Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results
Discussion
Conclusions
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(13):3707-3721. doi:10.7150/thno.25166 This issue Cite
Research Paper
AP-2β inhibits hepatocellular carcinoma invasion and metastasis through Slug and Snail to suppress epithelial-mesenchymal transition
Liu Yang1, Junlu Qiu1, Yuzhong Xiao2, Xiang Hu1,3, Qing Liu1, Li Chen1, Wenhuan Huang1, Xinxin Li1, Limin Li4, Jian Zhang1,5, Xiaofeng Ding1,3,5 ![]() , Shuanglin Xiang1,3,5
, Shuanglin Xiang1,3,5 ![]()
1. Key Laboratory of Protein Chemistry and Development Biology of State Education Ministry of China, College of Life Science, Hunan Normal University, Changsha, 410081, P.R. China
2. Department of Endocrinology, Endocrinology Research Center, Xiangya Hospital of Central South University, Changsha, 410011, P.R. China
3. State Key Laboratory of Developmental Biology of Freshwater Fish, College of Life Science, Hunan Normal University, Changsha, 410081, P.R. China
4. College of Engineering and Design, Hunan Normal University, Changsha, 410081, P.R. China
5. The National & Local Joint Engineering Laboratory of Animal Peptide Drug development, College of Life Science, Hunan Normal University, Changsha, 410081, P.R. China
Received 2018-1-26; Accepted 2018-5-18; Published 2018-6-12
Abstract

Transcription factor AP-2β plays an important role in human cancer, but its clinical significance in hepatocellular carcinogenesis is largely unknown.
Methods: AP-2β expression was detected in human hepatocellular cancer (HCC) tissues and cell lines. The effects of AP-2β on HCC proliferation, migration, invasion, tumor formation and metastasis were evaluated by MTT, colony formation and transwell assays in vitro and mouse experiments in vivo. The association between AP-2β and miR-27a/EMT markers in HCC cell lines and tissues was analyzed.
Results: AP-2β expression was decreased in HCC tissues and cell lines. Reduced expression of AP-2β was significantly associated with more advanced tumor stages and larger tumor sizes. The overexpression of AP-2β reduced HCC proliferation, migration, invasion, tumor formation and metastasis in vitro and in vivo. Additionally, AP-2β overexpression increased the sensitivity of HCC cells to cisplatin. Moreover, AP-2β modulates the levels of EMT markers through Slug and Snail in HCC cell lines and tissues. Furthermore, oncogenic miR-27a inhibits AP-2β expression by binding to the AP-2β 3′ untranslated region (UTR) and reverses the tumor suppressive role of AP-2β.
Conclusion: These results suggested that AP-2β is lowly expressed in HCC by inhibiting EMT signaling to regulate HCC cell growth and migration. Therefore, AP-2β in the novel miR-27a/AP-2β/Slug/EMT regulatory axis enhances the chemotherapeutic drug sensitivity of HCC and might represent a potential target for evaluating the treatment and prognosis of human HCC.
Keywords: AP-2β, hepatocellular carcinoma, epithelial-to-mesenchymal transition (EMT), Slug, miR-27a
Introduction
Hepatocellular carcinoma (HCC) accounts for more than 5% of all cancer types and is the fifth leading cause of cancer mortality worldwide with an extremely poor prognosis [1]. Early stage patients are standardly treated with surgical resection, local ablation and liver transplantation, but most patients at an advanced stage preclude these treatments, the 5-year survival rate is 25-39%, and the recurrence rate is as high as 80% [2]. Sorafenib, a kinase inhibitor with antiangiogenic and antiproliferative properties, is the only targeted drug approved by the FDA to prolong the overall survival of patients with advanced HCC, but the response rate of sorafenib is as low as 2.2% [3]. Currently, none of the new drugs tested have shown positive results in first-line or second-line settings after sorafenib progression [4]. Therefore, the molecular events underlying hepatocarcinogenesis should largely be addressed. The identification of the key genes underlying HCC progression and metastasis and the development of more effective strategies for future therapeutic intervention are urgently required.
Transcription factor AP-2β serves as an oncogene or tumor suppressor. In one respect, AP-2β activates the human telomerase reverse transcriptase (hTERT) promoter, enhances telomerase activity, and functions as a novel tumor marker in lung cancer [5]. AP-2β also mediates ERK/p38, caspase/cytochrome-c and VEGF/PEDF-dependent signaling pathways and promotes tumor growth with a poor prognosis in human lung adenocarcinoma [6]. AP-2β interacts with the glioma amplified sequence 41 (GAS41), which is amplified in 23% of glioblastomas and 80% of grade I astrocytomas, suggesting their potential significance in glioma [7]. In the other respect, AP-2β was highly methylated in esophageal squamous cell carcinoma (ESCC) cell lines compared with non-cancerous esophageal mucosae [8]. Moreover, the CpG methylation of AP-2β is significantly associated with an unfavorable prognosis as well as adverse patient outcome in primary neuroblastoma [9]. AP-2β exerts no effects on HPV16/18 E6/E7 promoters [10], but enhances β-catenin degradation, inhibits the Wnt/ β-catenin pathway, and suppresses the proliferation of cervical cancer cells [11]. Some studies have shown that AP-2β was more highly expressed in lobular samples than in ductal breast carcinomas, AP-2β inhibition diminished the proliferation of lobular breast cancer (BC) cell lines and controlled cell growth in this slow-growing BC subtype [12, 13]. In contrast, normal and pure ductal carcinoma in situ (DCIS) samples expressed higher levels of AP-2β than invasive tumors, AP-2β might be correlated with less aggressive features and better patient outcomes in invasive breast cancer [14]. These data emphasized the complexity and importance of AP-2β in carcinogenesis.
AP-2β plays an important role in different types of human cancer, while the role of AP-2β in HCC and its associated clinicopathological features are not well investigated. In the present study, we showed the downregulation of AP-2β in human HCC tissues and cell lines. Moreover, the tumor suppressive effects of AP-2β were exerted through the inhibition of EMT signaling pathway via Slug and Snail, which was reversed by oncogenic miR-27a, it binds to the 3′ UTR of AP-2β and inhibits its expression. AP-2β overexpression sensitized HCC cells to cisplatin treatment. Therefore, AP-2β is negatively associated with HCC malignancies, and markedly inhibits HCC growth and metastasis. AP-2β will likely serve as an important therapeutic target in future applications to evaluate HCC patient prognosis and AP-2β agonist or recombinant proteins combined with chemotherapy drug maybe effective tools in confronting HCC.
Methods
Human tissues
70 liver cancer specimens and 10 adjacent non-tumorous tissues were used in this study (Table 1). The experiment was approved by Human Ethics Committee of Hunan Normal University and informed consent was obtained from all patients. The immunohistochemical (IHC) analysis was performed on polyformalin-fixed and paraffin-embedded tissues [11]. Briefly, sections (5 µm) were deparaffinized by two 10-min washes in xylene, then rehydrated through successive graded ethanol solutions. Endogenous peroxidase was quenched with 3% H2O2 in methanol for 10 min and washed for 10 min in PBS. Antigen retrieval was achieved by microwaving sections in 0.01 M citrate buffer (pH 6.0) for 10 min at 800 W. The tissues were blocked in 10% bovine serum albumin (BSA) in PBS for 1 h before the addition of the mouse monoclonal antibodies against AP-2β (E-8) (1:200, Santa Cruz Biotechnology Inc., Santa Cruz, CA), E-cadherin (4A2), Slug (L40C6) and Snail (L70G2) (1:200, cell signaling technologies, MA, USA) or normal mouse IgG control (1:200, BD Biosciences, CA, USA) at 4 °C for overnight. The sections were incubated with HRP-conjugated goat anti-mouse secondary antibody (diluted 1:1000, Sigma-Aldrich, St. Louis, MO, USA) for 30 min and then with 3,3-diaminobenzidine (DAB)/H2O2 for 5 min. Sections were counterstained with hematoxylin, mounted and photographed using an optical microscope (Olympus CX41, Tokyo, Japan). The protein levels of AP-2β, E-cadherin, Slug and Snail were analyzed according to the following standard. The percentage of tumor cells stained is scored as: 0 (no cell staining), 1 (≤30%), 2 (31% to 60%) and 3 (61% to 100%). Staining between two score values is given 0.5.
