Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results
Discussion
Conclusion
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(13):3693-3706. doi:10.7150/thno.24364 This issue Cite
Research Paper
Precise theranostic nanomedicines for inhibiting vulnerable atherosclerotic plaque progression through regulation of vascular smooth muscle cell phenotype switching
Sai Ma1,2,#, Seyed Mohammad Motevalli3,#, Jiangwei Chen2,#, Meng-Qi Xu1, Yabin Wang1, Jing Feng4, Ya Qiu1, Dong Han2, Miaomiao Fan2, Meiling Ding5, Li Fan6, Weisheng Guo3 ![]() , Xing-Jie Liang3
, Xing-Jie Liang3 ![]() , Feng Cao1
, Feng Cao1 ![]()
1. Department of Cardiology & National Clinical Research Center of Geriatrics Disease, Chinese PLA General Hospital, Beijing 100853, China.
2. Department of Cardiology, Xijing Hospital, Fourth Military Medical University, Xi'an 710032, China.
3. Laboratory of Controllable Nanopharmaceuticals, Chinese Academy of Sciences (CAS) Center for Excellence in Nanoscience and CAS Key Laboratory for Biomedical Effects of Nanomaterials and Nanosafety, National Center for Nanoscience and Technology of China, Beijing 100190, China.
4. Department of Emergency Medicine, Jinling Hospital, Nanjing 210000, China.
5. State Key Laboratory of Cancer Biology, National Clinical Research Center for Digestive Disease and Xijing Hospital of Digestive Diseases, Fourth Military Medical University, Xi'an 710032, China.
6. Department of Geriatric Cardiology & National Clinical Research Center of Geriatric Disease, Chinese PLA General Hospital, Beijing 100853, China
# These authors contributed equally to this work.
Received 2017-12-24; Accepted 2018-5-22; Published 2018-6-12
Abstract

Coronary heart disease is a prevalent and fatal killer caused by vulnerable atherosclerotic plaques (VASPs). However, the precise detection and treatment of VASPs remains a difficult challenge. Here, we present the development of noninvasive human serum albumin (HSA)-based theranostic nanomedicines (NMs) for the specific diagnosis and effective therapy of VASPs.
Methods: The ICG/SRT@HSA-pept NMs were formulated to contain payloads of the near-infrared (NIR) fluorescent dye indocyanine green (ICG) and the sirtuin 1 (Sirt1) activator SRT1720, and modified with a peptide moiety targeting osteopontin (OPN). The in vivo atherosclerotic mouse model was established with the high-fat diet (HFD). The in vitro vascular smooth muscle cells (VSMCs) phenotypic switching was induced using the ox-LDL stimulation.
Results: Due to the overexpression of OPN in activated VSMCs and VASPs, the targeted NMs specifically accumulated within the VASPs region after intravenous injection into the atherosclerotic mice, achieving the precise detection of VASPs. In addition, in the presence of SRT1720, the NMs could activate intracellular Sirt1 and activate an antiatherogenesis effect by inhibiting the phenotypic switching of VSMCs, which is an essential contributor to the vulnerability and progression of atherosclerotic plaques. After therapeutic administration of the ICG/SRT@HSA-pept NMs for two weeks, the physiological sizes and plaque compositions of VASPs were markedly improved. Furthermore, ICG/SRT@HSA-pept NMs-treated mice presented a more favorable plaque phenotype than that was observed in free SRT1720-treated mice, suggesting the enhanced delivery of pharmaceutical agents to the atherosclerotic lesions and improved therapeutic efficacy of NMs compared with free SRT1720.
Conclusions: The theranostic ICG/SRT@HSA-pept NMs showed great potential for the precise identification and targeted treatment of atherosclerotic diseases.
Keywords: atherosclerosis, theranostic nanomedicines, sirtuin 1, osteopontin, vascular smooth muscle cells
Introduction
Atherosclerosis (AS) is an essential cause of major cardiovascular events, accounting for 31% of total deaths worldwide [1]. AS is a silent and threatening killer, and the development of AS is a chronic process, characterized by the asymptomatic build-up of plaques over decades that can result in the sudden occurrence of fatal cardiovascular events [2, 3]. Annually, over 500,000 cardiac deaths occur without warning in the United States [4]. Therefore, it is of critical importance to detect AS during its dormant stage and to perform proactive therapy to prevent the occurrence of life-threatening events.
The rupture of vulnerable atherosclerosis plaques (VASPs) is the primary cause of coronary events and sudden deaths [5]. Currently, several medical imaging technologies have been employed in clinics to visualize the structural and anatomic features of atherosclerotic plaques, including magnetic resonance imaging (MRI), ultrasound (US), computed tomography (CT), and optical coherence tomography (OCT) [6, 7]. However, these techniques have notable limitations for characterizing the pathological features of plaque instability and, thus, fail to precisely differentiate VASPs from stable lesions. The precise identification of VASPs is very important for making decisions based on current therapeutic methodologies. The current clinical therapies for developing AS primarily rely on systemic medications, but these therapies suffer from systemic adverse effects and poor specific affinity to the AS lesions; invasive treatments are widely employed for advanced and late-stage AS stenosis, which might cause security risk issues with a large expense. Therefore, it is of great significance in the clinic to achieve a precise diagnosis and to develop targeted therapeutics for VASPs.
Nanoparticles (NPs) have received much attention in the field of AS detection and treatment by targeting the AS plaque and enhancing the therapeutic efficacy of anti-atherogenic drugs [8-11]. Over the last several decades, great efforts have been made to improve the ability of NPs to identify targets at the atherosclerotic lesions [12]. For this purpose, the surfaces of NPs could be decorated with the targeting ligands, including antibodies and peptides, to improve the sensitivity and specificity of AS NPs [13]. It was demonstrated that vascular smooth muscle cells (VSMCs) within VASPs will undergo phenotypic switching from a contractile phenotype to a synthetic phenotype, which remarkably facilitates the progression of AS and results in the overexpression of synthetic markers, such as osteopontin (OPN) [14, 15]. These findings make OPN a potential molecular target for the imaging of atherosclerotic lesions.
Inspired by the above issues and knowledge, we constructed a type of human serum albumin (HSA)-based theranostic nanomedicine (NM), aimed at achieving precise VASPs identification in a noninvasive manner, as well as delivering a mild yet effective therapy against AS that prevents the pathological development of AS. The theranostic NMs were denoted as ICG/SRT@HSA-pept NMs and were formulated from a peptide moiety targeting OPN and encapsulated payloads of the near-infrared (NIR) fluorescent dye indocyanine green (ICG) and the sirtuin 1 (Sirt1) activator SRT1720. Due to the overexpression of OPN in the VSMCs within VASPs, the NMs specifically accumulated within the VASPs region after intravenous injection into the high-fat diet (HFD)-induced atherosclerotic mouse model, achieving the precise recognition of VASPs. In addition, the NMs containing SIRT1720 could activate the intracellular Sirt1 and trigger an anti-atherosclerotic effect by inhibiting the phenotype transition of VSMCs. After systemic administration of the NMs for over two weeks, the sizes and compositions of VASPs were markedly improved, suggesting the efficient targeted therapeutic efficacy against VASPs. Taken together, the reported theranostic ICG/SRT@HSA-pept NMs showed great potential for the precise identification and targeted treatment of atherosclerotic diseases.
Methods
Synthesis and characterization of ICG/SRT@HSA-pept NMs
The fabrication of the fluorescein isothiocyanate (FITC)-labeled ICG/SRT@HSA-pept NMs began with the preparation of HSA-FITC and HSA-peptide conjugates. The HSA-FITC was prepared by following the protein labeling protocol of the amine-reactive FITC. Typically, 1 mL of fluorescein-5-isothiocyanate solution (1 mg/mL in DMSO) was added to 20 mL of HSA solution (2 mg/mL in 0.1 M sodium borate, pH 8.7), and the obtained mixture was maintained at room temperature for two hours under stirring. Then, the HSA-FITC conjugate was purified three times with an Amicon centrifugal device (30 kDa cut off) to remove the excess free FITC molecules. The purified HSA-FITC conjugate was finally dissolved into PBS (1X) at a fixed concentration and stored at 4 ℃.
The sequence of the targeting peptide used in the current study was Leu-Arg-Ser-Lys-Ser-Arg-Ser-Phe-Gln-Val-Ser-Asp-Glu-Gln-Tyr-Pro-Asp-Ala-Thr-Asp-Glu, which enabled the NMs to target the overexpressed OPN in synthetic VSMCs and atherosclerotic lesions. The HSA-peptide conjugate was synthesized in two steps. First, 50 mg of peptide was thiolated with Traut reagent (5.6 mg) in 5 mL of sodium borate (0.2 M, pH 8) at a molar ratio of 1:0.9 for 1 h at room temperature. In the meantime, 5 mL of PBS containing 40 mg of HSA was mixed with 0.4 mL of the Sulfo-SMCC solution (5 mg/mL in DMSO). After a 2 h reaction at room temperature, the excess Sulfo-SMCC was removed with a centrifugal filter device (MWCO = 30 kDa) to purify the SMCC-activated HSA. The SMCC-activated HSA and the thiolated peptide were mixed in PBS (1X, pH 7.4) at a molar ratio of 1:10. After an overnight reaction at 4 °C, excess OPN peptide was removed using the Amicon centrifugal device (MWCO = 30 kDa) and the obtained HSA-peptide was stored at 4 °C.
