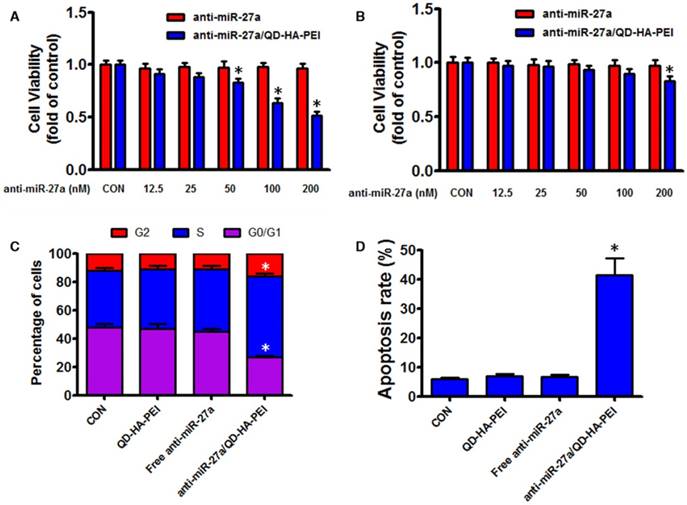
As the results above suggested that anti-miR-27a/QD-HA-PEI could specifically deliver anti-miR-27a into HepG2 cells, we hypothesized that anti-miR-27a/QD-HA-PEI would result in selective anti-cancer activity due to the CD44 receptor-mediated endocytosis and pH-dependent drug release. As shown in Figure 5A-B, anti-miR-27a/QD-HA-PEI exhibited markedly higher cell killing activity with a dose-dependent manner in HepG2 cells that was higher than that in HL-7702 cells. On the other hand, neither free anti-miR-27a nor QD-HA-PEI affected cell viability even at high concentrations (Figure 5A-B and Figure S11). Given that miR-27a can modulate a variety of cellular pathways associated with cell proliferation and apoptosis, results from cell cycle assays (Figure 5C) demonstrated that anti-miR-27a/QD-HA-PEI could drastically decrease the percentage of cells in the G1/G0 peak while increasing the percentage of cells in the S peak than other groups. Apoptosis assays (Figure 5D) also showed that both free anti-miR-27a and QD-HA-PEI groups had little influence on apoptosis, while this was much more obvious after cells were treated with anti-miR-27a/QD-HA-PEI.
Anti-tumor effects of anti-miR-27a/QD-HA-PEI in vitro. Cell viability of (A) HepG2 cells and (B) HL-7702 cells incubated with various concentrations of anti-miR-27a/QD-HA-PEI for 24 h. FACS analysis of the (C) cell cycle and (D) cell apoptosis of anti-miR-27a/QD-HA-PEI-treated HepG2 cells. These data represent three separate experiments and are presented as the mean values ± SD. *P < 0.05 versus anti-miR-27a group.
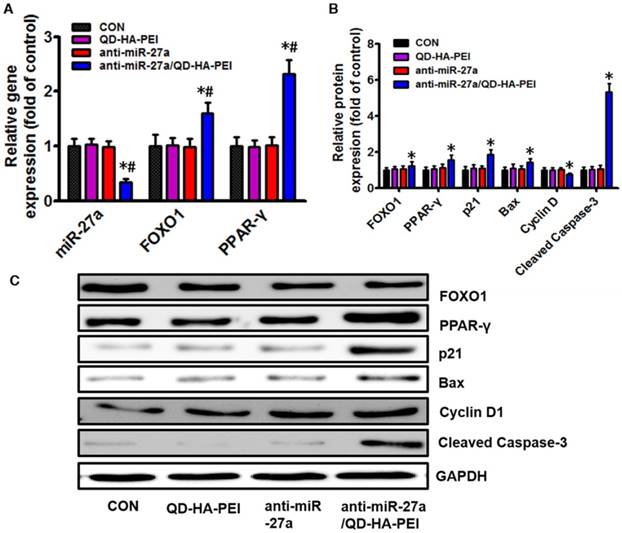
To further clarify the molecular mechanism of anti-miR-27a/QD-HA-PEI-mediated anti-cancer cell effect, the intracellular inhibition of tumor enhancer miR-27a was quantified by real-time PCR. As shown in Figure 6A, over a 3-fold decrease in miR-27a was observed in HepG2 cells treated with anti-miR-27a/QD-HA-PEI compared with untreated cells as well as those treated with free anti-miR-27a or QD-HA-PEI. We then assessed the effects of a decreased miR-27a expression level on the regulation of its target genes. Since transcriptional factor FOXO1 and PPAR-γ are promising downstream targets of miR-34a according to our previous report, their expressions were then examined by RT-PCR and western-blot. As shown in Figure 6A-C, both mRNA and protein expressions of FOXO1 and PPAR-γ were observed in the anti-miR-27a/QD-HA-PEI-treated group compared with the other three groups. We subsequently studied the expression of downstream targets of FOXO1 and PPAR-γ (Figure 6B-C). As expected, western-blot results revealed that Cyclin D1 protein level was downregulated while p21, Bax and caspase-3 protein levels were upregulated in anti-miR-27a/QD-HA-PEI-treated groups.