AP-2β expression and clinical characteristics.
| Features | Total | Overexpression | Low expression | P value |
|---|---|---|---|---|
| Sex | ||||
| Male | 62 | 24 | 38 | |
| Female | 8 | 3 | 5 | 0.376 |
| Age (median, 48 years) | ||||
| <48 years | 31 | 8 | 23 | |
| ≥48 years | 39 | 19 | 20 | 0.084 |
| Tumor size (cm) | ||||
| ≤5 | 47 | 24 | 23 | |
| >5 | 23 | 3 | 20 | 0.004 |
| Cell differentiation | ||||
| Well/ Moderately | 53 | 25 | 28 | |
| Poor | 17 | 2 | 15 | 0.009 |
| Tumor stage | ||||
| I/II | 47 | 24 | 23 | |
| III/IV | 23 | 3 | 20 | 0.004 |
HCC cell lines
SMMC7721, MHCC97H, Hep3B, LO2, HepG2, HEK293 and 293T cells were cultured in DMEM medium (Gibco BRL, Grand Island, NY, USA) supplemented with 10% fetal calf serum (FBS, Hyclone, Australia), and 1% penicillin/streptomycin (Gibco). All cells were maintained in a humidified atmosphere containing 5% CO2 at 37 °C.
Plasmid construction
Plasmid pCMV-Myc-AP-2β was described previously [15]. The lentiviral vectors pGC-FU-AP-2β-3Flag-IRES-Puromycin and pGC-FU-3Flag-IRES-Puromycin were constructed as described previously [11]. The miR-27a stem-loop sequence was inserted into plasmid pEZX-MR03 (GeneCopoeia, Guangzhou, China). The 3′ UTR of the AP-2β gene was obtained from the TargetScan database (http://www.targetscan.org). The 3′ UTR of AP-2β was amplified from gDNA of LO2 cells and inserted into the pmirGLO vector (Promega Corporation, Madison, WI, USA). Site-directed mutagenesis of the AP-2β 3′ UTR was performed by overlapping PCR to mutate potential miR-27a-binding sites as described previously [16]. Hsa-miR-27a mimics were synthesized by GenePharma Company (Shanghai, China).
The regulatory regions of Slug and Snail were predicted by Proscan and AP-2 binding sites were predicted by the JASPAR software. Genomic regions upstream of translation start codon (ATG) were amplified from human genomic DNA using primer pairs (Table S1) and inserted into pGL3-Basic vector (Promega), denoted pSlug(-973/+225)-Luc and pSnail(-1000/+267)-Luc. All constructs have been sequenced for verification.
AP-2β overexpression lentivirus generation
Lentiviral particles were prepared as described in our previous work [11]. Briefly, the lentivirus expression plasmids and packaging plasmids (pHelper 1.0 and pHelper 2.0) were cotransfected into 293T cells, supernatants were harvested 48 h after transfection and filtered through a 0.45-μm pore size filter (Millipore, Billerica, MA, USA) and concentrated by ultracentrifugation. The infectious titer was determined using hole-by-dilution titer assay. SMMC7721 and MHCC97H cells were infected with AP-2β-Flag-lentivirus or negative control (NC)-Flag-lentivirus at the multiplicity of infection (MOI) of 5-10 with 5 μg/mL of polybrene (Sigma) and detected on the 4th day by an invert fluorescence microscope (Zeiss Axioskop 2, LLC, US) followed by the screening of 1.5 μg/mL of puromycin for stable cell lines.
Proliferation assays
For MTT assay, 5,000 HCC cells per well were plated in 48-well plates untreated or treated with 5-30 μM of cisplatin (DDP) or 0.9% NaCl (NS) for 24 h. On day 1 to 5, cells were analyzed with 1 mg/mL 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetra zolium bromide (MTT, Sigma) at 37 °C for 4 h. Then 100 μL dimethyl sulfoxide (DMSO) per well was added to dissolve the formazan crystals. The absorbency at 490 nm was measured with a spectrophotometer (UV-2102C, Changsha, China). Liquid colony formation was performed as described previously [17]. Briefly, 1,000 HCC cells were seeded in triplicate in 6-well plates and grown for over 12 days. Colonies were fixed with methanol, stained with Giemsa (BBI International, Cardiff, UK) and photographed with the digital camera (Olympus). Only colonies containing over 30 cells were counted. All experiments were carried out for at least three times.
Migration and invasion assays
For wound-healing assay, HCC cells were cultured in 6-well plates until over 95% confluence. A 100-μL pipette tip was used to generate wounds. After wound creation, the medium was changed to remove cellular debris. Three wounded areas in each well were photographed at 1 and 3 days with the invert microscope (Zeiss Axioskop 2). The transwell cell migration and invasion assays were performed in polyethylene terephthalate (PET)-based migration chambers and BD BioCoat Matrigel Invasion Chambers (BD Biosciences, Bedford, MA) with 8 μm porosity as described previously [18]. HCC cells (2×104) in serum-free DMEM/F12 were seeded onto uncoated or Matrigel-coated filters in the upper chambers. DMEM/F12 containing 15% FBS was added to the lower chambers. After 48 h of incubation, cells on the upper surface of the filters were removed with a cotton swab, and the filters were fixed with 100% methanol and stained with Giemsa. The migration and invasive ability of HCC cells was calculated as the mean number of cells in all fields and expressed as the relative ratio compared with control cells. The experiments were carried out three times independently.
In vivo functional assays
The mouse experiments were performed according to the ethical guidelines for laboratory animal use and approved by the Ethics Committee of Hunan Normal University. The in vivo tumorigenic ability was performed in a xeno-graft mouse model. Approximately 1×107 of lentivirus-infected HCC cells in 0.2 mL of sterile PBS were injected subcutaneously into the left and right dorsal regions of 5-week-old female nude mice (n=6 mice/group), respectively. Mice were checked every 2 days; the formed tumors were measured as described previously [19] and analyzed by IHC. After 45 days, mice were sacrificed and tumors were excised, weighed and photographed. In vivo metastatic ability was estimated by tail vein injection, two groups of 6 mice each were given intravenous injections of 2.5×106 HCC cells, respectively. After 6 weeks the mice were sacrificed and the tumor nodules formed on the lung and liver surfaces were counted. Lungs and livers were excised and embedded in paraffin for conventional Hematoxylin and Eosin (HE) staining.
Quantitative real-time PCR
Total RNA from HCC cell lines was extracted using TRIzol reagent (Invitrogen, Carlsbad, CA), and reverse transcription was performed using a first strand cDNA synthesis kit (Roche Diagnostics, IN) according to the manufacturer's instructions. Real-time PCR analysis was performed using SYBR Green kit (Takara Bio Inc., Shiga, Japan) on 7900HT Fast Real-time PCR system (Applied biosystems, Weiterstadt, Germany). The reactions were incubated in a 384-well plate at 95 °C for 10 min followed by 40 cycles of 95 °C for 15 s and 60 °C for 30 s. The designed primers were listed in Table S1. The Ct value was measured during the exponential amplification phase. The relative expression levels of target genes were given by 2-∆∆Ct and log2 values were indicated as the relative difference compared to the controls.
Luciferase reporter assays
HEK293 cells were cultured in 12-well plates and transfected with reporter plasmids and pCMV-Myc-AP-2β plasmid or miR-27a mimics using Lipofectamine 2000 as described previously [20, 21].
Immunofluorescence analysis
HCC cells were cultured on glass coverslips in a 12-well plate and grown to 70% confluence. After 24 h, cells were treated as described previously [15]. The primary antibodies used were rabbit polyclonal anti-Slug and Snail antibodies (AbClonal Technology, USA) while the secondary antibodies were Alexa 488 goat anti-Rabbit antibodies (Molecular Probes, USA). The nucleus was stained with Hoechst 33258 (Sigma). The fluorescence signals were collected using an upright fluorescence microscope (Zeiss Axioskop 2).