The ICG/SRT@HSA-pept NMs were prepared by ICG- and SRT1720-induced self-assembly of albumin proteins. Typically, 0.2 mL of HSA-peptide stock (10 mg/mL) and 0.2 mL of HSA-FITC stock (10 mg/mL) were mixed in water with a final fixed volume of 2 mL. Then, 20 μL of DMSO containing 0.3 mg of SRT1720 and 0.5 mg of ICG was added dropwise into the aqueous protein solution under vigorous stirring. Afterward, 100 μL of the glutaraldehyde aqueous solution (0.25%, v/v) was added to the mixture to increase the stability of the protein nanoparticle. After a 3 h reaction under stirring, the obtained ICG/SRT@HSA-pept NMs were purified three times with PBS (0.1 M, pH 7.4) using an Amicon centrifugal filter device (MWCO = 100 kDa) and dispersed into PBS at a fixed concentration for further use. The ICG/SRT@HSA NMs were fabricated following the same procedures as described above, except that the HSA-peptide conjugate was replaced with unlabeled HSA protein.
The size distributions of ICG/SRT@HSA-pept NMs and ICG/SRT@HSA NMs were determined using a Zetasizer Nano ZS (Malvern Instruments, Worcestershire, UK). The nanoparticles were stained with uranyl acetate (1% wt) before transmission electron microscopic (TEM) characterization using a JEM-200CX (JEOL, Tokyo, Japan). The absorption spectra and the fluorescence spectra of the NPs were recorded by a visible spectrophotometer (Lambda 950, PerkinElmer, USA) and a fluorescence spectrophotometer (HITACHI F-4600, Tokyo, Japan), respectively.
Cell culture and in vitro experimental protocols
A mouse vascular smooth muscle MOVAS cell line (VSMCs) was incubated with DMEM (Gibco, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (FBS, HyClone, Logan, Utah, USA) and 1% Penicillin-Streptomycin (HyClone, Logan, Utah, USA). The VSMCs were maintained in a 5% CO2 incubator at 37 °C. For in vitro AS simulation, VSMCs were treated with ox-LDL (Shanghai Leuven Biological Technology, Shanghai, China) at a concentration of 50 mg/mL for 24 h. For in vitro SRT1720 treatment, cells were treated with SRT1720 at a concentration of 10 μM for 4 h. For NMs-treated cells, cells were incubated with ICG/SRT@HSA-pept NMs or ICG/SRT@HSA NMs (at a dilution factor of 100) for 24 h. For the blocking group, OPN antibody was added 30 min prior to treatment with the NMs.
In vitro cytotoxicity assessment of NMs
The thiazolyl blue tetrazolium bromide (MTT) assay was used to evaluate the cytotoxic effects of synthesized nanoparticles on VSMCs. After the incubation of VSMCs with ox-LDL or NMs in 96-well plates (n=6), the media was removed. Then, 50 μL of serum-free media and 50 μL of MTT solution (ab211091, Abcam, Cambridge, MA) were added to each well. After incubation at 37 °C for 3 h, 150 μL of MTT solvent was added to each well. The absorbance at OD=590 nm was read at time points of 15 min and 1 h.
In vitro cellular imaging
VSMCs were seeded onto glass-bottom confocal dishes. For in vitro AS simulation, VSMCs were treated with ox-LDL at a concentration of 50 mg/mL for 24 h. Cells were then incubated with ICG/SRT@HSA-pept NMs or ICG/SRT@HSA NMs (dilution factor of 100) respectively for 24 h. For the blocking group, OPN antibody was added 30 min prior to adding the NMs. After treatments, the VSMCs were rinsed three times with PBS to remove the unbound probes and fixed with 4% paraformaldehyde for 20 min. Cell nuclei were counterstained with DAPI (5 μg/mL) for 5 min. Fluorescence imaging was performed with the Olympus FV1200 (Olympus, Tokyo, Japan).
In vivo study design
ApoE-/- mice (male, eight-week-old, 15-25 g), purchased from Animal Center of Fourth Military Medical University, were initially fed with a standard laboratory chow diet for one week. The mice fed with the standard laboratory diet served as negative controls (low-fat diet, LFD). Atherosclerosis (AS)-model mice were fed a high-fat diet (containing 15% fat and 0.25% cholesterol) for 12 weeks (high-fat diet, HFD). After the diet period, the mice received an intravenous injection of ICG/SRT@HSA-pept NMs, ICG/SRT@HSA NMs or PBS (100 μL/per mouse), through the tail vein (n=10). In vivo NIR fluorescence imaging (n=10) was performed before and 1 h, 4 h, 12 h and 24 h after the injection. The mice were sacrificed at 24 h and 48 h post-injection (p.i), and the aortas, liver, lung, heart, spleen and kidney were collected and analyzed with ex vivo NIR fluorescence imaging to analyze the bio-distribution of the NMs (n=6 at 24 h p.i. and n=4 at 48 h p.i.). Immunofluorescence analysis of aortic arteries was performed to confirm the accumulation of ICG/SRT@HSA-pept NMs in atherosclerotic plaques (n=6).
In the therapeutic study, the mice were divided into four groups (n=15): the LFD group (LFD), the HFD+ PBS group (Vehicle), the HFD+ ICG/SRT@HSA-pept NMs group (NMs), and the HFD+ free SRT1720 group (free SRT). AS-model mice fed a HFD were injected with ICG/SRT@HSA-pept NMs or an equivalent volume of PBS every other day for two weeks, starting at three months of age. For free SRT1720-treated mice, animals were treated with intraperitoneal injections of SRT1720 at a dose of 50 mg/kg/d every other day for two weeks, starting at three months of age, according to previous studies [16-18]. Four months after the beginning of the experiment, mice were sacrificed, and aortas were collected for further examination. In each group, several entire aortas (n=5) were excised for en face Oil Red O staining; for the remaining therapeutically treated mice (n=10), aortic root tissues were used for morphological staining, including Oil Red O, H&E, and Masson staining, and the remaining aortic tissues were used for Western blot analysis.
Mice were maintained in an animal facility with controlled temperature (22-24 °C) and 12-hour light/dark cycles. Animals had free access to food and water during the whole experimental process. All animal study procedures were performed in accordance with the Chinese National Institutes of Health. The experimental protocol was approved by the Fourth Military Medical University Committee on Animal Care (XJYYLL-2014251).
In vivo NIR fluorescence imaging
To evaluate the in vivo distribution of NMs, solutions of PBS, ICG/SRT@HSA-pept NMs or ICG/SRT@HSA NMs, at a dose of 100 μL, were intravenously injected into mice through the tail vein (n=10). At time points of 1 h, 4 h, 12 h and 24 h after injection, the mice were euthanized and examined using an IVIS Lumina XR System (Caliper Life Sciences, Hopkinton, USA) with the following parameters: exposure time 10 s, f/stop 2, binning 4, and field of view 12.8.
Ex vivo NIR fluorescence imaging
The mice were sacrificed at 24 h p.i (n=6) and 48 h p.i. (n=4) to collect aortas and major organs, including the liver, lung, heart, spleen and kidney. Organs were visualized ex vivo using an IVIS Lumina XR System (Caliper Life Sciences, Hopkinton, Massachusetts, USA) with the following parameters: exposure time 10 s, f/stop 2, binning 4, and field of view 12.8. For aortas, the arteries were washed with saline three times and visualized using an IVIS Lumina XR System. The ex vivo NIR signal intensities of whole carotid arteries were quantified using Living Image version 4.4 software (Caliper Life Sciences, Hopkinton, USA).
Immunofluorescence microscopy
To verify the location of NMs within plaques, the carotid arteries of mice were harvested 24 h after the injection of PBS, ICG/SRT@HSA-pept NMs or ICG/SRT@HSA NMs (n=6) in HFD mice. Slides of arteries containing plaques were fixed with 4% paraformaldehyde for 15 min at 4 °C. After washing with PBS three times, cells were treated with 1% Triton and blocked with 2% horse serum for 1 h at room temperature. Then, the slides were incubated with primary antibody (VSMCs marker SM α-actin, 1/100 dilution, Abcam, Cambridge, MA, USA) at 4 °C, overnight. After being washed with PBS three times, the samples were incubated with Rhodamine-conjugated secondary antibody for two hours at room temperature. After a final wash, the slides were visualized under the Olympus FV1200 system (Olympus, Tokyo, Japan).