Molecular mechanisms of the anti-tumor effect of anti-miR-27a/QD-HA-PEI. (A) qRT-PCR analysis of miR-27a, FOXO1 and PPAR-γ expression in anti-miR-27a/QD-HA-PEI-treated HepG2 cells. (B-C) Western blot analysis of FOXO1, PPAR-γ, p21, Bax, CyclinD and cleaved-Caspase-3 expression in anti-miR-27a/QD-HA-PEI-treated HepG2 cells. These data represent three separate experiments and are presented as the mean values ± SD. *P < 0.05 versus anti-miR-27a group.
In vivo targeted NIR fluorescence imaging and biodistribution of anti-miR-27a/QD-HA-PEI
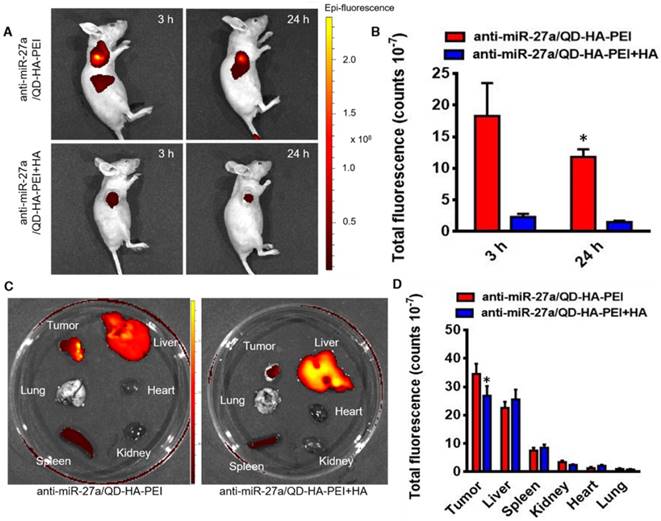
To examine the CD44 receptor-targeted effects of anti-miR-27a/QD-HA-PEI in vivo, the nanoparticles were injected intravenously into nude mice bearing HepG2 xenografts and NIR fluorescence in the tumor was non-invasively monitored at various time points (3 and 24 h). Additionally, a competitive inhibition study was performed to investigate the tumor-targeting mechanism of anti-miR-27a/QD-HA-PEI through intravenously pre-injecting HA polymer to block CD44-mediated binding of anti-miR-27a/QD-HA-PEI at the tumor site. As shown in Figure 7A-B, the NIR fluorescence signal in the tumor site was quite strong at 3 h and decreased at 24 h. Importantly, mice treated with anti-miR-27a/QD-HA-PEI had strong NIR fluorescence at the tumor site, while pre-injected free HA could significantly reduce the fluorescence intensity. To evaluate the in vivo biodistribution of anti-miR-27a/QD-HA-PEI, main organs of mice were collected at 24 h post-injection and ex vivo NIR fluorescence images were obtained (Figure 7C-D). For both anti-miR-27a/QD-HA-PEI and HA plus anti-miR-27a/QD-HA-PEI groups, strong fluorescence signals were observed in the liver and spleen, which may be due to accumulation of anti-miR-27a/QD-HA-PEI in the mononuclear phagocyte system and uptake by cells expressing HARE receptor. As expected, the strongest intensity of anti-miR-27a/QD-HA-PEI was observed at the tumor and the intensity was approximately 2-fold higher than that of HA+anti-miR-27a/QD-HA-PEI.
Bioimaging and biodistribution of anti-miR-27a/QD-HA-PEI. (A) HepG2 tumor-bearing mice were intravenously injected with free anti-miR-27a/QD-HA-PEI or anti-miR-27a/QD-HA-PEI plus free HA for in vivo imaging at different time points. (B) Quantitation of fluorescence intensity at the tumor site of each group for up to 24 h. (C) Ex vivo NIR imaging of anti-miR-27a/QD-HA-PEI in tumor, liver, lung, heart, spleen and kidney. (D) Quantitation of fluorescence intensity of NIR QD signal ex vivo. The values represent mean values ± SD, n=6. *P < 0.05 versus anti-miR-27a/QD-HA-PEI group.