Western blotting
HCC cells were lysed in RIPA buffer as described previously [11, 15]. Western blotting was performed according to the standard protocol. Information of antibodies was followed, rabbit polyclonal antibodies against MMP9 and AP-2β, mouse monoclonal antibodies against cyclin D1 (CCND1), Vimentin, β-catenin, α-catenin, Flag, β-actin and GAPDH were from Santa Cruz Biotechnology. Mouse monoclonal antibodies against AKT and phosphorylated AKT473, ERK, p-ERK, p-GSK-3β were from Cell Signaling Technology. Rabbit polyclonal antibodies against Ki67, Slug and Snail were from AbClonal Technology. Rabbit polyclonal antibodies against E-cadherin and N-cadherin were from Sangon Biotech (Shanghai, China). HRP-conjugated goat anti-rabbit and goat anti-mouse secondary antibodies were from Sigma.
Hsa-miR-27a overexpression lentivirus generation
The sequence of miR-27a pre-miRNA was cloned to pLenti-GFP lentiviral vector pEZX-MR03, recombinant and control vectors were then transfected into HEK293T cells with the Lenti-Pac HIV Packaging Mix (GeneCopoeia), and viral supernatants were collected 48 h after transfection. The lentivirus particles were purified and stored at -80 °C. After 96 h infection, stable miR-27a-infected HCC cells were screened at 1 μg/mL of puromycin.
Statistical analysis
Statistical analyses were performed using the SPSS 16.0 (SPSS Inc., Chicago, IL, USA), GraphPad software (SanDiego, California, USA) and SigmaPlot 12.5 (Systat Software Inc., San Jose, CA, USA). The Pearson's χ2 test was used to analyze the association of AP-2β expression with clinicopathologic characteristics. The expression levels of AP-2β or target genes in HCC tumor tissues and adjacent normal tissues were compared using a paired Student's t-test. Differences between gene expression were assessed by Fisher׳s exact test. Survival analyses were assessed by Kaplan-Meier plotter. Data are shown as mean ± SD from at least 3 independent experiments. Results were considered statistically significant when P < 0.05.
Results
Downregulation of AP-2β in HCC tissues and cell lines
To determine the clinical significance of AP-2β in HCC, the expression levels of AP-2β in 10 normal and 70 HCC liver tissue samples were examined by IHC staining. AP-2β was highly expressed in 70% of normal liver tissues. Strong AP-2β staining (2.5/3+) was detected in 7 (10%) of the 70 HCC tissue samples, while moderate staining (1.5/2+) was detected in 26 (37%) HCC tissue samples, and weak or negative AP-2β staining (0~1+) was detected in 37 (53%) HCC samples (Figure 1A-B). Low AP-2β expression was observed in low-grade hepatocellular cancers (I and II) and especially in high grade HCCs (III and IV) compared with that in normal liver tissues (P<0.001, paired Student's t-test; Figure 1B). Clinicopathological association analyses of the 70 HCCs revealed that AP-2β expression was significantly associated with advanced clinical stage and HCC tumor size (Pearson's χ2 test, P=0.004; Table 1). Moreover, AP-2β expression was negatively correlated with tumor grade (Pearson's correlation coefficient, -0.415, P=0.01; Figure 1C). Therefore, AP-2β expression was significantly lower in high-grade HCC tissues than in normal liver tissues.
The expression levels of AP-2β in hepatocellular cancer tissues and cell lines. (A) AP-2β expression was examined by immunohistochemical analysis in 70 hepatocellular tumors and 10 adjacent normal tissues. Strong nucleic expression of AP-2β (brown staining) was detected in normal tissues and stage I/II hepatocellular cancer cells, the nucleus was stained blue with hematoxylin. (B) Immunohistochemical score of AP-2β expression in hepatocellular cancers and normal tissues. The staining intensity was scored with grades 0-3. Each symbol represents an individual sample. Statistical comparisons between hepatocellular cancers and normal tissues were performed using SPSS and GraphPad software. **, p<0.01. (C) The correlation of AP-2β expression and tumor grade was analyzed by SigmaPlot. (D) AP-2β protein levels in different hepatocellular cancer cell lines were detected by Western blotting. β-actin was used as an internal control.
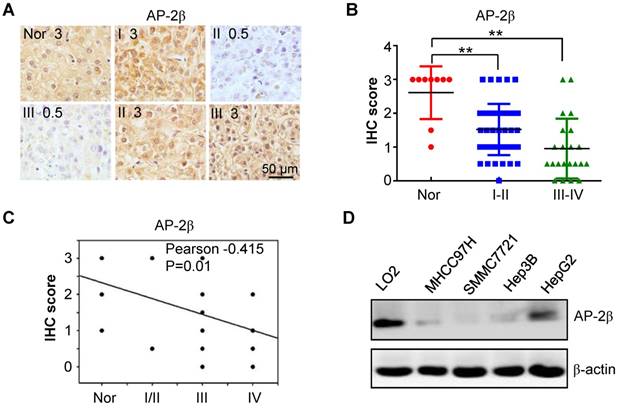
We next analyzed the expression of AP-2β proteins in human HCC lines. Low expression or the loss of AP-2β proteins was detected in human hepatoma cell lines MHCC97H, SMMC7721, Hep3B and HepG2, while high AP-2β expression was found in the normal human hepatic cell line LO2 (Figure 1D). Thus, low metastatic HCC cell line SMMC7721 and high metastatic HCC cell line MHCC97H were further selected to overexpress AP-2β proteins, not non-metastatic cell lines Hep3B and HepG2.
AP-2β overexpression inhibits HCC cell proliferation and tumorigenicity
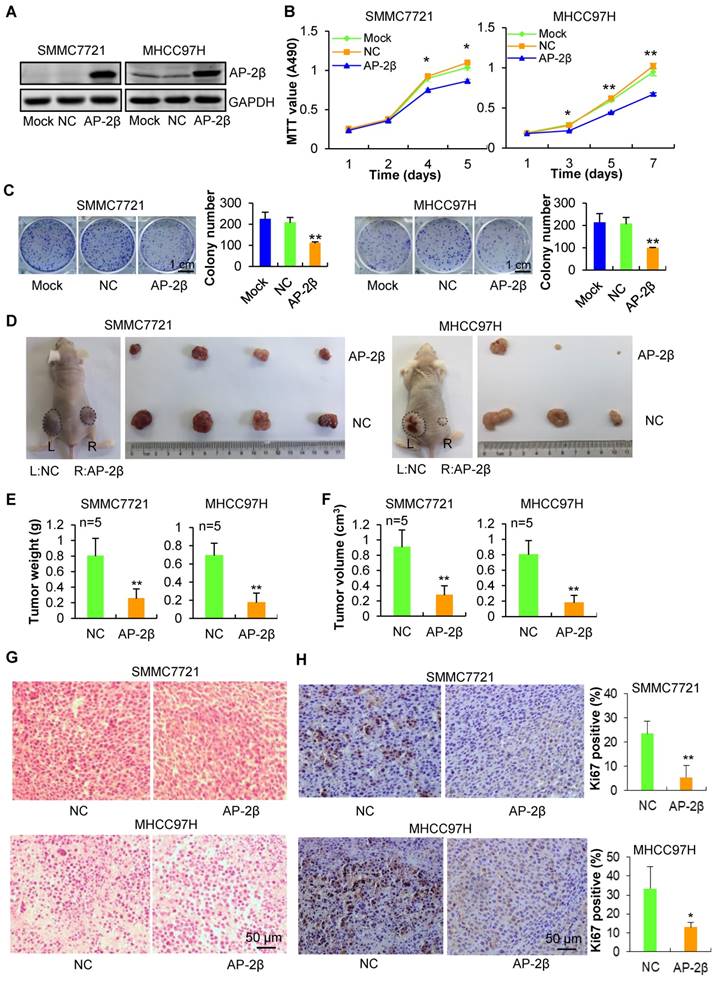
To explore the role of AP-2β in human HCC, AP-2β was cloned into the lentiviral vector pGC-FU-3Flag-IRES-Puromycin, and stable HCC cell lines with AP-2β overexpressed or not (pFLAG-AP-2β and pFLAG-NC) were screened and established. The overexpression of AP-2β decreased the viability, colony number and size of HCC cells (Figure 2A-C), revealing the strong anti-tumorigenic ability of AP-2β. To further examine the effect of AP-2β on the in vivo tumorigenicity of HCC cells, AP-2β LV-infected and control NC-infected HCC cells were subcutaneously injected into the left and right dorsal flanks of nude mice, respectively. Within 45 days, the body weight of mice showed no obvious change (data not shown), but the average tumor weight and tumor volume of the AP-2β overexpressed group were markedly reduced when compared with the controls (Figure 2D-F). H&E staining confirmed that cells are more loosely arranged in the AP-2β group (Figure 2G). Ki67 IHC staining further confirmed that AP-2β overexpression inhibits proliferation marker Ki67 expression in HCC (Figure 2H). These results indicated that AP-2β overexpression markedly inhibits the tumorigenic ability of HCC cells.