Tissue collection and morphological staining
To examine the atherosclerotic model and therapeutic effects of free SRT1720 and ICG/SRT@HSA-pept NMs, the total carotid artery and descending aorta were removed from mice after euthanasia, followed by several morphological staining protocols, including Oil Red O, H&E, Sirius Red, Masson, and immunohistochemistry staining. Several arteries (n=4) were frozen in Optimum Cutting Temperature (OCT) compound. Frozen aortic sections (8 μm) were stained with Oil Red O for ten minutes, followed by counterstaining with hematoxylin for two minutes at room temperature. Several arteries (n=6) were fixed in a 4% paraformaldehyde solution, embedded in paraffin, and sectioned at 5 mm for H&E and Masson staining. The sections were stained with hematoxylin-eosin (H&E) or Masson trichrome staining, and images of the stained sections were visualized using a light microscope (Olympus, Japan). The sizes of the plaque areas were quantified by delineating plaque areas in 3-4 root H&E stained sections/per mouse, and the data are presented as the mean plaque area. The necrotic core area was defined as the area that was negative for H&E staining within each plaque. The plaque composition was evaluated by the combination of necrotic core area and collagen fraction (Masson staining), according to previous reports [19].
En face Oil Red O staining
En face Oil Red O staining was performed to evaluate the plaque area of lipid deposits. Briefly, the entire aortas of mice in all groups (n=5) were dissected, and the adventitial fat was removed carefully. After being opened longitudinally with the luminal surface toward the outside, aortas were stained with Oil Red O solution for 4 h and then differentiated with 60% propylene glycol. The stained aortas were then placed on a black pad and images were captured.
Quantitative real-time PCR
Total RNA was isolated from MOVAS cells or primary VSMCs using a NucleoSpin RNA II kit (Macherey-Nagel GmbH, Mannheim, Germany). The cDNA was synthesized with the QuantiTect reverse transcription kit (Qiagen, Hiden, Germany). Quantitative Real-time PCR was performed using the KAPA SYBR fast qPCR kit (KAPA Biosystems, Woburn, MA, USA). The primer sequences used were designed according to previous reports (sequences are shown in Table S1 and Table S2) [14] and synthesized commercially by TAKARA (TAKARA Biotechnology Co., Dalian, Liaoning, China). The amplification for genes was performed with the following settings: 94 °C for 30 s, followed by 40 cycles of the amplification step (94 °C for 30 s, 60 °C for 60 s and 72 °C for 1 min). Relative mRNA expression levels were calculated by the ΔΔCT method, with GAPDH as an internal standard, using 7500 System SDS Software Version 1.2.1.22 (Applied Biosystems, Thermo Fisher Scientific, Waltham, Massachusetts, USA).
Western blot assay
Tissue or cellular proteins were extracted using RIPA solution and separated with SDS-PAGE gels. Samples were then transferred to polyvinylidene difluoride membrane (Millipore, Burlington, Massachusetts, USA) and incubated overnight (4 °C) with primary antibodies against several targets, including OPN, smooth muscle α-actin (SM α-actin), smooth muscle-myosin heavy chain (SM-MHC), calponin, vimentin, Sirt1 and GAPDH (Cell Signaling Technology, MA, USA, 1:1,000 dilution). Membranes were then washed with TBST and incubated with corresponding secondary antibodies for one hour at 37 °C. Finally, the bands were visualized with a chemiluminescence detection kit (Thermo Electron Corp., Rockford, USA) and analyzed with the Imagelab software system (Bio-Rad, CA, USA).
Statistical analysis
All analyses were performed with SPSS 20.0 software (SPSS Inc., Chicago, IL, USA). Data are presented as the mean ± S.D. The normality of the data distribution was tested by using the Shapiro-Wilk test. The multi-group comparisons were made with a one-way ANOVA analysis, followed by Dunnett's post hoc test. Comparisons between two groups were made with Student's t tests, assuming a Gaussian distribution. Values of P < 0.05 were considered to be statistically significant differences.
Results
Elevated OPN expression within atherosclerotic lesions and synthetic-phenotype VSMCs
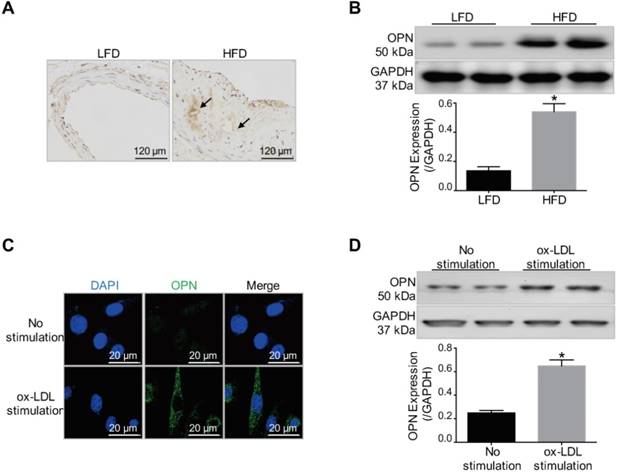
First, we investigated whether OPN is a potential target for atherosclerotic plaque imaging. As was revealed by immunohistochemistry staining and Western blot results (Figure 1A-B), OPN expression was markedly increased in AS plaques in HFD mice compared with control vessel tissue in LFD mice (P < 0.05 HFD group vs. LFD group). The expression of OPN in VSMCs during phenotype transition induced by ox-LDL stimulation was also examined. The expression of OPN was significantly increased in ox-LDL-treated VSMCs (P < 0.05 ox-LDL stimulation group vs. No stimulation group), as demonstrated by immunofluorescence staining and Western blot analysis (Figure 1C-D). These experimental observations revealed the elevated expression of OPN in activated VSMCs and atherosclerotic plaques, indicating OPN to be a promising molecule for targeted imaging.
Increased OPN expression in atherosclerotic plaques and ox-LDL-stimulated VSMCs. (A) Immunohistochemistry staining revealed increased OPN expression in atherosclerotic lesions in low-fat diet (LFD) mice compared with control vessels in high-fat diet (HFD) mice (arrows: OPN positive staining). (B) Western blot results indicated that OPN expression was increased within atherosclerotic plaques compared to control vessels (P < 0.05). *P < 0.05 vs. LFD group. (C) Immunofluorescence images showed that ox-LDL induced OPN expression in VSMCs. (D) Increased OPN expression in synthetic VSMCs stimulated by ox-LDL treatment (P < 0.05). *P < 0.05 vs. No stimulation group.
Sirt1 activator SRT1720 inhibits phenotypic switching in VSMCs
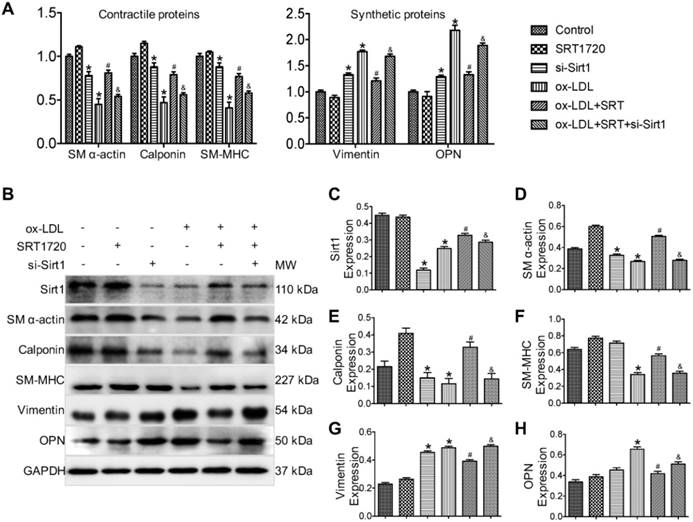
Next, the therapeutic efficacy of the Sirt1 activator SRT1720 against phenotypic switching in VSMCs was evaluated in vitro. As was revealed by real-time PCR results (Figure 2A), ox-LDL-stimulated VSMCs that represented the synthetic phenotype displayed decreased expression levels of contractile phenotype markers (SM α-actin, SM-MHC and Calponin) and increased expression levels of synthetic phenotype markers (Vimentin and OPN). SRT1720 treatment significantly attenuated the changes induced by ox-LDL (P < 0.05 ox-LDL group vs. ox-LDL+SRT1720 group). However, the effect of SRT1720 was diminished by siRNA targeting Sirt1, indicating that the function of SRT1720 was dependent on Sirt1 (P < 0.05 ox-LDL+SRT1720 group vs. ox-LDL+SRT1720+si-Sirt1 group). The beneficial effects of SRT1720 against phenotypic switching were further confirmed in primary VSMCs cells (Figure S1). In accordance with the real-time PCR results, Western blot analysis further confirmed the inhibitory effects of SRT1720 against ox-LDL induced phenotypic switching in VSMCs (Figure 2B-H). siRNA targeting Sirt1 decreased the expression levels of contractile phenotype markers (SM α-actin, SM-MHC and calponin) and increased the expression levels of synthetic phenotype markers (vimentin and OPN); however, notably, the differences are not significant for SM-MHC and OPN. Moreover, SRT1720 treatment significantly attenuated the changes induced by ox-LDL (P < 0.05 ox-LDL group vs. ox-LDL+SRT1720 group), which demonstrates that the effect was dependent on Sirt1 (P < 0.05 ox-LDL+SRT1720 group vs. ox-LDL+SRT1720+si-Sirt1 group).