In vivo antitumor activity of anti-miR-27a/QD-HA-PEI
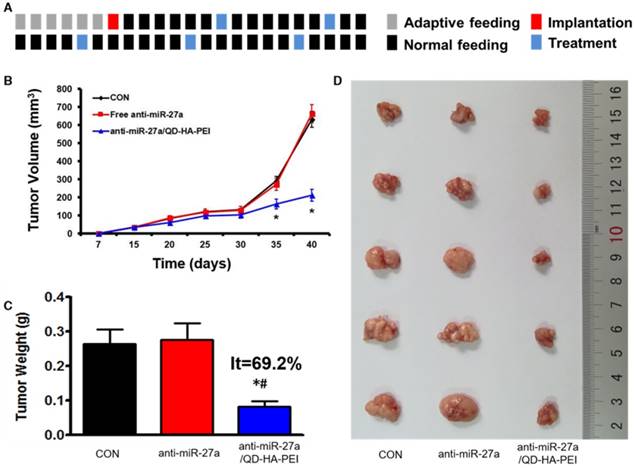
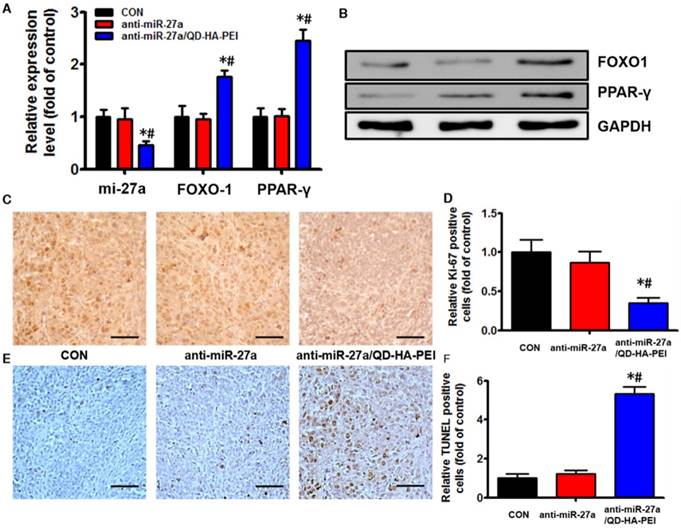
Antitumor efficacy of anti-miR-27a/QD-HA-PEI was studied in nude mice bearing HepG2 tumor xenografts for 40 days (Figure 8A). As shown in Figure 8B, anti-miR-27a/QD-HA-PEI treatment markedly inhibited tumor growth when compared with the control group and free anti-miR-27a group. At the end of the experiment, tumors were excised and photographed (Figure 8C-D). The tumor inhibition rate in the anti-miR-27a/QD-HA-PEI group (69.23%) was significantly higher than that in the free anti-miR-27a group. To further confirm the mechanism of miR-27a-mediated antitumor activity in vivo, mRNA expression of miR-27a and protein expressions of FOXO1 and PPAR-γ in tumor sites were evaluated at the end of treatment (Figure 9A-B). Significant downregulation of miR-27a and upregulation of FOXO1 and PPAR-γ were found in the tumors treated with anti-miR-27a/QD-HA-PEI when compared with the other two groups, indicating the successful delivery of functional anti-miR-27a into the tumor site. Immuno-histochemical staining of Ki-67 and TUNEL in the tumor tissue was also performed to study anti-proliferative and pro-apoptotic activity, respectively. As shown in Figure 9C-D, anti-miR-27a/QD-HA-PEI treatment increased the number of apoptotic cancer cells, as indicated by a higher percentage of TUNEL positive cells (532.7%) when compared with control and free anti-miR-27a. Anti-miR-27a/QD-HA-PEI treatment also resulted in the lowest proliferation rate (35.1%) in tumors compared to control and free anti-miR-27a (Figure 9E -F)
In vivo anti-tumor effect of anti-miR-27a/QD-HA-PEI. (A) Dosage regimen of in vivo treatment for 46 days. (B) Tumor growth curves, (C) tumor weights and (D) tumor photographs of HepG2 tumor-bearing mice in each group. The values represent mean values ± SD, n=6. *P < 0.05 versus anti-miR-27a group.
In vivo anti-tumor mechanisms of anti-miR-27a/QD-HA-PEI. (A) miR-27a and miRNA of FOXO1 and PPAR-γ expression, (B) protein expressions of FOXO1 and PPAR-γ, (C) Ki-6-stained images and (D) quantitation, (E) TUNNEL-strained images and (F) quantitation of tumor sites in each group. The values represent mean values ± SD, n=6. *P < 0.05 versus anti-miR-27a group. Scale bars represent 100 µm.