Effects of AP-2β overexpression on HCC proliferation and growth. (A) Western blots of NC/Flag and AP-2β/Flag expression in HCC cells using anti-Flag antibodies. GAPDH was served as a loading control. (B) MTT assays of mock or infected HCC cells. 3,000 cells were plated in 96-well plates and grown in DMEM with 10% FBS. The absorbance was analyzed for 1-7 days. (C) Liquid colony formation analysis of mock or infected HCC cells. 1,000 cells were seeded in 6-well plates, and grown for over 10 days. Colonies were fixed with methanol, stained with Giemsa, photographed and counted. (D) AP-2β overexpression results in decreased cell proliferation of HCC cells in vivo. About 1×107 of lentivirus-infected cells were injected s.c. into the left and right back of female nude mice (BALB/c) (n=6 per group). After 45 days, tumors were excised, photographed, and weighed (E-F). The weight and volume of the tumors excised are mean ± SD in three independent experiments. (G) H&E staining was performed on serial sections of mouse tumors from HCC cells. (H) IHC analysis of Ki67 expression and qualification of Ki67-positive cells in mouse tumors. These data represent at least three independent experiments with similar results. Statistical analysis was performed using SPSS software. *, p<0.05, **, p<0.01, compared with controls.
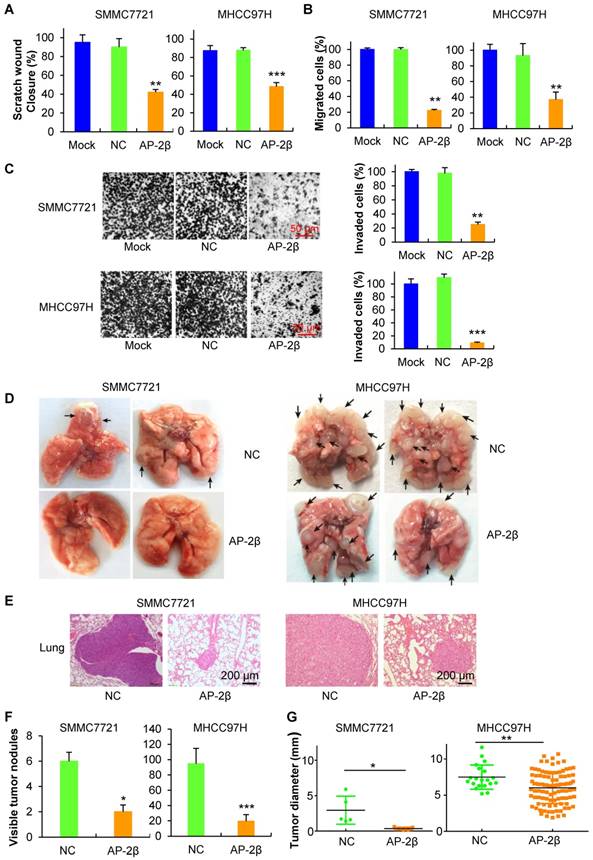
AP-2β suppresses HCC cell migration and metastasis
To further investigate the effects of AP-2β on HCC cell invasion and metastasis, the in vitro invasion and in vivo metastasis assays were performed. Wound-healing assays showed that AP-2β overexpression inhibits cell migration approximately 50% at the edge of exposed regions in HCC cells (Figure 3A). Transwell migration assays indicated that overexpression of AP-2β leads to a marked decrease in cell motility (Figure 3B). Moreover, Matrigel invasion assays revealed that AP-2β overexpressed cells exhibit a significantly slower rate of cell invasion than control cells (Figure 3C). These results suggested that AP-2β decreases cell invasion, which was further demonstrated in vivo. The pFLAG-AP-2β cells and control cells were injected into 6-week-old nude mice via tail-vein injection to mimic cell metastasis through circulation. Six weeks after injection, the metastatic nodules that formed on the surface of the lungs were counted (Figure 3D). The metastatic lesions on the surfaces of the mouse lungs were demonstrated by H&E staining (Figure 3E). The mice in the control group had more and larger lung metastatic nodules, whereas fewer and smaller metastatic nodules were observed in mice injected with pFLAG-AP-2β cells (P<0.05, Student's t-test; Figure 3F-G). The liver cells had no obvious morphological changes (data not shown). Taken together, these findings demonstrated that AP-2β inhibits HCC cell invasion and metastasis in vitro and in vivo.
AP-2β enhances HCC cell migration and invasion. (A) Wound-healing assays were performed at indicated time following initiation of the scratch in lentivirus-infected and parental HCC cells. These areas were calculated by ImageJ software. (B) Effect of AP-2β overexpression on cell migration by transwell assays. The relative proportion of migrated cells through the PET membrane (pore size: 8 μm) is shown. (C) Effect of AP-2β overexpression on cell invasion through Matrigel. Examples of cells migrated through Matrigel-coated transwell are shown in the left panel. Relative invasion proportion of cells is shown in the right panel. (D) Pictures of metastatic lung nodules in nude mice by tail-vein injection of HCC cells. The arrows indicate the metastatic tumor on the surface of the lung. (E) H&E staining was performed on serial sections of the lung issue. The number (F) and the diameter (G) of nodules were quantified on lungs of nude mice (n=6 per group) 6 weeks after tail-vein injection of HCC cells. Statistical analysis was performed using SPSS and GraphPad software. *, p<0.05, **, p<0.01.
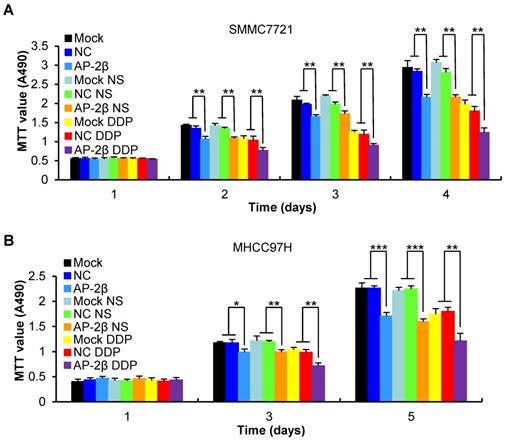
AP-2β increases the sensitivity of HCCs to cisplatin
Because of a tendency of metastasis and recurrence of HCC, post-operation chemotherapy is necessary; but, drug resistance is easily induced in HCC [22]. We investigated the influence of AP-2β on the chemosensitivity of HCC cells. A 35% reduction of MTT absorbance was detected in AP-2β overexpressed cells compared with a 23% inhibition detected in control cells treated with DDP for 24 h (Figure 4A-B). These results showed that AP-2β could sensitize HCC cells to the cytotoxicity of cisplatin, consistent with the findings in endometrial cancer cells [23].
AP-2β increases the sensitivity of hepatocellular cancer cells to cisplatin. (A-B) Lentivirus-infected and parental HCC cells (SMMC7721 and MHCC97H) were treated with 5-30 μM of cisplatin or control agent, 0.9% NaCl (NS), for 24 h, and cell viability was measured by MTT assay. All results indicate the mean ± SD of triplicate experiments. Statistical analysis was performed using SPSS software. **, p< 0.01, ***, p<0.001.