Sirt1 activator SRT1720 inhibits VSMC phenotypical switching induced by ox-LDL stimulation. (A) Real-time PCR results demonstrated that ox-LDL-stimulated VSMCs, which represent the synthetic phenotype, displayed decreased expression levels of contractile phenotype markers (SM α-actin, SM-MHC and calponin) and increased expression levels of synthetic phenotype markers (vimentin and OPN). SRT1720 treatment significantly attenuated the changes induced by ox-LDL, and this effect was diminished by siRNA targeting Sirt1. (B-H) Western blot results further confirmed that SRT1720 treatment significantly attenuated the phenotype switching in VSMCs induced by ox-LDL, an effect that was dependent on Sirt1. *P < 0.05 vs. control group; #P < 0.05 vs. ox-LDL group; &P < 0.05 vs. ox-LDL+SRT1720 group.
Structure and characteristics of NMs
A schematic illustration of ICG/SRT@HSA-pept NMs (labeled as ICG/SRT@HSA-pept in the figures) and ICG/SRT@HSA NMs (labeled as ICG/SRT@HSA in the figures) is shown in Figure 3A. The theranostic ICG/SRT@HSA-pept NMs were formulated using a surface peptide moiety targeting OPN and encapsulated payloads of the NIR fluorescent dye ICG, the fluorescent dye FITC and the Sirt1 activator SRT1720. The control ICG/SRT@HSA NMs were not modified with the peptide targeting OPN. Figure S2 shows the colloidal stability of ICG/SRT@HSA-pept NMs, with no obvious precipitation after being maintained at room temperature for two weeks. In addition, the colloidal stability of the ICG/SRT@HSA-pept NMs in various media, including pure water, PBS (1X, pH 7.4), and aqueous FBS (10%), was monitored for 48 h. As shown in Figure 3B, the ICG/SRT@HSA-pept NMs showed negligible changes in the hydrodynamic diameters (HDs) during the incubation in pure water or PBS. When incubated in an FBS-containing solution, a slight increase in HDs was observed at the initial stage, which could be attributed to the absorbed protein corona at the NM surface. The dynamic laser scattering (DLS) test suggests a fairly good colloidal stability of the ICG/SRT@HSA-pept NMs, without aggregations.
Structure and characterization of NMs. (A) A schematic illustration of ICG/SRT@HSA-pept NMs (labeled as ICG/SRT@HSA-pept) and ICG/SRT@HSA NMs (labeled as ICG/SRT@HSA). (B) The colloidal stability of the ICG/SRT@HSA-pept NMs in various media, including pure water, PBS (1X, pH 7.4), and aqueous FBS (10%). (C) TEM images of spherical-shaped ICG/SRT@HSA-pept NMs and ICG/SRT@HSA NMs (Scale bar: 100 nm). (D) The ICG fluorescence signal intensity exhibited a positive correlation with the ICG/SRT@HSA-pept NMs concentration. (E-F) The fluorescence spectra and UV-Vis absorption spectra of ICG/SRT@HSA-pept NMs and ICG/SRT@HSA NMs. (G) The dynamic light scattering (DLS) results of ICG/SRT@HSA-pept NMs and ICG/SRT@HSA NMs. (H) The drug release curve of ICG/SRT@HSA-pept NMs in the physical media of PBS and FBS.
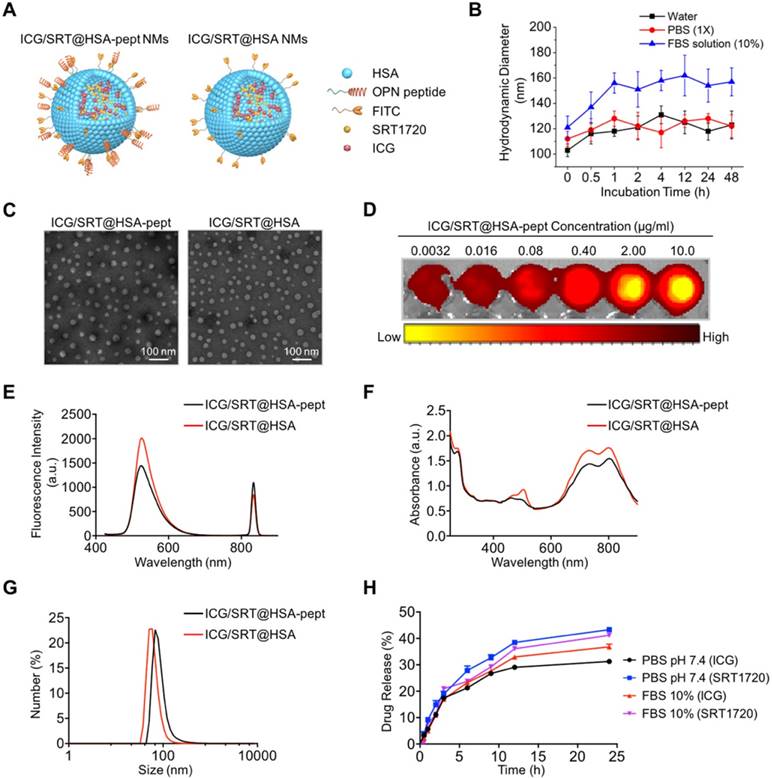
Figure 3C shows the TEM images of spherical-shaped ICG/SRT@HSA-pept NMs and ICG/SRT@HSA NMs (Table S2). The ICG fluorescence signal intensity showed a positive correlation with the ICG/SRT@HSA-pept NMs concentration (Figure 3D). Figures 3E-F reveal the fluorescence spectra and UV-Vis absorption spectra of ICG/SRT@HSA-pept NMs and ICG/SRT@HSA NMs, respectively. Figure 3G demonstrates the DLS results of ICG/SRT@HSA-pept NMs and ICG/SRT@HSA NMs. In addition, the drug release curve of ICG/SRT@HSA-pept NMs in the physical media of PBS and FBS is shown in Figure 3H.
Selectivity of ICG/SRT@HSA-pept NMs toward synthetic phenotype VSMCs
The cytotoxicity of synthesized NMs was evaluated using the MTT assay. After incubation with ICG/SRT@HSA-pept NMs or ICG/SRT@HSA NMs for 24 h, VSMCs did not show a marked decrease in viability (Figure S3A). The targeting properties of ICG/SRT@HSA-pept NMs were studied in vitro in ox-LDL-treated VSMCs. First, the cellular uptake of ICG/SRT@HSA-pept NMs was evaluated using immunofluorescence, which is shown in Figure 4A.
Selectivity of ICG/SRT@HSA-pept NMs toward synthetic phenotype VSMCs. (A) The ox-LDL-stimulated VSMCs, which represent the synthetic phenotype, displayed a strong FITC signal representing ICG/SRT@HSA-pept NMs in the cytoplasm. (B-H) After a 24 h incubation, ICG/SRT@HSA-pept NMs significantly elevated SIRT1 expression and inhibited VSMC phenotype switching markers in ox-LDL-treated VSMCs compared with the PBS and ICG/SRT@HSA NMs groups (*P < 0.05 ICG/SRT@HSA-pept group vs. PBS group; #P < 0.05, NMs+Block group vs. ICG/SRT@HSA-pept group).