Biosafety evaluation of anti-miR-27a/QD-HA-PEI
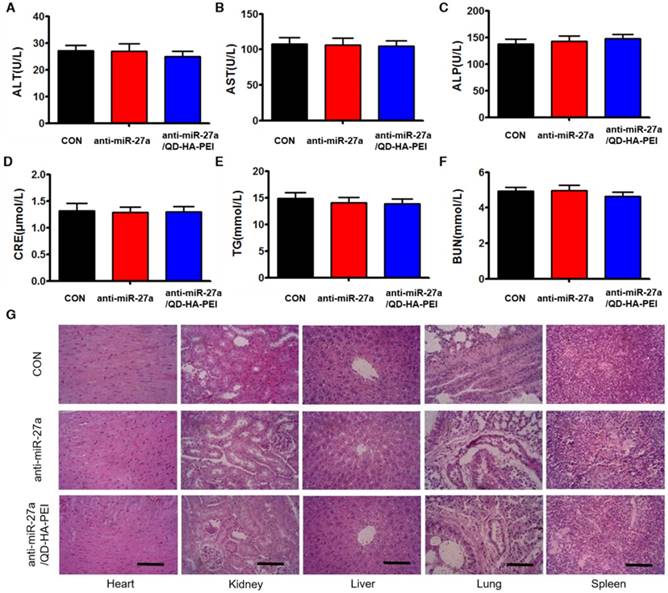
To further evaluate the systemic toxicity of anti-miR-27a/QD-HA-PEI, body weight was examined during the treatment. There was no observable weight loss in all treated groups compared with the control group (Figure S12). Serum alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase (ALP), serum blood urea nitrogen (BUN), creatinine (CRE) and total triglycerides (TG) were measured at the end of the experiment, which are critical biomarkers of liver, heart and renal damage, respectively. The results shown in Figure 10A-F demonstrated that serum ALT, AST, ALP, BUN, CRE and TG were not significantly different among all groups. Furthermore, H&E-stained images revealed no histopathological abnormalities, lesions or degenerations in the slices of heart, liver, spleen, lung and kidney of all groups (Figure 10G).
Assessment of the systemic side effects after treatment. Serum biochemistry indexes of mice in each group, including (A) ALT, (B) AST, (C) ALP, (D) CRE, (E) BUN, and (F) TG. (G) Representative H&E-stained images of heart, kidney, liver, lung and spleen from mice in each group; scale bars represent 100 µm.
Discussion
It has been proved by many reports that miRNAs play important roles in the development of tumors because abnormal expression of miRNAs usually results in a change of cell functions including proliferation, apoptosis, migration and autophagy. [12-15] Numerous reports have suggested that normalizing the expressions of certain miRNAs could be regarded as efficient gene therapy strategies for liver cancer. [16-20] Compared to siRNAs, perfect sequence pairing is not strictly required in gene silencing of miRNAs. This feature endows miRNAs the function of regulating multiple genes, which is quite practical in tumor therapy since more than one gene is responsible for liver cancer. Moreover, as a double-stranded RNA molecule is usually required in siRNA-induced gene silencing, the residual strand could also form a siRNA-induced silencing complex. In contrast, a single-stranded RNA molecule is enough for disabling the function of target miRNA, which lowers the risk of off-target effects. [45, 46] Therefore, therapeutic approaches to restoring homeostasis by altering miRNA expression have great potential to be more practical and effective strategies than silencing individual genes by siRNAs for liver cancer.
By suppressing the expressions of FOXO1 and PPAR-γ, miR-27a shoulders important responsibilities in the development of liver cancer. [20, 47] Since FOXO1 and PPAR-γ are both important positive regulators in cell proliferation and apoptosis, inhibition of their expressions will no doubt lead to unwanted increase of cells. The abundant miR-27a in HepG2 cells first interferes with the translation of mRNA related to FOXO1 and PPAR-γ, and then expressions of downstream proteins of those, including the negative regulation of p21, Bax and caspase-3, and the upregulation of cyclin D1. Changes in the expressions of these proteins collectively lead to the proliferation of HepG2 cells and finally result in worsening symptoms of liver cancer. However, when anti-miR-27a is incorporated into HepG2 cells, the life cycle of those cells shows a trend towards the regulated proliferation and apoptosis of normal cells, which is the result of inactivation of miR-27a. So, we deem that anti-miR-27a-induced inhibition of miR-27a could be considered an effective approach for therapy of liver cancer. It is worth noting that we did not detect phosphorylation of FOXO1 and PPAR-γ because miR-27a only inhibits the mRNA or protein expression of their targeted proteins rather than affecting their phosphorylation state [48-50]. Moreover, reports have demonstrated that miR-27a is involved in multiple drug resistance in various kinds of human cancers including gastric cancer, ovarian cancer, cervix carcinoma and liver cancer, [51-54] and its overexpression has been detected in serum and tumor tissues of liver cancer patients. Hence it can be concluded that miR-27a is an important target for cancer diagnosis and therapy.