AP-2β inhibits HCC epithelial-mesenchymal transition by binding to the 5′ regulatory regions of Slug and Snail
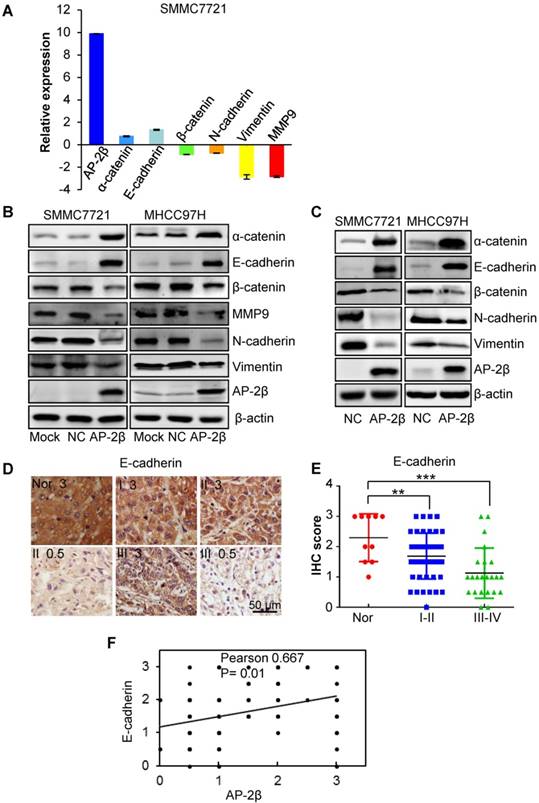
Since EMT is a key process in tumor invasion and metastasis, we next analyzed the effect of AP-2β on EMT by detecting the expression levels of EMT markers. The results of qRT-PCR showed the increased expression of epithelial markers (E-cadherin and α-catenin) with decreased expression of mesenchymal markers (Vimentin and N-cadherin) and MMP9 in AP-2β overexpressed SMMC7721 cells (Figure 5A). Western blotting further demonstrated that AP-2β increases the protein levels of the epithelial markers and decreases the protein levels of mesenchymal markers in HCC cell lines (Figure 5B) and mouse tumor tissues (Figure 5C), respectively. The correlation between AP-2β and E-cadherin was further analyzed by IHC staining in HCC samples. As reported, E-cadherin expression was significantly decreased in hepatocellular cancer tissues when comparing to that in normal liver tissues (paired Student's t-test; Figure 5D-E and Table S2) [24]. The expression of E-cadherin was positively correlated with AP-2β expression in HCC samples (Pearson correlation coefficient, 0.667, P=0.01; Figure 5F), and low levels of E-cadherin were primarily observed in HCCs with low AP-2β expression (Fisher's exact test, P<0.05; Table S3).
The effect of AP-2β on EMT genes. (A) qRT-PCR analysis of the influence of AP-2β on the expression of EMT markers. (B-C) Western blots were used to detect the expression of EMT markers in AP-2β-overexpressed HCC cells and mouse tumor tissues. (D) Immunohistochemistry analysis of E-cadherin expression was performed on the same hepatocellular tumors and normal tissues used for AP-2β detection. (E) Statistical analysis of the staining intensity of E-cadherin between hepatocellular cancers and normal tissues using GraphPad software. (F) The correlation of AP-2β and E-cadherin expression was analyzed using SigmaPlot. **, p<0.01, ***, p<0.001.
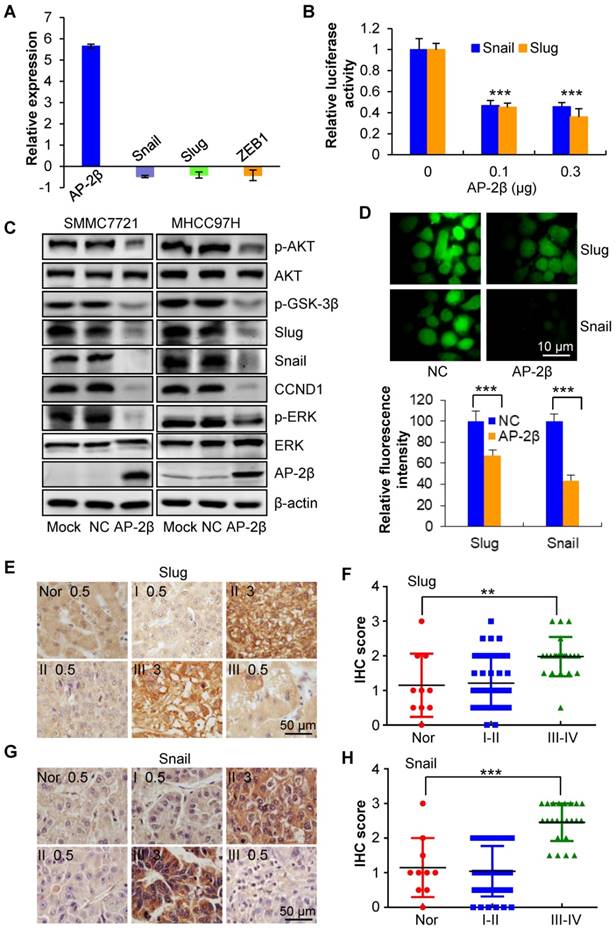
To investigate the regulatory relationship between AP-2β and EMT genes, three EMT-related transcription factors (Snail, Slug and ZEB1) were detected by qRT-PCR, and the results showed that AP-2β decreases the mRNA levels of these genes (Figure 6A). We next used the JASPAR database to search for potential AP-2 binding sites in the regulatory regions of these genes. Two AP-2 consensus binding sites (Slug, -783~-775 bp GCCCCTGGC and Snail -501~-493 GCCCGAGGC) were identified within the upstream regions of Slug and Snail. We first examined the effect of AP-2β on Slug and Snail transcription activities. The luciferase activities of pGL3-Slug (-943/+253) and pGL3-Snail (-1073/ +260) reporter plasmids were significantly decreased in AP-2β-overexpressed cells compared with control cells (Figure 6B). Furthermore, the expression of Slug and Snail was downregulated by AP-2β overexpression in HCC cell lines. Intriguingly, upstream phosphorylated AKT and GSK-3β as well as p-ERK and CCND1 were downregulated in AP-2β-overexpressed cells (Figure 6C). The fluorescence intensity of Slug and Snail was significantly decreased by AP-2β in MHCC97H cells (Figure 6D). Consistent with these findings, the correlation between AP-2β and Slug/Snail was further analyzed in HCC samples by IHC staining (paired Student's t-test; Figure 6E-H, Table S4, and Table S6). High levels of Slug were primarily observed in HCCs with low AP-2β expression, but this trend did not reach statistical significance (Fisher's exact test, P>0.05; Table S5). A significant inverse correlation between the expression of Snail and AP-2β was detected in HCC samples (Fisher's exact test, P<0.05; Table S7). Importantly, the negative correlation between AP-2β and Slug/Snail showed statistical significance (Fisher's exact test, P<0.05, Table 2). Therefore, AP-2β efficiently inhibits Slug and Snail gene expression.
The effect of AP-2β on EMT regulators. (A) Q-PCR analysis was used to detect EMT regulator expression in AP-2β-overexpressed SMMC7721 cells. (B) Luciferase assays were performed to detect the effects of AP-2β on Slug and Snail transcriptional activity. Western blotting (C) and immunofluorescent staining (D) confirmed the expression change of EMT regulators in AP-2β-overexpressed HCCs. (E) Immunohistochemistry analysis of Slug expression was performed on the same hepatocellular tumors and normal tissues used for detection of AP-2β. (F) Statistical analysis of the staining intensity of Slug between hepatocellular cancer and normal tissue samples using GraphPad software. (G) Immunohistochemistry analysis of Snail expression was performed on the same hepatocellular tumor and normal tissue samples used for detection of AP-2β. (H) Statistical analysis of the staining intensity of Snail between hepatocellular cancers and normal tissues using GraphPad software. **, p<0.01, ***, p<0.001.
Correlation between AP-2β expression and Slug/Snail expression in HCC samples by IHC analysis.
| AP-2β expression | Cases | Snail/Slug expression | p value* | |
|---|---|---|---|---|
| Low, No (%) | High, No (%) | |||
| Low (≤30%) | 43 | 12(27.9%) | 23(53.5%) | 0.008 |
| High (>30%) | 27 | 9(33.3%) | 7(25.9%) | |
| Total | 70 | 21(30%) | 30(42.9%) | |
All 70 samples were divided according to the proportion score to define AP-2β or Slug/Snail expression with low or high staining in HCCs.
*Fisher′s exact test.