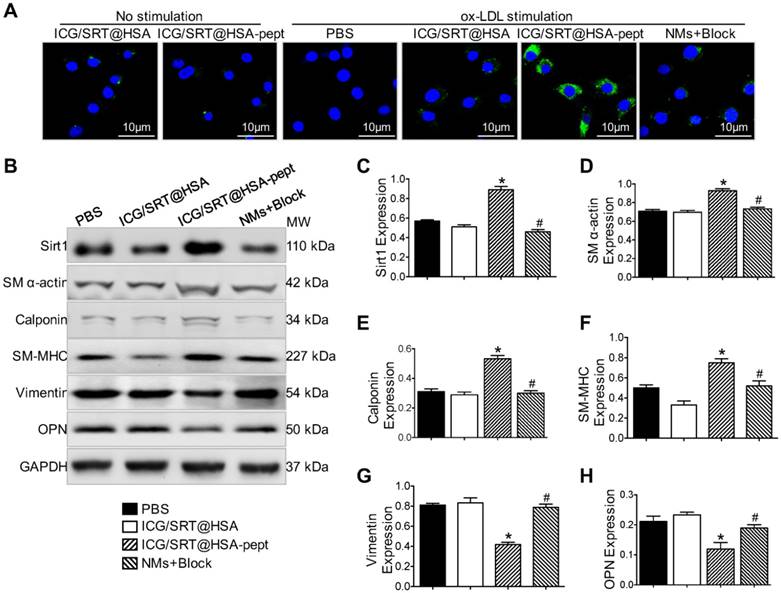
The ox-LDL-stimulated VSMCs, which represent synthetic phenotype VSMCs, displayed a strong FITC signal representing ICG/SRT@HSA-pept NMs in the cytoplasm. Interestingly, the FITC signal was markedly reduced with the addition of the OPN antibody in activated VSMCs. Contrarily, a weak signal was detected in VSMCs that were not treated with ox-LD and in the ICG/SRT@HSA NMs group. Upon 24 h of incubation, ICG/SRT@HSA-pept NMs significantly elevated Sirt1 expression and inhibited VSMC phenotype switching markers in ox-LDL treated VSMCs compared with the PBS and ICG/SRT@HSA NMs groups (Figure 4B-H), as demonstrated by the increased expression levels of contractile markers, including SM α-actin, calponin and SM-MHC, and the decreased expression levels of synthetic markers, including vimentin and OPN (P < 0.05 ICG/SRT@HSA-pept group vs. PBS group). Of importance, this effect was reduced by the addition of the OPN antibody (P < 0.05, NMs+Block group vs. ICG/SRT@HSA-pept group). Taken together, these results show that the ICG/SRT@HSA-pept NMs demonstrated a strong binding affinity and inhibitory effects for synthetic phenotype VSMCs.
Atherosclerotic plaque-targeting ability of ICG/SRT@HSA-pept NMs in mice
The histological results were observed in mice either one week or one month after intravenous administration of treatments. No abnormal histopathological gross lesions were observed in the treated mice liver, lung, testis, heart, brain, spleen or kidney, demonstrating that the repeated administration of ICG/SRT@HSA-pept NMs did not cause any apparent toxicity to these vital organs and indicating that the synthesized ICG/SRT@HSA-pept NMs possessed a safe profile for in vivo application (Figure S3B). Furthermore, the bio-distribution of ICG/SRT@HSA-pept NMs was evaluated through ex vivo NIR imaging 24 h and 48 h p.i. The ICG images (Figure S4A) showed that our synthesized ICG/SRT@HSA-pept NMs primarily accumulated in the liver at 24 h and 48 h p.i., indicating that the nanoparticles were cleared primarily by the liver.
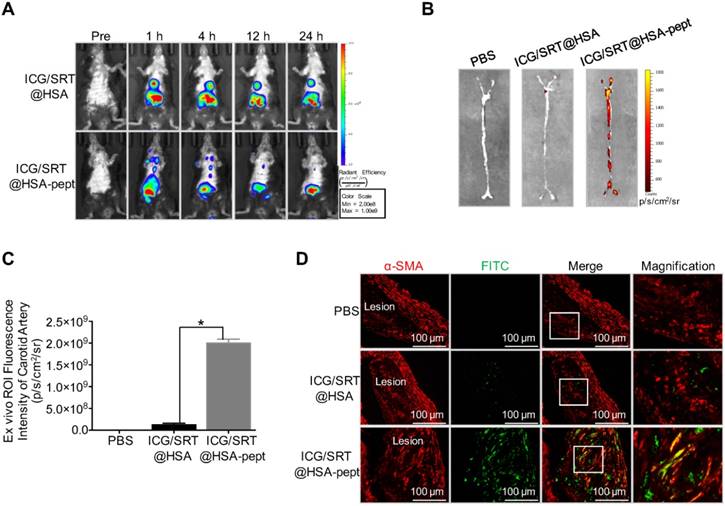
ApoE-/- mice fed with an HFD were used as an animal AS model for in vivo study (n=10). To evaluate the accumulation of ICG/SRT@HSA-pept NMs in vivo, NIR imaging was performed before and 1 h, 4 h, 12 h and 24 h after ICG/SRT@HSA-pept NMs or ICG/SRT@HSA NMs injection. As shown in Figure 5A, compared with the ICG/SRT@HSA NMs group, distinct fluorescence signals in the atherosclerotic lesions were observed in ICG/SRT@HSA-pept NMs-injected mice, with the strongest signals appearing at the time points of 1 h and 4 h. An ex vivo fluorescence imaging study was performed 24 h p.i. The ICG fluorescence signal of ICG/SRT@HSA-pept NMs in the isolated carotid artery was significantly increased compared with that of ICG/SRT@HSA NMs or PBS (P < 0.05 ICG/SRT@HSA-pept group vs. ICG/SRT@HSA group, Figure 5B-C). To observe the detailed distribution of ICG/SRT@HSA-pept NMs in the atherosclerotic lesions, aortas containing plaques were assessed with immunofluorescence staining (n=6). As expected, no significant green signals were detected from the aortas of PBS and ICG/SRT@HSA NMs-treated mice. However, strong signals were detected in ICG/SRT@HSA NMs-injected atherosclerotic mice, and the co-localization of the VSMCs marker α-SMA and ICG/SRT@HSA-pept NMs confirmed the accumulation of the targeting NMs in the AS lesions (Figure 5D). Overall, these results implied that the ICG/SRT@HSA-pept NMs could effectively accumulate within atherosclerotic lesions after intravenous injection.
Atherosclerotic plaque-targeting ability of ICG/SRT@HSA-pept NMs in mice. (A) Compared with the ICG/SRT@HSA NMs group, a distinct fluorescence signal in the atherosclerotic lesion was observed in ICG/SRT@HSA-pept NMs-injected mice, with the strongest signal appearing at the time points of 1 h and 4 h after injection. (B-C) The ICG fluorescence signal of ICG/SRT@HSA-pept NMs in an isolated carotid artery was significantly higher than that of ICG/SRT@HSA NMs or PBS (*P < 0.05 ICG/SRT@HSA-pept group vs. ICG/SRT@HSA group). (D) The colocalization of the VSMC marker α-SMA and ICG/SRT@HSA-pept NMs confirmed the accumulation of the targeting nanomedicines in the AS lesions.
ICG/SRT@HSA-pept NMs inhibit plaque progression in atherosclerotic mice
To test the therapeutic efficacy, we studied the effects of ICG/SRT@HSA-pept NMs in HFD-induced atherosclerotic mice and compared the effects with those of free SRT1720 administration (n=15). First, we examined the effect of SRT1720 on Sirt1 activation within atherosclerotic lesions. As revealed by Western blot results, ICG/SRT@HSA-pept NMs markedly increased the expression of Sirt1 in the plaque area (P < 0.05 NMs group vs. PBS group, Figure 6A). Notably, compared with free SRT1720 administration, ICG/SRT@HSA-pept NMs treatment resulted in a greater increase in Sirt1 expression (P < 0.05 NMs group vs. free SRT group), indicating the in vivo targeted delivery of the loaded drug SRT1720 to atherosclerotic lesions.
ICG/SRT@HSA-pept NMs inhibit plaque progression in atherosclerotic mice. (A) ICG/SRT@HSA-pept NMs markedly increased the expression of Sirt1 in the plaque area (P < 0.05). (B-C) The en face Oil Red O staining of treated mice aortas revealed decreased plaque areas in ICG/SRT@HSA-pept NMs-treated mice compared with AS mice and free SRT1720-treated mice (P < 0.05). (D-F) ICG/SRT@HSA-pept NMs-treated mice displayed the most favorable plaque phenotypes, in which the lesions had significantly smaller plaque sizes (P < 0.05) with higher collagen contents (P < 0.05). Notably, ICG/SRT@HSA-pept NMs treatment induced more favorable therapeutic effects than free SRT1720 treatment. *P < 0.05 NMs group vs. PBS group; #P < 0.05, NMs group vs. free SRT group.