Although QD-HA-PEI is negatively charged, its PEI branches are free to interact with miRNAs as they are not coated. Interaction between QD-HA-PEI and miRNA is closely related to the attraction between negatively charged RNAs and positively charged PEI branches, forming a spherical structure with a HA “shell” and PEI-miRNA “core” by self-assembly [32]. As is mentioned above, an amide bond is formed between HA and PEI through the EDC reaction, which not only functionalizes HA for miRNA loading, but also generates a new emission peak at 460 nm. This unexpected fluorescence peak makes the quick confirmation of functionalized HA possible. Moreover, this newly formed fluorescence peak offers the possibility of real-time monitoring of miRNA release in cells through tracking the fluorescence change. Interestingly, we found that the intrinsic fluorescence of HA-PEI quenches when the nanoparticles are incubated in acidic solution. We assumed that this phenomenon might partly be due to instability of the amide bonds as acidic environments also led to the rapid release of miRNAs loaded in QD-HA-PEI. Nevertheless, the mechanism remains unclear, and further study is required. Regardless, by studying the fluorescence intensity at 460 nm, the structural change of the HA-based nanosystem could be confirmed, which indicated the release of RNA. Collectively, this “on/off” effect provides a convenient way to determine the intracellular behaviors of the vector and its contents in vitro.
Interactions between carriers of nucleic acids and the living body, including therapeutic and toxic effects, always attract attention because their concentrations are usually higher than the nucleic acids loaded on or in them. At first, we thought QD-HA-PEI might have biological effects in some respects since the best weight ratio between it and anti-miR-27a was 9:1, as described above. However, neither therapeutic nor toxic effects were observed. Results of molecule mechanism studies showed that QD-HA-PEI had no effect on the expression of miR-27a or the pathway it regulated, and it did not impact cell cycle or apoptosis. Moreover, even though QDs were used for in vivo imaging, it could still be demonstrated that the whole nanosystem is safe. We attributed these findings mainly to the limited use of QDs. Because the fluorescence efficiency of QDs is quite high, only a small number of QDs is required to acquire satisfying fluorescence signal. Furthermore, cell apoptosis that is induced by quantum dots is dose-dependent [55]; the concentration of QDs in this work was obviously not high enough. What's more, as anti-miR-27a/QD-HA-PEI is CD-44-targeted, more nanoparticles could accumulate in tumor sites. The pH-responsive RNA release feature also makes the nanosystem safer because the cytoplasm in tumor cells is always acidic. Apart from our work, well-designed nucleic acid delivery systems developed by other researchers have shown similar properties [56,57]. Taken together, our results demonstrate that anti-miR-27a works well both in vitro and in vivo, and QD-HA-PEI is a promising candidate for targeted delivery of anti-miR-27a to achieve efficient and safe liver cancer theranostics.
In conclusion, a dual-fluorescence nanosystem that possesses the features of “ON/OFF” fluorescence, pH-sensitive drug release, CD44 receptor-targeted gene delivery and real-time fluorescence tracking was established. The fluorescence monitoring showed that upon intracellular reduction with low pH, the self-assembled nanosystem undergoes degradation to release anti-miR-27a. Successful delivery of anti-miR-27a by these particles was subsequently confirmed by the decreased intracellular miR-27a level and thereby normalized expressions of FOXO-1 and PPAR-γ and their downstream proteins including p21, Bax, cyclin D1 and capsase-3, resulting in cell proliferation inhibition and apoptosis. Moreover, in vivo tumor targeting was further confirmed by bioimaging, indicating that this system could target tumor site, and the delivery procedure could be monitored. Anti-miR-27a/QD-HA-PEI has proved itself to be an effective and safe tool for liver cancer therapy both in vitro and in vivo. In general, the self-assembled dual fluorescence nanosystem show great promise as a dual-function delivery vector of therapeutic miRNA, providing a novel, simple and elegant approach to the design of the next generation of nanomedicine liver cancer theranostics.
Abbreviations
ALP: alkaline phosphatase; ALT: alanine aminotransferase; AST: aspartate aminotransferase; BUN: blood urea nitrogen; CLSM: confocal fluorescence microscope; CRE: creatinine; EDC: 1-Ethyl-(3-dimethylaminopropyl) carbodiimide hydrochloride; FACS: fluorescence-activated cell sorter; FBS: fetal bovine serum; FTIR: Fourier transform infrared; HA: sodium hyaluronate; miRNA: micro RNA; N.C.: negative control; NHS: N-hydroxysuccinimide; NIR: near-infrared; NPs: nanoparticles; PAA: polyacrylic acid; PEI: polyethyleneimine; QDs: quantum dots; SEM: scanning electron microscope; siRNA: small interfering RNA; TEM: transmission electron microscope; TG: total triglycerides; TUNEL: transferase-mediated deoxyuridine triphosphate-biotin nick end labeling.