MiR-27a targets the 3′ UTR of AP-2β, inhibits AP-2β expression, subsequently decreases the inhibitory effects of AP-2β in HCC
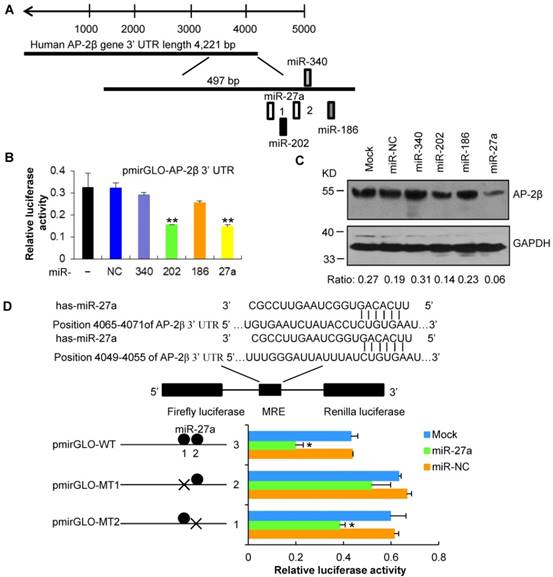
To identify miRNAs that regulate AP-2β, we used four computational programs (TargetScan, RNA22, miRWalk and miRanda) to search potential miRNAs binding to the 3′ UTR of AP-2β gene and identified the conserved miR-27a (Figure 7A). The wild-type or mutated 3′ UTR of AP-2β was co-transfected with miR-27a mimics into HEK293 cells. As shown in Figure 7B, the overexpression of miR-27a suppressed the firefly luciferase activities of the 3′ UTR of AP-2β. Furthermore, the miR-27a repressed AP-2β protein expression (Figure 7C). However, the luciferase activity was restored by using MRE-mutated MT1 of AP-2β, but not MT2 (Figure 7D), suggesting that the predicted MT1 mediates the binding of the miR-27a to AP-2β. These data indicated that miR-27a binds to the 3′ UTR of AP-2β and negatively regulates AP-2β expression.
miR-27a targets AP-2β. (A) Putative binding sites of miR-27a in the AP-2β 3′ UTR region as analyzed by TargetScan. (B) miR-27a regulates the luciferase activity of the AP-2β 3′ UTR. The firefly luciferase reporter activity was significantly reduced when pmirGLO-AP- 2β was cotransfected with miR-27a mimics. Firefly luciferase activity was normalized based on Renilla luciferase activity. (C) Western blot analysis of AP-2β expression in miR-27a-transfected HEK293 cell lines. The cells were collected at 48 h after miR-27a transfection. (D) The predicted wild-type or MRE-mutated AP-2β 3′ UTRs were cotransfected with miR-27a mimics. The luciferase activity was analyzed as described above. Statistical analysis was performed using SPSS software. *, p<0.05, **, p<0.01.
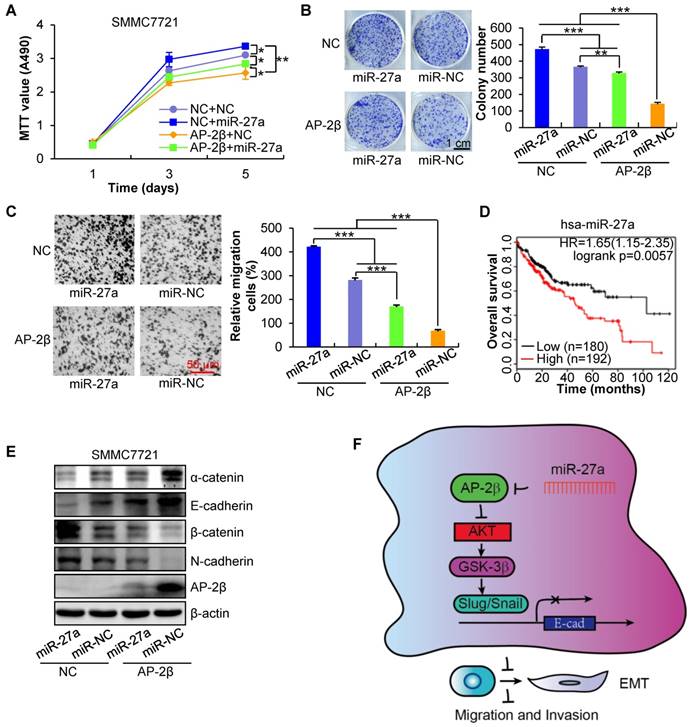
To verify the crucial role of miR-27a in AP-2β-regulated HCC cell proliferation and invasion, AP-2β-overexpressed HCC cells were infected with miR-27a LV. The results showed that miR-27a could dramatically rescue the inhibited cell proliferation (Figure 8A-B) and migration (Figure 8C) of AP-2β-overexpressed HCCs. Additionally, miR-27a is negatively associated with the overall survival of patients from the Cancer Genome Atlas (TCGA) data (Figure 8D) [25]. Moreover, miR-27a also reversed the EMT transition by downregulating epithelial marker expression and upregulating mesenchymal marker expression (Figure 8E). These results showed that oncogenic miR-27a reverses the inhibitory effects of AP-2β in HCC.
miR-27a reverses the effects of AP-2β in SMMC7721 cells. (A) MTT assays of miR-27a-infected AP-2β-overexpressing HCC cells. (B) Liquid colony formation analysis of miR-27a-infected AP-2β-overexpressing HCC cells. (C) Effect of AP-2β overexpression on miR-27a-infected HCC cell migration. Relative migration proportion of cells is shown in the right panel. (D) The correlation of miR-27a expression and overall survival in HCC samples was determined from TCGA data. (E) Western blotting was used to detect the effect of miR-27a on the expression of EMT markers in AP-2β-overexpressed HCCs. (F) Schematic presentation of the mechanism underlying AP-2β-suppressed HCC metastasis. Statistical analysis was performed using SPSS and GraphPad software. *, p<0.05, **, p<0.01, ***, p<0.001.
Discussion
Hepatocarcinogenesis is a multi-factor and multi-step complex process, associated with genetic alterations that involve dominant gain-of-function mutations, the amplification and/or overexpression of oncogenes as well as the recessive loss-of-function, deletion and/or epigenetic silencing of tumor suppressor genes [26-28]. AP-2β, first identified in embryogenesis [29], has been associated with tumor malignancy in lung cancer [5], and higher differentiation and a better clinical outcome in breast cancer, and neuroblastoma [9, 14]. In the present study, we observed that the expression level of AP-2β was decreased in HCC tissues, low AP-2β expression was detected in 53% of HCC samples, and the AP-2β expression was negatively associated with tumor size and tumor grade (p=0.004). The correlation between AP-2β and E-cadherin, Slug/Snail was further confirmed in HCC tissues and cell lines. Moreover, 55.8% of patients with decreased AP-2β expression displayed decreased E-cadherin levels, while 53.5% of patients with decreased AP-2β expression revealed enhanced Slug and Snail levels, and only 32.6% of patients with decreased AP-2β/E-cadherin expression showed enhanced Slug and Snail levels (p<0.05, Table 3). These results indicated that the combination of AP-2β with target gene expression signatures might provide novel molecular approaches for the diagnosis and clinical therapy of HCC.
Correlations among AP-2β, E-cadherin, Slug/Snail expression in HCC samples by IHC analysis.
| AP-2β expression | Cases | E-cadherin expression | p value* | |||
|---|---|---|---|---|---|---|
| Low (≤30%) | High (>30%) | |||||
| Low (≤30%) | 43 | 24(55.8%) | Snail/Sluglow 7(16.3%) | 19(44.2%) | Snail/Sluglow 5(11.6%) | 0.027 |
| Snail/Slughigh 14(32.6%) | Snail/Slughigh 9(20.9%) | |||||
| High (>30%) | 27 | 4(14.8%) | Snail/Sluglow 0(0%) | 23(85.2%) | Snail/Sluglow 9(33.3%) | |
| Snail/Slughigh 2(7.4%) | Snail/Slughigh 5(18.5%) | |||||
| Total | 70 | 28(40%) | 42(60%) | |||
All 70 samples were divided according to the proportion score to define AP-2β, E-cadherin or Slug/Snail expression with low or high staining in HCCs.
*ANOVA.