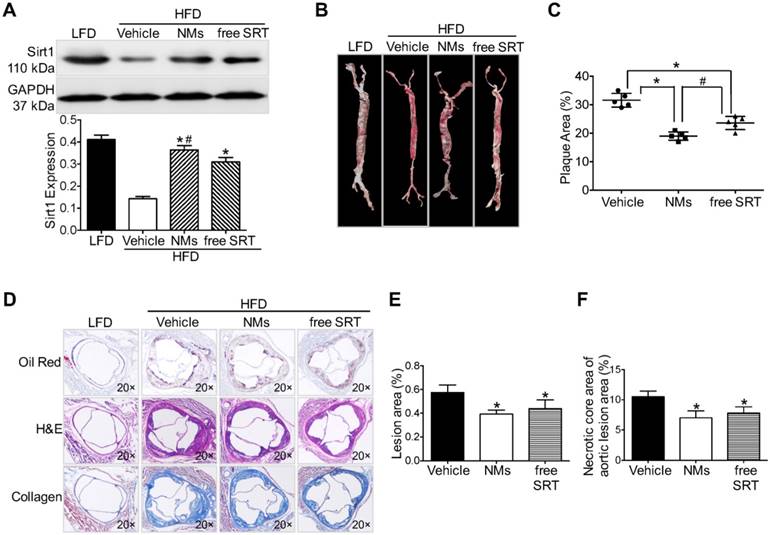
The en face Oil Red O (ORO) staining of treated mice aortas (Figure 6B) and the quantitative analysis of the ORO-positive area of aortas (Figure 6C) revealed a decrease in the plaque area in ICG/SRT@HSA-pept NMs-treated mice compared with untreated AS mice and free SRT1720-treated mice (P < 0.05, PBS group: 31.60±2.408; NMs group: 19.00±1.458; free SRT group: 23.60±2.302 group). Histological cross-sections were stained with H&E, Masson, and ORO to quantitatively assess the therapeutic efficacy of ICG/SRT@HSA-pept NMs on plaque area and plaque composition (collagen). As shown in Figures 6D-F, ICG/SRT@HSA-pept NMs-treated mice displayed the most favorable plaque phenotypes, in which the lesions were significantly smaller (decreased plaque size observed in Figure 6E, P < 0.05 NMs groups vs. PBS group) with a higher collagen content, as observed in Figure 6F (P < 0.05 NMs groups vs. PBS group). Notably, ICG/SRT@HSA-pept NMs treatment induced more favorable therapeutic effects than free SRT1720 treatment, as demonstrated by the slightly smaller plaque size and better composition (necrotic core area and collagen content) of ICG/SRT@HSA-pept NMs-treated mice when compared with the free SRT1720 treated group, although the difference was not significant. Our findings suggested that the ICG/SRT@HSA-pept NMs were capable of the enhanced delivery of Sirt1 activator SRT1720 to atherosclerotic lesions with a better therapeutic efficacy when compared to free SRT1720 treatment.
Discussion
In the present study, we formulated theranostic ICG/SRT@HSA-pept NMs for the in vivo targeting and treatment of atherosclerotic plaques. Due to the overexpression of OPN in activated VSMCs and VASPs, the ICG/SRT@HSA-pept NMs specifically accumulated within the VASPs after intravenous injection, achieving the precise detection of VASPs. Furthermore, with systemic therapy, the ICG/SRT@HSA-pept NMs presented antiatherogenesis effects by inhibiting phenotypic switching of VSMCs through the function of the loaded SRT1720. Collectively, the targeting ICG/SRT@HSA-pept NMs functioned as efficient theranostic NMs for atherosclerotic plaques.
Over the last two decades, the increased insight into the role of VSMCs in the pathophysiology of atherosclerosis has shown promising potential for the development of pharmaceutical interventions for atherosclerosis treatment. The historical view that VSMCs have athero-protective plaque-stabilizing properties and are “entirely beneficial” has been challenged [20]. In normal arteries, VSMCs express a range of contractile protein markers, including SM α-actin, calponin, SM-MHC, and smoothelin. However, within atherosclerotic lesions, VSMCs reduce the expression levels of these conventional VSMCs markers and acquire the expression of synthetic proteins, including vimentin and OPN. This “phenotypic switching” of VSMCs has been considered to be an important contributor to the vulnerability and progression of atherosclerotic plaques [21, 22]. Recently, several studies demonstrated that the phenotypic modulation of VSMCs played an essential role in atherosclerotic plaque pathogenesis [14, 23]. For instance, in a recent study by Shankman et al., they demonstrated that the contribution of VSMCs to atherosclerotic plaques had been greatly underestimated and that the phenotypic switching of VSMCs was critical to lesion pathogenesis [23]. Similarly, Ding Y et al. reported that AMPK-alpha 2 deletion induced phenotypic switching in VSMCs and promoted features of atherosclerotic plaque instability [14]. These key breakthroughs motivated our present attempt to regulate phenotypic switching in VSMCs as a therapeutic intervention for atherosclerosis. In our current study, we found that both free SRT1720 and ICG/SRT@HSA-pept NMs could reverse the ox-LDL-induced phenotypic switching in VSMCs in vitro and could inhibit the atherosclerotic plaque progression in vivo, supporting the idea that modulating the VSMCs phenotype could be a therapeutic approach for atherosclerosis.
The therapeutic efficacy of NMs is primarily determined by the loaded medications. Sirt1, a nicotinamide adenine dinucleotide (NAD+)-dependent deacetylase, has been proposed to exert a beneficial role in aging and age-associated diseases [24, 25]. Previous studies have shown that Sirt1 expression was reduced in atherosclerotic plaques, and pharmacological interventions to activate Sirt1 could result in multiple benefits during atherosclerosis [26, 27]. It was also demonstrated that Sirt1 activators provided athero-protection in ApoE-/- mice through the regulation of lipid metabolism, endothelial function and inflammatory responses within atherosclerotic lesions [28-30]. However, the role of Sirt1 in the regulation of phenotypic switching in VSMCs during atherosclerosis is only partially known. Here, we aimed to investigate the phenotypic changes of VSMCs upon Sirt1 activation. Although resveratrol has been regarded to be a conventional Sirt1 activator, recent studies revealed that it might have multiple additional targets, such as COX, PDEs, PI3K, ERα/β or p70S6K, which were not dependent on Sirt1 [31]. Unlike resveratrol, chemical activators, such as SRT1720, appear to be a better choice for Sirt1 activation. SRT1720 (C25H23N7OS.HCl) is a specific Sirt1 activator, which can increase the expression and activity of Sirt1 without affecting other sirtuin proteins. The beneficial effects of SRT1720, through Sirt1 activation, in cardiovascular and metabolic diseases have been addressed in multiple studies [24, 32, 33]. Furthermore, the small molecular weight of SRT1720 made it an ideal loaded drug for NMs. As was shown in the Results section, we demonstrated that the Sirt1 activator SRT1720 robustly increased Sirt1 expression and inhibited the phenotypic transition of VSMCs in vitro. This conclusion was drawn based on evidence from both MOVAS cell lines and primary VSMCs. In the next step, we examined the effect of SRT1720 in atherosclerosis ApoE-/- mice and found that the systemic treatment of SRT1720 decreased plaque sizes and improved plaque composition with decreased necrotic core areas and increased collagen content. These results demonstrate that the Sirt1 activator SRT1720 is capable of regulating VSMC plasticity and inhibiting the ox-LDL-induced phenotypic switch, both in vitro and in vivo, making SRT1720 a promising pharmaceutical agent for antiatherogenesis treatment. In accordance with other studies, our present study provides a better understanding of the modulation of Sirt1 for the treatment of atherosclerotic plaque development [29, 34, 35].
NMs have shown inspiring outcomes in clinical studies [36, 37]. Targeted NPs that are designed to accumulate in desired tissues are considered to be promising NP-based therapies with improved pharmacokinetic profiles and drug efficiencies [38, 39]. NPs modified with specific antibodies or peptides to target atherosclerotic lesions have been reported to present more efficient pharmaceutical benefits with reduced toxicities. OPN is a secretory protein, which is highly expressed in atherosclerotic lesions rather than normal vessels. In addition, OPN is associated with enhanced inflammatory responses, contributing to the vulnerability of atherosclerotic plaques. These properties make OPN a promising biomarker for VASPs [40-42]. Specifically, the elevated expression of OPN is a hallmark for VSMC phenotypic switching, making OPN an effective molecular target for synthetic VSMCs within atherosclerotic lesions. Our group previously developed an OPN-antibody modified Fe3O4 NP for the imaging of vulnerable atherosclerotic plaques and observed the accumulation of the NP in atherosclerotic lesions [6]. As therapeutic delivery systems, NPs should have specific physicochemical properties, including a smaller size, increased stability and improved bio-distribution [43]. To fulfill this goal, OPN-targeting peptide-modified NMs, based on HSA, were developed in this study. As expected, our HSA-based ICG/SRT@HSA-pept NMs presented favorable stability, biocompatibility and bio-distribution, which was in accordance with previous studies that reported that HSA-based NPs have improved biological characteristics [44, 45]. Furthermore, the successful delivery of NMs also requires an effective targeting strategy. For this purpose, the surface of ICG/SRT@HSA-pept NMs were conjugated with a predesigned peptide, which enabled the targeted ICG/SRT@HSA-pept NMs to bind to the overexpressed OPN in synthetic VSMCs and atherosclerotic lesions. As was revealed in the in vitro imaging results, the ICG/SRT@HSA-pept NMs presented selectivity toward synthetic VSMCs stimulated by ox-LDL, which was blocked by the addition of an OPN antibody. After intravenous injections into HFD ApoE-/- mice, ICG/SRT@HSA-pept NMs showed obvious accumulations in atherosclerotic lesions. Interestingly, the immunofluorescence results further confirmed the colocalization of ICG/SRT@HSA-pept NMs with VSMCs within atherosclerotic plaques. Interestingly, several studies have selected other molecular targets for atherosclerotic plaques, including vascular cell adhesion protein-1 (VCAM-1), αvβ3, intercellular adhesion molecule-1 (ICAM-1) and other AS-associated biomarkers [46, 47]. In our present study, the ICG/SRT@HSA-pept NMs exhibited atherosclerotic plaque-targeting ability, which is an essential characteristic for targeted NMs.