Acknowledgements
Prof. Chungang Wang, Dr. Yong Jia and Dr. Shuang Li are acknowledged for their help in preparing the paper. This work is sponsored by the National Natural Science Foundation of China (81371681 and 81571199), the Science and technology development project of Jilin Province (No. 20160101203JC), the Education Department of Jilin Province (No. [2016] 452), the Graduate Innovation Fund of Jilin University (2016218), and the Opening Project of the State Key Laboratory of Supramolecular Structure and Materials of Jilin University under Grant No. SKLSSM 201605, 201713 and 201832.
Supplementary Material
Supplementary figures.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bruix J, Gores GJ, Mazzaferro V. Hepatocellular carcinoma: clinical frontiers and perspectives. Gut. 2014;63:844-855
2. Maluccio M, Covey A. Recent progress in understanding, diagnosing, and treating hepatocellular carcinoma. CA Cancer J Clin. 2012;62:394-399
3. Akinyemiju T, Abera S, Ahmed M, Alam N, Alemayohu MA, Allen C. et al. The Burden of Primary Liver Cancer and Underlying Etiologies From 1990 to 2015 at the Global, Regional, and National Level: Results From the Global Burden of Disease Study 2015. JAMA Oncol. 2017;3:1683-1691
4. Oh YK, Park TG. siRNA delivery systems for cancer treatment. Adv Drug Deliv Rev. 2009;61:850-862
5. Zuckerman JE, Davis ME. Clinical experiences with systemically administered siRNA-based therapeutics in cancer. Nat Rev Drug Discov. 2015;14:843-856
6. Llovet JM, Villanueva A, Lachenmayer A, Finn RS. Advances in targeted therapies for hepatocellular carcinoma in the genomic era. Nat Rev Clin Oncol. 2015;12:408-424
7. Huang S, He X. The role of microRNAs in liver cancer progression. Br J Cancer. 2011;104:235-240
8. Ladeiro Y, Couchy G, Balabaud C, Bioulac-Sage P, Pelletier L, Rebouissou S. et al. MicroRNA profiling in hepatocellular tumors is associated with clinical features and oncogene/tumor suppressor gene mutations. Hepatol. 2008;47:1955-1963
9. Rossi JJ. New hope for a microRNA therapy for liver cancer. Cell. 2009;137:990-992
10. Murakami Y, Yasud T, Saigo K, Urashima T, Toyoda H, Okanoue T. et al. Comprehensive analysis of microRNA expression patterns in hepatocellular carcinoma and non-tumorous tissues. Oncogene. 2006;25:2537-2545
11. Ji J, Shi J, Budhu A, Yu Z, Forgues M, Roessler S. et al. MicroRNA expression, survival, and response to interferon in liver cancer. N Engl J Med. 2009;361:1437-1447
12. Gramantieri L, Fornari F, Ferracin M, Veronese A, Sabbioni S, Calin GA. et al. MicroRNA-221 targets Bmf in hepatocellular carcinoma and correlates with tumor multifocality. Clin Cancer Res. 2009;15:5073-5081
13. Qiu D, Chen J, Liu J, Luo Z, Jiang W, Huang J. et al. Expression of microRNA let-7a positively correlates with hepatitis B virus replication in hepatocellular carcinoma tissues. Exp Biol Med. 2017;242:939-944
14. Gougelet A, Sartor C, Bachelot L, Godard C, Marchiol C, Renault G. et al. Antitumour activity of an inhibitor of miR-34a in liver cancer with β-catenin-mutations. Gut. 2016;65:1024-1034
15. Callegari E, Gramantieri L, Domenicali M, D'abundo L, Sabbioni S, Negrini M. MicroRNAs in liver cancer: a model for investigating pathogenesis and novel therapeutic approaches. Cell Death Differ. 2015;22:46-57
16. Wang Y, Chen F, Zhao M, Yang Z, Zhang S, Ye L. et al. MiR-107 suppresses proliferation of hepatoma cells through targeting HMGA2 mRNA 3′ UTR. Biochem Biophys Res Commun. 2016;480:455-460
17. Ge W, Yu DC, Li QG, Chen X, Zhang CY, Ding YT. Expression of serum miR-16, let-7f, and miR-21 in patients with hepatocellular carcinoma and their clinical significances. Clin Lab. 2014;60:427-434
18. Chen Z, Ma T, Huang C, Zhang L, Lv X, Xu T. et al. MiR-27a modulates the MDR1/P-glycoprotein expression by inhibiting FZD7/β-catenin pathway in hepatocellular carcinoma cells. Cell Signal. 2013;25:2693-2701