HCC invasion is a key step that leads to metastasis and poor prognosis [30]. Functional assays confirmed that AP-2β suppresses the cell proliferation, migration and invasion of hepatocellular cancer in vitro. These effects were efficiently reversed by oncogenic miR-27a molecules [31], which target the 3′ UTR of AP-2β. The role of AP-2β in inhibiting HCC cell migration and invasion was further identified in a mouse model of pulmonary metastasis. As expected, AP-2β inhibited cell motility in vivo. Moreover, AP-2β mediated the suppression of angiogenesis during corneal development [32]. As reported, HCC is one of the most chemoresistant cancers to systemic chemotherapy, with a response rate varying from 0% to 20% [33]. However, AP-2β overexpression significantly enhanced the cisplatin-induced death of HCC cells, suggesting that AP-2β has strong tumor inhibition ability and functions as an auxiliary enhancer of chemotherapeutic drugs.
EMT is well considered as a central mechanism for tumor invasion and metastasis including HCC [34-36]. The downregulation of E-cadherin is the key step of EMT. Several transcription factors that can repress E-cadherin expression, including Snail, Slug, ZEB-1/2, Twist and E47 proteins, have been identified [37]. Cancer-associated cascades, which include phosphoinositide 3-kinase (PI3K)/Akt-, Wnt-, Notch-, Hedgehog- or NF-kB-dependent pathways, have emerged as important regulatory signaling for EMT [38]. Based on the function of AP-2β in carcinogenesis, we proposed that AP-2β participates in HCC epithelial-to-mesenchymal transition and cancer metastasis. Here, we found that AP-2β overexpression exerts a significant effect on EMT by increasing the mRNA and protein levels of epithelial markers and decreasing the expression of mesenchymal markers. These results were further supported by Western blot analysis of cultured HCC cells and immunohistochemical staining of HCC tissue samples. AP-2β inhibited the metastatic potential of HCC cells and led to fewer and smaller tumor nodules in the lungs of nude mice. Moreover, AP-2β overexpression decreased the levels of phosphorylated Akt, which correlated with decreased levels of phosphorylated GSK-3β and the EMT regulators Snail and Slug as reported [39, 40]. However, the effects of AP-2β could be reversed by oncogenic miR-27a (Figure 8F). AP-2β was previously reported to decrease the phosphorylation of AKT and insulin receptor substrate 1 (IRS-1) in preadipocytes [41], but it remains unknown how AP-2β inhibits the versatile protein AKT in HCC. Additionally, AP-2β is involved in the cross-talk of other signaling pathways, which could decrease the levels of phosphorylated ERK and β-catenin as the same in cervical cancer [11], and CCND1 as downstream targets of ERK and Wnt signaling. Taken together, these data showed that AP-2β regulates HCC cell proliferation and migration, at least in part, by affecting EMT-related cell environment.
The relationship between obesity-induced fatty liver and HCC development has long been determined [42, 43], and some studies have showed that obesity increases cancer risk from elevated insulin and insulin-like growth factor 1 (IGF-1), sex steroids and cytokines in adipose tissues [44]. Obesity-promoted HCC development was dependent on the enhanced production of the tumor-promoting cytokines IL-6 and TNF [45]. Several studies have revealed that AP-2β enhances lipid accumulation and insulin resistance [41] by directly inhibiting the secretion of insulin [46] and the expression of adiponectin and leptin in adipocytes [47], enhancing the expression of inflammatory adipokines IL-6 and MCP-1 [48], and decreasing the tyrosine phosphorylation of IR and IRS-1 [49], which suggests the enhancing role of AP-2β in obesity and inflammation. AP-2β might enhance obesity and fat liver inflammation by IL-6 in obese patients. Thus, AKT inactivation is unlikely to enhance HCC development in obese persons, and there may be a negative feedback mechanism or epigenetic change for AP-2β to activate AKT and enhance HCC development. Similarly, rapamycin is a universal inhibitor of mTORC1-dependent S6K1 phosphorylation, but the strong negative feedback loop from S6K1 to AKT signaling could promote cell survival and chemoresistance [50]. Thus, it would be interesting to elucidate the interplay of AP-2β with inflammatory cytokines, immune cells and other critical proteins in high-fat diet (HFD)-induced obesity, fat liver and hepatocellular carcinogenesis, which will expand the current knowledge of AP-2β and reveal the molecular importance of AP-2β from obesity to HCC for the development of pharmaceutical agents.
Conclusions
AP-2β functions as a novel tumor suppressor to inhibit the proliferation, migration, and invasion of HCCs by decreasing the expression of EMT regulators and markers. AP-2β could increase the cytotoxicity of chemotherapeutic drugs in HCCs. Additionally, the downregulated AP-2β expression in high-grade HCC suggests it might serve as a marker for diagnosis and combined therapy. Therefore, understanding the underlying mechanisms of AP-2β in regulating HCC has new implications for the future intervention and inhibition of HCC metastasis.
Abbreviations
CCND1: Cyclin D1; DCIS: ductal carcinoma in situ; DDP: cisplatin; DMSO: dimethyl sulfoxide; EMT: epithelial-mesenchymal transition; HCC: hepatocellular cancer; H&E: hematoxylin and eosin; HFD: high-fat diet; IGF-1: insulin-like growth factor 1; IHC: immunohistochemistry; IRS-1: insulin receptor substrate 1; LV: lentivirus; MOI: multiply of infection; MRE: miRNA response element; MTT: 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; NC: negative control; NS: NaCl; PI3K: phosphoinositide 3-kinase; qRT-PCR: Quantitative real-time PCR; TCGA: the Cancer Genome Atlas; 3′ UTR: 3′ untranslated region.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 81770389 and 81272190), Distinguished Youth Foundation of Hunan Province (No. 2015JJ1011), State key laboratory of developmental biology of freshwater fish, Youth innovation and entrepreneurship platform of Hunan Province (No. 1), and the Cooperative Innovation Center of Engineering and New Products for Developmental Biology of Hunan Province (20134486).
Supplementary Material
Supplementary tables.
Competing Interests
The authors have declared that no competing interest exists.
References
1. El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132:2557-76
2. Thomas MB, Zhu AX. Hepatocellular carcinoma: the need for progress. J Clin Oncol. 2005;23:2892-9
3. Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF. et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378-90
4. Llovet JM, Hernandez-Gea V. Hepatocellular carcinoma: reasons for phase III failure and novel perspectives on trial design. Clin Cancer Res. 2014;20:2072-9
5. Deng WG, Jayachandran G, Wu G, Xu K, Roth JA, Ji L. Tumor-specific activation of human telomerase reverses transcriptase promoter activity by activating enhancer-binding protein-2beta in human lung cancer cells. J Biol Chem. 2007;282:26460-70
6. Fu L, Shi K, Wang J, Chen W, Shi D, Tian Y. et al. TFAP2B overexpression contributes to tumor growth and a poor prognosis of human lung adenocarcinoma through modulation of ERK and VEGF/PEDF signaling. Mol Cancer. 2014;13:89
7. Fischer U HD, Michel A, Janka M, Hulsebos T, Meese E. Cloning of a novel transcription factor-like gene amplified in human glioma including astrocytoma grade I. Hum Mol Genet. 1997;6:1817-22
8. Takahashi T, Matsuda Y, Yamashita S, Hattori N, Kushima R, Lee YC. et al. Estimation of the fraction of cancer cells in a tumor DNA sample using DNA methylation. PLoS One. 2013;8:e82302
9. Ikram F, Ackermann S, Kahlert Y, Volland R, Roels F, Engesser A. et al. Transcription factor activating protein 2 beta (TFAP2B) mediates noradrenergic neuronal differentiation in neuroblastoma. Mol Oncol. 2016;10:344-59