In the next step, we applied the ICG/SRT@HSA-pept NMs in HFD ApoE-/- mice for therapeutic purposes. As shown by the histological results, ICG/SRT@HSA-pept NMs-treated mice displayed the most favorable plaque phenotypes, with decreased plaque sizes and enhanced stability (decreased necrotic core areas and higher collagen contents). Interestingly, the SRT1720-encapsulated ICG/SRT@HSA-pept NMs presented significantly increased therapeutic efficacy than free SRT1729 treatment, with enhanced Sirt1 expression and improved plaque composition. In this context, the ICG/SRT@HSA-pept NMs- mediated delivery of SRT1720 can facilitate the targeted delivery and improve the therapeutic efficacy of SRT1720. Conventional pharmaceutical agents are primarily delivered systemically, suffering from the rapid clearance from plasma and the lack of accumulation in targeted tissues, which could limit the therapeutic efficacy of drugs in chronic conditions such as AS [48]. In contrast, NP-based nanotherapies could facilitate the delivery of drugs to the target lesions, decrease off-target drug toxicity and improve antiatherogenic efficacy. For instance, Mikyung Yu and Xueqing Zhang et al. recently reported a new form of nanoparticles capable of the enhanced delivery of liver X receptor (LXR) agonists to atherosclerotic lesions, without altering hepatic metabolism, which is the undesirable adverse side effect of LXR activation [39, 49]. Concurrently, several formulated NPs from other research teams have also shown favorable targeting properties and antiatherogenic effects in animal models [19, 50-52]. Together with these inspiring findings, our novel ICG/SRT@HSA-pept NMs showed great therapeutic efficacy for atherosclerotic plaques, shedding light on the potential for nanomedicines in atherosclerosis.
However, there are several limitations to the current study. First, in the therapeutic study, we demonstrated the anti-atherogenic effects of ICG/SRT@HSA-pept NMs and free SRT1720 on decreasing plaque sizes and improving plaque composition based on the evidence from H&E, Masson, and ORO staining. Due to the limited number of mice, the macrophage content and VSMC content of plaques were not analyzed. Therefore, the therapeutic efficacy of ICG/SRT@HSA-pept NMs and free SRT1720 on atherosclerotic plaque vulnerability was not evaluated. Second, the current conclusions were based on data in mice, and more data from rabbit models or large animal models would be more convincing.
Conclusion
In the current study, we developed highly biocompatible HSA-based NMs as imaging and therapy vectors for atherosclerosis. In our nanoformulation, OPN, which is highly expressed in activated VSMCs and atherosclerotic lesions, serves as the targeting molecule for VASPs. ICG/SRT@HSA-pept NMs were preferentially uptaken by VSMCs undergoing phenotypic switching in vitro and displayed high selectivity toward atherosclerotic lesions in vivo. Furthermore, the SRT1720-encapsulated ICG/SRT@HSA-pept NMs showed greater therapeutic efficacy for the inhibition of atherosclerotic plaque progression than systemic SRT1720 treatment. Collectively, the novel theranostic NMs show great potential for the precise identification and targeted treatment of atherosclerosis, which is an encouraging step toward the clinical application of nanotherapeutics for cardiovascular diseases.
Abbreviations
AS: atherosclerosis; CT: computed tomography; DLS: dynamic light scattering; FBS: fetal bovine serum; FITC: fluorescein isothiocyanate; HFD: high-fat diet; HSA: human serum albumin; ICG: indocyanine green; ICG/SRT@HSA NMs: ICG/SRT@HSA nanomedicines; ICG/SRT@HSA-pept NMs: ICG/SRT@HSA-peptide nanomedicines; LFD: low-fat diet; MRI: magnetic resonance imaging; NAD+: nicotinamide adenine dinucleotide; NIR: near infrared; NMs: nanomedicines; NPs: nanoparticles; OCT: optical coherence tomography; OPN: osteopontin; Sirt1: sirtuin 1; TEM: transmission electron microscopy; US: ultrasound; VASPs: vulnerable atherosclerotic plaques; VSMCs: vascular smooth muscle cells.
Acknowledgements
This work was supported by the National Key Research Program of China (No. 2016YFA0100900, 2016YFC1300301), the National Fund for Distinguished Young Scientists of China (No. 81325009), the National Nature Science Foundation of China (No 81530058, and 81570272), and the Beijing Natural Science Foundation (No. 7152131).
Supplementary Material
Supplementary figures and tables.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Anker S, Asselbergs FW, Brobert G, Vardas P, Grobbee DE, Cronin M. Big data in cardiovascular disease. Eur Heart J. 2017;38:1863-5
2. Zahradka P. Inhibition of nadph oxidase by vaspin may prevent progression of atherosclerosis. Acta Physiol (Oxf). 2013;209:195-8
3. Polonsky TS, Ning H, Daviglus ML, Liu K, Burke GL, Cushman M. et al. Association of cardiovascular health with subclinical disease and incident events: The multi-ethnic study of atherosclerosis. J Am Heart Assoc. 2017;6(3):e004894
4. Kong MH, Fonarow GC, Peterson ED, Curtis AB, Hernandez AF, Sanders GD. et al. Systematic review of the incidence of sudden cardiac death in the united states. J Am Coll Cardiol. 2011;57:794-801
5. Stefanadis C, Antoniou CK, Tsiachris D, Pietri P. Coronary atherosclerotic vulnerable plaque: Current perspectives. J Am Heart Assoc. 2017;6(3):e005543
6. Qiao H, Wang Y, Zhang R, Gao Q, Liang X, Gao L. et al. Mri/optical dual-modality imaging of vulnerable atherosclerotic plaque with an osteopontin-targeted probe based on fe3o4 nanoparticles. Biomaterials. 2017;112:336-45
7. Rafailidis V, Chryssogonidis I, Tegos T, Kouskouras K, Charitanti-Kouridou A. Imaging of the ulcerated carotid atherosclerotic plaque: A review of the literature. Insights Imaging. 2017;8:213-25
8. Anwaier G, Chen C, Cao Y, Qi R. A review of molecular imaging of atherosclerosis and the potential application of dendrimer in imaging of plaque. Int J Nanomedicine. 2017;12:7681-93
9. MacRitchie N, Grassia G, Noonan J, Garside P, Graham D, Maffia P. Molecular imaging of atherosclerosis: Spotlight on raman spectroscopy and surface-enhanced raman scattering. Heart. 2018;104(6):460-467
10. Vaidyanathan K, Gopalakrishnan S. Nanomedicine in the diagnosis and treatment of atherosclerosis - a systematic review. Cardiovasc Hematol Disord Drug Targets. 2017;17(2):119-131
11. Su T, Wang YB, Han D, Wang J, Qi S, Gao L. et al. Multimodality imaging of angiogenesis in a rabbit atherosclerotic model by gebp11 peptide targeted nanoparticles. Theranostics. 2017;7:4791-804
12. Lindsay AC, Choudhury RP. Form to function: Current and future roles for atherosclerosis imaging in drug development. Nat Rev Drug Discov. 2008;7:517-29
13. Lobatto ME, Fuster V, Fayad ZA, Mulder WJ. Perspectives and opportunities for nanomedicine in the management of atherosclerosis. Nat Rev Drug Discov. 2011;10:835-52
14. Ding Y, Zhang M, Zhang W, Lu Q, Cai Z, Song P. et al. Amp-activated protein kinase alpha 2 deletion induces vsmc phenotypic switching and reduces features of atherosclerotic plaque stability. Circ Res. 2016;119:718-30
15. Zhang MJ, Zhou Y, Chen L, Wang YQ, Wang X, Pi Y. et al. An overview of potential molecular mechanisms involved in vsmc phenotypic modulation. Histochem Cell Biol. 2016;145:119-30