19. Sun BZ, Li J, Shao D, Pan Y, Chen Y, Li S. et al. Adipose tissue-secreted miR-27a promotes liver cancer by targeting FOXO1 in obese individuals. Onco Targets Ther. 2015;8:735-744
20. Li S, Li J, Fei BY, Shao D, Pan Y, Mo ZH. et al. MiR-27a promotes hepatocellular carcinoma cell proliferation through suppression of its target gene peroxisome proliferator-activated receptor γ. Chin Med J. 2015;128:941-947
21. Cheng CJ; Bahal R, Babar IA, Pincus Z, Barrera F, Liu C. et al. MicroRNA silencing for cancer therapy targeted to the tumour microenvironment. Nature. 2015;518:107-110
22. Kasinski A; Slack FJ. MicroRNAs en route to the clinic: progress in validating and targeting microRNAs for cancer therapy. Nat Rev Cancer. 2011;11:849-864
23. Vickers KC, Palmisano BT, Shoucri BM, Shamburek RD, Remaley AT. MicroRNAs are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nat Cell Biol. 2011;13:423-433
24. Pereira DM, Rodrigues PM, Borralho PM, Rodrigues CM. Delivering the promise of miRNA cancer therapeutics. Drug Discov Today. 2013;18:282-289
25. Muthiah M, Park IK, Cho CS. Nanoparticle-mediated delivery of therapeutic genes: focus on miRNA therapeutics. Expert Opin Drug Deliv. 2013;10:1259-1273
26. Chen Y, Gao DY, Huang L. In vivo delivery of miRNAs for cancer therapy: challenges and strategies. Adv Drug Deliv Rev. 2015;81:128-141
27. Zhang Y, Wang Z, Gemeinhart RA. Progress in microRNA delivery. J Control Release. 2013;172:962-974
28. Lin LZ, Fan Y, Gao F, Jin LF, Li D, Sun WJ. et al. UTMD-Promoted Co-Delivery of Gemcitabine and miR-21 Inhibitor by Dendrimer-Entrapped Gold Nanoparticles for Pancreatic Cancer Therapy. Theranostics. 2018;8:1923-1939
29. Yang T, Zhao PX, Rong Z, Li B, Xue HY, You J. et al. Anti-tumor efficiency of lipid-coated cisplatin nanoparticles co-loaded with microRNA-375. Theranostics. 2016;6:142-154
30. Wang H, Jiang Y, Peng H, Chen Y, Zhu P, Huang Y. Recent progress in microRNA delivery for cancer therapy by non-viral synthetic vectors. Adv Drug Deliv Rev. 2015;81:142-160
31. Sun SY, Wang YL Zhou R, Deng ZC Han Y, Han X et al. Targeting and regulating of an oncogene via nanovector delivery of MicroRNA using patient-derived xenografts. Theranostics. 2017;7:677-693
32. Ganesh S, Iyer AK, Morrissey DV, Amiji MM. Hyaluronic acid based self-assembling nanosystems for CD44 target mediated siRNA delivery to solid tumors. Biomaterials. 2013;34:3489-3502
33. Tian H, Lin L, Chen J, Chen X, Park TG, Maruyama A. RGD targeting hyaluronic acid coating system for PEI-PBLG polycation gene carriers. J Control Release. 2011;155:47-53
34. Zhong Y, Goltsche K, Cheng L, Xie F, Meng F, Deng C. et al. Hyaluronic acid-shelled acid-activatable paclitaxel prodrug micelles effectively target and treat CD44-overexpressing human breast tumor xenografts in vivo. Biomaterials. 2016;84:250-261
35. Jain S, Kumar S, Agrawal A, Thanki K, Banerjee U. Hyaluronic acid-PEI-cyclodextrin polyplexes: implications for in vitro and in vivo transfection efficiency and toxicity. RSC Adv. 2015;5:41144-41154
36. Shao D, Li J, Xiao XA, Zhang M, Pan Y, Li S. et al. Real-time visualizing and tracing of HSV-TK/GCV suicide gene therapy by near-infrared fluorescent quantum dots. ACS Appl Mater Interfaces. 2014;6:11082-11090
37. Shao D, Li J, Pan Y, Zhang X, Zheng X, Wang Z. et al. Noninvasive theranostic imaging of HSV-TK/GCV suicide gene therapy in liver cancer by folate-targeted quantum dot-based liposomes. Biomater Sci. 2015;3:833-841
38. Jiang G, Park K, Kim J, Kim KS, Hanhn SK. Target Specific Intracellular Delivery of siRNA/PEI-HA Complex by Receptor Mediated Endocytosis. Mol Pharm. 2009;6:727-737
39. Shao D, Li J, Zheng X, Pan Y, Wang Z, Zhang M. et al. Janus “nano-bullets” for magnetic targeting liver cancer chemotherapy. Biomaterials. 2016;100:118-133