10. Beger M, Butz K, Denk C, Williams T, Hurst HC, Hoppe-Seyler F. Expression pattern of AP-2 transcription factors in cervical cancer cells and analysis of their influence on human papillomavirus oncogene transcription. J Mol Med (Berl). 2001;79:314-20
11. Wang F, Huang W, Hu X, Chen C, Li X, Qiu J. et al. Transcription factor AP-2beta suppresses cervical cancer cell proliferation by promoting the degradation of its interaction partner beta-catenin. Mol Carcinog. 2017;56:1909-23
12. Korkola JE, DeVries S, Fridlyand J, Hwang ES, Estep AL, Chen YY. et al. Differentiation of lobular versus ductal breast carcinomas by expression microarray analysis. Cancer Res. 2003;63:7167-75
13. Raap M, Gronewold M, Christgen H, Glage S, Bentires-Alj M, Koren S. et al. Lobular carcinoma in situ and invasive lobular breast cancer are characterized by enhanced expression of transcription factor AP-2beta. Lab Invest. 2018;98:117-29
14. Gee JM, Robertson JF, Ellis IO, Nicholson RI, Hurst HC. Immunohistochemical analysis reveals a tumour suppressor-like role for the transcription factor AP-2 in invasive breast cancer. J Pathol. 1999;189:514-20
15. Ding X, Fan C, Zhou J, Zhong Y, Liu R, Ren K. et al. GAS41 interacts with transcription factor AP-2beta and stimulates AP-2beta-mediated transactivation. Nucleic Acids Res. 2006;34:2570-8
16. Ding X, Yang Z, Zhou F, Wang F, Li X, Chen C. et al. Transcription factor AP-2alpha regulates acute myeloid leukemia cell proliferation by influencing Hoxa gene expression. Int J Biochem Cell Biol. 2013;45:1647-56
17. Ding X, Zhou F, Wang F, Yang Z, Zhou C, Zhou J. et al. Eps8 promotes cellular growth of human malignant gliomas. Oncology reports. 2013;29:697-703
18. Huang W, Chen C, Liang Z, Qiu J, Li X, Hu X. et al. AP-2alpha inhibits hepatocellular carcinoma cell growth and migration. Int J Oncol. 2016;48:1125-34
19. Chen C, Liang Z, Huang W, Li X, Zhou F, Hu X. et al. Eps8 regulates cellular proliferation and migration of breast cancer. Int J Oncol. 2015;46:205-14
20. Li X, Chen C, Wang F, Huang W, Liang Z, Xiao Y. et al. KCTD1 suppresses canonical Wnt signaling pathway by enhancing beta-catenin degradation. PLoS One. 2014;9:e94343
21. Ding X, Yang Z, Zhou F, Wang F, Li X, Chen C. et al. Transcription factor AP-2alpha regulates acute myeloid leukemia cell proliferation by influencing Hoxa gene expression. Int J Biochem Cell Biol. 2013;45:1647-56
22. Zhang YS, Yuan FJ, Jia GF, Zhang JF, Hu LY, Huang L. et al. CIK cells from patients with HCC possess strong cytotoxicity to multidrug-resistant cell line Bel-7402/R. World journal of gastroenterology. 2005;11:3339-45
23. Wu Y, Xiao Y, Ding X, Zhuo Y, Ren P, Zhou C. et al. A miR-200b/200c/429-binding site polymorphism in the 3' untranslated region of the AP-2alpha gene is associated with cisplatin resistance. PloS one. 2011;6:e29043
24. Endo K, Ueda T, Ueyama J, Ohta T, Terada T. Immunoreactive E-cadherin, alpha-catenin, beta-catenin, and gamma-catenin proteins in hepatocellular carcinoma: relationships with tumor grade, clinicopathologic parameters, and patients' survival. Human pathology. 2000;31:558-65
25. Li S, Li J, Fei BY, Shao D, Pan Y, Mo ZH. et al. MiR-27a promotes hepatocellular carcinoma cell proliferation through suppression of its target gene peroxisome proliferator-activated receptor gamma. Chin Med J (Engl). 2015;128:941-7
26. Tanaka S, Sugimachi K, Maehara S, Harimoto N, Shirabe K, Wands JR. et al. Oncogenic signal transduction and therapeutic strategy for hepatocellular carcinoma. Surgery. 2002;131:S142-7
27. Teufel A, Staib F, Kanzler S, Weinmann A, Schulze-Bergkamen H, Galle PR. Genetics of hepatocellular carcinoma. World journal of gastroenterology. 2007;13:2271-82
28. Martin J, Dufour JF. Tumor suppressor and hepatocellular carcinoma. World journal of gastroenterology. 2008;14:1720-33
29. Moser M, Pscherer A, Roth C, Becker J, Mucher G, Zerres K. et al. Enhanced apoptotic cell death of renal epithelial cells in mice lacking transcription factor AP-2beta. Genes Dev. 1997;11:1938-48
30. Kraljevic Pavelic S, Sedic M, Bosnjak H, Spaventi S, Pavelic K. Metastasis: new perspectives on an old problem. Mol Cancer. 2011;10:22
31. Chen Z, Ma T, Huang C, Zhang L, Lv X, Xu T. et al. MiR-27a modulates the MDR1/P-glycoprotein expression by inhibiting FZD7/beta-catenin pathway in hepatocellular carcinoma cells. Cell Signal. 2013;25:2693-701
32. Chen L, Martino V, Dombkowski A, Williams T, West-Mays J, Gage PJ. AP-2beta Is a Downstream Effector of PITX2 Required to Specify Endothelium and Establish Angiogenic Privilege During Corneal Development. Invest Ophthalmol Vis Sci. 2016;57:1072-81
33. Nerenstone SR, Ihde DC, Friedman MA. Clinical trials in primary hepatocellular carcinoma: current status and future directions. Cancer Treat Rev. 1988;15:1-31
34. Thiery JP. Epithelial-mesenchymal transitions in development and pathologies. Current opinion in cell biology. 2003;15:740-6
35. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nature reviews Molecular cell biology. 2014;15:178-96
36. Yang MH, Chen CL, Chau GY, Chiou SH, Su CW, Chou TY. et al. Comprehensive analysis of the independent effect of twist and snail in promoting metastasis of hepatocellular carcinoma. Hepatology. 2009;50:1464-74
37. Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer. 2007;7:415-28
38. Huber MA, Kraut N, Beug H. Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr Opin Cell Biol. 2005;17:548-58
39. Bachelder RE, Yoon SO, Franci C, de Herreros AG, Mercurio AM. Glycogen synthase kinase-3 is an endogenous inhibitor of Snail transcription: implications for the epithelial-mesenchymal transition. J Cell Biol. 2005;168:29-33
40. Zhou BP, Deng J, Xia W, Xu J, Li YM, Gunduz M. et al. Dual regulation of Snail by GSK-3beta-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat Cell Biol. 2004;6:931-40
41. Tao Y, Maegawa H, Ugi S, Ikeda K, Nagai Y, Egawa K. et al. The transcription factor AP-2beta causes cell enlargement and insulin resistance in 3T3-L1 adipocytes. Endocrinology. 2006;147:1685-96
42. Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S. et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature. 2013;499:97-101
43. Caldwell SH, Crespo DM, Kang HS, Al-Osaimi AM. Obesity and hepatocellular carcinoma. Gastroenterology. 2004;127:S97-103
44. Calle EE, Kaaks R. Overweight, obesity and cancer: epidemiological evidence and proposed mechanisms. Nat Rev Cancer. 2004;4:579-91
45. Park EJ, Lee JH, Yu GY, He G, Ali SR, Holzer RG. et al. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell. 2010;140:197-208
46. Tsukada S, Kobayashi MA, Omori S, Unoki H, Maeda S. Transcription factor AP-2beta inhibits glucose-induced insulin secretion in cultured insulin-secreting cell-line. Diabetes Res Clin Pract. 2009;85:279-85
47. Fuke T, Yoshizaki T, Kondo M, Morino K, Obata T, Ugi S. et al. Transcription factor AP-2beta inhibits expression and secretion of leptin, an insulin-sensitizing hormone, in 3T3-L1 adipocytes. Int J Obes (Lond). 2010;34:670-8
48. Ikeda K, Maegawa H, Ugi S, Tao Y, Nishio Y, Tsukada S. et al. Transcription factor activating enhancer-binding protein-2beta. A negative regulator of adiponectin gene expression. J Biol Chem. 2006;281:31245-53
49. Meng X, Kondo M, Morino K, Fuke T, Obata T, Yoshizaki T. et al. Transcription factor AP-2beta: a negative regulator of IRS-1 gene expression. Biochem Biophys Res Commun. 2010;392:526-32
50. Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12:9-22
Author contact
![]() Corresponding authors: Tel: 86 731 88872916; Fax: 86 731 88872792; E-mail address: fengxiaodingcom (Xiaofeng Ding) and xshlinedu.cn (Shuanglin Xiang)
Corresponding authors: Tel: 86 731 88872916; Fax: 86 731 88872792; E-mail address: fengxiaodingcom (Xiaofeng Ding) and xshlinedu.cn (Shuanglin Xiang)