16. Feige JN, Lagouge M, Canto C, Strehle A, Houten SM, Milne JC. et al. Specific sirt1 activation mimics low energy levels and protects against diet-induced metabolic disorders by enhancing fat oxidation. Cell Metab. 2008;8:347-58
17. Yao H, Chung S, Hwang JW, Rajendrasozhan S, Sundar IK, Dean DA. et al. Sirt1 protects against emphysema via foxo3-mediated reduction of premature senescence in mice. J Clin Invest. 2012;122:2032-45
18. Kulkarni SR, Soroka CJ, Hagey LR, Boyer JL. Sirtuin 1 activation alleviates cholestatic liver injury in a cholic acid-fed mouse model of cholestasis. Hepatology. 2016;64:2151-64
19. Beldman TJ, Senders ML, Alaarg A, Perez-Medina C, Tang J, Zhao Y. et al. Hyaluronan nanoparticles selectively target plaque-associated macrophages and improve plaque stability in atherosclerosis. ACS Nano. 2017;11:5785-99
20. Grootaert MOJ, Moulis M, Roth L, Martinet W, Vindis C, Bennett MR. et al. Vascular smooth muscle cell death, autophagy and senescence in atherosclerosis. Cardiovasc Res. 2018;114(4):622
21. Bennett MR, Sinha S, Owens GK. Vascular smooth muscle cells in atherosclerosis. Circ Res. 2016;118:692-702
22. Alexander MR, Owens GK. Epigenetic control of smooth muscle cell differentiation and phenotypic switching in vascular development and disease. Annu Rev Physiol. 2012;74:13-40
23. Shankman LS, Gomez D, Cherepanova OA, Salmon M, Alencar GF, Haskins RM. et al. Klf4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat Med. 2015;21:628-37
24. Mitchell SJ, Martin-Montalvo A, Mercken EM, Palacios HH, Ward TM, Abulwerdi G. et al. The sirt1 activator srt1720 extends lifespan and improves health of mice fed a standard diet. Cell Rep. 2014;6:836-43
25. Watroba M, Dudek I, Skoda M, Stangret A, Rzodkiewicz P, Szukiewicz D. Sirtuins, epigenetics and longevity. Ageing Res Rev. 2017;40:11-9
26. Brandes RP. Activating sirt1: A new strategy to prevent atherosclerosis? Cardiovasc Res. 2008;80:163-4
27. Yu W, Fu YC, Chen CJ, Wang X, Wang W. Sirt1: A novel target to prevent atherosclerosis. J Cell Biochem. 2009;108:10-3
28. Gorenne I, Kumar S, Gray K, Figg N, Yu H, Mercer J. et al. Vascular smooth muscle cell sirtuin 1 protects against DNA damage and inhibits atherosclerosis. Circulation. 2013;127:386-96
29. Miranda MX, van Tits LJ, Lohmann C, Arsiwala T, Winnik S, Tailleux A. et al. The sirt1 activator srt3025 provides atheroprotection in apoe-/- mice by reducing hepatic pcsk9 secretion and enhancing ldlr expression. Eur Heart J. 2015;36:51-9
30. Chen ML, Yi L, Jin X, Liang XY, Zhou Y, Zhang T. et al. Resveratrol attenuates vascular endothelial inflammation by inducing autophagy through the camp signaling pathway. Autophagy. 2013;9:2033-45
31. Kulkarni SS, Canto C. The molecular targets of resveratrol. Biochim Biophys Acta. 2015;1852:1114-23
32. Chen YX, Zhang M, Cai Y, Zhao Q, Dai W. The sirt1 activator srt1720 attenuates angiotensin ii-induced atherosclerosis in apoe(-)/(-) mice through inhibiting vascular inflammatory response. Biochem Biophys Res Commun. 2015;465:732-8
33. Gano LB, Donato AJ, Pasha HM, Hearon CM Jr, Sindler AL, Seals DR. The sirt1 activator srt1720 reverses vascular endothelial dysfunction, excessive superoxide production, and inflammation with aging in mice. Am J Physiol Heart Circ Physiol. 2014;307:H1754-63
34. D'Onofrio N, Servillo L, Balestrieri ML. Sirt1 and sirt6 signaling pathways in cardiovascular disease protection. Antioxid Redox Signal. 2017:10 28 (8): 711-732
35. Vikram A, Kim YR, Kumar S, Li Q, Kassan M, Jacobs JS. et al. Vascular microrna-204 is remotely governed by the microbiome and impairs endothelium-dependent vasorelaxation by downregulating sirtuin1. Nat Commun. 2016;7:12565
36. Davis ME, Chen ZG, Shin DM. Nanoparticle therapeutics: An emerging treatment modality for cancer. Nat Rev Drug Discov. 2008;7:771-82
37. Sheridan C. Proof of concept for next-generation nanoparticle drugs in humans. Nat Biotechnol. 2012;30:471-3
38. Bourzac K. Nanotechnology: Carrying drugs. Nature. 2012;491:S58-60
39. Yu M, Amengual J, Menon A, Kamaly N, Zhou F, Xu X. et al. Targeted nanotherapeutics encapsulating liver x receptor agonist gw3965 enhance antiatherogenic effects without adverse effects on hepatic lipid metabolism in ldlr-/- mice. Adv Healthc Mater. 2017;6(20):1700313
40. O'Brien ER, Garvin MR, Stewart DK, Hinohara T, Simpson JB, Schwartz SM. et al. Osteopontin is synthesized by macrophage, smooth muscle, and endothelial cells in primary and restenotic human coronary atherosclerotic plaques. Arterioscler Thromb. 1994;14:1648-56
41. Cho HJ, Cho HJ, Kim HS. Osteopontin: A multifunctional protein at the crossroads of inflammation, atherosclerosis, and vascular calcification. Curr Atheroscler Rep. 2009;11:206-13
42. Bruemmer D, Collins AR, Noh G, Wang W, Territo M, Arias-Magallona S. et al. Angiotensin ii-accelerated atherosclerosis and aneurysm formation is attenuated in osteopontin-deficient mice. J Clin Invest. 2003;112:1318-31
43. Zhang L, Gu FX, Chan JM, Wang AZ, Langer RS, Farokhzad OC. Nanoparticles in medicine: Therapeutic applications and developments. Clin Pharmacol Ther. 2008;83:761-9
44. Wosikowski K, Biedermann E, Rattel B, Breiter N, Jank P, Loser R. et al. In vitro and in vivo antitumor activity of methotrexate conjugated to human serum albumin in human cancer cells. Clin Cancer Res. 2003;9:1917-26
45. Chen Q, Liang C, Wang C, Liu Z. An imagable and photothermal "abraxane-like" nanodrug for combination cancer therapy to treat subcutaneous and metastatic breast tumors. Adv Mater. 2015;27:903-10
46. Qiao R, Qiao H, Zhang Y, Wang Y, Chi C, Tian J. et al. Molecular imaging of vulnerable atherosclerotic plaques in vivo with osteopontin-specific upconversion nanoprobes. ACS Nano. 2017;11:1816-25
47. Bruckman MA, Jiang K, Simpson EJ, Randolph LN, Luyt LG, Yu X. et al. Dual-modal magnetic resonance and fluorescence imaging of atherosclerotic plaques in vivo using vcam-1 targeted tobacco mosaic virus. Nano Letters. 2014;14:1551-8
48. Kamaly N, Fredman G, Fojas JJ, Subramanian M, Choi WI, Zepeda K. et al. Targeted interleukin-10 nanotherapeutics developed with a microfluidic chip enhance resolution of inflammation in advanced atherosclerosis. ACS Nano. 2016;10:5280-92
49. Zhang XQ, Even-Or O, Xu X, van Rosmalen M, Lim L, Gadde S. et al. Nanoparticles containing a liver x receptor agonist inhibit inflammation and atherosclerosis. Adv Healthc Mater. 2015;4:228-36
50. Wei X, Ying M, Dehaini D, Su Y, Kroll AV, Zhou J. et al. Nanoparticle functionalization with platelet membrane enables multifactored biological targeting and detection of atherosclerosis. ACS Nano. 2018;12:109-16
51. Gao W, Sun Y, Cai M, Zhao Y, Cao W, Liu Z. et al. Copper sulfide nanoparticles as a photothermal switch for trpv1 signaling to attenuate atherosclerosis. Nat Commun. 2018;9:231
52. Winter PM, Caruthers SD, Zhang H, Williams TA, Wickline SA, Lanza GM. Antiangiogenic synergism of integrin-targeted fumagillin nanoparticles and atorvastatin in atherosclerosis. JACC Cardiovasc Imaging. 2008;1:624-34
Author contact
![]() Corresponding authors: Feng Cao: wind8828com; Weisheng Guo: tjuguoweishengcom; Xing-Jie Liang: liangxjcn
Corresponding authors: Feng Cao: wind8828com; Weisheng Guo: tjuguoweishengcom; Xing-Jie Liang: liangxjcn