40. Shao D, Zhang X, Liu W, Zhang F, Zheng X, Qiao P. et al. Janus silver-mesoporous silica nanocarriers for SERS traceable and pH-sensitive drug delivery in cancer therapy. ACS Appl Mater Interfaces. 2016;8:4303-4308
41. Wang Z, Chang Z, Lu M, Shao D, Yue J, Yang D. et al. Janus Silver/Silica Nanoplatforms for Light-Activated Liver Cancer Chemo/Photothermal Therapy. ACS Appl Mater Interfaces. 2017;9:30306-30317
42. Endo K, Terada T. Protein expression of CD44 standard and variant isoforms. in hepatocellular carcinoma: relationships with tumor grade, clinicopathologic parameters, p53 expression, and patient survival. J Hepatol. 2000;32:78-84
43. Zhao QF, Liu J, Zhu WQ, Sun CS, Di DH, Zhang Y. et al. Dual-stimuli responsive hyaluronic acid-conjugated mesoporous silica for targeted delivery to CD44-overexpressing cancer cells. Acta Biomater. 2015;23:147-156
44. Zhao QF, Wang SY, Yang Y, Li X, Di DH, Zhang CG. et al. Hyaluronic acid and carbon dots-gated hollow mesoporous silica for redox and enzyme-triggered targeted drug delivery and bioimaging. Mater Sci Eng C. 2017;78:475-484
45. Meister G, Tuschl T. Mechanisms of gene silencing by double-stranded RNA. Nature. 2004:431 7006, 343-349
46. Carthew RW, Sontheimer EJ. Origins and mechanisms of miRNAs and siRNAs. Cell. 2009;136:642-655
47. Zhu H, Wu H, Liu X, Evans BR, Medina DJ, Liu CG. et al. Role of MicroRNA miR-27a and miR-451 in the regulation of MDR1/P-glycoprotein expression in human cancer cells. Biochem Pharmacol. 2008;76:582-588
48. Guttilla IK, White BA. Coordinate regulation of FOXO1 by miR-27a, miR-96, and miR-182 in breast cancer cells. J Biol Chem. 2009;284:23204-23216
49. Kim SY, Kim AY, Lee HW, Son YH, Lee GY, Lee JW. et al. miR-27a is a negative regulator of adipocyte differentiation via suppressing PPARγ expression. Biochem Biophys Res Commun. 2010;392:323-328
50. Yu Y, Du HW, Wei SN, Feng LJ, Li JN, Yao F. et al. Adipocyte-Derived Exosomal MiR-27a Induces Insulin Resistance in Skeletal Muscle Through Repression of PPARγ. Theranostics. 2018;8:2171-2188
51. Zhang H, Li M, Han Y, Hong L, Gong T, Sun L. et al. Down-regulation of miR-27a might reverse multidrug resistance of esophageal squamous cell carcinoma. Dig Dis Sci. 2010;55:2545-2551
52. Zhao X, Yang L, Hu J. Down-regulation of miR-27a might inhibit proliferation and drug resistance of gastric cancer cells. J Exp Clin Cancer Res. 2011;30:55
53. Feng DD, Zhang H, Zhang P, Zheng YS, Zhang XJ, Han BW. et al. Down-regulated miR-331-5p and miR-27a are associated with chemotherapy resistance and relapse in leukaemia. J Cell Mol Med. 2011;15:2164-2175
54. Li J, Wang Y, Song Y, Fu Z, Yu W. miR-27a regulates cisplatin resistance and metastasis by targeting RKIP in human lung adenocarcinoma cells. Mol Cancer. 2014;13:19
55. Shao D, Li J, Guan FY, Pan Y, Xiao XA, Zhang M. et al. Selective inhibition of liver cancer growth realized by the intrinsic toxicity of a quantum dot-lipid complex. Int J Nanomedicine. 2014;9:5753-5369
56. Wang YY, Chen XJ, Tian BQ, Liu JF, Yang L, Zeng LL. et al. Nucleolin-targeted extracellular vesicles as a versatile platform for biologics delivery to breast cancer. Theranostics. 2017;7:1360-1372
57. Qin YY, Chen WL, Liu BJ, Zhou L, Deng L, Niu WX. et al. MiR-200c inhibits the tumor progression of glioma via targeting moesin. Theranostics. 2017;7:1663-1673
Author contact
![]() Corresponding authors: Li Chen, Tel: +86-0431-85619799, E-mail: chenledu.cn; Dan Shao, Tel: +86-0431-85619799, E-mail: stanauagatecom; Jing Li, Tel: +86-0431-85619799, E-mail: lijingedu.cn.
Corresponding authors: Li Chen, Tel: +86-0431-85619799, E-mail: chenledu.cn; Dan Shao, Tel: +86-0431-85619799, E-mail: stanauagatecom; Jing Li, Tel: +86-0431-85619799, E-mail: lijingedu.cn.